
Cationic octahedral molybdenum cluster complexes functionalized with mitochondria-targeting ligands: photodynamic anticancer and antibacterial activities†
Kaplan
Kirakci
 *a,
Jaroslav
Zelenka
b,
Michaela
Rumlová
c,
Josef
Cvačka
d,
Tomáš
Ruml
b and
Kamil
Lang
*a
*a,
Jaroslav
Zelenka
b,
Michaela
Rumlová
c,
Josef
Cvačka
d,
Tomáš
Ruml
b and
Kamil
Lang
*a
aInstitute of Inorganic Chemistry of the Czech Academy of Sciences, 250 68 Husinec-Řež, Czech Republic. E-mail: lang@iic.cas.cz
bDepartment of Biochemistry and Microbiology, University of Chemistry and Technology Prague, Technická 5, 166 28 Praha 6, Czech Republic
cDepartment of Biotechnology, University of Chemistry and Technology Prague, Technická 5, 166 28 Praha 6, Czech Republic
dInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, 166 10 Praha 6, Czech Republic
First published on 8th January 2019
Abstract
Octahedral molybdenum cluster complexes have recently come forth as pertinent singlet oxygen photosensitizers towards biological applications. Still, their phototoxic efficiency in the absence of nanocarriers remains limited due to their poor cellular uptake. Here, two cationic octahedral molybdenum cluster complexes, bearing carboxylate ligands with triphenylphosphonium (1) or N-methyl pyridinium (2) mitochondria-targeting terminal functions, have been designed and synthesized. Their photophysical properties in water and in vitro biological activity were investigated in the context of blue-light photodynamic therapy of cancer and photoinactivation of bacteria. Upon blue light irradiation, complex 1 displays red luminescence with a quantum yield of 0.24 in water, whereas complex 2 is much less emissive (ΦL < 0.01). Nevertheless, both complexes efficiently produce singlet oxygen, O2(1Δg). Complex 1 is rapidly internalized into HeLa cells and accumulated in mitochondria, followed by relocation to lysosomes and clearance at longer times. In contrast, the more hydrophilic 2 is not internalized into HeLa cells, highlighting the effect of the apical ligands on the uptake properties. The treatment with 1 results in an intensive phototoxic effect under 460 nm irradiation (IC50 = 0.10 ± 0.02 μM), which exceeds by far those previously reported for octahedral cluster-based molecular photosensitizers. The ratio between phototoxicity and dark toxicity is approximately 50 and evidences a therapeutic window for the application of 1 in blue-light photodynamic therapy. Complex 1 also enters and efficiently photoinactivates Gram-positive bacteria Enterococcus faecalis and Staphylococcus aureus, documenting its suitability as a blue-light photosensitizer for antimicrobial applications.
Introduction
The photodynamic killing of tumor cells, bacteria, or viruses has attracted widespread interest during the last few decades.1,2 One of the most potent mediators of phototoxicity is singlet oxygen O2(1Δg), which is typically generated by energy transfer from the excited triplet states of a photosensitizer (PS) to molecular oxygen.3 This feature, coupled with the limited diffusion length of O2(1Δg) in cells (d ∼ 150–190 nm)4 due to its short lifetime, leads to a high selectivity in the annihilation of targeted problematic cells. Among the variety of PSs, those activated by blue light are generally used in dermatology to treat actinic keratosis, basal cell carcinoma, squamous cell carcinoma, and acne.5 In addition, these PSs can also be employed in the photoinactivation of antibiotic-resistant and antibiotic-susceptible bacteria. Because O2(1Δg) interacts with several cell structures and interferes with different metabolic pathways, it prevents the development of resistance to photodynamic treatment.6In this respect, octahedral molybdenum cluster (Mo6) complexes are relevant PSs with attractive properties. The complexes can be depicted as an octahedron of molybdenum atoms surrounded by eight strongly bonded inner ligands, usually halogen atoms, and six labile inorganic/organic apical ligands (Fig. 1). Upon excitation from the UVA to the green spectral region, these complexes form long-lived triplet states that relax via red-NIR luminescence.7,8 This luminescence is efficiently quenched by oxygen, leading to the formation of O2(1Δg) in high yields, making these clusters not only good PSs but also relevant luminescent probes for in situ monitoring of oxygen levels.8 In contrast to commonly used organic PSs such as porphyrins, which lose their photosensitizing activity upon aggregation mostly due to π–π stacking interactions, these complexes remain good O2(1Δg) photosensitizers even in their aggregated form. In general, the coordination of carboxylate ligands to the {Mo6I8}4+ core provides complexes with suitable photophysical properties and has the potential for additional functionalization.9–11
 | ||
| Fig. 1 Schematic representation of the molecular structures of 1 and 2. Color coding: Molybdenum (blue), iodine (magenta), caboxylated ligands (green), and hydrogen atoms are omitted for clarity. | ||
In aqueous media, especially at physiological pH, the complexes generally undergo a hydrolytic process characterized by the replacement of apical ligands by hydroxyl groups. This results in a change of the photophysical and chemical properties of the complexes that can be detrimental to biological applications. The stabilization against hydrolysis can be achieved by immobilization in inorganic or organic nanocarriers.12–16 This strategy led to promising results in blue-light photodynamic therapy (PDT)17–21 or photoinactivation of bacteria.22,23 The applicability of Mo6 complexes as PSs in their molecular form still remains a challenge as the only phototoxic effects of molecular clusters were recently reported for [Re6Q8(CN)6]4− (Q = S or Se) at high concentrations (>100 μM).24 We also reported the absence of phototoxic effects of water soluble Mo6 complexes bearing azide and isothiocyanate apical ligands on the HeLa and HEK-293T cell lines.25 This fact was explained by the poor cellular uptake attributable to the anionic and hydrophilic character of these complexes. The research on novel Mo6 complexes with positive charges appears to be a relevant direction towards biological applications. Indeed, cationic PSs are generally well internalized into cells and have a tendency to accumulate into mitochondria, whose damage has become a popular cancer therapeutic strategy for PSs.26,27 Mitochondria are energy sources for cells and play a critical role in the initiation of cellular apoptosis when their integrity is disrupted. Due to the negative potential and lipophilicity of the mitochondrial membrane, most of the mitochondria targeted PSs are lipophilic cations. Among the organic functions providing a positive charge, triphenylphosphonium and N-methyl pyridinium moieties are commonly used for the functionalization of typical PSs such as porphyrin or phtalocyanines.28,29
Herein, we report on the luminescence properties, singlet oxygen photogeneration, water stability, toxicity, cellular uptake, and in vitro photodynamic toxicity of two cationic cluster complexes based on the {Mo6I8}4+ core associated with (4-carboxybutyl)triphenylphosphonium (1) and 4-carboxy-1-methylpyridinium (2) apical ligands (Fig. 1). Both HeLa cancer cells and bacteria, namely Gram-negative Pseudomonas aeruginosa and Escherichia coli and Gram-positive Enterococcus faecalis and Staphylococcus aureus strains, were utilized in this investigation.
Results and discussion
Synthesis and characterization
Compounds [Mo6I8(OCOC4H8PPh3)6]Br4 (1) and [Mo6I8(OCOC5H4NMe)6]Cl4 (2) were obtained by a method derived from a previously published procedure.30 In brief, precursor Na2[Mo6I8(OMe)6] was allowed to react overnight with six equivalents of (4-carboxybutyl)triphenylphosphonium bromide or 4-carboxy-1-methylpyridinium chloride in dimethylsufoxide (DMSO). 1H NMR of 1 and 2 in d6-DMSO revealed the disappearance of the 1H signals at 12.09 and 13.65 ppm, belonging to the COOH protons of the starting (4-carboxybutyl)triphenylphosphonium bromide and 4-carboxy-1-methylpyridinium chloride, respectively. It evidences the coordination of the ligands to the {Mo6I8}4+ core through their carboxylate functions (Fig. S1†). When compared with the free ligands, the 1H NMR signals of the aliphatic protons in 1 exhibited upfield shifts (−0.34, −0.08, −0.06, and −0.06 ppm), whereas the aromatic signals belonging to the phenyl groups are not shifted. For 2, the signals of the aromatic protons are significantly shifted upfield (−0.3 and −0.28 ppm), whereas the aliphatic protons of the methyl groups are less affected (−0.12 ppm). The high-resolution electrospray ionization mass spectrometry of 1 and 2 revealed peaks with m/z values of 943.8846 and 1317.3861, corresponding to [Mo6I8(OCOC4H8PPh3)6]4+ and {[Mo6I8(OCOC5H4NMe)6]Cl6}2− for 1 (Fig. S2†) and 2 (Fig S3†), respectively. C, H, and N elemental analysis confirmed the purity of the bulk material used for the measurement of the photophysical properties and the evaluation of the biological activity.Photophysical properties and singlet oxygen productivity
Complexes 1 and 2 are not readily soluble in water, and therefore, their water solutions were prepared by adding small aliquots of highly concentrated DMSO solutions into deionized water. As shown in Fig. 2A, fresh solutions of 1 and 2 exhibited broad unresolved absorption bands in the UV and visible regions with an onset approximately at 550 nm, which are typical spectral features of [Mo6I8(OCO-R)6]2− complexes.8 Upon excitation at 400 nm, deaerated solutions of 1 and 2 displayed broad emission bands with maxima at 700 and 685 nm, respectively (Table 1). Complexes considerably differed in luminescence quantum yields, being 0.24 for 1 and less than 0.01 for 2. Thus, complex 1 has the highest luminescence efficiency ever reported for an octahedral transition metal cluster complex dissolved in water. The luminescence quantum yields of complexes increased to 0.50 and 0.21 in deaerated DMSO, for 1 and 2, respectively, substantiating, especially for 2, the quenching of the excited triplet states by water through coupling with O–H vibrational modes. The luminescence intensities (Fig. 2B and C) and the corresponding lifetimes (Fig. S4,†Table 1) were considerably quenched by oxygen, suggesting that both complexes can produce O2(1Δg), similarly to already reported molybdenum cluster complexes.25 It is worth mentioning that the luminescence decay curves of both complexes are purely monoexponential under oxygen-free conditions, whereas the decay curves become biphasic in the presence of oxygen. These features can be interpreted by the contribution of two luminescent populations to the overall luminescence, i.e., monomeric species and cluster aggregates, differing in diffusion parameters and oxygen accessibility.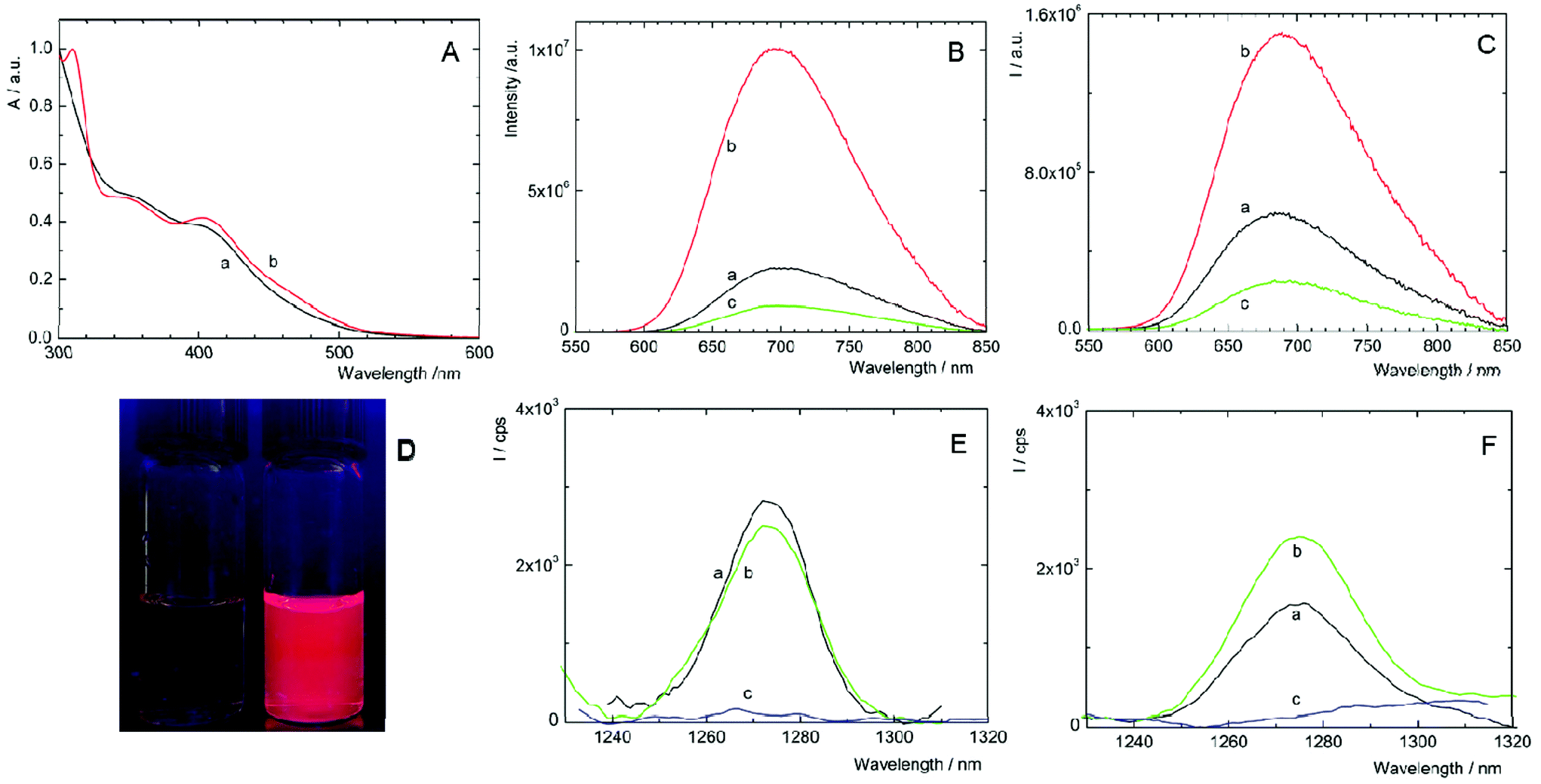 | ||
| Fig. 2 Spectral and photophysical properties of 1 and 2. (A) Normalized absorption spectra of fresh aqueous solutions of 1 (a) and 2 (b). Luminescence spectra of 1 (B) and 2 (C) in air- (a), Ar- (b) and oxygen-saturated (c) water. The samples were excited at 400 nm. (D) Luminescence of oxygen-saturated (left) and argon-saturated (right) water solution of 1 under 365 nm excitation. Luminescence emission band of O2(1Δg) produced by 1 (E) and 2 (F) in oxygen-saturated, fresh aqueous solution (a), and the same solution after 11 days (b). The same spectral region was recorded in fresh, oxygen-free solution (c). The excitation wavelength was 400 nm. | ||
| λ L/nm | τ T0/μs | τ air/μs | τ O2/μs | Φ L | |
|---|---|---|---|---|---|
| a λ L is the maximum of the luminescence emission band; τT0, τair, and τO2 are the amplitude average lifetimes of the triplet states in oxygen-free, air-, and oxygen-saturated water, respectively (recorded at 700 (1) and 690 nm (2), excited at 390 nm); ΦL is the luminescence quantum yield in oxygen-free water solution (excitation wavelengths were from 320 to 440 nm). All data are recorded in fresh solutions and in the same solutions after 11 days. | |||||
| 1, fresh | 700 | 119 | 76 | 54 | 0.24 |
| 1, 11 days | 700 | 58 | 42 | 37 | 0.08 |
| 2, fresh | 685 | 22 | 6.7 | 2.0 | <0.01 |
| 2, 11 days | 695 | 51 | 8.6 | 2.5 | 0.07 |
Changes in the luminescence properties can serve as an indicator of the stability of the cluster complexes in a water environment, which is essential for their application as photosensitizers for PDT. For this purpose, the luminescence properties of freshly prepared water solutions were compared with 11-day old ones (i.e., when the photophysical properties stabilized at constant levels). The water-induced changes of 1 are indicated by the considerable decrease of luminescence quantum yield and luminescence lifetimes in older solutions (Table 1, Fig. S5A†). In the case of 2, the changes are in the opposite direction; the lifetimes and ΦL increase, accompanied by a red shift of the emission band (Table 1, Fig. S5B†). All the changes indicate some instability of 1 and 2 in water, associated with the progressive replacement of one or more apical ligands by hydroxyl groups as already observed for some [Mo6I8L6]2− complexes.14,16,25 A more precise evaluation of the kinetics of formation of the hydrolyzed species can be obtained from the evolution of the ΦL values over time. It is clear that the hydrolysis process is slow in the time frame of a typical biological experiment with the majority of the cluster complexes remaining intact after 24 h (Fig S6†). It is noteworthy that the addition of PBS and irradiation for 15 min with a 460 nm LED source did not affect the kinetics of the formation of hydrolyzed species. These changes pose a relevant question in the context of photodynamic applications using Mo6 clusters. How these water-induced processes affect the productivity of cytotoxic O2(1Δg)?
The photosensitization of O2(1Δg) by 1 and 2 in fresh water solutions was directly evidenced by the appearance of the characteristic O2(1Δg) phosphorescence band around 1270 nm in air- and oxygen-saturated solutions and its disappearance in argon-saturated solutions (Fig. 2E and F). The quantum yields of the O2(1Δg) formation in water are 0.41 ± 0.05 and 0.23 ± 0.03 for 1 and 2, respectively, and document quite high efficiency of the O2(1Δg) production in water. As shown in Fig. 2E, the integral production of O2(1Δg) mediated by 1 is lower by approximately 4% after 11 days, indicating that the water-induced changes lead to a species with slightly smaller O2(1Δg) productivity, when compared to fresh 1 solutions. In contrast, the O2(1Δg) production of 2 is favorably affected by the hydrolytic process as evidenced by an approximately 60% increase of the intensity of O2(1Δg) luminescence after 11 days. The presented results demonstrate that hydrolyzed cluster complexes can be comparable or even better PSs of O2(1Δg) than the original compounds. This is an important finding in the area of transition-metal cluster complexes since it was supposed that hydrolysis diminishes both luminescence and O2(1Δg) production.
Uptake and toxicity in cancer cell lines
The cellular uptake of 1 and 2 in HeLa cells was quantitatively analyzed by flow cytometry in the concentration range of 0.5–20 μM. The luminescence intensities of cells after incubation with 2 did not exceed the level of the control experiments for all tested concentrations and incubation times, indicating no uptake of this complex by HeLa cells (Fig. S7C†). On the other hand, compound 1 was quickly internalized in a dose-dependent manner within 30 min of incubation with only a slight increase at longer incubation times (Fig. 3A and B, Fig. S7A and B†). The cellular accumulation of 1 decreased after a 22 h washout in the fresh medium suggesting the clearance of the complex from cells (Fig S8A†). There was only a negligible effect of fetal bovine serum on the uptake of 1, probably due to the positive charge of the complex that limits the interaction with serum proteins (Fig S9A†). The experiments were also conducted with 11-day old solutions of 1, in order to evaluate the effects of the hydrolysis on the uptake features. In this case, hydrolyzed 1 is not internalized (Fig. S7D†). The lack of the cellular uptake of 2 and hydrolyzed 1 probably originates from their hydrophilic character, similar to the behavior of [Mo6I8(NCS)6]2− and [Mo6I8(N3)6]2− cluster complexes.25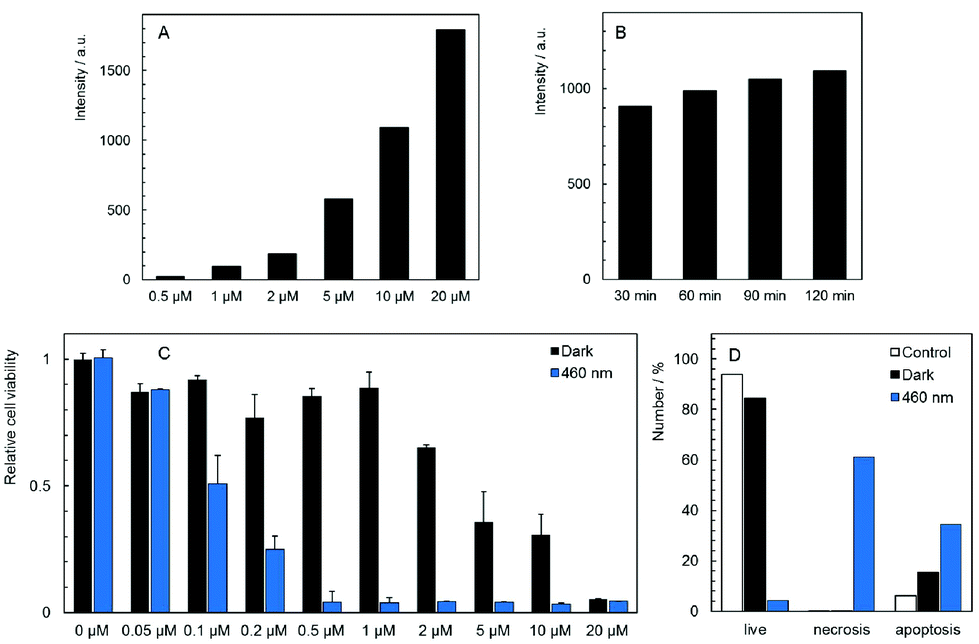 | ||
| Fig. 3 Uptake, toxicity, and phototoxicity of 1 against HeLa cells. (A) Dose-dependent uptake of 1 after 120 min incubation with indicated concentrations of 1. The y axis represents the fluorescence intensity of cells minus the fluorescence intensity from the control experiment. (B) Time-dependent uptake of 1 after the indicated time of incubation with 10 μM 1. The y axis represents the fluorescence intensity of cells minus the fluorescence intensity from the control experiment. (C) Dark toxicity of 1 measured 24 h after incubation compared with the phototoxicity of 1 measured 24 h after incubation and irradiation (460 nm, 20 mW cm−2, 15 min). The bar labeled 0 μM belongs to the control experiment, i.e., cells were irradiated in the absence of 1. (D) Percentage of apoptotic and necrotic cells measured 4 h after incubation with 5 μM 1 in the dark or after incubation with 1 μM 1 followed by irradiation (460 nm, 20 mW cm−2, 15 min). The empty bars are for control experiments performed in the absence of 1 and in the dark. | ||
The subcellular localization of 1 in HeLa cells was investigated using a spinning-disc confocal microscope. An intense red intracellular luminescence originating from 1 was observed within the cells after incubation with this complex (Fig. 4). Staining of HeLa cells with MitoTracker Green and LysoTracker Green indicated co-localization of 1 with mitochondria after 2 h of incubation (Pearson coefficient = 0.58, Manders coefficient = 0.56), followed by the clearance (Pearson coefficient = −0.06, Manders coefficient = 0.15) and relocation of 1 to lysosomes after incubation for 6 h (Pearson coefficient = 0.53, Manders coefficient = 0.62). The presented results suggest that 1 can reach mitochondria far before its considerable hydrolysis.
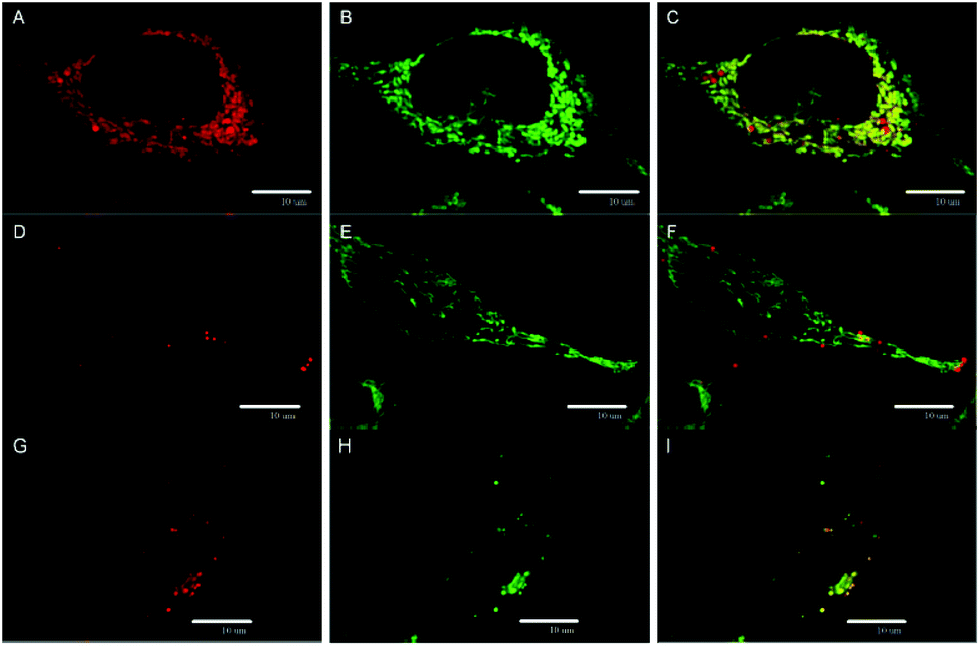 | ||
| Fig. 4 Localization of 1 in HeLa cells. Confocal sections showing colocalization of 1 with mitochondria after 2 h of incubation: (A) localization of 1 (red luminescence); (B) mitochondria stained with the mitochondrial marker MitoTracker Green (green); (C) merging of the panels (A) and (B). Confocal sections documenting the redistribution of 1, 6 h of incubation: (D) and (G) localization of 1 (red luminescence); mitochondria (E) and lysosomes (H) stained with the mitochondrial marker MitoTracker Green (green) and lysosomal marker LysoTracker Green (green), respectively; (F) and (I) merging of two corresponding panels on the left. Scale bars: 10 μm. | ||
The dark toxicity of 1 and 2 against HeLa cells was evaluated at pharmacologically relevant concentrations ranging from 0.05 to 20 μM. Analysis of the results from these experiments showed that 1 displays a moderate cytotoxic effect on HeLa cells with an IC50 value of 5.3 ± 1.5 μM, whereas compound 2 is non-toxic at all concentrations (Fig. S10†). It is noteworthy that several cationic PSs were shown to induce mitochondrial toxicity.26 Thus, the dark toxicity of 1 could originate from the interaction between the complex and mitochondria as evidenced by microscopy. Analysis of the cellular death pathway reveals that cells incubated with 1 mostly undergo apoptosis, consistent with a toxic effect on mitochondria which serve as a principal trigger of apoptosis (Fig. 3D).31
Photodynamic toxicity was evaluated by irradiation of HeLa cells treated for 2 h with 1 and 2 in the concentration range of 0.02 to 20 μM. Cells were irradiated for 15 min with a 460 nm LED source and their viability was analyzed after 24 h. As expected, there was no phototoxic effect of 2, in accordance with the absence of the cellular uptake (Fig. S10†). On the other hand, HeLa cells incubated with 1 exhibited a strong dose-dependent phototoxicity with an IC50 value of 0.10 ± 0.02 μM (Fig. 3C), which is at least three orders of magnitude lower than the value for Na4[Re6Se8(CN)6], the most efficient M6 cluster molecular PS reported so far.24 The phototoxicity of 1 was reduced by the 22 h washout in the fresh medium as expected from the clearance of the complex from cells (Fig. S8B†). The presence of fetal bovine serum only slightly decreased the phototoxicity of 1 which is an important feature for possible translation to in vivo experiments (Fig. S9B†). Interestingly, the cellular death pathway after irradiation of cells incubated with 1 was assigned mainly to necrosis, differing from the cellular death pathway observed in the case of the dark toxicity (Fig. 3D). The ratio between phototoxicity and dark toxicity is approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 and points out to a therapeutic window for the use of 1 in blue-light PDT. Phototoxicity can be attributed to the localization of 1 within mitochondria that are highly sensitive to ROS, especially in cancer cells.31 In contrast, hydrolyzed 1 showed no phototoxicity in the studied concentration range (Fig. S11†), in accordance with the lack of cellular uptake evidenced by flow cytometry (Fig. S7D†). It points out to the dramatic effect of the hydrolytic process on the biological activity of 1. Therefore, the phototoxicity of 1 at different incubation times was compared (Fig. S12†). The 24 h incubation which led to a slightly better cellular accumulation than the 2 h incubation also resulted in a moderately higher phototoxicity. It indicates that the effect of the hydrolysis process on the biological activity of 1 can be considered as negligible within 24 h.
:50 and points out to a therapeutic window for the use of 1 in blue-light PDT. Phototoxicity can be attributed to the localization of 1 within mitochondria that are highly sensitive to ROS, especially in cancer cells.31 In contrast, hydrolyzed 1 showed no phototoxicity in the studied concentration range (Fig. S11†), in accordance with the lack of cellular uptake evidenced by flow cytometry (Fig. S7D†). It points out to the dramatic effect of the hydrolytic process on the biological activity of 1. Therefore, the phototoxicity of 1 at different incubation times was compared (Fig. S12†). The 24 h incubation which led to a slightly better cellular accumulation than the 2 h incubation also resulted in a moderately higher phototoxicity. It indicates that the effect of the hydrolysis process on the biological activity of 1 can be considered as negligible within 24 h.
Antibacterial effects
In vitro studies of antimicrobial photodynamic inactivation with 1 and 2 were performed with several strains of opportunistic pathogens, namely, Gram-negative Pseudomonas aeruginosa and Escherichia coli and Gram-positive Enterococcus faecalis and Staphylococcus aureus. Under the experimental conditions employed, only negligible effects of DMSO, blue LED light (460 nm, 20 mW cm−2, 30 min), and compounds 1 or 2 were observed when used separately.Irradiation of Gram-negative bacteria preincubated with up to 50 μM of compound 1 by 460 nm light showed no effect on viability (Fig. 5A and B). In contrast, Gram-positive bacteria were effectively inactivated under these conditions (Fig. 5C and D). Interestingly, no significant phototoxicity was observed for 2 and for hydrolyzed 1 (Fig. S13†). We determined the uptake of 1 and 2 with bacteria using flow cytometry and found significant uptake of 1 for Gram-positive bacteria and much lower uptake for Gram-negative bacteria, while no uptake was observed for 2 on both bacteria types (Fig. 6 and Fig. S14†). Next, we inspected the bacteria stained with 1 using a confocal microscope and found that only Gram-positive bacteria are stained, whereas Gram-negative strains showed no cluster luminescence probably because the luminescence intensity was below the detection limit (Fig. 7). These results are in line with the generally higher resistance of Gram-negative bacteria to antiseptics, disinfectants, and antibiotics due to different compositions of cell walls, which limits the uptake of bactericidal molecules.32
 | ||
| Fig. 5 Photoinactivation of Gram-negative Escherichia coli (A) and Pseudomonas aeruginosa (B) and Gram-positive Enterococcus faecalis (C) and Staphylococcus aureus (D) by 1 of indicated concentrations upon 460 nm light irradiation. Dark controls are represented with black bars. Bars labeled 0 μM are control experiments in the absence of 1. | ||
 | ||
| Fig. 6 Uptake of 10 μM 1 or 2 into Gram-positive Enterococcus faecalis and Gram-negative Escherichia coli measured using flow cytometry after 1 h incubation. Data are expressed as medians of cellular fluorescence intensity with subtracted medians of control (unloaded) cells. | ||
 | ||
| Fig. 7 Bright field images of E. coli (A) and E. faecalis (B) merged with confocal images of 1 (red) after 30 min incubation with 50 μM 1. The white bar represents 10 μm. | ||
Next, the Gram-positive strains were incubated in decreasing concentrations of 1 to determine IC50 values. Interestingly, the antimicrobial photodynamic efficacy of 1 significantly depended on the metabolic state of cells. The IC50 was 2.0 ± 0.5 μM for well-fed E. faecalis freshly grown on plates. In contrast, the IC50 value decreased to 0.15 ± 0.10 μM for the same cells stored on plates for 1 month at 4 °C. In the case of S. aureus, the phototoxic IC50 was 1.0 ± 0.5 μM for fed cells and 0.08 ± 0.04 μM for starving cells. This effect may be important for the application of 1 in antimicrobial photodynamic inactivation, since well-fed bacteria are expected in wounds and on catheter surfaces, whereas starving bacteria are preferentially present in air-conditioning units and sanitized surfaces. These effects are in clear contrast to the efficacy of many other disinfectants and antibiotics which show lower efficiency for starved cells due to the upregulation of resistance mechanisms and formation of biofilms.33,34 Since the phototoxicity of 1 is related to oxidative damage by reactive oxygen species, we may hypothesize that the higher sensitivity of starving cells is due to the lack of substrates for the regeneration of glutathione and other thiols, which are critical components of the antioxidant system.31
Conclusion
We have designed and prepared two new cationic octahedral molybdenum cluster complexes bearing (4-carboxybutyl)triphenylphosphonium (1) and 4-carboxy-1-methylpyridinium (2) mitochondria-targeting apical ligands. Complex 1 is highly emissive in water solutions with a quantum yield of 0.24, whereas 2 displays low luminescence quantum efficiency due to the quenching of the triplet states by the solvent. The triplet states of both complexes interact with oxygen and produce O2(1Δg). The studied complexes undergo hydrolysis in water solutions, which changes the luminescence quantum yields; however, the O2(1Δg) production is affected to a lesser extent. Investigation on the biological activity of 1 revealed its efficient mitochondria-targeting activity which resulted in a moderate dark toxicity and a strong phototoxicity upon 460 nm irradiation with an IC50 value of 100 ± 20 nM, unmatched for PDT systems based on Mo6 cluster-based molecular PSs. On the other hand, neither 2 nor hydrolyzed 1 showed a phototoxic effect because of the absence of cellular internalization, illustrating the dramatic influence of the apical ligands on the uptake features. It is worth mentioning that a well-timed deactivation of the phototoxic effect of a PS can be advantageous in the context of PDT in order to avoid post-treatment photosensitization issues. Complex 1 also showed efficient photoinactivation of Gram-positive bacteria E. faecalis and S. aureus, whereas 2 and hydrolyzed 1 were inactive. Overall, this study highlights the importance of the apical ligands of these complexes in their photophysical properties in aqueous media and biological activities against cancer cells and bacteria and gives a bright prospective towards the use of these complexes as molecular PSs or luminescent probes for biological applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (No. 18-05076S), the Operative Program Prague – Competitiveness (OPPC CZ.2.16/3.1.00/21537, OPPC CZ.2.16/3.1.00/24503), and the National Program of Sustainability (NPU I LO1601). The authors thank Ms Dominika Fečková and Ms Veronika Sucháňová for their experimental work.References
- S. Singh, A. Aggarwal, N. V. S. D. K. Bhupathiraju, G. Arianna, K. Tiwari and C. M. Drain, Chem. Rev., 2015, 115, 10261 CrossRef CAS.
- J. Hu, Y. Tang, A. H. Elmenoufy, H. Xu, Z. Cheng and X. Yang, Small, 2015, 11, 5860 CrossRef CAS.
- K. Lang, J. Mosinger and D. M. Wagnerová, Coord. Chem. Rev., 2004, 248, 321 CrossRef CAS.
- M. Westberg, M. Bregnhøj, C. Banerjee, A. Blázquez-Castro, T. Breitenbach and P. R. Ogilby, Methods, 2016, 109, 81 CrossRef CAS.
- D. van Straten, V. Mashayekhi, H. S. de Bruijn, S. Oliveira and D. J. Robinson, Cancers, 2017, 9, 19 CrossRef.
- M. Wainwright and K. B. Crossley, Int. Biodeterior. Biodegrad., 2004, 53, 119 CrossRef CAS.
- A. W. Maverick, J. S. Najdzionek, D. Mackenzie, D. G. Nocera and H. B. Gray, J. Am. Chem. Soc., 1983, 105, 1878 CrossRef CAS.
- K. Kirakci, P. Kubát, J. Langmaier, T. Polívka, M. Fuciman, K. Fejfarová and K. Lang, Dalton Trans., 2013, 42, 7224 RSC.
- Y. Molard, F. Dorson, V. Cîrcu, T. Roisnel and F. Artzner, Angew. Chem., Int. Ed., 2010, 49, 3351 CrossRef CAS.
- K. Kirakci, P. Kubát, M. Dušek, K. Fejfarová, V. Šícha, J. Mosinger and K. Lang, Eur. J. Inorg. Chem., 2012, 2012, 3107 CrossRef CAS.
- M. N. Sokolov, M. A. Mihailov, E. V. Peresypkina, K. A. Brylev, N. Kitamura and V. P. Fedin, Dalton Trans., 2011, 40, 6375 RSC.
- T. Aubert, F. Cabello-Hurtado, M.-A. Esnault, C. Neaime, D. Lebret-Chauvel, S. Jeanne, P. Pellen, C. Roiland, L. Le Polles, N. Saito, K. Kimoto, H. Haneda, N. Ohashi, F. Grasset and S. Cordier, J. Phys. Chem. C, 2013, 117, 20154 CrossRef CAS.
- O. A. Efremova, M. A. Shestopalov, N. A. Chirtsova, A. I. Smolentsev, Y. V. Mironov, N. Kitamura, K. A. Brylev and A. J. Sutherland, Dalton Trans., 2014, 43, 6021 RSC.
- K. Kirakci, V. Šícha, J. Holub, P. Kubát and K. Lang, Inorg. Chem., 2014, 53, 13012 CrossRef CAS PubMed.
- C. Neaime, M. Amela-Cortes, F. Grasset, Y. Molard, S. Cordier, B. Dierre, M. Mortier, T. Takei, K. Takahashi, H. Haneda, M. Verelst and S. Lechevallier, Phys. Chem. Chem. Phys., 2016, 18, 30166 RSC.
- K. Kirakci, P. Kubát, K. Fejfarová, J. Martinčík, M. Nikl and K. Lang, Inorg. Chem., 2016, 55, 803 CrossRef CAS.
- E. V. Svezhentseva, A. O. Solovieva, Y. A. Vorotnikov, O. G. Kurskaya, K. A. Brylev, A. R. Tsygankova, M. V. Edeleva, S. N. Gyrylova, N. Kitamura, O. A. Efremova, M. A. Shestopalov, Y. V. Mironova and A. M. Shestopalov, New J. Chem., 2017, 41, 1670 RSC.
- A. M. Cheplakova, A. O. Solovieva, T. N. Pozmogova, Y. A. Vorotnikov, K. A. Brylev, N. A. Vorotnikova, E. V. Vorontsova, Y. V. Mironov, A. F. Poveshchenko, K. A. Kovalenko and M. A. Shestopalov, J. Inorg. Biochem., 2017, 166, 100 CrossRef CAS.
- A. O. Solovieva, Y. A. Vorotnikov, K. E. Trifonova, O. A. Efremova, A. A. Krasilnikova, K. A. Brylev, E. V. Vorontsova, P. A. Avrorov, L. V. Shestopalova, A. F. Poveshchenko, Y. V. Mironov and M. A. Shestopalov, J. Mater. Chem. B, 2016, 4, 4839 RSC.
- N. Brandhonneur, T. Hatahet, M. Amela-Cortes, Y. Molard, S. Cordier and G. Dollo, Eur. J. Pharm. Biopharm., 2018, 125, 95 CrossRef CAS PubMed.
- K. Kirakci, J. Zelenka, M. Rumlová, J. Martinčík, M. Nikl, T. Ruml and K. Lang, J. Mater. Chem. B, 2018, 6, 4301 RSC.
- A. Beltrán, M. Mikhailov, M. N. Sokolov, V. Pérez-Laguna, A. Rezusta, M. J. Revillo and F. Galindo, J. Mater. Chem. B, 2016, 4, 5975 RSC.
- C. Felip-León, C. Arnau del Valle, V. Pérez-Laguna, M. I. Millán-Lou, J. F. Miravet, M. Mikhailov, M. N. Sokolov, A. Rezusta-López and F. Galindo, J. Mater. Chem. B, 2017, 5, 6058 RSC.
- A. O. Solovieva, K. Kirakci, A. A. Ivanov, P. Kubát, T. N. Pozmogova, S. M. Miroshnichenko, E. V. Vorontsova, A. V. Chechushkov, K. E. Trifonova, M. S. Fufaeva, E. I. Kretov, Y. V. Mironov, A. F. Poveshchenko, K. Lang and M. A. Shestopalov, Inorg. Chem., 2017, 56, 13491 CrossRef CAS PubMed.
- K. Kirakci, P. Kubát, M. Kučeráková, V. Šícha, H. Gbelcová, P. Lovecká, P. Grznárová, T. Ruml and K. Lang, Inorg. Chim. Acta, 2016, 441, 42 CrossRef CAS.
- W. Xu, Z. Zeng, J.-H. Jiang, Y.-T. Chang and L. Yuan, Angew. Chem., Int. Ed., 2016, 55, 13658 CrossRef CAS PubMed.
- R. Hilf, J. Bioenerg. Biomembr., 2007, 39, 85 CrossRef CAS PubMed.
- J. Zielonka, J. Joseph, A. Sikor, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043 CrossRef CAS PubMed.
- J. L. Reedy, D. K. Hedlund, M. T. Gabr, G. M. Henning, F. C. Pigge and M. K. Schult, Bioconjugate Chem., 2016, 27, 2424 CrossRef CAS PubMed.
- K. Kirakci, K. Fejfarová, M. Kučeráková and K. Lang, Eur. J. Inorg. Chem., 2014, 14, 2331 CrossRef.
- J. Zelenka, M. Koncošová and T. Ruml, Biotechnol. Adv., 2018, 36, 583 CrossRef CAS.
- G. McDonnell and A. D. Russell, Clin. Microbiol. Rev., 1999, 12, 147 CrossRef CAS.
- C. Cherchi and A. Z. Gu, Water Sci. Technol., 2011, 64, 7 CrossRef CAS.
- P. K. Taylor, A. T. Yeung and R. E. Hancock, J. Biotechnol., 2014, 191, 121 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of complexes, experimental section, luminescence properties, flow cytometry histograms, cell viability, and photoinactivation. See DOI: 10.1039/c8bm01564c |
| This journal is © The Royal Society of Chemistry 2019 |