
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid ligands with calixarene and thiodigalactoside groups: galectin binding and cytotoxicity†‡
Hao Zhang§
a,
Hans Ippelb,
Michelle C. Millerc,
Tse J. Wongd,
Arjan W. Griffioend,
Kevin H. Mayoc and
Roland J. Pieters *a
*a
aDepartment of Chemical Biology & Drug Discovery, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, P.O. Box 80082, NL-3508 TB Utrecht, The Netherlands. E-mail: r.j.pieters@uu.nl
bDepartment of Biochemistry and the Cardiovascular Research Institute Maastricht (CARIM), Maastricht University, Maastricht, The Netherlands
cDepartment of Biochemistry, Molecular Biology & Biophysics, University of Minnesota, Minneapolis, MN 55455, USA
dAngiogenesis Laboratory, Amsterdam University Medical Center, location VUMC, Cancer Center Amsterdam, Amsterdam, The Netherlands
First published on 12th July 2019
Abstract
Galectins have diverse functions and are involved in many biological processes because of their complex intra- and extracellular activities. Selective and potent inhibitors for galectins will be valuable tools to investigate the biological functions of these proteins. Therefore, we describe here the synthesis of galectin inhibitors with a potential “chelate effect”. These compounds are designed to bind to two different binding sites on galectins simultaneously. In this paper a series of asymmetric “hybrid” compounds are prepared, which combine two galectin ligands (1) a substituted thiodigalactoside derivative and (2) an antagonist calixarene-based therapeutic agent. NMR spectroscopy was used to evaluate the interactions of these compounds with Galectin-1 and -3. In addition, cellular experiments were conducted to compare the cytotoxic effects of the hybrids with those of a calixarene derivative. While only the thiodigalactoside part of the hybrids showed strong binding, the calixarene part was responsible for observed cytoxoxicity effects, suggesting that the calixarene moiety may also be addressing a non-galectin target.
Introduction
As a subfamily of lectins, galectins generally recognize β-galactosides, among other types of saccharides and polysaccharides.1 Galectins contain conserved carbohydrate recognition domains (CRDs) that consist of about 135 amino acids responsible for carbohydrate binding.2 They also display an intriguing combination of intra- and extracellular activities and are involved in many biological process like cell growth, cell adhesion and signaling.3,4 In addition, galectins play a profound role in many diseases, like inflammation, cancer and fibrosis.5–8 To understand the mechanism of action of galectins and their relevant therapeutic potential, there is a clear need for selective and potent inhibitors targeting different galectins.The last two decades have witnessed a growing number of galectin inhibitors derived from carbohydrates and peptides.9–12 The majority of the galectin inhibitors are carbohydrate-based, most often modifications of the β-galactoside lactose, a naturally galectin-binding ligand. Typically, those inhibitors bind to the carbohydrate binding site. Furthermore, a significant increase in binding affinity for certain galectins, like galectin-3 (Gal-3), was found when an aromatic group is introduced at the C3 position of the galactose moiety.13–15 Subsequently, a new scaffold, thiodigalactoside (TDG, Kd = 49 μM for Gal-3, Kd = 24 μM for Gal-1), produced more potent inhibitors in addition to enhancing glycolytic stability, while maintaining a similar binding mode compared to lactose.13,16–18
Compared to carbohydrates, peptides are easier to synthesize. For carbohydrate-binding proteins, some peptides may prevent carbohydrate binding either by directly occupying the carbohydrate-binding site (“carbohydrate mimetic”) or by simply occluding access to the carbohydrate binding site.19–23 For peptide-based galectin inhibitors, both modes of action have been suggested; however, the exact binding modes were not always elucidated.21,24 The best-studied peptide-based galectin inhibitor is anginex,25 a potent anti-angiogenic and anti-tumor peptide that can bind to Gal-1, -2, -7, -8N and -9N, but apparently not to Gal-4N, -4C and -9C.6,26 Intriguingly, anginex was also shown to significantly enhance the binding of a similar list of galectins, such as Gal-1, -2, -7, -8N, -8C, -9N and not -9C, -3, -4N and -4C to certain glycans (though not lactose or LacNAc).27 In attempts to produce simplified analogues of anginex, various calix[4]arene derivatives, such as calixarene 0118 (Scheme 2), were found to have an anti-angiogenic effect like anginex.28 Considering the functional similarity, Gal-1 was assumed to be the target of 0118. Subsequent studies using 15N–1H HSQC NMR spectroscopy provided support for this hypothesis, and showed that 0118 binds to Gal-1 at a site distant from the galectin's carbohydrate binding site with a Kd of around 30 μM and that it attenuated Gal-1 binding to lactose.29 Although the Gal-1 binding of 0118 in a cellular environment has yet to be directly shown, there is evidence that 0118 targets Gal-1 in vivo.29,30
Using CuAAC (Copper-catalyzed azide–alkyne cyclo-addition) to functionalize TDG-based ligands strongly increases the affinity for certain galectins.17,31,32 In addition, among the symmetrical TDG derivatives, the inclusion of various aromatic groups can direct the selectivity between Gal-1 and Gal-3.31 So far, however, there has been little discussion about non-symmetrical TDG compounds for galectin inhibition. The recent published work on 0118 and the available data on TDG-based inhibitors lead us to consider the creation of hybrid or hetero-bivalent galectin inhibitors that contain both a calix[4]arene ligand and a TDG ligand connected via a tether, to make a series of non-symmetrical galectin inhibitors. These bivalent inhibitors may potentially interact simultaneously at two different sites on galectins.32 To this end, we set out to make a series of such non-symmetrical “hybrid” compounds that combine the two core structures of TDG and 0118. NMR spectroscopy was used to demonstrate interactions between these compounds and Gal-1 and Gal-3. All compounds were characterized in terms of cytotoxicity and anti-proliferative effects on HUVEC cells (human umbilical venous endothelial cells), HT-29 cells (Human colorectal adenocarcinoma grade II) and on Caki-2 cells (Human Renal Cancer Cell Line), along with TDG and calixarene control compounds.
Results and discussion
Design and synthesis of hybrid galectin ligands
The synthetic design of hybrid galectin ligands 1–3 was based on retrosynthetic analysis shown in Scheme 1. To obtain the target molecules, precursors based on calixarenes 5a–c and the precursor based on thiodigalactoside 4 needed to be synthesized. The synthetic route towards 5a–5c involved a direct CuAAC reaction of the calixarene analogue with dipropargylated polyethylene glycol.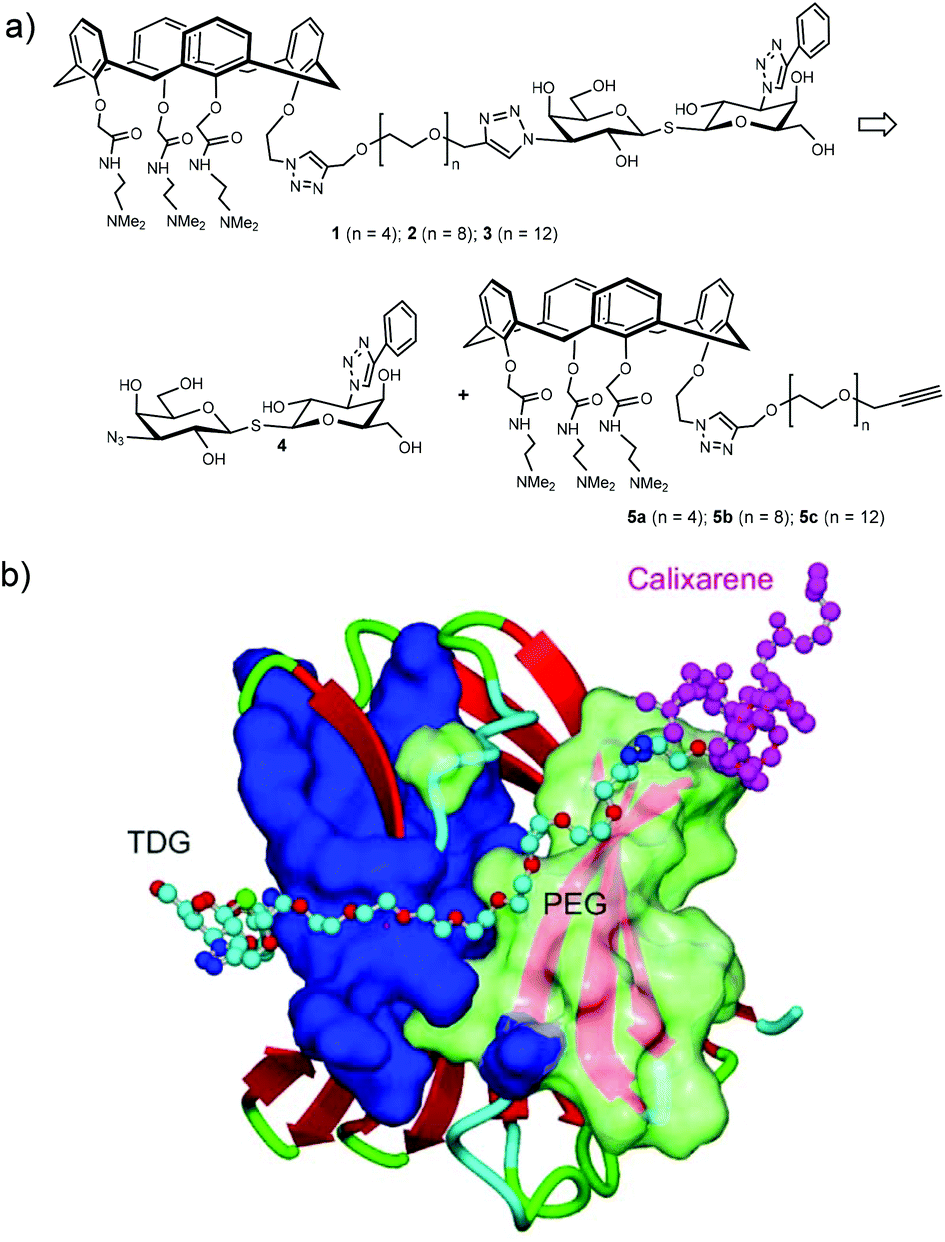 | ||
| Scheme 1 (a) Structure and retrosynthetic analysis of hybrid galectin ligands. (B) Proposed binding mode of hybrids galectin ligand to Gal-3 CRD. | ||
Synthesis of the calixarene precursor proceeded as shown in Scheme 2. Based on a published procedure, compound 7 was obtained by mono-alkylating 6 with 1-azido-2-iodoethane.33 Subsequent treatment with an excess of ethyl bromoacetate in the presence of K2CO3 yielded compound 8. It should be noted that compound 8 contained some non-cone conformers as revealed by NMR with respect to the rotation of the benzene rings in calixarene. In any event, this product was used in the next reaction without further purification. Previously synthesized 9–11 were separately reacted with 8, and the resulting products were reacted with N,N-dimethylethylene-diamine to yield 5a–c.34,35 After preparative HLPC purification, NMR and mass spectrometry showed that the isolated compounds were in the “cone” conformation.
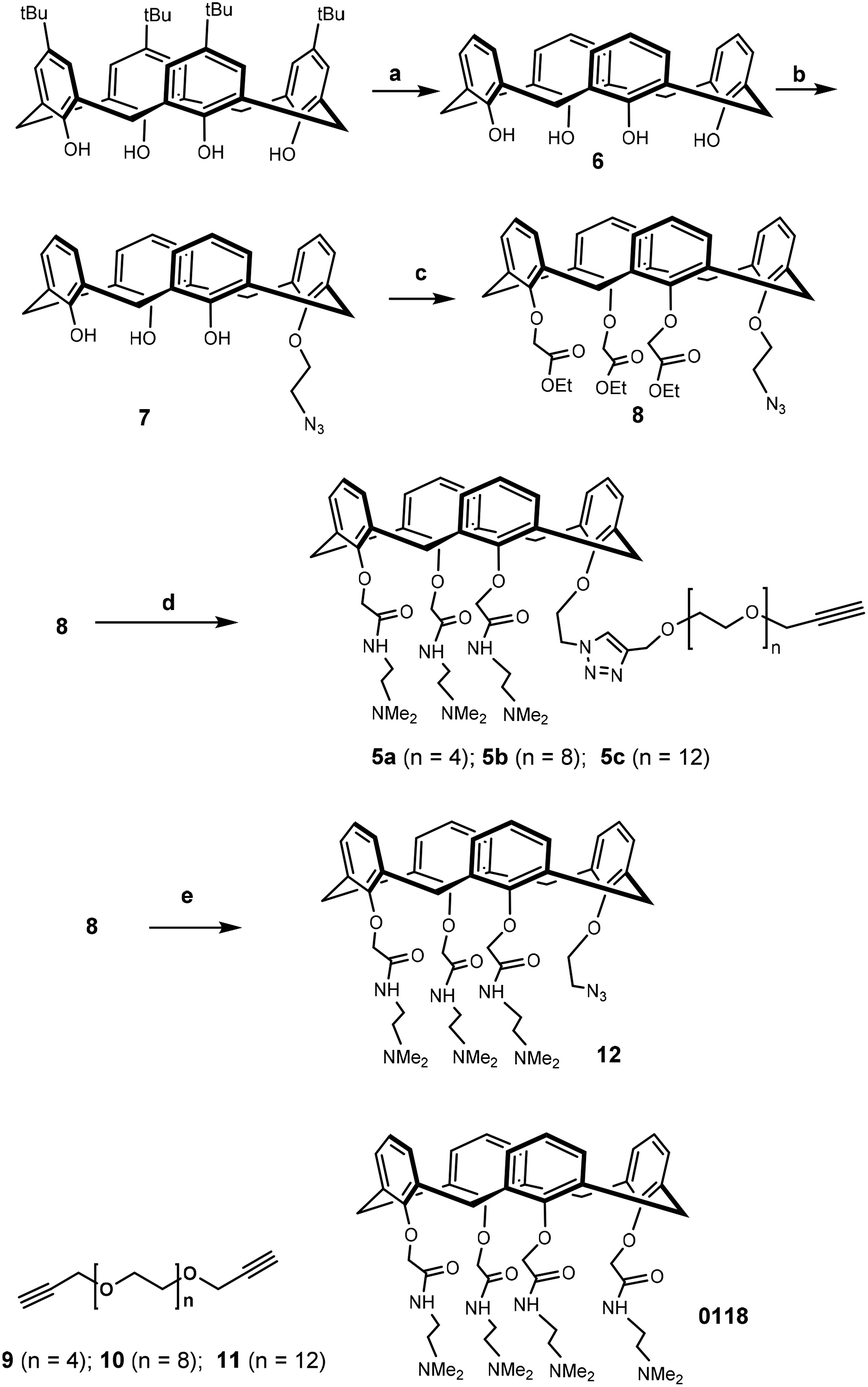 | ||
| Scheme 2 Synthesis of targets 5a–c. Reaction conditions: (a) AlCl3, phenol, toluene, 98%; (b) 1-azido-2-iodoethane, NaOMe, CH3CN, 85 °C, 67%; (c) ethyl bromoacetate, K2CO3, CH3CN, 85 °C; 65%; (d), (i) compound 9–11, CuI, CH3CN, 80 °C, microwave; (ii) N,N-dimethylethylenediamine, 50 °C, 10% in two steps (5a), 23% in two steps (5b), 27% in two steps (5c); (e) N,N-dimethylethylenediamine, 50 °C, 39%. | ||
To construct the required TDG derivatives, the tri-isopropylsilyl thio-glycoside 17 and the glycosyl halide 15 were required. The TDG synthesis was based on a published method that consists of a one-pot de-silylation and glycosyl thiol alkylation with a glycosyl halide (Scheme 3).36 The synthesis benefited from previous work, in which compound 18 was obtained.37 After removing the acetyl protecting group of 18, the resulting crude 4 was used for “click” with 5a–c without further purification. Aside for this, CuAAC modification of compound 18 with phenylacetylene and compound 11 gave the corresponding products that, following deprotection, yielded control compounds 19 and 20.
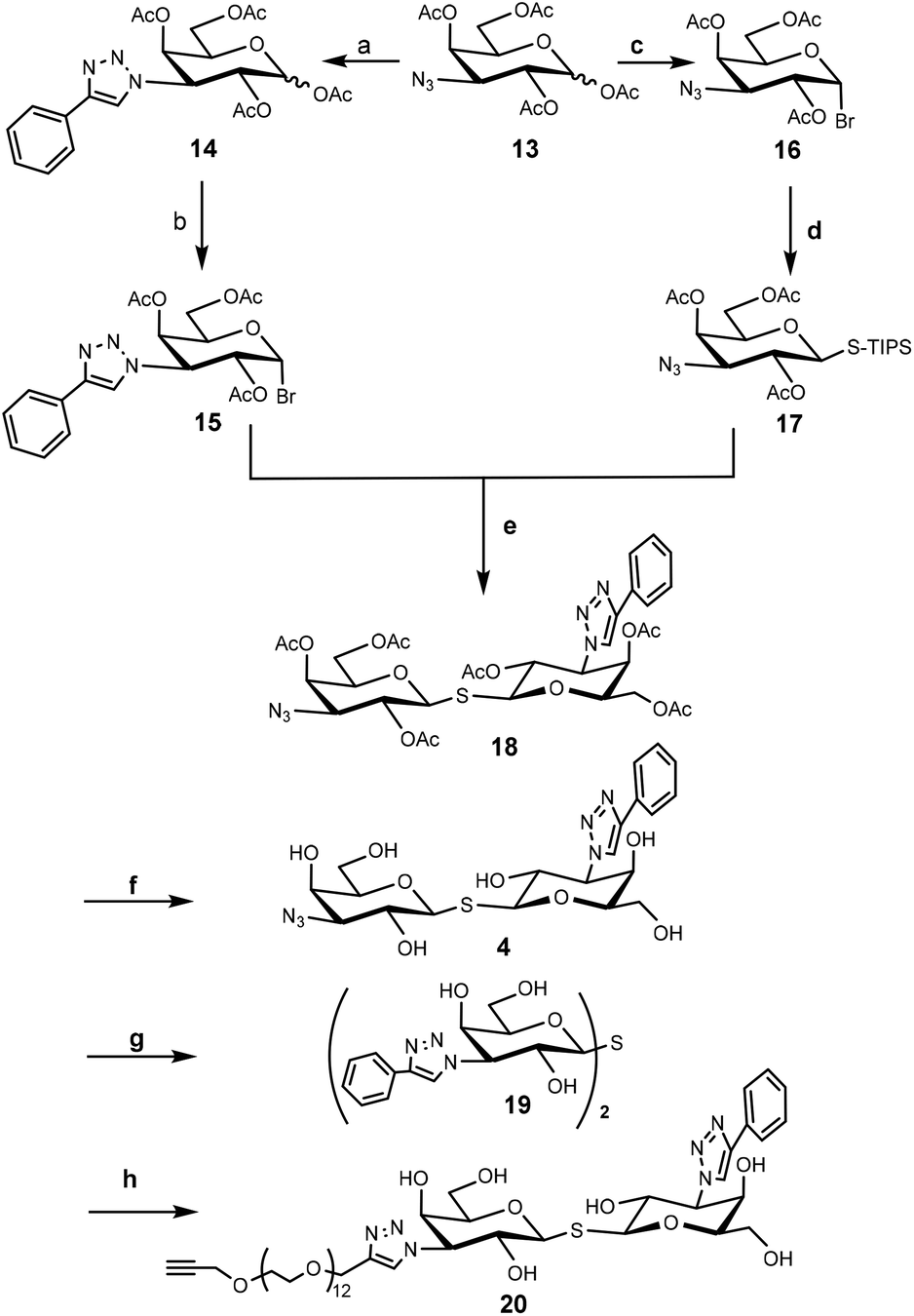 | ||
| Scheme 3 Synthesis of TDG component. Reaction conditions: (a) Phenylacetylene, CuSO4, sodium ascorbate, DMF/H2O, 80 °C, microwave; (b) HBr, CH2Cl2, 79% in two steps; (c) TiBr4, CH2Cl2/EtOAc, 67%; (d) TIPSSH, K2CO3, CH3CN, 30%; (e) TBAF, CH3CN, 62%; (f) NaOMe, CH3OH; (g), (i) Phenylacetylene, CuSO4, sodium ascorbate, DMF/H2O, 80 °C, microwave; (ii) NaOMe, CH3OH, 23% in two steps; (h), (i) compound 11, CuI, CH3OH, 80 °C, microwave; (ii) NaOMe, CH3OH, 5.0% in two steps. | ||
Having assembled the important intermediates, calixarene parts 5a–c and TDG, the next objective was CuAAC conjugation and purification of the resulting product to yield the final hybrid compounds (Scheme 4). Taking hybrid 3 as an example, compounds 5c and 4 were coupled using CuAAC under microwave irradiation to give the target compound (12 mg, 22% yield) after purification by preparative HPLC. Via analysis of the NMR spectrum, the calixarene part of hybrid 3 was confirmed to be in the “cone” conformation (i.e. identical to calixarene 0118 (ref. 35)) and was used for biological testing and binding studies.
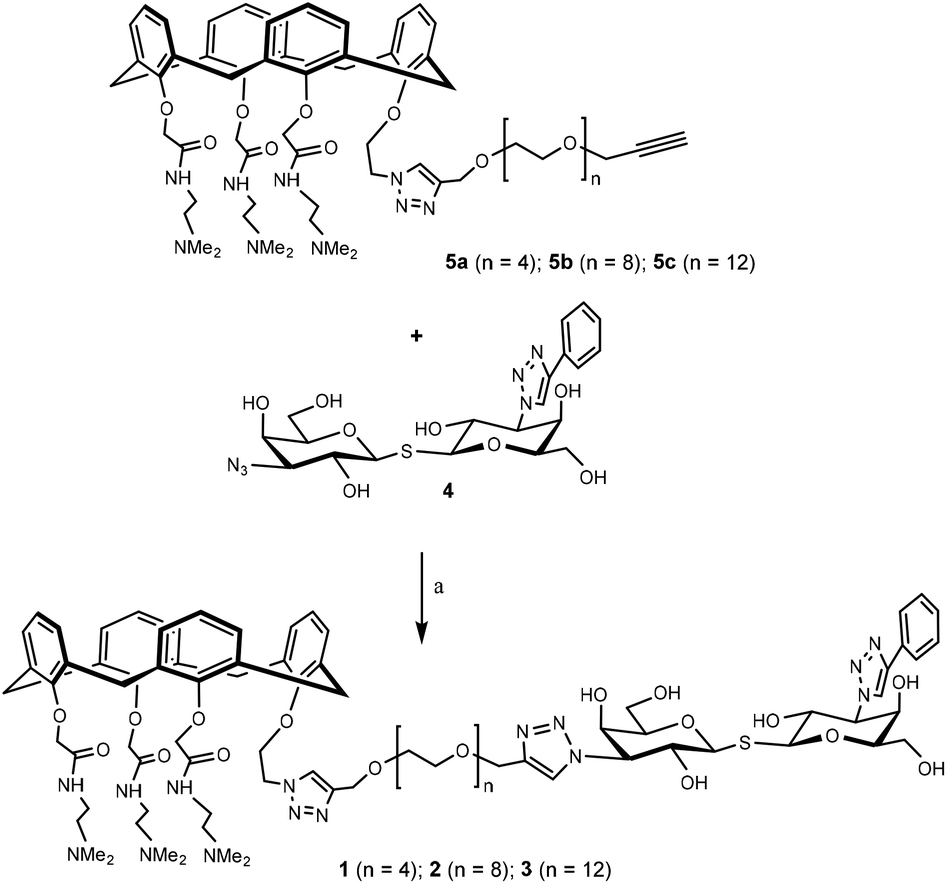 | ||
| Scheme 4 Synthesis of hybrid 1–3. Hybrid 1–3 were synthesized in a similar fashion starting from the corresponding precursor. Reaction conditions: CuI, CH3OH, 80 °C, microwave, 90 min. | ||
Conformation analysis of hybrid galectin ligands
Calix[4]arenes are not planar and can adopt four different stable conformations: cone, partial cone (paco), 1,2-alternate (1,2-alt), and 1,3-alternate (1,3-alt). As the phenolic hydroxyls in the lower rim are replaced by bulky groups, rotation of the aromatic rings are prevented, and stable conformations are produced. A method to determine the conformation has been published,38,39 in which the 13C-NMR chemical shift of the bridged methylene carbons that connect each pair of aromatic rings is observed at 30–31 ppm when neighbouring aromatic rings are oriented on the same side of the cone and at 37–40 ppm when they point in opposite directions (Fig. S1A‡). In the NMR spectra of our calixarene analogues, the 13C signal for the bridging methylene carbon can be observed, and its chemical shift is found at 30–31 ppm, indicating that these calixarene compounds adopted the “cone” conformation. In addition, NOESY spectra were acquired for information on the proximities of various protons (Fig. S1B‡). All NOESY cross-peaks from aromatic protons from the calixarene moiety in hybrids 1–3 only appear from the bridged methylene protons, and none are observed from substituents of the lower rim which are more remote in the cone conformation.NMR spectroscopy to demonstrate the interaction between ligands with Gal-1 and Gal-3
To illustrate effects to Gal-3 induced by the binding of the hybrid compounds, perturbation maps are shown in which chemical shift differences (± compound) of protein backbone NH resonances in the complex are plotted vs. the Gal-3 amino acid sequence (Fig. 1).40,41 These chemical shift maps provide insight into which protein residues interact with the ligand. For example, Gal-3 backbone amide 1H–15N signals in the presence and absence of hybrids 2 and 3 (protein![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand ratios of 1:1.2) are shown in Fig. 1A and B, respectively. In both instances, the ligand binds to the protein, with the amplitude and pattern of chemical shift perturbations throughout the Gal-3 amino acid sequence remaining basically the same. TDG control compound 20 and calixarene control compound 12 were measured under the same conditions (Fig. 1C and D). In Fig. 1C, 20 that contains TDG and the linker, exhibits a similar interaction pattern with Gal-3 compared to hybrids 2 and 3 (compare Fig. 1A and B). In contrast, 12 induces minimal perturbations to Gal-3 resonances, even when the ligand concentration is increased to 4 mM (Fig. 1D). In addition, residues involved in interactions between 12 and Gal-3 are different from those involved in binding TDG. This observation is consistent with our previous finding that another calixarene compound (called 0118) also binds at a non-canonical site on Gal-1, as observed here with Gal-3.29,30 Furthermore, while the binding affinity between Gal-3 and calix-azide 12 is not easily saturated, maximum chemical shift perturbations are quite large (∼0.4 ppm) through Gal-3 residues 200–220. These chemical shift maxima are similar to those observed with the TDG moiety. The binding phenomena observed here are consistent with known literature values for TDG's (low nanomolar)17 and mono-substituted TDG's (low micromolar or better),37 in that they all indicate relatively tight binding in the NMR slow exchange regime on the chemical sift time scale. For the calixarene derivatives, 4 mM was required to achieve nearly saturable binding, indicating that the equilibrium dissociation constant, KD, should be ∼0.75–1 mM.
:ligand ratios of 1:1.2) are shown in Fig. 1A and B, respectively. In both instances, the ligand binds to the protein, with the amplitude and pattern of chemical shift perturbations throughout the Gal-3 amino acid sequence remaining basically the same. TDG control compound 20 and calixarene control compound 12 were measured under the same conditions (Fig. 1C and D). In Fig. 1C, 20 that contains TDG and the linker, exhibits a similar interaction pattern with Gal-3 compared to hybrids 2 and 3 (compare Fig. 1A and B). In contrast, 12 induces minimal perturbations to Gal-3 resonances, even when the ligand concentration is increased to 4 mM (Fig. 1D). In addition, residues involved in interactions between 12 and Gal-3 are different from those involved in binding TDG. This observation is consistent with our previous finding that another calixarene compound (called 0118) also binds at a non-canonical site on Gal-1, as observed here with Gal-3.29,30 Furthermore, while the binding affinity between Gal-3 and calix-azide 12 is not easily saturated, maximum chemical shift perturbations are quite large (∼0.4 ppm) through Gal-3 residues 200–220. These chemical shift maxima are similar to those observed with the TDG moiety. The binding phenomena observed here are consistent with known literature values for TDG's (low nanomolar)17 and mono-substituted TDG's (low micromolar or better),37 in that they all indicate relatively tight binding in the NMR slow exchange regime on the chemical sift time scale. For the calixarene derivatives, 4 mM was required to achieve nearly saturable binding, indicating that the equilibrium dissociation constant, KD, should be ∼0.75–1 mM.
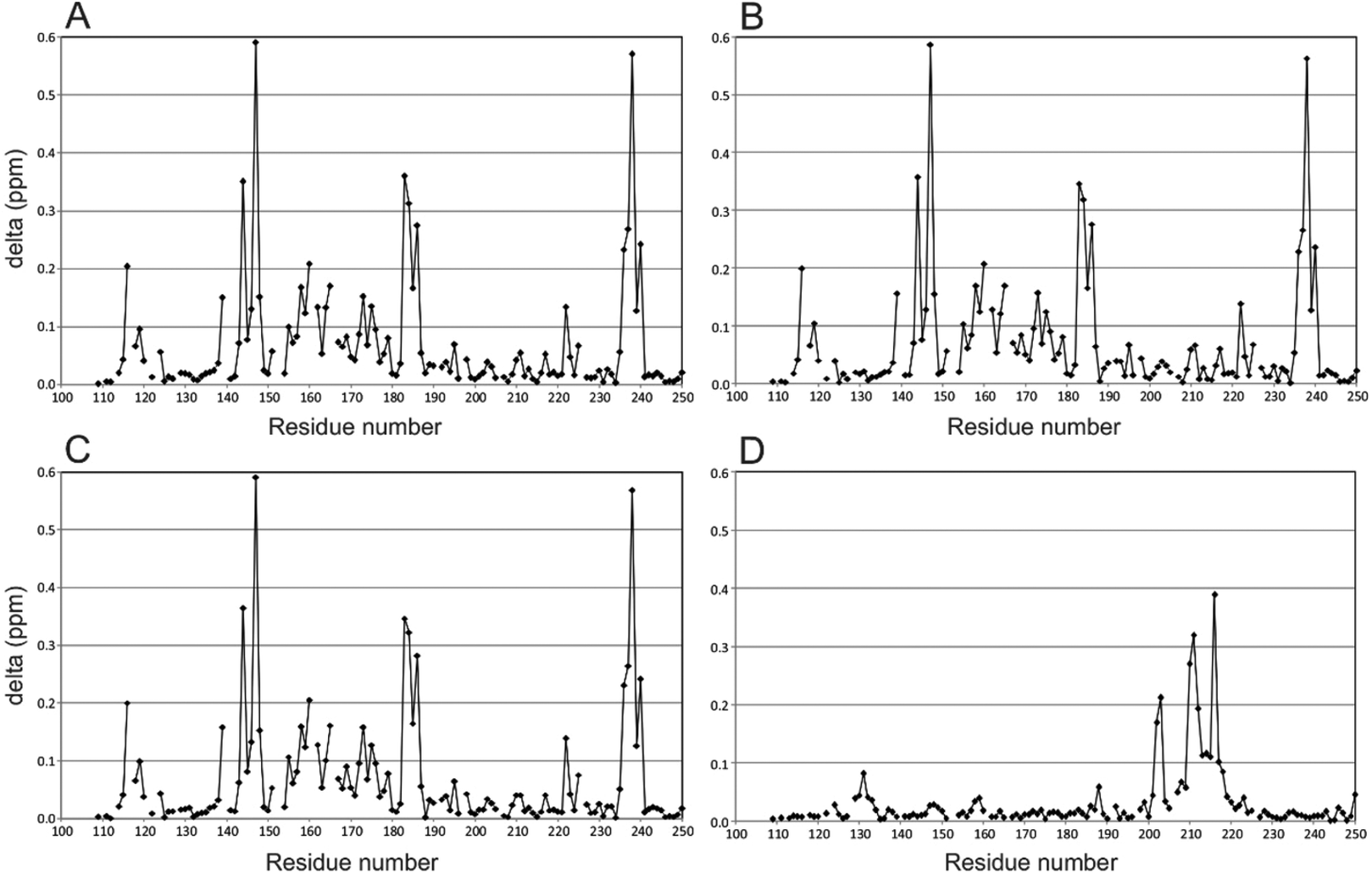 | ||
| Fig. 1 Chemical shift perturbations observed for Gal-3 CRD (108–250), 50 mM in the presence of ligands are shown vs. the residue number of Gal-3. A: Gal-3 CRD in complex with 2, protein (50 μM):ligand (60 μM) ratio of 1:1.2. B: Gal-3 CRD in complex with 3 protein (50 μM):ligand (63 μM) ratio of 1:1.25. C: Gal-3 CRD with 20 protein (50 μM):ligand (60 μM ratio of 1:1.2). D: Gal-3 CRD with 12, protein (50 μM):ligand (4 mM) ratio of 1:80. Large perturbation values of delta chemical shifts reflect large conformational changes of the protein backbone upon ligand binding. Delta (ppm) is defined as the sum of square root [0.25 × Δ15N]2 and square root [Δ1H]2. | ||
Overall, our NMR binding data support the idea that the hybrid compounds interact with these galectins primarily via the TDG moiety. Hybrids 2 and 3 bind to Gal-1 and Gal-3 with high affinity, which corroborates our previous findings.31,37,42 Even though we did not observe synergistic binding with these TDG-linked-calixarene hybrids, there are a few plausible explanations for this. For one, it may be that modifying one of the hydrophilic, positively charged arms of the calixarene reduces binding. Alternatively, it could be that the PEG linker in 2 or 3 is too short to wrap around the Gal-3 CRD from S-face to F-face, making it sterically impossible to simultaneously interact with both sites on Gal-3.
Cytotoxicity studies
Even though calixarene 0118 effectively inhibits cell proliferation,29 there is little information on the biological effects from TDG analogues. In this regard, evaluation of these hybrid compounds in terms of cytotoxicity and inhibition of cell proliferation is of interest. Here, we screened these new compounds in a colorimetric cell viability assay. As shown in Fig. 2, dose–response curves obtained post-incubation of our compounds with HUVEC, HT-29, Caki-2 and HEK293T cells are expressed as the percentage of cell survival relative to control vehicle alone. | ||
| Fig. 2 Dose–response curves obtained from cell viability assays43 after incubation of human umbilical vein endothelial cells (HUVEC) (A), human colorectal carcinoma HT-29 cells (B), human renal cell carcinoma Caki-2 cells (C), and human embryo kidney HEK293T cells (D), with increasing concentrations of the compounds. | ||
All of our compounds that have a calixarene moiety (compound 12, hybrid 2 and hybrid 3) inhibited cell survival, with similar potency compared to parent calixarene 0118. TDG derivatives (19 and 20), however, exhibited no effect on cell viability. The cell types investigated were chosen in an attempt to correlate cell viability with expression of Gal-1 and -3. The strongest cell killing was observed with HT29 cells, known to express Gal-3, but not Gal-1.44 Moderate cell killing was seen with HUVECs that express both galectins.45 Similarly, CAKI-2 cells were only moderately killed, and these cells are at least known to express Gal-3.46 No killing was observed with HEK293T cells that express neither galectin.44 Taken together, these results demonstrate that our hybrid compounds containing calixarene moieties exhibit significant effects on cell viability. However, there was no clear correlation between cell killing and galectin expression, even though Gal-3 was expressed in all instances where cell death occurred.
Conclusions
Here, we described the design and synthesis of a series of galectin inhibitors that were engineered to simultaneously bind to two different epitopes on Gal-1 and -3. TDG derivatives, validated as galectin inhibitors, target the galectin carbohydrate recognition domain. Calixarene 0118, whose discovery was inspired by the peptide-based galectin inhibitor anginex, binds to galectins at a site that is distinct from that of TDG. We thus report here on the synthesis of a non-symmetrical TDG-calixarene conjugate with the prospect of creating bivalent or hybrid ligands to target galectins and antagonize their function. This approach is considerably different from that with other calixarenes outfitted with sugars to bind to galectins.47,48 To obtain the desired compounds, TDG and calixarene-containing precursors were synthesized separately and linked through CuAAC. Using our compounds, NMR spectroscopy was used to study the interaction between the hybrid ligands and Gal-1 and Gal-3. Our results showed that these compounds strongly bind to these galectins, primarily via the TDG moiety. In addition, cytotoxicity studies were performed to evaluate inhibition of cell proliferation by the hybrid ligands. In comparison with parent TDG and calixarene 0118, we found that the hybrid compounds significantly affected cell viability, and this time through the calixarene moiety. A clear correlation between cell killing and galectin expression cannot be made, although Gal-3 expression was present in all cases where cell killing occurred.Overall, we find that both components of our hybrid compounds are fully functional, as evidenced by galectin binding and viability studies. Because calixarene-based effects on biological systems are intriguing and may involve other proteins in addition to galectins, further study is warranted.
Experimental section
Reagents and general methods
TLC analysis was performed on Merck precoated silica gel 60 F-254 plates. Spots were visualized with UV-light, ninhydrin stain (1.5 g ninhydrin and 3.0 mL acetic acid in 100 mL n-butanol), potassium permanganate (1.5 g KMnO4, 10 g K2CO3, and 1.25 mL 10% NaOH in 200 mL H2O) and sulfuric acid (10% sulfuric acid in methanol). Column chromatography was performed using Silica-P Flash silica gel (60 Å, particle size 40–63 μm) from Silicycle (Canada). Microwave reactions were carried out in a Biotage Initiator (300 W) reactor. Lyophilization was performed on a Christ Alpha 1–2 apparatus. 1H and 13C NMR spectroscopy was carried on an Agilent 400-MR spectrometer operating at 400 MHz for 1H and 101 MHz for 13C HSQC, TOCSY and NOESY (500 MHz) were performed with a VARIAN INOVA-500. Complexes between uniformly Gal3-[15N] CRD and various TDG and calixarene ligands were studied on a Bruker Avance III HD 700 MHz spectrometer, equipped with a TCI cryoprobe. Amide proton assignments of apo Gal-3 CRD were taken from a previous research.49Electrospray mass spectrometry was performed using a Shimadzu LCMS QP-8000. High resolution mass spectrometry (HRMS) analysis was recorded using a Bruker ESI-Q-TOF II. Analytical LC-MS (electrospray ionization) was performed using a Thermo-Finnigan LCQ Deca XP Max with the same buffers and protocol as described for analytical HPLC. Analytical HPLC was performed on a Shimadzu-10AVP (Class VP) system using a Phenomenex Gemini C18 column (110 Å, 5 μm, 250 × 4.60 mm) at a flow rate of 1 mL min. The used buffers were 0.1% trifluroacetic acid in CH3CN:H2O = 5:95 (buffer A) and 0.1% trifluroacetic acid in CH3CN:H2O = 95:5 (buffer B). Runs were performed using a standard protocol: 100% buffer A for 2 min, then a linear gradient of buffer B (0–100% in 38 min) and UV-absorption was measured at 214 and 254 nm. Purification using preparative HPLC was performed on an Applied Biosystems workstation with a Phenomenex Gemini C18 column (10 μm, 110 Å, 250 × 21.2 mm) at a flow rate of 6.25 mL min−1. Runs were performed by a standard protocol: buffer A for 5 min followed by a linear gradient of buffer B (0–100% in 70 min) with the same buffer as described for the analytical HPLC.
Synthetic procedures and compound characterization
1H NMR (400 MHz, DMSO-d6) δ 10.00 (d, J = 35.1 Hz, 3H), 8.60–8.36 (m, 3H), 8.17 (s, 1H), 7.05–6.91 (m, 4H), 6.89–6.78 (m, 2H), 6.35 (m, 6H),4.41–4.26 (m, 9H), 4.24 (s, 2H), 4.13 (d, J = 2.4 Hz, 1H), 3.26–3.19 (m, 6H), 3.19–3.07 (m, 5H), 2.81 (s, 13H), 2.77 (s, 6H), 5.05 (q, J = 6.9 Hz, 2H), 4.73 (d, J = 4.1 Hz, 2H), 4.64 (d, J = 13.4 Hz, 2H), 4.53 (d, J = 4.4 Hz, 2H), 3.60–3.39 (m, 16H). 13C NMR (101 MHz, DMSO-d6) δ 170.69, 169.56, 159.15, 158.82, 157.24, 156.28, 154.70, 154.65, 144.19, 144.15, 135.90, 135.80, 135.39, 135.34, 133.57, 133.54, 133.37, 133.30, 129.49, 129.27, 129.23, 128.35, 124.87, 123.44, 123.20, 123.09, 77.53, 74.36, 74.14, 71.89, 70.16, 70.13, 70.11, 70.07, 70.03, 69.88, 69.45, 69.37, 68.92, 63.88, 63.84, 57.90, 56.16, 56.13, 55.90, 49.37, 49.32, 42.85, 42.82, 40.25, 40.05, 34.30, 34.26, 30.68. HRMS (EI, m/z): calculated for C62H85N9O12H+ ([M + H]+): 1148.6390, found 1148.6360.
:CH3OH = 10:1). To the compound (70 mg) N,N-dimethylethylenediamine (5.0 mL) was added under nitrogen. The mixture was stirred for 24 h at 50 °C. The excess of N,N-dimethylethylenediamine was removed by evaporation under reduced pressure. The mixture was dissolved in CH3CN (20 mg mL−1) and purified by preparative HPLC using the standard protocol. Fractions containing the product (tR = 40 min, broad peak) were pooled and the buffer was removed by freeze-drying to obtain the pure compound 5b as a clear oil (30 mg, 0.023 mmol, 23% in two steps). 1H NMR (500 MHz, DMSO-d6) δ 9.59–9.55 (m, 3H, –NHMe2), 8.50–8.34 (m, 3H, –C(O)NH), 8.17 (s, 1H, CH triazole), 7.02 (t, J = 6.0 Hz, 4H, Ar–H), 6.86 (t, J = 7.6 Hz, 2H, Ar–H), 5.05 (t, J = 7.7 Hz, 2H, –CH2N triazole), 4.73 (s, 2H, Ar–OCH2C(O)), 6.39 (t, J = 7.6 Hz, 2H, Ar–H), 6.30 (dd, J = 20.4, 7.5 Hz, 4H, Ar–H), 4.63 (d, J = 13.4 Hz, 2H, Ar2CH2), 4.53 (s, 2H, –CH2O–), 4.38 (d, J = 14.0 Hz, 2H, Ar–OCH2C(O)), 4.32 (t, J = 7.6 Hz, 2H, Ar–OCH2CH2), 4.28–4.23 (m, 3H, Ar–OCH2C(O) and Ar2CH2), 4.13 (d, J = 2.2 Hz, 2H, –CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 3.59–3.52 (m, 6H, –NHCH2), 3.43–3.33 (m, –OCH2CH2O–), 3.23 (d, J = 13.5 Hz, 5H, Ar2CH2), 3.14 (s, 6H, –CH2NMe2), 2.80 (s, –N(CH3)2), 2.77 (s, –N(CH3)2). 13C NMR (126 MHz, DMSO-d6, extracted from HSQC) δ 123.87, 128.60, 128.45, 128.30, 122.37, 122.11, 127.40, 127.33, 127.38, 48.66, 74.01, 29.86, 62.77, 73.76, 73.20, 29.79, 57.80, 70.45, 68.51, 32.86, 33.44, 33.44, 69.71, 68.20, 68.24, 29.25, 55.51, 55.05, 56.48, 54.98, 42.77, 39.64, 40.78, 42.19, 43.86, 38.99. HRMS (EI, m/z): calculated for C70H101N9O16H+ ([M + H]+): 1324.7439, found 1324.7462.:CH3OH = 10:1). To the compound (120 mg) N,N-dimethylethylenediamine (5.0 mL) was added under nitrogen. The mixture was stirred for 24 h at 50 °C. The excess of N,N-dimethylethylenediamine was removed by evaporation under reduced pressure. The mixture was dissolved in CH3CN (20 mg mL−1) and purified by preparative HPLC using the standard protocol. Fractions containing the product (tR = 40 min, broad peak) were pooled and the water was removed by freeze-drying to obtain the compound 5c as a clear oil (40 mg, 0.027 mmol, 27% in two steps). 1H NMR (500 MHz, DMSO-d6) δ 9.75–9.59 (m, 3H, –NHMe2), 8.49–8.38 (m, 3H, –C(O)NH), 8.18 (s, 1H, CH triazole), 7.01 (dd, J = 7.6, 4.0 Hz, 4H, Ar–H), 6.85 (t, J = 7.5 Hz, 2H, Ar–H), 6.39 (t, J = 7.5 Hz, 2H, Ar–H), 6.31 (dd, J = 20.1, 7.5 Hz, 4H, Ar–H), 5.05 (t, J = 7.6 Hz, 2H, –CH2N triazole), 4.74 (s, 2H, Ar–OCH2C(O)), 4.64 (d, J = 13.4 Hz, 2H, Ar2CH2), 4.53 (s, 2H, –CH2O–), 4.38 (d, J = 14.0 Hz, 2H, Ar–OCH2C(O)), 4.33 (t, J = 7.6 Hz, 2H, Ar–OCH2CH2), 4.30–4.25 (m, 3H, Ar–OCH2C(O) and Ar2CH2), 4.14 (d, J = 2.3 Hz, 2H, –CH2CCH), 3.56–3.52 (m, 6H, –C(O)NHCH2), 3.46–3.27 (m, –OCH2CH2O–), 3.23 (d, J = 13.6 Hz, 5H, Ar2CH2), 3.17–3.10 (m, 6H, –CH2N(CH3)2), 2.80 (s, –N(CH3)2), 2.77 (s, –N(CH3)2). 13C NMR (126 MHz, DMSO-d6, extracted from HSQC) δ 124.87, 129.23, 129.48, 123.97, 123.33, 123.11, 128.38, 128.46, 49.02, 74.50, 30.26, 63.79, 70.14, 74.17, 71.64, 74.19, 30.28, 56.98, 66.46, 72.30, 70.73, 70.13, 30.21, 43.14, 44.01. HRMS (EI, m/z): calculated for C78H117N9O20H+([M + H]+): 1500.8488, found 1500.8480.
CH), 3.59–3.52 (m, 6H, –NHCH2), 3.43–3.33 (m, –OCH2CH2O–), 3.23 (d, J = 13.5 Hz, 5H, Ar2CH2), 3.14 (s, 6H, –CH2NMe2), 2.80 (s, –N(CH3)2), 2.77 (s, –N(CH3)2). 13C NMR (126 MHz, DMSO-d6, extracted from HSQC) δ 123.87, 128.60, 128.45, 128.30, 122.37, 122.11, 127.40, 127.33, 127.38, 48.66, 74.01, 29.86, 62.77, 73.76, 73.20, 29.79, 57.80, 70.45, 68.51, 32.86, 33.44, 33.44, 69.71, 68.20, 68.24, 29.25, 55.51, 55.05, 56.48, 54.98, 42.77, 39.64, 40.78, 42.19, 43.86, 38.99. HRMS (EI, m/z): calculated for C70H101N9O16H+ ([M + H]+): 1324.7439, found 1324.7462.:CH3OH = 10:1). To the compound (120 mg) N,N-dimethylethylenediamine (5.0 mL) was added under nitrogen. The mixture was stirred for 24 h at 50 °C. The excess of N,N-dimethylethylenediamine was removed by evaporation under reduced pressure. The mixture was dissolved in CH3CN (20 mg mL−1) and purified by preparative HPLC using the standard protocol. Fractions containing the product (tR = 40 min, broad peak) were pooled and the water was removed by freeze-drying to obtain the compound 5c as a clear oil (40 mg, 0.027 mmol, 27% in two steps). 1H NMR (500 MHz, DMSO-d6) δ 9.75–9.59 (m, 3H, –NHMe2), 8.49–8.38 (m, 3H, –C(O)NH), 8.18 (s, 1H, CH triazole), 7.01 (dd, J = 7.6, 4.0 Hz, 4H, Ar–H), 6.85 (t, J = 7.5 Hz, 2H, Ar–H), 6.39 (t, J = 7.5 Hz, 2H, Ar–H), 6.31 (dd, J = 20.1, 7.5 Hz, 4H, Ar–H), 5.05 (t, J = 7.6 Hz, 2H, –CH2N triazole), 4.74 (s, 2H, Ar–OCH2C(O)), 4.64 (d, J = 13.4 Hz, 2H, Ar2CH2), 4.53 (s, 2H, –CH2O–), 4.38 (d, J = 14.0 Hz, 2H, Ar–OCH2C(O)), 4.33 (t, J = 7.6 Hz, 2H, Ar–OCH2CH2), 4.30–4.25 (m, 3H, Ar–OCH2C(O) and Ar2CH2), 4.14 (d, J = 2.3 Hz, 2H, –CH2CCH), 3.56–3.52 (m, 6H, –C(O)NHCH2), 3.46–3.27 (m, –OCH2CH2O–), 3.23 (d, J = 13.6 Hz, 5H, Ar2CH2), 3.17–3.10 (m, 6H, –CH2N(CH3)2), 2.80 (s, –N(CH3)2), 2.77 (s, –N(CH3)2). 13C NMR (126 MHz, DMSO-d6, extracted from HSQC) δ 124.87, 129.23, 129.48, 123.97, 123.33, 123.11, 128.38, 128.46, 49.02, 74.50, 30.26, 63.79, 70.14, 74.17, 71.64, 74.19, 30.28, 56.98, 66.46, 72.30, 70.73, 70.13, 30.21, 43.14, 44.01. HRMS (EI, m/z): calculated for C78H117N9O20H+([M + H]+): 1500.8488, found 1500.8480.Preparation of galectins
Galectins used here were supplied by Prof. Tai Guihua. Briefly, uniformly 15N-enriched human galectins were expressed using BL21(DE3) cells grown in minimal media, purified over a lactose affinity column, and fractionated on a FPLC column, as described using a slightly modified protocol provided in Nesmelova et al.50 Typically, 50 milligrams of purified protein were obtained from 1 liter of cell culture. Protein purity was checked by using SDS PAGE and NMR spectroscopy.Heteronuclear NMR spectroscopy
Uniformly 15N-labeled recombinant galectins were dissolved in 20 mM potassium phosphate buffer at pH 7.0, 0.1 mM EDTA, 8 mM DTT, made up using a 98% H2O/2% D2O mixture. NMR experiments of the titrations were typically carried out at 50 μM at 30 °C on a Bruker Ascend III 700 MHz HD spectrometer equipped with a cryogenic triple-channel TCI probe. 1H–15N HSQC NMR gradient sensitivity-enhanced, flip-back experiments were used to investigate binding of inhibitors to 15N-labeled galectins. 1H and 15N resonance assignments for the galectins investigated have been previously reported.36,37 NMR spectra were processed using the program Topspin 3.2 (Bruker, Rheinstetten, Germany), and after conversion of 2D spectra analyzed by Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco).Cell viability assay
Cell viability and migration assays were performed as previously described.51 Cells, both human umbilical cord endothelial cells (HUVEC), as well as Caki-1 human renal cell carcinoma, HT29 human colorectal carcinoma cells and human embryonic kidney HEK293T cells, were seeded in a 96-well culture plate at a density of 2.5–10 × 103 cells per well. Cells were incubated with drugs for 72 h (for drug acquisition and cells and culture conditions, see ESI section‡). Drugs were premixed in culture medium and applied at the appropriate doses. Cell viability was assessed using the CellTiter-Glo luminescence assay (Promega, Madison, WI, USA). Maximum DMSO concentration in cultures was 0.28% and displayed no measurable activity in cell assays. Results are expressed as mean viability (±SEM) of two independent experiments carried out in triplicate.Conflicts of interest
None of the authors have any conflicts of interest to report.Acknowledgements
Hao Zhang gratefully acknowledge the financial support by a scholarship (file no. 201306210049) from the China Scholarship Council (http://www.csc.edu.cn/). We are very grateful to Prof. Guihua Tai for supplying us with isotopically-enriched galectins used in this study. We are also grateful to the Minnesota NMR Center and funding for NMR instrumentation from the Office of the Vice President for Research, the Medical School, the College of Biological Science, NIH, NSF, and the Minnesota Medical Foundation.References
- M. C. Miller, A. Klyosov and K. H. Mayo, Glycobiology, 2009, 19, 1034–1045 CrossRef CAS
.
- N. W. D. Cooper, Biochim. Biophys. Acta, Gen. Subj., 2002, 1572, 209–231 CrossRef
- F. T. Liu, R. J. Patterson and J. L. Wang, Biochim. Biophys. Acta, Gen. Subj., 2002, 1572, 263–273 CrossRef CAS
- N. L. Perillo and L. G. Baum, J. Mol. Med., 1998, 76, 402–412 CrossRef CAS
- F. T. Liu and G. A. Rabinovich, Nat. Rev. Cancer, 2005, 5, 29–41 CrossRef CAS
- V. L. J. L. Thijssen, R. Postel, R. J. M. G. E. Brandwijk, R. P. M. Dings, I. Nesmelova, S. Satijn, N. Verhofstad, Y. Nakabeppu, L. G. Baum, J. Bakkers, K. H. Mayo, F. Poirier and A. W. Griffioen, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15975–15980 CrossRef CAS
- P. G. Traber and E. Zomer, PLoS One, 2013, 8, e83481 CrossRef
- F. Liu and G. A. Rabinovich, Ann. N. Y. Acad. Sci., 2010, 1183, 158–182 CrossRef CAS PubMed
- H. Blanchard, X. Yu, P. M. Collins and K. Bum-Erdene, Expert Opin. Ther. Pat., 2014, 24, 1053–1065 CrossRef CAS
- A. Mackinnon, W.-S. Chen, H. Leffler, N. Panjwani, H. Schambye, T. Sethi and U. J. Nilsson, in Topics in Medicinal Chemistry, 2014, vol. 12, pp. 95–121 Search PubMed
- H. Blanchard, K. Bum-Erdene, M. H. Bohari and X. Yu, Expert Opin. Ther. Pat., 2016, 26, 537–554 CrossRef CAS PubMed
- R. J. Pieters, ChemBioChem, 2006, 7, 721–728 CrossRef CAS
- I. Cumpstey, A. Sundin, H. Leffler and U. J. Nilsson, Angew. Chem., Int. Ed., 2005, 44, 5110–5112 CrossRef CAS
- P. Sörme, Y. Qian, P. G. Nyholm, H. Leffler and U. J. Nilsson, ChemBioChem, 2002, 3, 183–189 CrossRef
- D. Giguere, R. Patnam, M. A. Bellefleur, C. St-Pierre, S. Sato and R. Roy, Chem. Commun., 2006, 2379–2381 RSC
- H. Leffler and S. H. Barondes, J. Biol. Chem., 1986, 261, 10119–10126 CAS
- T. Delaine, P. Collins, A. Mackinnon, G. Sharma, J. Stegmayr, V. K. Rajput, S. Mandal, I. Cumpstey, A. Larumbe, B. A. Salameh, B. Kahl-Knutsson, H. VanHattum, M. VanScherpenzeel, R. J. Pieters, T. Sethi, H. Schambye, S. Oredsson, H. Leffler, H. Blanchard and U. J. Nilsson, ChemBioChem, 2016, 17, 1759–1770 CrossRef CAS
- S. Martín-Santamaría, S. André, E. Buzamet, R. Caraballo, G. Fernández-Cureses, M. Morando, J. P. Ribeiro, K. Ramírez-Gualito, B. De Pascual-Teresa, F. J. Cañada, M. Menéndez, O. Ramström, J. Jiménez-Barbero, D. Solís and H. J. Gabius, Org. Biomol. Chem., 2011, 9, 5445–5455 RSC
- T. Matsubara, J. Nucleic Acids, 2012, 2012, 1–15 CrossRef
- M. Simon-haldi, N. Mantei, J. Franke, H. Voshol and M. Schachner, J. Neurochem., 2002, 83, 1380–1388 CrossRef CAS
- J. Zou, V. V. Glinsky, L. A. Landon, L. Matthews and S. L. Deutscher, Carcinogenesis, 2005, 26, 309–318 CrossRef CAS
- L. A. Landon and S. L. Deutscher, J. Cell. Biochem., 2003, 90, 509–517 CrossRef CAS
- S. D. Burke, Q. Zhao, M. C. Schuster, L. L. Kiessling and R. V. January, J. Am. Chem. Soc., 2000, 122, 4518–4519 CrossRef CAS
- D. W. J. van der Schaft, R. P. M. Dings, Q. G. de Lussanet, L. I. van Eijk, A. W. Nap, R. G. H. Beets-Tan, J. C. A. Bouma-Ter Steege, J. Wagstaff, K. H. Mayo and A. W. Griffioen, FASEB J., 2002, 16, 1991–1993 CrossRef CAS
- A. W. Griffioen, D. W. J. Van der Schaft, A. F. Barendsz-Janson, A. Cox, H. A. J. Struijker Boudier, H. F. P. Hillen and K. H. Mayo, Biochem. J., 2001, 354, 233–242 CrossRef CAS
- K. H. Mayo, J. Haseman, E. Ilyina and B. Gray, Biochim. Biophys. Acta, Gen. Subj., 1998, 1425, 81–92 CrossRef CAS
- E. Salomonsson, V. L. Thijssen, A. W. Griffioen, U. J. Nilsson and H. Leffler, J. Biol. Chem., 2011, 286, 13801–13804 CrossRef CAS
- R. P. M. Dings, X. Chen, D. M. E. I. Hellebrekers, L. I. van Eijk, Y. Zhang, T. R. Hoye, A. W. Griffioen and K. H. Mayo, J. Natl. Cancer Inst., 2006, 98, 932–936 CrossRef CAS
- R. P. M. Dings, M. C. Miller, I. Nesmelova, L. Astorgues-Xerri, N. Kumar, M. Serova, X. Chen, E. Raymond, T. R. Hoye and K. H. Mayo, J. Med. Chem., 2012, 55, 5121–5129 CrossRef CAS
- R. P. M. Dings, J. I. Levine, S. G. Brown, L. Astorgues-Xerri, J. R. MacDonald, T. R. Hoye, E. Raymond and K. H. Mayo, Invest. New Drugs, 2013, 31, 1142–1150 CrossRef CAS
- H. Van Hattum, H. M. Branderhorst, E. E. Moret, U. J. Nilsson, H. Leffler and R. J. Pieters, J. Med. Chem., 2013, 56, 1350–1354 CrossRef CAS PubMed
- M. van Scherpenzeel, E. E. Moret, L. Ballell, R. M. J. Liskamp, U. J. Nilsson, H. Leffler, R. J. Pieters and M. van Scherpenzee, ChemBioChem, 2009, 10, 1724–1733 CrossRef CAS
- T. Läppchen, R. P. M. Dings, R. Rossin, J. F. Simon, T. J. Visser, M. Bakker, P. Walhe, T. van Mourik, K. Donato, J. R. van Beijnum, A. W. Griffioen, J. Lub, M. S. Robillard, K. H. Mayo and H. Grüll, Eur. J. Med. Chem., 2015, 89, 279–295 CrossRef
- S. R. Sung, S. C. Han, S. Jin and J. W. Lee, Bull. Korean Chem. Soc., 2011, 32, 3933–3940 CrossRef CAS
- L. H. Bryant, A. T. Yordanov, J. J. Linnoila, M. W. Brechbiel and J. Frank, Angew. Chem., Int. Ed., 2000, 39, 1641–1643 CrossRef CAS
- S. Mandal and U. J. Nilsson, Org. Biomol. Chem., 2014, 12, 4816–4819 RSC
- H. Zhang, D. Laaf, L. Elling and R. J. Pieters, Bioconjugate Chem., 2018, 29, 1266–1275 CrossRef CAS
- C. Jaime, J. De Mendoza, P. Prados, P. M. Nieto and C. Sanchez, J. Org. Chem., 1991, 56, 3372–3376 CrossRef CAS
- J. O. Magrans and J. De Mendoza, J. Org. Chem., 1997, 3263, 4518–4520 CrossRef
- M. C. Miller, Y. Zheng, Y. Zhou, G. Tai and K. H. Mayo, Glycobiology, 2019, 29, 74–84 CrossRef PubMed
- J. Rahkila, F. S. Ekholm, A. Ardá, S. Delgado, J. Savolainen, J. Jiménez-Barbero and R. Leino, ChemBioChem, 2019, 20, 203–209 CrossRef CAS
- M. P. Williamson, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 73, 1–16 CrossRef CAS
- P. Nowak-Sliwinska, K. Alitalo, E. Allen, A. Anisimov, A. C. Aplin, R. Auerbach, H. G. Augustin, D. O. Bates, J. R. van Beijnum, R. H. F. Bender, G. Bergers, A. Bikfalvi, J. Bischoff, B. C. Böck, P. C. Brooks, F. Bussolino, B. Cakir, P. Carmeliet, D. Castranova, A. M. Cimpean, O. Cleaver, G. Coukos, G. E. Davis, M. De Palma, A. Dimberg, R. P. M. Dings, V. Djonov, A. C. Dudley, N. P. Dufton, S. M. Fendt, N. Ferrara, M. Fruttiger, D. Fukumura, B. Ghesquière, Y. Gong, R. J. Griffin, A. L. Harris, C. C. W. Hughes, N. W. Hultgren, M. L. Iruela-Arispe, M. Irving, R. K. Jain, R. Kalluri, J. Kalucka, R. S. Kerbel, J. Kitajewski, I. Klaassen, H. K. Kleinmann, P. Koolwijk, E. Kuczynski, B. R. Kwak, K. Marien, J. M. Melero-Martin, L. L. Munn, R. F. Nicosia, A. Noel, J. Nurro, A. K. Olsson, T. V. Petrova, K. Pietras, R. Pili, J. W. Pollard, M. J. Post, P. H. A. Quax, G. A. Rabinovich, M. Raica, A. M. Randi, D. Ribatti, C. Ruegg, R. O. Schlingemann, S. Schulte-Merker, L. E. H. Smith, J. W. Song, S. A. Stacker, J. Stalin, A. N. Stratman, M. Van de Velde, V. W. M. van Hinsbergh, P. B. Vermeulen, J. Waltenberger, B. M. Weinstein, H. Xin, B. Yetkin-Arik, S. Yla-Herttuala, M. C. Yoder and A. W. Griffioen, Angiogenesis, 2018, 21, 425–532 CrossRef PubMed
- A. Satelli, P. S. Rao, P. K. Gupta, P. R. Lockman, K. S. Srivenugopal and U. S. Rao, Oncol. Rep., 2008, 19, 587–594 CAS
- V. L. Thijssen, S. HuIsmans and A. W. Griffioen, Am. J. Pathol., 2008, 172, 545–553 CrossRef CAS
- Y. Xu, C. Li, J. Sun, J. Li, X. Gu and W. Xu, Exp. Biol. Med., 2016, 241, 1365–1373 CrossRef CAS
- S. André, C. Grandjean, F. M. Gautier, S. Bernardi, F. Sansone, H. J. Gabius and R. Ungaro, Chem. Commun., 2011, 47, 6126–6128 RSC
- S. André, F. Sansone, H. Kaltner, A. Casnati, J. Kopitz, H. J. Gabius and R. Ungaro, ChemBioChem, 2008, 9, 1649–1661 CrossRef
- H. Ippel, M. C. Miller, M. A. Berbis, S. Andre, T. M. Hackeng, F. J. Canada, C. Weber, H. Gabius, J. Jimenez-Barbero and K. H. Mayo, Biomol. NMR Assignments, 2015, 9, 59–63 CrossRef CAS
- I. V. Nesmelova, M. Pang, L. G. Baum and K. H. Mayo, Biomol. NMR Assignments, 2008, 2, 203–205 CrossRef CAS
- P. Nowak-Sliwinska, J. R. Van Beijnum, A. Casini, A. A. Nazarov, G. Wagnières, H. Van Den Bergh, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS
Footnotes |
| † Dedication: This paper is dedicated to Professor Julius Rebek Jr, on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qo00810a |
| § Present address: College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, PR China. |
| This journal is © the Partner Organisations 2019 |