
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitching of the mechanism of charge transport induced by phase transitions in tunnel junctions with large biomolecular cages†
Nipun Kumar
Gupta‡
ab,
Rupali Reddy
Pasula‡
c,
Senthil Kumar
Karuppannan‡
 a,
Zhang
Ziyu
a,
Anton
Tadich
d,
Bruce
Cowie
d,
Dong-Chen
Qi
e,
Peter
Bencok
f,
Sierin
Lim
*cg and
Christian A.
Nijhuis
*abh
a,
Zhang
Ziyu
a,
Anton
Tadich
d,
Bruce
Cowie
d,
Dong-Chen
Qi
e,
Peter
Bencok
f,
Sierin
Lim
*cg and
Christian A.
Nijhuis
*abh
aDepartment of Chemistry, National University of Singapore, 3 Science Drive, Singapore 117543, Singapore
bCentre for Advanced 2D Materials, National University of Singapore, 6 Science Drive 2, Singapore 117546, Singapore
cSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 70 Nanyang Drive, Singapore 637457, Singapore
dAustralian Synchrotron Clayton, Victoria 3168, Australia
eCentre for Materials Science, School of Chemistry and Physics, Queensland University of Technology, Brisbane, Queensland 4001, Australia
fI10, Diamond Light Source Ltd, Diamond House, Harwell Science & Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
gNTU-Northwestern Institute for Nanomedicine, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: slim@ntu.edu.sg
hHybrid Materials for Opto-Electronics Group, Department of Molecules and Materials, Faculty of Science and Technology, University of Twente, 7500 AE Enschede, The Netherlands. E-mail: c.a.nijhuis@utwente.nl
First published on 8th March 2021
Abstract
Tunnel junctions based on Fe storing globular proteins are an interesting class of biomolecular tunnel junctions due to their tunable Fe ion loading, symmetrical structure and thermal stability, and are therefore attractive to study the mechanisms of charge transport (CT) at the molecular level. This paper describes a temperature-induced change in the CT mechanism across junctions with large globular (∼25 nm in diameter) E2-proteins bioengineered with Fe-binding peptides from ferritin (E2-LFtn) to mineralise Fe ions in the form of iron oxide nanoparticles (NPs) inside the protein's cavity. The iron oxide NPs provide accessible energy states that support high CT rates and shallow activation barriers. Interestingly, the CT mechanism changes abruptly, but reversibly, from incoherent tunnelling (which is thermally activated) to coherent tunnelling (which is activationless) across the E2-LFtn-based tunnel junctions with the highest Fe ion loading at a temperature of 220–240 K. During this transition the current density across the junctions increases by a factor of 13 at an applied voltage of V = −0.8 V. X-ray absorption spectroscopy indicates that the iron oxide NPs inside the E2-LFtn cages undergo a reversible phase transition; this phase transition opens up new a tunnelling pathway changing the mechanism of CT from thermally activated to activationless tunnelling despite the large size of the E2-LFtn and associated distance for tunnelling.
Introduction
In biological processes including photosynthesis,1,2 enzymatic catalysis,3 DNA transcription or repair,4,5 charge transport (CT) is highly efficient in nature, but the reasons why this is so are unclear. One of the key challenges is to identify the conduction pathways across proteins which can be realised by incorporating proteins in tunnel junctions and to study how CT depends, on for example, site-selective modifications of the proteins,6,7 mechanical deformation of proteins,8,9 orientation of the protein with respect to the electrode,10,11 or the role of the cofactors.12–16 Specifically, it is challenging to distinguish between the contributions to the mechanism of CT from the protein shell and the co-factors or (multiple) heme groups.7,12,17–20 Cahen and Sheves and co-workers demonstrated that replacement of the native metal ion co-factor with another metal ion changes the mechanism of CT from coherent tunnelling (which is activationless) to incoherent tunnelling (which is thermally activated and also called hopping).12,21 The mechanism of CT also changes from coherent to incoherent tunnelling when the metal centre of azurin (Az),12 or the ferrihydrite core of ferritin,15 was removed. Here, we report a change in the mechanism of CT as a function of the Fe ion loading and temperature in tunnel junctions with large (diameter of 25 nm obtained from its crystal structure22) globular E2 proteins bioengineered with Fe binding sites from ferritin, E2-LFtn, allowing us to the control the iron oxide content inside the cavity ranging from 500 to 3000Fe ions per E2-LFtn cage. Depending on the Fe ion loading, the mechanism of CT switches from incoherent to coherent tunnelling caused by a thermally induced phase transition of the iron oxide NPs inside the E2-LFtn. The mechanism of CT across junctions with the E2-LFtn lacking the iron oxide NPs (i.e., apo-E2-LFtn) is incoherent tunnelling. These results give us new insights into the conduction pathways of tunnel junctions with large proteins.Various mechanisms have been proposed to describe CT across proteins, which can be roughly divided into two classes: coherent tunnelling which is independent of temperature and incoherent tunnelling which depends on temperature. The general tunnelling equation (eqn (1)) shows how fast the tunnelling current density (J in A cm−2) decays (determined by the tunnelling decay coefficient β in nm−1) as a function of the tunnelling barrier width d (in nm) where J0 (in A cm−2) is the pre-exponential factor.
| J = J0e−βd | (1) |
 | (2) |
Incoherent tunnelling has been observed in tunnel junctions with various proteins including bacteriorhodopsin (bR),13 cytochrome-C (Cyt-C)17 and in proteins without the co-factor, e.g., apo-Az,12 apo-ferritin15 and apo-PS-I10 (but the same junctions with the co-factor showed thermally activated CT). Remarkably, for a few systems a change in incoherent tunnelling regime (each characterised by a different value of Ea) has been observed as a function of T or d. For example, in bR junctions where the π-conjugated retinal co-factor is decoupled from the bR protein (reconstituted bR), the value of Ea = 490 meV at high T (T = 340–260 K) is substantially larger than the value of Ea = 180 meV at low T (T = 260–200 K).13 Similarly, in Cyt-C junctions with the Fe ion removed from the porphyrin ring, a low Ea = 105 meV for T = 285–200 K while a high Ea = 550 meV for T = 335–285 K was observed.17 A transition from coherent to incoherent tunnelling with d has been observed for conjugated molecular wires,28,35 but a transition from coherent tunnelling with large β to low β with increasing d has been reported for junctions with long molecular wires27,28,35 or ferritin.15 These examples highlight the different types of CT regimes that can be accessed in (bio)molecular tunnel junction although the underlying reasons why the mechanism of CT changes are not fully understood.
The E2 protein is a chemically and thermally stable (up to 85 °C) cage-like globular protein which naturally occurs as a part of the pyruvate hydrogenase multi-enzyme complex in the thermophile Geobacillus stearothermophilus and is responsible for linking glycolysis and the Krebs cycle.36 The E2 protein consists of 60 identical subunits self-assembled into a hollow spherical architecture with an external diameter of 24 nm and an internal diameter of 12 nm (Fig. 1). The cage structure has 12 openings, each with a diameter of 5 nm. The E2 protein has been bioengineered with frog M-ferritin mimicking peptides to form E2-LFtn,22 which can mineralise FeII into iron oxide NPs in a similar fashion to ferritin.15,37,38 The structure, magnetic behaviour and composition of the iron oxide NPs in ferritin is a function of the Fe ion loading/cage and its mineralisation conditions have been studied in detail. The E2-LFtn stores the Fe ions as iron oxide NPs (3–6 nm in size) in its hollow core.22 Amongst the large variety of iron oxides known, FeII always exists in an octahedral (Oh) symmetry state while FeIII can exist in both Oh and tetrahedral (Td) symmetry states.39,40 The Fe ions are stored predominately in the form of ferrihydrite in ferritin, but other iron oxide phases can be present in varying degrees depending on the preparation methods, Fe ion loading and types of ferritin.41,42 Iron oxides readily undergo thermally-induced phase transitions. For instance, phase transitions of ferrihydrite to other iron oxide phases (including lepidocrocite (γ-FeIIIOOH), goethite (α-FeIIIOOH),43 haematite (α-Fe2O3)44 and magnetite45 (Fe3O4) in NPs have been investigated in detail. We note that it has been reported that FeII ions can assist in phase transitions involving ferrihydrite.43,45–47 These examples highlight the complexity of iron oxide chemistry which, as we show here, can lead to interesting CT phenomena across biomolecular junction with E2-LFtn.
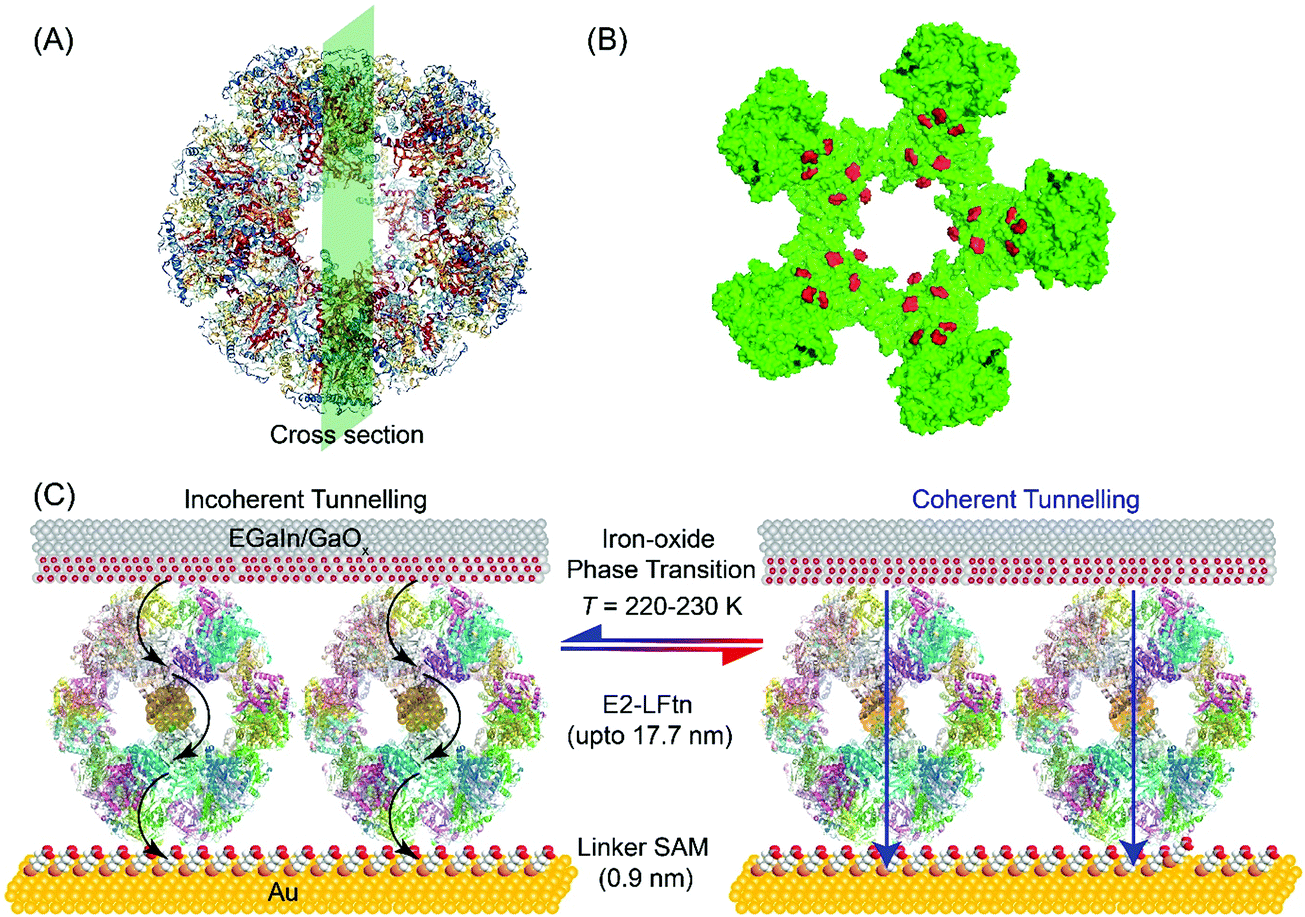 | ||
| Fig. 1 (A) PyMOL representation of apo-E2-LFtn and (B) cross-section where the Fe ion binding sites are highlighted in red colour. (C) Schematic illustration of the Au-linker-E2-LFtn//GaOx/EGaIn tunnel junction. Here, “//” represents a van der Waals contact, “/” represents the contact between the GaOx and EGaIn, “-” represents a chemical contact, the black arrows represent incoherent tunnelling, and the blue arrow represents coherent tunnelling. | ||
We report a thermally induced reversible and irreversible switching of the mechanism of CT across biomolecular tunnel junctions with bioengineered E2-LFtn. The mechanism of CT across apo-E2-LFtn is characterised by two distinct incoherent tunnelling regimes with high (254 ± 32 meV) and low (24 ± 1 meV) values of Ea, while the mechanism of CT across E2-LFtn is characterised by a single incoherent tunnelling regime with an intermediate Ea (35–61 meV) depending on the iron oxide loading. Thus, we conclude that the iron oxide NPs provide accessible energy states that support an alternate incoherent tunnelling pathway. For junctions with E2-LFtn with a large Fe ion loading (3000Fe), reversible conduction switching was observed at T = 220–240 K which was accompanied by a change in the CT mechanism from incoherent tunnelling in the low conduction state to coherent tunnelling in the high conduction state. Temperature-dependent X-ray absorption spectroscopy (XAS) indicates a reversible phase transition of the iron oxide NPs inside the E2-LFtn. These results suggest that the mechanism of CT can be switched as a function of temperature and iron oxide loading in E2-LFtn biomolecular tunnel junctions. Our findings give new insights in the conduction pathways of biomolecular junctions defined by the protein cage and the quantity of co-factors present inside the protein and demonstrate how these conduction pathways can be controlled.
Results and discussion
Design of the junctions
Fig. 1A shows the PyMOL representation of apo-E2-LFtn, showing the globular shape and the cavity with the structural information obtained from Protein Data Bank with identification number 1B5S. The FeII binding chemistry of the frog M-ferritin mimicking peptides in E2-LFtn has been reported previously.22,37 Each of the 60 subunits of the E2 cage has been modified with the catalytic binding sites from frog M-ferritin making it possible to load the E2 cage with iron oxide NPs (see ref. 22 for all details and characterisation of E2-LFtn). The Fe ion content of E2-LFtn can be controlled similar to that reported for controlled loading of ferritin.15,48 In this study, we loaded E2-LFtn with 500–3000 Fe ions/cage (at higher loadings, the iron oxide precipitated from solution). Fig. 1B shows the corresponding cross-section of apo-E2-LFtn with the Fe binding sites highlighted in red. Fig. 1C illustrates the Au-linker-E2-LFtn//GaOx/EGaIn junction and illustrates the iron oxide phase transition inside the E2-LFtn as a function of temperature, which, in turn, modulates the possible mechanism of CT and induces conductance switching. The preparation and characterisation of apo-E2-LFtn and E2-LFtn loaded with iron oxide NPs are briefly described in Sections S1–S3, ESI†). Before using E2-LFtn, we characterised the size of the proteins and confirmed that E2-LFtn retained its globular structure after mineralisation with dynamic light scattering technique, as shown in Fig. S1A (ESI†). The actual Fe ion content present in the E2-LFtn was characterised with inductively coupled plasma atomic emission spectroscopy (see Fig. S1B, ESI†).The smooth Au bottom electrodes were obtained with template-stripping (Section S4, ESI†), on which we formed the self-assembled monolayers (SAMs) of E2-LFtn using a linker SAM (Section S5, ESI†).15 The linker SAM was derived from 6-mercaptohexanoic acid and was activated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC); these activated carboxyl groups react with the NH2 groups located at the periphery of E2-LFtn to form covalent amide bonds ensuring the proteins are firmly anchored to the Au surface. After monolayer formation, the EGaIn top electrode was introduced using cone-shaped EGaIn tips49 or with EGaIn stabilised in a microchannel perpendicularly aligned over an array of micropores in Al2O3 fabricated on Au.50
Characterisation of the E2-LFtn monolayers
Fig. 2A shows an atomic force microscope (AFM) image of a bare Au substrate (see Section S6 for experimental details, ESI†) with a root mean square (rms) surface roughness of 0.49 nm measured over an area of 1.0 × 1.0 μm2, which is similar to previously reported values.49Fig. 2B shows an AFM image of a densely packed monolayer of 1000Fe ion loaded E2-LFtn on the Au-linker surface with a rms surface roughness of 2.6 nm (measured over an area of 1.0 × 1.0 μm2). The line scans are presented in Fig. S2 (ESI†). To determine whether E2-LFtn retained its globular structure upon adsorption, we used tapping mode AFM to determine the height of individual proteins from the AFM height profiles recorded for sub-monolayers of E2-LFtn on Au-linker. Fig. 2C shows an example of an AFM image for a sub-monolayer of apo-E2-LFtn where individual proteins can be seen (see Section S6 for additional images, ESI†), can be seen. We used a similar procedure to obtain AFM height profiles of E2-LFtn as a function of Fe ion loading. Fig. 2D shows the histograms along with Gaussian fits to these histograms of the heights of individual E2-LFtn proteins, dE2-LFtn (in nm), as a function of Fe ion loading, and the dE2-LFtn shows the linear dependence with Fe ion loading (Fig. 2E). The apo-E2-LFtn forms thin layers with a measured height of 6.9 ± 0.3 nm, which is much smaller than the hydrodynamic diameter of the protein measured in solution, dHD,E2-LFtn = 25 nm, from which we conclude that it does not retain it globular structure and likely forms dense, flattened monolayers (since these junctions do not short, see below). In contrast, the E2-LFtn loaded with iron show larger diameter for loading as low as 500Fe E2-LFtn the value of dE2-LFtn of 11.0 ± 0.3 nm indicating that, although the protein flattens, the globular structure is preserved. The highest value of dE2-LFtn of 17.7 ± 0.4 nm for maximum Fe ion loading which is close to dHD,E2-LFtn = 25 nm. This observation can be attributed to compression induced by the AFM tip or loss of water from the cavity.51,52 We note that these observations are consistent with previously reported thickness values of ferritin Aftn-AA, dAftn, on the same Au-linker surface as a function of Fe ion loading.15 The ratio between maximum protein height obtained from the AFM image (dAftn = 10.9 nm) and hydrodynamic radius (obtained from dynamic light scattering measurements for ferritin Aftn-AA dHD,Aftn = 12.1 nm) is dAftn/dHD,Aftn = 0.90, which is closer to unity than that of E2-LFtn of dE2-LFtn/dHD,E2-LFtn = 0.70, from which we conclude that the E2-LFtn is more dynamic and flexible than ferritin Aftn-AA. | ||
| Fig. 2 AFM images of the bare template-stripped Au surface (A), 1000Fe E2-LFtn monolayer on Au (B), and a sub-monolayer of apo-E2-LFtn on Au (C). (D) Histograms of dE2-LFtn as a function of Fe ion loading along with Gaussian fits these histograms. (E) The value of dE2-LFtnvs. Fe ion loading (error bars represent 95% confidence levels), where the red dashed line indicates the correlation between dE2-LFtn and the Fe loading (R2 = 0.985). | ||
Characterisation of iron oxide NPs inside E2-LFtn
To investigate the composition of the iron oxide NPs inside the E2-LFtn cages immobilised on the Au-linker surface as a function of temperature, we measured the Fe L2,3 edge XAS (Section S7, ESI†). This technique resolves the presence of FeII and FeIII ions along with their atomistic symmetry states. Fig. 3 shows the Fe L3 edge XAS spectra of the monolayers of 3000 and 2000Fe E2-LFtn recorded at three different temperatures of T = 300 K, followed by cooling down to 120 K, and after heating back to 300 K. The peak observed at 707.5 eV is assigned to FeII. The peak at 709.2 eV is assigned to FeIII and corresponds to Fe cations in both Oh and Td symmetries. The ratio (in both Td and Oh geometries) of FeII and FeIII were obtained from modelling of the XAS data with the CTM4XAS software53 (see Section S7 for details, ESI†) and Fig. 3 shows the results. Interestingly, the iron oxide NPs inside E2-LFtn are dominated by FeII rather than FeIII ions as reported for ferritin.54,55 This observation suggests that the iron mineralisation process in E2-LFtn differs from that of ferritin which could be related to the differences in cavity size and spatial arrangement of the M-ferritin mimicking catalytic sites.22 The ratio of the peak areas (RI = FeII/FeIII) for 3000Fe E2-LFtn decreases with decreasing T from 1.25 at 300 K to 0.50 at 120 K (Fig. 3A, B and Table 1). On heating back to 300 K, Fig. 3C shows that the RI ratio increases to 1.00 which is nearly its original value indicating that this phase transition is reversible. In contrast, for 2000Fe E2-LFtn, initially no signal could be recorded at 300 K (Fig. 3D) but a signal at the Fe L3 edge was recovered at 300 K (RI = 5.00, Fig. 3F) after cooling to 120 K (RI = 2.00, Fig. 3E). This observation can be explained as follows. Initially, the iron oxide NPs are buried in the protein cavity hampering their detection, but (partial) denaturation of the protein induced by cooling to low T exposes the iron oxide nanoparticles and, consequently, increases the XAS signals. Denaturation also explains the irreversible nature of the corresponding junction (discussed below). We also note that 3000Fe E2-LFtn has a higher Fe ion content than 2000Fe E2-LFtn, which is apparently high enough to conduct XAS measurements around the Fe L3 edge despite the iron oxide nanoparticles being inside the protein cavity and thus shielded by a ∼6 nm thick protein shell. We confirmed the presence of iron oxide at 300 K for intact protein using a beam line with a larger beam flux (Section S8, ESI†). These results suggest that Fe ion loading stabilises the E2-LFtn which agrees with the larger value of dE2-LFtn = 17.7 ± 0.4 nm for 3000Fe E2-LFtn than dE2-LFtn = 16.3 ± 0.4 nm for 2000Fe E2-LFtn indicating that flattening of 2000Fe E2-LFtn is more significant than for 3000Fe E2-LFtn. We note that Rakshit et al.52 reported an increase in the Young's modulus of ferritin as a function of Fe ion loading. From these results we conclude that 3000Fe E2-LFtn forms stable monolayers supporting a reversible phase transition of the iron oxide NPs inside the E2-LFtn cavity. | ||
| Fig. 3 XAS data around the Fe L3 edge recorded from a monolayer of 3000Fe E2-LFtn on Au at 300 K (A), 120 K (B) and after heating back to 300 K (C), and the same but for a monolayer of 2000Fe E2-LFtn 300 K (D), 120 K (E) and after heating to 300 K (F). The solid lines are fits as explained in the text. | ||
| Sample | R I at 300 K (initial) | R I at 120 K | R I at 300 K (final) |
|---|---|---|---|
| 2000Fe | — | 2.00 | 5.00 |
| 3000Fe | 1.25 | 0.50 | 1.00 |
Charge transport measurements
To investigate the mechanism of CT across E2-LFtn, we formed electrical contacts to the SAMs of E2-LFtn with cone-shaped tips of EGaIn (Fig. 1C) and measured the J(V) characteristics as a function of Fe ion loading. We followed previously reported procedures for the junction formation, data collection and statistical data analysis (see Section S8 for details, ESI†).49 We recorded 400–440 J(V) curves for each type of junction and plotted all values of log10|J| for each measured applied bias voltage in histograms to determine the Gaussian average value of log10|J|, 〈log10|J|〉G, and the Gaussian log-standard deviation, σlog. Fig. 4A shows the 〈log10|J|〉Gvs. V curves as a function of the Fe ion loading and Fig. 4B shows the corresponding current decay plot at −0.5 V. The value of J exponentially decays with the value of dE2-LFtn and fitting the data to eqn (1) yields β = 0.39 ± 0.02 nm−1. It is noteworthy that the CT distance is at 11.0 nm for 500Fe E2-LFtn, and nearly 17.7 nm for 3000Fe E2-LFtn meaning these proteins are highly conductive (see below). We note that the values of J for junctions with apo-E2-LFtn are similar to the junctions of 500Fe and 1000Fe E2-LFtn. This observation can be explained by the notion that the shorter dE2-LFtn of the apo-E2-LFtn junction (6.9 nm vs. 11–12 nm for junctions with 500Fe or 1000Fe E2-LFtn) compensates for the lack of tunnelling sites associated with the iron oxide particles. | ||
| Fig. 4 Plots of 〈log10|J|〉Gvs. V (A and B) 〈ln|J|〉G at −0.5 V vs. dE2-LFtn (error bars in J and dE2-LFtn represent the 95% confidence levels, solid line is a fit to eqn (1)). (C) Arrhenius plots of the values of J measured at −0.5 V and T = 240–340 K for junctions with E2-LFtn with different Fe ion loadings. (D) Arrhenius plot for junctions with apo-E2-LFtn at −0.5 V in the range of T = 230–340 K. Error bars represent the standard deviation obtained from 3 different junctions and the solid lines in panels C and D are fits to eqn (2). | ||
Temperature-dependent CT
To investigate the mechanism of CT in more detail, we performed J(V,T) measurements for the E2-LFtn junctions. The proteins were immobilised on Au (following the same procedure as described above) inside micropores AlOx over which microchannels filled with EGaIn were aligned following previously reported methods, and the fabrication and experimental details are presented in Sections S9 and S10 (ESI†).50Fig. 4C shows the Arrhenius plots at V = −0.5 V for junctions with monolayers of E2-LFtn with 1500Fe, 2000Fe and 3000Fe loading along with fits to eqn (2) to determine Ea. The mechanism of CT is incoherent tunnelling with small values of Ea ranging from 35 ± 3 to 61 ± 10 meV. This incoherent tunnelling mechanism explains the small value of β (Fig. 4B).Fig. 4D shows the Arrhenius plots at V = −0.5 V for junctions with apo-E2-LFtn along with fits to eqn (2). Remarkably, two distinct incoherent tunnelling regimes can be observed with a value of Ea of 254 ± 32 meV for T = 310–340 K and Ea = 24 ± 1 meV for T = 230–300 K. The transition temperature, TT, is around 300 K. A similar transition (TT = 290 K) between two incoherent tunnelling regimes (Ea = 550 meV to Ea = 105 meV) has been reported for junctions with apo-Cyt-C (where the Fe ion was removed from the porphyrin).17 In contrast, for junctions with holo-Cyt-C proteins, the Fe ion increased the CT rates by a factor of two and only one incoherent tunnelling regime with Ea = 105 meV dominates CT indicating that the Fe ion opens up more efficient CT pathways.17 Thus, our junctions show a similar behaviour where two thermally activated CT pathways are observed for junctions with apo-E2-Lftn. As discussed above in the AFM characterisation section, apo-E2-LFtn immobilised on the surface does not retain its cage-like structure and forms a disordered protein layer where charges must hop between different disorganised protein segments which constitute relatively large incoherent tunnelling barriers. This incoherent tunnelling mode is only active at relatively high temperatures (T > TT). In contrast, only one thermally activated CT pathway with relatively low Ea is observed for junctions with holo-E2-LFtn where the iron oxide NPs likely provide (multiple) low energy incoherent tunnelling sites. Here, the charges cross the protein segments of an organised monolayer where the globular tertiary structure of the protein is preserved. This difference in structure of the junctions, in conjunction with low energy incoherent tunnelling sites provided by the iron oxide NPs, could explain the large observed Ea for apo-E2-LFtn and low Ea for holo-E2-LFtn. At T < TT, incoherent tunnelling across segments becomes energetically unfavourable and a different pathway dominates CT with low Ea. Often, low Ea values are associated with “efficient” incoherent tunnelling, but here the currents at low T are small (since the primary conduction channel at high T is turned off). Therefore, at low T another mechanism is at work where incoherent tunnelling may, for instance, involve nuclear tunnelling,56 partial loss of coherence,57 or voltage induced changes to electronic states.32
Controlled switching of CT
Fig. 5 shows the switching behaviour of representative junctions with 3000Fe and 2000Fe E2-LFtn (see Section S11 for additional data sets, ESI†). As we have reported before, the shape of the J(V) curves recorded with junctions with flat EGaIn stabilised in microchannels are, unlike rough cone-shaped EGaIn tips, not obscured by artefacts caused by the high surface roughness of cone-shape EGaIn tips.50,58Fig. 5A shows the J(V) traces recorded during cooling of the 3000Fe E2-LFtn junction from 340 to 150 K and Fig. 5B shows the same during warming up back to 340 K. Fig. 5C shows the values of J at V = −0.8 V for the cooling and heating cycles. We observe that the junction undergoes a reversible transition at 220 K from a low to a high conduction state with 13-fold increase in the J. The value of J restores to its original value at 240 K during the heating cycle, but a small hysteretic behaviour is visible. This behaviour coincides with the reversible phase transition of the iron oxide NPs inside 3000Fe E2-LFtn observed with XAS as described above. Therefore, we conclude that the phase transition of the iron oxide NPs modulates the conduction across the E2-LFtn tunnel junctions. | ||
| Fig. 5 Temperature-dependent J(V) measurements of cooling (A) and heating of a 3000Fe E2-LFtn junction, and that of (D–E) 2000 Fe E2-LFtn junction in the range of T = 150–340 K. The temperature dependency of |J| at −0.8 V for 3000Fe E2-LFtn (C) and 2000Fe E2-LFtn (F) junctions. | ||
Fig. 5D–F shows the same for a 2000Fe E2-LFtn junction, but here an irreversible transition occurs at 220 K during cooling with a factor of 40 increase in J at V = −0.8 V, which is likely result of (partial) protein denaturation of 2000Fe E2-LFtn upon cooling. This behaviour resembles the irreversible phase transition of the iron oxide NPs inside 2000Fe E2-LFtn as observed with XAS as described above (see Fig. 3). Junctions with low Fe ion loading (i.e., 1500Fe E2-LFtn) and apo-E2-LFtn shorted at T = 240–230 K. These observations suggest that the current switching observed in 2000Fe E2-LFtn junctions is caused by partial failure due to denaturation of the protein. Denaturation of 2000Fe E2-LFtn also could explain the temperature variable XAS described above reinforcing this interpretation of the J(V,T) data.
Interestingly, CT across 3000Fe E2-LFtn junctions in the high temperature regime (T = 220–340 K) is thermally activated with Ea = 55 ± 8 eV at −0.8 V during cooling but not in the low temperature regime (T = 150–220 K; Fig. 5C). In contrast, the corresponding J(V) curves recorded from 2000Fe E2-LFtn junctions are thermally activated with Ea = 82 ± 17 mV and 61 ± 3 mV at −0.8 V, in both the low and the high conduction states, respectively. Our data suggest that the phase transition in the iron oxide NPs triggers a change in CT from incoherent to coherent tunnelling. The arrows in Fig. 5A and B indicate a sudden change in the slope of the J(V) curve at high T (with a blue arrow that identifies the ‘cusp’) which disappears at low T accompanied by an increase of the tunnelling current which indicates that a different, more efficient coherent tunnelling pathway is available at low T.
Comparison of E2-LFtn with other types of proteins and molecular wires
Fig. 6 shows how the J values of the junctions with E2-LFtn compares to other types of molecular junctions with biomolecules, saturated alkanethiolate SAMs and long conjugated molecular wires to visualise the efficiency of the CT process across the E2-LFtn. Considering the relatively low β of 0.39 nm−1 and relatively high J values, which are considerably greater than that of other proteins (including PS-1, ferritin Aftn-AA, bR and Cyt-C), we observe that E2-LFtn is a highly conductive protein. By comparing the CT rates for E2-LFtn with other molecules with comparable d, we note that only oligo-tetrathiafulvalene-pyromellitic-diimideimine (OPTI)59 junctions support higher CT rates. Fig. 6 also includes our previously reported data on junctions with ferritin which are characterised by two distinct coherent tunnelling regimes with values of β = 1.30 nm−1 for 500Fe to 3000Fe loading, and β = 0.28 nm−1 for 3000Fe to 4800Fe loading,15 whilst for E2-LFtn only thermally activated regime is observed (see Fig. 4B). Based on our current study and results reported by others, we conclude that Fe ion binding proteins are highly conductive class of biomolecules.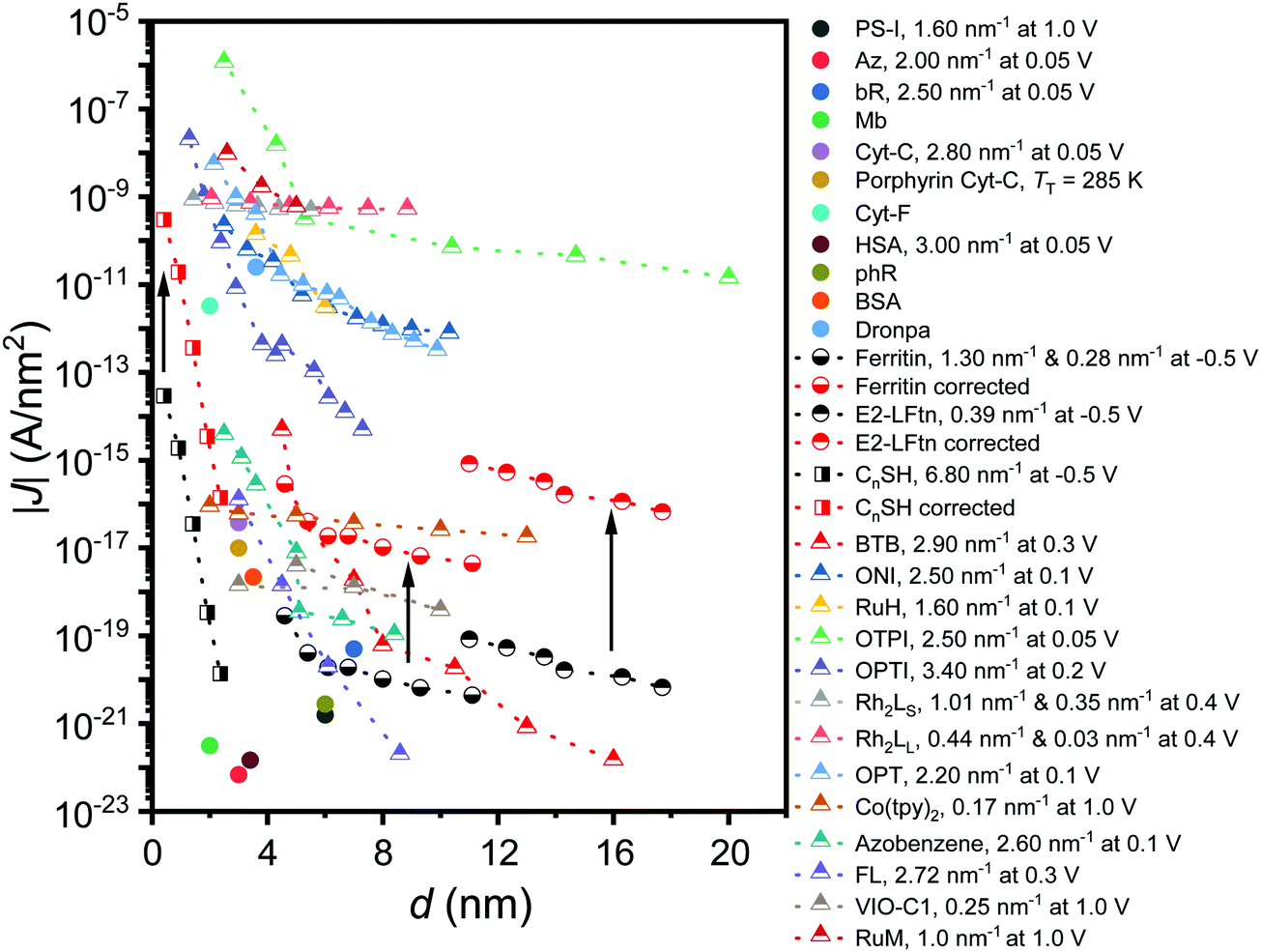 | ||
| Fig. 6 J at 0. 1 V as a function of the d for alkanethiols (squares) taken from ref. 60. The proteins (circles) presented are PS-I,10 bR,13,23 ferritin,15 Cyt-C and Cyt-C with Fe ions removed (porphyrin-Cyt-C),17,23 halorhodopsin (phR),14 Mb,61 Az, cytochrome-F (Cyt-F), dronpa, bovine serum albumin (BSA) and human serum albumin (HSA).23 The conjugated molecular wires (triangles) presented here are bis-thienylbenzene (BTB),28 1-(4-aminophenyl)-1′-methyl-4,4′-bipyridinium bis-hexafluorophosphate (VIO-C1),27 OPTI,59 oligo-tetrathiafulvalene-pyromelliticdiimide-imine (OTPI),62 oligonaphthalenefluoreneimine (ONI),63 copper bipyridine (Co(tpy)2),64 oligophenylenetriazole (OPT),65 fluorene (FL),66 Ru-bis(σ-arylacetylide) wires with –CH2– termini (RuM) and –O(CH2)6– termini (RuH),67 azobenezene,68 and [Rh2(O2CCH3)4] (Rh2) wires with linear with pyrazine (LS) (Rh2LS) and 1,2-bis(4-pyridyl)ethene (LL) (Rh2LL) N,N0-bidentate pyrazine ligands.69 The β value at the reported V is shown in the legend, and two β values are reported for molecules that undergo a transition in the mechanism of CT as a function of d. For porphyrin-Cyt-C, the TT is reported instead of the β.17 A correction factor for effective electrical contact area of 104 is taken from ref. 58 and is applied to EGaIn junctions from ref. 15 and 60; the solid black arrows link the uncorrected (black-coloured) and corrected (red-coloured) datasets. The opensource software WebPlotDigitizer (link: https://automeris.io/WebPlotDigitizer/index.html) was utilised to extract numerical data from graphs of the respective publications where needed. | ||
Conclusions
We demonstrated a transition in the mechanism of CT from incoherent to coherent tunnelling across ∼17.7 nm in bioengineered E2-LFtn based biomolecular tunnel junctions. The investigation of the CT across the E2-LFtn junction as a function of dE2-LFtn and T confirms that the mechanism of the CT is incoherent tunnelling for junctions with apo-E2-LFtn and E2-LFtn. In addition, we further report conductance switching in E2-LFtn biomolecular tunnel junctions, and the reversibility of the conductance switching is a function of the Fe ion loading/cage. Temperature dependent XAS data indicate that this switching is caused by a phase transition in the iron oxide NPs inside E2-LFtn at TT = 220–240 K. Remarkably, this phase transition changes the mechanism where it transitions from coherent tunnelling at T < TT to incoherent tunnelling for T > TT for junctions with 3000Fe loading. In contrast, in junctions with 2000Fe ion loading, the transition is irreversible. Our findings suggest that the iron oxide NPs offer accessible energy states that are separated by a low energy barrier and provide multiple incoherent tunnelling sites resulting in highly conductive CT channels that mediate long-range CT. Our results also highlight the importance of stability as junctions with high Fe ion loadings are more stable than those junctions with low Fe iron loadings. Structural order plays a pivotal role as for protein structures with disorder charge have to hop across segments with high energy barriers while CT across ordered protein structures involves incoherent tunnelling with low energy barriers. The results reported here improve our understanding of the mechanisms of CT across biomolecular junctions. The E2 protein cages are highly conductive, and combined with our previous studies on ferritin, leading us to conclude that iron-binding globular proteins are highly conductive in nature, and can undergo long range coherent activationless CT, and thus, may have potential applications in biomolecular electronic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Diamond Light Source and Australian Synchrotron (ANSTO) are acknowledged for assistance in characterisation of the SAMs. D. Q. acknowledges the support of the Australian Research Council (Grant No. FT160100207). We acknowledge the Ministry of Education (MOE) for supporting this research under award no. MOE2019-T2-1-137. Prime Minister's Office, Singapore, under its Medium-sized centre program is also acknowledged for supporting this research. Prof. Jean-Christophe Lacroix are acknowledged for providing us with the CT data from ref.27References
- G. S. Engel, T. R. Calhoun, E. L. Read, T.-K. Ahn, T. Mančal, Y.-C. Cheng, R. E. Blankenship and G. R. Fleming, Nature, 2007, 446, 782–786 Search PubMed.
- E. Collini, C. Y. Wong, K. E. Wilk, P. M. G. Curmi, P. Brumer and G. D. Scholes, Nature, 2010, 463, 644–647 Search PubMed.
- M. L. Ghirardi, A. Dubini, J. Yu and P.-C. Maness, Chem. Soc. Rev., 2009, 38, 52–61 Search PubMed.
- M. A. Grodick, N. B. Muren and J. K. Barton, Biochemistry, 2015, 54, 962–973 Search PubMed.
- A. R. Arnold, M. A. Grodick and J. K. Barton, Cell Chem. Biol., 2016, 23, 183–197 Search PubMed.
- N. Amdursky, D. Ferber, C. A. Bortolotti, D. A. Dolgikh, R. V. Chertkova, I. Pecht, M. Sheves and D. Cahen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5556–5561 Search PubMed.
- M. P. Ruiz, A. C. Aragonès, N. Camarero, J. G. Vilhena, M. Ortega, L. A. Zotti, R. Pérez, J. C. Cuevas, P. Gorostiza and I. Díez-Pérez, J. Am. Chem. Soc., 2017, 139, 15337–15346 Search PubMed.
- C. Romero-Muñiz, M. Ortega, J. G. Vilhena, I. Diéz-Pérez, J. C. Cuevas, R. Pérez and L. A. Zotti, Biomolecules, 2019, 9, 506 Search PubMed.
- J. Zhao, J. J. Davis, M. S. P. Sansom and A. Hung, J. Am. Chem. Soc., 2004, 126, 5601–5609 Search PubMed.
- O. E. Castañeda Ocampo, P. Gordiichuk, S. Catarci, D. A. Gautier, A. Herrmann and R. C. Chiechi, J. Am. Chem. Soc., 2015, 137, 8419–8427 Search PubMed.
- M. Ortega, J. G. Vilhena, L. A. Zotti, I. Díez-Pérez, J. C. Cuevas and R. Pérez, Biomolecules, 2019, 9, 611 Search PubMed.
- N. Amdursky, L. Sepunaru, S. Raichlin, I. Pecht, M. Sheves and D. Cahen, Adv. Sci., 2015, 2, 1400026 Search PubMed.
- L. Sepunaru, N. Friedman, I. Pecht, M. Sheves and D. Cahen, J. Am. Chem. Soc., 2012, 134, 4169–4176 Search PubMed.
- S. Mukhopadhyay, S. Dutta, I. Pecht, M. Sheves and D. Cahen, J. Am. Chem. Soc., 2015, 137, 11226–11229 Search PubMed.
- K. S. Kumar, R. R. Pasula, S. Lim and C. A. Nijhuis, Adv. Mater., 2016, 28, 1824–1830 Search PubMed.
- B. Zhang, W. Song, J. Brown, R. Nemanich and S. Lindsay, J. Am. Chem. Soc., 2020, 142, 6432–6438 Search PubMed.
- N. Amdursky, I. Pecht, M. Sheves and D. Cahen, J. Am. Chem. Soc., 2013, 135, 6300–6306 Search PubMed.
- C. Romero-Muñiz, M. Ortega, J. G. Vilhena, I. Díez-Pérez, J. C. Cuevas, R. Pérez and L. A. Zotti, Phys. Chem. Chem. Phys., 2018, 20, 30392–30402 Search PubMed.
- Y. Li, J. M. Artés, B. Demir, S. Gokce, H. M. Mohammad, M. Alangari, M. P. Anantram, E. E. Oren and J. Hihath, Nat. Nanotechnol., 2018, 13, 1167–1173 Search PubMed.
- J. M. A. Vivancos, J. Hihath and I. Díez-Pérez, Molecular Electronics: An Experimental and Theoretical Approach, Pan Stanford Publishing Pte. Ltd, 2016, pp. 281–323 Search PubMed.
- L. Sepunaru, I. Pecht, M. Sheves and D. Cahen, J. Am. Chem. Soc., 2011, 133, 2421–2423 Search PubMed.
- T. Peng, D. Paramelle, B. Sana, C. F. Lee and S. Lim, Small, 2014, 10, 3131–3138 Search PubMed.
- N. Amdursky, D. Marchak, L. Sepunaru, I. Pecht, M. Sheves and D. Cahen, Adv. Mater., 2014, 26, 7142–7161 Search PubMed.
- S. K. Karuppannan, E. H. L. Neoh, A. Vilan and C. A. Nijhuis, J. Am. Chem. Soc., 2020, 142, 3513–3524 Search PubMed.
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Chem. Rev., 2016, 116, 4318–4440 Search PubMed.
- L. Luo, S. H. Choi and C. D. Frisbie, Chem. Mater., 2011, 23, 631–645 Search PubMed.
- Q. Van Nguyen, P. Martin, D. Frath, M. L. Della Rocca, F. Lafolet, S. Bellinck, P. Lafarge and J.-C. Lacroix, J. Am. Chem. Soc., 2018, 140, 10131–10134 Search PubMed.
- H. Yan, A. J. Bergren, R. McCreery, M. L. Della Rocca, P. Martin, P. Lafarge and J. C. Lacroix, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5326–5330 Search PubMed.
- J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2014, 136, 2930–2939 Search PubMed.
- Y. Zhang, C. Liu, A. Balaeff, S. S. Skourtis and D. N. Beratan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10049–10054 Search PubMed.
- C. Joachim and M. A. Ratner, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8801–8808 Search PubMed.
- K. Michaeli, D. N. Beratan, D. H. Waldeck and R. Naaman, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5931–5936 Search PubMed.
- C. D. Bostick, S. Mukhopadhyay, I. Pecht, M. Sheves, D. Cahen and D. Lederman, Rep. Prog. Phys., 2018, 81, 026601 Search PubMed.
- M. Buttiker, IBM J. Res. Dev., 1988, 32, 63–75 Search PubMed.
- S. Ho Choi, B. Kim and C. D. Frisbie, Science, 2008, 320, 1482–1486 Search PubMed.
- T. Peng, H. Lee and S. Lim, Biomacromolecules, 2012, 13, 699–705 Search PubMed.
- T. Tosha, M. R. Hasan and E. C. Theil, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18182–18187 Search PubMed.
- G. Jutz, P. van Rijn, B. Santos Miranda and A. Böker, Chem. Rev., 2015, 115, 1653–1701 Search PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2003 Search PubMed.
- J.-P. Jolivet, C. Chanéac and E. Tronc, Chem. Commun., 2004, 477–483 Search PubMed.
- N. Gálvez, B. Fernández, P. Sánchez, R. Cuesta, M. Ceolín, M. Clemente-León, S. Trasobares, M. López-Haro, J. J. Calvino, O. Stéphan, J. M. Domínguez-Vera, N. Gálvez, B. Fernández, P. Sánchez, R. Cuesta, M. Ceolín, M. Clemente-León, S. Trasobares, M. López-Haro, J. J. Calvino, O. Stéphan and J. M. Domínguez-Vera, J. Am. Chem. Soc., 2008, 130, 8062–8068 Search PubMed.
- M. G. Walls, C. Cao, K. Yu-Zhang, J. Li, R. Che and Y. Pan, Microsc. Microanal., 2013, 19, 835–841 Search PubMed.
- D. D. Boland, R. N. Collins, C. J. Miller, C. J. Glover and T. D. Waite, Environ. Sci. Technol., 2014, 48, 5477–5485 Search PubMed.
- S. Das, M. J. Hendry and J. Essilfie-Dughan, Environ. Sci. Technol., 2011, 45, 268–275 Search PubMed.
- L. Yang, C. I. Steefel, M. A. Marcus and J. R. Bargar, Environ. Sci. Technol., 2010, 44, 5469–5475 Search PubMed.
- C. Liu, Z. Zhu, F. Li, T. Liu, C. Liao, J. J. Lee, K. Shih, L. Tao and Y. Wu, Chem. Geol., 2016, 444, 110–119 Search PubMed.
- H. Liu, P. Li, B. Lu, Y. Wei and Y. Sun, J. Solid State Chem., 2009, 182, 1767–1771 Search PubMed.
- X. Liu, W. Jin and E. C. Theil, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3653–3658 Search PubMed.
- L. Yuan, L. Jiang, B. Zhang and C. A. Nijhuis, Angew. Chem., Int. Ed., 2014, 53, 3377–3381 Search PubMed.
- S. K. Karuppannan, H. Hongting, C. Troadec, A. Vilan and C. A. Nijhuis, Adv. Funct. Mater., 2019, 1904452, 1904452 Search PubMed.
- J. Zhang, C. Cui and X. Zhou, Chinese Sci. Bull., 2009, 54, 723–726 Search PubMed.
- T. Rakshit, S. Banerjee, S. Mishra and R. Mukhopadhyay, Langmuir, 2013, 29, 12511–12519 Search PubMed.
- E. Stavitski and F. M. F. de Groot, Micron, 2010, 41, 687–694 Search PubMed.
- M. Koralewski, L. Balejčíková, Z. Mitróová, M. Pochylski, M. Baranowski and P. Kopčanský, ACS Appl. Mater. Interfaces, 2018, 10, 7777–7787 Search PubMed.
- Y. H. Pan, K. Sader, J. J. Powell, A. Bleloch, M. Gass, J. Trinick, A. Warley, A. Li, R. Brydson and A. Brown, J. Struct. Biol., 2009, 166, 22–31 Search PubMed.
- K. Asadi, A. J. Kronemeijer, T. Cramer, L. Jan Anton Koster, P. W. M. Blom and D. M. de Leeuw, Nat. Commun., 2013, 4, 1710 Search PubMed.
- X. Chen, T. Salim, Z. Zhang, X. Yu, I. Volkova and C. A. Nijhuis, ACS Appl. Mater. Interfaces, 2020, 12, 45111–45121 Search PubMed.
- S. Mukhopadhyay, S. K. Karuppannan, C. Guo, J. A. Fereiro, A. Bergren, V. Mukundan, X. Qiu, O. E. Castañeda Ocampo, X. Chen, R. C. Chiechi, R. McCreery, I. Pecht, M. Sheves, R. R. Pasula, S. Lim, C. A. Nijhuis, A. Vilan and D. Cahen, iScience, 2020, 23, 101099 Search PubMed.
- C. E. Smith, S. O. Odoh, S. Ghosh, L. Gagliardi, C. J. Cramer and C. D. Frisbie, J. Am. Chem. Soc., 2015, 137, 15732–15741 Search PubMed.
- L. Jiang, C. S. S. Sangeeth, L. Yuan, D. Thompson and C. A. Nijhuis, Nano Lett., 2015, 15, 6643–6649 Search PubMed.
- S. Raichlin, I. Pecht, M. Sheves and D. Cahen, Angew. Chem., Int. Ed., 2015, 54, 12379–12383 Search PubMed.
- S. H. Choi and C. D. Frisbie, J. Am. Chem. Soc., 2010, 132, 16191–16201 Search PubMed.
- S. H. Choi, C. Risko, M. C. R. Delgado, B. Kim, J.-L. Brédas and C. D. Frisbie, J. Am. Chem. Soc., 2010, 132, 4358–4368 Search PubMed.
- Q. van Nguyen, U. Tefashe, P. Martin, M. L. Della Rocca, F. Lafolet, P. Lafarge, R. L. McCreery and J. C. Lacroix, Adv. Electron. Mater., 2020, 6, 1–11 Search PubMed.
- L. Luo and C. D. Frisbie, J. Am. Chem. Soc., 2010, 132, 8854–8855 Search PubMed.
- A. Morteza Najarian and R. L. McCreery, ACS Nano, 2017, 11, 3542–3552 Search PubMed.
- L. Luo, A. Benameur, P. Brignou, S. H. Choi, S. Rigaut and C. D. Frisbie, J. Phys. Chem. C, 2011, 115, 19955–19961 Search PubMed.
- A. Santos, U. M. Tefashe, R. L. McCreery and P. R. Bueno, Phys. Chem. Chem. Phys., 2020, 22, 10828–10832 Search PubMed.
- D. Bu, Y. Xiong, Y. N. Tan, M. Meng, P. J. Low, D.-B. Kuang and C. Y. Liu, Chem. Sci., 2018, 9, 3438–3450 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc05773h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |