
In vivo assessment of triazine lipid nanoparticles as transfection agents for plasmid DNA†
David
Nardo
,
Michelle G.
Pitts
,
Rupinder
Kaur
and
Vincent J.
Venditto
 *
*
Department of Pharmaceutical Sciences, University of Kentucky College of Pharmacy, Lexington, KY 40536, USA. E-mail: Vincent.venditto@uky.edu
First published on 7th October 2022
Abstract
Non-viral vectors for in vivo delivery of plasmid DNA rely on optimized formulations to achieve robust transgene expression. Several cationic lipids have been developed to deliver nucleic acids, but most recent literature has focused on mRNA due to its increased expression profile and excluded plasmid DNA, which may have the advantage of being less immunogenic. In this study, we describe the in vivo evaluation of cationic triazine based lipids, previously prepared by our group. We identify one lipid with limited in vivo toxicity for studies to optimize the lipid formulations, which include an evaluation of the influence of PEG and helper lipids on transgene expression. We then demonstrate that lipoplexes, but not lipid nanoparticles, formed from triazine lipids achieve similar transgene expression levels as AAV vectors and offer enhanced expression as compared to a commercially available cationic lipid, DOTAP. Importantly, the lipid nanoparticles and lipoplexes induce minimal antibody profiles toward the expressed protein, while serving as a platform to induce robust antibody responses when directly delivering the protein. Collectively, these data demonstrate the potential for triazine based lipids as non-viral vectors for gene delivery, and highlights the need to optimize each formulation based on the exact contents to achieve enhanced transgene expression with plasmid DNA constructs.
Introduction
Lipid-based non-viral vectors for nucleic acid delivery have advanced significantly over the past 30 years leading to two approved COVID-19 mRNA vaccines and a number of other approved therapies based on antisense oligonucleotides, siRNA, and mRNA.1,2 Additionally, several other therapies are currently in pre-clinical and clinical development to treat various diseases and infections.3 The advances made to achieve clinical success have focused on nucleic acid stability, cationic lipid development, formulation composition, and manufacturing.3 Nevertheless, evaluation of new lipid constructs are needed to identify platforms with improved efficacy and reduced toxicity as non-viral delivery vehicles for nucleic acid therapeutics.4The design criteria for novel lipid structures are largely based on structure activity relationships of lipid libraries that have converged on lead derivatives for nucleic acid complexation and eventual release from the endosomal compartment.5–7 Some of the lead lipids include monovalent cationic lipids such as 1,2-di-O-octadecenyl-3 trimethylammonium propane (DOTAP),8 DLin-MC3-DMA (MC3),9 and others with similar structural motifs.3 Additionally, cationic lipids with aromatic head groups have been shown to complex plasmid DNA more efficiently, but exhibit mixed results between in vitro and in vivo transfection efficiency.10,11 Building upon these data, we previously reported the synthesis of a novel class of triazine (TZ) lipids, based on cyanuric chloride, that demonstrate potential for nucleic acid delivery due to the aromatic linker with appended cationic moieties.12 Like the dimerizable, redox-sensitive lipids reported by Candiani et al. and the compounds published recently by Pennetta et al., these lipids show strong potential for gene delivery.13,14
The leading cationic and ionizable lipids for gene delivery have been further optimized by including them in specific formulations containing helper lipids (e.g. DOPE, DSPE-PEG, cholesterol) to achieve maximal transfection efficiency of siRNA and mRNA in vitro and in vivo.15 Much of the subsequent work with novel lipids have utilized similar formulations to deliver RNA, but the generalizability across lipid architectures and in combination with RNA or DNA constructs are necessary to ensure optimal transfection efficiency in vivo.5 Furthermore, the resultant immune responses induced with different lipid constructs are critical to ensure that minimal anti-transgene immune responses are observed when not being used as vaccine carriers.16
To build upon our previous work evaluating a series of TZ lipids in vitro, we sought to extend the evaluation of cationic lipids as non-viral vectors for plasmid DNA delivery by assessing toxicity, transfection efficiency, and transgene immunogenicity. To this end, lipoplexes (LPs) and microfluidic lipid nanoparticles (LNPs) prepared with TZ lipids or 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) were used to deliver an AAV-based GFP plasmid and a lentiviral human alpha-1 antitrypsin (hAAT) plasmid.17 The LNPs were formulated using the lipids shown in Fig. 1 based on an optimized lipid composition reported in the literature.17 Herein, we demonstrate the ability of TZ lipids to improve in vivo plasmid transfection beyond that achieved with DOTAP formulations and explore the immunologic response induced against the expressed transgenes.
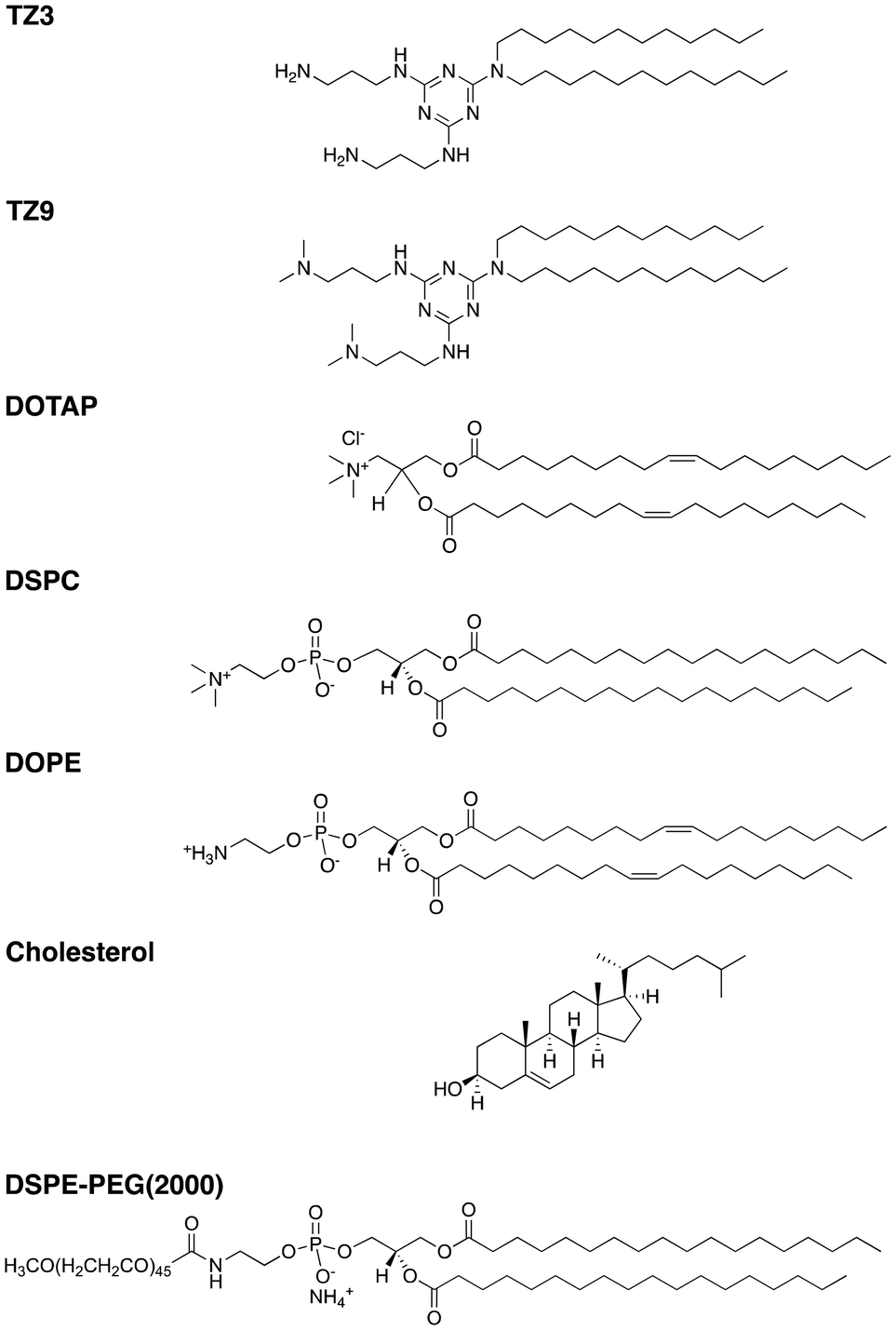 | ||
| Fig. 1 Structure of triazine lipids and other lipids used in plasmid formulations. | ||
Experimental methods
Development of lipid nanoparticles
Two types of nanoparticles were used for experiments: liposomes and plasmid lipid nanoparticles (LNPs). In both cases, the lipids used were dissolved in chloroform, mixed at the ratio described in each figure legend, dried into a thin lipid film by rotary evaporation, and placed under house vacuum overnight before use. To form liposomes, the dried lipids were rehydrated in HEPES buffered saline (20 mM HEPES, 145 mM NaCl, pH 4) (HBS) and sonicated until translucent at 60 °C before being mixed with HBS to the final concentration of 10 mM at pH 7.4. Lipoplexes were formed from liposomes by mixing liposomes and DNA at a ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 positive to negative charges in Opti-MEM (for cells) or pH 7.4 HBS (for mice) and incubated at room temperature for at least 12 minutes prior to administration.
:1 positive to negative charges in Opti-MEM (for cells) or pH 7.4 HBS (for mice) and incubated at room temperature for at least 12 minutes prior to administration.
To form LNPs, dried lipids were rehydrated to a concentration of 10 mM in ethanol with 10 μL of 5 M HCl per mL of ethanol and mixed with a solution of DNA at 40 ng μL−1 of DNA in 300 mM citric acid, pH 4. The ethanol and aqueous solutions were mixed into LNPs using the Ignite microfluidic system (Precision NanoSystems, Inc.) at a ratio of 1:3 ethanol to aqueous, at a rate of 12 mL min−1. The LNPs were then transferred into 3 mL Slide-A-Lyzer cassettes (ThermoFisher # PI87732) and stirred at 200 rpm in 1.5 L of a 300 mM citric acid, pH 4 solution for three hours, followed by three hours in 1.5 L of HBS buffer, pH 4 (145 mM NaCl and 20 mM HEPES), before being moved overnight to a 1.5 L solution of HBS, pH 7.4.
Characterization of nanoparticles
Nanoparticle size was determined using a Zetasizer Nano ZS (Malvern Panalytical) with the following settings: four measurements of fifteen, five second runs detected at a backscatter angle of 173° at room temperature. The zeta potential for the liposomes was determined in a DTS1070 folded capillary zeta cell using the following settings: four measurements of at least 50 runs modelled with the Smoluchowski equation at room temperature using the automatic settings from the instrument. The concentration of DNA in LNPs after dialysis was quantified using an AccuClear® Ultra High Sensitivity dsDNA Quantification Kit (Biotium # 31027) and quantified on a BioTek Synergy H1 plate reader. Encapsulation efficiencies were determined by comparing the amount of DNA in the LNP solution vs. the DNA solution used to make them, after disrupting the LNPs with 0.5% C12E10 (Abcam # ab146563) and adjusting for volume differences (i.e.: excess volume added during dialysis and dilution volume during ethanol mixture).Optimization of LNP formulation in vitro
To measure transfection efficiency and subsequent green fluorescent protein (GFP) expression in vitro, 5 × 104 HEK293T or J774A.1 cells, or 1 × 105 mature dendritic cells, were plated in 96-well flat-bottom sterile cell culture plates and allowed to become confluent or adhere overnight. The next day, the cells were treated with 200 ng of pDNA encoding for GFP (Addgene product number 37825), delivered via nanoparticles, and incubated overnight with the nanoparticles. The media was changed at 24 hours, and after 72 total hours, cells were trypsinized briefly and transferred to a round bottom 96 well plate for flow cytometric analysis of viability and GFP expression. Live/dead staining was performed using Zombie viability dye (Biolegend) according to manufacturer instructions. Cells were washed and resuspended in FACS buffer (Mg2+/Ca2+-free Hanks’ balanced salt solution, 2 mM EDTA, 25 mM HEPES and 1% FBS) for fluorescence measurement. The gating schemes used for all flow cytometry are shown in Fig. S4.† For generating images, 1 × 105 HEK 293T cells per well were seeded into a black 96-well plate and treated with 100 ng of GFP plasmid delivered via LNPs of varying length for 6 hours, incubated for another 48 hours in phenol free media, stained with RedDot™ 1 nuclear stain (Biotium) and imaged with a BioTek Cytation 7 (Fig. S4G†).Uptake of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD) liposome was assessed 24 hours after liposome treatment after washing cells three times with PBS to remove free liposomes prior to trypsinization and staining as described above.
Cells
HEK293T cells, kindly donated by Dr Gregory Graf of the University of Kentucky College of Pharmacy, J774A.1 macrophages (ATCC TIB-67) or bone marrow derived dendritic cells were used for cell experiments and maintained at 37 °C with 5% CO2.Differentiation of bone marrow derived dendritic cells (BMDCs)
Mature murine dendritic cells were obtained by culture of bone marrow monocytes as described previously18 using recombinant murine GM-CSF (Biolegend). On day 10 of culture, lightly adherent cells were detached with gentle washing and moved to a 96-well flat bottom cell culture plate at a density of 100000 cells per well, in triplicate, for experiments.
Mice
Mice were purchased from Jackson Labs at 5–6 weeks of age and used in experiments at 7–9 weeks. C57BL/6J (#000664) mice were used for toxicity experiments shown in Fig. 2 and Fig. S1,† while BALB/cJ (#000651) were used for transfections in all other figures. Equal numbers of male and female mice were used in each experiment. Mice were sedated using isoflurane gas prior to blood collection by saphenous vein puncture or intraperitoneal (i.p.) injections. Baseline serum levels of all experimental parameters were established one week prior to injections. Mice were housed in a specific-pathogen free facility at the University of Kentucky, and all experimental procedures were approved by the University of Kentucky Institutional Animal Care and Use Committee #2020-3523.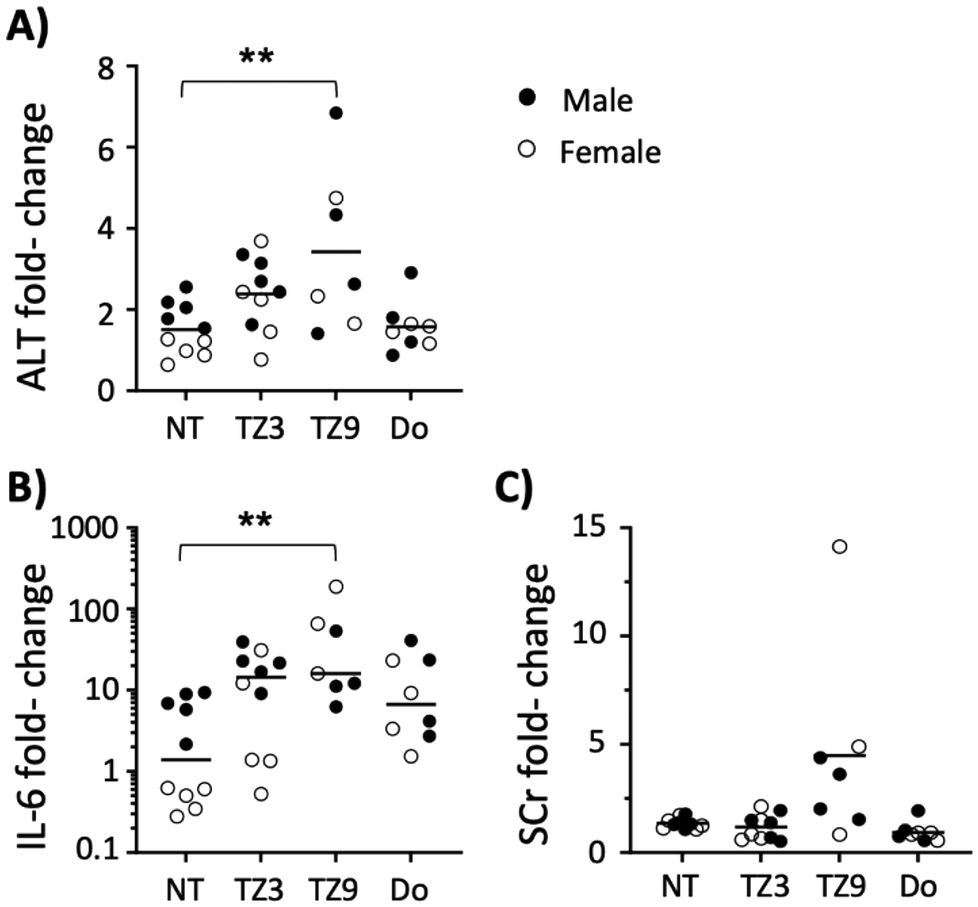 | ||
| Fig. 2 TZ3 does not result in significant in vivo toxicity at 20 mM. Equal numbers of male and female seven-week-old C57BL/6J mice were administered 100 μL of 20 mM cationic lipid (TZ3, TZ9 or DOTAP (Do)) nanoparticles i.p. in HEPES buffered saline (10 total mice per group – 5 male, 5 female). (A–C) Serum alanine aminotransferase (ALT), serum creatinine (SCr), and interleukin-6 (IL-6) levels were measured 48 hours after treatment. Fold-change from baseline measurements drawn one week prior were compared with those of untreated animals (NT). Lines represent mean, and dots represent individual animals. The TZ9 group represents only the 7 surviving animals. Significance was compared using one way ANOVA and Dunnett's (A) or Kruskal Wallis (B and C) tests; only significant comparisons are shown. | ||
Evaluation of TZ lipid toxicity in vivo
Mice were administered 0.1 mL of the liposomal solution i.p. at a concentration of 10 mM or 20 mM. Forty-eight hours later, the mice were bled for evaluation of serum creatinine (SCr; Crystal Chem no. 80350), alanine aminotransferase (ALT; AAT Bioquest no. 13803) and interleukin-6 (IL-6; Biolegend no. 431304) according to manufacturer instructions.Evaluation of GFP transfection in the liver
For in vivo evaluation of GFP transfection, mice were administered GFP plasmid (Addgene no. 37825) i.p. using LNPs or LPs at a dose of 10 μg of DNA or AAV8 at a dose of 2 × 109 genome copies per mouse (equating to approximately 200 ng of DNA) and serum was collected 3 days later to evaluate ALT levels as described above. Seven days after transfection, mice were euthanized by CO2 inhalation and perfused with 10 mL of Ca2+/Mg2+-free HBSS followed by 10 mL of HBSS containing 1 mg mL−1 type IV collagenase (MP Biomedicals) via the hepatic portal vein. Livers were excised, diced with a scalpel, and incubated for 30 minutes at 37 °C in RPMI containing collagenase at 1 mg mL−1 and 50 μg mL−1 DNAse (MP Biomedicals). Digested liver fragments were gently pressed through a 0.22 μm mesh filter and the cells were collected, centrifuged at 50g for 3 minutes with the supernatant discarded, and then washed twice more with phosphate buffered saline. The remaining cell suspension (50 μL) from each liver was then moved to polycarbonate tubes and diluted 1:10 in FACS buffer containing anti-mouse CD16/32 to block Fc receptors. After blocking, samples were incubated with fluorescent antibodies directed against mouse CD45 and CD146 for 30 minutes at 4 °C. After 30 minutes, the cells were washed twice in FACS buffer before being resuspended for fluorescence measurement.
All flow cytometry antibodies, as well as viability dyes, were purchased from Biolegend. Fluorescence measurement was performed using an Attune NxT flow cytometer (ThermoFisher).
Evaluation of hAAT transfection efficiency in vivo
To assess hAAT transfection efficiency in vivo, mice were administered 10 μg of DNA per mouse in either LPs or LNPs, or 25 μg of hAAT protein in saline or liposomal solution. Seventy-two hours after injection the mice were bled for assessment of ALT levels (assessed as described in the toxicity section) and hAAT expression. hAAT levels were determined via ELISA using serum diluted 1:1 in PBS containing 0.05% casein (PBS-C; 124250; Beantown Chemical), as described previously.19
Quantification of anti-hAAT antibody titers and determination of subclass ratios
To assess the presence of antibodies toward hAAT, 50 μL of hAAT (OriGene #RG202082) was plated at 2 μg mL−1 in carbonate buffer, pH 9.7, on a 96 well high binding plate (Greiner #82050-720) and incubated overnight at 4 °C. The next day, the plates were washed with PBS containing 0.1% Tween-20 (PBS-T) and blocked for 1 hour with PBS-C at 37 °C. After blocking, serum samples were plated at dilutions ranging from 1:100 to 1:1000000 and incubated at 37 °C for 2 hours. Secondary antibody (goat anti-mouse IgG HRP; Invitrogen #16066) diluted 1:2000 was applied for 30 min at 37 °C, followed by a 30 min incubation with tetramethylbenzidine (Rockland). Absorbance at 450 nm was recorded using a BioTek Synergy H1 microplate reader. Reciprocal endpoint titers were determined by plotting A450 values versus known dilutions, calculating the slope of that line, and dividing the slope by two times the average of the blank (no serum) wells.
Anti-hAAT IgG subclass ratios were assessed as described above, using a single 1:100 sample dilution and the following detection antibodies: goat anti-mouse IgG1-HRP (Abcam ab98693) at 1:10000, IgG2a-HRP (Abcam ab98698) at 1:5000, IgG2b-HRP (Abcam ab98703) at 1:10000 and IgG3 (Jackson 115-035-209) at 1:5000. Subclass ratios were calculated by dividing the absorbance of each subclass by that of IgG1 for each individual mouse.
Data analysis and statistics
Data were organized and analysed using Graph Pad Prism v.9 for Windows. Data in each experiment were assessed for normality, followed y ANOVA or non-parametric tests (described in each figure legend), as appropriate, to assess statistically significant differences among them; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. In all figures, only statistically significant comparisons are shown.Results and discussion
In vivo toxicity of TZ lipids
Two triazine lipids that previously demonstrated efficacy in vitro (triazine lipids 3 and 9, or TZ3 and TZ9)12 were first evaluated for their in vivo toxicity before analysing their transfection efficiency. Groups of male and female C57BL/6J mice were administered TZ LNPs at 10 and 20 mM intraperitoneally (i.p.) in HEPES buffered saline and forty-eight hours later serum levels of alanine aminotransferase (ALT), interleukin 6 (IL-6), and creatinine (SCr) were tested and compared with baseline levels drawn one week prior. As Fig. 2 demonstrates, TZ3 treatment at 20 mM led to an upward, but insignificant trend in ALT and IL-6 levels, with no changes observed in SCr. TZ9 lipid, however, led to significant elevations in ALT and IL-6, as well as a trend toward higher SCr levels. Additionally, three of the ten mice treated with this compound died after of exposure. At 10 mM, TZ9 also induced statistically significant increases in ALT and IL-6, with one mouse dying in this treatment group (Fig. S1†).The toxicity of TZ9 in vivo was unexpected as in vitro studies, published previously, indicated TZ9 to be less toxic than TZ3.12 The discordant results between in vitro and in vivo studies suggest that the cause of toxicity is more complex than simple cytotoxicity; however, the exact physiologic mechanism of toxicity remains unclear. Based on the exposure-induced increase in a limited number of toxicity indicators and the visual inspection of some of the mice, it seems likely that a multi-system inflammatory response occurred following i.p. treatment with TZ9, thus highlighting the need for more extensive characterization of these compounds and their potential mechanisms of toxicity.20,21 Regardless, because of these findings, only TZ3 was chosen for further evaluation in transfection experiments.
LNP formulation containing DSPE-PEG2000 and DSPC fails to transfect mice in vivo
We have previously shown that TZ lipids can deliver human alpha-1 antitrypsin (hAAT) plasmid in vitro.12 This protease inhibitor has significant relevance in gene therapy as it is associated with severe lung damage and other sequelae in deficient patients, in addition to showing promise in other diseases due to its anti-inflammatory activity.22,23 Furthermore, research by Song et al. shows AAV8 delivery of hAAT into mice can prevent the antibody development associated with delivery of the protein, a major concern with biologic therapeutics.24–32Previous efforts to achieve in vivo hAAT plasmid delivery using liposomal vectors have resulted in modest outcomes in animals.33 Additionally, in a phase I clinical study, the hAAT levels achieved with this method were subtherapeutic.25,34 In recent years, the field of liposomal vaccines has demonstrated that mRNA can increase transgene expression and immunogenicity, compared with plasmids.35 However, the immunogenic potential of mRNA can deter its use in gene replacement, where anti-transgene antibodies lead to therapeutic failure. For this reason, we decided to pursue TZ lipid transfection efficiency with plasmids, rather than mRNA.24,36–38
Most modern liposomal gene therapy uses lipid nanoparticles (LNPs) made via ethanol injection in microfluidic devices, as these can improve delivery by better entrapping the nucleic acid and preventing the aggregation which can occur with lipoplexes (LPs).5,39–42 LNPs are often prepared using a molar ratio of 50% cationic lipid, which ionically binds nucleic acid, 10% 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 38.5–39% cholesterol, both of which stabilize the lipid bilayer, and 1–1.5% of lipid conjugated to PEG2000, usually 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) or 1,2-dimyristoyl-rac-glycero-3-methoxy polyethylene glycol-2000 (DMG-PEG2000).43–46 To investigate the utility of TZ3 in LNP plasmid delivery, TZ3, DSPC, cholesterol, and DSPE-PEG2000 were combined at a 50:10:39:1 molar ratio and compared to DOTAP LNPs. The resulting nanoparticles ranged in size between 70–80 nm in diameter, with zeta potentials between 8–16 mV, and encapsulation efficiency above 70% (Table S1†).47 However, when administered to mice via tail vein injection, these formulations failed to elicit detectable hAAT protein levels (Fig. S2†).
In vitro optimization of LNP formulation using GFP plasmid
In trying to understand the reason behind the failure of these LNPs a thorough evaluation of the literature was made. Among the components of LNPs, PEG content has been tied to reduced transfection efficiency, as the polymer can inhibit interactions between the cationic nanoparticles and cells.48–50 However, PEG has been shown to be necessary for improving circulation half-life and providing stability to nanoparticles in vivo.48,50–53 Therefore, rather than removing the 1% DSPE-PEG2000 in the LNPs, we investigated the effect of reducing PEG length on in vitro transfection efficiency. LNP formulations were made with TZ3 and different lengths of DSPE-PEG to encapsulate a GFP plasmid.Dynamic light scattering analysis of the initial nanoparticles made with PEG2000 and hAAT plasmid for the data presented in Fig. S2† exhibit similar characteristics to those described in the literature for plasmid-based nanoparticles (Table S1†).47,50,54 However, the nanoparticles made with shorter PEG chains were larger and more polydisperse, a trend that has been reported previously with the reduction of PEG2000 concentration.47,50,54 DOPE also increased size and polydispersity compared with DSPC. This change could possibly be attributed to the increased rigidity of the stearoyl tails of DSPC compared with DOPE's oleyl tails but has not been previously noted to the best of our knowledge. Finally, TZ3, while successful at encapsulating DNA, trended toward lower encapsulation efficiencies as compared to DOTAP, generally encapsulating 60–70% of DNA vs. DOTAP's 70–80% encapsulation.
Confluent HEK293T cells were then transfected with 200 ng of plasmid per well. Because HEK293T cells aggregate, initial microscopic imaging was inconclusive (see Fig. S4G†), therefore GFP quantification, following transfection, was performed via flow cytometry using the gating scheme shown in Fig. S4A.†55,56 As shown in Fig. 3A, increased PEG length correlated with a decrease in GFP expression. The nanoparticles made without PEG or with PEG550 yielded the highest GFP expression. However, PEG-free LNPs were unstable and formed aggregates. Consequently, formulations with PEG550 were chosen for further evaluation.
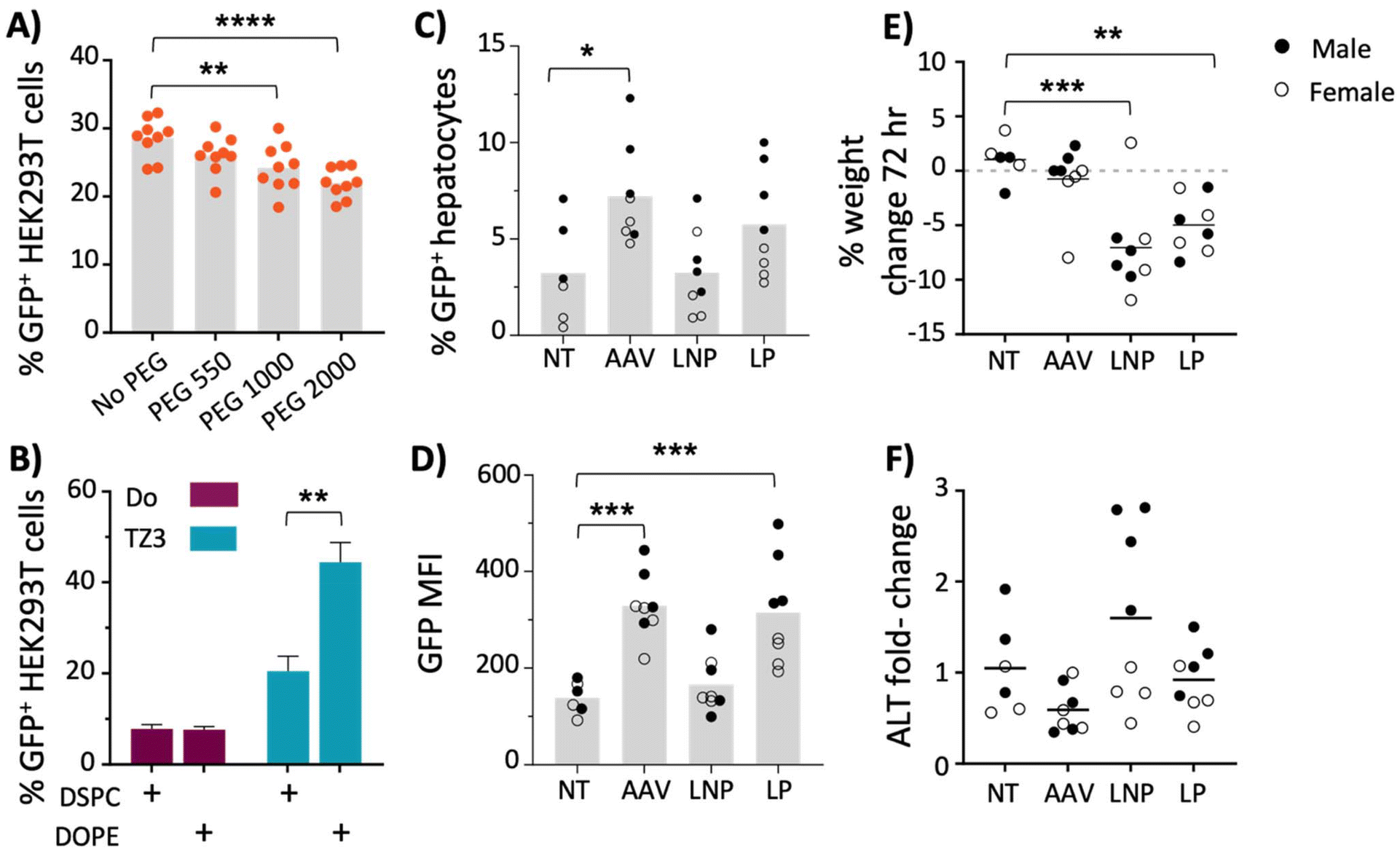 | ||
| Fig. 3 PEG550, DOPE, and TZ3 yield improved transfection with LNP. (A and B) HEK293T cells were transfected with 200 ng GFP plasmid per well using LNPs and analyzed three days later for GFP expression by flow cytometry. (A) LNPs formulated with 50% TZ3, 10% DPSC, 39% cholesterol and 1% DSPE-PEG(550–2000), or 40% cholesterol and no PEG. (B) LNPs formulated with 50% DOTAP (Do) or TZ3, 10% DSPC or DOPE, 39% cholesterol and 1% DSPE-PEG550. Pooled data from three independent experiments are shown; n = 3 transfected wells per group per experiment. (C–F) Equal numbers of male and female BALB/c mice (6 total mice in untreated group (NT), 8 total mice in treated groups) were administered 1 × 109 genome copies of AAV8-GFP or 10 μg of GFP plasmid in either LNPs made with 50% TZ3, 10% DOPE, 39% cholesterol and 1% DSPE-PEG550 or LPs made with 50% TZ3 and 50% DOPE. One week post administration, hepatocytes were evaluated for percent GFP positive cells (C) or mean fluorescence intensity (MFI; D). Percent weight change (E) and serum ALT (F) were evaluated at 72 hours post-administration. Bars indicate mean transfection efficiency; dots represent individual transfection wells (A) or mice (C–F). Data were compared with one-way ANOVA and Dunnett's post-hoc test (A and C–F) or Sidak's test (B); comparisons shown in (A) are to No PEG and in (C–F) to untreated (NT), only significant comparisons are shown. | ||
Another LNP alteration that has been shown to improve transfection is the use of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), rather than DSPC, as a helper lipid.45,47,57 To further optimize transfection, LNP formulations containing DSPE-PEG550 was tested using DSPC or DOPE as helper lipids, with either TZ3 or DOTAP as the cationic component. As shown in Fig. 3B, the nanoparticles containing both TZ3 and DOPE significantly increased transfection efficiency. While the attributes of the nanoparticles can likely be improved by further altering multiple parameters such as cholesterol content, no additional alterations were made and further evaluation of TZ3 was pursued using PEG550 and DOPE.58,59
In vivo transfection with GFP plasmid
Upon optimization of a LNP formulation containing PEG550 and DOPE, 10 μg of GFP plasmid was delivered to BALB/c mice via either LNP or our previously reported LPs made with a 1:1 TZ3:DOPE ratio and compared to AAV8 delivery at 2 × 109 GC per mouse.12 The LNPs were over 200 nm in diameter (Table S1†) and non-PEGylated formulations exhibited a propensity to aggregate, so delivery was made by i.p. injection to prevent potential harm to the animals from particle aggregation after intravenous (i.v.) administration. Furthermore, this route has been shown to yield similar results as i.v. administration.41,60–62
Because of our interest in antitrypsin, which is expressed primarily in the liver, and in comparing the nanoparticles to AAV8, which has liver tropism, GFP expression was quantified only in the liver.63,64 However, it is important to note that lipid nanoparticles have been shown to distribute to, and very effectively transfect, various other tissues, particularly the lungs and spleen.46,65–69
While these tissues were not evaluated in the experiments carried out in this manuscript, future experiments that characterize the biodistribution of TZ lipid nanoparticles and their transgenes are warranted. As shown in Fig. 3C and D, transfection with LNPs was less efficient than AAV8 transduction with a low viral dose of 2 × 109 GC per mouse (∼200 ng of DNA). LP transfection led to similar GFP expression as AAV, with average mean fluorescence intensity (MFI) of approximately 300. However, the fluorescence was heterogeneous with males trending higher than females (Fig. 3D).
In addition to being less efficacious, the LNPs also showed greater toxicity than AAV8 transduction, as mice treated with LNPs and LPs lost 1–12% of their body weight at 72 hours and those treated with LNPs had slight (non-significant) elevations in ALT levels at the same timepoint, while AAV8-treated mice displayed neither sign of toxicity (Fig. 3E and F).
In vivo transfection with hAAT plasmid
Based on these data, we then sought to re-evaluate hAAT transfection, using both LNPs and LPs containing 10 μg of hAAT DNA. Since we were interested in the immune responses generated against the protein, a group of control mice was also given 25 μg of hAAT protein in saline.32,70,71 Additionally, since lipids themselves can increase protein immunogenicity, separate groups of mice also received hAAT with 1 mM TZ3, DOTAP, or 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) as a natural phospholipid control.72Following transfection, the optimized LNPs led to detectable serum hAAT in some of the mice (Fig. 4), but as with GFP, LPs led to higher transfection efficiency. In mice treated with LPs, hAAT levels were, on average, 9.5 ng mL−1 for TZ3 and 3 ng mL−1 for DOTAP, which were similar to the concentrations reported by Crepso et al. and Aliño et al.33,70 In light of previous literature comparing LNPs to LPs, these finding are surprising, but could be related to the remaining PEG in the LNP formulation, which can hinder transduction with both LNPs and LPs.5,40,73 To our knowledge most reports suggest that LNPs formed through microfluidics should outperform LPs. One report by Meisel et al. using LPs formed by sonication (as with our LPs) showed improved transfection with these nanoparticles compared with LPs made with DMSO or ethanol, especially when these contained DOPE. However, their lipoplexes were formed with DMSO or ethanol and both contained organic solvents but did not contain PEG.74
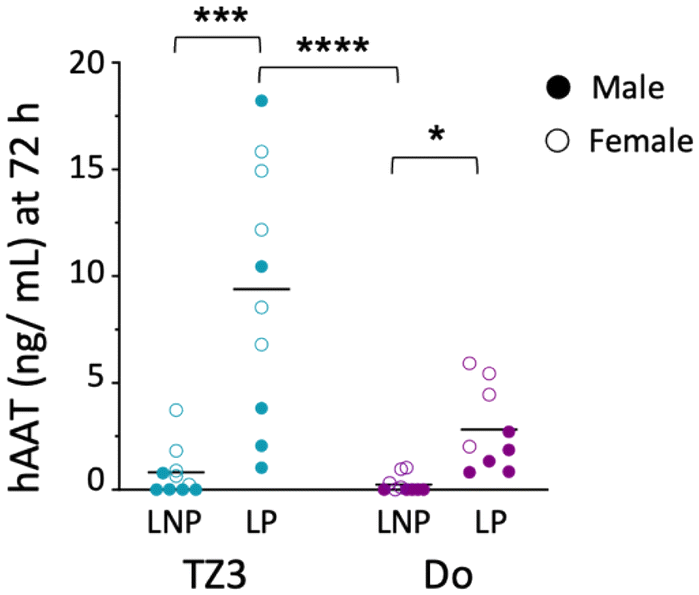 | ||
| Fig. 4 TZ3 LP transfection is more efficient in vivo than DOTAP or LNP. Five male (filled circles) and five female (open circles) BALB/c mice were administered 10 μg of hAAT DNA with LNPs made with 50% TZ3 or DOTAP (Do), 10% DOPE, 39% cholesterol and 1% DSPE-PEG550 or LPs made with 50% TZ3 and 50% DOPE. Seventy-two hours later, protein expression in the serum was assessed via ELISA. Lines represent mean hAAT concentration; dots represent individual animals. Data were compared with one-way ANOVA and Kruskal–Wallis test, only significant comparisons are shown. | ||
In the mice given hAAT protein with individual lipids, serum hAAT levels at 72 hours were detectable but overall lower than expected based on the reported half-life.75,76 However, DMPC produced an intriguing protein increase in females that was not detected in males (Fig. S3A†), perhaps due to the mass difference in the assessed via ALT quantification. As shown in Fig. S3B,† ALT levels in LNP and LP-treated mice rose 2–6 times above baseline at 48 hours. As with GFP transfection, toxicity was also observed, while direct protein administration did not exhibit signs of toxicity, suggesting that the toxicity is associated with liposomal transfection, but not the lipids themselves.
Finally, hAAT levels persisted at 4 weeks after treatment, but only in animals treated with TZ3 lipoplexes, highlighting the efficacy of this transfection strategy compared with LNPs and free protein (Fig. S3C†).
Immune response to hAAT in vivo
Human alpha-1 antitrypsin has previously been reported to be immunogenic in mice.24,77 Therefore, anti-hAAT reciprocal endpoint titers (RETs) were assessed two weeks after hAAT transfection (day 14) and compared with the response induced by the protein. As Fig. 5A shows, the LNP transgenes did not induce a significant increase of anti-hAAT IgG titers while LP transfection led to titers similar to those of the free protein in saline.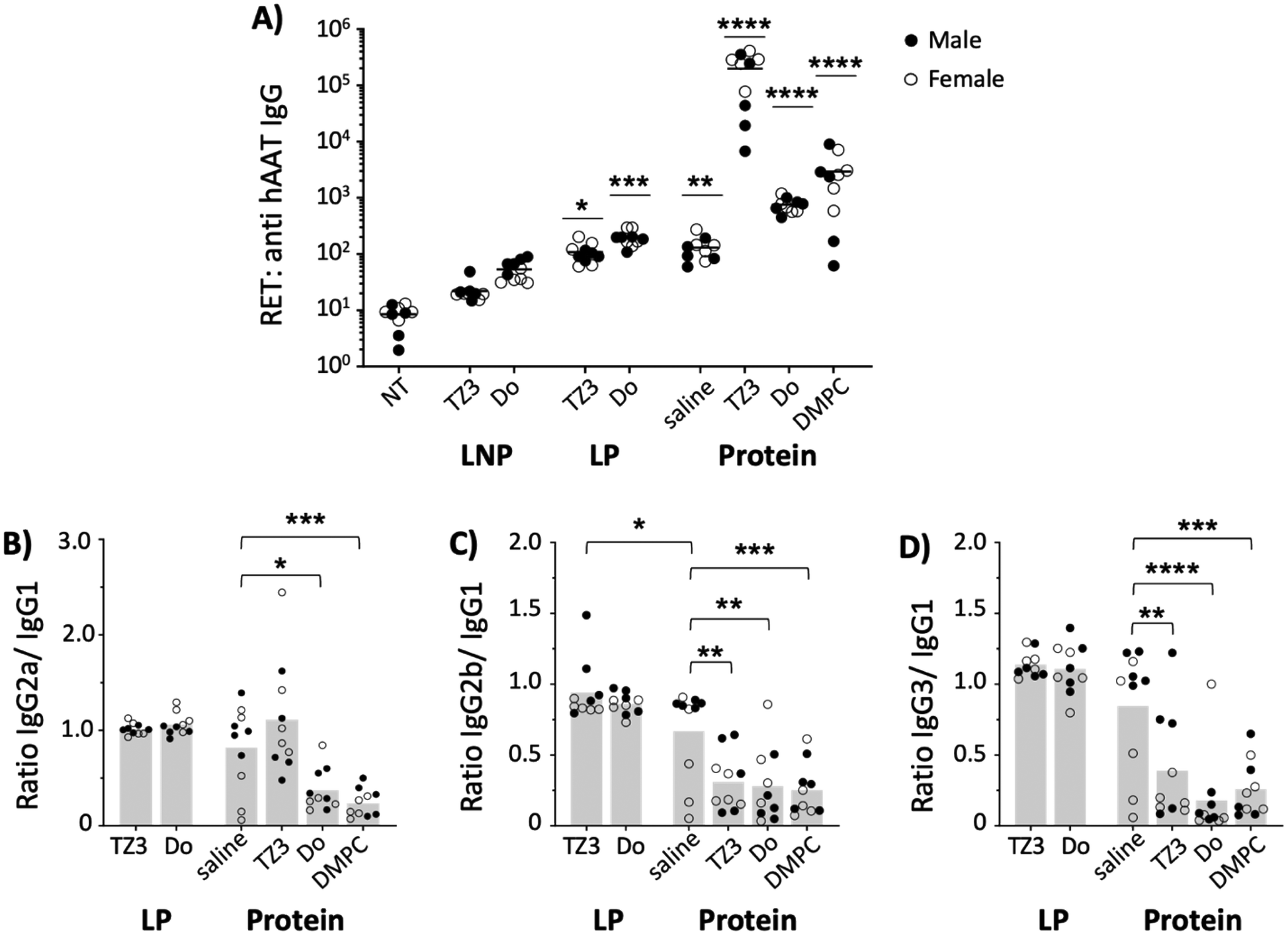 | ||
| Fig. 5 Transgene expression using TZ3 as a delivery vector elicits minimal antibody responses, while administration of hAAT protein with TZ3 results in significant immunogenicity and a Th1 bias. BALB/c mice were administered 10 μg of hAAT DNA with LNPs made with 50% TZ3 or DOTAP (Do), 10% DOPE, 39% cholesterol and 1% DSPE-PEG550 or LPs made with 50% TZ3 and 50% DOPE; or 25 μg of hAAT protein in saline or 1 mM lipid solution (10 mice per group – 5 male, 5 female). (A) Fourteen days after administration, anti-hAAT IgG reciprocal endpoint titers (RET) were assessed via serum ELISA. Significance determined by Kruskal–Wallis test; comparisons made to untreated animals (NT). Ratios of IgG2a/IgG1 (B), IgG2b/IgG1 (C), and IgG3/IgG1 (D) were assessed at the same timepoint for treatment groups that had significantly higher RET than untreated. Bars indicate mean, while dots indicate individual animals. Significance as compared to protein delivered in saline was determined by one-way ANOVA in (B–D). | ||
While this difference could be accounted for by the difference in protein expression between the two groups (Fig. 4), previous literature shows that protein concentrations do not necessarily correlate with titer development45 and that proteins need not achieve quantifiable serum levels for protein to induce robust immunity.78
In addition to serving as gene delivery vehicles, cationic lipids can also display adjuvant properties. For this reason, the hAAT protein was also administered with 1 mM TZ3 liposomes, using DOTAP and DSPC as cationic and neutral lipid controls. As expected, DOTAP and DSPC increased titers by 10- and 100-fold, respectively. TZ3 administration with hAAT protein also led to an elevation in titers that was surprisingly 1000-fold higher than the saline control, suggesting a potential role for TZ lipids in the setting of protein immunizations.79,80
Since shifts in the Th1- or Th2-type response against a protein can affect how it is processed following administration, the anti-hAAT subclass composition was also assessed via ELISA and the ratios of IgG2a, 2b, and 3 to IgG1 were determined (Fig. 5B–D) in all but LNP samples, which did not achieve a sufficient antibody response.81–85 Based on the subclass ratios, both TZ3 and DOTAP-delivered transgenes led to a balanced Th1/Th2 response, similar to hAAT protein in saline. However, when the protein was delivered with lipids, a strong Th2 response was observed, as indicated by ratios lower than 1.0. This response is similar to that induced with Freund's incomplete adjuvant, suggesting that the lipids behave similarly to an oil in water emulsion.82,86 Interestingly, the response to hAAT protein delivered with TZ3 led to a more balanced IgG2a/IgG1 ratio than DOTAP and DMPC, an observation that could have clinically relevant implications that will be explored in future experiments.
In vitro uptake of liposomes in antigen presenting cells is reduced by PEG
Lu et al. previously showed that the development of anti-hAAT antibodies following transfection was dependent on whether the AAV serotype used (AAV1 vs. AAV8) was able to transfect antigen presenting cells (APCs).77 Since PEGylation of nanoparticles reduces uptake by APCs,87,88 it was hypothesized that PEG550 in the LNPs was accountable for the difference in antibody responses toward hAAT. To test this hypothesis, liposomes containing the fluorescent lipophilic dye DiD were made with or without DSPE-PEG2000 and incubated with J774 macrophages or bone marrow derived dendritic cells (DC). After 18 hours, cells were washed to remove free liposomes and assayed for DiD fluorescence by flow cytometry. As expected, the addition of PEG resulted in lower DiD fluorescence in both cell types, but most prominently in DCs, which showed a more than 60% reduction in DiD fluorescence when PEG was included as a LNP component (Fig. 6A).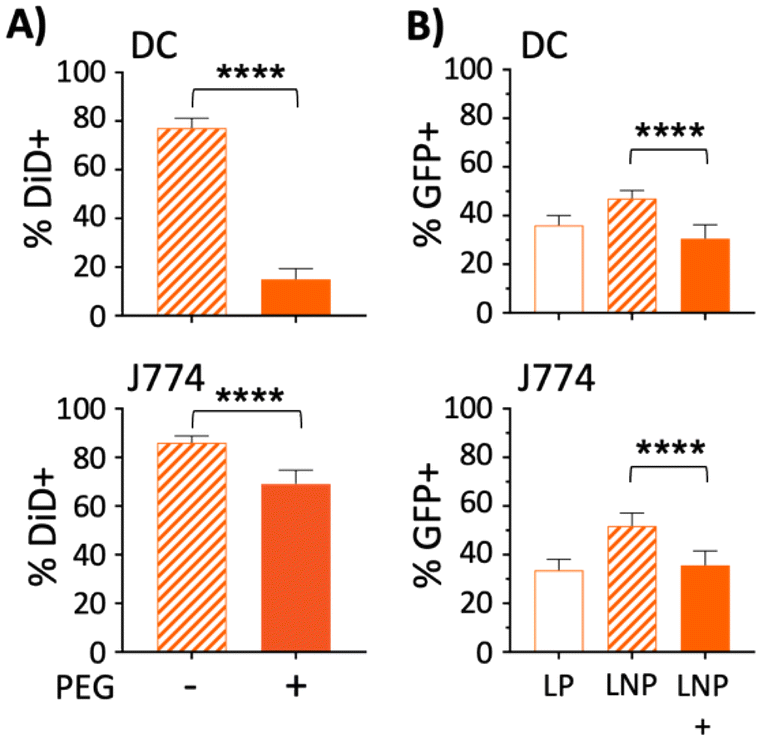 | ||
| Fig. 6 PEGylation decreases LNP uptake by antigen presenting cells. (A) Bone marrow-derived dendritic cells (DC) or J774 macrophages were incubated for 18 hours with LNPs made with 5% DiD and DSPE-PEG2000, or PEG-free liposomes. The percentage of cells positive for DiD fluorescence by flow cytometry is shown; data represent pooled results from three independent experiments, N = 3 wells per treatment. (B) Bone marrow-derived dendritic cells or J774 macrophages were transfected with 200 ng of GFP DNA delivered with LPs made of 50% TZ3 and 50% DOPE; LNPs made with 50% TZ3, 10% DOPE, 40% cholesterol; or LNPs made with 50% TZ3, 10% DOPE, 39% cholesterol and 1% DSPE-PEG550. Seventy-two hours after transfection, the cells were analyzed by flow cytometry for GFP expression; data represent pooled results from three independent experiments, N = 3 wells per treatment. Bars indicate mean ± SD. Only statistically significant comparisons are shown. Significance determined by one-sample T-test (A) or one-way ANOVA (B). | ||
To test whether the PEG550 included in our optimized LNPs had an effect on GFP expression in APCs,87 both DCs and macrophages (J774) were treated with 200 ng GFP plasmid in LPs, or LNPs containing either PEG550 or no PEG. In both cell types, the addition of PEG to LNPs decreased GFP positivity by more than 15%, compared with PEG-free LNPs (Fig. 6B). GFP expression in DCs was also slightly more efficient (∼6%) with LP treatment than PEGylated LNPs; however, this pattern was not observed in J774 macrophages. These studies collectively suggest that the addition of PEG to nanoparticles may have an advantage in reducing the immunogenicity of liposomal DNA transgenes, with a concomitant reduction in transfection efficiency. Furthermore, the differences in transfection between the PEG-free LNPs and LPs, compared with PEGylated LNPs, could partially explain the observed in vivo disparities between the two, as PEGylation of lipoplexes has also been reported to hamper transfection.73 Based on this finding, it is possible that non-PEGylated LNPs made by microfluidics could outperform LPs, although this will require further optimization of the LNP preparation to inhibit their aggregation in the absence of PEG.
Conclusions
The present manuscript highlights the utility of cationic triazine lipids as tools for in vivo research. Evaluation of in vivo toxicity of the compounds showed that TZ3 produced comparable toxicity to DOTAP, a commonly used cationic lipid. Furthermore, TZ3 demonstrated an increase in transfection efficacy compared with DOTAP, both in vivo and in vitro. These results concur with the findings of Martinez-Negro et al. and Candiani et al. showing improved transfection efficacy in cationic lipids containing aromatic moieties.13,89 While the role of the triazine ring of the lipids described here and their interaction with plasmid DNA have not been determined, others have indicated that the aromatic rings improve interactions with DNA base pairs through π–π stacking and intercalation for improved binding.90,91While LP transfection achieved serum hAAT levels similar to those reported previously,33 lipid-based plasmid delivery systems were not able to achieve the concentrations observed with viral vectors.24,92 The plasmid used in the studies described here is based on a lentiviral system reported by Wilson et al.92 where viral transfection led to protein concentrations of about 50 μg mL−1, on average. Similar concentrations were achieved by Akbar et al. with AAV.24 While further optimization of the lipids or nanoparticle formulation may improve transfection, it is also possible that modifications to the plasmids that increase protein levels or improve nuclear translocation may also be useful.35,47
Higher protein levels may also be attained with mRNA transfection; however, plasmids offer certain advantages including longer stability and lower resultant immunogenicity toward transgenes.35,36,93 In a recent report, Karadagi et al. showed that mRNA transfection could in fact increase hAAT protein in the liver, but the authors did not quantify serum levels, nor did they explore the immune response to the transgene.94 Additionally, evaluation of the more permanent transfection systems, such as CRISPR, could allow for more effective and long lasting outcomes in the setting of gene replacement.95,96
In addition to advances in mRNA delivery, much of the recent literature using LNPs for gene delivery takes advantage of ionizable lipids in formulations optimized primarily for siRNA delivery.5,9,48 While these compounds are successful and offer many advantages to gene delivery, we have shown here that formulations containing triazine lipids can provide a successful tool for plasmid delivery. Furthermore, we have shown that formulations containing DOPE and PEG550, rather than DSPC and PEG2000, can enhance the efficacy of plasmid delivery both in cells and in mice. Particularly interesting was the finding that LNPs, which contained PEG, reduced titers against the transgene compared with LPs without PEG. While the antibody response to hAAT is relatively low, these data suggest a need for further interrogation of the role of PEG in cationic lipid vaccines. Although we have shown that PEG can reduce nanoparticle uptake and transfection in antigen presenting cells (APCs), PEG is recognized by B cells in vivo,97 which could help increase uptake and expression of antigens in B cells that recognize the polymer as an epitope and counter the reduced uptake by professional phagocytes. Another confounding factor for our evaluation of these findings is that differences in nanoparticle size can affect titers generated by mRNA vaccines, a hypothesis that was not evaluated in the present manuscript.98
In addition to the modest increase in immunogenicity toward the transgene when delivered as a lipoplex, TZ3 induced a robust antibody induction (RET > 105) when used as an adjuvant with hAAT protein. This is not altogether surprising, as cationic lipids are known to possess immunomodulatory properties72,80,99 and serve as adjuvants,79 but the magnitude of the induction with TZ3 was unexpected, thus warranting additional studies to assess the mechanism and extent of this property in a vaccine relevant model. Overall, the findings presented in this manuscript highlight the potential of TZ based lipids and suggest the need for further investigation into their structure activity relationship and evaluation in biological systems.
Author contributions
DN developed the experimental design, conducted experiments, analysed data, generated figures, and wrote the first draft of the manuscript. MGP assisted with experimental design, performing toxicity experiments, formatted figures and edited the manuscript for publication. RK assisted with IV administration of nanoparticles and editing the manuscript. VJV provided supervision, funding acquisition, preparation of figures for publication and review & editing the manuscript.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work was supported by National Institute of Health grants including the Kentucky Center for Clinical and Translational Science (UL1 TR001998; DN), R01 HL152081 (VJV), and the Center of Biomedical Research Excellence (COBRE) in Pharmaceutical Research and Innovation (CPRI, P20 GM130456; VJV), and the University of Kentucky College of Pharmacy. We also thank the Pharmacy Practice and Science Core Lab for use of use the BioTek Synergy H1 plate reader and Attune NxT flow cytometer, and Younsoo Bae for use of the Malvern Zetasizer Nano ZS.References
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, Nat. Nanotechnol., 2021, 16, 630–643 CrossRef CAS PubMed.
- J. Zabner, Adv. Drug Delivery Rev., 1997, 27, 17–28 CrossRef CAS PubMed.
- A. Gupta, J. L. Andresen, R. S. Manan and R. Langer, Adv. Drug Delivery Rev., 2021, 178, 113834 CrossRef CAS PubMed.
- E. Dolgin, Science, 2022, 376, 680–681 CrossRef CAS PubMed.
- P. R. Cullis and M. J. Hope, Mol. Ther., 2017, 25, 1467–1475 CrossRef CAS PubMed.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- A. Hirko, F. Tang and J. A. Hughes, Curr. Med. Chem., 2003, 10, 1185–1193 CrossRef CAS PubMed.
- S. Zhang, Y. Xu, B. Wang, W. Qiao, D. Liu and Z. Li, J. Controlled Release, 2004, 100, 165–180 CrossRef CAS PubMed.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda, J. K. Narayanannair, K. G. Rajeev, I. M. Hafez, A. Akinc, M. A. Maier, M. A. Tracy, P. R. Cullis, T. D. Madden, M. Manoharan and M. J. Hope, Angew. Chem., Int. Ed., 2012, 51, 8529–8533 CrossRef CAS PubMed.
- B. Wang, R. M. Zhao, J. Zhang, Y. H. Liu, Z. Huang, Q. Y. Yu and X. Q. Yu, Eur. J. Med. Chem., 2017, 136, 585–595 CrossRef CAS PubMed.
- T. Ren, G. Zhang, Y. K. Song and D. Liu, J. Drug Targeting, 1999, 7, 285–292 CrossRef CAS PubMed.
- D. Nardo, C. M. Akers, N. E. Cheung, C. M. Isom, J. T. Spaude, D. W. Pack and V. J. Venditto, RSC Adv., 2021, 11, 24752–24761 RSC.
- G. Candiani, M. Frigerio, F. Viani, C. Verpelli, C. Sala, L. Chiamenti, N. Zaffaroni, M. Folini, M. Sani, W. Panzeri and M. Zanda, ChemMedChem, 2007, 2, 292–296 CrossRef CAS PubMed.
- C. Pennetta, N. Bono, F. Ponti, M. C. Bellucci, F. Viani, G. Candiani and A. Volonterio, Bioconjugate Chem., 2021, 32, 690–701 CrossRef CAS PubMed.
- A. Algarni, E. H. Pilkington, E. J. A. Suys, H. Al-Wassiti, C. W. Pouton and N. P. Truong, Biomater. Sci., 2022, 10, 2940–2952 RSC.
- R. Dingman and S. V. Balu-Iyer, J. Pharm. Sci., 2019, 108, 1637–1654 CrossRef CAS PubMed.
- J. A. Kulkarni, P. R. Cullis and R. van der Meel, Nucleic Acid Ther., 2018, 28, 146–157 CrossRef CAS PubMed.
- K. Inaba, W. J. Swiggard, R. M. Steinman, N. Romani, G. Schuler and C. Brinster, Curr. Protoc. Immunol., 2009, 86, 3.7.1–3.7.19 Search PubMed.
- M. R. Ehlers, Biol. Chem., 2014, 395, 1187–1193 CAS.
- S. A. D. Hoekstra, Mol. Membr. Biol., 2001, 18, 129–143 CrossRef.
- M. A. Dobrovolskaia, Curr. Pharm. Des., 2017, 23, 3134–3141 CrossRef CAS PubMed.
- S. Song, Chronic Obstruct. Pulm. Dis., 2018, 5, 289–301 Search PubMed.
- M. R. Ehlers, Biol. Chem., 2014, 395, 1187–1193 CAS.
- M. A. Akbar, J. J. Cao, Y. Lu, D. Nardo, M. J. Chen, A. S. Elshikha, R. Ahamed, M. Brantly, L. S. Holliday and S. Song, Hum. Gene Ther., 2016, 27, 679–686 CrossRef CAS PubMed.
- A. R. Tonelli and M. L. Brantly, Ther. Adv. Respir. Dis., 2010, 4, 289–312 CrossRef CAS PubMed.
- T. R. Flotte, B. C. Trapnell, M. Humphries, B. Carey, R. Calcedo, F. Rouhani, M. Campbell-Thompson, A. T. Yachnis, R. A. Sandhaus, N. G. McElvaney, C. Mueller, L. M. Messina, J. M. Wilson, M. Brantly, D. R. Knop, G. J. Ye and J. D. Chulay, Hum. Gene Ther., 2011, 22, 1239–1247 CrossRef CAS PubMed.
- K. A. Oppelt, J. G. Kuiper, Y. Ingrasciotta, V. Ientile, R. M. C. Herings, M. Tari, G. Trifirò and U. Haug, Front. Oncol., 2021, 11, 630456 CrossRef PubMed.
- D. C. P. Selinger, D. I. Carbery and D. J. Al-Asiry, Br. J. Hosp. Med., 2018, 79, 686–693 CrossRef PubMed.
- M. C. McGregor, J. G. Krings, P. Nair and M. Castro, Am. J. Respir. Crit. Care Med., 2019, 199, 433–445 CrossRef CAS PubMed.
- V. Jawa, F. Terry, J. Gokemeijer, S. Mitra-Kaushik, B. J. Roberts, S. Tourdot and A. S. De Groot, Front. Immunol., 2020, 11, 1301 CrossRef CAS PubMed.
- S. Sethu, K. Govindappa, M. Alhaidari, M. Pirmohamed, K. Park and J. Sathish, Arch. Immunol. Ther. Exp., 2012, 60, 331–344 CrossRef CAS PubMed.
- R. Dingman and S. V. Balu-Iyer, J. Pharm. Sci., 2019, 108, 1637–1654 CrossRef CAS PubMed.
- S. F. Aliño, Biochem. Pharmacol., 1997, 54, 9–13 CrossRef.
- K. L. Brigham, K. B. Lane, B. Meyrick, A. A. Stecenko, S. Strack, D. R. Cannon, M. Caudill and A. E. Canonico, Hum. Gene Ther., 2000, 11, 1023–1032 CrossRef CAS PubMed.
- M. A. Liu, Vaccines, 2019, 7, 37 CrossRef CAS PubMed.
- A. J. Geall, A. Verma, G. R. Otten, C. A. Shaw, A. Hekele, K. Banerjee, Y. Cu, C. W. Beard, L. A. Brito, T. Krucker, D. T. O'Hagan, M. Singh, P. W. Mason, N. M. Valiante, P. R. Dormitzer, S. W. Barnett, R. Rappuoli, J. B. Ulmer and C. W. Mandl, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14604–14609 CrossRef CAS PubMed.
- M. D. Buschmann, M. J. Carrasco, S. Alishetty, M. Paige, M. G. Alameh and D. Weissman, Vaccines, 2021, 9, 65 CrossRef CAS PubMed.
- E. Kenjo, H. Hozumi, Y. Makita, K. A. Iwabuchi, N. Fujimoto, S. Matsumoto, M. Kimura, Y. Amano, M. Ifuku, Y. Naoe, N. Inukai and A. Hotta, Nat. Commun., 2021, 12, 7101 CrossRef CAS PubMed.
- B. A. Duguay, K. W.-C. Huang and M. Kulka, J. Leukocyte Biol., 2018, 104, 587–596 CrossRef CAS PubMed.
- M. Kulka and N. A. Puga, J. Immunol., 2020, 204, 159 CrossRef PubMed.
- E. Quagliarini, S. Renzi, L. Digiacomo, F. Giulimondi, B. Sartori, H. Amenitsch, V. Tassinari, L. Masuelli, R. Bei, L. Cui, J. Wang, A. Amici, C. Marchini, D. Pozzi and G. Caracciolo, Pharmaceutics, 2021, 13, 1292 CrossRef CAS PubMed.
- C. B. Roces, G. Lou, N. Jain, S. Abraham, A. Thomas, G. W. Halbert and Y. Perrie, Pharmaceutics, 2020, 12, 1095 CrossRef CAS PubMed.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda, J. K. Narayanannair, K. G. Rajeev, I. M. Hafez, A. Akinc, M. A. Maier, M. A. Tracy, P. R. Cullis, T. D. Madden, M. Manoharan and M. J. Hope, Angew. Chem., Int. Ed., 2012, 51, 8529–8533 CrossRef CAS PubMed.
- J. A. Kulkarni, D. Witzigmann, S. Chen, P. R. Cullis and R. van der Meel, Acc. Chem. Res., 2019, 52, 2435–2444 CrossRef CAS PubMed.
- A. K. Blakney, P. F. McKay, K. Hu, K. Samnuan, N. Jain, A. Brown, A. Thomas, P. Rogers, K. Polra, H. Sallah, J. Yeow, Y. Zhu, M. M. Stevens, A. Geall and R. J. Shattock, J. Controlled Release, 2021, 338, 201–210 CrossRef CAS PubMed.
- R. Zhang, R. El-Mayta, T. J. Murdoch, C. C. Warzecha, M. M. Billingsley, S. J. Shepherd, N. Gong, L. Wang, J. M. Wilson, D. Lee and M. J. Mitchell, Biomater. Sci., 2021, 9, 1449–1463 RSC.
- J. A. Kulkarni, J. L. Myhre, S. Chen, Y. Y. C. Tam, A. Danescu, J. M. Richman and P. R. Cullis, Nanomedicine, 2017, 13, 1377–1387 CrossRef CAS PubMed.
- B. L. Mui, Y. K. Tam, M. Jayaraman, S. M. Ansell, X. Du, Y. Y. C. Tam, P. J. Lin, S. Chen, J. K. Narayanannair, K. G. Rajeev, M. Manoharan, A. Akinc, M. A. Maier, P. Cullis, T. D. Madden and M. J. Hope, Mol. Ther.–Nucleic Acids, 2013, 2, e139 CrossRef CAS PubMed.
- S. J. Sung, S. H. Min, K. Y. Cho, S. Lee, Y. J. Min, Y. I. Yeom and J. K. Park, Biol. Pharm. Bull., 2003, 26, 492–500 CrossRef CAS PubMed.
- R. C. Ryals, S. Patel, C. Acosta, M. McKinney, M. E. Pennesi and G. Sahay, PLoS One, 2020, 15, e0241006 CrossRef CAS PubMed.
- E. Ambegia, S. Ansell, P. Cullis, J. Heyes, L. Palmer and I. MacLachlan, Biochim. Biophys. Acta, Biomembr., 2005, 1669, 155–163 CrossRef CAS PubMed.
- H. Tanaka, R. Miyama, Y. Sakurai, S. Tamagawa, Y. Nakai, K. Tange, H. Yoshioka and H. Akita, Pharmaceutics, 2021, 13, 2097 CrossRef CAS PubMed.
- V. Francia, R. M. Schiffelers, P. R. Cullis and D. Witzigmann, Bioconjugate Chem., 2020, 31, 2046–2059 CrossRef CAS PubMed.
- T. Nakamura, M. Kawai, Y. Sato, M. Maeki, M. Tokeshi and H. Harashima, Mol. Pharmaceutics, 2020, 17, 944–953 CrossRef CAS PubMed.
- S. Ansorge, S. Lanthier, J. Transfiguracion, Y. Durocher, O. Henry and A. Kamen, J. Gene Med., 2009, 11, 868–876 CrossRef CAS PubMed.
- M. S. Cho, H. Yee and S. Chan, J. Biomed. Sci., 2002, 9, 631–638 CrossRef CAS PubMed.
- Z. Du, M. M. Munye, A. D. Tagalakis, M. D. I. Manunta and S. L. Hart, Sci. Rep., 2014, 4, 7107 CrossRef PubMed.
- C. Webb, S. Khadke, S. T. Schmidt, C. B. Roces, N. Forbes, G. Berrie and Y. Perrie, Pharmaceutics, 2019, 11, 653 CrossRef CAS PubMed.
- G. Basha, T. I. Novobrantseva, N. Rosin, Y. Y. C. Tam, I. M. Hafez, M. K. Wong, T. Sugo, V. M. Ruda, J. Qin, B. Klebanov, M. Ciufolini, A. Akinc, Y. K. Tam, M. J. Hope and P. R. Cullis, Mol. Ther., 2011, 19, 2186–2200 CrossRef CAS PubMed.
- A. Albanese and W. C. Chan, ACS Nano, 2011, 5, 5478–5489 CrossRef CAS PubMed.
- A. Radomski, P. Jurasz, D. Alonso-Escolano, M. Drews, M. Morandi, T. Malinski and M. W. Radomski, Br. J. Pharmacol., 2005, 146, 882–893 CrossRef CAS PubMed.
- N. Pardi, S. Tuyishime, H. Muramatsu, K. Kariko, B. L. Mui, Y. K. Tam, T. D. Madden, M. J. Hope and D. Weissman, J. Controlled Release, 2015, 217, 345–351 CrossRef CAS PubMed.
- J. A. Carlson, B. B. Rogers, R. N. Sifers, H. K. Hawkins, M. J. Finegold and S. L. Woo, J. Clin. Invest., 1988, 82, 26–36 CrossRef CAS PubMed.
- C. Zincarelli, S. Soltys, G. Rengo and J. E. Rabinowitz, Mol. Ther., 2008, 16, 1073–1080 CrossRef CAS PubMed.
- J. Di, Z. Du, K. Wu, S. Jin, X. Wang, T. Li and Y. Xu, Pharm. Res., 2022, 39, 105–114 CrossRef CAS PubMed.
- A. Bakhtiar, A. S. Neah, K. Y. Ng and E. H. Chowdhury, J. Pharm. Invest., 2022, 52, 95–107 CrossRef CAS.
- A. K. O. Åslund, R. J. Vandebriel, F. Caputo, W. H. de Jong, C. Delmaar, A. Hyldbakk, E. Rustique, R. Schmid, S. Snipstad, I. Texier, K. Vernstad and S. E. F. Borgos, Drug Delivery Transl. Res., 2022, 12, 2114–2131 CrossRef PubMed.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Nat. Nanotechnol., 2020, 15, 313–320 CrossRef CAS PubMed.
- S. Liu, Q. Cheng, T. Wei, X. Yu, L. T. Johnson, L. Farbiak and D. J. Siegwart, Nat. Mater., 2021, 20, 701–710 CrossRef CAS PubMed.
- J. Crepso, C. Blaya, A. Crespo and S. F. Aliño, Biochem. Pharmacol., 1996, 51, 1309–1314 CrossRef CAS PubMed.
- J. Stolk, N. Tov, K. R. Chapman, P. Fernandez, W. MacNee, N. S. Hopkinson, E. Piitulainen, N. Seersholm, C. F. Vogelmeier, R. Bals, G. McElvaney and R. A. Stockley, Eur. Respir. J., 2019, 54, 1900673 CrossRef CAS PubMed.
- A. G. Allison and G. Gregoriadis, Nature, 1974, 252, 252 CrossRef CAS PubMed.
- T. Gjetting, N. S. Arildsen, C. L. Christensen, T. T. Poulsen, J. A. Roth, V. N. Handlos and H. S. Poulsen, Int. J. Nanomed., 2010, 5, 371–383 CAS.
- J. W. Meisel and G. W. Gokel, Sci. Rep., 2016, 6, 27662 CrossRef CAS PubMed.
- M. Campos, F. Kueppers, J. Stocks, C. Strange, J. Chen, R. Griffin, L. Wang-Smith, M. Cruz, P. Vandeberg and M. Brantly, Eur. Respir. J., 2013, 42(Suppl. 57), 4151 Search PubMed.
- R. Vidal Pla, N. Padullés Zamora, F. Sala Piñol, R. Jardí Margaleff, F. Rodríguez Frías and J. B. Montoro Ronsano, Arch. Bronconeumol., 2006, 42, 553–556 CrossRef PubMed.
- Y. Lu and S. Song, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17158 CrossRef CAS PubMed.
- A. F. Ogata, C.-A. Cheng, M. Desjardins, Y. Senussi, A. C. Sherman, M. Powell, L. Novack, S. Von, X. Li, L. R. Baden and D. R. Walt, Clin. Infect. Dis, 2021, 1, 715–718 Search PubMed.
- S. Tandrup Schmidt, C. Foged, K. Smith Korsholm, T. Rades and D. Christensen, Pharmaceutics, 2016, 8, 7 CrossRef PubMed.
- D. Christensen, K. S. Korsholm, P. Andersen and E. M. Agger, Expert Rev. Vaccines, 2011, 10, 513–521 CrossRef CAS PubMed.
- B. D. Freimark, H. P. Blezinger, V. J. Florack, J. L. Nordstrom, S. D. Long, D. S. Deshpande, S. Nochumson and K. L. Petrak, J. Immunol., 1998, 160, 4580–4586 CAS.
- L. Habjanec, B. Halassy and J. Tomašić, Int. Immunopharmacol., 2010, 10, 751–759 CrossRef CAS PubMed.
- L. Krishnan and G. D. Sprott, Vaccine, 2008, 26, 2043–2055 CrossRef CAS PubMed.
- M. Rao, G. R. Matyas, T. C. Vancott, D. L. Birx and C. R. Alving, Immunol. Cell Biol., 2004, 82, 523–530 CrossRef CAS PubMed.
- C. R. Alving, Biochim. Biophys. Acta, 1992, 1113, 307–322 CrossRef CAS.
- R. D. Weeratna, M. J. McCluskie, Y. Xu and H. L. Davis, Vaccine, 2000, 18, 1755–1762 CrossRef CAS PubMed.
- S. M. Moghimi and J. Szebeni, Prog. Lipid Res., 2003, 42, 463–478 CrossRef CAS PubMed.
- M. L. Immordino, F. Dosio and L. Cattel, Int. J. Nanomed., 2006, 1, 297–315 CrossRef CAS PubMed.
- M. Martinez-Negro, A. L. Barran-Berdon, C. Aicart-Ramos, M. L. Moya, C. T. de Ilarduya, E. Aicart and E. Junquera, Colloids Surf., B, 2018, 161, 519–527 CrossRef CAS PubMed.
- P. T. Wong and S. K. Choi, Int. J. Mol. Sci., 2015, 16, 1772–1790 CrossRef CAS PubMed.
- J. Mohanty, N. Barooah, V. Dhamodharan, S. Harikrishna, P. I. Pradeepkumar and A. C. Bhasikuttan, J. Am. Chem. Soc., 2013, 135, 367–376 CrossRef CAS PubMed.
- A. A. Wilson, G. J. Murphy, H. Hamakawa, L. W. Kwok, S. Srinivasan, A. H. Hovav, R. C. Mulligan, S. Amar, B. Suki and D. N. Kotton, J. Clin. Invest., 2010, 120, 379–389 CrossRef CAS PubMed.
- H. Huysmans, Z. Zhong, J. De Temmerman, B. L. Mui, Y. K. Tam, S. Mc Cafferty, A. Gitsels, D. Vanrompay and N. N. Sanders, Mol. Ther.–Nucleic Acids, 2019, 17, 867–878 CrossRef CAS PubMed.
- A. Karadagi, A. G. Cavedon, H. Zemack, G. Nowak, M. E. Eybye, X. Zhu, E. Guadagnin, R. A. White, L. M. Rice, A. L. Frassetto, S. Strom, C. Jorns, P. G. V. Martini and E. Ellis, Sci. Rep., 2020, 10, 7052 CrossRef CAS PubMed.
- C. A. Lino, J. C. Harper, J. P. Carney and J. A. Timlin, Drug Delivery, 2018, 25, 1234–1257 CrossRef CAS PubMed.
- T. Wei, Q. Cheng, Y.-L. Min, E. N. Olson and D. J. Siegwart, Nat. Commun., 2020, 11, 3232 CrossRef CAS PubMed.
- G. Besin, J. Milton, S. Sabnis, R. Howell, C. Mihai, K. Burke, K. E. Benenato, M. Stanton, P. Smith, J. Senn and S. Hoge, ImmunoHorizons, 2019, 3, 282 CrossRef CAS PubMed.
- K. J. Hassett, J. Higgins, A. Woods, B. Levy, Y. Xia, C. J. Hsiao, E. Acosta, Ö. Almarsson, M. J. Moore and L. A. Brito, J. Controlled Release, 2021, 335, 237–246 CrossRef CAS PubMed.
- D. K. Hong, S. Chang, C. M. Botham, T. D. Giffon, J. Fairman and D. B. Lewis, J. Virol., 2010, 84, 12691–12702 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01289h |
| This journal is © The Royal Society of Chemistry 2022 |