
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanomaterials for photocatalytic hydrogen production: from theoretical perspectives
Mahesh Datt Bhatt and
Jae Sung Lee*
and
Jae Sung Lee*
School of Energy & Chemical Engineering, Ulsan National Institute of Science & Technology (UNIST), Ulsan, 689-798, Republic of Korea. E-mail: jlee1234@unist.ac.kr
First published on 12th July 2017
Abstract
To overcome the increasing demand of energy worldwide and global warming due to CO2 emissions from the use of traditional fuel sources, renewable and clean energy sources are in high demand. Solar energy is one of the important renewable energy sources since it can be converted into hydrogen fuel via water splitting. To produce highly efficient and low-cost H2 from the reduction of water and CO2, photocatalytic materials have been developed. Presently, the efficiency of H2 production using transition metal-oxide and non-metal oxide photocatalysts via water splitting is quite low. The main issues reported are low light absorption and poor charge separation. The reasons for these two issues are the large band gaps and band edge positions of the present photocatalysts used for H2 production. To produce H2 to a larger extent from the decomposition of water, the present photocatalysts have been modified by co-catalysts or dopants using different techniques, including the reduction of the band gap and adjustment of the morphology, band edge positions, crystallinity, surface structure, etc., such that these photocatalysts can absorb sufficient light in the visible-light region. This type of modified nanostructured photocatalysts (both oxide and non-oxide) can enhance the efficiency of H2 production via absorbing sufficient light in the visible-light region of the solar spectrum and improving the charge separation by suppressing charge recombination. In this regard, we reviewed both UV- and visible-light active nanostructured photocatalysts and modified photocatalysts reported in theoretical studies.
1. Introduction
The use of traditional energy sources, such as fossil and mineral fuels and nuclear and hydroelectric sources, causes global warming due to the emission of CO2, methane, CO, etc. that can cause serious disasters in various places worldwide. Moreover, there is a continuous depletion of traditional energy sources. To solve these issues, alternative energy sources, such as renewable energy sources, have been developed by researchers worldwide to meet the world's energy needs as well as to decrease pollution.Various types of renewable energy sources are under development or have been developed. Among these, hydrogen can be a promising potential candidate and its production from renewable energy sources is CO2-free. In this regard, hydrogen can be considered as a clean energy carrier that can address the current energy and environmental issues.1,2
Hydrogen can be produced from primary energy sources including coal, natural gas, waste, biomass, solar, wind, hydropower, nuclear power, and geothermal power. For example, commercial hydrogen can be produced via steam reforming of methane3,4 and also from coal using gasification technology; however, both these methods result in CO2 emissions. Hydrogen can be stored in its gaseous, liquid, or metal hydride form and can be distributed over large distances via pipelines or tankers. Numerous studies have been carried out to discuss the key role of hydrogen in sustainable development.5–10
The abundant supply of water and sunlight offers us an affordable alternative source to produce hydrogen. Therefore, photocatalytic water splitting is one of the potential techniques for clean solar hydrogen production and has been utilized in small- to large-scale hydrogen generators.11 In 1972, Honda and Fujishima12 first investigated water splitting using a single TiO2 crystal as a photoanode and Pt as a cathode. Subsequently, numerous studies on water splitting have been conducted13–19 with the exploration of more than 100 different new catalysts including multi-component oxides, sulfides, nitrides, and carbides. All of them are concentrated on the various factors affecting the conversion efficiency such as how much sunlight is absorbed, the exciton generation, the separation/recombination of holes and electrons received by dissociation of excitons, and the charge collection by the respective electrodes to produce hydrogen and oxygen. Some other practical factors are also important for photocatalytic hydrogen production such as stability and resistance under visible light, reduced cost, and non-toxicity of utilized materials.20–23 In this regard, we speculated that photocatalytic hydrogen could be a commercial fuel in future; however, significant research efforts should be devoted in this direction, addressing the abovementioned key issues for its industrial production.
Nanomaterials such as CdS, SiC, CuInSe2, and TiO2 can be utilized to produce cheap and clean photocatalytic hydrogen24–27 due to the fact that these nanomaterials show better photocatalytic properties as compared to their bulk counterparts. Currently, many other nanomaterials such as Nb2O5,28 Ta2O5,29 α-Fe2O3,30,31 ZnO,32,33 TaON,34 BiVO4,35,36 and WO3 have been explored.37 In most of the photocatalysts, band gap limitation is a key issue that results in low H2 production.38 To solve this issue, some photocatalysts have been modified via noble metal doping, ion doping, sensitization, and metal-ion implantation. In noble metal doping, Pt has been found to be the best noble metal; however, it is too expensive. Thus, other efficient and cheaper metals such as Ni, Cu, Ag, Ru, Pd, and Ir have been explored.39–47 Ion doping involves transition metal and rare-earth metal ions, anions of nitrogen and sulphur, etc. Sensitization means dye-sensitization and coupling of semiconductors. Among the abovementioned modification techniques, metal-ion implantation and dye-sensitization have been found to be the most effective photoanode surface modification techniques. Moreover, researchers are interested in exploring co-catalysts with photocatalyst nanomaterials that can be used for photocatalytic hydrogen production.
The theoretical and computational description of many body systems is still one of the biggest challenges in solid state science although significant progress has been made in this regard. The majority of theoretical and computational studies on photocatalytic nanomaterials have used density functional theory (DFT) because DFT predicts the ground-state properties in terms of electronic density. The band gap of the materials is often significantly underestimated by DFT,48 computed using Kohn–Sham method by introducing an exchange correlation (XC) functional such as local density approximation (LDA)49 or the generalized gradient approximation (GGA).50 The band gap of the materials can be improved by applying several approaches. For example, a hybrid functional can improve the accuracy by combining the approximate DFT functionals (LDA or GGA) with the exact Hartree–Fock (HF) exchange energy.51 Alternatively, the DFT + U method with the Hubbard band U term can provide more accurate results.52 The accuracy of these two approaches depends on the selection of several empirical parameters. Moreover, the parameter-free GW method yields an accurate description of the electronic structure of the materials, but at a huge computational expense.53 Therefore, the use of GW method is not more reliable for complex structures.
Herein, the study was concentrated on modeling a nanostructured photocatalyst and modified nanophotocatalysts at the atomic level to provide information on the key atomic level structures, processes, and parameters that control the behavior of photocatalytic materials during their applications in photocatalytic H2 production. In particular, substantial contributions have been made by modeling methods in the study of the electronic states, defects, and surface properties of nanophotocatalysts for H2 production.
2. Theoretical achievements for the development of nanophotocatalysts and modified photocatalysts for hydrogen production via water splitting
Many nanophotocatalysts have been developed to split water into H2 and O2 under UV and visible-light illumination. The industrial production of clean and recyclable H2 via the direct splitting of water using a particulate photocatalyst could be the best method.54–57 However, there is a lack of suitable materials with appropriate band gap positions for overall water splitting, and stability is required for practical applications. The reason is that most metal oxides have optical band gaps that lie outside the visible-light range (Eg > 3 eV); thus, even if they are catalytically active for a given reaction, they cannot make efficient use of the solar spectrum. For example, TiO2 has a wide band gap (Eg = 3.2 eV), which is limited to a 1% solar-to-hydrogen (STH) conversion efficiency for the water splitting process, whereas for a smaller band gap material such as Fe2O3 (Eg = 2.2 eV), the theoretical efficiency increased to 15%.58 The STH of 10% is required for the solar cell-powered electrolysis of water. Therefore, the development of highly active photocatalysts should be explored for large-scale photocatalytic H2 production via water splitting.59–62 There are two types of nanostructured photocatalyst materials for H2 production via water splitting.(i) Metal-oxide photocatalysts and modified metal-oxide photocatalysts (with hybrid systems)
Although there have been many experimental investigations reported on metal oxide nanophotocatalysts for H2 production via water splitting under UV- and visible-light irradiation, very few theoretical investigations based on the DFT methods have been reported. Among these, most theoretical investigations have been reported on nanostructured TiO2 and modified TiO2 photocatalysts. For example, Kaur et al.63 investigated amorphous TiO2 as a photocatalyst for H2 production using the DFT methods. The authors claimed via the analysis of the electronic properties that amorphous TiO2 may act as a cheaper, more abundant, but somewhat less efficient photocatalyst as compared to crystalline TiO2. To improve the photocatalytic activity of the TiO2 nanophotocatalyst, the doping of TiO2 was carried out using different techniques via narrowing the band gap of TiO2 to absorb more visible-light in the solar spectrum as well the dependence of charge separation and recombination on the distance between the catalytic core and semiconductor surface. For example, Reynal et al.64 reported that the photoinduced reduction of H2 on a Co electrocatalyst immobilized on TiO2 was 104 times faster than the reverse charge recombination. The authors claimed that both processes show an exponential dependence on the distance between the semiconductor and the catalytic core, as shown in Fig. 1(a)–(d).64 As shown in Fig. 1, the authors computed the charge separation and recombination processes when a semiconductor (TiO2) was functionalized with three related cobalt electrocatalysts, whose molecular structure varied the physical separation between the catalytic core and the semiconductor surface. In this hybrid system, the semiconductor acted as a light harvester and H2 evolution was driven by the anchored molecular catalyst. Some theoretical investigations have shown that BaTiO3 (ref. 65 and 66) and PbTiO3 (ref. 67–69) nanowires exhibit ferroelectricity and Bi2Ti2O7,70 SrTiO3,71 and CaTiO3 (ref. 72) nanowires exhibit photocatalytic activity towards water splitting. Bandura et al.73 demonstrated the photocatalytic activity of SrTiO3 nanowires and concluded that via comparison of the band edge positions and Fermi levels relative to the levels of the reduction H+/H2 and oxidation O2/H2O potentials as well as analysis of the band gap widths, the probable SrTiO3 nanowire configurations and SrTiO3 nanotube74 configurations can be suggested for application in the photocatalytic splitting of water molecules under solar irradiation in the visible-light range.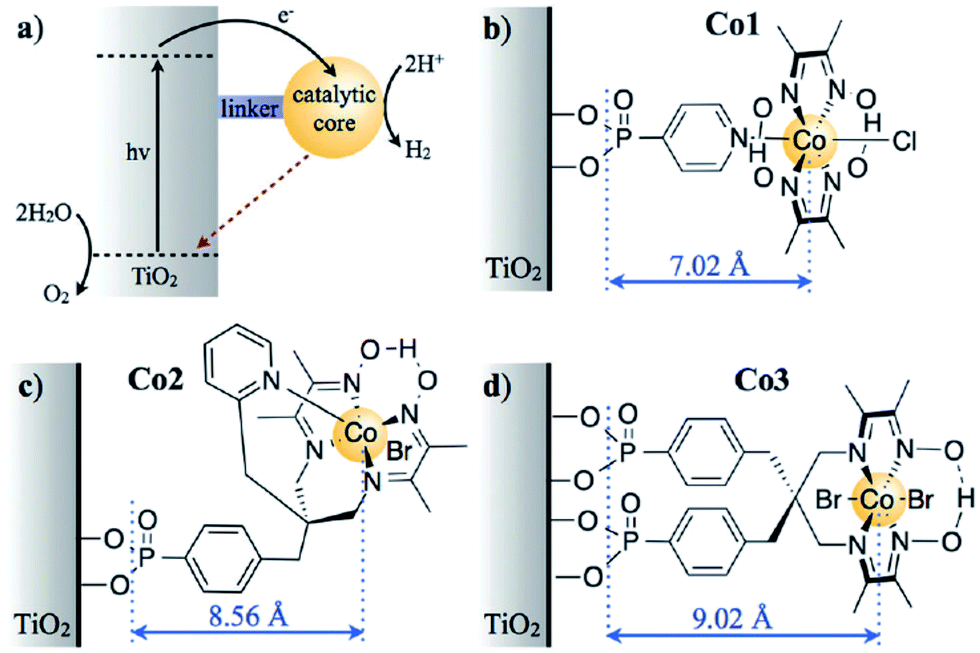 | ||
| Fig. 1 (a) The electron transfer processes in TiO2 functionalised with a molecular catalyst for H2 production after UV-light excitation. The solid black and dashed red arrows indicate the charge separation and recombination, respectively. The molecular structures of the catalysts for H2 reduction are shown in (b) for Co1, (c) for Co2, and (d) for Co3 (the charges have been omitted for clarity). The blue arrows indicate the distance between the anchoring groups and the catalyst metal centre (r, A), as determined by the energy minimised DFT calculations. Reprinted with permission of ref. 64. Copyright 2014 Royal Society of Chemistry. | ||
In the case of TiO2, the phase of the photocatalyst is a key factor for its photocatalytic activity. For example, although the rutile phase exhibits a narrower band gap (approx. 3.0 eV) than the anatase phase (approx. 3.2 eV),75–77 anatase is generally considered to exhibit superior photocatalytic activity due to its larger surface area and thus higher activity.78 The third metastable phase is brookite that has also been reported to exhibit photocatalytic activity,79,80 and mixtures of anatase, rutile, and brookite have been reported to exhibit significant photocatalytic activity.81,82 However, this phase is of lesser interest due to the complexity of its synthesis. The doping of TiO2 can provide several outcomes: formation of new valence states in TiO2,83–86 creation of charge carrier trapping sites,87–90 band gap reduction,91–93 control of phase transformation behavior,94–97 and surface enhancement.96,98 The effects of dopants on the anatase to rutile phase transformation have been comprehensively reviewed elsewhere.99 The phase transformation mainly occurs due to the atomic rearrangement involved in the transformation, which is often a result of an increase in the density of the anion vacancies. This may occur when cationic dopants of low valence substitute Ti in the anatase lattice.79,99,100 Hanaor et al.100 performed DFT calculations for the relative stability of anatase and rutile polymorphs of TiO2. The rutile phase exhibited a stability of 3 meV as compared to the anatase phase in pristine TiO2. The authors also demonstrated the doping of TiO2 with Si, Al, Fe, and F atoms and found that the anatase phase stabilized the rutile phase in the dopant order F > Si > Fe > Al obtained via comparison of ΔF (a-TiO2:F) with ΔF (r-TiO2:F), which provided their formation energies as 84.78, 32.43, 18.46, and 17.68 meV, respectively. As the authors considered only flourine anion dopant in their study, the inhibition of rutile formation via F doping was supported by previously reported experimental results.101,102 A schematic of the interstitial and substitutional dopant positions in anatase and rutile TiO2 is shown in Fig. 2(a) and (b).100
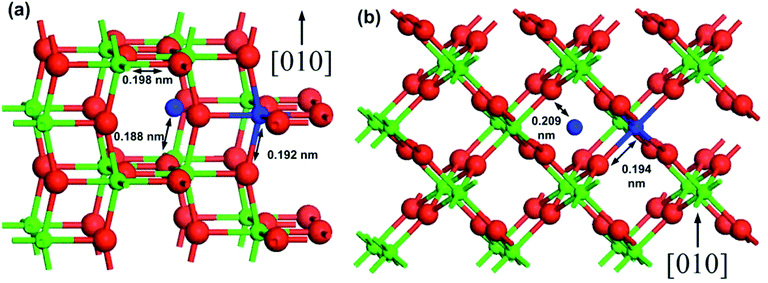 | ||
| Fig. 2 A schematic of the interstitial and substitutional dopant positions in (a) anatase and (b) rutile TiO2 with the arrows showing the cation-dopant to oxygen distances. Reprinted with permission of ref. 100. Copyright 2012 Springer-Verlag Berlin Heidelberg. | ||
In general, efforts to modify TiO2 to enhance the visible-light absorption have been focused on the substitution of metal cations and/or non-metal anions.103–111 In some cases, the choice of DFT methodology can have an influence on the reduction of the band gap to the visible-light region of the solar spectrum.109 Co-doping with compensating cation–anion pairs from DFT simulations has been reported,112,113 along with a recent experimental study of Mo–C co-doped TiO2.114 Key parameters such as stability, solubility, and reproducibility are also important along with the band gap reduction as well as the method of doping and/or the DFT methodology to enhance the photocatalytic activity of TiO2 due to minimum charge recombination. Moreover, nanostructure engineering of TiO2 can reduce the charge recombination.115–119 In this regard, heterostructure techniques of TiO2 with other oxides can enhance the photocatalytic activity to tune the structure to increase the absorption of visible-light.120–133 For example, heterostructures such as Bi4Ti3O12–TiO2,124 MgO–TiO2,134 and Ga2O3–TiO2 (ref. 134) can reduce the band gap, leading to visible-light photoactivity, efficient charge separation, and improved photocatalytic activity as compared to pure TiO2.
In the case of the Bi4Ti3O12–TiO2 heterostructure, Bi 6s mainly contributes to the valence band, whereas the Bi 6p state contributes to the conduction band, which result in the reduction of the band gap of Bi4Ti3O12 (2.5 eV) as compared to that of TiO2 (3.2 eV). This leads to large absorption of visible-light. A schematic showing the energy band structure and electron–hole pair separation in the Bi4Ti3O12–TiO2 heterostructure is shown in Fig. 3.124 Similarly, in the case of MgO–TiO2 and Ga2O3–TiO2 heterostuctures, the author claimed the presence of two states in the band gap, as shown in Fig. 4.134 The first state comes from an occupied Ti3+ state at 0.9 eV and 1.0 eV above the valence band edge in the MgO–TiO2 and Ga2O3–TiO2 heterostuctures, respectively. The second state comes from an unoccupied O 2p state (oxygen hole polaron), which lies 1.8 eV and 1.5 eV above the valence band of the MgO–TiO2 and Ga2O3–TiO2 heterostuctures, respectively. These results show that the excitonic electrons and holes are localized on the TiO2 and the metal oxide nanostructures, which lead to a reduction in charge recombination and an enhancement in the photocatalytic activity.
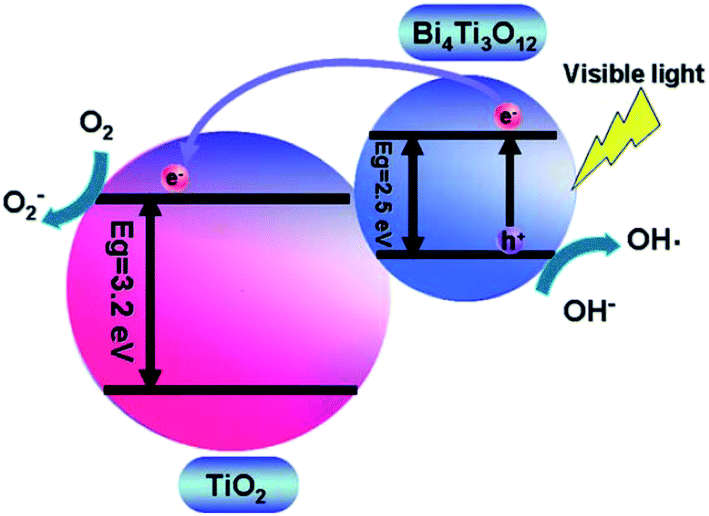 | ||
| Fig. 3 A schematic showing the energy band structure and electron–hole pair separation in the Bi4Ti3O12/TiO2 heterostructure. Reprinted with permission of ref. 124. Copyright 2011 Royal Society of Chemistry. | ||
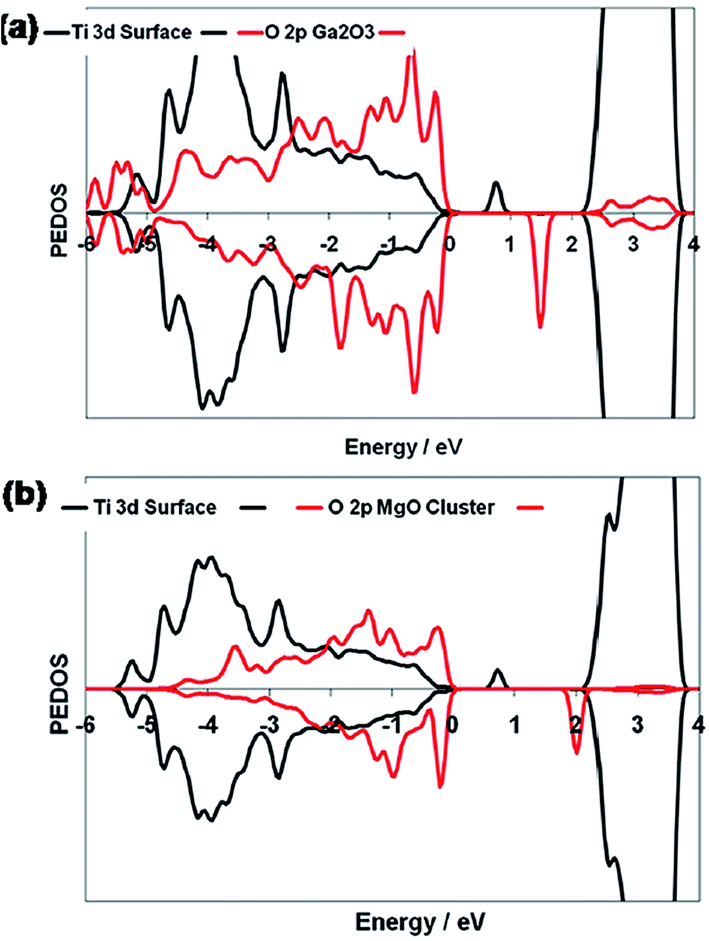 | ||
| Fig. 4 Ti 3d and O 2p PEDOS of the (Ga2O3)2 and (MgO)4 clusters for the relaxed triplet excited states of (a) Ga2O3-modified TiO2 and (b) MgO-modified TiO2. The zero of energy is the top of the valence band. Reprinted with permission of ref. 134. Copyright 2012 American Chemical Society. | ||
In addition, TiO2 nanoparticles can attach materials such as zeolites,135 silica,136 activated carbon137–142 or carbon nanotubes.143–149 Recently, graphene has attracted significant interest150–154 due to its large surface area and potentially higher photocatalytic efficiency.155 However, very few reports are available for theoretical investigations on the interaction of nanoparticles and graphene sheets, and the mechanism of charge transfer from graphene to the nanoparticles. The decorated surface of graphene with both TiO2 nanofibers155 and Fe-doped TiO2 nanofibers156 in supercritical carbon dioxide exhibits high photocatalytic activity. Das et al.157 deposited different semiconductor nanoparticles, such as TiO2 and ZnO, and some magnetic nanoparticles, such as Fe3O4 and Ni, on graphene. They calculated their electronic structure using DFT methods and found that charge transfer occurred between graphene and the deposited nanoparticles.
Among all the metal oxides, WO3 has attracted significant interest due to its photosensitivity,158–160 good electron transport properties,161 and stability against photocorrosion.162,163 Moreover, its smaller band gap (approx. 2.8 eV) than other oxides such as TiO2 makes it suitable for the absorption of visible-light. However, the gap of WO3 is still too large for sufficient visible-light absorption. Experimental results have shown that the conduction band minimum (CBM) of bulk WO3 is about 0.4 eV below the hydrogen redox potential.164,165 For the WO3 surface, the CBM lies 0.31 eV above the hydrogen redox potential, which is still low for hydrogen production.166
Doping and co-doping are good ways to tailor the electronic structure and photocatalytic properties of semiconducting oxides. For example, Mo-doped WO3 nanowires showed an enhancement in visible-light photoactivity, as the band gap of the MoxW1−xO3 solid solutions was narrowed by 0.48 eV upon increasing the Mo content from 0 to 0.75 .167,168 The effect of several transition metals on the photocatalytic activity under UV irradiation was also studied, suggesting maximum H2 production via Ni doping with WO3.169 Doping with other metals (Ti, Zn, Dy, Te, Ta, V, Cu, Ag, and Ce) has also been reported in the literature,170–178 with some claims of improved photocatalytic activity. For example, Wang et al.179 studied Mo, Cr, Ti, Zr, and Hf-doped WO3 and found that in Hf-doped WO3, the oxygen vacancies have a negative formation energy, which leads to a shift in both the VB and CB to higher energies and a reduction of the band gap, with potential benefits for photocatalytic H2 production, as shown in Fig. 5.179
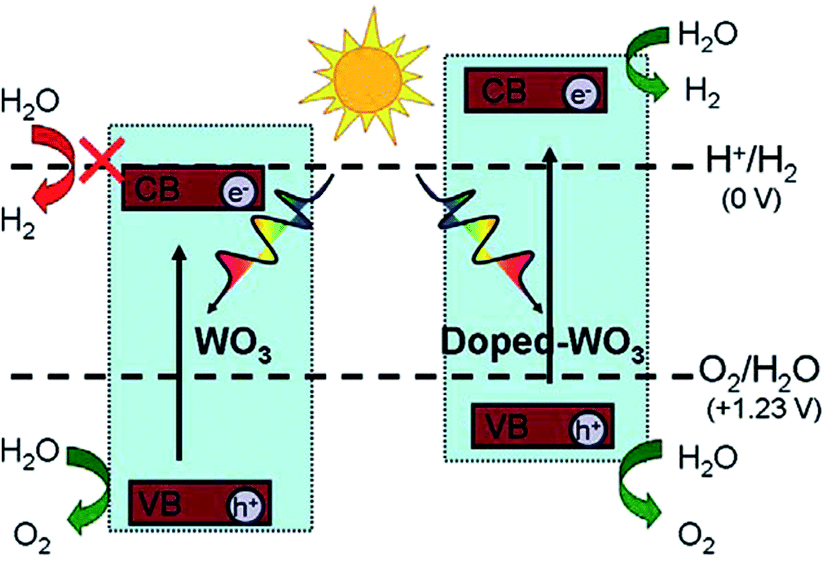 | ||
| Fig. 5 A schematic of the superior photocatalytic activity of metal-doped WO3 for H2 production as compared to that of WO3. Reprinted with permission of ref. 179. Copyright 2012 American Chemical Society. | ||
In previous studies, it has been reported that perovskite-type alkali tantalates, ATiO3 (A = Li, Na, and K) show significant photocatalytic activity under UV irradiation. However, these are less active under visible-light irradiation.180,181 Transition metal ions were found to be attractive dopants for the effective modification of photocatalysts, which can play multiple roles ranging from reducing the band gap to efficient electron transfer due to their multiple oxidation states.182,183 Electron transitions from the valence band to the dopant level or from the dopant level to the conduction band can effectively red shift the band edge absorption threshold.
For example, Liu et al.184 demonstrated M-doped NaTaO3 (M = V, Cr, Mn, Fe, Ni, Cu, and Zn) nanoparticles using both experimental and DFT techniques. The authors found that the substitution of Ta3+ by metal ions results in the formation of an intermediate band, which is due to the metal 3d state. The supercell of M-doped NaTaO3, total and projected DOS of pure and V-doped NaTaO3, and intermediate band formation by the d-orbitals of the doping metal ions are shown in Fig. 6(a), (b) and (c), respectively.184
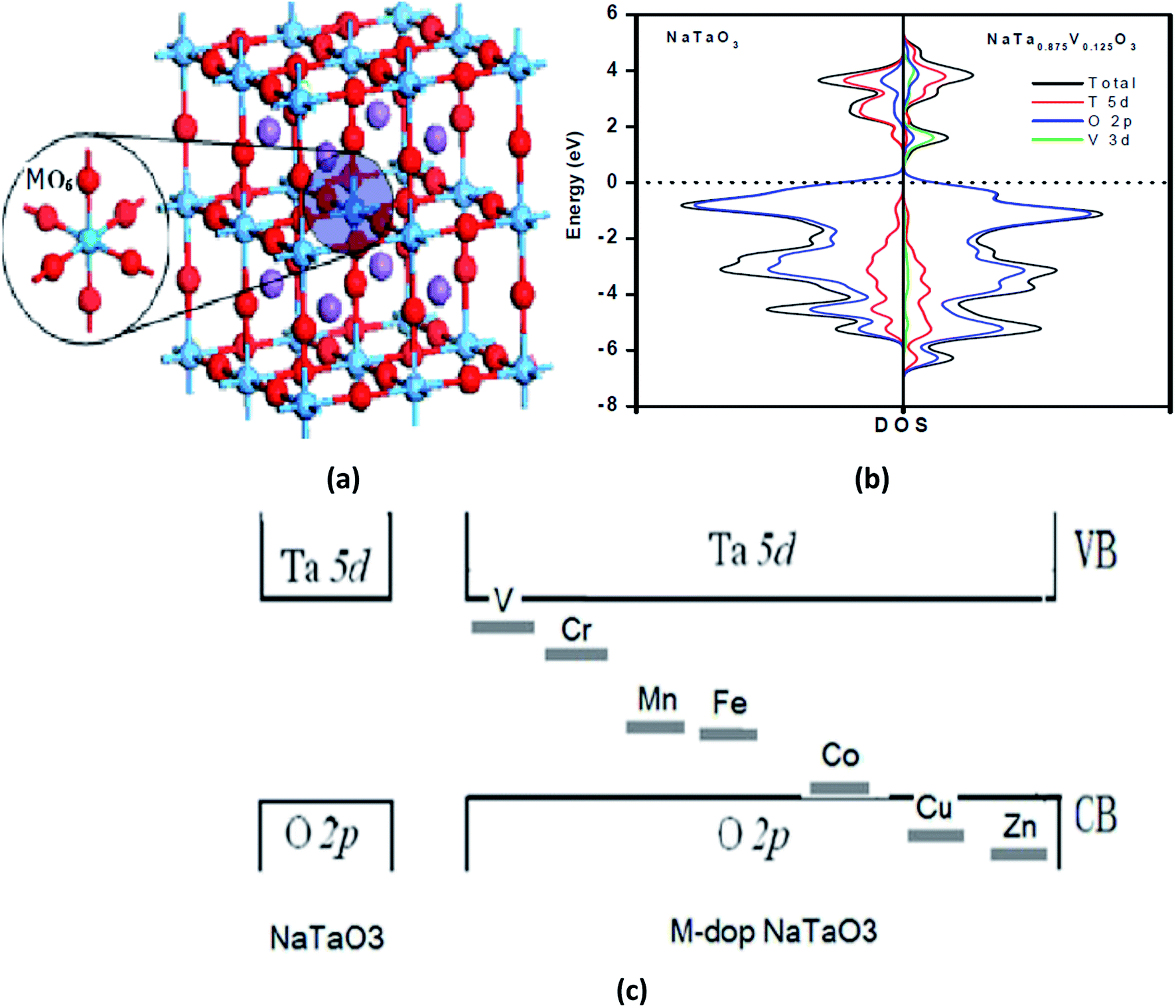 | ||
| Fig. 6 (a) A supercell of M-doped NaTaO3 2 × 2 × 2 (40-atoms), (b) the total density of states and projected density of states of the pure and V-doped NaTaO3, and (c) intermediate band formation by the d-orbital of the doped metal ions. Reprinted with permission of ref. 184. Copyright 2012 IACSIT Press, Singapore. | ||
Moreover, a ZnO/graphene hybrid nanostructure was reported to have excellent potential for application in electronic devices.185–190 Most studies on the ZnO/graphene hybrid structures have been focused on their structural morphology and electronic properties.191,192 Recently, Xiang et al.193 first proposed a three-component composite, TiO2/graphene/MoS2, containing 0.25 wt% graphene. The results showed that graphene acts as an electron reservoir and MoS2 can act as a source of active adsorption sites to achieve a highly efficient synergetic H2 evolution at the rate of 165.3 μmol h−1, which is 17 times that of TiO2/MoS2.194 Wu et al.195 studied ZnO-dotted porous ZnS cluster microspheres for highly efficient, Pt-free photocatalytic H2 evolution.
(ii) Non-metal oxide photocatalysts and modified non-metal oxide photocatalysts (with hybrid systems)
The surface modification of oxide photocatalysts, particularly TiO2, via doping with N, C or S, or metal nanoparticles cannot provide significant photocatalytic activity in the modified materials in the visible-light region of the solar spectrum.196,197 Therefore, researchers have been interested in exploring alternative materials for nanophotocatalysts and modified photocatalysts for photocatalysis and solar energy conversion. As a result, the large-scale production of stable visible-light active photocatalysts remains a challenge for industrial applications.198 For example, polymeric carbon nitride has been found to be an efficient photocatalyst that produces H2 from water under visible-light irradiation; however, in this case, a sacrificial donor is required.199 Similarly, a class of metal-free photocatalysts, including elemental boron,200 sulfur,201 and phosphor,202 as well as the binary carbon nitride203 and boron carbide, has emerged.204 Conjugated polymer nanostructures have emerged as alternate materials for applications in solar energy conversion. However, there is a lack of photocatalytic studies of these conjugated polymers. Semiconductor nanostructures modified with conducting polymers have been studied for photocatalytic applications. Two-dimensional materials such as graphene205 and layered hexagonal (h-BN) are promising photocatalysts.206–208 However, graphene (monolayer) exhibits a zero band gap, whereas h-BN exhibits a wide band gap (∼5.5 eV). These materials (say ternary B–C–N compounds) can constitute the desired medium band gap semiconductors via adjustment of the band gap and absolute energy levels via chemical variations.209,210 Similarly, graphene oxide based on a carbon support may improve the charge separation and spontaneous redox processes.211,212 In recent years, graphene-based heterogenous photocatalytic nanomaterials have attracted significant attention due to their unique sp2 hybrid carbon networks, exhibiting ultra-fast electron mobility at room temperature, conductivity, large theoretical surface area, high work function, etc.213–215 Huang et al.215 demonstrated a ternary structure of B–C–N nanosheets that has the functionality to catalyze H2 and O2 evolution from water as well as CO2 reduction under visible-light illumination. The plane-wave DFT calculations of the electronic structure of h-BN are shown in Fig. 5(a) and (b).215 As shown in Fig. 7(a) and (b), for a B11C12N9 compound, the gap is significantly reduced from 4.56 eV to 2.00 eV. The partial DOS, as shown in Fig. 7(b), signifies that the VB and CB edges of B11C12N9 mainly comprise C 2p orbitals, which is different from that of pure h-BN (Fig. 7(a)).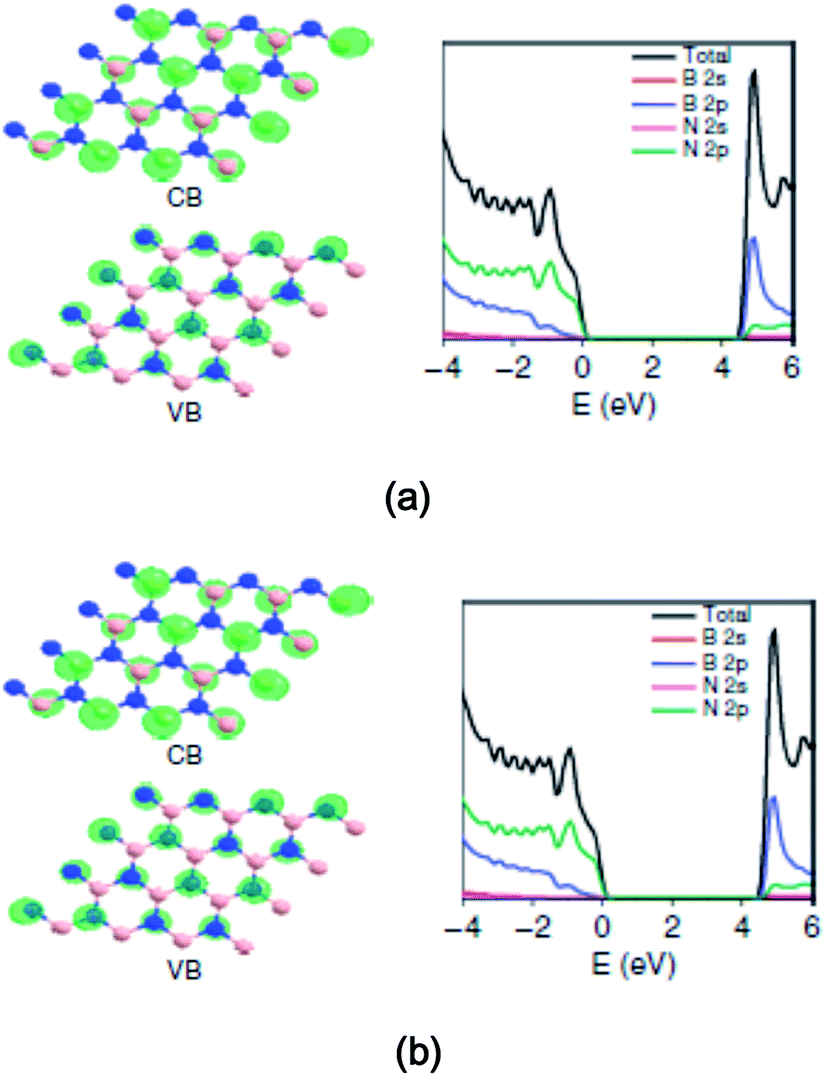 | ||
| Fig. 7 The plane-wave DFT calculations of the electronic structure of h-BN and h-BCN. The optimized structure of B16N16 with the corresponding valence band (VB)/conduction band (CB) and the corresponding total and ion-decomposed electronic density of states (a and b). Reprinted with permission of ref. 215. Copyright 2015 Macmillan Publishers Limited. | ||
Metal chalcogenides have been found to be promising photocatalyst materials for photocatalytic H2 production due to their appropriate band gap width and band edge position.216 In particular, CdS has been found to be an efficient semiconductor H2-production photocatalyst.217 However, some key issues such as the quick recombination of photogenerated charge carriers and photocorrosion under visible-light irradiation still exist and prohibit the wide application of CdS to a large extent. To solve these issues, forming Zn1−xCdxS solid solutions is a viable method because ZnS possesses the same coordination mode with CdS,218–220 and the band gap width and band edge position of the Zn1−xCdxS solid solutions can be tuned by changing the molar ratio of ZnS and CdS.221,222 For example, Li et al.223 demonstrated the visible-light photocatalytic H2 production activity of Zn1−xCdxS solid solutions with different molar ratios of ZnS and CdS using both experimental and DFT methods. The authors found that for Zn/Cd with a molar ratio equal to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the Zn0.5Cd0.5S solid solution exhibits the highest H2-production rate of 7.42 mmol h−1 g−1, exceeding that of the pure CdS and ZnS samples by more than 24 and 54 times, respectively. The supercell models for bulk ZnS and CdS, the geometry of Zn0.5Cd0.5S, the band structures of the Zn1−xCdxS systems for various Zn/Cd molar ratios and the conduction and valence band edge potentials of the samples (x = 0, 0.1, 0.3, 0.5, 0.7, 0.9, and 1.0) are shown in Fig. 8(a), (b) and (c), respectively.223 Fig. 8(d) shows that the band gap of the Zn1−xCdxS samples gradually becomes narrower from 1.921 to 1.141 eV upon increasing the Cd content, which is well consistent with the experimental results.
:1, the Zn0.5Cd0.5S solid solution exhibits the highest H2-production rate of 7.42 mmol h−1 g−1, exceeding that of the pure CdS and ZnS samples by more than 24 and 54 times, respectively. The supercell models for bulk ZnS and CdS, the geometry of Zn0.5Cd0.5S, the band structures of the Zn1−xCdxS systems for various Zn/Cd molar ratios and the conduction and valence band edge potentials of the samples (x = 0, 0.1, 0.3, 0.5, 0.7, 0.9, and 1.0) are shown in Fig. 8(a), (b) and (c), respectively.223 Fig. 8(d) shows that the band gap of the Zn1−xCdxS samples gradually becomes narrower from 1.921 to 1.141 eV upon increasing the Cd content, which is well consistent with the experimental results.
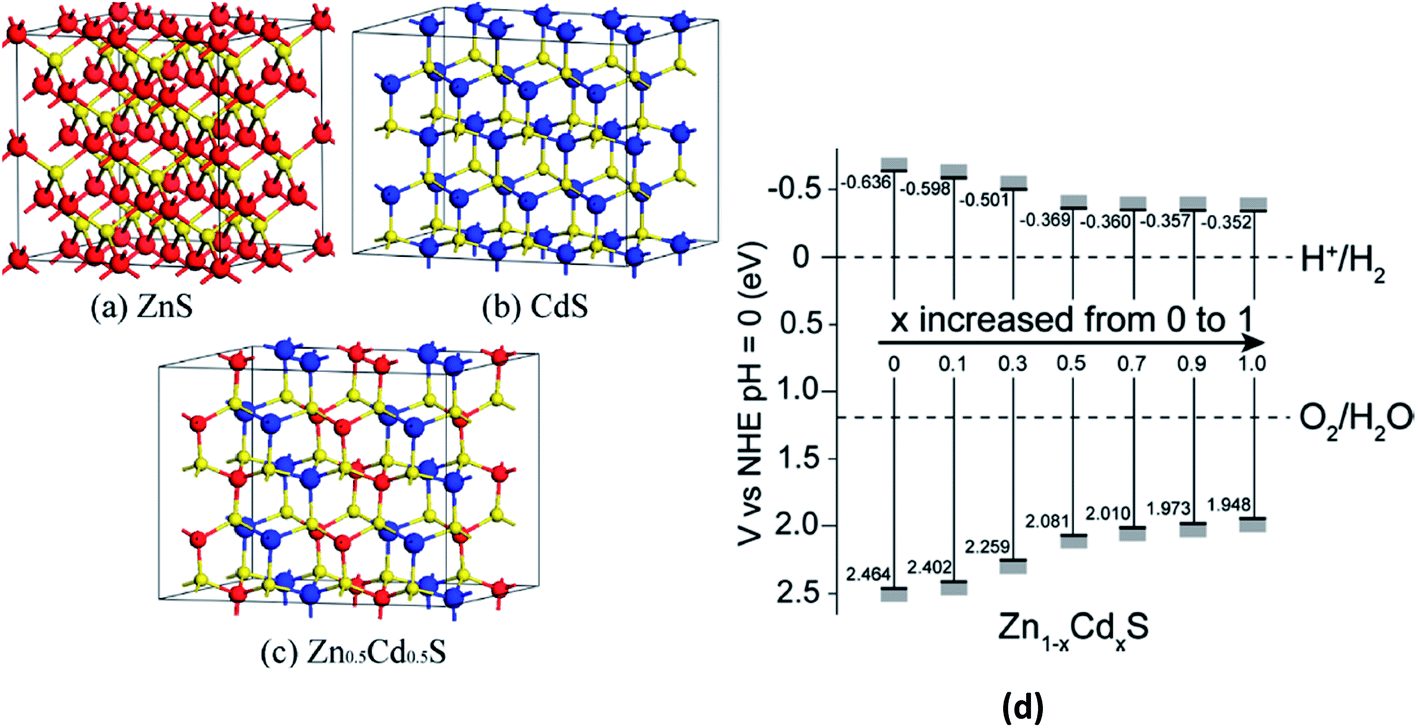 | ||
| Fig. 8 The supercell models for bulk (a) ZnS (64 atoms) and (b) CdS (64 atoms). The red, yellow, and blue spheres represent the Zn, S, and Cd atoms, respectively. (c) The geometry model for Zn0.5Cd0.5S and (d) conduction and valence band edge potentials of the Zn1−xCdxS samples (x = 0, 0.1, 0.3, 0.5, 0.7, 0.9, and 1.0). Reprinted with permission of ref. 223. Copyright 2013 American Chemical Society. | ||
Many studies have been conducted on the development of semiconducting material/graphene hybrid structures using either gas phase223–226 or liquid phase techniques.185,186,191,227 Hou et al.228 have developed a CdS QDs/graphene/ZnIn2S4 system that exhibits highly efficient H2 production due to the high hydrothermal stability and efficient electron transfer. Graphene can also play a role as a co-catalyst and constitutes a synergistic effect with the other co-catalyst to improve the photocatalytic efficiency.229–231 Similarly, Zhu et al.230 demonstrated ZnS loaded with 0.25 wt% graphene and 2 atom% MoS2 that yielded a high H2 production rate of 2258 μmol h−1, which is 2 times of that observed with ZnS only.
Consequently, there is a huge scarcity of theoretical investigations based on the DFT methods on nanophotocatalysts (both metal-oxide and non-metal oxide) and modified photocatalysts for photocatalytic H2 production via water splitting. In the case of metal-oxide photocatalysts, few studies were reported on pure TiO2 and modified TiO2, which were focused on the electronic structures of modified TiO2, signifying the gap reduction and efficient charge separation due to doping of TiO2 to enhance the photocatalytic H2 production. Moreover, very few studies have been reported for graphene and metal chalcogenides in the form of B–C–N ternary compounds and multicomponent heterostructures (metal oxide/graphene/metal chalcogenides) for efficient photocatalytic H2 production due to the synergistic effect of graphene and MoS2. There is a big opportunity for theoretical researchers to conduct DFT calculations on metal oxide and non-metal oxide photocatalysts and their modified forms for highly efficient photocatalytic H2 production. Therefore, more DFT investigations should be devoted to either experimentally explore photocatalysts and modified photocatalysts or on the exploration of new photocatalytic materials for highly efficient H2 production to a large extent.
3. Summary and perspectives
Herein, we reviewed nanostructured photocatalysts (both transition metal oxides and non-metal oxides) and modified photocatalysts for highly efficient H2 production via water splitting under both UV and visible-light irradiation investigated via DFT techniques. However, the current lack of industrial applications of current semiconductor-based photocatalytic H2 production is mainly due to two reasons: the low photocatalytic efficiency and lack of extensive studies for successful industrial applications of photocatalytic H2 production experimentally as well as the lack of DFT studies to investigate the factors responsible for the enhancement of the efficiency of H2 production. The current H2 production yield is quite low, which is far from the targeted quantitative efficiency of 30% at 600 nm for practical applications.232 The critical light conversation efficiency for photocatalytic H2 production via water splitting is about 15%.233 In this regard, the present research does not explore the highly efficient and cost-effective photocatalysts and modified photocatalysts for H2 production via water splitting. The reason is that the present metal oxide and non-metal oxide photocatalysts have wide band gaps, which do not absorb sufficient sunlight in the visible-light region. To reduce the band gaps towards visible-light absorption and photogenerated charge separation in photocatalysis, the photocatalysts have been modified using different techniques in different forms. However, the modified photocatalysts do not enhance the efficiency of H2 production from the decomposition of water under visible-light irradiation. Moreover, the fast charge recombination and fast back reactions are also drawbacks for the solar water splitting system.234–236 DFT investigations have reported the control of band gap and band edge positions via variations of the morphology, composition, crystallinity, and surface structure of photocatalysts for efficient H2 or O2 evolution from water splitting.Consequently, more theoretical studies should be devoted towards the exploration of highly efficient, stable, eco-friendly and cost-effective photocatalysts, and modified photocatalysts via the continual addition of electron donors to overcome the key issues of current solar water splitting systems in the production of H2 to a larger extent.
Acknowledgements
This work was supported by the Korea Center for Artificial Photosynthesis (KCAP, 2009-0093880, 2009-0093886), Basic Science Research Program (No. 2012-017247), BK Plus Program and A3 Foresight Program, all funded by the Ministry of Science, ICT and Future Planning through the National Research Foundation (NRF) Korea.References
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed.
- C. Liao, C. Huang and J. C. S. Wu, Catalysts, 2012, 2, 490 CrossRef CAS.
- S. C. Tsang, J. B. Claridge and M. L. H. Green, Catal. Today, 1995, 23, 3 CrossRef CAS.
- G. A. Olah and A. Molnar, Hydrocarbon chemistry, Wiley Interscience, 1995 Search PubMed.
- L. G. Thomas and A. K. Nelson, Int. J. Hydrogen Energy, 2010, 35, 900 CrossRef.
- L. Barelli, G. Bidini, F. Gallorini and A. Ottaviano, Int. J. Hydrogen Energy, 2011, 36, 3206 CrossRef CAS.
- M. Ni, M. K. H. Leung and D. Y. C. Leung, Energy Convers. Manage., 2007, 48, 1525 CrossRef CAS.
- I. Dincer, Int. J. Energy Res., 2007, 31, 29 CrossRef CAS.
- A. Saeed, M. Ali and S. Mahrokh, Int. J. Hydrogen Energy, 2010, 35, 9283 CrossRef.
- T. A. H. Ratlamwala, A. L. El-Sinawi, M. A. Gadalla and A. Aidan, Int. J. Energy Res., 2012, 36, 1121 CrossRef CAS.
- A. Kudo, Pure Appl. Chem., 2007, 79, 1917 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- Y. Li and J. Z. Zhang, Laser Photonics Rev., 2010, 4, 517 CrossRef CAS.
- Z. Chen, T. F. Jaramillo, T. G. Deutsch, A. Kleiman-Shwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske, M. Sunkara, E. W. McFarland, K. Domen, E. L. Miller, J. A. Turner and H. N. Dinh, J. Mater. Res. Technol., 2010, 25, 3 CrossRef CAS.
- L. J. Minggu, W. R. W. Daud and M. B. Kassim, Int. J. Hydrogen Energy, 2010, 35, 5233 CrossRef CAS.
- R. van de Krol and Y. S. Liang, J. Mater. Chem., 2008, 18, 2311 RSC.
- B. D. Alexander, P. J. Kulesza, I. Rutkowska, R. Solarska and J. Augustynski, J. Mater. Chem., 2008, 18, 2298 RSC.
- K. J. Rajeshwar, J. Appl. Electrochem., 2007, 37, 765 CrossRef CAS.
- V. M. Aroutiounian, V. M. Arakelyan and G. E. Shahnazaryan, Sol. Energy, 2005, 78, 581 CrossRef CAS.
- A. Steinfeld, Int. J. Hydrogen Energy, 2002, 27, 611 CrossRef CAS.
- I. Akkerman, M. Janssen, J. Rocha and R. H. Wijffels, Int. J. Hydrogen Energy, 2002, 27, 1195 CrossRef CAS.
- D. Das and T. N. Veziroglu, Int. J. Hydrogen Energy, 2008, 33, 6046 CrossRef CAS.
- Y. F. Guan, M. C. Deng, X. J. Yu and W. Zhang, Biochem. Eng. J., 2004, 1, 69 CrossRef.
- J. S. Jang, H. G. Kim, U. A. Joshi, J. W. Jang and J. S. Lee, Int. J. Hydrogen Energy, 2008, 33, 5975 CrossRef CAS.
- P. J. Sebastian, R. Castaneda, L. Ixtlilco, R. Mejia, J. Pantoja and A. Olea, Proc. SPIE, 2008, 7044, 704405 CrossRef.
- L. A. Silva, S. Y. Ryu, J. Choi, W. Choi and M. R. Hoffmann, J. Phys. Chem. C, 2008, 112, 12069 CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401 CrossRef CAS.
- X. Chen, T. Yu, X. Fan, H. Zhang, Z. Li, J. Ye and Z. Zou, Appl. Surf. Sci., 2007, 253, 8500 CrossRef CAS.
- Y. Takahara, J. Konde, N. T. Takata, D. Lu and K. Domen, Chem. Mater., 2001, 13, 1194 CrossRef CAS.
- J. Y. Kim, J. W. Jang, D. H. Youn, G. Magesh and J. S. Lee, Adv. Energy Mater., 2014, 4, 1400476 CrossRef.
- K. Sivula, F. Le Formal and M. Gratzel, ChemSusChem, 2011, 4, 432 CrossRef CAS PubMed.
- A. I. Hochbaum and P. Yang, Chem. Rev., 2010, 110, 527 CrossRef CAS PubMed.
- Y. J. Lin, G. B. Yaun, R. Liu, S. Zhou, S. W. Sheehan and D. W. Wang, Chem. Phys. Lett., 2011, 507, 209 CrossRef CAS.
- G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Chem. Commun., 2002, 1698 RSC.
- A. Kudo, K. Ueda, H. Kato and I. Mikami, Catal. Lett., 1998, 53, 229 CrossRef CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459 CrossRef CAS.
- X. Liu, F. Wang and Q. Wang, Phys. Chem. Chem. Phys., 2012, 14, 7894 RSC.
- E. Baniasadi, I. Diner and G. F. Naterer, Chem. Eng. Sci., 2012, 84, 638 CrossRef CAS.
- N. L. Wu and M. S. Lee, Int. J. Hydrogen Energy, 2004, 29, 1601 CrossRef CAS.
- S. Sakthivel, M. V. Shankar, M. Palanichamy, B. Arabindoo, D. W. Bahnemann and V. Murugesan, Water Res., 2004, 38, 3001 CrossRef CAS PubMed.
- S. Kim and W. Choi, J. Phys. Chem. B, 2002, 106, 13311 CrossRef CAS.
- S. Jin and F. Shiraishi, Chem. Eng. J., 2004, 97, 203 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. Kamat, J. Am. Chem. Soc., 2004, 126, 4943 CrossRef CAS PubMed.
- I. H. Tseng, J. C. S. Wu and H. Y. Chou, J. Catal., 2004, 221, 432 CrossRef CAS.
- S. X. Liu, Z. P. Qu, X. W. Han and C. L. Sun, Catal. Today, 2004, 93–95, 877 CrossRef CAS.
- P. V. Kamat, M. Flumiani and A. Dawson, Colloids Surf., A, 2002, 202, 269 CrossRef CAS.
- E. S. Bardos, H. Czili and A. Horvath, J. Photochem. Photobiol., A, 2003, 154, 195 CrossRef.
- K. A. Johnson and N. W. Asheroft, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 15548 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- (a) J. P. Perdew and W. Yue, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8800 CrossRef; (b) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982 CrossRef CAS.
- I. A. Vladimir, F. Aryasetiawan and A. I. Lichtenstein, J. Phys.: Condens. Matter, 1997, 9, 767 CrossRef.
- (a) F. Aryastiawan and O. Gunnarsson, Rep. Prog. Phys., 1998, 61, 237 CrossRef; (b) C. Franchini, R. Kovaik, M. Marsman, S. S. Murthy, J. He, C. Ederer and G. Kresse, J. Phys.: Condens. Matter, 2012, 24, 235 Search PubMed; (c) J. Y. Gou, J. W. Benett, H. Takenaka and A. M. Rappe, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 205115 CrossRef.
- M. Law, L. E. Greene, J. C. Johnson, R. Saykally and P. D. Yang, Nat. Mater., 2005, 4, 455 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. S. Guo and S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- Y. Moriya, T. Takatab and K. Domen, Coord. Chem. Rev., 2013, 257, 1957 CrossRef CAS.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141 CrossRef CAS.
- O. Akhavan and R. Azimirad, Appl. Catal., A, 2009, 369, 62 CrossRef.
- X. Li, Y. Hou, Q. Zhao, W. Teng, X. Hu and G. Chen, Chemosphere, 2011, 82, 581 CrossRef CAS PubMed.
- E. Zintl and L. Varino, Process for the manufacture of pure bismuth vanadate, German Patent, 422947, 1925.
- J. Tang, Z. Zou and J. Ye, Angew. Chem., Int. Ed., 2004, 43, 4463 CrossRef CAS PubMed.
- J. Tang, Z. Zou and J. Ye, Chem. Mater., 2004, 16, 1644 CrossRef CAS.
- K. Kaur and C. V. Singh, Energy Procedia, 2012, 29, 291 CrossRef CAS.
- A. Reynal, J. Willkomm, N. M. Muresan, F. Lakadamyali, M. Planells, E. Reisner and J. R. Durrant, Chem. Commun., 2014, 50, 12768 RSC.
- G. Geneste, E. Bousquet, J. Junquera and P. Ghosez, Appl. Phys. Lett., 2006, 88, 112906 CrossRef.
- G. Pilania, S. P. Alpay and R. Ramprasad, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 014113 CrossRef.
- T. Shimada, S. Tomoda and T. Kitamura, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 024102 CrossRef.
- G. Pilania and R. Ramprasad, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 155442 CrossRef.
- G. Pilania and R. Ramprasad, J. Mater. Sci., 2012, 47, 7580 CrossRef CAS.
- Q. Fu, T. He, J. L. Li and G. W. Yang, J. Appl. Phys., 2012, 111, 124306 CrossRef.
- Q. Fu, T. He, J. L. Li and G. W. Yang, J. Appl. Phys., 2012, 112, 104322 CrossRef.
- Q. Fu, T. He, J. L. Li and G. W. Yang, J. Appl. Phys., 2013, 113, 104303 CrossRef.
- A. V. Bandura, R. A. Evarestov and Y. F. Zukovskii, RSC Adv., 2015, 5, 24115 RSC.
- Y. F. Zhukovskii, S. Piskunov, J. Begens, J. Kazerovskis and O. Lisovski, Phys. Status Solidi B, 2013, 250, 793 CrossRef CAS.
- N. Daude, C. Gout and C. Jouanin, Phys. Rev. B: Solid State, 1977, 15, 3229 CrossRef CAS.
- K. M. Reddy, S. V. Manorama and A. R. Reddy, Mater. Chem. Phys., 2003, 78, 239 CrossRef.
- T. Morikawa, R. Asahi, T. Ohwaki, K. Aoki and Y. Taga, Jpn. J. Appl. Phys., Part 2, 2001, 40, 561 Search PubMed.
- A. Sclafani and J. M. Herrmann, J. Phys. Chem., 1996, 100, 13655 CrossRef CAS.
- H. Kominami, Y. Ishii, M. Kohno, S. Konishi, Y. Kera and B. Ohtani, Catal. Lett., 2003, 91, 41 CrossRef CAS.
- B. Ohtani, J. Handa, S. Nishimoto and T. Kagiya, Chem. Phys. Lett., 1985, 120, 292 CrossRef CAS.
- H. Hu, H. L. Tsai and C. L. Huang, J. Eur. Ceram. Soc., 2003, 23, 691 CrossRef.
- H. Wang and J. P. Lewis, J. Phys.: Condens. Matter, 2006, 18, 421 CrossRef CAS.
- D. Hanaor, M. Michelazzi, J. Chenu, C. Leonelli and C. C. Sorrell, J. Eur. Ceram. Soc., 2011, 31, 2877 CrossRef CAS.
- G. Liu, L. Wang, H. G. Yang, H. M. Cheng and G. Q. M. Lu, J. Mater. Chem., 2009, 20, 831 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2008, 38, 253 RSC.
- A. Burns, W. Li, C. Baker and S. I. Shah, Mater. Res. Soc. Symp. Proc., 2002, 703, 5.2.1 Search PubMed.
- M. Batzill, E. H. Morales and U. Diebold, Phys. Rev. Lett., 2006, 96, 26103 CrossRef PubMed.
- B. Xin, Z. Ren, P. Wang, L. Jing, H. Fu and J. Liu, Appl. Surf. Sci., 2007, 253, 4390 CrossRef CAS.
- B. Sun, A. V. Vorontsov and P. G. Smirniotis, Langmuir, 2003, 19, 3151 CrossRef CAS.
- K. Nagaveni, M. S. Hegde, N. Ravishankar, G. N. Subbanna and G. Madrass, Langmuir, 2004, 20, 2900 CrossRef CAS PubMed.
- N. Serpone, J. Phys. Chem. B, 2006, 110, 24287 CrossRef CAS PubMed.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, Appl. Phys. Lett., 2002, 81, 454 CrossRef CAS.
- K. V. Baiju, C. P. Sibu, K. Rajesh, P. K. Pillai, P. Mukundan, K. G. K. Warrier and W. Wunderlich, Mater. Chem. Phys., 2005, 90, 123 CrossRef CAS.
- D. Kim, T. Kim and K. Hong, Mater. Res. Bull., 1999, 34, 771 CrossRef CAS.
- J. Kim, K. C. Song, S. Foncillas and S. Pratsinis, J. Eur. Ceram. Soc., 2001, 21, 2863 CrossRef CAS.
- D. J. Reidy, J. D. Holmes, C. Nagle and M. A. Morris, J. Mater. Chem., 2005, 3494 RSC.
- S. D. Sharma, D. Singh, K. Saini, C. Kant, V. Sharma, S. C. Jain and C. P. Sharma, Appl. Catal., A, 2006, 314, 40 CrossRef.
- D. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855 CrossRef CAS.
- K. J. D. Mackenzie, Trans. J. Br. Ceram. Soc., 1975, 74, 77 CAS.
- D. A. H. Hanaor, M. H. N. Asadi, S. Li, A. Yu and C. S. Sorrell, Comput. Mech., 2012, 50, 185 CrossRef.
- Y. Takashi and Y. Mastuoka, J. Mater. Sci., 1988, 23, 2259 CrossRef.
- J. C. Yu, J. Yu, W. Ho, Z. Jiang and L. Zhang, Chem. Mater., 2002, 14, 3808 CrossRef CAS.
- C. Di Valentin, G. Pacchioni, H. Onishi and H. Kudo, Chem. Phys. Lett., 2009, 469, 166 CrossRef CAS.
- C. Di Valentin, E. Finazzi, G. Pacchioni, A. Selloni, S. Livarghi, M. C. Paganini and E. Giamello, Chem. Phys., 2007, 339, 44 CrossRef CAS.
- J. G. Yu, Q. J. Xiang, M. H. Zhou and X. Y. Zhou, Appl. Catal., B, 2009, 90, 595 CrossRef CAS.
- R. Long and N. J. English, J. Phys. Chem. C, 2010, 114, 11984 CAS.
- J. W. Zheng, A. Bhattcahrayya, P. Wu, Z. Chen, J. Highfield, Z. L. Dong and R. Xu, J. Phys. Chem. C, 2010, 114, 7063 CAS.
- X. L. Nie, S. P. Zhou, G. Maeng and K. Sohlberg, Int. J. Photoenergy, 2009, 294042 Search PubMed.
- Y. Cui, H. H. Du and L. S. Wen, J. Mater. Sci. Technol., 2008, 24, 675 CAS.
- H. W. Peng, J. B. Li, S. S. Li and J. B. Xia, J. Phys.: Condens. Matter, 2008, 20, 125207 CrossRef.
- A. Iwaszuk and M. Nolan, J. Phys. Chem. C, 2011, 115, 12995 CAS.
- M. Liu, L. Piao, L. Zhao, S. Ju, Z. Yan, T. He, C. Zhou and W. Wang, Chem. Commun., 2010, 46, 1664 RSC.
- W. Yang, J. Li, Y. Wang, F. Zhu, W. Shi, F. Wan and D. Xu, Chem. Commun., 2011, 47, 1809 RSC.
- L. Gu, J. Wang, H. Cheng, Y. Du and X. Han, Chem. Commun., 2012, 48, 6978 RSC.
- J. Qiu, S. Zhang and H. Zhao, J. Hazard. Mater., 2012, 211–212, 381 CrossRef CAS PubMed.
- C. Aprile, A. Corma and H. Garcia, Phys. Chem. Chem. Phys., 2008, 10, 769 RSC.
- C. X. Kronawitter, L. Vayssieres, S. Shen, L. Guo, D. A. Wheeler, J. Z. Zhang, B. R. Antoun and S. S. Mao, Energy Environ. Sci., 2011, 4, 3889 CAS.
- F. A. Frame, T. K. Townsend, R. L. Chamousis, E. M. Sabio, Th. Dittrich, N. D. Browning and F. E. Osterloh, J. Am. Chem. Soc., 2011, 133, 7264 CrossRef CAS PubMed.
- M. Law, L. E. Greene, J. C. Johnson, R. Saykally and P. Yang, Nat. Mater., 2005, 4, 455 CrossRef CAS PubMed.
- Y. Cao, T. He, Y. Chen and Y. Cao, J. Phys. Chem. C, 2010, 114, 3627 CAS.
- L. Kong, Z. Jiang, T. Xiao, L. Lu, M. Jones and P. P. Edwards, Chem. Commun., 2011, 47, 5512 RSC.
- H. Cheng, B. Huang, Y. Dai, X. Qin and X. Zhang, Langmuir, 2010, 26, 6618 CrossRef CAS PubMed.
- S. Hong, S. Lee, J. Jang and J. Lee, Energy Environ. Sci., 2011, 4, 1781 CAS.
- T. Cao, Y. Li, C. Wang, Z. Zhang, M. Zhang, C. Shao and Y. Liu, J. Mater. Chem., 2011, 21, 6922 RSC.
- V. Bharat, R. Boppana and R. F. Lobo, ACS Catal., 2011, 1, 923 CrossRef.
- J. Libera, J. Elam, N. Sather, T. Rajh and N. M. Dimitrijevic, Chem. Mater., 2010, 22, 409 CrossRef CAS.
- H. Tada, Q. Jin, H. Nishijima, H. Yamamoto, M. Fujishima, S.-I. Okuoka, T. Hattori, Y. Sumida and H. Kobayashi, Angew. Chem., Int. Ed., 2011, 50, 3501 CrossRef CAS PubMed.
- Q. Jin, M. Fujishima and H. Tada, J. Phys. Chem. C, 2011, 115, 6478 CAS.
- Q. Jin, Y. Ikeda, M. Fujishima and H. Tada, Chem. Commun., 2011, 47, 8814 RSC.
- M. Fujishima, Q. Jin, H. Yamamoto, H. Tada and M. Nolan, Phys. Chem. Chem. Phys., 2012, 14, 705 RSC.
- Q. Jin, M. Fujishima, M. Nolan, A. Iwaszuk and H. Tada, J. Phys. Chem. C, 2012, 116, 12621 CAS.
- M. Nolan, Phys. Chem. Chem. Phys., 2011, 13, 18194 RSC.
- A. Iwaszuk and M. Nolan, Phys. Chem. Chem. Phys., 2011, 13, 4963 RSC.
- M. Nolan, ACS Appl. Mater. Interfaces, 2012, 4, 5863 CAS.
- E. P. Reddy, L. Davydov and P. Smirniotis, Appl. Catal., B, 2003, 42, 1 CrossRef CAS.
- M. Hirano, K. Ota and H. Iwata, Chem. Mater., 2004, 16, 3725 CrossRef CAS.
- W. Wang, C. G. Silva and J. L. Faria, Appl. Catal., B, 2007, 70, 470 CrossRef CAS.
- T. Torimoto, Y. Okawa, N. Takeda and H. Yoneyama, J. Photochem. Photobiol., A, 1997, 103, 153 CrossRef CAS.
- C. H. Ao and S. C. Lee, Appl. Catal., B, 2003, 44, 191 CrossRef CAS.
- J. Arana, J. M. Doña-Rodríguez, E. Tello Rendón, C. Garriga, I. Cabo, O. González-Díaz, J. A. Herrera-Melián, J. Pérez-Peña, G. Colón and J. A. Navío, Appl. Catal., B, 2003, 44, 161 CrossRef CAS.
- P. Fu, Y. Luan and X. Dai, J. Mol. Catal. A: Chem., 2004, 221, 81 CrossRef CAS.
- X. Zhang, M. Zhou and L. Lei, Carbon, 2005, 43, 1700 CrossRef CAS.
- A. Jitianu, T. Cacciaguerra, R. Benoit, S. Delpeux, F. Béguin and S. Bonnamy, Carbon, 2004, 42, 1147 CrossRef CAS.
- H. Huang, W. K. Zhang, X. P. Gan, C. Wang and L. Zhang, Mater. Lett., 2007, 61, 296 CrossRef CAS.
- I. Moriguchi, R. Hidaka, H. Yamada, T. Kudo, H. Murakami and N. Nakashima, Adv. Mater., 2006, 18, 69 CrossRef CAS.
- S. R. Jang, R. Vittal and K.-J. Kim, Langmuir, 2004, 20, 9807 CrossRef CAS PubMed.
- Y. Yao, G. Li, S. Ciston, R. M. Lueptow and K. A. Gray, Environ. Sci. Technol., 2008, 42, 4952 CrossRef CAS PubMed.
- B. Liu and H. C. Zeng, Chem. Mater., 2008, 20, 2711 CrossRef CAS.
- B. Gao, G. Z. Chen and P. G. Li, Appl. Catal., B, 2009, 89, 503 CrossRef CAS.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233 CrossRef CAS.
- D. Wang, D. Choi, J. Li, Z. Yang, Z. Nie, R. Kou, D. Hu, C. Wang, L. V. Saraf and J. Zhang, ACS Nano, 2009, 3, 907 CrossRef CAS PubMed.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2009, 4, 380 CrossRef PubMed.
- Y.-B. Tang, et al., ACS Nano, 2010, 4, 3482 CrossRef CAS PubMed.
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car and R. K. Prud’homme, Chem. Mater., 2007, 19, 4396 CrossRef CAS.
- N. Farhangi, Y. Medina-Gonzalez, R. R. Chowdhury and P. A. Charpentier, Nanotechnology, 2012, 23, 294005 CrossRef PubMed.
- N. Farhangi, R. R. Chowdhury, Y. Medina-Gonzalez, M. B. Ray and P. A. Charpentier, Appl. Catal., B, 2011, 110, 25 CrossRef CAS.
- B. Das, B. Choudhury, A. Gomathi, A. K. Manna, S. K. Pati and C. N. R. Rao, ChemPhysChem, 2011, 12, 937 CrossRef CAS PubMed.
- S. K. Deb, Sol. Energy Mater. Sol. Cells, 2008, 92, 245 CrossRef CAS.
- S. K. Deb, Appl. Opt., 1969, 3, 192 CrossRef.
- S. K. Deb, Philos. Mag., 1973, 27, 801 CrossRef CAS.
- J. M. Berek and J. Sienko, J. Solid State Chem., 1970, 2, 109 CrossRef.
- M. A. Butler, R. D. Nasby and R. K. Quinn, Solid State Commun., 1976, 19, 1011 CrossRef CAS.
- D. E. Scaife, Sol. Energy, 1980, 25, 41 CrossRef CAS.
- A. J. Nozik, Annu. Rev. Phys. Chem., 1978, 29, 189 CrossRef CAS.
- G. R. Bamwenda and H. Arakawa, Appl. Catal., A, 2001, 210, 181 CrossRef CAS.
- L. Weinhardt, M. Blum, M. Bär, C. Heske, B. Cole, B. Marsen and E. L. Miller, J. Phys. Chem. C, 2008, 112, 3078 CAS.
- X. C. Song, E. Yang, G. Liu, Y. Zhang, Z. S. Liu, H. F. Chen and Y. Wang, J. Nanopart. Res., 2010, 12, 2813 CrossRef CAS.
- L. Zhou, J. Zhu, M. Yu, X. Huang, Z. Li, Y. Wang and C. Yu, J. Phys. Chem. C, 2010, 114, 20947 CAS.
- A. Hameed, M. A. Gondal and Z. H. Yamani, Catal. Commun., 2004, 5, 715 CrossRef CAS.
- M. Radecka, P. Sobas, M. Wierzbicka and M. Rekas, Phys. B, 2005, 364, 85 CrossRef CAS.
- X. F. Cheng, W. H. Leng, D. P. Liu, J. Q. Zhang and C. N. Cao, Chemosphere, 2007, 68, 1976 CrossRef CAS PubMed.
- H. Liu, T. Peng, D. Ke, Z. Peng and C. Yan, Mater. Chem. Phys., 2007, 104, 377 CrossRef CAS.
- B. Yang and V. Luca, Chem. Commun., 2008, 4454–4456 RSC.
- A. Enesca, A. Duta and J. Schoonman, Phys. Status Solidi A, 2008, 205, 2038 CrossRef CAS.
- K. M. Karuppasamy and A. Subrahmanyam, J. Phys. D: Appl. Phys., 2008, 41, 035302 CrossRef.
- P. Maruthamuthu, M. Ashokkumar, K. Gurunathan, E. Subramanian and M. V. C. Sastri, Int. J. Hydrogen Energy, 1989, 14, 525 CrossRef CAS.
- W. Erbs, J. Desilvestro, E. Borgarello and M. Gräzel, J. Phys. Chem., 1984, 88, 4001 CrossRef CAS.
- X. Chang, S. Sun, Y. Zhou, L. Dong and Y. Yin, Nanotechnology, 2011, 22, 265603 CrossRef PubMed.
- F. Wang, C. Di Valentin and G. Pacchioni, J. Phys. Chem. C, 2012, 116, 8901 CAS.
- C. Zhou, G. Chen, Y. X. Li, H. J. Zhang and J. Pei, Int. J. Hydrogen Energy, 2009, 34, 2113 CrossRef CAS.
- R. Shi, J. Lin, Y. J. Wang, J. Xu and Y. F. Zhu, J. Phys. Chem. C, 2010, 114, 6472 CAS.
- D. W. Hwang, H. G. Kim, J. S. Lee, J. Kim, W. Li and S. H. Oh, J. Phys. Chem. B, 2005, 109, 2093 CrossRef CAS PubMed.
- C. Diaz-Guerra, P. Umek, A. Gloter and J. Piqueras, J. Phys. Chem. C, 2010, 114, 8192 CAS.
- X. Liu, Y. Meng and X. Wang, IPCBEE, 2012, 38, 115 CAS.
- J. M. Lee, Y. B. Pyun, J. Yi, J. W. Choung and W. I. Park, J. Phys. Chem. C, 2009, 113, 19134 CAS.
- W. T. Song, J. Xie, S. Y. Liu, Y. X. Zheng, G. S. Cao and T. J. Zhu, et al., Int. J. Electrochem. Sci., 2012, 7, 2164 CAS.
- G. Ambrozic, S. D. Škapin, M. Zigon and Z. C. Orel, J. Colloid Interface Sci., 2010, 346, 317 CrossRef CAS PubMed.
- D. Fan, R. Zhang, X. Wang, S. Huang and H. Peng, Phys. Status Solidi A, 2012, 209, 335 CrossRef CAS.
- S. Music, A. Šarić and S. Popović, Ceram. Int., 2010, 36, 1117 CrossRef CAS.
- T. Singh, D. K. Pandya and R. Singh, Thin Solid Films, 2012, 520, 4646 CrossRef CAS.
- N. F. Ahmad, N. L. Rusli, M. R. Mahmood, K. Yasui and A. M. Hashim, Nanoscale Res. Lett., 2014, 9, 83 CrossRef PubMed.
- X. Pengtao, T. Qing and Z. Zhen, Nanotechnology, 2013, 24, 1 Search PubMed.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575 CrossRef CAS PubMed.
- S. Kanda, T. Akita, M. Fujishima and H. Tada, J. Colloid Interface Sci., 2011, 354, 607 CrossRef CAS PubMed.
- A. Wu, L. Jing, J. Wang, Y. Qu, Y. Xie, B. Jiang, C. Tian and H. Fu, Sci. Rep., 2015, 5, 88581 Search PubMed.
- E. Grabowska, et al., J. Phys. Chem. C, 2013, 117, 1955 CAS.
- X. Wang, et al., Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC.
- G. Liu, L. C. Yin, P. Niu, W. Jiao and H. M. Cheng, Angew. Chem., Int. Ed., 2013, 52, 6242 CrossRef CAS PubMed.
- G. Liu, P. Niu, L. Yin and H. M. Cheng, J. Am. Chem. Soc., 2012, 134, 9070 CrossRef CAS PubMed.
- F. Wang, et al., Appl. Catal., B, 2012, 111–112, 409 CrossRef CAS.
- X. Wang, et al., Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- J. Liu, et al., Angew. Chem., Int. Ed., 2013, 52, 3241 CrossRef CAS PubMed.
- K. S. Novoselov, et al., Science, 2004, 306, 666 CrossRef CAS PubMed.
- N. G. Chopra, et al., Science, 1995, 269, 966 CrossRef CAS PubMed.
- K. Watanabe, T. Taniguchi and H. Kanda, Nat. Mater., 2004, 3, 404 CrossRef CAS PubMed.
- L. Song, et al., Adv. Mater., 2012, 24, 4878 CrossRef CAS PubMed.
- L. Ci, et al., Nat. Mater., 2010, 9, 430 CrossRef CAS PubMed.
- J. Lu, et al., Nat. Commun., 2013, 4, 2681 Search PubMed.
- X. Li, J. Zhao and J. Yang, Sci. Rep., 2013, 3, 1858 CrossRef PubMed.
- L. Song, et al., Nano Lett., 2010, 10, 3209 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- X. An and J. C. Yu, RSC Adv., 2011, 1, 1426 RSC.
- H. Tang, C. M. Hessel, J. Wang, N. Yang, R. Yu, H. Zhao and D. Wang, Chem. Soc. Rev., 2014, 43, 4281 RSC.
- C. Huang, C. Chen, M. Zhang, L. Lin, X. Ye, S. Lin, M. Antonietti and X. Wang, Nat. Commun., 2015, 6, 76981 Search PubMed.
- (a) S. Kanda, T. Akita, M. Fujishima and H. Tada, J. Colloid Interface Sci., 2011, 354, 607 CrossRef CAS PubMed; (b) J. Zhang, J. G. Yu, Y. M. Zhang, Q. Li and J. R. Gong, Nano Lett., 2011, 11, 4774 CrossRef CAS PubMed; (c) J. Zhang, S. W. Liu, J. G. Yu and M. Jaroniec, J. Mater. Chem., 2011, 21, 14655 RSC; (d) J. Zhang, J. G. Yu, M. Jaroniec and J. R. Gong, Nano Lett., 2012, 12, 4584 CrossRef CAS PubMed.
- (a) M. Matsumura, S. Furukawa, Y. Saho and H. Tsubomura, J. Phys. Chem., 1985, 89, 1327 CrossRef CAS; (b) J. G. Yu, Y. F. Yu and B. Cheng, RSC Adv., 2012, 2, 11829 RSC; (c) Q. J. Xiang, B. Cheng and J. G. Yu, Appl. Catal., B, 2013, 138–139, 299 CrossRef CAS.
- J. G. Yu, J. Zhang and M. Jaroniec, Green Chem., 2010, 12, 1611 RSC.
- X. X. Yu, J. G. Yu, B. Cheng and B. B. Huang, Chem.–Eur. J., 2009, 15, 6731 CrossRef CAS PubMed.
- L. Wang, W. Z. Wang, M. Shang, W. Z. Yin, S. M. Sun and L. Zhang, Int. J. Hydrogen Energy, 2010, 35, 19 CrossRef CAS.
- C. Wu, J. Zheng, C. L. Zacherl, P. Wu, Z. K. Liu and R. Xu, J. Phys. Chem. C, 2011, 115, 19741 Search PubMed.
- C. J. Xing, Y. J. Zhang, W. Yan and L. Guo, Int. J. Hydrogen Energy, 2006, 31, 2018 CrossRef CAS.
- Q. Li, H. Meng, P. Zhou, Y. Zheng, J. Wang, J. Yu and J. Gong, ACS Catal., 2013, 3, 882 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782 RSC.
- N. S. A. Aziz, T. Nishiyama, N. I. Rusli, M. R. Mahmood, K. Yasui and A. M. Hashim, Nanoscale Res. Lett., 2014, 9, 337 CrossRef PubMed.
- N. S. A. Aziz, M. R. Mahmood, K. Yasui and A. M. Hashim, Nanoscale Res. Lett., 2014, 9, 95 CrossRef PubMed.
- M. Hilder, O. Winther-Jensen, B. Winther-Jensen and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2012, 14, 14034 RSC.
- J. Hou, C. Yang, H. Cheng, Z. Wang, S. Jiao and H. Zhu, Phys. Chem. Chem. Phys., 2013, 15, 15660 RSC.
- (a) J. M. Lee, Y. B. Pyun, J. Yi, J. W. Choung and W. I. Park, J. Phys. Chem. C, 2009, 113, 19134 CrossRef CAS; (b) S. Zhuang, X. Xu, B. Feng, J. Hu, Y. Pang, G. Zhou, L. Tong and Y. Zhou, ACS Appl. Mater. Interfaces, 2014, 6, 613 CrossRef CAS PubMed.
- B. L. Zhu, B. Z. Lin, Y. Zhou, P. Sun, Q. R. Yao, Y. L. Chen and B. F. Gao, J. Mater. Chem. A, 2014, 2, 3819 CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851 CAS.
- R. M. Navarro, M. C. Sanchez-Sanchez, M. C. Alvarez-Galvan, F. del Valle and J. L. G. Fierro, Energy Environ. Sci., 2009, 2, 35 CAS.
- X. Feng, W. Mao and W. Yan, Int. J. Hydrogen Energy, 2008, 33, 3644 CrossRef CAS.
- K. Domen, Y. Ebina, T. Sekine, A. Tanaka, J. Kondo and C. Hirose, Catal. Today, 1993, 16, 479 CrossRef CAS.
- Z. L. Jin and X. G. Lu, Energy Fuels, 2005, 19, 1126 CrossRef CAS.
- S. M. Ji, H. Jun, J. S. Jang, H. C. Son, P. H. Borse and J. S. Lee, J. Photochem. Photobiol., A, 2007, 189, 141 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |