
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadiolabelling small and biomolecules for tracking and monitoring
Martin R. Edelmann
 ab
ab
aDepartment of Pharmacy and Pharmacology, University of Bath, Bath BA2 7AY, UK. E-mail: martin.edelmann@roche.com
bRoche Pharma Research and Early Development, Roche Innovation Center Basel, Therapeutic Modalities, Small Molecule Research, Isotope Synthesis, F. Hoffmann-La Roche Ltd, CH-4070 Basel, Switzerland
First published on 11th November 2022
Abstract
Radiolabelling small molecules with beta-emitters has been intensively explored in the last decades and novel concepts for the introduction of radionuclides continue to be reported regularly. New catalysts that induce carbon/hydrogen activation are able to incorporate isotopes such as deuterium or tritium into small molecules. However, these established labelling approaches have limited applicability for nucleic acid-based drugs, therapeutic antibodies, or peptides, which are typical of the molecules now being investigated as novel therapeutic modalities. These target molecules are usually larger (significantly >1 kDa), mostly multiply charged, and often poorly soluble in organic solvents. However, in preclinical research they often require radiolabelling in order to track and monitor drug candidates in metabolism, biotransformation, or pharmacokinetic studies. Currently, the most established approach to introduce a tritium atom into an oligonucleotide is based on a multistep synthesis, which leads to a low specific activity with a high level of waste and high costs. The most common way of tritiating peptides is using appropriate precursors. The conjugation of a radiolabelled prosthetic compound to a functional group within a protein sequence is a commonly applied way to introduce a radionuclide or a fluorescent tag into large molecules. This review highlights the state-of-the-art in different radiolabelling approaches for oligonucleotides, peptides, and proteins, as well as a critical assessment of the impact of the label on the properties of the modified molecules. Furthermore, applications of radiolabelled antibodies in biodistribution studies of immune complexes and imaging of brain targets are reported.
 Martin R. Edelmann | Martin studied Biological Chemistry at the University of Applied Science Mannheim, Germany and completed his diploma thesis at Novartis in Switzerland in 2006. After 4 years of research in medicinal chemistry at Novartis, Martin moved to Roche's isotope synthesis laboratory. His responsibilities include radiolabelling small molecules, oligonucleotides, and proteins with carbon-14 and tritium. At the beginning of 2020, Martin started his PhD studies at the University of Bath, UK in the Department of Pharmacy and Pharmacology under supervision of Dr Ian S. Blagbrough and Prof. Stephen M. Husbands as well as Dr Michael B. Otteneder and Dr Filippo Sladojevich (Roche). |
Introduction
When people hear of radioactivity, they immediately think of scientists like Marie Curie, Henri Becquerel, or Wilhelm Conrad Röntgen. Marie Curie's topic for her doctoral thesis1 was inspired by new discoveries made by Becquerel and Röntgen. In December 1895, the German physicist Röntgen detected a type of rays that could penetrate solid wood or flesh and provide photographs of the bones of living people.2 These mysterious rays were called X-rays by Röntgen, where X stands for unknown. Early in 1896, just a few months after Röntgen's detection, the French physicist Henri Becquerel had reported to the French Academy of Sciences that uranium compounds, even when kept in the dark, emitted rays that fogged a photographic plate.3 This was the beginning of radiographic imaging as we know it today from hospitals.4 These X-rays, together with gamma rays and electron beams, are also used in radiation therapy to treat cancer or kill malignant cells.5 This review is about radiolabelling molecules that are used for tracking and monitoring to study their biological and physicochemical behaviour.Small molecules have been the dominant group of drugs since the early days of pharmaceutical development, but this is not a matter of course for the future. New therapeutic modalities are now filling the pipeline of the pharmaceutical industry. We need to expand the target space for targeting “undruggable” targets that are limited to reach with small molecules.6 New innovative biological modalities have emerged in the first two decades of the 21st century. Various formats of antibodies, bispecifics, antibody conjugates, peptides, modified RNA molecules such as antisense oligonucleotides (Fig. 1) are gaining popularity due to their potential to enter undruggable space.
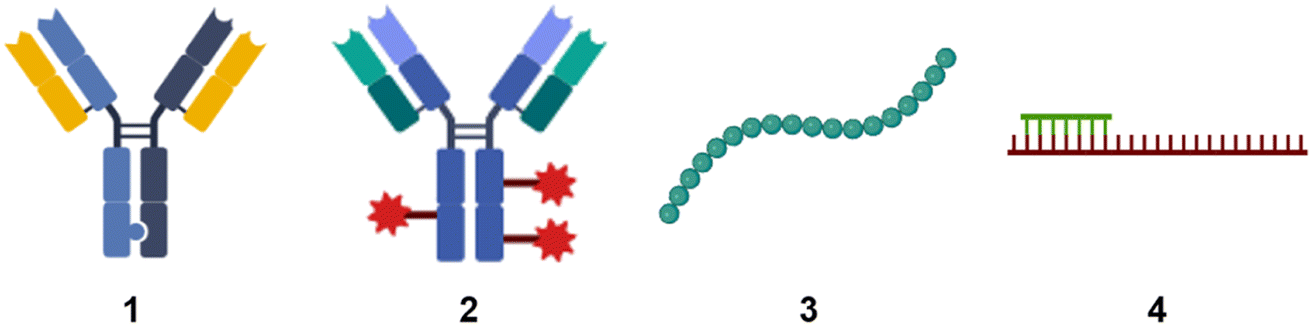 | ||
| Fig. 1 Schematic sketches of (1) bispecific antibody based on knobs-into-holes-technology;7 (2) antibody conjugates. The red star symbolises payloads such as a small molecule, peptide, or oligonucleotide; (3) peptide; (4) antisense oligonucleotide in green with mRNA in red. | ||
A major challenge with the novel modalities is the prediction of drug targeting, which together with solubility contribute to the ability of drugs and candidates to have overall exposure in the systemic circulation or tissue targeting like brain penetration.8 It is important to understand the fate of the potential drug candidate in humans and the relevance of the animal species used for preclinical toxicity and pharmacodynamic studies. In preclinical research, biotransformation, metabolic identification, or binding studies are required in order to obtain information about the pharmacokinetic (PK) behaviour or the efficacy of the drug candidates. Although state-of-the-art high-resolution LC/MS techniques are commonly used for these studies, radiolabelled molecules are often required for quantification of metabolites, biodistribution, or to assess retention and excretion of all drug-related components. The classical approaches for radiolabelling small molecule drug candidates with long-lived isotopes carbon-14 (14C; t1/2: 5730 year; molar activity: 62.4 mCi per mmol) and tritium (3H; t1/2: 12.3 year; molar activity: 26.6 Ci per mmol) are limited for novel and complex modalities. 14C-labelling is preferred for small molecule studies, but the molar activity is too low to be used for a corresponding study (in particular for imaging studies) with larger molecules (MW > 5000 Da). In contrast, due to its more than 400-fold higher molar activity, tritium is better suited for radioactive labelling of large molecules in preclinical research.
In addition to the long-lived isotopes, which are mainly used in the preclinical phase to characterise drug candidates, short-lived isotopes such as 11C (t1/2: 20 min) and 18F (t1/2: 110 min), are widley used in diagnostic imaging studies. In this respect, functional imaging such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT) has become essential for clinical decision-making in various areas of medicine.9–11 Due to their short half-life, these molecules are not used to induce a therapeutic effect, but they bind to their target with fast kinetics for imaging or they can be tracked live in the body. While 11C and 18F are mainly important in research with small molecule, metallic radionuclides are found in novel modalities.
Of the commonly available radionuclides for PET imaging, 68Ga (t1/2: 68 min) is best suited for radiolabelling peptides, as the physical half-life is similar to the biological half-life of peptides, with a significant prevalence of 18F and almost no 11C work reported in the literature.12 An issue for these radionuclides is that they require the incorporation of synthetic functional groups into the native peptide. The two most common strategies for introducing the radionuclide into the peptide of choice use either bifunctional chelating agents (BFCs, Fig. 2) or prosthetic groups. Prosthetic groups are small, reactive entities that are first radiolabelled before being coupled to the peptide. BFCs are metal-binding chelators that are conjugated to the peptide of choice and subsequently complex a radioactive metal.
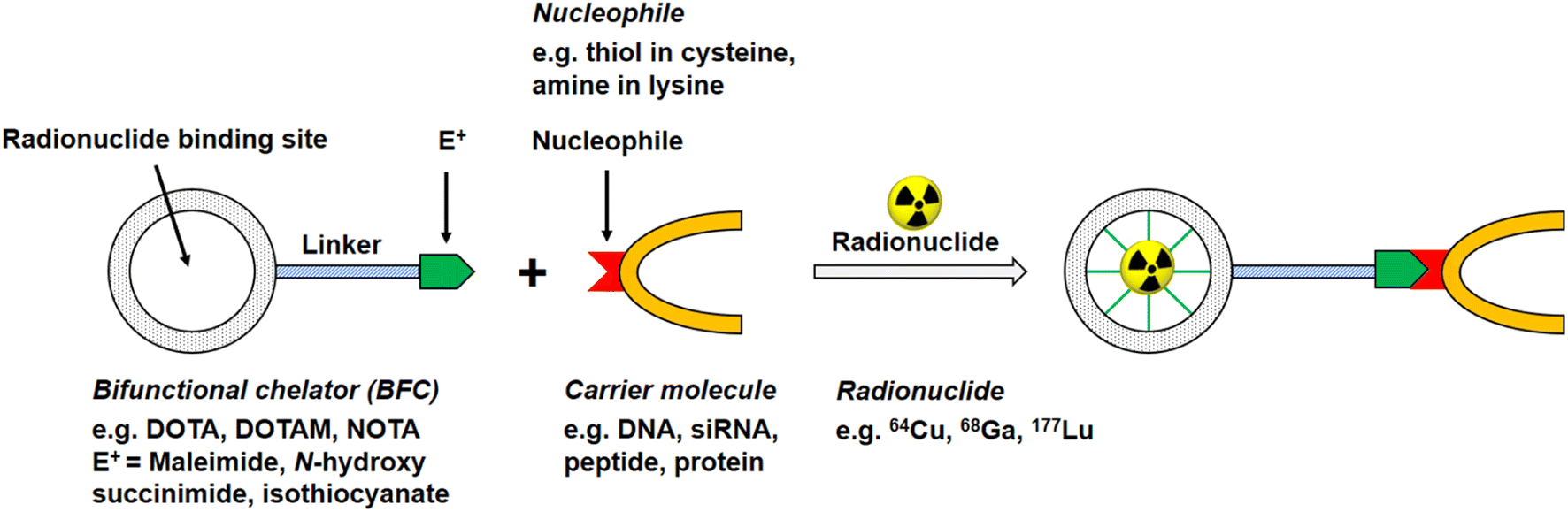 | ||
| Fig. 2 Schematic reaction illustration of a radionuclide complexing bifunctional chelator linked to a carrier molecule, e.g. oligonucleotide, peptide, antibody. | ||
With regard to the radiolabelling of monoclonal antibodies (mAbs), 89Zr (t1/2: 78 h) and 64Cu (t1/2: 13 h) for PET or 111In (t1/2: 168 h) for SPECT are the most frequently mentioned radionuclides in the literature for diagnostic imaging of mAbs.13,14 Similar to peptide radiolabelling, synthetic modifications using chelating agents or by introduction of prosthetic groups are required.
Currently, 99mTc (t1/2: 6 h) is used in most diagnostic SPECT-based imaging of nucleic acid-based medicines. In 2014, a direct labelling method with 99mTc was reported for the first time, in which the radionuclide was attached to the oligonucleotide without a chelator.15 Another method describes the indirect labelling of oligomers with 99mTc, a labelling strategy similar to that for antibodies and peptides, by using BFCs conjugated via a linker to the oligonucleotide that binds the radiometal.16
Regardless of whether radiolabelling is achieved by the introduction of a BFC or by conjugation with a radiolabelled prosthetic group, a modification of the molecular structure can lead to changes in molecular properties or behaviour. To date, it has not been thoroughly investigated whether the change has an impact on the physicochemical or biological properties of a target protein, so tracking and monitoring assays produce data that correspond to the unlabelled protein.
Radionuclide therapy
Another group of radiolabelled molecules are those that have a therapeutic effect. Of particular interest are radiotherapies using radionuclide-labelled peptides or antibodies, which make use of the therapeutic effect of ionising radiation in cancer therapy. Radioimmunotherapies (RIT) exploit an antibody as a carrier for radioactive isotopes to deliver a high radiation dose to a tumour.17–19 The choice of the optimal radionuclide for RIT depends both on its intended use and on practical considerations. From the plethora of existing radionuclides, the recently reported “Hopeful Eight” α-emitters20 have aroused great interest in therapeutic applications: 225Ac (t1/2: 10 day), 211At (t1/2: 7 day), 212Bi (t1/2: 1 h), 213Bi (t1/2: 46 min), 212Pb (t1/2: 11 h), 223Ra (t1/2: 11 day), 146Tb (t1/2: 4 h), and 227Th (t1/2: 19 day). Each of these radionuclides has its own physical properties that hold promise for targeted alpha-therapy. Currently, 225Ac apears to be the front-runner compared to other α-emitters. The interest in 225Ac is also due to its half-life of 10 days which allows centralised production at the country level. 213Bi was the first α-emitter to be clinically evaluated.21,22With respect to β-emitting radionuclides, 131I (t1/2: 8 day), 177Lu (t1/2: 7 day), and 90Y (t1/2: 3 day) are the most common in clinical RIT studies and represent the current standard in therapeutic beta-minus emitters.23 90Y-Ibritumomab tiuxetan was the first radioimmunotherapy drug approved by the FDA in 2002 to treat B-cell non-Hodgkin lymphoma.24 In 2018, Lutathera® was approved by the European Medicines Agency (EMA) as the first radiopharmaceutical peptide for radionuclide therapy.25 Lutathera® uses the ionising radiation of 177Lu for the treatment of gastroenteropancreatic neuroendocrine tumours.
Tritium-labelled oligonucleotides
Many disease targets are considered “undruggable” because they are located intracellularly or lack a specific binding pocket. Oligonucleotide therapeutics can precisely and efficiently modulate intracellular targets and they do not rely on the identification of a binding pocket, as is predominantly the case with small molecule-based therapeutics. The majority of oligonucleotide-based therapeutics address gene silencing,26 although other strategies are also pursued, including splicing modulation and gene activation.27 This expands the range of potential targets beyond what is typically accessible to traditional pharmaceutical modalities. As a result, an increasing number of nucleic acid-based therapeutics have been approved to treat or prevent diseases that previously could not be addressed.28–32In preclinical research, radiolabelled drug candidates can provide a reliable quantitative tool for distribution, mass balance, metabolite profiling, and identification studies. In the case of nucleic acid-based medicines, several aspects need to be considered for planning and execution of radiolabelling. Such considerations include the choice of radioisotope, the feasibility of the labelling methodology, and metabolic stability. However, to benefit from metabolite quantification by radiolabelling, the radiolabel must be confined to a specific position in the molecule. Random labelling in the oligonucleotide sequence, leading to multiple incorporation of radioisotopes, is a major disadvantage, as the molar activity of each metabolite is different depending on the number of radioisotopes remaining in its sequence, making structural analysis for quantification almost impossible.
Three isotopes (3H, 14C, 35S) for radiolabelling of oligonucleotides are mainly discussed in the literature, and the most commonly used labelling positions with their isotopes are shown in Fig. 3.
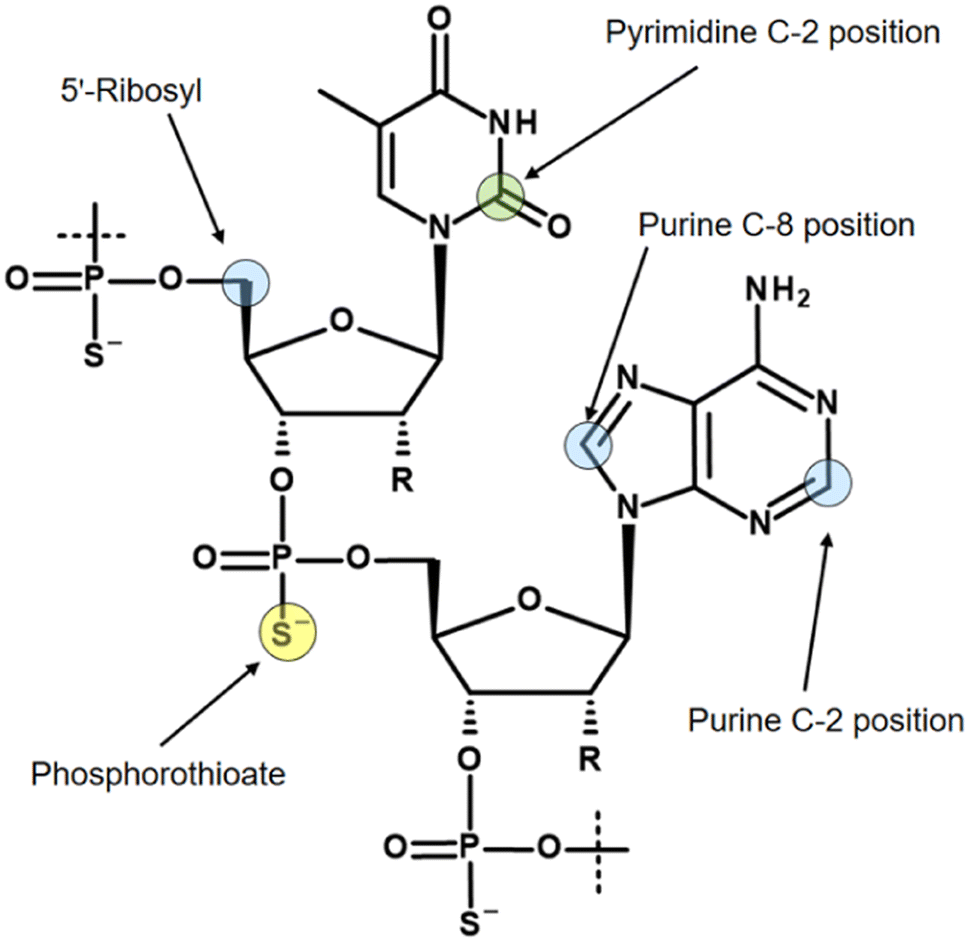 | ||
| Fig. 3 Literature reported positions for radiolabelling of oligonucleotides in example of thymidine and adenosine. Colouring reflects the corresponding radioisotopes. Blue: 3H; green: 14C; yellow: 35S. | ||
Due to the relatively long terminal elimination half-life of oligonucleotides, in some cases 30–60 days or even longer,33,34 several properties of the most commonly used radioisotopes, 3H, 14C, and 35S, need to be considered. The use of a 35S label in the phosphorothioate backbone of drug candidates requires a timely study design due to the physical half-life of 35S (87.5 days). Replication of a study with identical material is nearly impossible due to rapid isotopic decay.
As already mentioned, the molar activity of 14C is the limiting factor for imaging of oligonucleotides. If a 14C-based study requirement permits a low molar activity, for example using accelerator mass spectrometry or cavity-ring down spectroscopy for quantification,35 then the 14C incorporation usually takes place in the C-2 position of pyrimidine bases.36 However, in studies with 14C-labelled oligonucleotides, it has been observed that pyrimidine bases account for a significant portion of metabolism, ultimately leading to loss of the label in the expired air as 14CO2.37
When a tritium atom is to be introduced into an oligonucleotide, the labelling position must be carefully considered. In the literature, three positions with different labelling methods are mainly described. Probably the easiest way to introduce a tritium atom into an oligonucleotide is a hydrogen/tritium exchange at the C-8 position of purine bases by using tritiated water without any catalyst.38 Although the C-8 position of purines has long been known to pose a risk of tritium-hydrogen back-exchange by formation of tritiated water, particularly under alkaline conditions,39 oligonucleotides are still tritiated using this approach.40 Recently, a ruthenium nanoparticle catalyst was reported that allows hydrogen-tritium exchange at both the C-8 position of purines and the C-2 position.41 The C-2 position is significantly more stable to back-exchange. However, this labelling concept relies on randomised labelling, which, as mentioned earlier, makes a quantitative metabolic study almost impossible.
The most commonly used approach to tritium-labelled oligonucleotides, which consist of a phosphorothioate backbone, is based on a methodology that dates back to 1995 and is still state-of-the-art.42 The chemical approach requires a pre-synthesis of [3H]-nucleoside phosphoramidite monomer by oxidation of the 5′-primary alcohol of ribose, followed by reduction with NaBT4 to introduce a tritium atom at the 5′-carbon. Due to the instability of the [3H]-nucleoside phosphoramidite caused by radiolysis, the monomer cannot be stored for the subsequent solid-phase synthesis, but has to be freshly prepared in a complex, multi-step synthesis. In addition, high molar activity cannot be achieved because the tritium-labelled nucleoside phosphoramidite must be diluted with non-radioactive monomer to avoid strong radiolysis. These issues lead to a high expenditure of time, a high level of radioactive waste, and thus to high costs. A different approach was reported by Ledoan and co-workers.43 An amine-functionalised linker was placed terminally at the ASO-sequence for post-conjugation with [3H]succinimidyl propionate ([3H]NSP, Fig. 4).
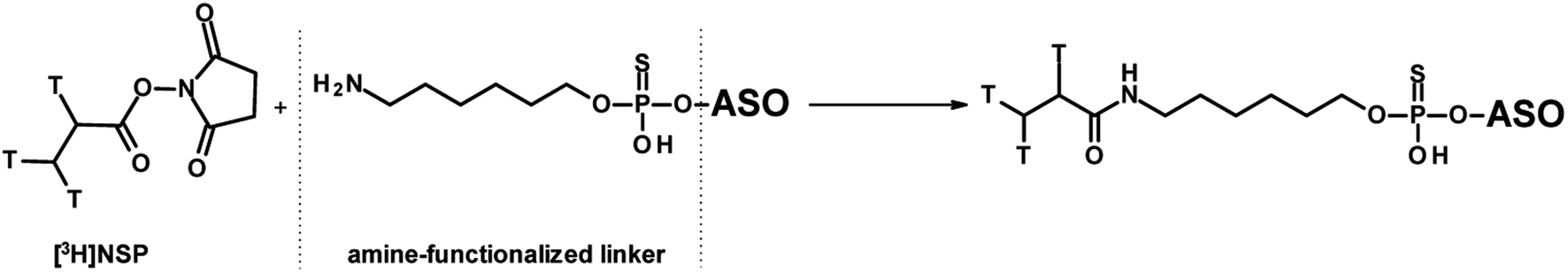 | ||
| Fig. 4 General synthetic route to ASOs containing tritium labelled propionate functionalisation on C6-amine linker. | ||
Several aspects have to be taken into account: (1) the stability of the label and (2) since the original chemical structure has been changed, the PK and pharmacodynamic behaviour should be compliant with that of the parent drug.
Synthesis of tritium-labelled maleimides for bioconjugation
An alternative to NSP and amine linkers is a 1,4-Michael-type addition of a maleimide derivative, such as N-ethylmaleimide (NEM), to a thiol linker. However, bioconjugation with the corresponding [3H]NEM is challenging. Commercially available [3H]NEM is usually supplied in a pentane solution. For conjugations in aqueous buffer, a solvent exchange to a water-miscible solvent must be carried out beforehand. Due to the high volatility of NEM, solvent evaporation is not possible. This issue can be addressed using a silica gel-based cartridge. After removing the pentane, [3H]NEM can be eluted from the cartridge with e.g. DMSO. This solvent exchange process can lead to a loss of the radioactive NEM since syntheses with tritium-labelled compounds are usually on the microgram scale. A general disadvantage of NEM is that the tritium compound can only be obtained in a complex, multi-stage synthesis in a molar activity range of 20–55 Ci per mmol. Tritium analogues with high molar activity (>75 Ci per mmol) are critical for accurate quantification in nanomolar ligand binding affinity studies as well as for imaging in vivo biodistribution by autoradiography. Thus, there is room for improvement in the development of tritiated maleimides for use in bioconjugation that exhibit low volatility and enable high molar activity.The incorporation of tritium into a maleimide derivative can be achieved by two general routes, namely by metal-catalysed hydrogen/tritium exchange (HTE) of the final compound or by the use of a precursor (Fig. 5). Metal-catalytic activation of aromatic C(sp2)–H for HTE using tritium gas requires directing groups such as ketones, sulfonic acids, carboxylic acids, or amides.44–47 The requirements for a successful HTE assume a “complex” molecular structure of the maleimide derivative. However, the modification of the tritium-labelled maleimide derivative should be minimal and the introduction of an additional catalyst-directing moiety containing functional groups that can form hydrogen bonds should be avoided in order to preserve the biological behaviour after conjugation with the oligonucleotide, and this limits the choice of a maleimide precursor. A recent method for HTE is based on photoredox-mediated hydrogen-atom transfer. Tritium can be installed in a single step using tritiated water as the source of 3H targeting aliphatic-amino C(sp3)–H bonds.48 Studies have shown that maleimides containing amines in N-alkyl chains promote intramolecular base-catalysed hydrolysis, which results in ring opening.49 Another approach to introduce tritium atoms into a molecule is based on the palladium-catalysed reduction of double/triple-bonds or halogen/tritium exchange using tritium gas. This is not possible with a maleimide derivative as the double bond of the maleimide core is also reduced and a subsequent conjugation to a thiol linker can no longer be carried out.50 The introduction of a tritium-methyl group, for example using [3H]-methyl nosylate, seems to be a successful approach as three tritium atoms are incorporated simultaneously into a maleimide derivative, resulting in a molar activity of >75 Ci per mmol. Therefore, the maleimide precursor for tritium labelling has to meet the following properties: minimal modification at the N-moiety; few (better no) additional hydrogen bonds; no basic N-alkyl group; contain a target for SN2-based methylation.
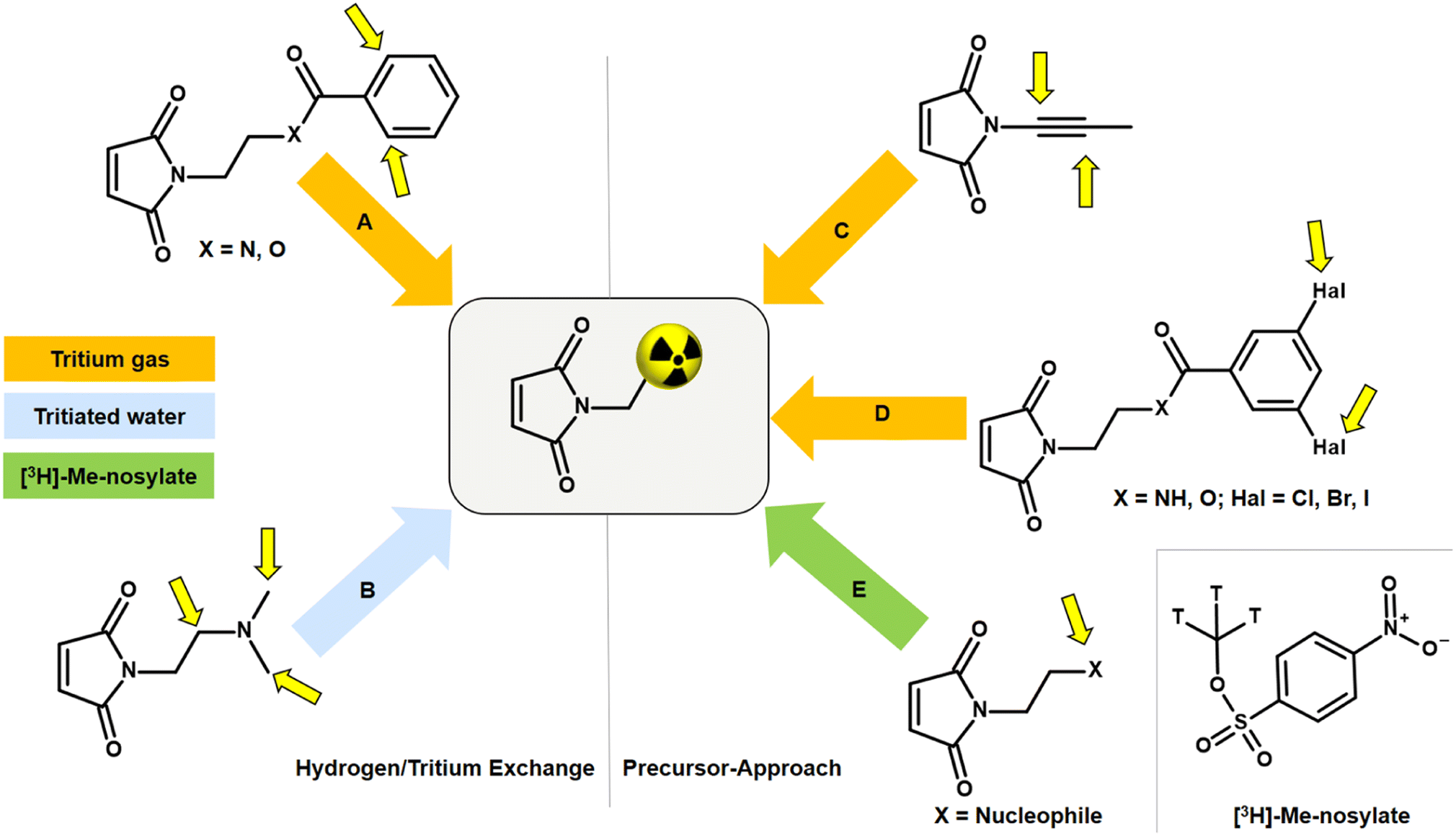 | ||
| Fig. 5 Synthesis routes to tritium-labelled maleimide derivative. (A) Metal-catalytic C–H activation followed by hydrogen/tritium exchange using tritium gas. (B) Photoredox-mediated hydrogen/tritium exchange using tritiated water. (C) Reduction of double/triple-bonds. (D) Palladium-catalysed halogen/tritium exchange. (E) Methylation on nucleophilic residue using [3H]-methyl nosylate. Yellow arrows indicate possible labelling positions. | ||
Peptide labelling
Investigation of peptide receptors requires biologically active peptides containing fluorescent51 or radioactive labels. The radioactive labelling of peptides depends on the choice of isotope. In the case of 14C labelling, 14C containing amino acids are incorporated into the sequence via peptide synthesis.52 Due to the low molar activity of 14C isotopes, this is a limitation for some studies, especially in imaging. Higher specific activities can be achieved by radioiodination. Depending on the physical and chemical properties of the different iodine isotopes, different synthetic routes are possible.53 The most commonly used iodine isotopes with their half-life and applications are: 123I (t1/2: 13 h; SPECT), 124I (t1/2: 4 day; PET), 125I (t1/2: 59 day; SPECT, in vitro bioassays), 131I (t1/2: 8 day; SPECT, radiotherapy). Radioiodination is generally performed on tyrosine (Tyr) or histidine (His) residues, in which an aromatic Tyr or His proton is replaced by the electrophilic radioiodine (I+). For this purpose, the radioiodide is generated from I− in situ with an oxidising agent.54,55Peptides can also be labelled with metallic radionuclides in combination with bifunctional chelating agents. In the example of the approved peptide radiotherapeutic Lutathera®, the same chelating agent was used to complex once with the diagnostic radionuclide 68Ga and thereafter with the therapeutic 177Lu. This has the advantage that with 68Ga a tumour-imaging biomarker is available and in a further step with 177Lu a therapeutic radiation dose can be delivered to the target tumour or metastasis.25 These molecules, which have a close connection to diagnostics and also to related therapy, are sometimes termed theranostics. This personalised therapy could become a major advantage in cancer treatment, and it is therefore to be expected that targeted radionuclide therapy will play a significant role in clinical nuclear medicine in the future.56
Tritium-labelled peptides
Different synthetic routes are available for labelling peptides with tritium (Fig. 6). A novel labelling method based on photocatalytic HTE using T2O as the tritium source was recently reported.57 This is a promising approach to tritium labelling of peptides, as it allows fast access to radioactive peptides. However, so far only low molar activities (2–3 Ci per mmol) are achievable with this approach and there is still room for improvement. Significantly, higher specific activities can be achieved by solid-phase peptide synthesis using 3H-labelled amino acids from amino acid precursors. This is reported in the literature for smaller peptide sequences of only a few amino acids.58 As an option for larger peptides, it is reported to incorporate the amino acid precursor into the sequence by solid phase synthesis. The most commonly used precursor amino acids are halogenated aromatic α-amino acids or synthetic-α-amino acids containing double or triple bonds. In the next step, the halogen is replaced by tritium by metal catalysis or the double/triple bond is reduced with tritium gas.59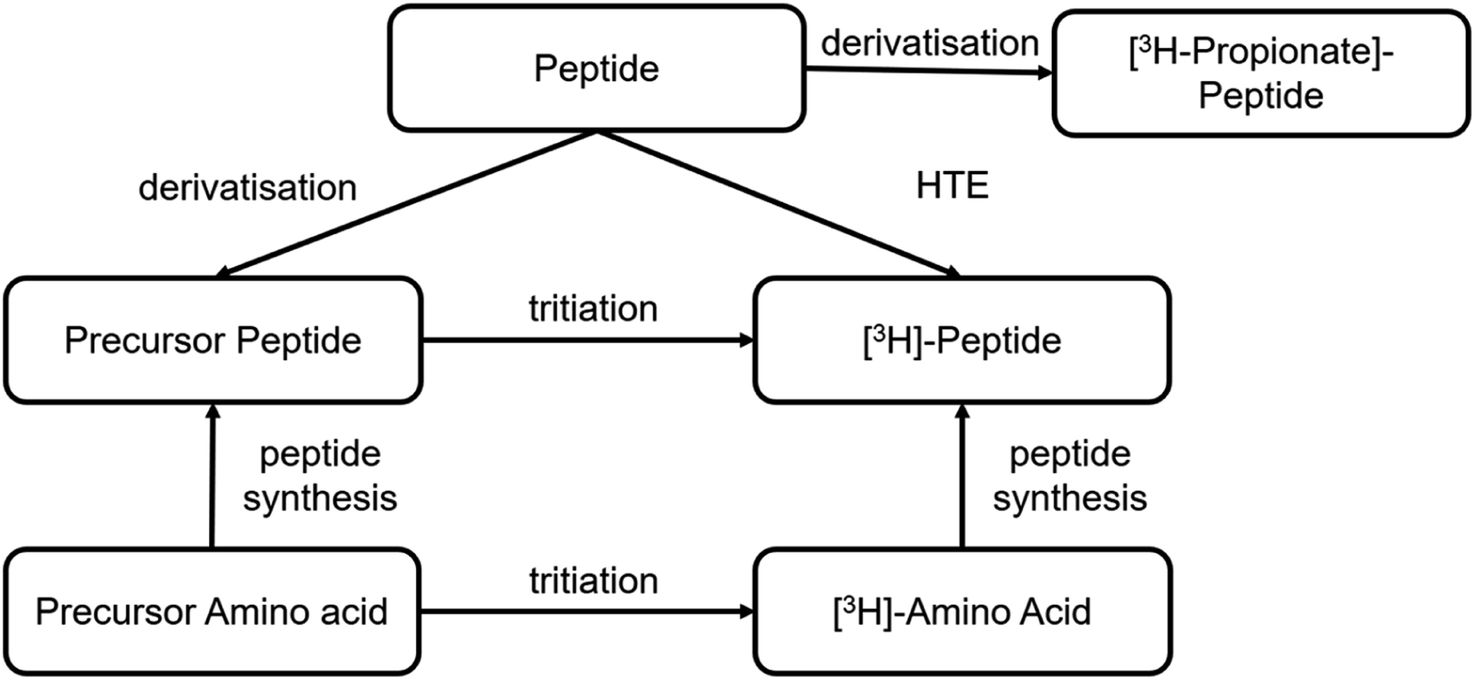 | ||
| Fig. 6 Current synthetic routes to tritium-labelled peptides. | ||
A stable precursor can also be obtained analogous to radioiodination with sodium iodide and an oxidising agent starting from the parent peptide, in which case the non-radioactive iodide (127I) is used. The iodine on the Tyr residues can be catalytically exchanged in the presence of tritium gas to obtain the tritiated peptide.60
Furthermore, radioactive labelling of a peptide by conjugating N-succinimidyl-[2,3-3H3]-propionate ([3H]NSP) to accessible primary amines in the sequence is possible, which derivatives the peptide.61,62 These amines can be located at the N-terminus, at lysine residues, or they can be preceded by a lysine-like linker at the terminal guanidine nitrogen atom of arginine residues.63 In this case, it must be taken into account that the chemical structure differs from the native peptide because of this derivatisation, which may result in changes in the biological and physicochemical properties, especially as Arg and Lys being basic and therefore protonated amino acid residues, may well play a significant role.
Labelling of proteins and its consequences
Antibodies exhibit complex pharmacokinetics (PK) due to their large size, long circulating half-life, and immunogenic responses, making predictions about the distribution of monoclonal antibodies extremely difficult. Therefore, robust characterisation of the PK of novel next-generation antibodies, such as antibody-drug conjugates, bispecific antibodies, and other antibody constructs,64–66 in preclinical and clinical research can support the development of drug candidates. Conventional techniques for determining antibody PK include plasma clearance measurements using ELISA or LC-MS and tissue distribution using immunohistochemistry or immunofluorescence.67 These bioanalytical methods, most of which require a labelled secondary antibody, are limited to ligand-binding assays. In vivo imaging or tracking of drug-related material at low concentrations is nearly impossible without a radioactive label.Many different labelling concepts are reported in the literature. These include radiometals complexed with conjugated chelating agents,68 labelling with radioactive iodine similar to peptide iodination,69 or tritiation with an activated tritiated propionic acid.61 In addition, labelling with fluorescent dyes by post-modifying chemical syntheses on mAbs has been widely reported, as well as dual labelling of radionuclide and dye on antibodies.70–73
As with peptides, modifying a protein with a fluorescent or radioactive tag bears the risk of altering its biological behaviour. The degree of labelling, which corresponds to the number of labels per protein, can also change the biological properties of the antibody.71 It has also been shown that both the label as well as the labelling chemistry can influence binding to the neonatal Fc receptor (FcRn).74 A lower affinity to FcRn results in a shorter half-life of the antibody. Modification of the framework with fluorescent dyes or chelators carrying carboxylic acid or sulfonic acid residues leads to a lower isoelectric point (pI), mainly due to a more negative charge. Charge-mediated interactions may be associated with non-specific interactions with cell surfaces that trigger enhanced pinocytosis. Vascular endothelial cells are coated with highly negatively charged oligosaccharides such as heparan and heparin sulfates.75 Excessive binding to these cell surface structures may increase the risk of pinocytotic uptake and consequently lead to more rapid proteolytic degradation. Non-specific binding to cell surfaces or extracellular matrix structures is considered to be one of the main factors for differences in the PK of antibodies.76 Biodistribution experiments confirmed that the variant with a high pI was catabolised in the liver and spleen. These results indicate that antibody loading may have an effect on PK through changes in antibody catabolism independent of FcRn-mediated recycling.77 There are only a few studies in the literature that compare different labelling methods with regard to their influence on PK or affinity properties.71,74,78–80 An in vitro assessment platform that allows predicting a change in biological behaviour after antibody modification using common labelling methods has not yet been reported. A chromatography-based platform should be able to detect changes in surface charges,81 affinity to FcRn, as well as high and low molecular weight impurities. A combination of size-exclusion chromatography (SEC), FcRn affinity, and heparin affinity chromatography can assess whether modification has affected protein properties. This would also avoid animal testing done with “damaged” labelled antibodies and thus corresponds to the 3R (replace, reduce, refine) principles. Fig. 7 shows a proposed integrated quality assessment in a typical antibody labelling workflow.
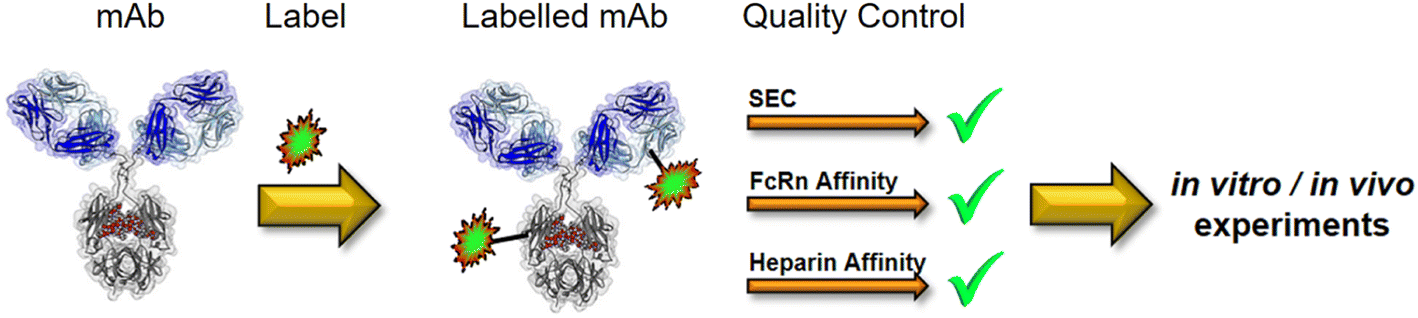 | ||
| Fig. 7 Quality control has to be integrated into a typical labelling process for mAbs used in biological studies. | ||
Monitoring of drug/anti-drug immune complexes
The choice of radionuclides for labelling proteins depends on the research objective. In particular, for long-term investigations such as biodistribution, biotransformation82 or metabolite identification studies, short-lived radioisotopes are not suitable. In these cases, incorporation of the long-lived isotope tritium into the protein is preferred, as subsequent analyses can be carried out several times for quantification after the studies without significant loss of radioactive radiation. In general, tritium labels offer excellent opportunities for monitoring PK behaviour of novel antibodies in preclinical research.As previously mentioned, a critical aspect of antibody PK is the immunogenic response. It is known that all therapeutic antibodies, even those that are fully human, are immunogenic.83 This means that they can trigger an immune response via an anti-drug antibody (ADA) and forming drug-ADA immune complexes (ICs). In this case, the therapeutic antibody can be considered as an antigen for the ADA. The ADA recognises the therapeutic mAb over a large area of its surface and binds to it through weak non-covalent interactions, such as electrostatic interactions, hydrogen bonds, van der Waals forces, and hydrophobic interactions. Depending on the mechanism of action as shown in Fig. 8, neutralising (binding to Fab region) or non-neutralising (binding to Fc domain), the presence of ICs can affect the concentration and function of the therapeutic antibody in the body, e.g. changing the biological effectiveness or even the safety profile.84,85 As a result, there may be reduced efficacy with neutralising ADAs or accelerated drug clearance with non-neutralising antibodies. Understanding the immunogenicity of biological therapies and how to manage them is important for the development of drug candidates.86 Immunogenicity can be influenced by several factors, which can be clustered into three main categories: treatment-related, patient-related, and drug-property-related factors.87 Many of these factors are not yet well understood. Furthermore, predicting an undesired immunogenicity of a therapeutic biomolecule is still difficult.
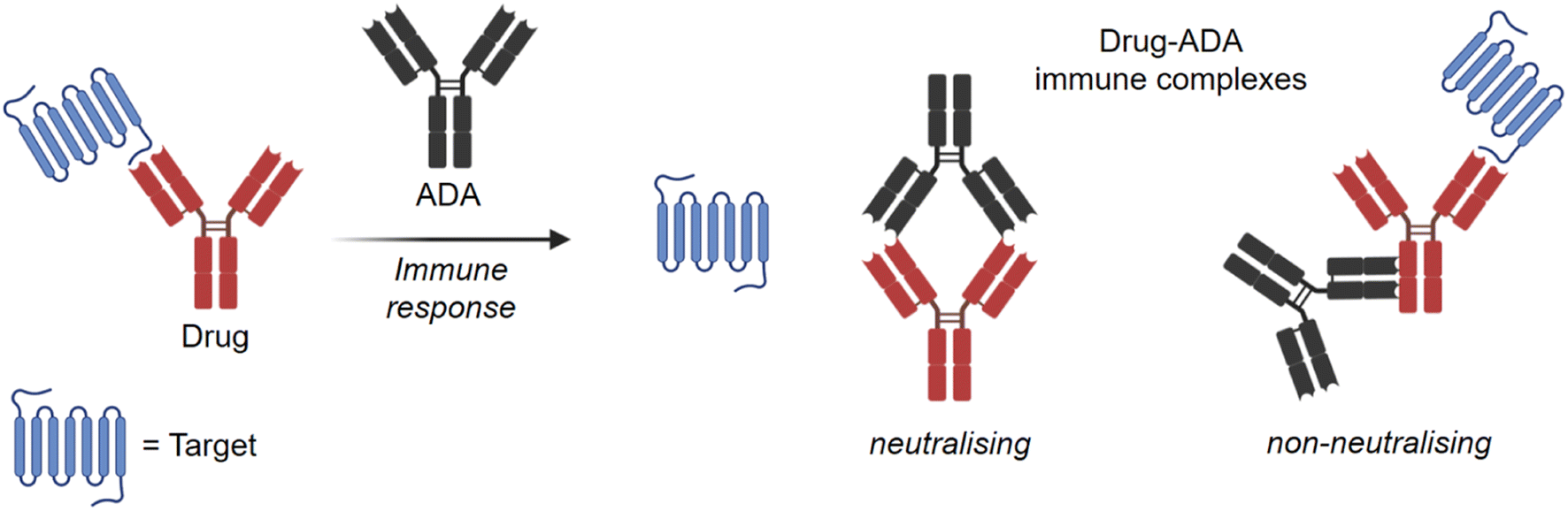 | ||
| Fig. 8 Neutralising and non-neutralising drug-ADA immune complexes. | ||
Currently, several in silico, in vitro, and in vivo approaches have been reported to predict immunogenicity in the very early stages of drug development.88–90 However, it has not yet been possible to reconcile these predictions with the immunogenicity observed in clinical practice. In particular, no preclinical in vivo approach to predict immunogenicity has been able to correlate with the results of clinical therapy.91,92 Although the detailed interaction or formation between ADA and drug is understood93 and according to the literature, large immune complexes degrade faster than smaller ICs,94 quantitative information on the exact size and structure of the immune complexs and their actual impact on drug PK is not yet available. Van Meer and co-workers accessed several market-registered mAbs to determine the relative immunogenicity of mAbs in non-human primates (NHPs) and humans. The results confirm that the ability to compare the relative immunogenicity of mAbs in NHPs and humans is low, with only 59% of cases.95 Topp and co-workers have provided a comprehensive overview of the immunogenicity of therapeutic protein aggregates with a focus on immune mechanisms and their relationship to aggregate properties.96 In conclusion, no preclinical in vivo approach to predict immunogenicity that correlates with outcomes of clinical therapy has been reported to date. This represents a gap to quantify the PK behaviour of immune complexes and consequently to be able to optimise the PK properties of therapeutic proteins that can influence the immune response. Therefore, the use of radiolabelled IC can provide a valuable contribution. In this way, the concentrations of ICs in different organs or tissues can be monitored, providing information about the biodistribution of the complexes by tracking or by imaging studies.
Positron emission tomography using small molecule tracer
Standard imaging techniques such as X-ray, computed tomography (CT), and magnetic resonance imaging (MRI) allow healthy and diseased tissue to be visualised with great detail. However, certain diseases do not have structural anatomical abnormalities or only appear at later stages. Therefore, functional PET imaging can complement structural modalities to overcome some of the shortcomings. As an imaging technique based on positron-emitting molecules, PET is widely used in clinical diagnostics, quantification of pharmacological processes, and drug development.97 A positron is the anti-particle of an electron and has all the properties of an electron except for the positive electric charge it possesses. Thus, a positron can be considered as an electron with a positive charge. A classical PET study uses small molecules in micromolar or nanomolar concentrations that have been labelled with short-lived positron-emitting isotopes such as 11C (t1/2: 20 min) and 18F (t1/2: 110 min). The development of molecular imaging approaches aimed at identifying pathologies in living patients is a very active area of research with the goal of distinguishing such disorders as early as possible and supporting the development of disease-modifying therapies. A PET tracer routinely used in the clinic represents the radiolabelled glucose analogue [18F]-fluorodeoxyglucose ([18F]FDG), which has revolutionised the imaging assessment of a variety of diseases and cancer in particular. Almost all PET examinations are now carried out in combination with CT. PET-CT imaging with [18F]FDG is now a common tool for diagnosis, initial staging, assessment of response to therapy, radiotherapy planning, restaging, and prognosis for many cancer types.98–100PET has also been able to provide unique and powerful insights into normal brain function as well as dysfunctions associated with various neurological disorders and diseases,101,102 focused on imaging six categories of brain proteins: G-protein coupled receptors (GCPR), membrane transporters, ligand gated ion channels (LGIC), tryptophan-rich sensory proteins (TSPO), enzymes, and misfolded proteins.103 Small molecule-based positron-emitting inhibitors, which bind to the target molecule in the manner of receptor-ligand interaction, are often used as PET tracer for imaging and concentration assessment of brain-enzymes, such as monoamine oxidase A and B (MAO-A and MAO-B), acetylcholine esterase, fatty acid amide hydrolase (FAAH), or monoacylglycerol lipase (MAGL).
Monitoring of brain-MAGL inhibitors
MAGL is the major enzyme involved in the endocannabinoid 2-arachidonoylglycerol (2-AG) catabolism and has received significant attention as a therapeutic target for neurological disorders.104,105 The development of a PET radioligand with high target specificity for the preclinical and clinical characterisation of MAGL inhibitor drug candidates is therefore of great interest. However, a good drug candidate may not necessarily be a good candidate for CNS-PET imaging. While drug candidates with long target residence times and high metabolic stability foster therapeutic efficacy in treatment, PET tracer candidates with these properties can result in slow PK, which generally require longer scan times and this generates more variable outcome measurements.106 The first set of five PET tracers for MAGL developed by Hicks et al. were published in 2014 and labelled with carbon-11.107 These compounds have been reported to have moderate blood–brain barrier (BBB) penetration. The lack of MAGL-specific brain imaging led to the conclusion that none of the tracers was suitable for in vivo brain-MAGL quantification. The next generation of MAGL radioligands (Fig. 9) are mainly irreversible, like [11C]SAR127303 (ref. 108) as well as [11C]PF-06809247 (ref. 109) and [18F]PF-06795071 (ref. 110) developed by Pfizer. However, irreversible PET tracers could hardly provide a comprehensive quantification of the drug-target interaction. Compared to irreversible MAGL radioligands,111 in which the formation of covalent bonds is a major contributor to binding, a reversible PET tracer probe requires additional moieties to interact non-covalently with residues of the binding site.112 This usually leads to increased molecular weight and therefore adds several obstacles to crossing the BBB through active transport. To date, only a few radioligands have been described as reversible MAGL tracers. The reported examples [11C]PAD and [18F]MAGL-4-11 (ref. 113) showed low brain uptake and were recognised by ATP-binding cassette (ABC) transporters expressed in the BBB. While their in vivo specificities were confirmed in peripheral MAGL-expressing organs, no specific interaction could be demonstrated in rat brain. [18F]T-401 and [18F]MAGL-2102 are recently reported reversible MAGL radioligands114 that showed specificity for CNS-MAGL in vivo. However, low to moderate brain uptake was observed for [18F]T-401 and a slow reversible binding mechanism was revealed for [18F]MAGL-2102. In summary, novel reversible MAGL-PET radioligands with improved brain uptake and suitable PK are highly desirable for clinical practice. These PET tracers would enable the development of new brain-MAGL inhibitors and monitor the effectiveness of the drugs.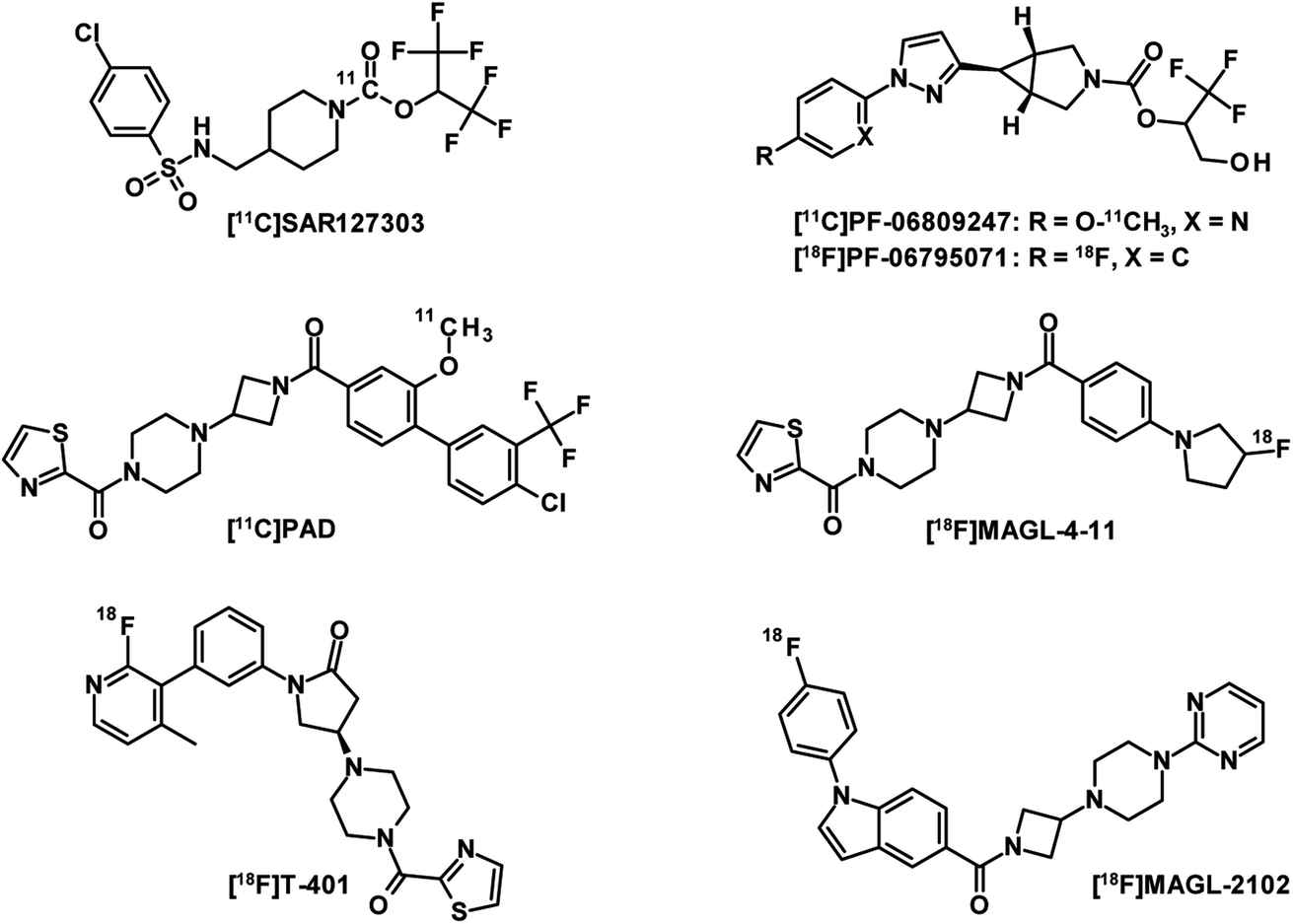 | ||
| Fig. 9 Chemical structures of irreversible and reversible MAGL-PET tracers. | ||
Synthesis of tetrazines for bioorthogonal click-reactions
In the early 2000s, click reactions were introduced by Sharpless and co-workers.115 Lang and Chin summarised in a paper the 20 most commonly reported bioorthogonal reactions, reactions that do not disrupt biological processes.116 The Staudinger ligation117 was one of the earliest reported bioorthogonal reactions. In Staudinger ligation, an azide reacts with phosphines with good selectivity. However, the main disadvantage of Staudinger ligation is its slow reaction kinetics which lead to inefficient labelling. Other types of bioorthogonal reactions include the [3 + 2] cycloaddition, such as the 1,3-dipolar azide-alkyne Huisgen cycloaddition118 and the strain-promoted azide-alkyne cycloaddition (SPAAC).119 Furthermore, isocyanides can undergo [4 + 1] cycloadditions with tetrazines.120 The most attractive reaction for a bioorthogonal click reaction is the current inverse electron-demand Diels–Alder (IEDDA) ligation between tetrazine (Tz) derivatives and trans-cyclooctene (TCO) functionalisations (Fig. 10).121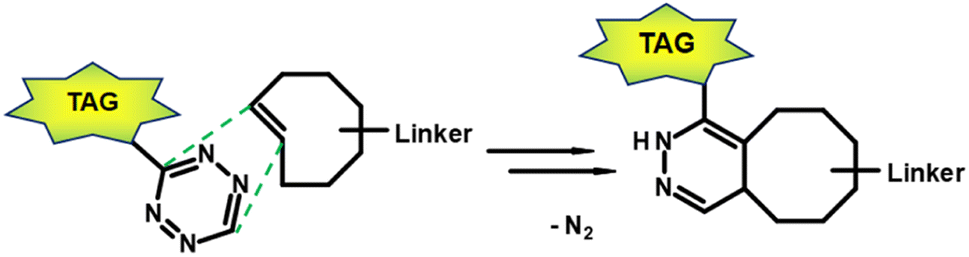 | ||
| Fig. 10 Inverse electron-demand Diels–Alder ligation between a TCO linked to a carrier moitey and a tagged Tz derivative. | ||
PET-nuclide labelled Tzs for the use of a bioorthogonal click reaction and pretargeted imaging can in principle be considered as small molecule PET tracers. In the case of Tzs, the target is not a receptor or enzyme, but a TCO-functionalised agent, such as antibodies,122 antibodies,123 or diabodies.124 A variety of Tzs have been developed for preclinical or diagnostic imaging. For this purpose, Tz derivatives were modified with fluorescent dyes125 or chelators capable of complexing the corresponding PET radionuclides. 111In and 68Ga are given as common radionuclides for Tzs.126 A variant with 44Sc (t1/2: 4 h) was also developed to evaluate its application for pretargeting PET images.127 Currently, 11C and 18F-labelled Tzs are predominantly reported, in which the PET nuclide is incorporated through a covalent bond rather than using a chelator.128,129 In a reverse approach, a TCO derivative is labelled with a PET nuclide and undergoes a click reaction with a Tz-modified counterpart.130–132 Although [18F]TCO can be considered a useful tracer for radiolabelling Tz-modified targeting molecules, studies indicate that its utility for in vivo chemistry and pretargeted imaging will be limited. [18F]TCO derivatives, while clearly reaching the brain, are rapidly metabolised with non-specific accumulation of radioactivity in brain and bone.133
While a conventional PET tracer should bind to its target as quickly as possible, the challenge with the click-reaction approach is that the Tz should react with the TCO moiety as quickly as possible. A key factor in reaction rates is the chemical environment of the Tz. Fast IEDDA-reaction rates often come at the expense of probe stability.134 In general, methyl-substituted Tzs showed the highest stability, but also had the lowest reaction kinetics. Electron-withdrawing substituents increase the click reactivity of Tzs with TCO, but these substituents also induce their decomposition. Electron-donating substituents show a higher stability, but also suffer from slow reaction kinetics (Fig. 11).135
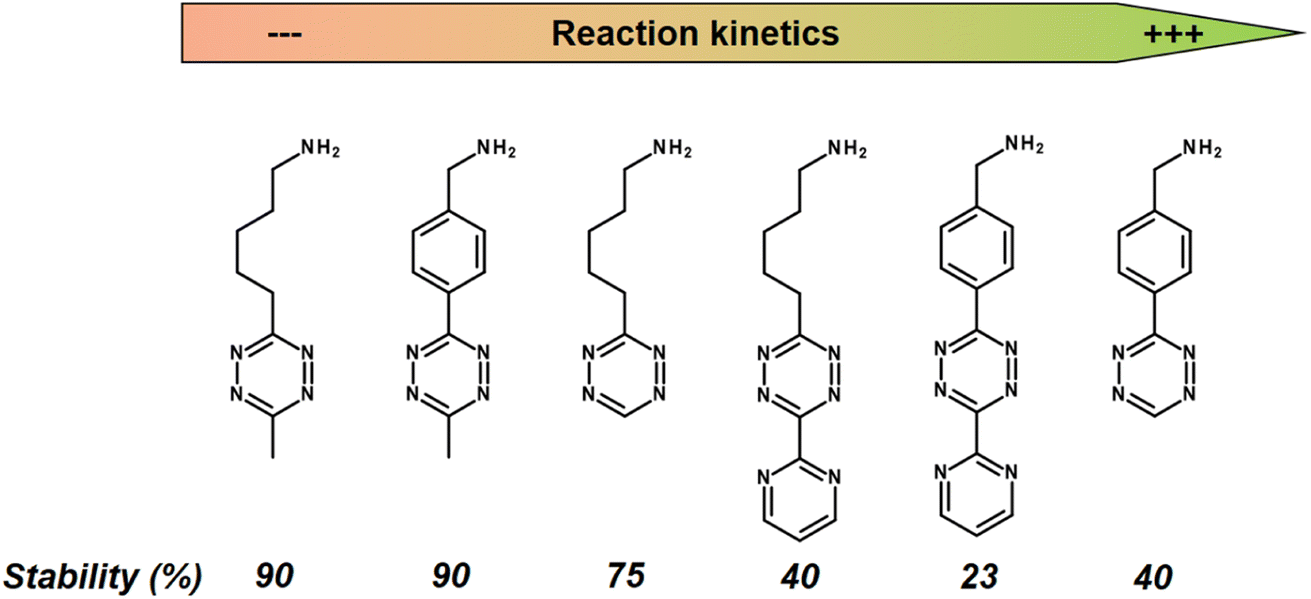 | ||
| Fig. 11 Reaction kinetics from slow (left) to high (right) of selected Tzs with TCO in PBS at 37 °C and corresponding stability assessed in PBS at 37 °C for 10 h. | ||
The IEDDA reaction kinetics also strongly depend on the choice of TCO derivatives, and therefore the activity of various dienophiles has been extensively investigated in recent years. Sauer studied the reactivity of substituted dienophiles and summarized several basic rules: (1) dienophiles with electron-rich substituents are favorable for fast reaction kinetics; (2) strained dienophiles are more reactive; (3) an increase in steric effect hinders reactivity.136 Researchers have therefore focused on improving the reaction kinetics of Tz-TCO binding (Fig. 12). A commonly used dienophile for IEDDA-based pretargeting studies is TCO, functionalised with a hydroxyl group at the 5-position (OH-TCO) to allow further conjugation to a molecule of interest. In this case, there are two stereoisomers (equatorial and axial) with a higher reaction rate from the axial isomer.137 Despite the fast kinetics observed with these TCOs, however, it has been observed that the TCO-construct is partially deactivated in vivo by isomerisation to a slowly reactive cis-cyclooctene (CCO).137 Based on Sauer's hypothesis that strained dienophiles are more reactive, Fox and co-workers developed TCO analogues in which the strained TCO ring was forced to adopt a half-chair conformation.138,139 In the example of dioxolane-fused TCO (d-TCO) and strained TCO (s-TCO), the energy was predicted to be 5.6–5.9 kcal mol−1 higher than for the more stable crown conformation from the OH-TCO scaffolds and exhibit significantly higher bimolecular rate constants.140 Recently, trans-cycloheptene (TCH) analogues have been reported as dienophiles in IEDDA cycloadditions with tetrazines. While TCH is readily isomerised under ambient conditions, the incorporation of an endocyclic silicon atom, e.g. in sila-trans-cycloheptene (SiTCH), stabilises the seven-membered cycloalkene. Similar to Tzs, it was observed that the high reaction rate leads to a decrease in stability.141
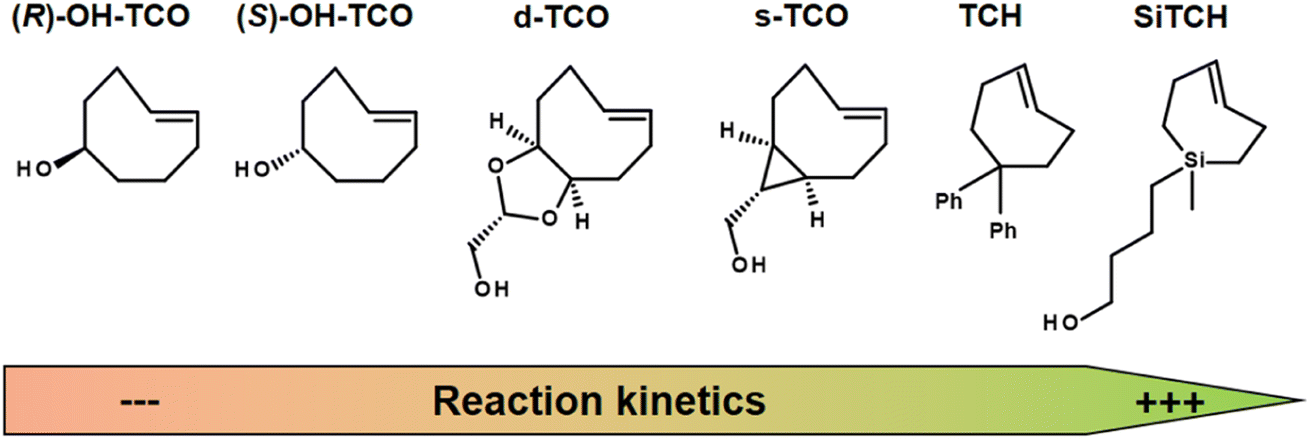 | ||
| Fig. 12 Examples of strained dienophiles for IEDDA reactions, from left to right with slow to high reaction kinetics. | ||
The challenge for effective labelling in in vivo systems is to achieve an optimal balance between reactivity and stability. It is of comparable complexity to deliver a radiolabelled Tz into the brain while maintaining its stability and reactivity. While there are many examples in the literature of an IEDDA click reaction in the periphery,127,142,143 there are only a few reports of Tzs penetrating the brain and serving as PET-nuclide carriers,128 which represents a gap in nuclear medicine diagnostics of brain targets.
Monitoring of misfolded proteins
PET imaging of misfolded proteins, such as for amyloid-β (Aβ) plaques and tau fibrils in Alzheimer's disease (AD) or α-synuclein in Parkinson's disease (PD), is an extremely valuable neuroimaging tool and supports ongoing global efforts to better understand brain function.144–149 Although the use of small molecules as PET tracers for misfolded protein aggregates has been reported,145,150 the use of small molecules has some limitations. The pathological accumulation of misfolded proteins can vary in composition and isoform. In addition, the morphology of the protein construct changes between disease states.151 Therefore, different binding sites can arise and the affinities of tracers can vary significantly between isoforms and morphologies of protein aggregates. Currently, no well-characterised, highly selective small molecule-based PET tracers are available for use in competitive binding studies that would allow the determination of Aβ, tau fibrils, or α-synuclein-specific signalling over the dynamic range of disease progression.Instead of small molecules, monoclonal antibodies (mAbs) with their high specificity, high affinity, and serum stability can also be used for molecular imaging to target radionuclides in vivo on protein aggregates of interest. Many studies have demonstrated the use of mAb-targeted imaging (Immuno-PET) for a variety of purposes, e.g., monitoring disease progressions and the effect on a therapeutic agent in pharmaceutical research and development.152–155 Antibody-based radioligands have so far only been described for peripheral targets related to cancer diagnostics and theranostics, including some clinical applications.156,157 The main hurdle for an antibody-based radioligand to be delivered to the brain is the blood–brain barrier (BBB). In 1994, it was demonstrated that only 0.1% of a peripherally administered immunoglobulin G (IgG) reaches the brain.158 However, the BBB uses natural receptors expressed on brain endothelial cells (BECs) for active transport purposes. The transferrin receptor (TfR) has, due to its expression on the BBB, an interesting role as a BBB delivery receptor. A Brain Shuttle (BS) technology was developed to allow the delivery of mAbs with fully functional bivalent IgG antigen bindings, anti-brain target and anti-TfR.159 This is achieved by fusing the BS module to the C-terminal part of the heavy chain (Fc) constant region. Therefore, these BS-antibodies could allow their use as PET radioligands within the CNS.160
While antibodies allow prolonged circulation in the body due to their molecular size and structure, but even more due to their interaction with the neonatal Fc receptor, which protects IgGs from degradation,161 this prolonged half-life is a limitation in molecular imaging due to higher background signals when using PET radionuclide-labelled mAbs. Compared to conventional imaging techniques in which a molecule is directly labelled with a radionuclide, the pretargeting approach offers an uncoupling of the PK half-life of the biological molecule of interest from the physical half-life of the radionuclide (Fig. 13).
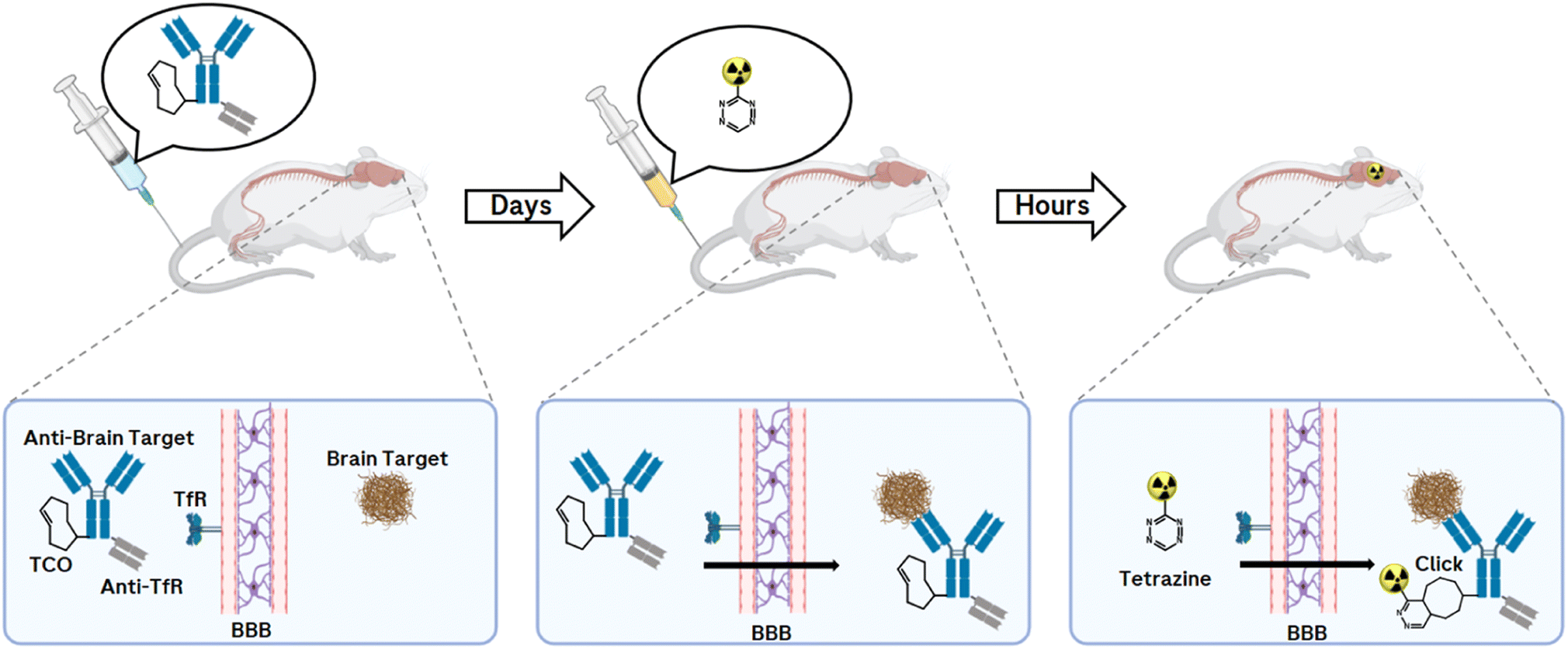 | ||
| Fig. 13 Pretargeting workflow. In the first step, a BBB-penetrating TCO-modified antibody is injected. The mAb is actively transported across the BBB and binds to a target. In the second step, a PET nuclide-labelled Tz is injected and clicks with the TCO-mAb to generate PET-images of misfolded proteins. | ||
The pretargeting approach allows an antibody to be prelocalised to a target before injecting after 1–3 days a small radiolabelled effector to recognise and specifically covalently bind to it via an in vivo click reaction.162 Applications are found, for example, in cancer imaging and targeted radionuclide therapy.163 So far, an in vivo bioorthogonal click reaction approach of a TCO-modified antibody with a PET nuclide-labelled Tz in the CNS has not been reported. The requirement for a successful click response is a brain-penetrating Tz that is stable and can react quickly with the pretargeted TCO-mAb. With this technique and the resulting imaging of misfolded proteins within the CNS, valuable information can be provided to accelerate drug development, monitor disease progression, and offer great benefit to patient care.
Conclusions
Radioactive labelling of molecules from novel modalities, such as oligonucleotides, peptides, and antibodies, is often a major challenge. The established labelling methods used for small molecules have limited applicability for large, polar, highly charged molecules. The choice of radionuclide depends not only on the type of study, but also on molecular properties such as biological half-life, hydrodynamic radius, molecular surface charge, or target binding. This review provides an overview of current labelling methods for oligonucleotides, peptides, and antibodies for use in tracking, monitoring, and imaging studies. In conclusion, there is an urgent need for new and efficient labelling concepts for molecules such as oligonucleotides, peptides, antibodies, and their characterisation in terms of a label impact on the molecular properties.Conflicts of interest
The author is a full-time employee of the company F. Hoffmann-La Roche AG.Acknowledgements
I thank Dr Ian S. Blagbrough and Prof Stephen M. Husbands from the University of Bath as well as Dr Michael B. Otteneder and Dr Filippo Sladojevich from Roche for their helpful comments and for their interest in these studies.References
- R. L. Wolke, Marie Curie's doctoral thesis: prelude to a Nobel Prize, J. Chem. Educ., 1988, 65, 561–573 CrossRef CAS.
- W. C. Röntgen, On a new kind of rays, Science, 1896, 3, 227–231 CrossRef PubMed.
- H. Becquerel, The radio-activity of matter, Nature, 1901, 63, 396–398 CrossRef.
- R. R. Carlton, A. M. Adler and V. Balac, Principles of radiographic imaging: an art and a science, Cengage Learning, 2019 Search PubMed.
- W. R. Hendee, G. S. Ibbott and E. G. Hendee, Radiation therapy physics, John Wiley & Sons, 2013 Search PubMed.
- J. W. Scannell, A. Blanckley, H. Boldon and B. Warrington, Diagnosing the decline in pharmaceutical R&D efficiency, Nat. Rev. Drug Discovery, 2012, 11, 191–200 CrossRef CAS PubMed.
- C. Klein, W. Schaefer and J. T. Regula, The use of CrossMAb technology for the generation of bi-and multispecific antibodies, mAbs, 2016, 8, 1010–1020 CrossRef CAS PubMed.
- G. Caron and G. Ermondi, Molecular descriptors for polarity: the need for going beyond polar surface area, Future Med. Chem., 2016, 8, 2013–2016 CrossRef CAS PubMed.
- W. Wei, E. B. Ehlerding, X. Lan, Q. Luo and W. Cai, PET and SPECT imaging of melanoma: the state of the art, Eur. J. Nucl. Med. Mol. Imaging, 2018, 45, 132–150 CrossRef CAS PubMed.
- V. Valotassiou, J. Malamitsi, J. Papatriantafyllou, E. Dardiotis, I. Tsougos, D. Psimadas, S. Alexiou, G. Hadjigeorgiou and P. Georgoulias, SPECT and PET imaging in Alzheimer's disease, Ann. Nucl. Med., 2018, 32, 583–593 CrossRef CAS PubMed.
- S. Vandenberghe, P. Moskal and J. S. Karp, State of the art in total body PET, EJNMMI Phys., 2020, 7, 1–33 CrossRef PubMed.
- I. M. Jackson, P. J. Scott and S. Thompson, Clinical applications of radiolabeled peptides for PET, Semin. Nucl. Med., 2017, 47, 493–523 CrossRef PubMed.
- S. Heskamp, R. Raavé, O. Boerman, M. Rijpkema, V. Goncalves and F. Denat, 89Zr-immuno-positron emission tomography in oncology: state-of-the-art 89Zr radiochemistry, Bioconjugate Chem., 2017, 28, 2211–2223 CrossRef CAS PubMed.
- M. Mosley, J. B. Torres, D. Allen and B. Cornelissen, Immuno-imaging of ICAM-1 in tumours by SPECT, Nucl. Med. Biol., 2020, 84, 73–79 CrossRef PubMed.
- C. R. Correa, A. L. B. de Barros, C. de Aguiar Ferreira, A. M. de Goes, V. N. Cardoso and A. S. R. de Andrade, Aptamers directly radiolabeled with technetium-99m as a potential agent capable of identifying carcinoembryonic antigen (CEA) in tumor cells T84, Bioorg. Med. Chem. Lett., 2014, 24, 1998–2001 CrossRef CAS PubMed.
- Y. Liu, G. Liu and D. J. Hnatowich, A brief review of chelators for radiolabeling oligomers, Materials, 2010, 3, 3204–3217 CrossRef CAS.
- S. M. Larson, J. A. Carrasquillo, N.-K. V. Cheung and O. W. Press, Radioimmunotherapy of human tumours, Nat. Rev. Cancer, 2015, 15, 347–360 CrossRef CAS PubMed.
- S. Vallabhajosula, A. Nikolopoulou, Y. S. Jhanwar, G. Kaur, S. T Tagawa, D. M Nanus, N. H Bander and S. J Goldsmith, Radioimmunotherapy of metastatic prostate cancer with; 177Lu-DOTAhuJ591 anti prostate specific membrane antigen specific monoclonal antibody, Curr. Radiopharm., 2016, 9, 44–53 CrossRef CAS PubMed.
- K. J. Allen, R. Jiao, M. E. Malo, C. Frank, D. R. Fisher, D. Rickles and E. Dadachova, Comparative radioimmunotherapy of experimental melanoma with novel humanized antibody to melanin labeled with 213bismuth and 177lutetium, Pharmaceutics, 2019, 11, 348 CrossRef CAS PubMed.
- R. Eychenne, M. Chérel, F. Haddad, F. Guérard and J.-F. Gestin, Overview of the most promising radionuclides for targeted alpha therapy: The “hopeful eight”, Pharmaceutics, 2021, 13, 906 CrossRef CAS PubMed.
- S. Poty, L. C. Francesconi, M. R. McDevitt, M. J. Morris and J. S. Lewis, α-Emitters for radiotherapy: from basic radiochemistry to clinical studies—part 1, J. Nucl. Med., 2018, 59, 878–884 CrossRef CAS PubMed.
- S. Poty, L. C. Francesconi, M. R. McDevitt, M. J. Morris and J. S. Lewis, α-emitters for radiotherapy: from basic radiochemistry to clinical studies—part 2, J. Nucl. Med., 2018, 59, 1020–1027 CrossRef CAS PubMed.
- G. Sgouros, L. Bodei, M. R. McDevitt and J. R. Nedrow, Radiopharmaceutical therapy in cancer: clinical advances and challenges, Nat. Rev. Drug Discovery, 2020, 19, 589–608 CrossRef CAS PubMed.
- T. E. Witzig, Yttrium-90-ibritumomab tiuxetan radioimmunotherapy: a new treatment approach for B-cell non-Hodgkin's lymphoma, Drugs Today, 2004, 40, 111–120 CrossRef CAS PubMed.
- U. Hennrich and K. Kopka, Lutathera®: the first FDA-and EMA-approved radiopharmaceutical for peptide receptor radionuclide therapy, Pharmaceuticals, 2019, 12, 114 CrossRef CAS PubMed.
- C. Kay, N. Skotte, A. Southwell and M. Hayden, Personalized gene silencing therapeutics for Huntington disease, Clin. Genet., 2014, 86, 29–36 CrossRef CAS PubMed.
- C. Rinaldi and M. J. Wood, Antisense oligonucleotides: the next frontier for treatment of neurological disorders, Nat. Rev. Neurol., 2018, 14, 9–21 CrossRef CAS PubMed.
- S. T. Crooke, J. L. Witztum, C. F. Bennett and B. F. Baker, RNA-targeted therapeutics, Cell Metab., 2018, 27, 714–739 CrossRef CAS PubMed.
- A. Khvorova and J. K. Watts, The chemical evolution of oligonucleotide therapies of clinical utility, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed.
- T. C. Roberts, R. Langer and M. J. Wood, Advances in oligonucleotide drug delivery, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- I. S. Blagbrough and C. Zara, Animal models for target diseases in gene therapy—using DNA and siRNA delivery strategies, Pharm. Res., 2009, 26, 1–18 CrossRef CAS PubMed.
- I. S. Blagbrough and A. A. Metwally, Chapter 7: siRNA and Gene Formulation for Efficient Gene Therapy, in Gene Therapy-Tools and Potential Applications, ed. F. M. Molina, IntechOpen, 2013, pp. 135–173 Search PubMed.
- R. S. Geary, B. F. Baker and S. T. Crooke, Clinical and preclinical pharmacokinetics and pharmacodynamics of mipomersen (Kynamro®): a second-generation antisense oligonucleotide inhibitor of apolipoprotein B, Clin. Pharmacokinet., 2015, 54, 133–146 CrossRef CAS PubMed.
- A. Kilanowska and S. Studzińska, In vivo and in vitro studies of antisense oligonucleotides—a review, RSC Adv., 2020, 10, 34501–34516 RSC.
- N. A. Kratochwil, S. R. Dueker, D. Muri, C. Senn, H. Yoon, B.-Y. Yu, G.-H. Lee, F. Dong and M. B. Otteneder, Nanotracing and cavity-ring down spectroscopy: a new ultrasensitive approach in large molecule drug disposition studies, PLoS One, 2018, 13, e0205435 CrossRef PubMed.
- S. T. Crooke, L. R. Grillone, A. Tendolkar, A. Garrett, M. J. Fratkin, J. Leeds and W. H. Barr, A pharmacokinetic evaluation of 14C-labeled afovirsen sodium in patients with genital warts, Clin. Pharmacol. Ther., 1994, 56, 641–646 CrossRef CAS PubMed.
- A. A. Levin, A review of issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides, Biochim. Biophys. Acta, 1999, 1489, 69–84 CrossRef CAS.
- Y. Liu, Y.-c. Tseng and L. Huang, Biodistribution studies of nanoparticles using fluorescence imaging: a qualitative or quantitative method?, Pharm. Res., 2012, 29, 3273–3277 CrossRef CAS PubMed.
- K. R. Shelton and J. M. Clark Jr, A proton exchange between purines and water and its application to biochemistry, Biochemistry, 1967, 6, 2735–2739 CrossRef CAS PubMed.
- H. Wang and P. Guo, Radiolabeled RNA nanoparticles for highly specific targeting and efficient tumor accumulation with favorable in vivo biodistribution, Mol. Pharm., 2021, 18, 2924–2934 CrossRef CAS PubMed.
- A. Palazzolo, S. Feuillastre, V. Pfeifer, S. Garcia-Argote, D. Bouzouita, S. Tricard, C. Chollet, E. Marcon, D. A. Buisson, S. Cholet, F. Fenaille, G. Lippens, B. Chaudret and G. Pieters, Efficient access to deuterated and tritiated nucleobase pharmaceuticals and oligonucleotides using hydrogen-isotope exchange, Angew. Chem., 2019, 131, 4945–4949 CrossRef.
- W. Tan, R. P. Iyer, D. Yu and S. Agrawal, Site-specific synthesis of [3H] oligonucleotides in high specific activity through direct solid-phase redox chemistry, Tetrahedron Lett., 1995, 36, 3631–3634 CrossRef CAS.
- T. Ledoan, R. Auger, A. Benjahad and J.-P. Tenu, High specific radioactivity labeling of oligonucleotides with 3H-succinimidyl propionate, Nucleosides, Nucleotides Nucleic Acids, 1999, 18, 277–289 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, C–H functionalisation for hydrogen isotope exchange, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed.
- M. Valero, D. Becker, K. Jess, R. Weck, J. Atzrodt, T. Bannenberg, V. Derdau and M. Tamm, Directed iridium-catalyzed hydrogen isotope exchange reactions of phenylacetic acid esters and amides, Chem.–Eur. J., 2019, 25, 6517–6522 CrossRef CAS PubMed.
- M. Valero, R. Weck, S. Güssregen, J. Atzrodt and V. Derdau, Highly selective directed iridium-catalyzed hydrogen isotope exchange reactions of aliphatic amides, Angew. Chem., Int. Ed., 2018, 57, 8159–8163 CrossRef CAS PubMed.
- W. J. Kerr, R. J. Mudd, P. K. Owens, M. Reid, J. A. Brown and S. Campos, Hydrogen isotope exchange with highly active iridium (I) NHC/phosphine complexes: a comparative counterion study, J. Labelled Compd. Radiopharm., 2016, 59, 601–603 CrossRef CAS PubMed.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. MacMillan, Photoredox-catalyzed deuteration and tritiation of pharmaceutical compounds, Science, 2017, 358, 1182–1187 CrossRef CAS PubMed.
- J. M. Ravasco, H. Faustino, A. Trindade and P. M. Gois, Bioconjugation with maleimides: a useful tool for chemical biology, Chem.–Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- M. Krivec, M. Gazvoda, K. Kranjc, S. Polanc and M. Kocevar, A way to avoid using precious metals: the application of high-surface activated carbon for the synthesis of isoindoles via the Diels–Alder reaction of 2H-pyran-2-ones, J. Org. Chem., 2012, 77, 2857–2864 CrossRef CAS PubMed.
- J. Ast, A. Arvaniti, N. H. Fine, D. Nasteska, F. B. Ashford, Z. Stamataki, Z. Koszegi, A. Bacon, B. J. Jones and M. A. Lucey, Super-resolution microscopy compatible fluorescent probes reveal endogenous glucagon-like peptide-1 receptor distribution and dynamics, Nat. Commun., 2020, 11, 1–18 CrossRef PubMed.
- A. Dell'isola, R. T. Brown, S. Jones, S. L. Kitson, T. S. Moody, J. Syvret, B. Upeandran and W. H. Watters, Synthesis of carbon-14-labelled peptides, J. Labelled Compd. Radiopharm., 2019, 62, 713–717 CrossRef PubMed.
- M. C. Oliveira and J. D. Correia, Biomedical applications of radioiodinated peptides, Eur. J. Med. Chem., 2019, 179, 56–77 CrossRef CAS PubMed.
- S. Doll, K. Woolum and K. Kumar, Radiolabeling of a cyclic RGD (cyclo Arg-Gly-Asp-d-Tyr-Lys) peptide using sodium hypochlorite as an oxidizing agent, J. Labelled Compd. Radiopharm., 2016, 59, 462–466 CrossRef CAS PubMed.
- G. Vaidyanathan and M. R. Zalutsky, The Radiopharmaceutical Chemistry of the Radioisotopes of Iodine, in Radiopharmaceutical chemistry, ed. J. Lewis, A. Windhorst and B. Zeglis, Springer, Cham, 2019, pp. 391–408 Search PubMed.
- T. Langbein, W. A. Weber and M. Eiber, Future of theranostics: an outlook on precision oncology in nuclear medicine, J. Nucl. Med., 2019, 60, 13S–19S CrossRef CAS PubMed.
- F. Legros, P. Fernandez-Rodriguez, A. Mishra, R. Weck, A. Bauer, M. Sandvoss, S. Ruf, M. Méndez, H. Mora-Radó and N. Rackelmann, Photoredox-mediated hydrogen isotope exchange reactions of amino-acids, peptides, and peptide-derived drugs, Chem.–Eur. J., 2020, 26, 12738–12742 CrossRef CAS PubMed.
- J. A. Egan and C. N. Filer, High specific activity tritium labelling of biologically active small peptides and a related analogue, J. Radioanal. Nucl. Chem., 2016, 307, 549–553 CrossRef CAS.
- A. Jazayeri, M. Rappas, A. J. Brown, J. Kean, J. C. Errey, N. J. Robertson, C. Fiez-Vandal, S. P. Andrews, M. Congreve and A. Bortolato, Crystal structure of the GLP-1 receptor bound to a peptide agonist, Nature, 2017, 546, 254–258 CrossRef CAS PubMed.
- A. De Prins, C. Martin, Y. Van Wanseele, L. J. Skov, C. Tömböly, D. Tourwé, V. Caveliers, A. Van Eeckhaut, B. Holst and M. M. Rosenkilde, Development of potent and proteolytically stable human neuromedin U receptor agonists, Eur. J. Med. Chem., 2018, 144, 887–897 CrossRef CAS PubMed.
- M. Wang, Y. Zhan, S. P. O'Neil, S. Harris, C. Henson, A. McEwen, R. Webster and D. M. O'Hara, Quantitative biodistribution of biotherapeutics at whole body, organ and cellular levels by autoradiography, Bioanalysis, 2018, 10, 1487–1500 CrossRef CAS PubMed.
- G. Jacquot, P. Lecorché, J.-D. Malcor, M. Laurencin, M. Smirnova, K. Varini, C. Malicet, F. Gassiot, K. Abouzid and A. Faucon, Optimization and in vivo validation of peptide vectors targeting the LDL receptor, Mol. Pharm., 2016, 13, 4094–4105 CrossRef CAS PubMed.
- M. Keller, K. K. Kuhn, J. Einsiedel, H. Hübner, S. Biselli, C. Mollereau, D. Wifling, J. Svobodova, G. Bernhardt and C. Cabrele, Mimicking of arginine by functionalized N ω-carbamoylated arginine as a new broadly applicable approach to labeled bioactive peptides: high affinity angiotensin, neuropeptide Y, neuropeptide FF, and neurotensin receptor ligands as examples, J. Med. Chem., 2016, 59, 1925–1945 CrossRef CAS PubMed.
- S. Dickopf, G. J. Georges and U. Brinkmann, Format and geometries matter: Structure-based design defines the functionality of bispecific antibodies, Comput. Struct. Biotechnol. J., 2020, 18, 1221–1227 CrossRef CAS PubMed.
- G. J. Georges, S. Dengl, A. Bujotzek, F. Hesse, J. A. Fischer, A. Gärtner, J. Benz, M. E. Lauer, P. Ringler and H. Stahlberg, The Contorsbody, an antibody format for agonism: Design, structure, and function, Comput. Struct. Biotechnol. J., 2020, 18, 1210–1220 CrossRef CAS PubMed.
- S. Dengl, K. Mayer, F. Bormann, H. Duerr, E. Hoffmann, B. Nussbaum, M. Tischler, M. Wagner, A. Kuglstatter and L. Leibrock, Format chain exchange (FORCE) for high-throughput generation of bispecific antibodies in combinatorial binder-format matrices, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed.
- K. Im, S. Mareninov, M. Diaz and W. H. Yong, An introduction to performing immunofluorescence staining, in Biobanking, ed. W. H. Yong, Humana Press, New York, 2019, vol. 1897, pp. 299–311 Search PubMed.
- C. G. England, D. Jiang, E. B. Ehlerding, B. T. Rekoske, P. A. Ellison, R. Hernandez, T. E. Barnhart, D. G. McNeel, P. Huang and W. Cai, 89Zr-labeled nivolumab for imaging of T-cell infiltration in a humanized murine model of lung cancer, Eur. J. Nucl. Med. Mol. Imaging, 2018, 45, 110–120 CrossRef CAS PubMed.
- N. Chen, W. Wang, S. Fauty, Y. Fang, L. Hamuro, A. Hussain and T. Prueksaritanont, The effect of the neonatal Fc receptor on human IgG biodistribution in mice, mAbs, 2014, 6, 502–508 CrossRef PubMed.
- M. Saccomano, C. Dullin, F. Alves and J. Napp, Preclinical evaluation of near-infrared (NIR) fluorescently labeled Cetuximab as a potential tool for fluorescence-guided surgery, Int. J. Cancer, 2016, 139, 2277–2289 CrossRef CAS PubMed.
- C. Cilliers, I. Nessler, N. Christodolu and G. M. Thurber, Tracking antibody distribution with near-infrared fluorescent dyes: impact of dye structure and degree of labeling on plasma clearance, Mol. Pharm., 2017, 14, 1623–1633 CrossRef CAS PubMed.
- S. Lütje, M. Rijpkema, G. M. Franssen, G. Fracasso, W. Helfrich, A. Eek, W. J. Oyen, M. Colombatti and O. C. Boerman, Dual-modality image-guided surgery of prostate cancer with a radiolabeled fluorescent anti-PSMA monoclonal antibody, J. Nucl. Med., 2014, 55, 995–1001 CrossRef PubMed.
- H. J. Lee, E. B. Ehlerding, D. Jiang, T. E. Barnhart, T. Cao, W. Wei, C. A. Ferreira, P. Huang, J. W. Engle and W. Cai, Dual-labeled pertuzumab for multimodality image-guided ovarian tumor resection, Am. J. Cancer Res., 2019, 9, 1454 CAS.
- M. R. Edelmann, H. Kettenberger, A. Knaupp, T. Schlothauer and M. B. Otteneder, Radiolabeled IgG antibodies: Impact of various labels on neonatal Fc receptor binding, J. Labelled Compd. Radiopharm., 2019, 62, 751–757 CrossRef CAS PubMed.
- M. Bernfield, M. Götte, P. W. Park, O. Reizes, M. L. Fitzgerald, J. Lincecum and M. Zako, Functions of cell surface heparan sulfate proteoglycans, Annu. Rev. Biochem., 1999, 68, 729–777 CrossRef CAS PubMed.
- A. Datta-Mannan, J. Lu, D. R. Witcher, D. Leung, Y. Tang and V. J. Wroblewski, The interplay of non-specific binding, target-mediated clearance and FcRn interactions on the pharmacokinetics of humanized antibodies, mAbs, 2015, 7, 1084–1093 CrossRef CAS PubMed.
- B. Li, D. Tesar, C. A. Boswell, H. S. Cahaya, A. Wong, J. Zhang, Y. G. Meng, C. Eigenbrot, H. Pantua and J. Diao, Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge, mAbs, 2014, 6, 1255–1264 CrossRef PubMed.
- J. Chen, M. Wang, A. Joyce, D. DeFranco, M. Kavosi, X. Xu and D. M. O'Hara, Comparison of succinimidyl [125I] iodobenzoate with iodogen iodination methods to study pharmacokinetics and ADME of biotherapeutics, Pharm. Res., 2014, 31, 2810–2821 CrossRef CAS PubMed.
- Á. Szabó, T. Szendi-Szatmári, L. Ujlaky-Nagy, I. Rádi, G. Vereb, J. Szöllősi and P. Nagy, The effect of fluorophore conjugation on antibody affinity and the photophysical properties of dyes, Biophys. J., 2018, 114, 688–700 CrossRef PubMed.
- S. Vira, E. Mekhedov, G. Humphrey and P. S. Blank, Fluorescent-labeled antibodies: Balancing functionality and degree of labeling, Anal. Biochem., 2010, 402, 146–150 CrossRef CAS PubMed.
- T. E. Kraft, W. F. Richter, T. Emrich, A. Knaupp, M. Schuster, A. Wolfert and H. Kettenberger, Heparin chromatography as an in vitro predictor for antibody clearance rate through pinocytosis, mAbs, 2020, 12, 1683432 CrossRef PubMed.
- S. Schadt, S. Hauri, F. Lopes, M. R. Edelmann, R. F. Staack, R. Villaseñor, H. Kettenberger, A. B. Roth, F. Schuler and W. F. Richter, Are biotransformation studies of therapeutic proteins needed? Scientific considerations and technical challenges, Drug Metab. Dispos., 2019, 47, 1443–1456 CrossRef CAS PubMed.
- M. Krishna and S. G. Nadler, Immunogenicity to biotherapeutics–the role of anti-drug immune complexes, Front. Immunol., 2016, 7, 21 Search PubMed.
- W.-H. Boehncke and N. C. Brembilla, Immunogenicity of biologic therapies: causes and consequences, Expert Rev. Clin. Immunol., 2018, 14, 513–523 CrossRef CAS PubMed.
- K. P. Pratt, Anti-drug antibodies: emerging approaches to predict, reduce or reverse biotherapeutic immunogenicity, Antibodies, 2018, 7, 19 CrossRef CAS PubMed.
- G. R. Gunn, D. C. Sealey, F. Jamali, B. Meibohm, S. Ghosh and G. Shankar, From the bench to clinical practice: understanding the challenges and uncertainties in immunogenicity testing for biopharmaceuticals, Clin. Exp. Immunol., 2016, 184, 137–146 CrossRef PubMed.
- C. Pineda, G. Castañeda Hernández, I. A. Jacobs, D. F. Alvarez and C. Carini, Assessing the immunogenicity of biopharmaceuticals, BioDrugs, 2016, 30, 195–206 CrossRef CAS PubMed.
- M. Wadhwa, I. Knezevic, H.-N. Kang and R. Thorpe, Immunogenicity assessment of biotherapeutic products: an overview of assays and their utility, Biologicals, 2015, 43, 298–306 CrossRef CAS PubMed.
- L. I. Salazar-Fontana, D. D. Desai, T. A. Khan, R. C. Pillutla, S. Prior, R. Ramakrishnan, J. Schneider and A. Joseph, Approaches to mitigate the unwanted immunogenicity of therapeutic proteins during drug development, AAPS J., 2017, 19, 377–385 CrossRef CAS PubMed.
- F. Groell, O. Jordan and G. Borchard, In vitro models for immunogenicity prediction of therapeutic proteins, Eur. J. Pharm. Biopharm., 2018, 130, 128–142 CrossRef CAS PubMed.
- J. Gokemeijer, V. Jawa and S. Mitra-Kaushik, How close are we to profiling immunogenicity risk using in silico algorithms and in vitro methods?: an industry perspective, AAPS J., 2017, 19, 1587–1592 CrossRef CAS PubMed.
- G. Shankar, Current challenges in assessing immunogenicity, Bioanalysis, 2019, 11, 1543–1546 CrossRef CAS PubMed.
- E. Hoffmann, G. Jordan, M. Lauer, P. Ringler, E. A. Kusznir, A. C. Rufer, S. Huber, A. Jochner, G. Winter and R. F. Staack, Generation, characterization, and quantitative bioanalysis of drug/anti-drug antibody immune complexes to facilitate dedicated in vivo studies, Pharm. Res., 2019, 36, 1–15 CrossRef CAS PubMed.
- J. L. Rojko, M. G. Evans, S. A. Price, B. Han, G. Waine, M. DeWitte, J. Haynes, B. Freimark, P. Martin and J. T. Raymond, Formation, clearance, deposition, pathogenicity, and identification of biopharmaceutical-related immune complexes: review and case studies, Toxicol. Pathol., 2014, 42, 725–764 CrossRef CAS PubMed.
- P. J. Van Meer, M. Kooijman, V. Brinks, C. C. Gispen-de Wied, B. Silva-Lima, E. H. Moors and H. Schellekens, Immunogenicity of mAbs in non-human primates during nonclinical safety assessment, mAbs, 2013, 5, 810–816 CrossRef PubMed.
- E. M. Moussa, J. P. Panchal, B. S. Moorthy, J. S. Blum, M. K. Joubert, L. O. Narhi and E. M. Topp, Immunogenicity of therapeutic protein aggregates, J. Pharm. Sci., 2016, 105, 417–430 CrossRef CAS PubMed.
- F. Luzi, A. D. Gee and S. Bongarzone, Silicon compounds in carbon-11 radiochemistry: present use and future perspectives, Org. Biomol. Chem., 2021, 19, 6916–6925 RSC.
- D. Groheux, A. Cochet, O. Humbert, J.-L. Alberini, E. Hindié and D. Mankoff, 18F-FDG PET/CT for staging and restaging of breast cancer, J. Nucl. Med., 2016, 57, 17S–26S CrossRef CAS PubMed.
- K. Kitajima, M. Nakajo, H. Kaida, R. Minamimoto, K. Hirata, M. Tsurusaki, H. Doi, Y. Ueno, K. Sofue and Y. Tamaki, Present and future roles of FDG-PET/CT imaging in the management of gastrointestinal cancer: an update, Nagoya J. Med. Sci., 2017, 79, 527 Search PubMed.
- S. Sheikhbahaei, E. Mena, A. Yanamadala, S. Reddy, L. B. Solnes, J. Wachsmann and R. M. Subramaniam, The value of FDG PET/CT in treatment response assessment, follow-up, and surveillance of lung cancer, Am. J. Roentgenol., 2017, 208, 420–433 CrossRef PubMed.
- A. Montagne, D. A. Nation, J. Pa, M. D. Sweeney, A. W. Toga and B. V. Zlokovic, Brain imaging of neurovascular dysfunction in Alzheimer's disease, Acta Neuropathol., 2016, 131, 687–707 CrossRef CAS PubMed.
- D. Perani, L. Iaccarino, A. A. Lammertsma, A. D. Windhorst, P. Edison, R. Boellaard, O. Hansson, A. Nordberg, A. H. Jacobs, M. Bottlaender, D. Brooks, M. A. Carroll, S. Chalon, A. Gee, A. Gerhard, C. Halldin, K. Herholz, M. M. Herth, R. Hinz, G. M. Knudsen, B. Kuhnast, F. López-Picón, R. M. Moresco, S. Pappata, J. O. Rinne, E. Rodriguez-Vieitez, M. J. Santiago-Ribeiro, F. E. Turkheimer, K. V. Laere, A. Varrone, J. Vercouillie and A. Winkeler, A new perspective for advanced positron emission tomography-based molecular imaging in neurodegenerative proteinopathies, Alzheimer's Dementia, 2019, 15, 1081–1103 CrossRef PubMed.
- R. N. Gunn, M. Slifstein, G. E. Searle and J. C. Price, Quantitative imaging of protein targets in the human brain with PET, Phys. Med. Biol., 2015, 60, R363 CrossRef CAS PubMed.
- S. G. Kinsey and A. H. Lichtman, The Endogenous Cannabinoid System: A Cadre of Potential Therapeutic Targets, in Cannabis Use Disorders, Springer, 2019, pp. 21–29 Search PubMed.
- C. Granchi, I. Caligiuri, F. Minutolo, F. Rizzolio and T. Tuccinardi, A patent review of Monoacylglycerol Lipase (MAGL) inhibitors (2013-2017), Expert Opin. Ther. Pat., 2017, 27, 1341–1351 CrossRef CAS PubMed.
- S. P. McCluskey, C. Plisson, E. A. Rabiner and O. Howes, Advances in CNS PET: the state-of-the-art for new imaging targets for pathophysiology and drug development, Eur. J. Nucl. Med. Mol. Imaging, 2020, 47, 451–489 CrossRef CAS PubMed.
- J. W. Hicks, J. Parkes, J. Tong, S. Houle, N. Vasdev and A. A. Wilson, Radiosynthesis and ex vivo evaluation of [11C-carbonyl] carbamate-and urea-based monoacylglycerol lipase inhibitors, Nucl. Med. Biol., 2014, 41, 688–694 CrossRef CAS PubMed.
- L. Wang, W. Mori, R. Cheng, J. Yui, A. Hatori, L. Ma, Y. Zhang, B. H. Rotstein, M. Fujinaga and Y. Shimoda, Synthesis and preclinical evaluation of sulfonamido-based [11C-carbonyl]-carbamates and ureas for imaging monoacylglycerol lipase, Theranostics, 2016, 6, 1145 CrossRef CAS PubMed.
- L. Zhang, C. R. Butler, K. P. Maresca, A. Takano, S. Nag, Z. Jia, R. Arakawa, J. R. Piro, T. Samad and D. L. Smith, Identification and development of an irreversible monoacylglycerol lipase (MAGL) positron emission tomography (PET) radioligand with high specificity, J. Med. Chem., 2019, 62, 8532–8543 CrossRef CAS PubMed.
- Z. Chen, W. Mori, H. Fu, M. A. Schafroth, A. Hatori, T. Shao, G. Zhang, R. S. Van, Y. Zhang and K. Hu, Design, synthesis, and evaluation of 18F-labeled monoacylglycerol lipase inhibitors as novel positron emission tomography probes, J. Med. Chem., 2019, 62, 8866–8872 CrossRef CAS PubMed.
- L. A. McAllister, C. R. Butler, S. Mente, S. V. O'Neil, K. R. Fonseca, J. R. Piro, J. A. Cianfrogna, T. L. Foley, A. M. Gilbert and A. R. Harris, Discovery of trifluoromethyl glycol carbamates as potent and selective covalent monoacylglycerol lipase (MAGL) inhibitors for treatment of neuroinflammation, J. Med. Chem., 2018, 61, 3008–3026 CrossRef CAS PubMed.
- J. Aida, M. Fushimi, T. Kusumoto, H. Sugiyama, N. Arimura, S. Ikeda, M. Sasaki, S. Sogabe, K. Aoyama and T. Koike, Design, synthesis, and evaluation of piperazinyl pyrrolidin-2-ones as a novel series of reversible monoacylglycerol lipase inhibitors, J. Med. Chem., 2018, 61, 9205–9217 CrossRef CAS PubMed.
- Z. Chen, W. Mori, X. Deng, R. Cheng, D. Ogasawara, G. Zhang, M. A. Schafroth, K. Dahl, H. Fu, A. Hatori, T. Shao, Y. Zhang, T. Yamasaki, X. Zhang, J. Rong, Q. Yu, K. Hu, M. Fujinaga, L. Xie, K. Kumata, Y. Gou, J. Chen, S. Gu, L. Bao, L. Wang, T. L. Collier, N. Vasdev, Y. Shao, J.-A. Ma, B. F. Cravatt, C. Fowler and L. Josephson, Design, synthesis, and evaluation of reversible and irreversible monoacylglycerol lipase positron emission tomography (PET) tracers using a “tail switching” strategy on a piperazinyl azetidine skeleton, J. Med. Chem., 2019, 62, 3336–3353 CrossRef CAS PubMed.
- Y. Hattori, K. Aoyama, J. Maeda, N. Arimura, Y. Takahashi, M. Sasaki, M. Fujinaga, C. Seki, Y. Nagai and K. Kawamura, Design, synthesis, and evaluation of (4R)-1-{3-[2-(18F) fluoro-4-methylpyridin-3-yl] phenyl}-4-[4-(1, 3-thiazol-2-ylcarbonyl) piperazin-1-yl] pyrrolidin-2-one ([18F] T-401) as a novel positron-emission tomography imaging agent for monoacylglycerol lipase, J. Med. Chem., 2019, 62, 2362–2375 CrossRef CAS PubMed.
- H. C. Kolb, M. Finn and K. B. Sharpless, Click chemistry: diverse chemical function from a few good reactions, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Bioorthogonal reactions for labeling proteins, ACS Chem. Biol., 2014, 1, 16–20 CrossRef PubMed.
- C. Bednarek, I. Wehl, N. Jung, U. Schepers and S. Bräse, The Staudinger ligation, Chem. Rev., 2020, 120, 4301–4354 CrossRef CAS PubMed.
- R. Huisgen, Centenary Lecture: 1,3-Dipolar Cycloadditions, Proc. Chem. Soc., 1961, 357–396 Search PubMed.
- D. Zeng, B. M. Zeglis, J. S. Lewis and C. J. Anderson, The growing impact of bioorthogonal click chemistry on the development of radiopharmaceuticals, J. Nucl. Med., 2013, 54, 829–832 CrossRef CAS PubMed.
- H. Stöckmann, A. A. Neves, S. Stairs, K. M. Brindle and F. J. Leeper, Exploring isonitrile-based click chemistry for ligation with biomolecules, Org. Biomol. Chem., 2011, 9, 7303–7305 RSC.
- H. Wu and N. K. Devaraj, Inverse Electron-Demand Diels–Alder Bioorthogonal Reactions, in Cycloadditions in Bioorthogonal Chemistry. Topics in Current Chemistry Collections., ed. M. Vrabel and T. Carell, Springer Nature, Switzerland, 2016, pp. 109–130, DOI:10.1007/978-3-319-29686-9_6.
- M. A. Shah, X. Zhang, R. Rossin, M. S. Robillard, D. R. Fisher, T. Bueltmann, F. J. Hoeben and T. P. Quinn, Metal-free cycloaddition chemistry driven pretargeted radioimmunotherapy using α-particle radiation, Bioconjugate Chem., 2017, 28, 3007–3015 CrossRef CAS PubMed.
- M. Altai, A. Perols, M. Tsourma, B. Mitran, H. Honarvar, M. Robillard, R. Rossin, W. Ten Hoeve, M. Lubberink and A. Orlova, Feasibility of affibody-based bioorthogonal chemistry-mediated radionuclide pretargeting, J. Nucl. Med., 2016, 57, 431–436 CrossRef CAS PubMed.
- S. M. van Duijnhoven, R. Rossin, S. M. van den Bosch, M. P. Wheatcroft, P. J. Hudson and M. S. Robillard, Diabody pretargeting with click chemistry in vivo, J. Nucl. Med., 2015, 56, 1422–1428 CrossRef CAS PubMed.
- G. Beliu, A. J. Kurz, A. C. Kuhlemann, L. Behringer-Pliess, M. Meub, N. Wolf, J. Seibel, Z.-D. Shi, M. Schnermann and J. B. Grimm, Bioorthogonal labeling with tetrazine-dyes for super-resolution microscopy, Commun. Biol., 2019, 2, 1–13 CrossRef CAS PubMed.
- P. E. Edem, J. T. Jørgensen, K. Nørregaard, R. Rossin, A. Yazdani, J. F. Valliant, M. Robillard, M. M. Herth and A. Kjaer, Evaluation of a 68Ga-labeled DOTA-tetrazine as a PET alternative to 111In-SPECT pretargeted imaging, Molecules, 2020, 25, 463 CrossRef CAS PubMed.
- P. E. Edem, J.-P. Sinnes, S. Pektor, N. Bausbacher, R. Rossin, A. Yazdani, M. Miederer, A. Kjær, J. F. Valliant and M. S. Robillard, Evaluation of the inverse electron demand Diels–Alder reaction in rats using a scandium-44-labelled tetrazine for pretargeted PET imaging, EJNMMI Res., 2019, 9, 1–8 CrossRef CAS PubMed.
- E. J. L. Stéen, J. T. Jørgensen, I. N. Petersen, K. Nørregaard, S. Lehel, V. Shalgunov, A. Birke, P. E. Edem, E. T. L'Estrade and H. D. Hansen, Improved radiosynthesis and preliminary in vivo evaluation of the 11C-labeled tetrazine [11C] AE-1 for pretargeted PET imaging, Bioorg. Med. Chem. Lett., 2019, 29, 986–990 CrossRef PubMed.
- O. Keinänen, K. Fung, J. Pourat, V. Jallinoja, D. Vivier, N. K. Pillarsetty, A. J. Airaksinen, J. S. Lewis, B. M. Zeglis and M. Sarparanta, Pretargeting of internalizing trastuzumab and cetuximab with a 18F-tetrazine tracer in xenograft models, EJNMMI Res., 2017, 7, 1–12 CrossRef PubMed.
- H. Feng, H. Zhang, M. Wang, R. Vannam, H. Wang, X. Yan, W. Ouyang, X. Jia, J. M. Fox and Z. Li, Improving tumor-to-background contrast through hydrophilic tetrazines: The construction of 18F-labeled PET agents targeting nonsmall cell lung carcinoma, Chem.–Eur. J., 2020, 26, 4690–4694 CrossRef CAS PubMed.
- E. Ruivo, K. Adhikari, F. Elvas, J. Fissers, C. Vangestel, S. Staelens, S. Stroobants, P. Van der Veken, L. Wyffels and K. Augustyns, Improved stability of a novel fluorine-18 labeled TCO analogue for pretargeted PET imaging, Nucl. Med. Biol., 2019, 76, 36–42 CrossRef PubMed.
- M. Rashidian, E. J. Keliher, A. M. Bilate, J. N. Duarte, G. R. Wojtkiewicz, J. T. Jacobsen, J. Cragnolini, L. K. Swee, G. D. Victora and R. Weissleder, Noninvasive imaging of immune responses, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6146–6151 CrossRef CAS PubMed.
- D. Thomae, A.-M. Waldron, J. Fissers, S. Dedeurwaerdere, P. Van der Veken, J. Joossens, S. Stroobants, K. Augustyns and S. Staelens, In vivo evaluation of 18F-labeled TCO for pre-targeted PET imaging in the brain, Nucl. Med. Biol., 2014, 41, 513–523 CrossRef PubMed.
- S. S. Nguyen and J. A. Prescher, Developing bioorthogonal probes to span a spectrum of reactivities, Nat. Rev. Chem., 2020, 4, 476–489 CrossRef CAS PubMed.
- B. Oliveira, Z. Guo and G. Bernardes, Inverse electron demand Diels–Alder reactions in chemical biology, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- J. Sauer, Structure-reactivity problem in cycloaddition reactions to form heterocyclic compounds, Chem. Heterocycl. Compd., 1995, 31, 1140–1154 CrossRef.
- R. Rossin, S. M. Van Den Bosch, W. Ten Hoeve, M. Carvelli, R. M. Versteegen, J. Lub and M. S. Robillard, Highly reactive trans-cyclooctene tags with improved stability for Diels–Alder chemistry in living systems, Bioconjugate Chem., 2013, 24, 1210–1217 CrossRef CAS PubMed.
- A. Darko, S. Wallace, O. Dmitrenko, M. M. Machovina, R. A. Mehl, J. W. Chin and J. M. Fox, Conformationally strained trans-cyclooctene with improved stability and excellent reactivity in tetrazine ligation, Chem. Sci., 2014, 5, 3770–3776 RSC.
- J. G. O'Brien, S. R. Chintala and J. M. Fox, Stereoselective Synthesis of Bicyclo [6.1. 0] nonene Precursors of the Bioorthogonal Reagents s-TCO and BCN, J. Org. Chem., 2017, 83, 7500–7503 CrossRef PubMed.
- R. D. Bach, Ring strain energy in the cyclooctyl system. The effect of strain energy on [3 + 2] cycloaddition reactions with azides, J. Am. Chem. Soc., 2009, 131, 5233–5243 CrossRef CAS PubMed.
- Y. Fang, H. Zhang, Z. Huang, S. L. Scinto, J. C. Yang, C. W. Am Ende, O. Dmitrenko, D. S. Johnson and J. M. Fox, Photochemical syntheses, transformations, and bioorthogonal chemistry of trans-cycloheptene and sila trans-cycloheptene Ag (I) complexes, Chem. Sci., 2018, 9, 1953–1963 RSC.
- J.-P. Meyer, P. Kozlowski, J. Jackson, K. M. Cunanan, P. Adumeau, T. R. Dilling, B. M. Zeglis and J. S. Lewis, Exploring structural parameters for pretargeting radioligand optimization, J. Med. Chem., 2017, 60, 8201–8217 CrossRef CAS PubMed.
- M. F. García, X. Zhang, M. Shah, J. Newton-Northup, P. Cabral, H. Cerecetto and T. Quinn, 99mTc-bioorthogonal click chemistry reagent for in vivo pretargeted imaging, Bioorg. Med. Chem., 2016, 24, 1209–1215 CrossRef PubMed.
- A. L. Vāvere and P. J. Scott, Clinical applications of small-molecule PET radiotracers: current progress and future outlook, Semin. Nucl. Med., 2017, 47, 429–453 CrossRef PubMed.
- L. C. Gobbi, H. Knust, M. Körner, M. Honer, C. Czech, S. Belli, D. Muri, M. R. Edelmann, T. Hartung and I. Erbsmehl, Identification of three novel radiotracers for imaging aggregated tau in Alzheimer's disease with positron emission tomography, J. Med. Chem., 2017, 60, 7350–7370 CrossRef CAS PubMed.
- H. Quigley, S. J. Colloby and J. T. O'Brien, PET imaging of brain amyloid in dementia: a review, Int. J. Geriatr. Psychiatry, 2011, 26, 991–999 CrossRef PubMed.
- M. Verdurand, E. Levigoureux, W. Zeinyeh, L. Berthier, M. Mendjel-Herda, F. Cadarossanesaib, C. Bouillot, T. Iecker, R. l. Terreux and S. Lancelot, In silico, in vitro, and in vivo evaluation of new candidates for α-synuclein PET imaging, Mol. Pharm., 2018, 15, 3153–3166 CrossRef CAS PubMed.
- A. Maurer, A. Leonov, S. Ryazanov, K. Herfert, L. Kuebler, S. Buss, F. Schmidt, D. Weckbecker, R. Linder and D. Bender, 11C Radiolabeling of anle253b: a putative PET tracer for Parkinson's disease that binds to α-synuclein fibrils in vitro and crosses the blood–brain barrier, ChemMedChem, 2020, 15, 411–415 CrossRef CAS PubMed.
- X. Zhang, H. Jin, P. K. Padakanti, J. Li, H. Yang, J. Fan, R. H. Mach, P. Kotzbauer and Z. Tu, Radiosynthesis and in vivo evaluation of two PET radioligands for imaging α-synuclein, Appl. Sci., 2014, 4, 66–78 CrossRef PubMed.
- P. W Miller, A. J deMello and A. D Gee, Application of microfluidics to the ultra-rapid preparation of fluorine-18 labelled compounds, Curr. Radiopharm., 2010, 3, 254–262 CrossRef.
- L. Walker, K. E. McAleese, A. J. Thomas, M. Johnson, C. Martin-Ruiz, C. Parker, S. J. Colloby, K. Jellinger and J. Attems, Neuropathologically mixed Alzheimer's and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes, Acta Neuropathol., 2015, 129, 729–748 CrossRef CAS PubMed.
- J. Garousi, A. Orlova, F. Y. Frejd and V. Tolmachev, Imaging using radiolabelled targeted proteins: Radioimmunodetection and beyond, EJNMMI radiopharm. chem., 2020, 5, 1–26 CrossRef PubMed.
- N. Dammes and D. Peer, Monoclonal antibody-based molecular imaging strategies and theranostic opportunities, Theranostics, 2020, 10, 938 CrossRef CAS PubMed.
- K. S. Carmon and A. Azhdarinia, Application of immuno-PET in antibody-drug conjugate development, Mol. Imaging, 2018, 17, 1–10 CrossRef CAS PubMed.
- I. Colombo, M. Overchuk, J. Chen, R. M. Reilly, G. Zheng and S. Lheureux, Molecular imaging in drug development: Update and challenges for radiolabeled antibodies and nanotechnology, Methods, 2017, 130, 23–35 CrossRef CAS PubMed.
- Y. W. Jauw, C. W. Menke-van der Houven van Oordt, O. S. Hoekstra, N. H. Hendrikse, D. J. Vugts, J. M. Zijlstra, M. C. Huisman and G. A. van Dongen, Immuno-positron emission tomography with zirconium-89-labeled monoclonal antibodies in oncology: what can we learn from initial clinical trials?, Front. Pharmacol., 2016, 7, 131 Search PubMed.
- A. Niemeijer, D. Leung, M. Huisman, I. Bahce, O. Hoekstra, G. Van Dongen, R. Boellaard, S. Du, W. Hayes and R. Smith, Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer, Nat. Commun., 2018, 9, 1–5 CrossRef CAS PubMed.
- J. F. Poduslo, G. L. Curran and C. T. Berg, Macromolecular permeability across the blood–nerve and blood–brain barriers, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5705–5709 CrossRef CAS PubMed.
- F. Weber, B. Bohrmann, J. Niewoehner, J. A. Fischer, P. Rueger, G. Tiefenthaler, J. Moelleken, A. Bujotzek, K. Brady and T. Singer, Brain shuttle antibody for Alzheimer's disease with attenuated peripheral effector function due to an inverted binding mode, Cell Rep., 2018, 22, 149–162 CrossRef CAS PubMed.
- D. Sehlin and S. Syvänen, Engineered antibodies: new possibilities for brain PET?, Eur. J. Nucl. Med. Mol. Imaging, 2019, 46, 2848–2858 CrossRef CAS PubMed.
- M. J. Eigenmann, L. Fronton, H. P. Grimm, M. B. Otteneder and B.-F. Krippendorff, Quantification of IgG monoclonal antibody clearance in tissues, mAbs, 2017, 9, 1007–1015 CrossRef CAS PubMed.
- G. Liu, A revisit to the pretargeting concept – a target conversion, Front. Pharmacol., 2018, 9, 1476 CrossRef CAS PubMed.
- A. Rondon and F. Degoul, Antibody pretargeting based on bioorthogonal click chemistry for cancer imaging and targeted radionuclide therapy, Bioconjugate Chem., 2019, 31, 159–173 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |