
Recent progress and perspectives in photo-induced organic reactions of acylsilanes
Wan Pyo
Hong
 a,
Hee Nam
Lim
*b and
Inji
Shin
*c
a,
Hee Nam
Lim
*b and
Inji
Shin
*c
aDepartment of Chemistry, Gachon University, 1342, Seongnam-daero, Sujeong-gu, Seongnam-si, Gyeonggi-do, 13120, Korea
bDepartment of Chemistry, Yeungnam University, 280 Daehak-Ro, Gyeongsan, Gyeongbuk 38541, Korea
cDepartment of Fine Chemistry, Seoul National University of Science and Technology, 232 Gongneung-ro, Nowon-gu, Seoul, 01811, Korea. E-mail: inji@seoultech.ac.kr
First published on 9th January 2023
Abstract
Acylsilanes have been utilized in various transformations for the synthesis of functionalized organosilicon derivatives due to their importance in medicinal and materials science. This review summarizes recent photo-induced reactions of acylsilanes, mostly focused on nucleophilic siloxycarbene intermediates and categorizes them by reaction types, nucleophilic carbene addition, insertion reactions, and cyclization. Several photo-induced reactions using acylsilanes beyond the reactivity of siloxycarbenes have also been covered in the miscellaneous part. Although significant efforts have already been devoted to the use of acylsilanes, further synthetic applications of acylsilanes are likely to follow in the near future to prepare valuable building blocks for various fields such as medicinal and materials chemistry.
 Hee Nam Lim (left), Inji Shin (center), Wan Pyo Hong (right) | Hee Nam Lim (left) received his Ph.D. degree in chemistry (2013) from SUNY Stony Brook University (USA) under the supervision of Prof. Kathlyn A. Parker. He was a postdoctoral scholar in Prof. Guangbin Dong's group at the University of Chicago (USA). He is now an assistant professor at Yeungnam University (Korea). Currently, he is working on fluorination strategies for agrochemical synthesis. Inji Shin (center) received her Ph.D. degree in chemistry (2013) from the University of Pennsylvania (USA) under the supervision of Prof. Gary Molander. As a postdoctoral scholar, she worked in the research group of Prof. Michael Krische at UT Austin (USA). She is now an assistant professor at Seoul National University of Science and Technology (Korea). She is currently interested in developing new methods for C–Si bond activations. Wan Pyo Hong (right) received his Ph.D. degree in chemistry (2011) from Purdue University (USA) under the supervision of Prof. Philip L. Fuchs. He was a postdoctoral scholar in Prof. Shannon Stahl's group at the University of Wisconsin–Madison (USA). He worked as a principal researcher in LG Chem Research Park. He is now an assistant professor at Gachon University (Korea) and currently engaged in the discovery of optoelectronic materials. |
1. Introduction
Organosilicon compounds are important synthetic intermediates and building blocks in various fields of chemistry, including organic, medicinal, and materials chemistry.1 Due to their unique physical and chemical properties, research involving organosilicon compounds has become extensive, particularly for exploring novel chemical spaces in various fields. The silicon atom is tetravalent like a carbon atom and the carbon–silicon (C–Si) bond (1.86 Å) is longer than the carbon–carbon (C–C) bond (1.54 Å). The C–Si bond has a lower bond dissociation energy (451 kJ mol−1) than the C–C bond (607 kJ mol−1). Therefore, the C–Si bond is weaker than the C–C bond and slightly polarized.Compounds containing silicon–carbon or silicon–heteroatom bonds are ubiquitous in synthetic chemistry. Silicon derivatives are widely used for the protection of hydroxyl functional groups in organic chemistry.2 Due to the sensitivity of hydroxyl groups under various reaction conditions, such as oxidation, substitution, and acid–base reactions, their protection is required to temporarily prevent undesired reactions. The O–Si bond is generally susceptible to strongly acidic conditions or fluoride attack, thus allowing facile deprotection after chemoselective manipulation. Moreover, organosilicon compounds are used as coupling reagents in Hiyama-type reactions.3
Organosilicon analogs have become valuable structures in medicinal chemistry.4 Because of the similarity between silicon and carbon, much effort has been devoted to the synthesis of organosilicon compounds with improved drug-like physicochemical and pharmacokinetic properties.5 More recently, sila-substitution in drugs, carbon and silicon exchange, has been studied for enhancing lipophilicity and permeability, and for improving electronic properties (Fig. 1).6 In addition, carbon–silicon derivatives have been studied in materials science, being applicable in dielectric materials, microelectronic packaging devices, light-emitting diodes (LEDs), transistors, and optical fibers.7 Materials comprising triphenylsilyl units exhibit suppression of intermolecular interactions while facilitating electron transport in organic LEDs (OLEDs). Consequently, OLEDs based on triphenylsilyl-substituted materials exhibit a maximum external quantum efficiency of over 20%, along with a long operational lifetime (Fig. 2).8
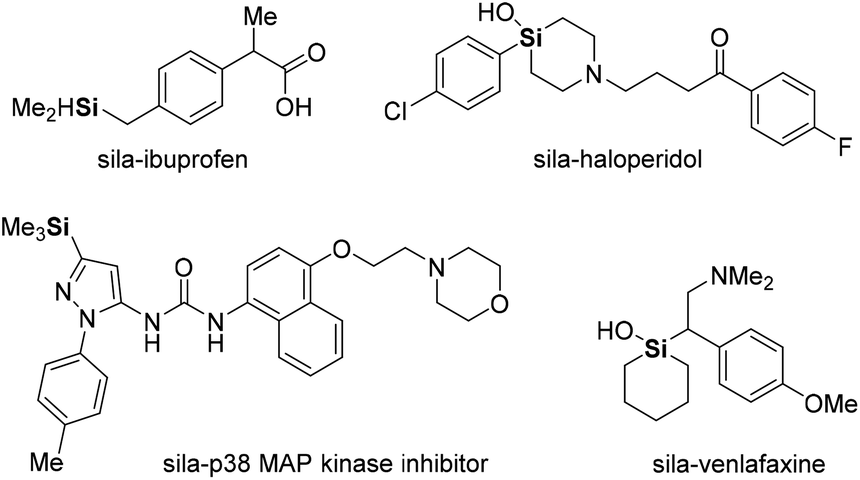 | ||
| Fig. 1 Examples of sila-substitution (C/Si exchange) in drugs. | ||
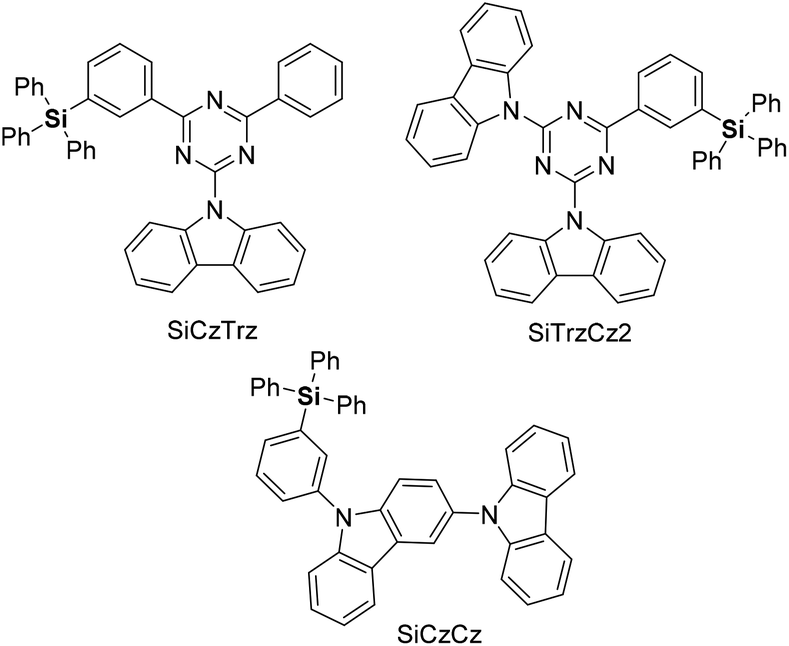 | ||
| Fig. 2 Examples of optoelectronic materials. | ||
Acylsilanes, carbonyl compounds with a silicon atom attached to the carbonyl carbon, exhibit interesting chemical and physical properties. When compared to analogous carbon derivatives, spectroscopic studies have shown that acylsilanes absorb infrared and ultraviolet spectral regions at longer wavelengths9 and the signal for their carbonyl carbon is shifted downfield in 13C NMR spectra.10 These findings suggest that the inductive effects of silicon affect the properties of the acylsilane carbonyl to a greater extent than their carbon counterparts. Moreover, in triphenylsilyl phenyl ketone, an even longer C–Si bond length of 1.93 Å was observed compared to the normal C–Si bond (1.86 Å) based on X-ray crystallographic analysis.11
Acylsilanes undergo a 1,2-shift of the silyl group under thermal or photoirradiation reaction conditions to generate siloxycarbene intermediates.12 Brook et al. first reported this transformation in 1957.13 The widely accepted mechanism for the generation of singlet-state siloxycarbene is presented in Scheme 1. Owing to the small energy gap between the non-bonding and anti-bonding orbitals of carbonyls, the excited singlet state of acylsilane 2S* is generated through relaxation from the vertically excited state 1* under thermal or photoirradiation conditions. The excited triplet state of acylsilane 2T* is formed from 2S*via intersystem crossing (ISC), followed by 1,2-Brook rearrangement, leading to the corresponding triplet state siloxycarbene intermediate 3T. Then, 3T undergoes ISC to generate singlet state siloxycarbene 3S.12a,14
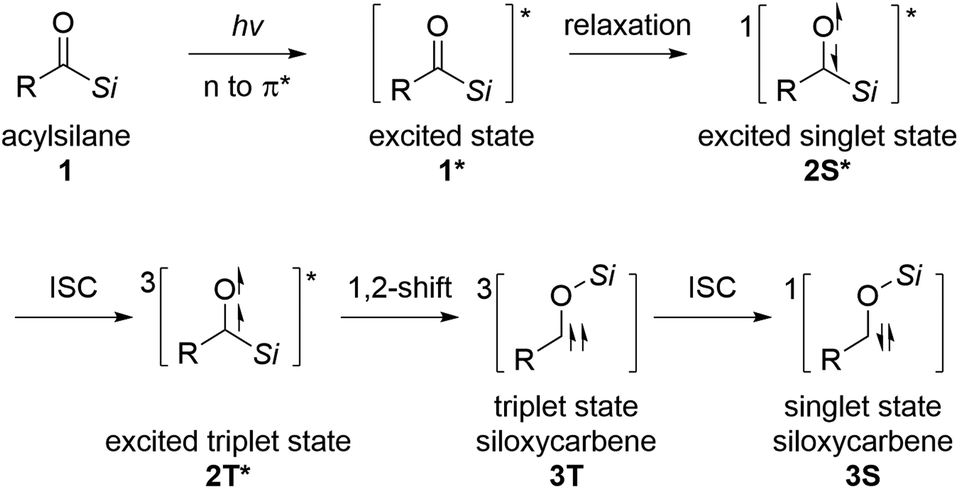 | ||
| Scheme 1 Profiles for the excitation of acylsilane to siloxycarbene. | ||
Carbene is a highly reactive divalent intermediate containing a neutral carbon atom with two unshared valence electrons, and in most cases, it is extremely short-lived. Depending on the electronic structure, carbenes can be either in the singlet or the triplet state.15 Singlet carbenes have two paired electrons (opposite spins), while triplet carbenes have two unpaired electrons (parallel spins). In general, singlet carbenes show both electrophilic and nucleophilic reactivity, and triplet carbenes possess diradical properties. Therefore, various types of organic transformations are possible with either singlet or triplet state carbenes.
Siloxycarbenes are known to exhibit nucleophilic reactivity in various organic reactions (Scheme 2). The nucleophilic addition of siloxycarbenes to various electrophiles, including carbonyl compounds, has been reported by several research groups (Scheme 2a).16 Moreover, the singlet state of the siloxycarbene species is capable of insertion into X–Y bonds, including C–H, B–H, and C–B bonds (Scheme 2b).17 Recently, studies on inter- and intramolecular addition reactions of nucleophilic siloxycarbenes to alkenes and alkynes have also been published (Scheme 2c).18
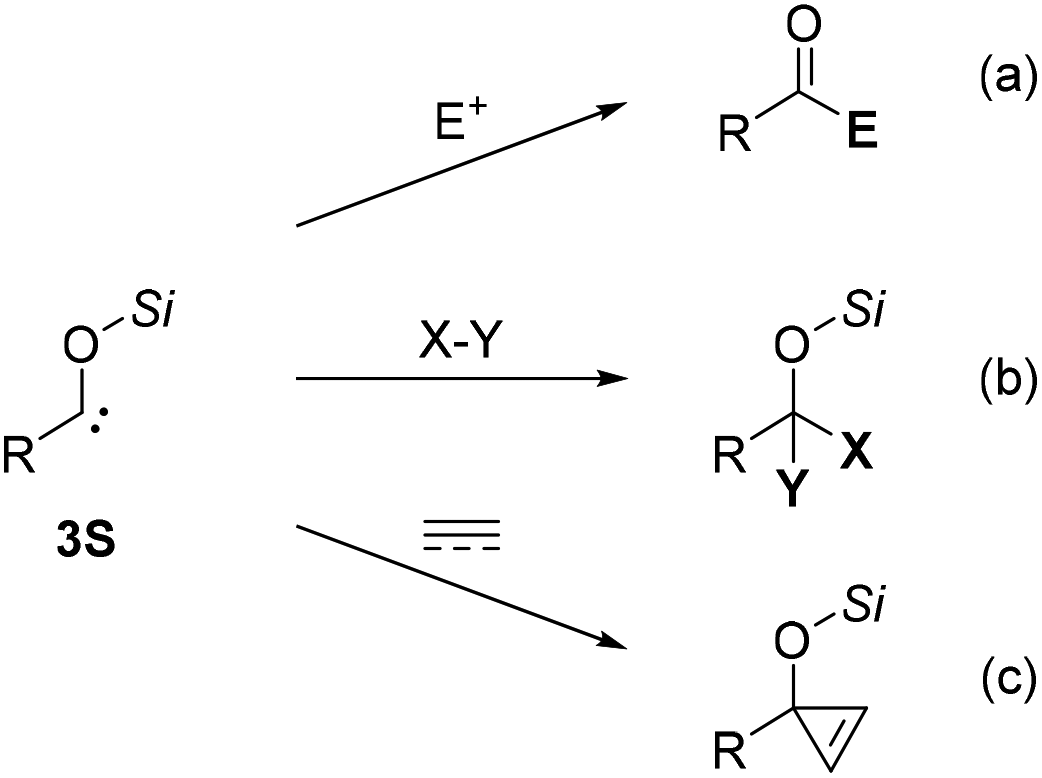 | ||
| Scheme 2 Representative reactions with acylsilanes (a) reaction with electrophiles, (b) X–Y insertion, (c) addition to unsaturated carbon–carbon bonds. | ||
Synthetic applications of acylsilanes in organic chemistry have been of significant interest since their first discovery by Brook,19 and several relevant reviews have been published. Rong et al. reviewed synthetic methods for accessing acylsilanes.20 Bolm's review discussed the preparation and comprehensive applications of acylsilanes in organic reactions developed until 2013.21 Priebbenow's review published in 2020 encompassed singlet carbene chemistry of siloxycarbenes generated from acylsilanes.22 Even though studies of siloxycarbenes from acylsilanes under photoirradiation have been reported for over 50 years, light-induced siloxycarbene chemistry is still of great interest to chemists and numerous impressive studies have been published more recently. Compared to thermal reactions of acylsilanes, the reaction conditions are milder with lower temperature and pressure in many cases.17d,e,18d,19 Moreover, the photo-induced synthetic methods of acylsilanes cost less than other transition-metal catalyzed photochemical reactions because there is no need to use expensive photocatalysts in many cases. By developing new synthetic methods using various acylsilanes, complex molecules such as heterocyclic compounds and acetals which are limited by the existing synthetic pathway can be readily accessed. Herein, we summarize the recent progress in photoirradiation methods involving acylsilanes made during the last decade from the perspectives of reaction mechanism, scope, and reaction systems.
2. Photo-induced reactions with siloxycarbenes as key intermediates
2.1 Nucleophilic carbene addition
In 2018, the synthesis of α-hydroxyketone via a Lewis acid-assisted intermolecular reaction between acylsilanes and aldehydes under photoirradiation conditions was reported (Scheme 3).16c Simple irradiation with a 500 W super-high-pressure Hg lamp did not induce sufficient reactivity in the acyclic siloxycarbene generated from the corresponding acylsilane 1a due to low nucleophilicity and/or short lifetime of the siloxycarbene. Instead, the electrophilicity of aldehyde 4 was enhanced in the presence of the Lewis acid, ZnI2, and as a result, the desired α-hydroxyketones 6 were obtained via intermediate 5 in moderate to excellent yields. Interestingly, the E/Z isomerization of trans-2-hexenal was not detected under photoirradiation conditions, which was unavoidable under thermal conditions. The reaction scope was broad, and two different band pass filters (365 or 436 nm) could be applied depending on the substituents on acylsilanes 1a or aldehydes 4.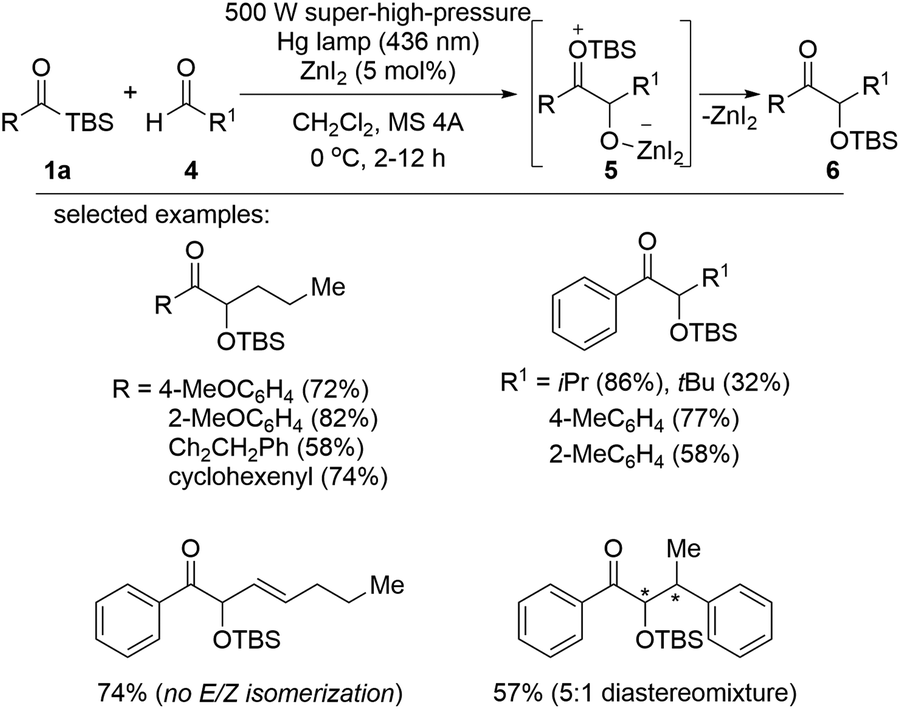 | ||
| Scheme 3 ZnI2-assisted benzoin-type condensation. | ||
The synthesis of α-hydroxyketones 7 from acylsilanes 1b and aldehydes 4 under visible light was also investigated by Huang et al. (Scheme 4).16d Low-energy blue LEDs (30 W, 425 nm) were effective in activating the acylsilanes to the corresponding carbene moieties. Acylsilanes bearing the trimethylsilyl (TMS) group afforded higher yields than those with other silyl groups, such as triethylsilyl (TES) and tert-butylmethylsilyl (TBS) groups, likely due to the increased steric hindrance in the latter. Aldehydes bearing aromatic rings with electron-deficient substituents displayed better reactivity than those with other functionalities. Based on several studies, a plausible mechanism was proposed. The 1,2-Brook rearrangement of acylsilane 1b upon photoirradiation provided carbene species I. This carbene intermediate reacted with aldehyde 4 to generate a highly reactive epoxide II, which was followed by a ring-opening reaction to afford the desired α-hydroxyketone 7 after an acidic work-up.
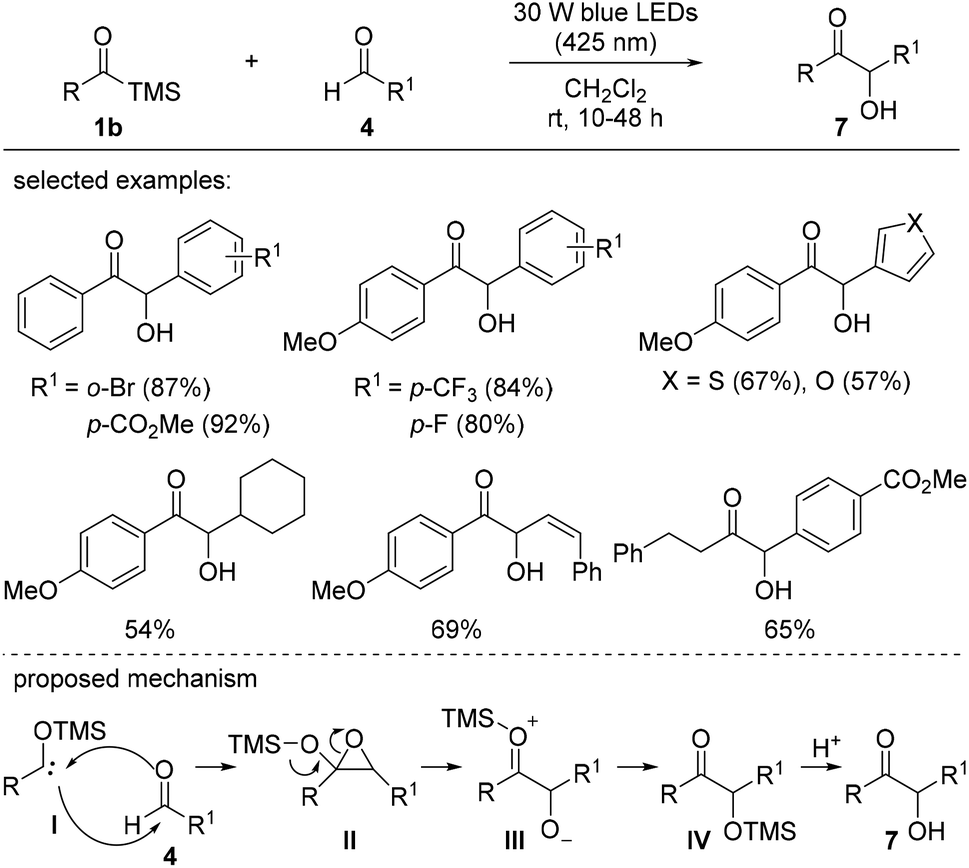 | ||
| Scheme 4 Synthesis of α-hydroxyketones via benzoin reactions. | ||
Very recently, Xi's group reported the visible-light-induced carboxylation of acylsilanes with carbon dioxide (Scheme 5).23 Acylsilanes possessing electron-donating and electron-neutral groups reacted smoothly to give the corresponding α-ketocarboxylates 8 in good yields. However, the reactions of acylsilanes bearing strongly electron-withdrawing substituents, such as p-CO2Me and -CF3, failed to afford the desired products, indicative of the electronically biased properties of the substrates, which dictate the nucleophilicity of the generated siloxycarbenes. In addition to substituted phenyl rings, other electron-rich heterocycles, such as thiophene and furan, were also applicable and afforded the desired carboxylated products, albeit in low yields.
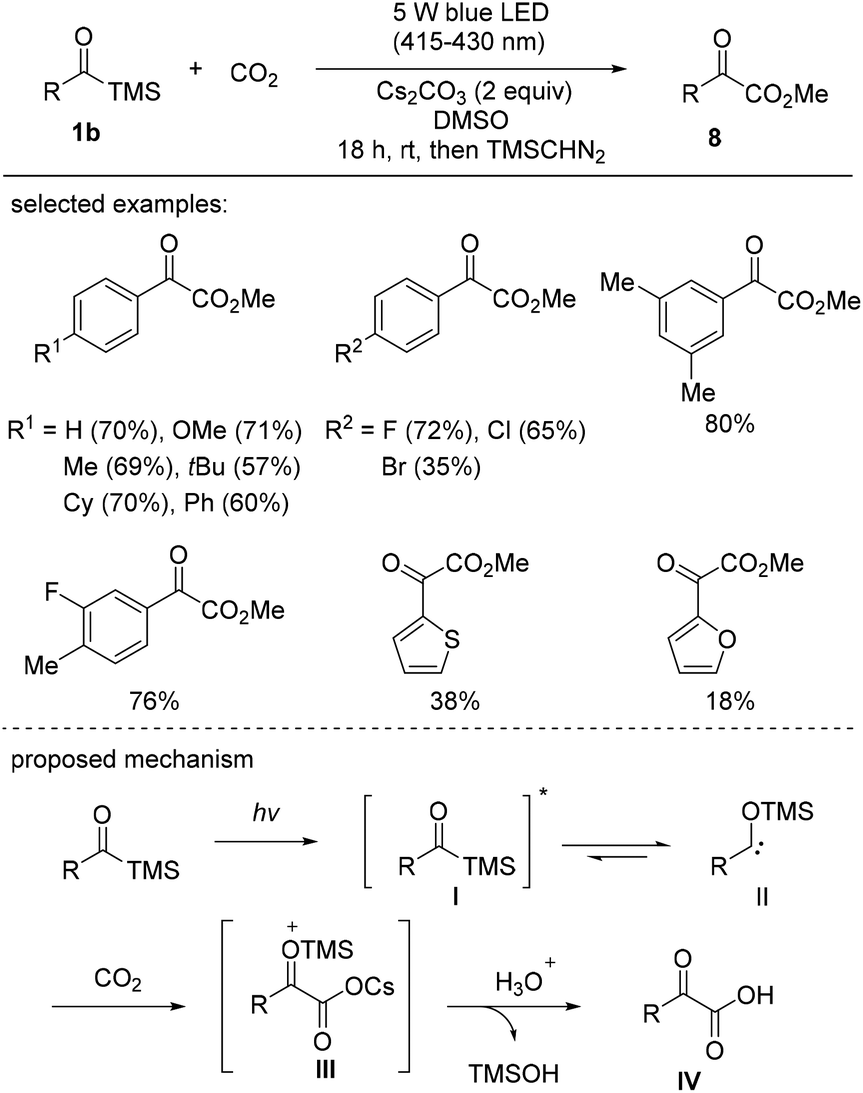 | ||
| Scheme 5 Synthesis of α-ketocarboxylates through the carboxylation of acylsilanes with carbon dioxide. | ||
The proposed reaction mechanism is presented in Scheme 5. Under photochemical conditions, the acylsilane is excited to its singlet excited state I, which undergoes a 1,2-silyl shift. The next step involves the trapping of singlet siloxycarbene II generated from the singlet excited state of acylsilane I to give the cesium carboxylate intermediate III, as confirmed by MALDI-TOF MS analysis. It should be noted that the mechanism involving cesium carboxylate III differs from that of a previous report, wherein the carboxylation was proposed to be realized via the 3-membered lactone.24 Hydrolysis resulted in the formation of the final product IV, and trimethyl silanol was released as a by-product.
In 2021, Priebbenow et al. reported the visible-light-induced 1,2-Brook rearrangement of benzoylsilanes to generate singlet nucleophilic carbenes, which then attacked trifluoroacetophenes 9 to afford fluorinated tertiary alcohol derivatives 10 (Scheme 6).25 Here, carbenes, generated through the irradiation of substituted benzoyl trimethylsilanes 1c with blue LEDs (427 nm, 40 W), effectively reacted with a variety of trifluoroacetophenone derivatives 9. Depending on the substituents on the aryl components, various fluorine-containing benzoin-type adducts 10 were obtained in good yields. Interestingly, in the reaction of benzoyl trimethylsilane with 2,2,2-trifluoroacetophenone under blue LED irradiation, a better yield (94%) and efficiency (15 min residence time) were achieved using a capillary flow reactor (4 mL volume) than under batch reaction conditions; the former protocol was performed on a gram scale, yielding 2.32 g of the desired fluorinated tertiary alcohol.
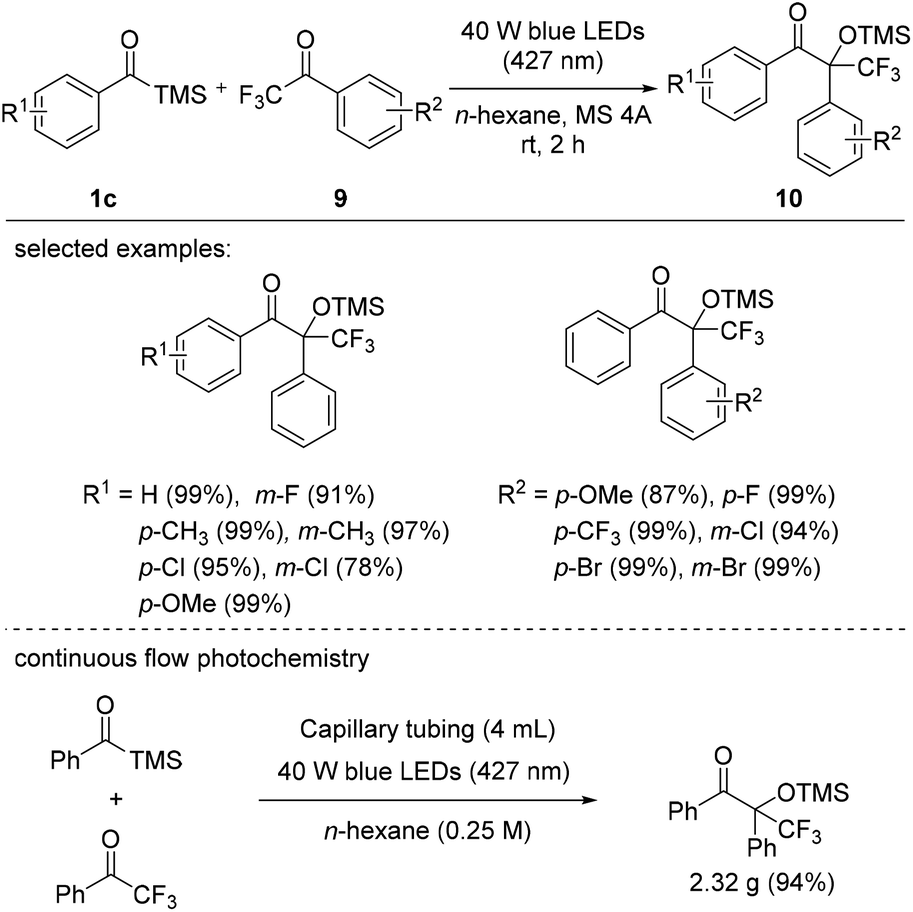 | ||
| Scheme 6 Photochemical reactions of acylsilanes with 2,2,2-trifluoroacetophenone derivatives. | ||
2.2 Carbene insertion reactions into X–Y (B–C, B–H, N–H, O–H or Si–H) bonds
Several studies have demonstrated that when nucleophilic siloxycarbenes are generated under photochemical conditions, carbene insertion into a wide range of X–H bonds occurs, including O–H, halogen–H, B–H, and Si–H.The Kusama group reported photoirradiated metal-free coupling reactions of acylsilanes with boron reagents (Scheme 7).26 Various ketones 12 were prepared using aroyl-, alkanoyl-, and alkenoylsilanes 1b and the organoboron reagent 11 employing a 500 W super-high-pressure Hg lamp. The reaction mechanism was elucidated based on the reaction intermediates, which were identified employing in situ NMR spectroscopy. First, the B–C insertion intermediate III was formed by the nucleophilic addition of I to boronate 11′ followed by 1,2-aryl migration on borate II. Then, the intermediate III further underwent a sequential rearrangement to afford 12′via the seven-membered cycle IV. In the case of aroylsilanes, only the seven-membered intermediate IV was observed. Further studies indicated that the rearrangement was affected by the electron density of substituents as well as the steric hindrance around the quaternary carbon center of the C–B intermediate III.
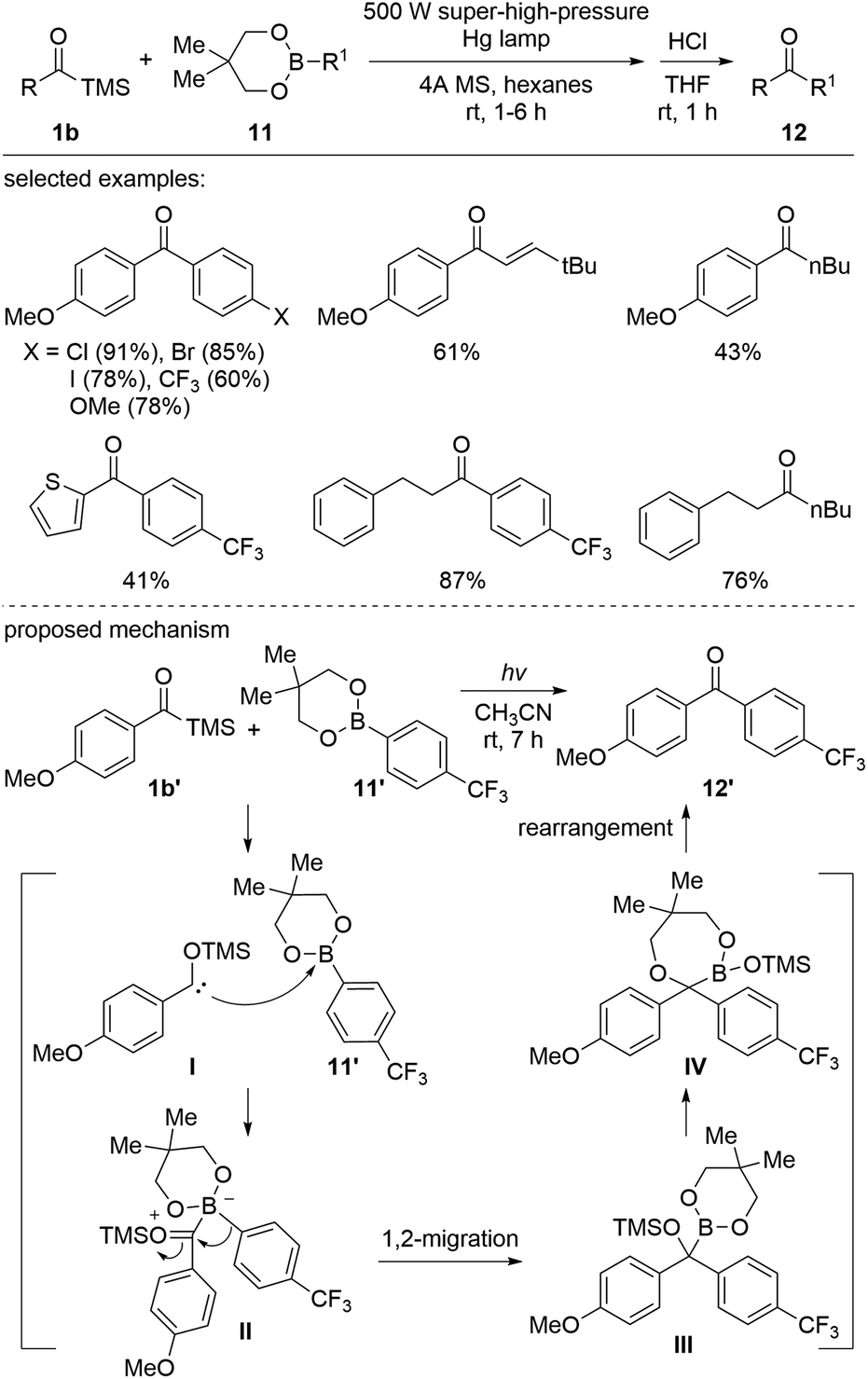 | ||
| Scheme 7 Transition-metal-free cross-coupling reactions between acylsilanes and organoboronic esters. | ||
Carbene insertion reactions into C–H bonds have not been studied as extensively as insertions into carbon–heteroatom bonds, such as N–H, O–H, S–H, and Si–H. Nucleophilic α-siloxycarbenes derived from acylsilanes upon photoirradiation and successful B–H insertion into pinacolborane (HBPin) were reported in 2019 by Glorius and co-workers (Scheme 8).14c The reaction between acylsilanes 1b and pinacolborane 13 gave α-alkoxyorganoboronate esters 14 after 12 hours of stirring under 3 W blue LED irradiation (420 nm) at room temperature. Regardless of the nature of the substrate, the desired products were obtained in quantitative yields (>99%) in most cases. Extensive mechanistic studies were conducted entailing control experiments and DFT calculations. Singlet α-siloxycarbene was generated from the acylsilane precursor under 420 nm blue LED light; however, the carbene intermediate could not be efficiently generated under 455 nm blue LED light without the use of a photosensitizer. The singlet carbene generated from acylsilanes 1b underwent an insertion into the B–H bond of pinacolborane 13 to provide the desired products 14.
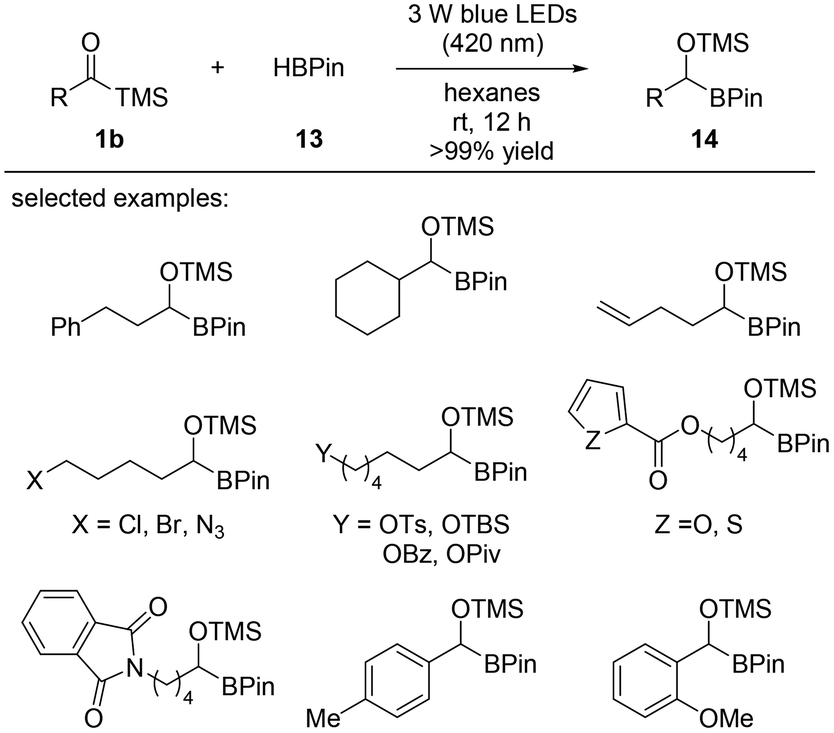 | ||
| Scheme 8 Metal-free carbene insertion into B–H bonds of pinacolborane. | ||
In 2020, Kusama's research group reported the generation of the triplet excited state of siloxycarbenes from alkanoylsilanes 1d under visible light, and the subsequent formation of a new carbon–carbon bond in the presence of boronic esters 15 or aldehydes 17 (Scheme 9).16b Compared to aroylsilanes, the utility of alkanoylsilanes 1d under photoirradiation conditions is limited primarily due to the formation of undesired products through the Norrish type fragmentation, resulting from the reaction of the singlet-excited state of the alkanoylsilanes rather than the triplet-excited state. To suppress this undesired process, a photosensitizer (PS) was adapted to absorb the triplet energy, and thereby prevent the direct photoexcitation of alkanoylsilanes. After absorption under photoirradiation, the PS was excited to its singlet excited state 1PS*, followed by conversion to its triplet excited state 3PS*. Through energy transfer from 3PS* to the ground state of acylsilane 1d, a triplet excited of acylsilane 31d* was generated, which is the precursor of siloxycarbene species I. Based on this method, carbon–carbon bond-forming reactions were developed using boronic ester 15 or aldehyde 17. The reaction with boronic ester 15 under visible light irradiation (436 nm) gave the desired product 16 after H2O2 oxidation. Moreover, α-siloxyketone 18 was prepared via the reaction between acylsilane 1d and aldehyde 17 in the presence of ZnI2. As previously proposed, the undesired Norrish-type fragmentation was successfully suppressed in most cases.
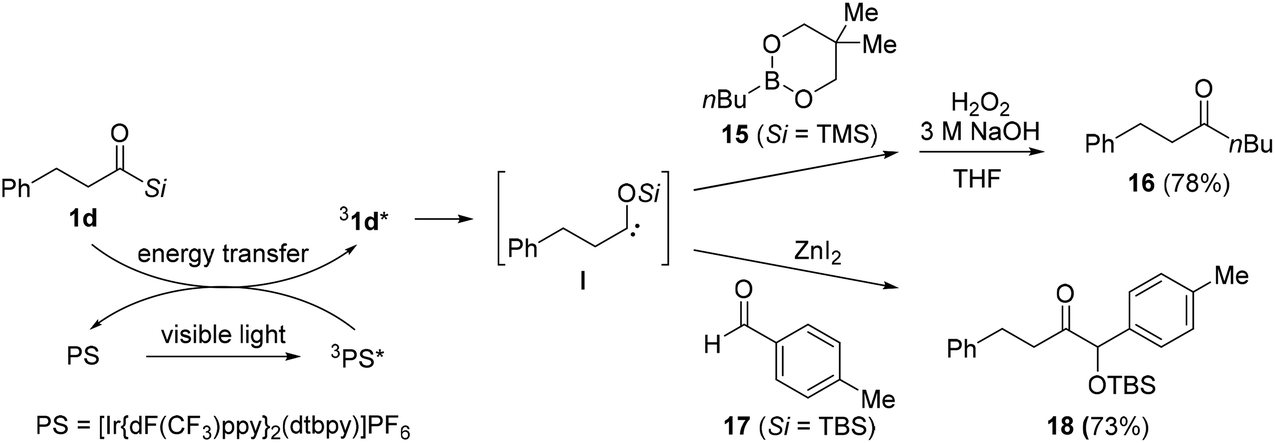 | ||
| Scheme 9 Reactions of alkyl-substituted siloxycarbenes with boronic esters or aldehydes. | ||
In 2021, the Studer group reported the insertion of siloxycarbenes 1b into the N–H bond of indole 19 to afford stabilized N,O-acetals 20 (Scheme 10).27 Ordinarily, N,O-acetals are structurally unstable, and thus prone to hydrolysis. However, the authors identified that aromatic N-heterocycles such as indole can form stable N,O-acetals 20via siloxycarbene addition to the N–H bond, likely due to the reduced electron-donating ability of the nitrogen atom of 20. The reaction was operated in a green fashion without the use of additional chemicals and was highly efficient; in most cases, quantitative yields were obtained using aryl acylsilanes. Adequate stability and complete conversion allow for clickable transformation in more complex systems. For example, the method has been applied to the conjugation of two biomolecules, 21 and 22 (Scheme 10a). In addition, the discovered siloxycarbene N–H insertion was orthogonally applied to the late-stage functionalization of polymeric material 24 entailing a copper-catalyzed click reaction between alkyne 25 and benzyl azide (Scheme 10b). Finally, a wavelength-responsive selective polymerization–depolymerization (27 to 29) was developed, which can be implemented in degradable polymer synthesis (Scheme 10c). Compound 29 formed after depolymerization and was further degraded by an acid. In the same year, the authors reported another application of N,O-acetal formation for tryptophan modification in oligopeptides.28 Various oligopeptide functional groups were tolerated under visible light conditions.
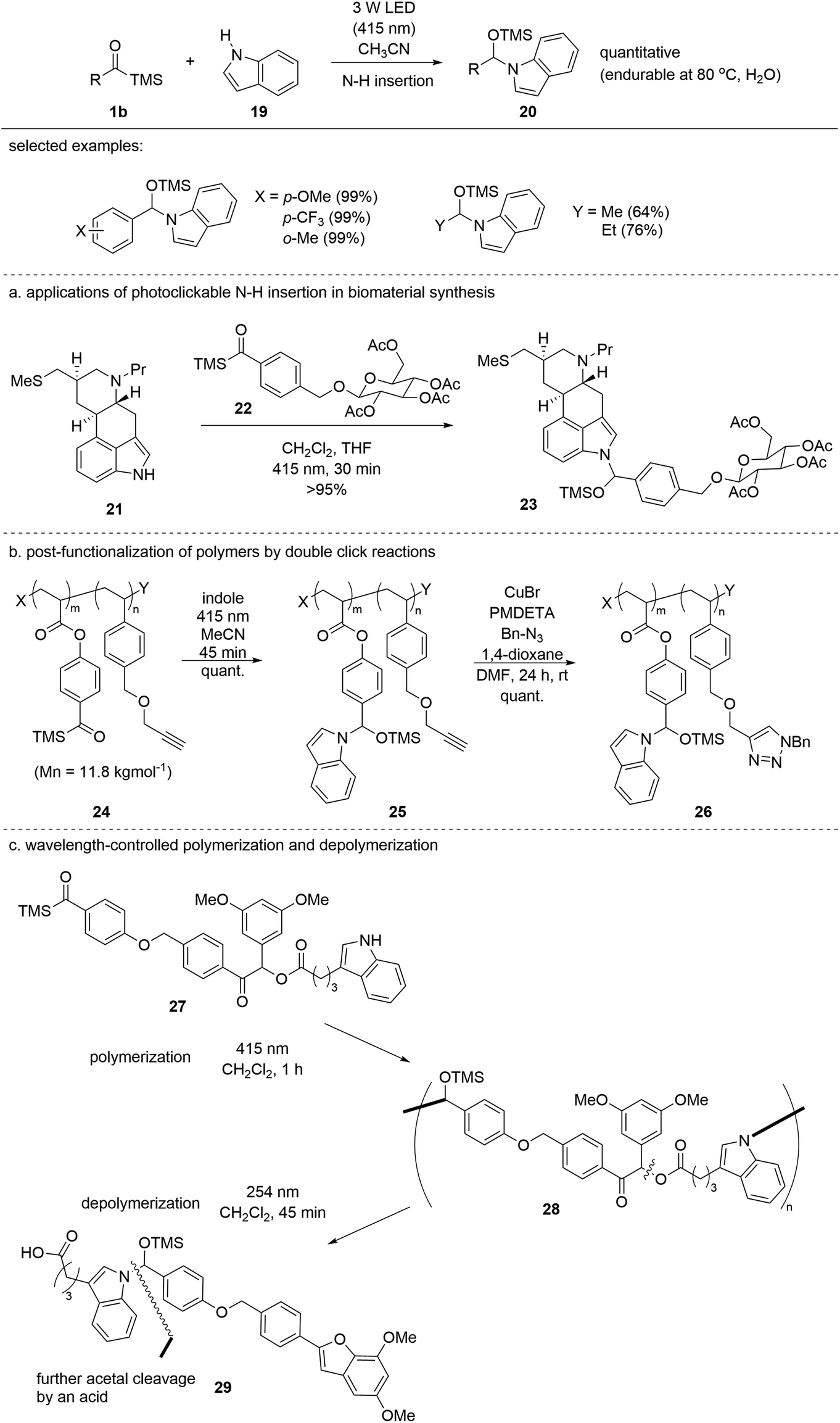 | ||
| Scheme 10 Siloxycarbene insertion into the indole N–H bond. | ||
Recently, Xuan et al. disclosed siloxycarbene oxime O–H insertion reactions. Building upon their previous work29 involving O-alkylation using stabilized diazo compounds as electrophilic carbene precursors for counter oxime substrates, the authors reported a simple protocol for oxime 30 O–H insertion, adopting acylsilanes 1e as nucleophilic carbene precursors (Scheme 11).30 The protocol was optimized to operate in THF under a 24 W blue LED light. The reaction scope exhibited broad functional group tolerance, including that towards alkyl, alkoxy, and halide arene substituents, as well as some heteroarenes, such as thiophene and pyridine. Furthermore, they demonstrated that other types of substituents on the silicon atom, TMS-, TIPS (triisopropyl)-, and TES groups, were all applicable in the reaction. The synthetic potential of this reaction protocol was further explored, and other types of X–H bonds, including non-oxime O–H, N–H and S–H bonds, were successfully converted into the corresponding mixed acetal structures 31.
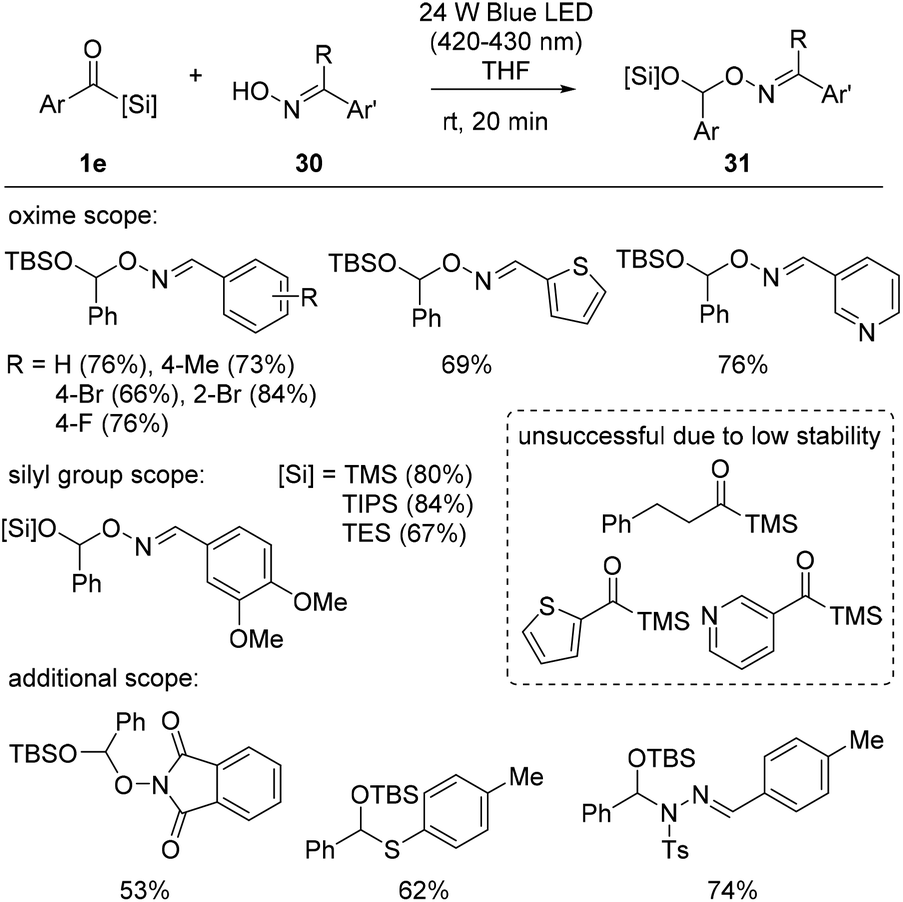 | ||
| Scheme 11 Acylsilane insertion into the O–H bond of oximes. | ||
2.3 Cyclization reactions
Despite the importance of trans-fused bicyclo skeletons, many synthetic methods of these moieties are limited due to harsh reaction conditions or the requirement of organometallic reagents. Very recently, Kusama's research group reported the photoinduced intramolecular cyclization of trans-fused bicyclo[n.3.0] compounds of acylsilanes bearing a boronate 1f (Scheme 12).31 The reactions proceed smoothly under a super-high pressure Hg lamp (365 nm band-pass filter) without any additional reagents. In particular, the stereospecific synthesis of a highly strained trans-fused cyclic system under photoirradiation conditions was achieved with trans-configuration at the ring junction of starting acylsilanes having a boronate prepared by the hydroboration of the corresponding cycloalkenes. The intramolecular nucleophilic attack of a siloxycarbene to the boronate moiety under light irradiation generates zwitterionic bicyclic intermediates II, followed by stereospecific 1,2-migration of the carbon substituent on the borate intermediate II providing the desired trans-fused cyclic products 32. The relative stereochemistry of the synthesized trans-fused compound was confirmed by X-ray analysis.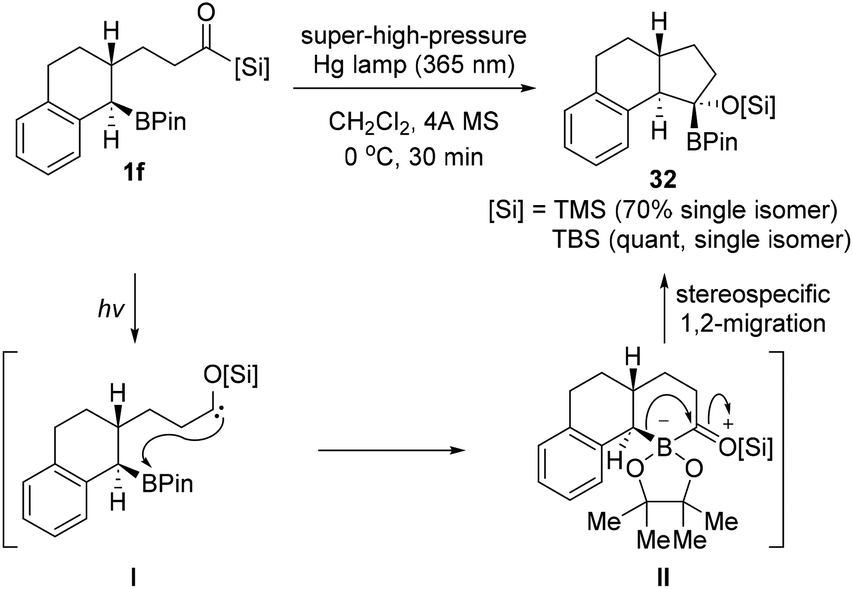 | ||
| Scheme 12 Intramolecular cyclization of acylsilanes bearing a boronate group. | ||
Bolm et al. reported the intermolecular silylacylation of electron-deficient internal alkynes 33 to afford vinylsilanes 34 in moderate to excellent yields (Scheme 13).18e The silylacylation mechanism was proposed to be initiated by the photochemical 1,2-Brook rearrangement of aroylsilanes to form a nucleophilic singlet carbene. Carbene addition into the alkynes then occurs to provide vinyl silanes 34via cyclopropanation and the subsequent 1,4-retro-Brook rearrangement. Electron-deficient internal alkynes afforded vinyl silanes in good yields. However, reactions using neutral internal alkynes, such as diethyl or diphenyl acetylene, were not successful, indicating the importance of the electronic properties of alkyne substrates. The reaction was sensitive to the steric hindrance of aroylsilanes; for example, bulky silyl groups (TES and TBS groups) resulted in low yields (31% and 44%, respectively).
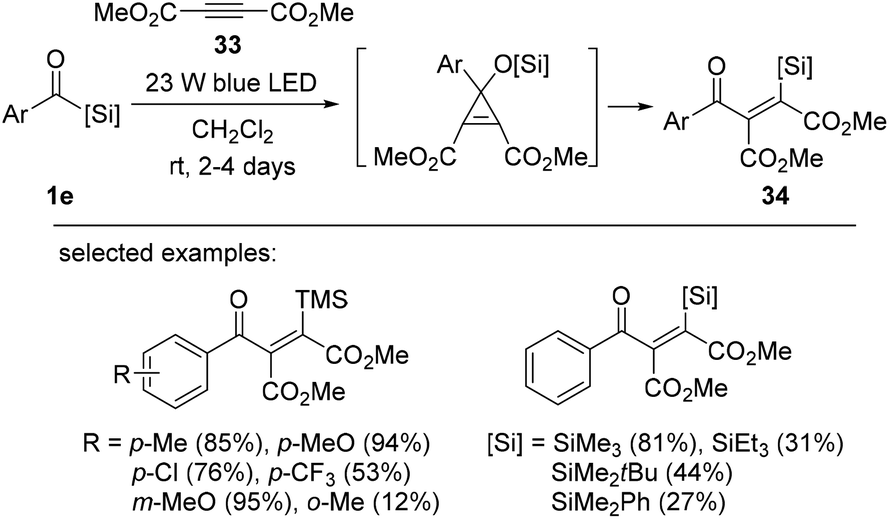 | ||
| Scheme 13 The intermolecular silylacylation of internal alkynes. | ||
In 2022, Zhou and Shen reported a novel [2 + 1] cycloaddition reaction under visible-light irradiation using acylsilane 1h and common alkynes 35 to afford cyclopropenols 37 (Scheme 14).32 It was discovered that the CF3 substituent of 1h is critical for effective cycloaddition; the donor–acceptor property of the generated carbene species imparts an ambiphilic nature. Notably, an aryl substituent on the silicon atom was required for achieving higher efficiencies than that in the case of trialkylsilanes. Two types of sensitizers could be used in the transformation as suitable photocatalysts for energy transfer: 4CzIPN (ET = 59.6 kcal mol−1), an organic compound, and the transition metal complex, FIrPic (ET = 61.1 kcal mol−1). Owing to the mild reaction conditions and high efficiency, the optimized protocol was broadly applicable to various functionalized alkynes. The reaction was operative irrespective of the electronic nature and substitution pattern of the alkynes. Finally, one-pot three sequential steps, visible-light-induced [2 + 1] cycloaddition, desilylation, and hydrogenation, afforded cyclopropanols 37 with unique substitution patterns. The hydrogenation of cyclopropenols is highly diastereoselective, likely due to the interaction between the hydroxyl group and the heterogeneous Pd catalyst to afford compound 38.
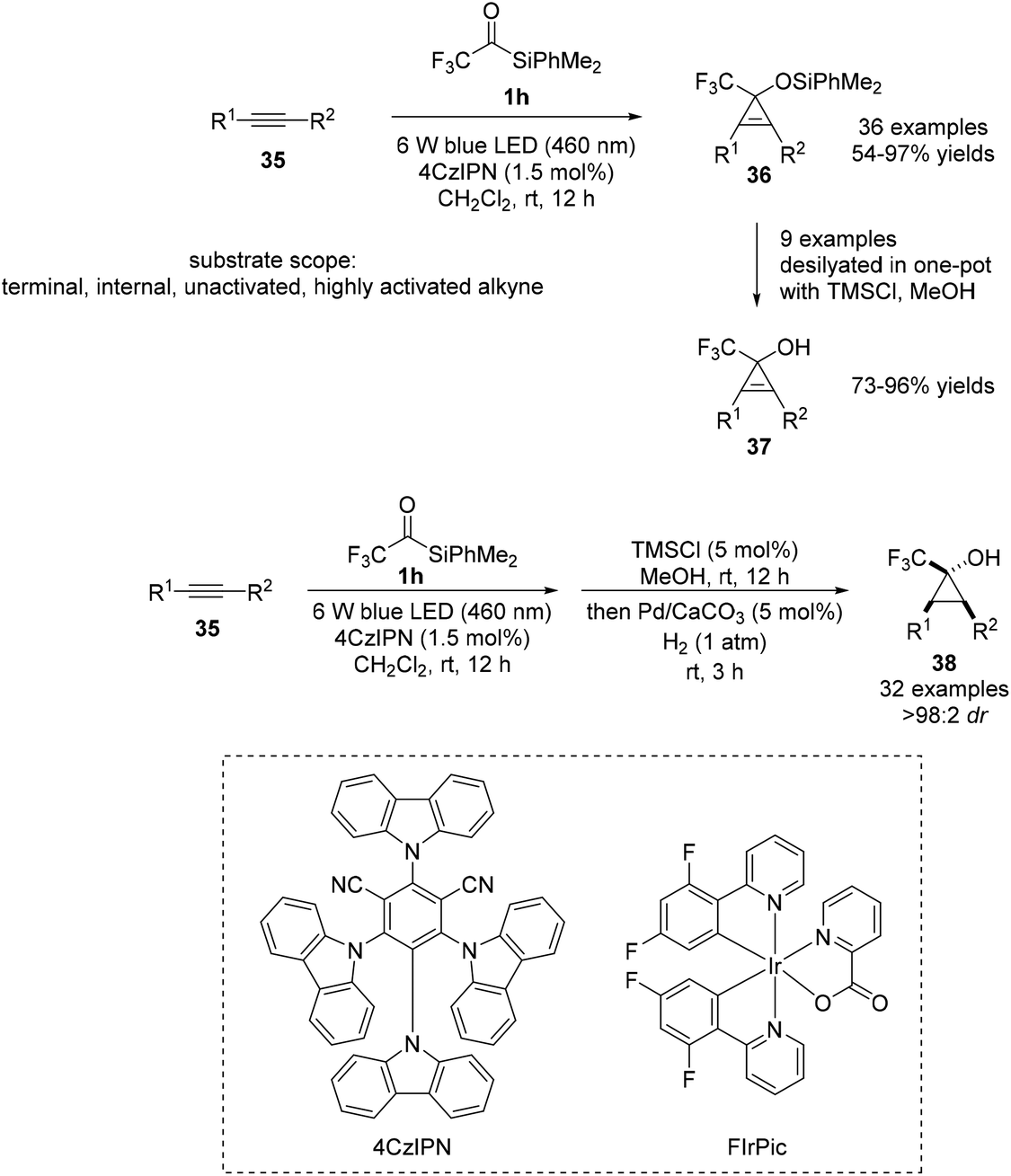 | ||
| Scheme 14 [2 + 1] cycloaddition between siloxycarbenes and alkynes. | ||
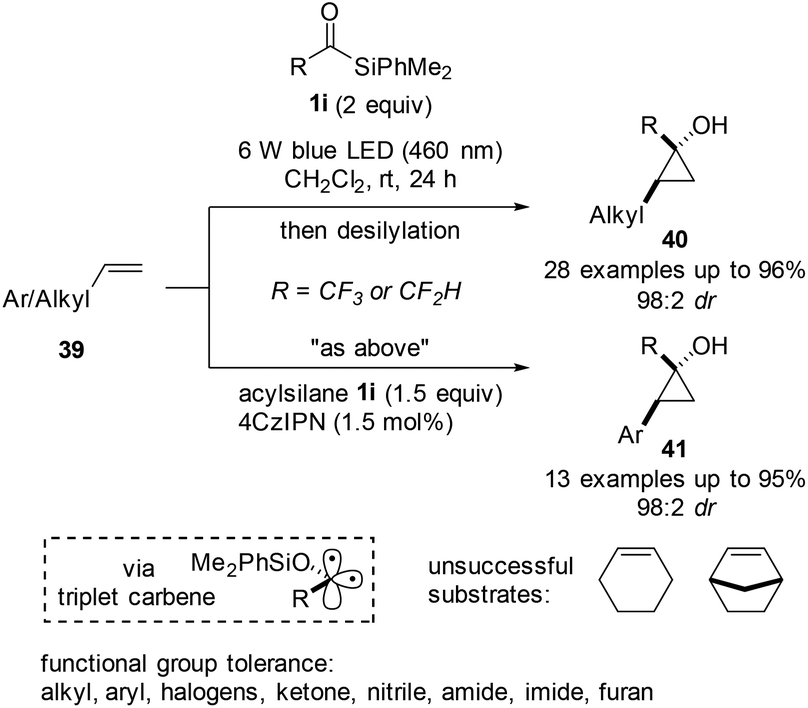
In the same year, the same group expanded the reaction to provide cyclopropanols (40 and 41) directly using acylsilanes 1i and simple alkenes 39 as coupling partners (Scheme 15).33 The authors reported that the siloxycarbene formed via photoactivation existed as a triplet state carbene, which was demonstrated by several control experiments. In contrast with the previous approach published34 by Tobisu et al. entailing cyclopropanation using Pd-carbene intermediates, the developed protocol tolerated halogens and proceeded with high diastereoselectivity. Notably, aliphatic alkenes participated in the reaction under irradiation without the photocatalyst, while arylalkenes required an excess amount of acylsilanes and the use of a photocatalyst. Although many functional groups, such as halogens, heterocycles, and carboxylic acid derivatives, were tolerated under mild reaction conditions, 1,2-dialkylolefins, such as cyclohexene and norbonene, did not react effectively, undergoing side reactions involving the allylic C–H bond. Overall, the designed reaction allowed for the successful conversion of trifluoromethyl- or difluoromethyl-substituted acylsilanes into highly strained cyclopropane rings. The reaction pathway and reasoning for the diastereoselectivity were proposed with the aid of computational studies.
 | ||
| Scheme 15 [2 + 1] cycloaddition between siloxycarbenes and alkenes. | ||
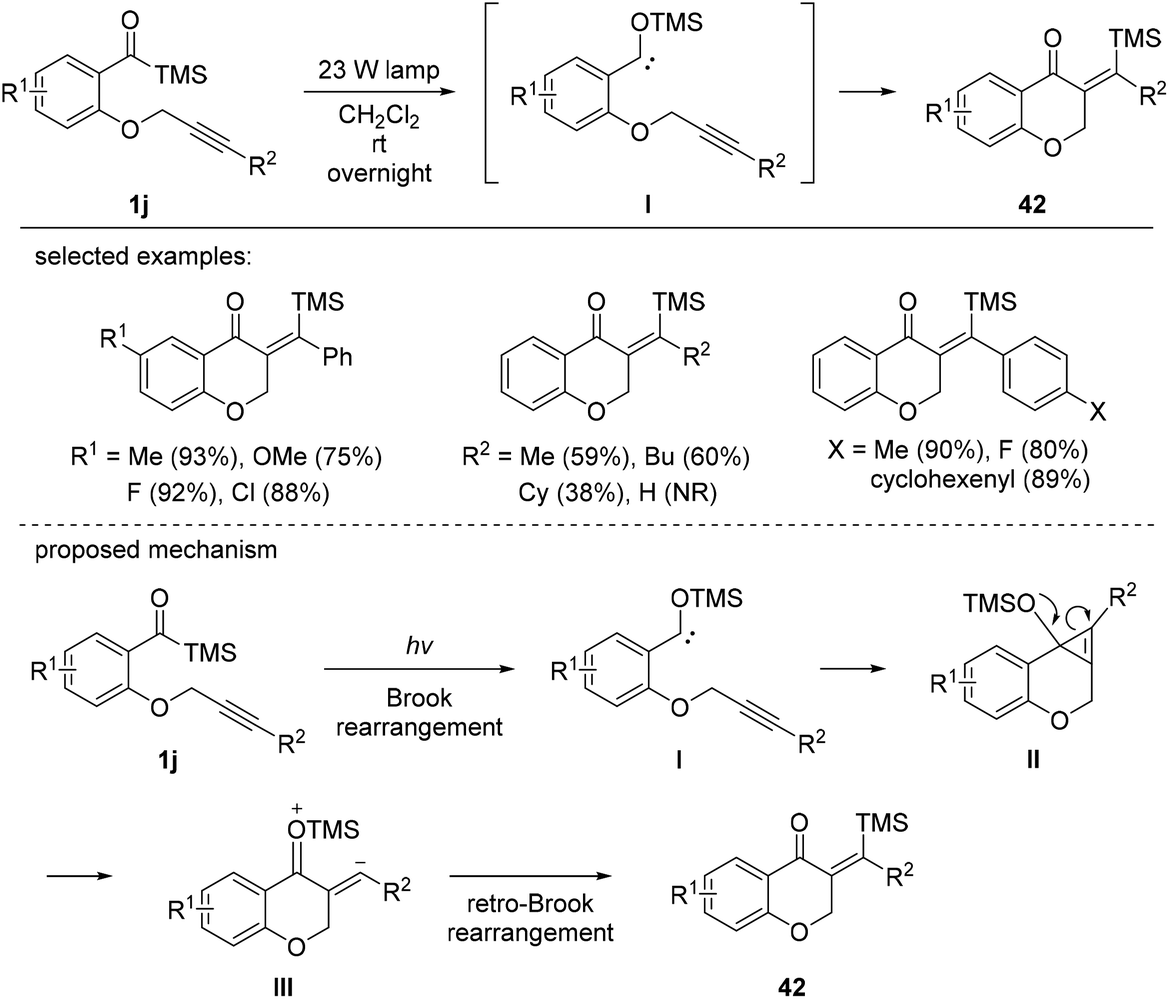
In 2012, Bolm et al. described the intramolecular silyl acylation of various alkynes (Scheme 16).18c The synthetic utility of this method was demonstrated by the synthesis of silylated chromone derivatives 42. Electronically varied substituents on the benzene ring were compatible, giving the corresponding chromone derivatives 42 in moderate to excellent yields. Aryl substituents on the alkynyl chain were also tolerated, whereas aliphatic moieties were less efficient. The reaction mechanism involved photo-induced Brook rearrangement to afford siloxycarbene I. Then, I participated in an intramolecular cyclization, followed by the cleavage of the intermediary cyclopropene II to afford intermediate III, which further underwent retro-Brook rearrangement to afford chromone products 42.
 | ||
| Scheme 16 The synthesis of chromones via photochemically induced silylacylations of alkynes with acylsilanes. | ||
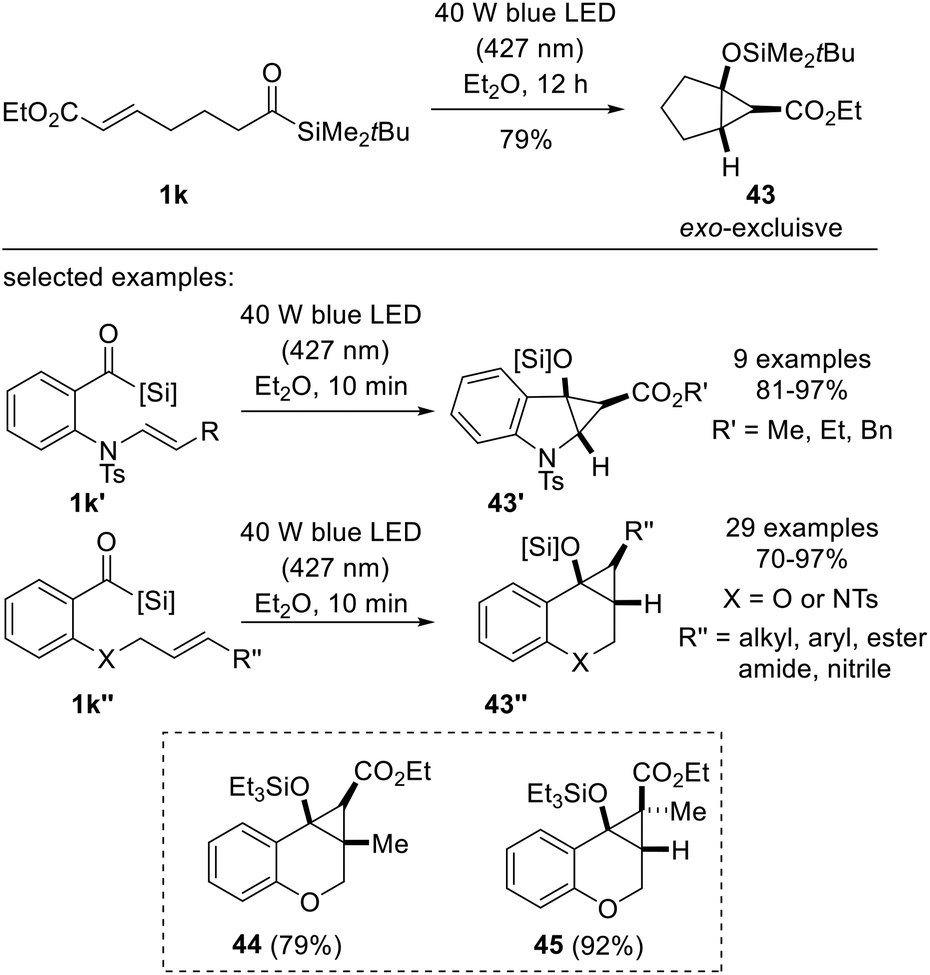
In 2022, Priebbenow and co-workers disclosed an intramolecular cyclopropanation method involving siloxycarbenes generated by visible-light (40 W LED, 427 nm) irradiation (Scheme 17).35 Building on Bolm's protocol where nucleophilic siloxycarbene addition across dimethyl acetylenedicarboxylate (DMAD) or alkynes afforded vinylsilanes,18c the authors aimed to discover visible-light-induced cyclopropanation between electron-poor alkenes and nucleophilic carbenes, which had not been reported until Shen's and Tobisu's reports on intermolecular cyclopropanation,31,32 with similar concepts being investigated around the same time. The desired reactivity was first demonstrated using substrate 1k, which contained acylsilane and acrylate moiety. The substrates used for the investigation of the scope were readily available oxygen- or sulfonamide-tethered alkenes 1k′ or 1k′′. They were smoothly converted to bicyclo[3.1.0] cyclohexane 43′ or bicyclo[4.1.0]cycloheptanes 43′′, respectively, which are medicinally significant scaffolds. Notably, the reaction tolerated various substituted alkenes, affording bicyclic compounds 44 and 45.
 | ||
| Scheme 17 Intramolecular [2 + 1] cycloaddition. | ||
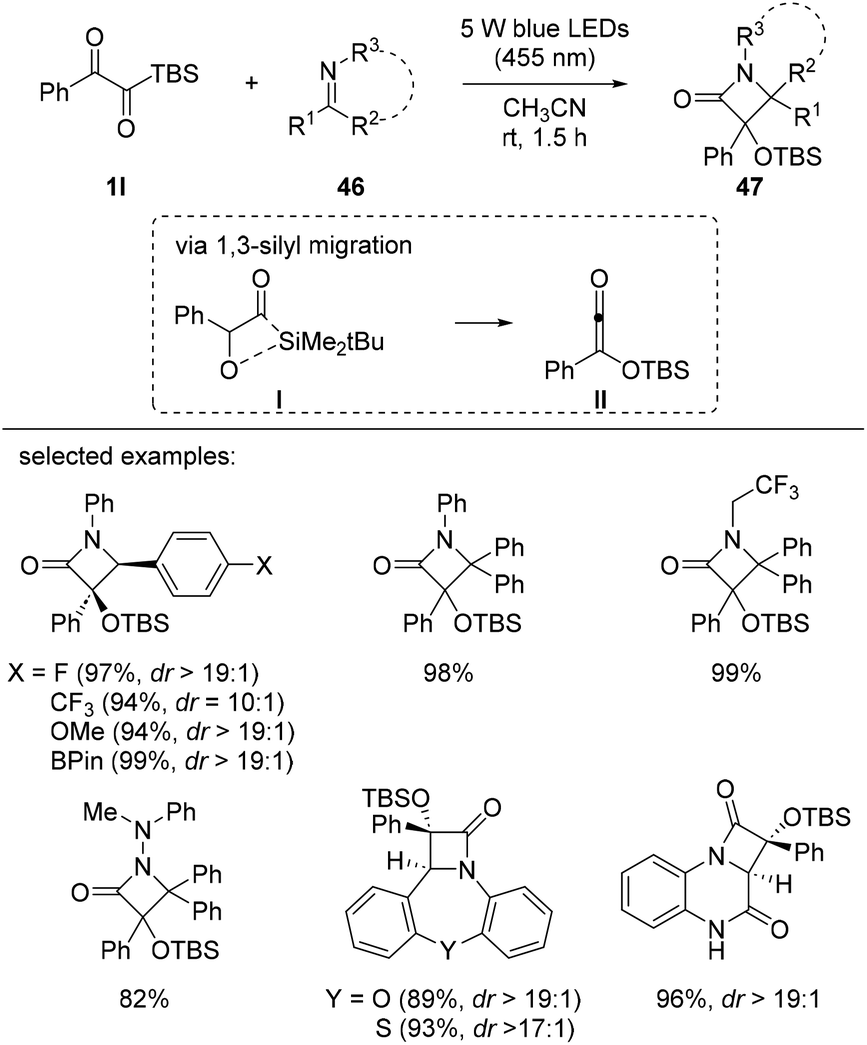
The synthesis of β-lactams 47 from α-ketoacylsilanes 1l was reported by Glorius et al. in 2021 (Scheme 18).36 Siloxyketenes II were generated from α-ketoacylsilanes 1lvia 1,3-silyl migration under visible light irradiation and reacted with various imine derivatives 46, such as aldimines, ketimines, hydrazones, and fused nitrogen heterocycles, to furnish β-lactams 47 with good diastereoselectivity. Computational mechanistic studies indicated that the 1,3-silyl migration of α-ketoacylsilane is favored over 1,2-silyl migration and the corresponding singlet siloxyketene intermediate II is generated which can participate in [2 + 2] ketene-imine cycloaddition reactions. The desired β-lactams 47 were obtained with excellent yields and diastereoselectivity, regardless of the electronic properties of the substrates.
 | ||
| Scheme 18 The synthesis of β-lactams via the cycloaddition of α-ketoacylsilanes and various imines. | ||
In 2019, the Afonso and Candeias group reported a unique synthesis of silacyclopentanols using cinnamyl silane substrates 1m, in which the cyclization mode mimics the UV-initiated Paternò–Büchi type [2 + 2] cycloaddition (Scheme 19).37 It is noteworthy that light irradiation did not initiate the formation of highly reactive siloxycarbenes. Instead, CO activation occurred selectively, enabling [2 + 2] cycloaddition upon reaction with alkenes. The use of dry aprotic solvents and the exclusion of free alcohol or amine functional groups are essential for successful reactions. Because intermediate 48 was unstable in silica gel, post treatment with an acid induced ring opening to deliver silacyclopentanols 49 in good to excellent yields. The high regioselectivity observed during the [2 + 2] cycloaddition was explained using DFT calculations, whereby the energies of the two possible biradical intermediates II and III were compared, and the stabilization effect of the aromatic ring was proposed to be a determining factor for the regioselectivity.
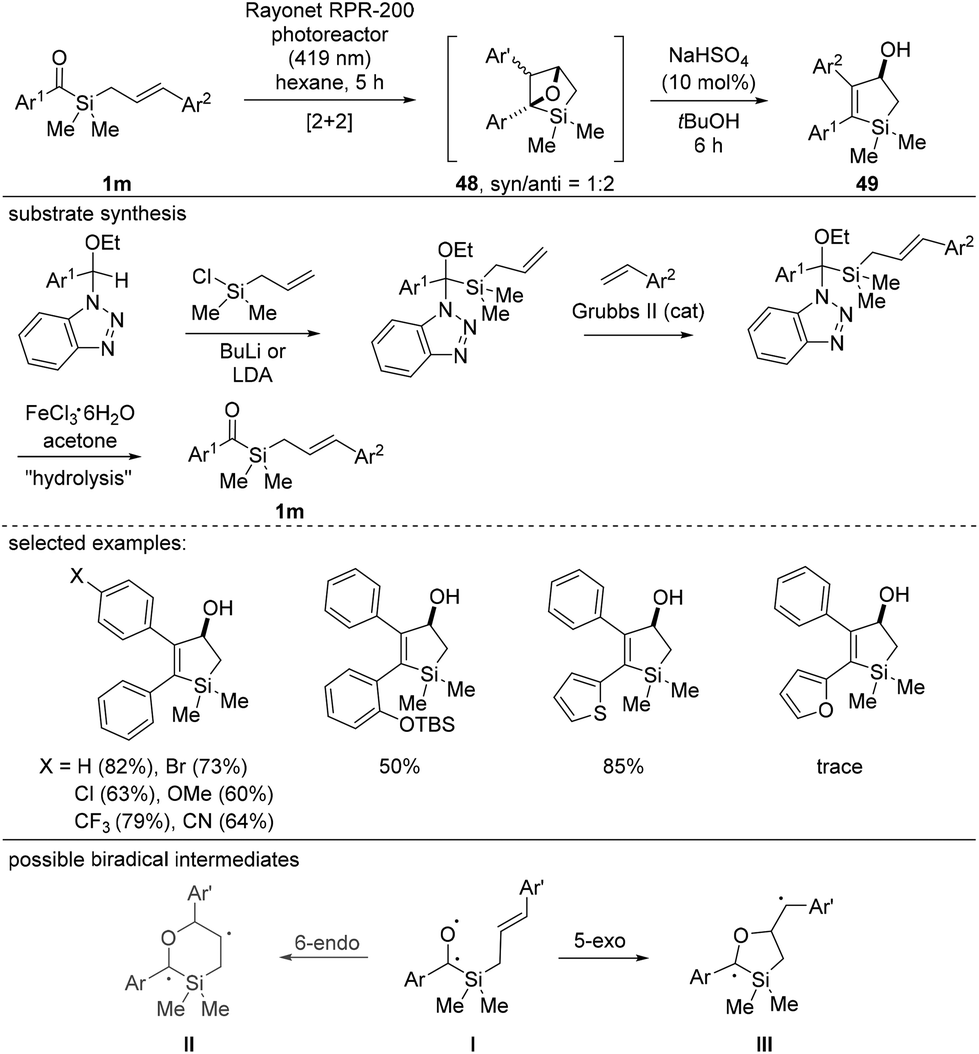 | ||
| Scheme 19 Intramolecular [2 + 2] cycloaddition between ketones and alkenes. | ||
In 2021, Kusama et al. reported a Cu-catalyzed [4 + 1] cycloaddition using Fischer-type copper carbene complex I and electron-rich dienes 50 (Scheme 20).38 This method provides functionalized cyclopentenes 51 with unique substitution patterns. Acylsilane 1n was first converted into siloxycarbene upon UV irradiation, a process known to be reversible at room temperature. The nucleophilic character of siloxycarbene enables complexation with a cationic Cu(I) salt, and the resulting Cu complex was unambiguously identified using in situ NMR and UV-Vis absorption studies. After complexation, siloxycarbene becomes electrophilic and thus, readily participates in cycloaddition with electron-rich siloxy dienes. The authors further developed a catalytic version of this cycloaddition.
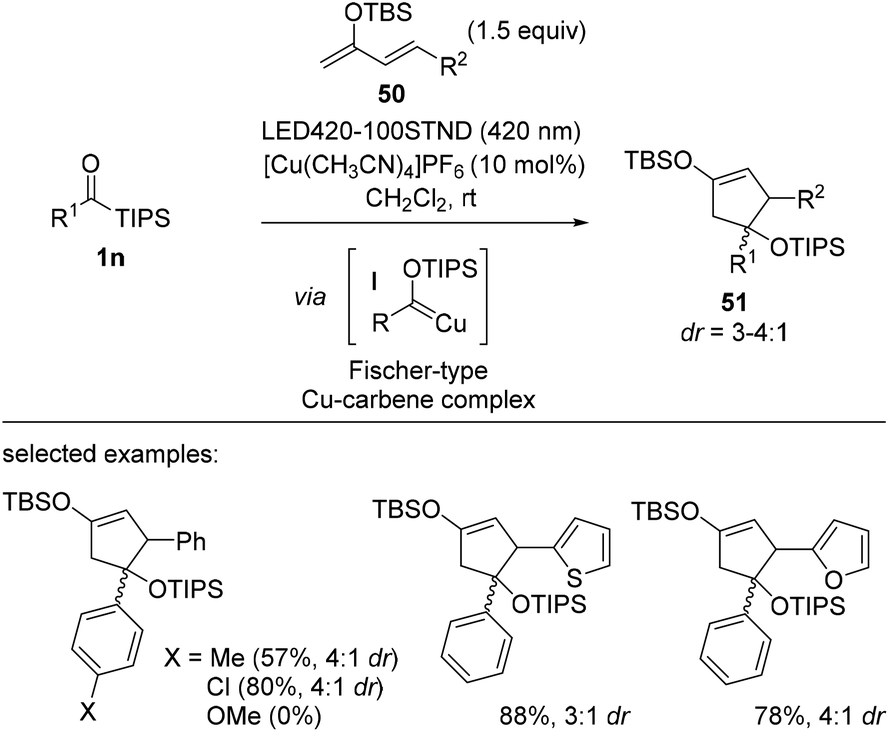 | ||
| Scheme 20 [4 + 1] Cycloaddition between siloxycarbenes and electron-rich dienes. | ||
Although the use of acylsilane-derived siloxycarbenes would provide access to highly substituted cyclopropanes, this variation remains challenging due to the presence of electron-withdrawing groups in the olefins as they facilitate the ring-opening of the resulting silyl ether cyclopropanes.39 This was conceptualized in the synthesis of cyclopentenes 53via cyclopropane intermediates I accomplished by tuning the electronic nature of the diene 52 (Scheme 21). In this study, the blue-light-induced cyclopropanation-vinyl cyclopropane rearrangement sequence, p-tolyltrimethylsilane and dienes in the presence of molecular sieves, provided cyclopropanes in 66% yield, which was increased to 74% using a TBS group. Electronically varied aryl substituents of benzoylsilane were compatible, providing a series of cyclopentenes in moderate to good yields. However, dienes with substituents on the aromatic ring proved to be less efficient. For example, highly electron-donating groups, such as dimethyl amine, were not compatible under the optimized conditions to give the desired cyclopentenes 53. The reaction mechanism involves C–C bond formation between siloxycarbenes and dienes to form the cyclopropane intermediate I. It is worth noting that all of the cyclopropane diastereoisomers have relatively similar energies, as determined by DFT calculations, indicating that the structural differences are insufficient for inducing diastereoselectivity.
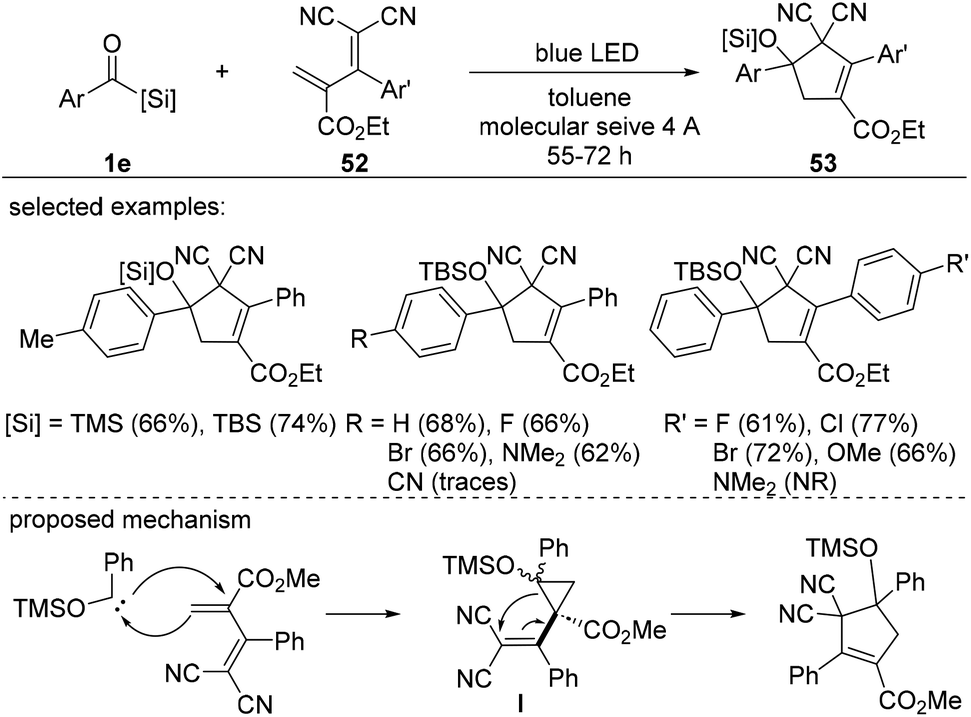 | ||
| Scheme 21 Synthesis of cyclopentene derivatives via formal [4 + 1] cycloaddition of siloxycarbenes from acylsilanes. | ||
Very recently, Shen et al. reported the synthesis of multiple-functionalized furans via the [4 + 1] cyclization–aromatization mode of acylsilane and α,β-unsaturated ketones (Scheme 22).40 A plausible reaction mechanism involves the addition of siloxycarbenes to form intermediate I, which is in equilibrium with intermediate II. Subsequently, intermediate II undergoes an intramolecular cyclization to afford 55 in 56–75% yield. The visible-light-induced reaction tolerated electron-rich and electron-deficient moieties in 1,4-diketones. The synthetic utility of this method was further demonstrated by varying the silyl and sulfonyl groups, affording multi-substituted furan derivatives.
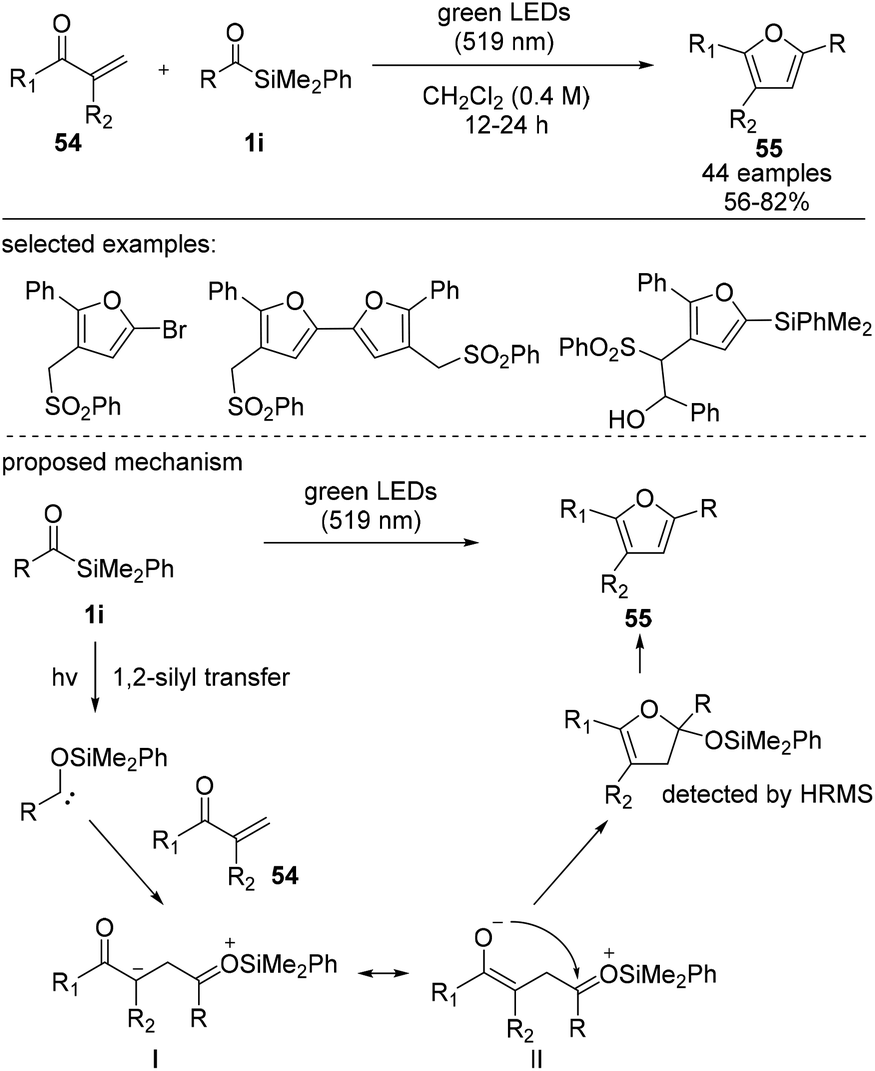 | ||
| Scheme 22 Synthesis of silyl furans through a sequential visible-light-induced [4 + 1] cyclization–aromatization of acylsilanes. | ||
In 2014, Bolm et al. reported the synthesis of several indanone derivatives 56via a cascade reaction entailing the photocatalyzed formation of siloxycarbene I and the subsequent intramolecular reaction with the pendant electron-poor alkene to afford indenone intermediate II (Scheme 23).18f They recognized that ortho-alkenylated aroylacylsilanes 1o could be excellent models for exploring the 6π-electrocyclization of siloxycarbenes. The conversion to 56 was accomplished via simple irradiation using a readily available 23 W energy-saver light bulb. All the tested substrates resulted in quantitative yields. Substrate synthesis using acylsilanes is another significant contribution to the field of arene ortho-alkenylation, whereby common functional groups, such as aldehydes and ketones were harnessed as directing groups. They optimized the reaction conditions based on Glorius's Rh(III)-catalyzed C–H functionalization method.41 Notably, higher reactivity was observed in acylsilanes compared to aldehydes, which was likely to be driven by the enhanced negative polarization at the carbonyl oxygen of acylsilanes.
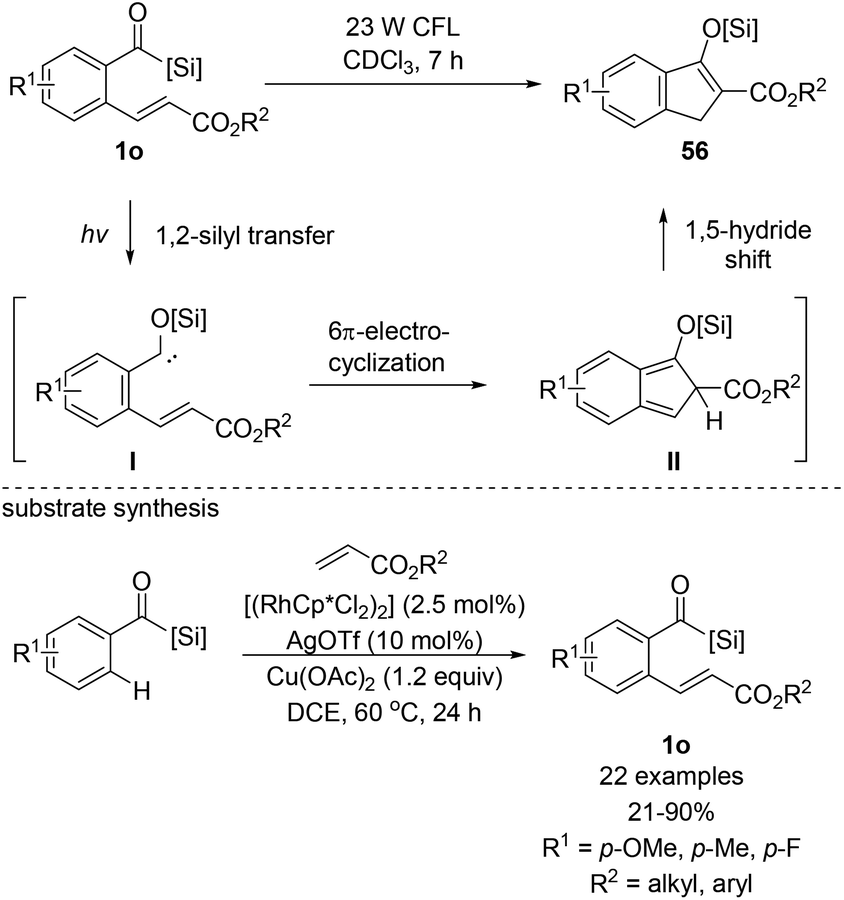 | ||
| Scheme 23 Photo-induced 6π-electrocyclization of acylsilanes containing acrylates. | ||
In 2015, Loh and coworkers reported an interesting example of 6π-electrocyclization of alkenylated indole substrates 1p (Scheme 24).42 When 1p were subjected to visible light, polycyclic indoles 57 were obtained in moderate yields. While rearomatization by 1,5-hydride shift was observed for the substrates 1o in Bolm's prior work for 6π-electrocyclization,18f 1,3-silicon shift in II occurred before rearomatization; this process resulted in structurally distinct α-silylated ketones 57. The overall mechanism for the transformation is firstly explained by the in situ generation of the alkenyl siloxycarbene intermediate I. Then, 6π-electrocyclization occurs to afford the intermediate II, which further undergoes the 1,3-silicon shift process to yield 57. The synthetic method for the preparation of substrates 1p was reported by the authors, which involves rhodium-catalyzed C–H alkenylation using acryloyl silanes. To enable this transformation, RhCp*(MeCN)3(SbF6)2 was an efficient catalyst in the presence of Cu(OAc)2 as the oxidant and TEMPO as the polymerization inhibitor.
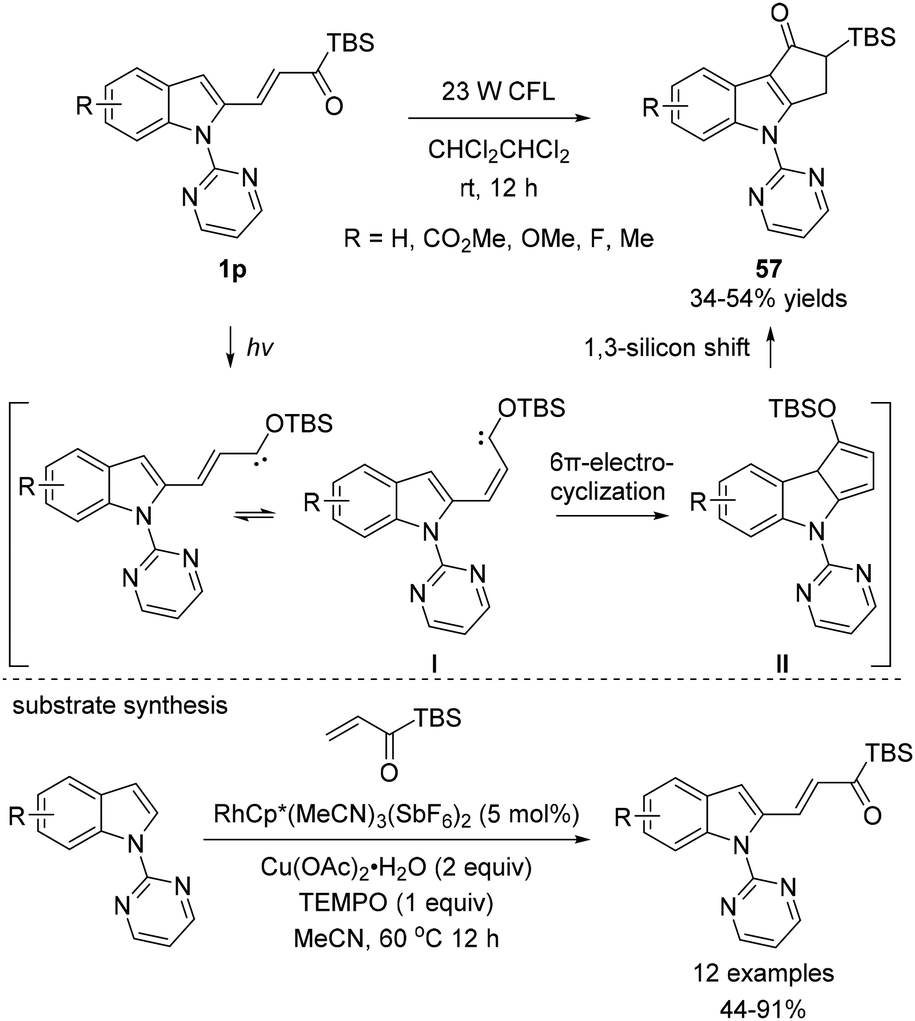 | ||
| Scheme 24 The synthesis of functionalized indole derivatives via electrocyclization. | ||
3. Miscellaneous
Although examples of singlet electron transfer between organometallic complexes and external substrates have been reported, C–C coupling reactions using metal-to-ligand charge transfer (MLCT) photoexcitation are still rare.43 Very recently, Hasegawa and Sawamura reported a photo-driven asymmetric allylic acylation that features charge transfer intermediate derived from a copper complex as a reactive intermediate (Scheme 25).44 Benzoyltrimethylsilanes bearing electron-donating and electron-withdrawing groups reacted smoothly to afford the products 59 in excellent yields with high enantioselectivities. Furthermore, an effort to engage an acylsilane derived from probenecid, a uricosuric drug, resulted in the product formation with 90% ee. Various allylic phosphates 58 are suitable coupling partners for the construction of tertiary stereogenic centers. Interestingly, the (S)-configuration was generated during the transformation. The reaction scope was usual and α-phenyl or α-cyclohexyl-branched ketones were prepared in good yields. The synthetic utility of this method was further demonstrated by the reaction of a γ-methyl-γ-prenylethyl-substituted allylic phosphate 58′ to generate a quaternary asymmetric center.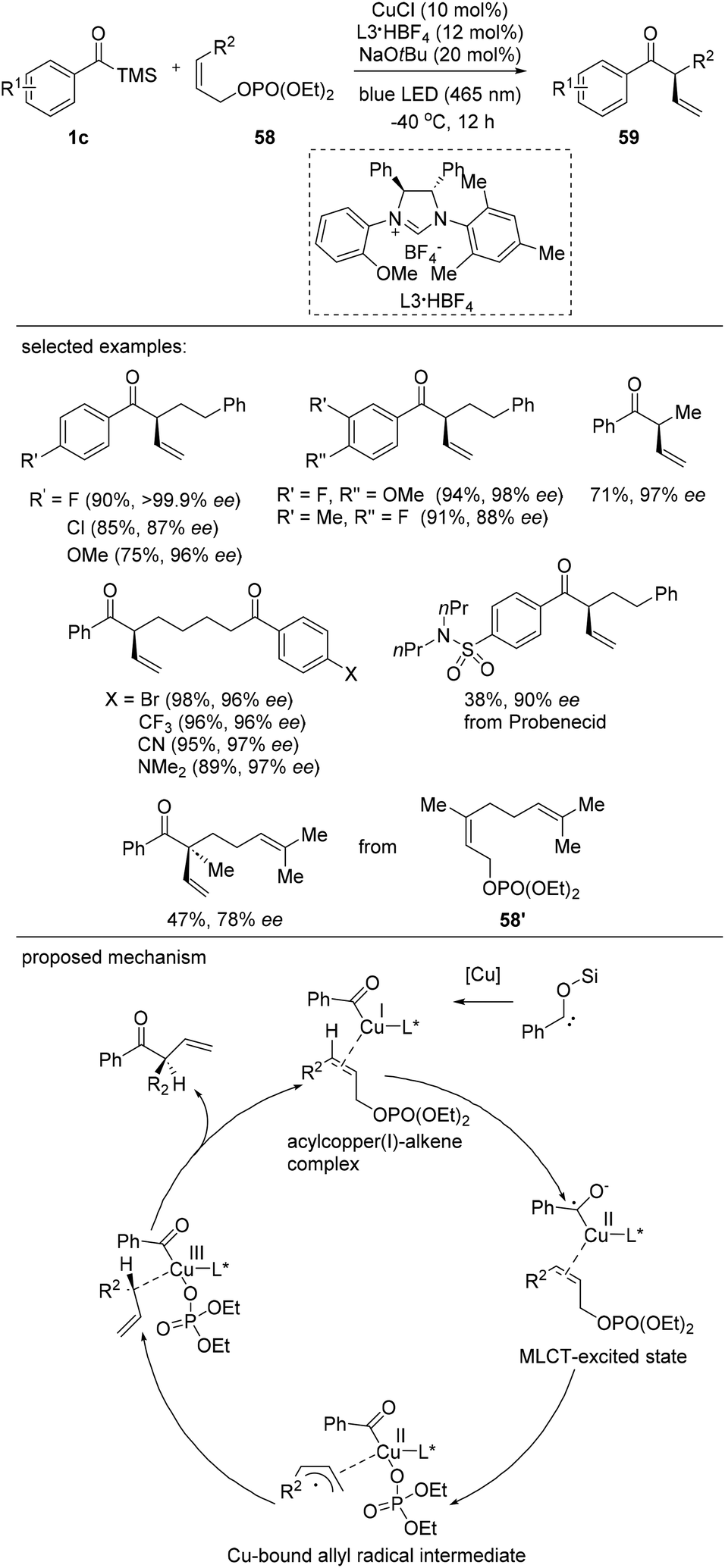 | ||
| Scheme 25 Photochemically induced copper-catalyzed asymmetric allylic acylation. | ||
The proposed mechanism is shown in Scheme 25. The mechanism of the photo-induced copper-catalyzed allylic acylation was proposed to be initiated by the photoexcitation of the acylsilane, which forms an acylcopper(I) intermediate. Next, a second photoexcitation occurs to afford a charge-separated species via metal–ligand charge transfer (MLCT). The resultant allylcopper(II) complex subsequently forms a copper(III) complex, which affords the enantioenriched product and regenerates the copper catalyst in the final step.
In 2018, Fagnoni and co-workers reported the photocatalyzed acyl radical formation and subsequent Michael-type addition reactions using acylsilanes 1b as acyl radical precursors (Scheme 26).45 While previously known acyl radical generation methods using light irradiation employed activated carboxylic acids, such as ketoacids,46 mixed anhydrides,47 and acyl halides,48 they utilized simple and stable acylsilanes as suitable alternatives for the generation of acyl radicals. However, the challenge was preventing the siloxycarbene formation, which is well-known to occur upon the UV irradiation of acylsilanes. Thus, reaction optimization was focused on identifying suitable light sources and catalysts; they demonstrated that the combinations of ammonium tungstate salt (TBADT) with a 310 nm light source and Acr+-Mes with a 410 nm light source enabled the targeted transformation. The proposed reaction mechanism is shown in Scheme 25. The activated photocatalyst (PC*) oxidizes substrate 1b to form the radical-cation intermediate I, in which the C–Si bond is readily cleaved to generate acyl radical II. Then, II likely adds to the Michael acceptor to generate the stabilized carbonyl α-radical III, which is then reduced and protonated, affording the desired products 60. The reaction scope revealed that acyl radicals readily add to various types of Michael acceptors.
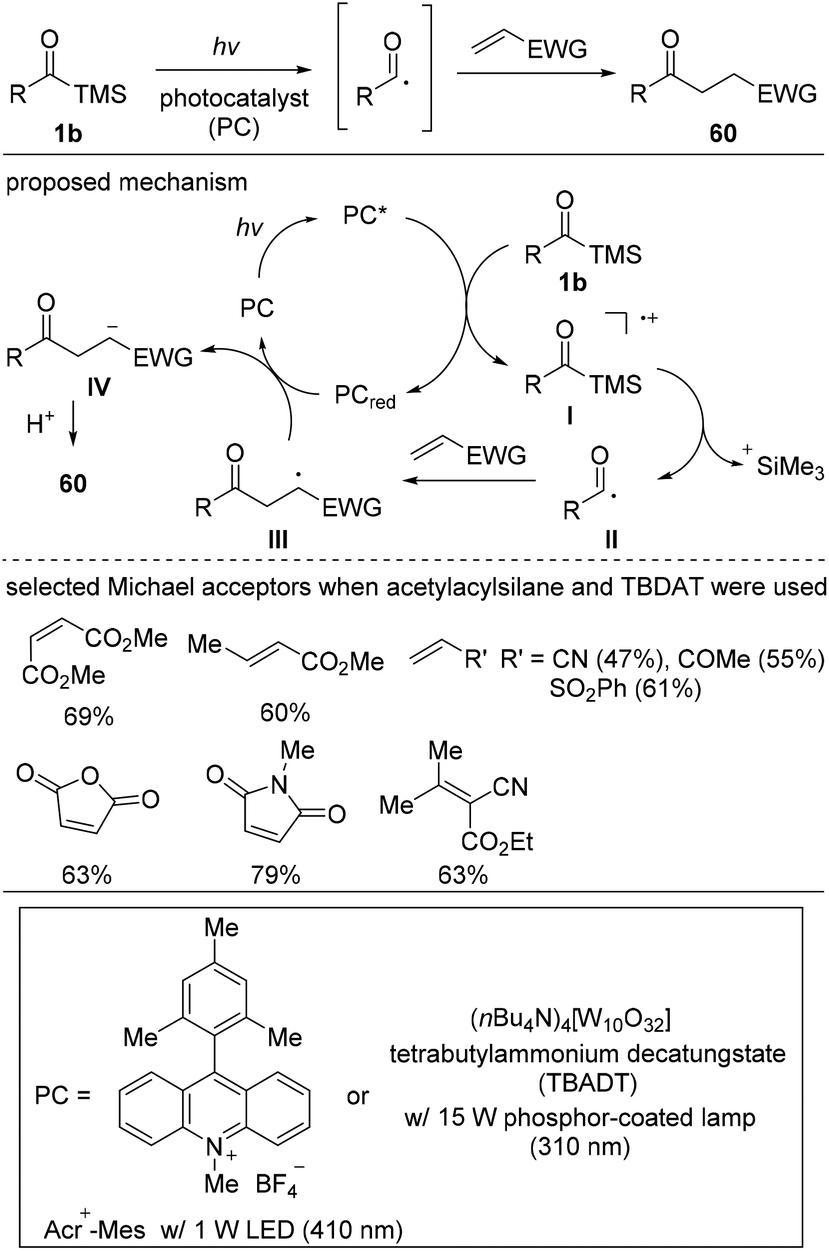 | ||
| Scheme 26 Acyl radical formation from acylsilanes and subsequent addition to Michael acceptors. | ||
4. Conclusions
Since the first synthesis of acylsilanes, a variety of methods entailing their utilization have been developed in various fields of chemistry. More recently, the activation of acylsilanes by photoirradiation to generate a reactive carbene moiety for further application has been reported by numerous research groups. This review is focused on recent advances in photo-induced organic transformations of various acylsilane derivatives. Depending on the reaction type of siloxycarbene intermediates, the content was mainly classified as carbene addition, carbene X–Y bond insertion, and cyclization employing various light sources, blue LEDs and Hg lamps. In most cases, the reactions did not require increased temperatures, and the desired products were obtained under photoirradiation conditions without the use of transition-metal catalysts.Siloxycarbenes derived from the corresponding acylsilanes by photoirradiation were nucleophilic; therefore, they readily reacted with carbonyl derivatives, including aldehydes, ketones, and carbon dioxide. Moreover, siloxycarbenes effected X–Y bond (in oximes, azides, indoles, and organoboron reagents) insertions to afford derivatives with newly formed carbon–carbon/heteroatom bonds. Recently, inter- and intramolecular ring-forming reactions have been reported. In particular, reactions with unsaturated carbon–carbon bonds (alkenes and alkynes) or carbon–nitrogen bonds (imines) provided valuable 3-, 4-, 5-, or 6-membered cyclic compounds. These newly formed ring systems, including fused rings and aromatic derivatives, could be further utilized in various fields of chemistry, such as medicinal and materials chemistry (Fig. 3).
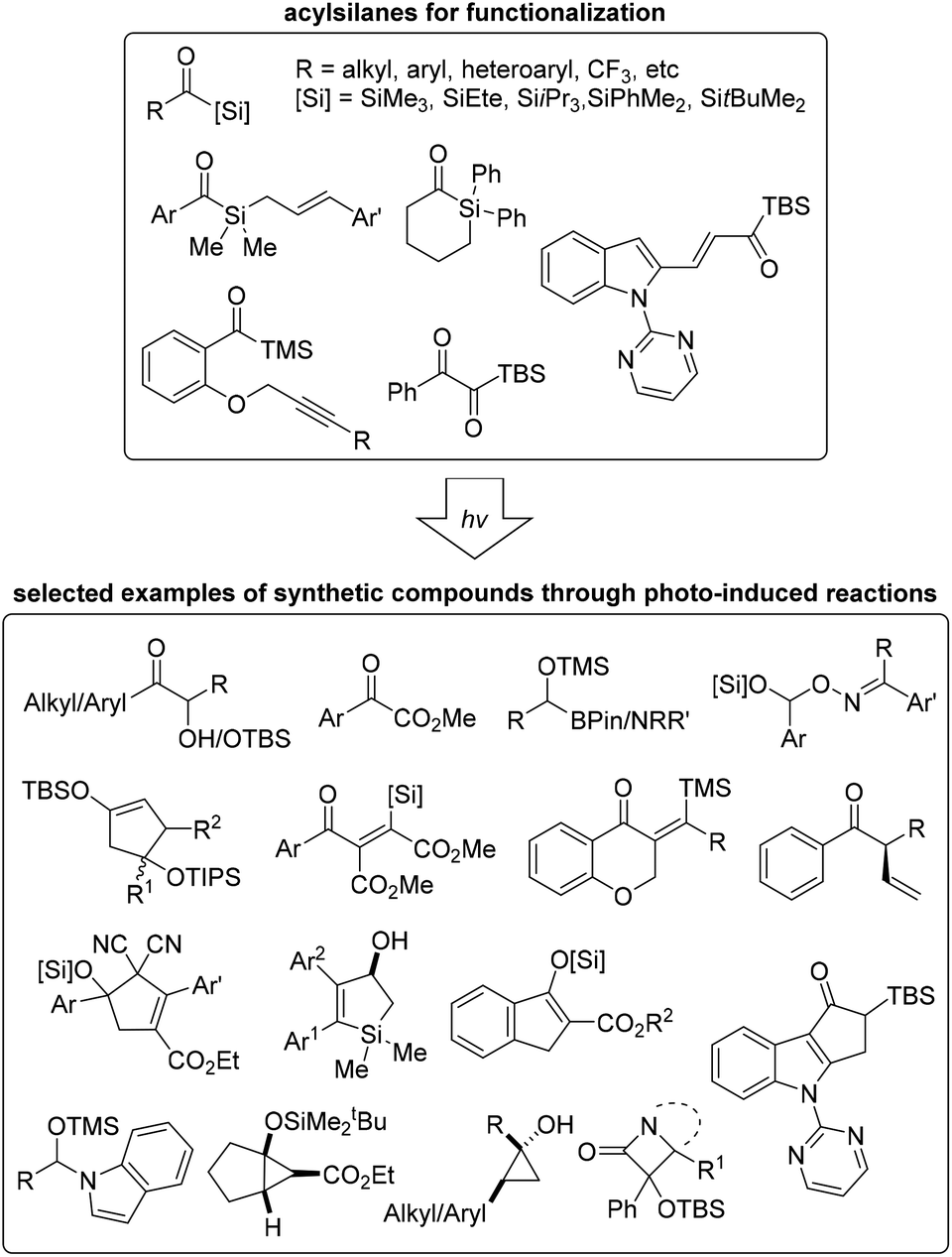 | ||
| Fig. 3 Various acylsilanes utilized in photo-induced organic reactions and synthesized building blocks. | ||
The photo-induced reactions with acylsilanes are extraordinarily simple and atom-economical compared with conventional synthetic methods, due to high conversion and clickable operation with less by-products. It is believed to provide complementarity to conventional approaches for the synthesis of the structurally novel compounds in Fig. 3. In particular, the products such as oxime-containing silyl acetal derivatives 31, cyclopentenols 37, cyclopropanols 38, 40 and 41 and β-lactams 47 were difficult to access because of limited synthetic methods. The present methods for the synthesis of unsaturated carbonyls with β-silyl groups such as 34 and 42 often require expensive catalytic systems using transition metal complexes.49 Moreover, synthetic methods for fully substituted alkenic silacycles 49 is scarce.50 The general methods for α-hydroxyketone derivatives requires external oxidants which potentially limits the reaction scope.51
Although considerable effort has been devoted to the synthetic applications of acylsilanes, there is still room for further development. During our review of the topic, new photo-induced chemical reactions of acylsilanes and synthetic methods have been reported by several research groups. We anticipate that a variety of synthetic strategies will be further investigated, and that an increasing number of valuable organic compounds will be prepared using acylsilane derivatives.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. P. Hong acknowledges financial support from the National Research Foundation of Korea (NRF-2020R1I1A3071821). H. N. Lim acknowledges financial support from the National Research Foundation of Korea (NRF-2022R1A2C1004866). I. Shin also acknowledges financial support from the National Research Foundation of Korea (NRF-2022R1H1A2011719).References
- (a) T. Hiyama and M. Oestreich, Organosilicon Chemistry: Novel Approaches and Reactions, Wiley-VCH, Weinheim, 2019 CrossRef; (b) T. Komiyama, Y. Minami and T. Hiyama, Recent Advances in Transition-Metal-Catalyzed Synthetic Transformations of Organosilicon Reagents, ACS Catal., 2017, 7, 631–651 CrossRef; (c) R. West and T. J. Barton, Organosilicon Chemistry: Part I, J. Chem. Educ., 1980, 57, 165–169 CrossRef.
- (a) P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 4th edn, 2006 CrossRef; (b) P. J. Kocienski, Protecting Groups, Thieme, New York, 2005 Search PubMed.
- (a) Y. Nakao and T. Hiyama, Silicon-Based Cross-Coupling Reaction: an Environmentally benign Version, Chem. Soc. Rev., 2011, 40, 4893–4901 RSC; (b) F. Foubelo, C. Najera and M. Yus, The Hiyama Cross-Coupling Reaction: New Discoveries, Chem. Rec., 2016, 16, 2521–2533 CrossRef PubMed; (c) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Palladium-Catalysed Cross-Coupling of Organosilicon Reagents, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC.
- (a) A. K. Franz and S. O. Wilson, Organosilicon Molecules with Medicinal Applications, J. Med. Chem., 2013, 56, 388–405 CrossRef PubMed; (b) C. G. Gadhe and S. J. Cho, Importance of Silicon Atom in the Drug Design Process, J. Chosun Nat. Sci., 2012, 5, 229–232 CrossRef; (c) P. Englebienne, A. V. Hoonacker and C. V. Herst, The Place of the Bioisosteric Sila-Substitution in Drug Design, Drug Des. Rev., 2005, 2, 467–483 CAS.
- (a) M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, John Wiley & Sons, New York, 2000 Search PubMed; (b) R. Tacke, V. I. Handmann, R. Bertermann, C. Burschka, M. Penka and C. Seyfried, Sila-Analogues of High-Affinity, Selective σ Ligands of the Spiro[indane-1,4′-piperidine] Type: Syntheses, Structures, and Pharmacological Properties, Organometallics, 2003, 22, 916–924 CrossRef CAS; (c) S. Gately and R. West, Novel Therapeutics with Enhanced Biological Activity Generated by the Strategic Introduction of Silicon Isosteres into Known Drug Scaffolds, Drug Dev. Res., 2007, 68, 156–163 CrossRef CAS; (d) G. A. Showell and J. S. Mills, Chemistry Challenges in Lead Optimization: Silicon Isosteres in Drug Discovery, Drug Discovery Today, 2003, 8, 551–556 CrossRef CAS.
- (a) F. Kleemiss, A. Justies, D. Duvinage, P. Watermann, E. Ehrke, K. Sugimoto, M. Fugel, L. A. Malaspina, A. Dittmer, T. Kleemiss, P. Puylaert, N. R. King, A. Staubitz, T. M. Tzschentke, R. Dringen, S. Grabowsky, J. Beckmann and T. Sila-Ibuprofen, J. Med. Chem., 2020, 63, 12614–12622 CrossRef CAS; (b) R. Ramesh and D. S. Reddy, Quest for Novel Chemical Entities through Incorporation of Silicon in Drug Scaffolds, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS PubMed; (c) M. J. Barnes, R. Conroy, D. J. Miller, J. S. Mills, J. G. Montana, P. K. Pooni, G. A. Showell, L. M. Walsh and J. B. H. Warneck, Trimethylsilylpyrazoles as Novel Inhibitors of p38 MAP Kinase: A New Use of Silicon Bioisosteres in Medicinal Chemistry, Bioorg. Med. Chem. Lett., 2007, 17, 354–357 CrossRef CAS PubMed; (d) R. Tacke, T. Heinrich, R. Bertermann, C. Burschka, A. Hamacher and M. U. Kassack, Sila-haloperidol: A Silicon Analogue of the Dopamine (D2) Receptor Antagonist Haloperidol, Organometallics, 2004, 23, 4468–4477 CrossRef CAS; (e) J. O. Daiss, C. Burschka, J. S. Mills, J. G. Montana, G. A. Showell, J. B. H. Warneck and R. Tacke, Sila-venlafaxine, a Sila-Analogue of the Serotonin/Noradrenaline Reuptake Inhibitor Venlafaxine: Synthesis, Crystal Structure Analysis, and Pharmacological Characterization, Organometallics, 2006, 25, 1188–1198 CrossRef CAS; (f) R. Tacke and H. Zilch, Sila-Substitution – a Useful Strategy for Drug Design?, Endeavour, 1986, 10, 191–197 CrossRef CAS.
- (a) N. Healy, U. Gibson and A. C. Peacock, A Review of Materials Engineering in Silicon-Based Optical Fibres, Semicond. Sci. Technol., 2018, 33, 023001 CrossRef; (b) G. Chandra, Organosilicon Materials, Springer Berlin, Heidelberg, 1997 CrossRef; (c) N. Auner and J. Weis, Organosilicon Chemistry V: From Molecules to Materials, John Wiley & Sons, New York, 2008 Search PubMed.
- (a) M. Jung, K. H. Lee, J. Y. Lee and T. Kim, A Bipolar Host Based High Triplet Energy Electroplex for an Over 10000 h Lifetime in Pure Blue Phosphorescent Organic Light-Emitting Diodes, Mater. Horiz., 2020, 7, 559–565 RSC; (b) J. Sun, H. Ahn, S. Kang, S.-B. Ko, D. Song, H. A. Um, S. Kim, Y. Lee, P. Jeon, S.-H. Hwang, Y. You, C. Chu and S. Kim, Exceptionally Stable Blue Phosphorescent Organic Light-Emitting Diodes, Nat. Photonics, 2022, 16, 212–218 CrossRef.
- (a) A. G. Brook, M. A. Quigley, G. J. D. Peddle, N. V. Schwartz and C. M. Warner, The Spectral and Chemical Properties of α-Silyl Ketones, J. Am. Chem. Soc., 1960, 82, 5102–5106 CrossRef; (b) A. G. Brook and G. J. D. Peddle, Dibenzoylsilanes and Substituted Benzoylsilanes, Can. J. Chem., 1963, 41, 2351–2356 CrossRef; (c) R. West, The Effect of Metalloid Substitution on the Electronic Spectra of Simple Chromophores, J. Organomet. Chem., 1965, 3, 314–320 CrossRef; (d) K. Yates and F. Agolini, The Importance of Inductive Effects in α-Silyl and α-Germyl Ketones, Can. J. Chem., 1966, 44, 2229–2231 CrossRef; (e) A. Yanagisawa, S. Habaue and H. Yamamoto, Regioselective Allylation and Propargylation using Acylsilanes: Facile Synthesis of PGE3 and F3α Methyl Ester, Tetrahedron, 1992, 48, 1969–1980 CrossRef CAS; (f) N. C. Baird and M. J. S. Dewar, Ground States of Sigma-Bonded Molecules. VI. Benzene and its Isomers. Inductive Effects in Benzene, J. Am. Chem. Soc., 1969, 91, 352–355 CrossRef.
- F. Bernardi, L. Lunazzi, A. Ricci, G. Seconi and G. Tonachini, A C-13 NMR and Theoretical Study of the Electronic Structure of Acylsilanes, Tetrahedron, 1986, 42, 3607–3610 CrossRef.
- P. C. Chieh and J. Trotter, The Structure of Acetyltriphenylsilane, J. Chem. Soc. A, 1969, 1778–1783 RSC.
- (a) D. L. Priebbenow, Insights into the Stability of Siloxy Carbene Intermediates and Their Corresponding Oxocarbenium Ions, J. Org. Chem., 2019, 84, 11813–11822 CrossRef; (b) W. Kirmse, M. Guth and S. Steenken, Production of α-Siloxycarbenium Ions by Protonation of Photochemically Generated α-Siloxycarbenes. Formation Mechanism and Reactivities with Nucleophiles, J. Am. Chem. Soc., 1996, 118, 10838–10849 CrossRef; (c) A. Holownia, C. N. Apte and A. K. Yudin, Acyl Metalloids: Conformity and Deviation from Carbonyl Reactivity, Chem. Sci., 2021, 12, 5346–5360 RSC.
- A. G. Brook, Triphenylsilyl Phenyl Ketone, J. Am. Chem. Soc., 1957, 79, 4373–4375 CrossRef CAS.
- (a) M. Trommer and W. Sander, Photochemistry and Photooxidation of Acylsilanes, Organometallics, 1996, 15, 189–193 CrossRef CAS; (b) P. C. B. Page, S. S. Klair and S. Rosenthal, Synthesis and Chemistry of Acyl Silanes, Chem. Soc. Rev., 1990, 19, 147–195 RSC; (c) J.-H. Ye, L. Quach, T. Paulisch and F. Glorius, Visible-Light-Induced, Metal-Free Carbene Insertion into B-H Bonds between Acylsilanes and Pinacolborane, J. Am. Chem. Soc., 2019, 141, 16227–16231 CrossRef CAS PubMed.
- (a) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Stable Carbenes, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed; (b) K. Hirai, T. Itoh and H. Tomioka, Persistent Triplet Carbenes, Chem. Rev., 2009, 109, 3275–3332 CrossRef CAS PubMed; (c) D. Zhu, L. Chen, H. Fan, Q. Yao and S. Zhu, Recent Progress on Donor and Donor–Donor Carbenes, Chem. Soc. Rev., 2020, 49, 908–950 RSC.
- (a) A. G. Brook, R. Pearce and J. B. Pierce, Nucleophilic Attack of Siloxycarbenes on Carbonyl Groups. The Formation of Oxiranes, Can. J. Chem., 1971, 49, 1622–1628 CrossRef CAS; (b) K. Ishida, H. Yamazaki, C. Hagiwara, M. Abe and H. Kusama, Efficient Generation and Synthetic Applications of Alkyl-Substituted Siloxycarbenes: Suppression of Norrish-Type Fragmentations of Alkanoylsilanes by Triplet Energy Transfer, Chem. – Eur. J., 2020, 26, 1249–1253 CrossRef CAS PubMed; (c) K. Ishida, F. Tobita and H. Kusama, Lewis Acid-Assisted Photoinduced Intermolecular Coupling between Acylsilanes and Aldehydes: A Formal Cross Benzoin-Type Condensation, Chem. – Eur. J., 2018, 24, 543–546 CrossRef CAS; (d) L. Ma, Y. Yu, L. Xin, L. Zhu, J. Xia, P. Ou and X. Huang, Visible Light Enabled Formal Cross Silyl Benzoin Reaction as an Access to α-Hydroxyketones, Adv. Synth. Catal., 2021, 363, 2573–2577 CrossRef CAS.
- (a) A. G. Brook and J. M. Duff, The Photochemistry of Silyl Ketones in Alcohol, J. Am. Chem. Soc., 1967, 89, 454–455 CrossRef CAS; (b) R. A. Bourque, P. D. Davis and J. C. Dalton, Mechanistic Photochemistry of Acylsilanes. 1. Reaction with Alcohols, J. Am. Chem. Soc., 1981, 103, 697–699 CrossRef CAS; (c) J. M. Duff and A. G. Brook, Photoisomerization of Acylsilanes to Siloxycarbenes, and Their Reactions with Polar Reagents, Can. J. Chem., 1973, 51, 2869–2883 CrossRef CAS; (d) C. Shih and J. S. Swenton, Thermal and Photochemical Rearrangement of (o-Tolylcarbonyl)trimethylsilane. A. 1,5-Shift of a Trimethylsilyl Group from Oxygen to Carbon, J. Org. Chem., 1982, 47, 2668–2670 CrossRef CAS; (e) Z. Shen and V. M. Dong, Benzofurans Prepared by C–H Bond Functionalization with Acylsilanes, Angew. Chem., Int. Ed., 2009, 48, 784–786 CrossRef CAS; (f) H. Watanabe, T. Kogure and Y. Nagai, The Photochemical Reaction of Benzoyltrimethylsilane with Organosilicon Hydrides, J. Organomet. Chem., 1972, 43, 285–291 CrossRef CAS; (g) H. Watanabe, N. Ohsawa, M. Sawai, Y. Fukusawa, H. Matsumoto and Y. Nagai, Photochemical Reaction of Benzoyltrimethylsilane with Substituted Phenylsilanes, J. Organomet. Chem., 1975, 93, 173–179 CrossRef CAS.
- (a) A. G. Brook, H. W. Kucera and R. Pearce, Photolysis of 1,1-Diphenylsilacyclohexanone in Diethyl Fumarate. The Trapping of a Siloxycarbene by an Electron-Deficient Olefin, Can. J. Chem., 1971, 49, 1618–1621 CrossRef CAS; (b) J. C. Dalton and R. A. Bourque, Mechanistic Photochemistry of Acylsilanes. 2. Reaction with Electron-Poor Olefins, J. Am. Chem. Soc., 1981, 103, 699–700 CrossRef CAS; (c) H.-J. Zhang, P. Becker, H. Huang, R. Pirwerdjan, F.-F. Pan and C. Bolm, Photochemically Induced Silylacylations of Alkynes with Acylsilanes, Adv. Synth. Catal., 2012, 354, 2157–2161 CrossRef CAS; (d) P. Becker, R. Pirwerdjan and C. Bolm, Acylsilanes in Iridium-Catalyzed Directed Amidation Reactions and Formation of Heterocycles via Siloxycarbenes, Angew. Chem., Int. Ed., 2015, 54, 15493–15496 CrossRef CAS PubMed; (e) P. Becker, D. L. Priebbenow, H.-J. Zhang, R. Pirwerdjan and C. Bolm, Photochemical Intermolecular Silylacylations of Electron-Deficient Internal Alkynes, J. Org. Chem., 2014, 79, 814–817 CrossRef CAS; (f) P. Becker, D. L. Priebbenow, R. Pirwerdjan and C. Bolm, Acylsilanes in Rhodium(III)-Catalyzed Directed Aromatic C–H Alkenylations and Siloxycarbene Reactions with C–C Double Bonds, Angew. Chem., Int. Ed., 2013, 53, 269–271 CrossRef.
- (a) A. G. Brook and J. M. Duff, Photolysis of Acylsilanes in Cyclohexane and Other Solvents, Can. J. Chem., 1973, 51, 352–360 CrossRef CAS; (b) A. G. Brook, J. W. Harris and A. R. Bassindale, Acylsilanes from the Pyrolysis of Silyl Esters of α-Ketoacids, J. Organomet. Chem., 1975, 99, 379–383 CrossRef CAS; (c) A. G. Brook, J. B. Pierce and J. M. Duff, Acylsilane Photolyses: 1,1-Diphenyl-1-Silacyclohexanone-2 in Cyclohexane, Can. J. Chem., 1975, 53, 2874–2879 CrossRef CAS; (d) A. G. Brook, One Thing Leads to Another - from Silylcarbinols to Silaethylenes, J. Organomet. Chem., 1986, 300, 21–37 CrossRef CAS; (e) A. G. Brook, J. W. Harris, J. Lennon and M. E. Sheikh, Relatively Stable Silaethylenes. Photolysis of Acylpolysilanes, J. Am. Chem. Soc., 1979, 101, 83–95 CrossRef CAS; (f) K. M. Baines and A. G. Brook, Photolysis of Acylpolysilanes Containing .alpha.-Hydrogens. Formation of Linear Head-to-Head Silene Dimers, Organometallics, 1987, 6, 692–696 CrossRef CAS; (g) A. R. Bassindale, A. G. Brook and J. Harris, Siloxycarbenes from the Thermolysis of Acylsilanes, J. Organomet. Chem., 1975, 90, C6–C8 CrossRef CAS.
- X. Wang, F. Liu, Y. Li, Z. Yan, Q. Qiang and Z.-Q. Rong, Recent Advances in the Synthesis of Acylsilanes, ChemCatChem, 2020, 12, 5022–5033 CrossRef CAS.
- H.-J. Zhang, D. L. Priebbenow and C. Bolm, Acylsilanes: Valuable Organosilicon Reagents in Organic Synthesis, Chem. Soc. Rev., 2013, 42, 8540–8571 RSC.
- D. L. Priebbenow, Silicon-Derived Singlet Nucleophilic Carbene Reagents in Organic Synthesis, Adv. Synth. Catal., 2020, 362, 1927–1946 CrossRef CAS.
- F. Zhengning, Y. Yaping, C. Shenhao and X. Chanjuan, Visible-Light-Induced Catalyst-Free Carboxylation of Acylsilanes with Carbon Dioxide, Org. Lett., 2021, 23, 2303–2307 CrossRef.
- (a) R. Wheland and P. D. Bartlett, .alpha.-Lactones from Diphenylketene and Di-tert-Butylketene, J. Am. Chem. Soc., 1970, 92, 6057–6058 CrossRef CAS; (b) W. W. Sander, Reaction of Diphenylmethylene with Carbon Dioxide. Matrix Isolation of Diphenyloxiranone, J. Org. Chem., 1989, 54, 4265–4267 CrossRef CAS.
- D. L. Priebbenow, R. L. Pilkington, K. N. Hearn and A. Polyzos, Fluorinated Ketones as Trapping Reagents for Visible-Light-Induced Singlet Nucleophilic Carbenes, Org. Lett., 2021, 23, 2783–2789 CrossRef.
- K. Ito, H. Tamashima, N. Iwasawa and H. Kusama, Photochemically Promoted Transition Metal-Free Cross-Coupling of Acylsilanes with Organoboronic Esters, J. Am. Chem. Soc., 2011, 133, 3716–3719 CrossRef CAS.
- C. Stuckhardt, M. Wissing and A. Studer, Photo Click Reaction of Acylsilanes with Indoles, Angew. Chem., Int. Ed., 2021, 60, 18605–18611 CrossRef CAS PubMed.
- J. Reimler and A. Studer, Visible-Light Mediated Modification in Oligopeptides Employing Acylsilanes, Chem. – Eur. J., 2021, 27, 15392–15395 CrossRef CAS PubMed.
- Q. Li, B.-G. Cai, L. Li and J. Xuan, Oxime Ether Synthesis through O–H Functionalization of Oximes with Diazo Esters under Blue LED Irradiation, Org. Lett., 2021, 23, 6951–6955 CrossRef CAS PubMed.
- B. G. Cai, Q. Li, L. Li and J. Xuan, Carbon-Oxygen Bond Formation via Visible-Light-Induced O–H Insertion between Acylsilanes and Oximes, Green Synth. Catal., 2022, 3, 194–197 CrossRef.
- K. Yamaguchi, T. Shimizu, A. Miura, K. Ishida and H. Kusama, Photoinduced Intramolecular Cyclization of Acylsilanes Bearing a Boronate Moiety: Construction of a Highly Strained trans-Fused Bicyclo[3.3.0]octane Skeleton, Org. Lett., 2022, 24, 5807–5811 CrossRef CAS PubMed.
- G. Zhou and X. Shen, Synthesis of Cyclopropenols Enabled by Visible-Light-Induced Organocatalyzed [2+1] Cyclization, Angew. Chem., Int. Ed., 2022, 61, e202115334 CAS.
- Y. Zhang, G. Zhou, X. Gong, Z. Guo, X. Qi and X. Shen, Diastereoselective Transfer of Tri(di)fluoroacetylsilanes-Derived Carbenes to Alkenes, Angew. Chem., Int. Ed., 2022, 61, e202202175 CAS.
- S. Sakurai, T. Inagaki, T. Kodama, M. Yamanaka and M. Tobisu, Palladium-Catalyzed Siloxycyclopropanation of Alkenes Using Acylsilanes, J. Am. Chem. Soc., 2022, 144, 1099–1105 CrossRef CAS.
- A. Bunyamin, C. Hua, A. Polyzos and D. L. Priebbenow, Intramolecular Photochemical [2+1]-Cycloadditions of Nucleophilic Siloxy Carbenes, Chem. Sci., 2022, 13, 3273–3280 RSC.
- J.-H. Ye, P. Bellotti, T. O. Paulisch, C. G. Daniliuc and F. Glorius, Visible-Light-Induced Cycloaddition of a-Ketoacylsilanes with Imines: Facile Access to β-Lactams, Angew. Chem., Int. Ed., 2021, 60, 13671–13676 CrossRef CAS.
- J. R. Vale, A. Valkonen, C. A. M. Afonso and N. R. Candeias, Synthesis of Silacyclopent-2-en-4-ols via Intramolecular [2+2] Photocycloaddition of Benzoyl(allyl)silanes, Org. Chem. Front., 2019, 6, 3793–3798 RSC.
- T. Takeuchi, T. Aoyama, K. Orihara, K. Ishida and H. Kusama, Visible-Light-Induced in situ Generation of Fischer-Type Copper Carbene Complexes from Acylsilanes and Its Application to Catalytic [4+1] Cycloaddition with Siloxydienes, Org. Lett., 2021, 23, 9490–9494 CrossRef PubMed.
- J. R. Vale, R. F. Gomes, C. A. M. Afonso and N. R. Candeias, Functionalized Cyclopentenes via the Formal [4+1] Cycloaddition of Photogenerated Siloxycarbenes from Acyl Silanes, J. Org. Chem., 2022, 87, 8910–8920 CrossRef PubMed.
- Z. Zhu, W. Zhang, Y. Zhang, S. Liu and X. Shen, Visible-Light-Induced [4+1] Cyclization-Aromatization of Acylsilanes and α,β-Unsaturated Ketones, CCS Chem., 2022 DOI:10.31635/ccschem.022.202202199 , just published.
- S. Rakshit, C. Grohmann, T. Besset and F. Glorius, Rh(III)-Catalyzed Directed C–H Olefination Using an Oxidizing Directing Group: Mild, Efficient, and Versatile, J. Am. Chem. Soc., 2011, 133, 2350–2353 CrossRef.
- P. Lu, C. Feng and T.-P. Loh, Divergent Functionalization of Indoles with Acryloyl Silanes via Rhodium-Catalyzed C–H Activation, Org. Lett., 2015, 17, 3210–3213 CrossRef PubMed.
- Y. Zhang, J.-J. Chen and H.-M. Huang, Radical Brook Rearrangements: Concept and Recent Developments, Angew. Chem., Int. Ed., 2022, 61, e202205671 Search PubMed.
- Y. Ueda, Y. Masuda, T. Iwai, K. Imaeda, H. Takeuchi, K. Ueno, M. Gao, J. Hasegawa and M. Sawamura, Photoinduced Copper-Catalyzed Asymmetric Acylation of Allylic Phosphates with Acylsilanes, J. Am. Chem. Soc., 2022, 144, 2218–2224 CrossRef.
- L. Capaldo, R. Riccardi, D. Ravelli and M. Fagnoni, Acyl Radicals from Acylsilanes: Photoredox-Catalyzed Synthesis of Unsymmetrical Ketones, ACS Catal., 2018, 8, 304–309 CrossRef.
- J. Liu, Q. Liu, H. Yi, C. Qin, R. Bai, X. Qi, Y. Lan and A. Lei, Visible-Light-Mediated Decarboxylation/Oxidative Amidation of α-Keto Acids with Amines under Mild Reaction Conditions Using O2, Angew. Chem., Int. Ed., 2014, 53, 502–506 CrossRef.
- G. Bergonzini, C. Cassani and C.-J. Wallentin, Acyl Radicals from Aromatic Carboxylic Acids by Means of Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2015, 54, 14066–14069 CrossRef.
- C.-G. Li, G.-Q. Xu and P.-F. Xu, Synthesis of Fused Pyran Derivatives via Visible-Light-Induced Cascade Cyclization of 1,7-Enynes with Acyl Chlorides, Org. Lett., 2017, 19, 512–515 CrossRef PubMed.
- (a) I. Ojima, P. Ingallina, R. J. Donovan and N. Clos, Silylformylation of 1-Hexyne Catalyzed by Rhodium-Cobalt Mixed-Metal Carbonyl Clusters, Organometallics, 1991, 10, 38–41 CrossRef; (b) T. Fujihara, Y. Tani, K. Semba, J. Terao and Y. Tsuji, Copper-Catalyzed Silacarboxylation of Internal Alkynes by Employing Carbon Dioxide and Silylboranes, Angew. Chem., Int. Ed., 2012, 51, 11487–11490 CrossRef; (c) B. Chen and X.-F. Wu, Palladium-Catalyzed Carbonylative Synthesis of Benzosilinones from (2-Iodophenyl)hydrosilanes and Terminal Alkynes, Adv. Synth. Catal., 2019, 361, 3441–3445 CrossRef.
- M. Onoe, K. Baba, Y. Kim, Y. Kita, M. Tobisu and N. Chatani, Rhodium-Catalyzed Carbon-Silicon Bond Activation for Synthesis of Benzosilole Derivatives, J. Am. Chem. Soc., 2012, 134, 19477–19488 CrossRef PubMed.
- J. Streuff, An Update on Catalytic Strategies for the Synthesis of α-Hydroxyketones, Synlett, 2013, 24, 276–280 CrossRef.
| This journal is © the Partner Organisations 2023 |