
Multifunctional silica nanoparticles as a promising theranostic platform for biomedical applications
Zhigang
Xu
 *abc,
Xiaoqian
Ma
abc,
Yong-E.
Gao
abc,
Meili
Hou
abc,
Peng
Xue
abc,
Chang Ming
Li
abc and
Yuejun
Kang
*abc
*abc,
Xiaoqian
Ma
abc,
Yong-E.
Gao
abc,
Meili
Hou
abc,
Peng
Xue
abc,
Chang Ming
Li
abc and
Yuejun
Kang
*abc
aInstitute for Clean Energy and Advanced Materials, Faculty of Materials and Energy, Southwest University, Chongqing, 400715, P. R. China. E-mail: zgxu@swu.edu.cn; yjkang@swu.edu.cn; Fax: +86-68253204; Tel: +86-68254056
bChongqing Key Laboratory for Advanced Materials and Technologies of Clean Energies, Chongqing 400715, P. R. China
cChongqing Engineering Research Centre for Micro-Nano Biomedical Materials and Devices, Chongqing 400715, P. R. China
First published on 31st May 2017
Abstract
Theranostic nanoplatforms represent an important tool in the diagnosis and therapy of many major diseases, especially in the field of cancer theranostics, for which a myriad of nanoplatforms, such as polymeric micelles, liposomes and inorganic nanoparticles (NPs), have been designed and developed in recent decades. Among these nanoplatforms, silica NPs provide many advantages over other similar systems for therapeutics, bioimaging, bio-adhesives and many other biomedical applications. Therapeutic nanoplatforms based on mesoporous silica NPs (typically of pore size 2–50 nm) have been reviewed extensively. Herein, we provide an overview of recent advances in the development of theranostic nanoplatforms based on nonporous silica NPs within a size range of 1–200 nm for many critical biomedical applications, particularly stimuli-triggered drug delivery, precise cancer diagnosis as well as tissue bio-adhesive materials.
 Zhigang Xu | Dr Zhigang Xu was born in Henan, China, in 1985. He received his doctorate from the College of Chemistry and Chemical Engineering in 2013 from Lanzhou University, China, under the supervision of Professor Haixia Zhang. After postdoctoral work at the Nanyang Technological University, Singapore, he joined the Faculty of Materials and Energy at Southwest University, China, in 2015 as an Associate Professor. His research interests include stimuli-responsive polymeric prodrugs and small-molecule prodrugs for drug delivery, bioimaging and theranostic applications. |
 Xiaoqian Ma | Xiaoqian Ma was born in Gansu, China, in 1992. She received her BS (2016) from the College of Chemistry and Chemical Engineering at Northwest Normal University, China. She is a postgraduate student at the Faculty of Materials and Energy of Southwest University, China. Her research is focused on the design and synthesis of star-like diblock copolymers for bioimaging and drug delivery. |
 Yong-E. Gao | Yong-E. Gao was born in Shanxi, China, in 1991. She received her BS (2015) from the School of Chemistry and Materials Science of Datong University, China. She is a postgraduate student at the Faculty of Materials and Energy of Southwest University, China. Her research is focused on the synthesis of ultra-pH responsive diblock copolymers as a theranostic nanoplatform for cancer therapy. |
 Meili Hou | Meili Hou was born in Shanxi, China, in 1993. She received her BS (2015) from the School of Chemistry and Materials Science of Shanxi Normal University, China. She is a postgraduate student at the Faculty of Materials and Energy of Southwest University, China. Her research is focused on the synthesis of stimuli-responsive drug delivery systems based on small-molecule prodrugs for cancer therapy. |
 Chang Ming Li | Professor Chang Ming Li received his doctorate from Wuhan University, China, in 1986. He is the Director of the Institute for Clean Energy and Advanced Materials, Southwest University, China. He has also worked as a Professor and the Head of Bioengineering at Nanyang Technological University, China, and a Science Advisory Board Associate for Motorola. His research interests cover nanomaterials and applications in green energies and biosensors. He has received the prestigious Motorola Master Innovator Award, Nanyang Innovation and Research Award, and China Overseas Innovation Award. He is a Fellow of the Royal Society of Chemistry and Fellow of the American Institute of Medical and Biological Engineering. |
 Yuejun Kang | Professor Yuejun Kang received his Bachelor of Engineering degree from the University of Science and Technology of China in 2000 and his doctorate from Vanderbilt University, USA, in 2008. He is a Professor of Bioengineering at the Institute for Clean Energy and Advanced Materials, Faculty of Materials and Energy at Southwest University, China. His research interests include bionanotechnology and lab-on-a-chip devices for biosensing, tissue engineering, drug delivery nanosystems and wearable therapeutic microdevices. |
1. Introduction
Motivated by the mission to maintain human health against major diseases, scientists from multiple fields have shown increasing interest in therapeutic platforms based on nanomedicine, which can effectively alter the pharmacokinetics and biodistribution of pharmacologically active agents, such as drugs, and thus significantly improve therapeutic efficacy.1–6 Ideally, drug delivery is desirable to be under precise control in a physiological environment for higher therapeutic effects. Although the latest development of nanotechnology can address some inherent limitations of therapeutic nanoplatforms (e.g., small molecular scale, non-specificity, low stability, fast excretion and high toxicity), drug delivery via these nanoscale agents is still difficult to control in complex biological environments.7,8 Several critical issues, including a lack of stability in blood circulation, undesirable side effects, insufficient drug efficacy, and costly synthetic processes, are among the major limitations that hinder the clinical applications of therapeutic nanoplatforms.To resolve these issues, various types of therapeutic agents based on organic nanoplatforms (e.g., liposomes, protein–drug conjugates, dendrimers, polymeric prodrugs), inorganic nanoplatforms (e.g., silicon or carbon nanoparticles (NPs), quantum dots (QDs), rare metal particles or other functionalized nanostructures) and organic/inorganic hybrid platforms (e.g., polymer- or small molecule-modified inorganic nanostructures) have been extensively utilized to construct stimuli-responsive nanoplatforms by harnessing specific endogenous stimuli (pH, redox potential and enzymes) or extracorporeal physical stimuli (light, temperature and magnetic field) to improve the therapeutic efficacy and to “tailor” pharmacokinetics.9–19 Among these platforms, silica NP-based therapeutic platforms exhibit preferential characteristics, such as highly controllable size and shape, low toxicity, excellent biocompatibility, stability in physiological conditions and biosafety, for clinical applications.16,17 Notably, the renal system can efficiently excrete the degradation products of silica NPs, namely orthosilicic acid, from the human body.20,21 As an important family of silica NPs, mesoporous silica NPs have a silica matrix containing a porous structure with diameters from 2 to 50 nm and can carry active agents by physical adsorption or chemical binding.15 In contrast, nonporous silica NPs carry drugs mainly through embedding or drug–silane conjugation, which exhibits better biological safety than their mesoporous analogs.17 It is encouraging that a class of ultrasmall nonporous silica NPs was approved by the US Food and Drug Administration (FDA) for clinical cancer imaging in 2011,22,23 indicating the promising potential of nonporous silica NPs as a safe therapeutic platform for clinical application.
Tang and Cheng contributed a systematic review in 2013 on the development of nonporous silica NP-based nanomedicine and applications for drug delivery and molecular imaging.17 Four years have passed since their insightful and prospective overview in this intriguing field, and there have been increasing efforts and exciting new advances in recent years on the design, synthesis and applications of silica NPs as a capable theranostic platform.
Therefore, in this review, we aim to highlight the significant progress made over the past 4 years in the field of multifunctional nonporous silica NPs within the size range of 1–200 nm for stimuli-responsive therapeutics, imaging and tissue bio-adhesives. To keep this topical review brief and concise, we do not cover the advances made in mesoporous silica NPs, rod-like or other non-spherical shaped silica NPs, or other drug–laden silica hybrid platforms, which have been reviewed elsewhere.15,17,24–27 This review is expected to assist the broad scientific community to understand the current progress and existing challenges of using nonporous silica NP platforms for various biomedical applications, and to inspire new research efforts and interdisciplinary collaboration utilizing this particular nanoplatform.
2. Functional properties of nonporous silica NPs
2.1 Size control
For applications in nanomedicine, many critical properties of a nanoplatform are size-dependent, which can profoundly affect the systemic and lymphatic distribution, tumor penetration and cellular internalization of the drug, and thus affect the subsequent therapeutic efficacy. Most reports have demonstrated that the size of tumoral inter-endothelial junctions range from 40 to 80 nm, whereas those of healthy tissues are smaller than 8 nm.4,28,29 The preferential uptake of drug carriers due to this drastic size difference between tumoral and healthy tissues and the defective lymphatic drainage of nanocomplexes make it possible for passive targeting of tumoral sites via the enhanced permeability and retention (EPR) effect.30,31 However, small-cargo molecules are usually featured by a molecular weight less than 1500 Da and the size smaller than 2 nm,32 thereby they can easily escape from healthy or tumoral tissues through blood or renal systems. It is necessary to develop a carrier that can prevent the cargos from “leaking” and improve their retention. Therefore, size control of the cargo carrier is a critical strategy during the design and synthesis of EPR-mediated therapeutic nanoplatforms for nanomedicine.33–35Since Stöber first proposed an efficient method to produce nonporous silica NPs with accurate size control down to 20 nm,36 many research efforts have been inspired to design and synthesize various nonporous silica NPs as multifunctional therapeutic platforms for drug delivery, bioimaging and other biomedical applications.17,37,38 In particular, the size and morphology of silica NPs play a key part in determination of the material properties required for specific biomedical applications. Traditional nonporous silica NPs prepared following the Stöber method usually employ tetraethylorthosilicate (TEOS) as the silicon source, ammonia, and water as the initiator in an alcoholic system (Fig. 1A). By adjusting the proportion of water, TEOS and ammonia in ethanol solution, respectively, the size of the obtained silica NPs can be controlled from tens to hundreds of nanometers, which covers the regular size range suitable for various applications (Scheme 1).
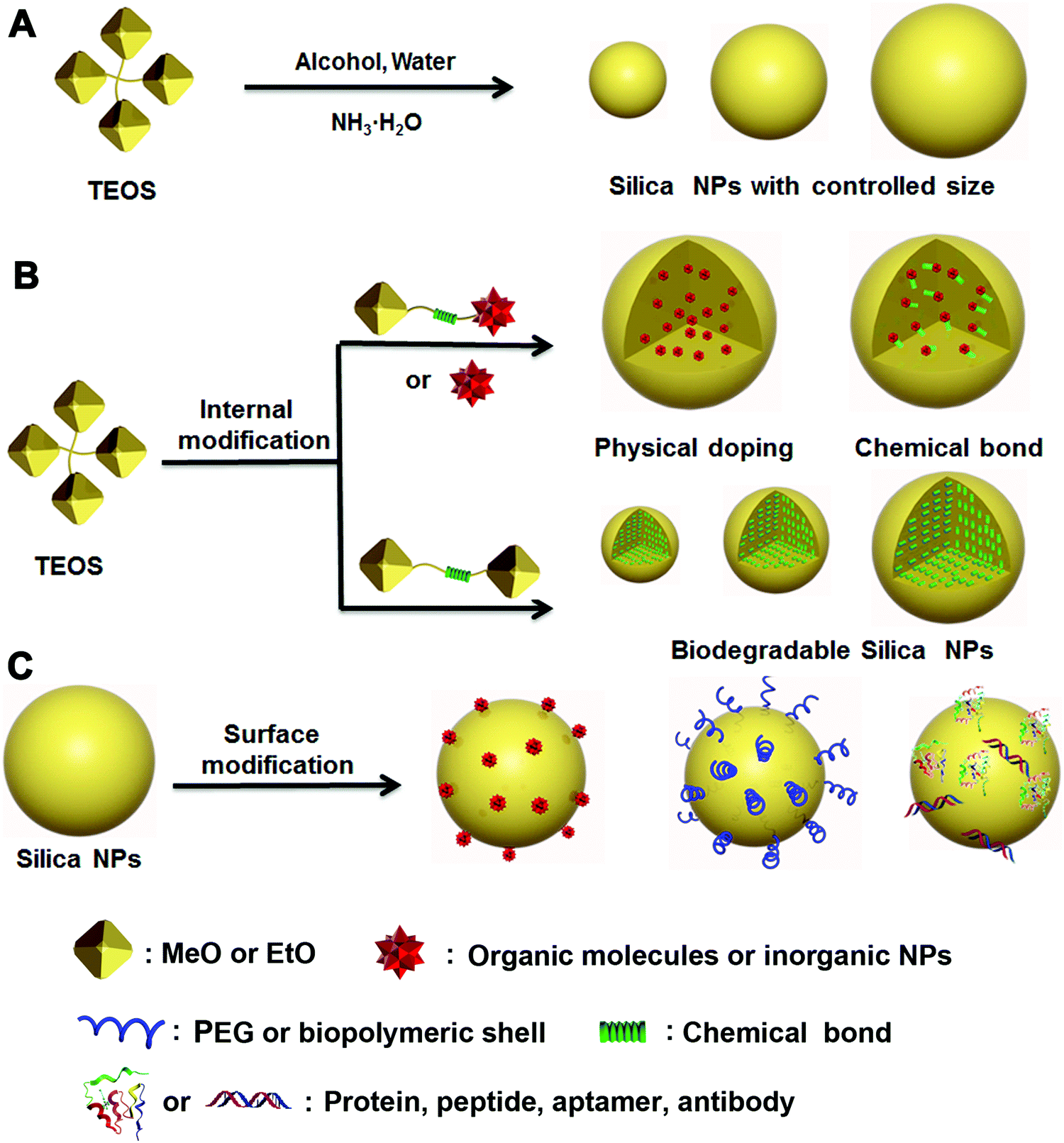 | ||
| Fig. 1 Schematic illustration of the design and synthesis of (A) traditional silica NPs prepared by the Stöber method using tetraethylorthosilicate (TEOS) as the lone silicon source; (B) silica NPs by internal modification using a modified version of the Stöber method that introduces cargo, such as organic or inorganic molecules and stimuli-responsive silane-bridging, within the silica NPs via physical doping or chemical bonding (e.g., disulfide bond and ester bond); (C) silica NPs with surface modification by silane chemistry via covalent conjugation or physical adsorption. | ||
 | ||
| Scheme 1 Schematic illustration of multifunctional nonporous silica nanoparticles for cargo release and cancer theranostics with on-demand function in one system. Silica NPs: silica nanoparticles; PEG: poly(ethylene glycol). | ||
To optimize the therapeutic nanoplatforms, it is important to understand the correlation between their physicochemical properties, especially size-dependent features, and the subsequent effect on biological systems. Recently, Cheng et al. systematically investigated the size effect (20, 50 and 200 nm) of a type of prodrug silica NPs for anticancer nanomedicine (Fig. 2A–D).39 The drug delivery was optimized through experiment and mathematical modelling. They investigated three NPs and found that the 50 nm prodrug silica NPs exhibited the highest retention rate in tumoral tissues and the highest efficacy against primary and metastatic tumours in vivo, which could be due to the cumulative contribution of efficient cancer cell internalization and deep penetration of tumoral tissue. However, it was still unclear if 50 nm was the optimal size for other similar polyethylene glycol (PEG)ylated nanoplatforms.
 | ||
| Fig. 2 (A) Schematic illustration of size-controlled prodrug NCs based on silica NPs using a silica prodrug monomer of 1. (B) A micro-PET/computed tomography imaging system was used to obtain whole-body images, and the yellow arrows and circles indicate the tumor site. (C) A model of a metastatic tumor was used to access size-dependent anticancer efficacy. (D) A clearance study of a MCF-7 tumor in vitro: Rhd-NCs of different sizes were first used to incubate MCF-7 tumor cells in opti-MEM in vitro for 24 h and then the clearance process of Rhd-NCs from the tumors was monitored after immersion in fresh opti-MEM. CLSM images of Rhd-NCs (red) in tumor sites after 48 h of incubation. Scale bar: 50 μm. Reprinted with permission from ref. 39. Copyright 2015 National Academy of Sciences. | ||
2.2 Functionalization
The modified Stöber method was later introduced to prepare functionalized silica NPs through two major strategies: internal and surface modifications (Fig. 1B and C). Various types of cargos, such as organic or inorganic molecules, can be introduced to the interior of silica NPs by “physical doping”, which is a simple and convenient method.40–46 However, the doped materials, especially small organic molecules, can easily escape from the interior of silica NPs, thereby limiting their drug-loading efficiency. To address this issue, researchers have modified small organic molecules with covalently conjugated triethoxysilane groups and then bonded them with TEOS during the doping process. The resultant silica NPs were highly stable and able to prevent the leakage of small molecules from within. As a more advanced strategy of internal functionalization, biodegradable silica NPs based on stimuli-responsive silane-bridging (Fig. 1C) were recently developed to realize controllable drug release.47Because the surface chemical and physical properties of NPs affect their interactions with biological systems, such as cellular internalization, trafficking and biodistribution of NPs,48–50 surface modification is usually essential to tailor the unique properties of silica NPs for specific biomedical applications. Specifically, the silica NP surface can be functionalized by covalent conjugation or physical adsorption (Fig. 1D) through silane chemistry.51–55 For example, native silica NPs usually have negative surface charge, but it can be converted to a positive surface charge by adding amine containing silane agents such as 3-aminopropyltriethoxysilane. In fact, many commercial silane agents could be employed to obtain silica NPs grafted with various functional ligands using these methods.56
Studies have shown that many organic molecules (e.g., drugs, dyes) and inorganic NPs (e.g., Au NPs, QDs) can be grafted onto the surface of silica NPs for functionalization.57–61 Francesca et al.48 found that silica NPs modified with amino and phosphate groups on the surface could mitigate the proinflammatory and immunomodulatory issues for the treatment of allergic airway inflammation. Silica NPs can also be coated with PEG, protein or other biopolymers by physical adsorption or chemical modification to improve their biocompatibility and degradation rate.62,63 Other advanced polymerization methods, such as reversible addition–fragmentation chain transfer (RAFT), atom-transfer radical polymerization (ATRP), click polymerization, and ring-opening polymerization (ROP), have been developed to prepare silica–polymer hybrids for various biomedical applications.64,65 For example, Li and co-workers grafted poly(N,N-dimethylaminoethyl methacrylate) onto the surface of silica NPs via tandem linking reaction and RAFT polymerization, for use as stimuli-responsive emulsifiers and antibacterial materials.64 Moreover, small biomolecules, such as peptides, aptamers and antibodies, can be conjugated onto the surface of silica NPs for diagnostic and therapeutic purposes.66–69
2.3 Biocompatibility and stability
Good biocompatibility and stability are among the most critical features of a therapeutic platform for safe and effective applications.70 Usually, surface shells comprising functional polymers, charged molecules or other biocompatible materials are grafted onto the NP surface to avoid aggregation and maintain stability.48,71–73 For example, the negatively charged surface of native silica NPs can be decorated with a positively charged shell to increase the dispersibility of silica NPs in aqueous environments.48 Functional polymeric shells such as PEG are also commonly used to improve biocompatibility and stability, and thus prolong the circulation time of silica NPs in peripheral blood.45The excretion pathway of a nanoplatform from the body after completion of drug release is another important consideration for nanomedicine. The threshold size for natural renal clearance ranges from 10 to 15 nm,74 which is a very narrow window for excretion of regular NP-based drug platforms. Although many research efforts have been inspired to design degradable therapeutic platforms, degradation of native silica NPs usually takes long time and may overload the metabolic system before they are harvested and excreted through renal clearance.21,75,76 In many recent applications for cancer therapy, scientists have attempted to optimize the degradation rate of drug carriers, and control the final products and their excretion pathways under specific stimuli.47,77 For instance, glutathione (GSH), a thiol-containing tripeptide, is the most abundant non-protein thiol in biological cells and can reduce disulfide bonds. GSH exhibits a significantly higher intracellular concentration in tumor cells (2–8 mM) than in plasma (1–2 mM) and thus can be used to control the degradation of GSH-responsive silica NPs.47
3. Applications in drug delivery
3.1 Drug-loading strategy
The essential features of a desirable drug delivery platform include controllable size, easy surface modification, excellent biocompatibility, low toxicity, high stability and sufficient biosafety in biological systems.78–81 Generally, cargos including drugs have been introduced into the interior or onto the surface of silica NPs by different strategies, based mainly on the weak binding mechanisms (e.g., hydrogen bond, intermolecular interaction) or strong binding mechanisms (e.g., chemical bond, coordination bond) depending on the particular feature of the cargos. These strategies have been used to design many silica-based therapeutic nanoplatforms with special functionality for drug delivery and cancer therapy.82–98 To reduce the side effects, stimuli-responsive chemical bonds (e.g., ester bond, hydrazone bond and disulfide bond) have been widely used to load cargo into or onto the mesoporous silica NPs,15 whereas the ester bond and amido bond have been applied mainly to nonporous silica NPs.85–87,94Generally, drugs can be loaded onto nonporous silica NPs following three methods (Fig. 3). The first method is based on physical incorporation (Fig. 3, type 1), by which the cargo (e.g., pristine small molecules of drug or dye) is introduced into the silica NPs during the particle formation process. Because NP formation usually involves a sol–gel process including hydrolysis and condensation reactions, the small drug molecules can be securely encapsulated into the interior of silica NPs by controlling the amount of TEOS. However, it is difficult for the cargo to be released from the interior of silica–cargo NPs under a physiological (pH = 7.4) condition or stimulation due to the strong and dense structure of silica gel. Therefore, this type of silica–cargo NPs is mainly used for imaging applications.22 The major limitation of such drug-silica complexes lies in the lack of controllability and specificity during drug release, which may cause considerable side effects or insufficient efficacy for administration of anticancer drugs.
 | ||
| Fig. 3 Schemes of the loading of drug cargos on nonporous silica NPs and the proposed mechanisms of cargo release. Type 1: physical incorporation; type 2: surface conjugation; type 3: modified silane chemistry by introduction of a cargo monomer. | ||
Surface conjugation is another important strategy to prepare silica–cargo NPs (Fig. 3, type 2). Typically, cargos are modified with siloxane groups and then conjugated onto the surface of silica NPs. The chemical bond between the cargo molecules and siloxane group can be adjusted to maintain the integrity of the complex in physiological conditions and prevent the cargo from shedding from the silica NPs. If the chemical bond is stimuli-responsive (e.g., ester bond, disulfide bond and hydrazine bond), the loaded cargo can be released in a controlled fashion from the surface of the NP carrier. Although surface drug loading is relatively easier than the physical incorporation method, the major concern of this delivery system is the long-term stability under a complex physiological environment in vivo, which may cause undesired drug leakage and thereby reduce the therapeutic effect. Some biological polymers, such as PEG, have been applied to protect the functionalized surface of silica NPs,45 but it cannot guarantee the stability of silica NPs in vivo.
The third strategy takes advantage of both of the methods stated above by using modified silane chemistry during cargo encapsulation to prepare stimuli-responsive nonporous silica NPs (Fig. 3, type 3). Instead of physical incorporation of cargo molecules, stimuli-responsive cargo monomers with siloxane groups are designed first.82–85,97 After hydrolysis and condensation processes in the presence of TEOS, the cargo monomers are introduced uniformly during formation of silica NPs, resulting in stimuli-responsive silica prodrug NPs. Such formed nanocomplexes can effectively protect the encapsulated cargo whereas the carrier decomposes after the drug load is released. These silica–cargo NPs are very stable because the cargo is conjugated in the interior of the NPs by stimuli-responsive chemical bonds. In the presence of external stimuli (e.g., esterase for the ester bond, GSH for the disulfide bond and low pH for the hydrazone bond) that can diffuse into the interior of silica–cargo NPs, the chemical bond breaks, leading to decomposition of the silica–cargo NPs.47,86 This method can address the major issues of the other two methods mentioned above and provide a rational design to prepare stimuli-responsive therapeutic platforms for cancer therapy and other biomedical applications.
3.2 Non-stimuli-responsive drug silica NPs
Physical incorporation based on the conventional Stöber method is a common strategy that has been widely used to prepare various types of silica–drug NPs.96,98 This approach can work effectively with most types of cargos by introducing them into silica NPs. For example, Li and co-workers encapsulated native organic cargo molecules (doxorubicin hydrochloride (DOX) and methyl blue) into a “sloopy” silica–cargo nanostructure obtained by reducing the ratio of TEOS during synthesis (Fig. 4). The released cargo from the interior of silica NPs could induce decomposition of the carrier (Fig. 4A), which was verified by tracking the time-dependent morphological change of silica NPs using transmission electron microscopy (Fig. 4B). It was noted that the decomposition mechanism of the silica carrier was dependent on the release mode of encapsulated drug cargos. As shown in Fig. 4A and B, this unique structural feature exhibited a radial drug concentration gradient with highly concentrated drug molecules in the NP center, which led to diffusion-driven drug release and induced simultaneous carrier decomposition from center outwards. These silica–cargo NPs exhibited good cellular uptake efficiency in live H1299 cells through endocytosis (Fig. 4C). On the other hand, the drug release from such NPs is mainly dependent on the diffusion mechanism, which usually suffers from nonspecific cargo leakage during drug administration.96 | ||
| Fig. 4 (A) Schematic illustration of the structure of silica–cargo NPs and the proposed mechanism of drug release and self-decomposition of the carrier; (B) the morphological change of silica–MB NPs after drug release and self-decomposition imaged by TEM. The silica–MB NPs (a, c and e) were prepared fresh as the control and the morphological change of NPs after incubation in DI water for 48 h noted (b, d and f). Samples of (a–f) denote a different MB loading amount. The scale bars are 100 nm, 200 nm and 2 μm, respectively; (C) CLSM images of H1299 cells incubated with silica–MB NPs for 24 h (a) and 48 h (c). Transmittance images of (b) and (d) were consistent with the red fluorescence signal from MB. (e) The released amount of MB organic molecules in cell cytoplasm after incubation with dense SiO2–MB and self-decomposable NPs for 24 h and 48 h, respectively. Reprinted with permission from ref. 96. Copyright 2013 American Chemical Society. | ||
3.3 Stimuli-responsive prodrug silica NPs
Different from conventional drug delivery modalities relying mainly on passive diffusion, stimuli-responsive drug delivery systems work only under specific internal stimuli (e.g., pH, glutathione, enzymes) or external stimuli (e.g., light, temperature). Stimuli-responsive drug delivery systems have attracted broad attention recently due to many advantageous characteristics for controlled release, lower toxicity, biocompatibility and biodegradability.81 It has been found that tumoral tissues and cells usually exhibit a higher GSH concentration (2–10 mM) and lower pH (≈5.0) compared with healthy tissues or blood plasma, which have inspired drug delivery platforms that are sensitive to these particular physiological cues for efficient cancer therapy. Several stimuli-responsive nanoplatforms, including polymeric micelles and mesoporous silica NPs, have been reported for drug delivery and other biomedical applications.99–103 However, very few reports have studied similar functionalities of nonporous silica NPs.85,94,97 Usually, stimuli-responsive nanoplatforms based on nonporous silica NPs can be realized by surface modification or silane chemistry using stimuli-responsive cargo monomers.In the first strategy, the stimuli-responsive chemical bond is functionalized onto the surface of silica NPs and then grafted with the cargo molecules. Kong and co-workers reported a pH-responsive drug delivery system based on fluorescent nonporous silica NPs (DOX-Hyd@FSiNPs)94 which incorporated a fluorescein isothiocyanate (FITC) fluorophore within the NPs and conjugated DOX on the surface via hydrazone groups (Fig. 5). This system ensured that most of the loaded drugs could be selectively released under acidic environment (pH ≈ 5.0) instead of physiological condition (pH ≈ 7.4) due to the pH-responsive hydrazone bond. To further enhance drug-loading efficiency and biocompatibility, stimuli-responsive polymer-coated silica NPs have also been developed. For example, Bhosale et al. recently reported a type of magnetic silica NPs with a yoctowell cavity derived from porphyrin, which could realize pH-triggered release of mitoxantrone (MTZ) from yoctowell cavities and achieve targeted delivery due to the magnetic property.104
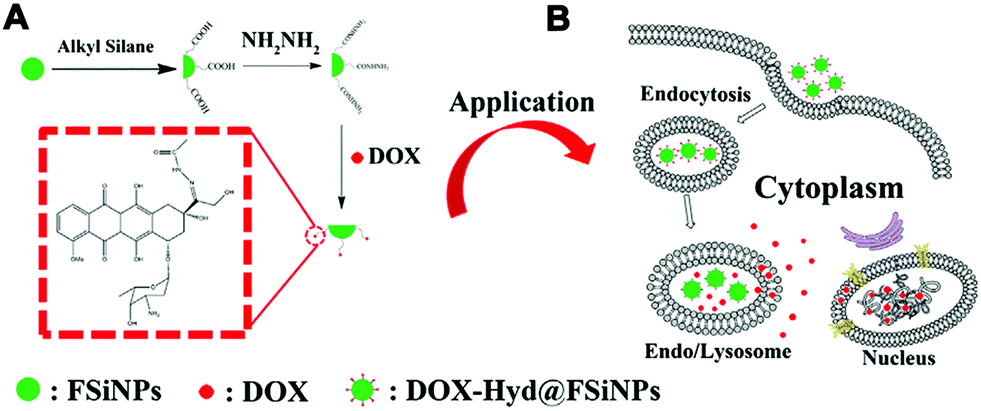 | ||
| Fig. 5 Schematic of (A) synthesis of DOX-Hyd@FSiNPs and (B) the intracellular release mechanism of DOX under an acidic environment in a cancer cell. Reprinted with permission from ref. 94. Copyright 2015 Elsevier. | ||
On the other hand, post-modification of cargo on the surface of silica NPs usually suffers from insufficient cargo grafting, lower drug loading capacity and stability. To address this issue, Cheng et al. developed monodisperse drug–silica nanoconjugates (NCs) of sizes from 20 to 200 nm using a series of cargo (drug or dye molecules) monomers with siloxane groups,82–86 which were introduced into silica NCs via an enzyme-responsive ester bond (Fig. 6). Further experiments showed that 20 nm NCs exhibited excellent tumor accumulation and cellular internalization, which was 2–5 and 10–20 folds higher than those of 50 nm and 200 nm NC analogs, respectively. Both in vitro and in vivo assays showed that the blank silica NPs induced minimal toxicity and the mice model injected with high dosages of NPs exhibited no mortality or deterioration, indicating the promising potential of this drug delivery platform. The efforts to optimize the size of silica nanocarriers are ongoing, which may favorably influence the performance of silica NP-based drug delivery platforms for nanomedicine.
 | ||
| Fig. 6 (A) The chemical structure of silica prodrug monomers (1, 2 and 3), silica dye monomers (4, 5 and 6), stimuli-responsive ester bond-bridged silane (7 and 8) and PEGylated silane (9) for the synthesis of various types of silica NPs. (B) Synthesis of silica–pyrene nanoconjugates (NCs) with controlled size (20 nm to 200 nm) using a silica monomer of 6. Different batches of silica–pyrene NCs with high consistence (denoted as PyrX) were synthesized with different sizes. (C) Preparation of different silica–drug/dye NCs (50 and 20 nm) using the silica drug/dye monomers stated above. (D) The size and monodispersity of NCs was characterized by scanning electron microscopy (SEM). Reprinted with permission from ref. 85. Copyright 2013 American Chemical Society. | ||
In addition to enzymes (e.g., esterase), there are many other tumor-specific environmental cues such as GSH-enrichment and acidic environment. Xu et al. synthesized a type of GSH-responsive silica prodrug NPs by introducing a disulfide bond silane (monomer 11 in Fig. 7A) into silica NPs with controllable size and excellent blood compatibility.47 They further synthesized single-hole hollow silica NPs using GSH-responsive silane, which could trigger drug release and carrier degradation in response to GSH and pH variation.105
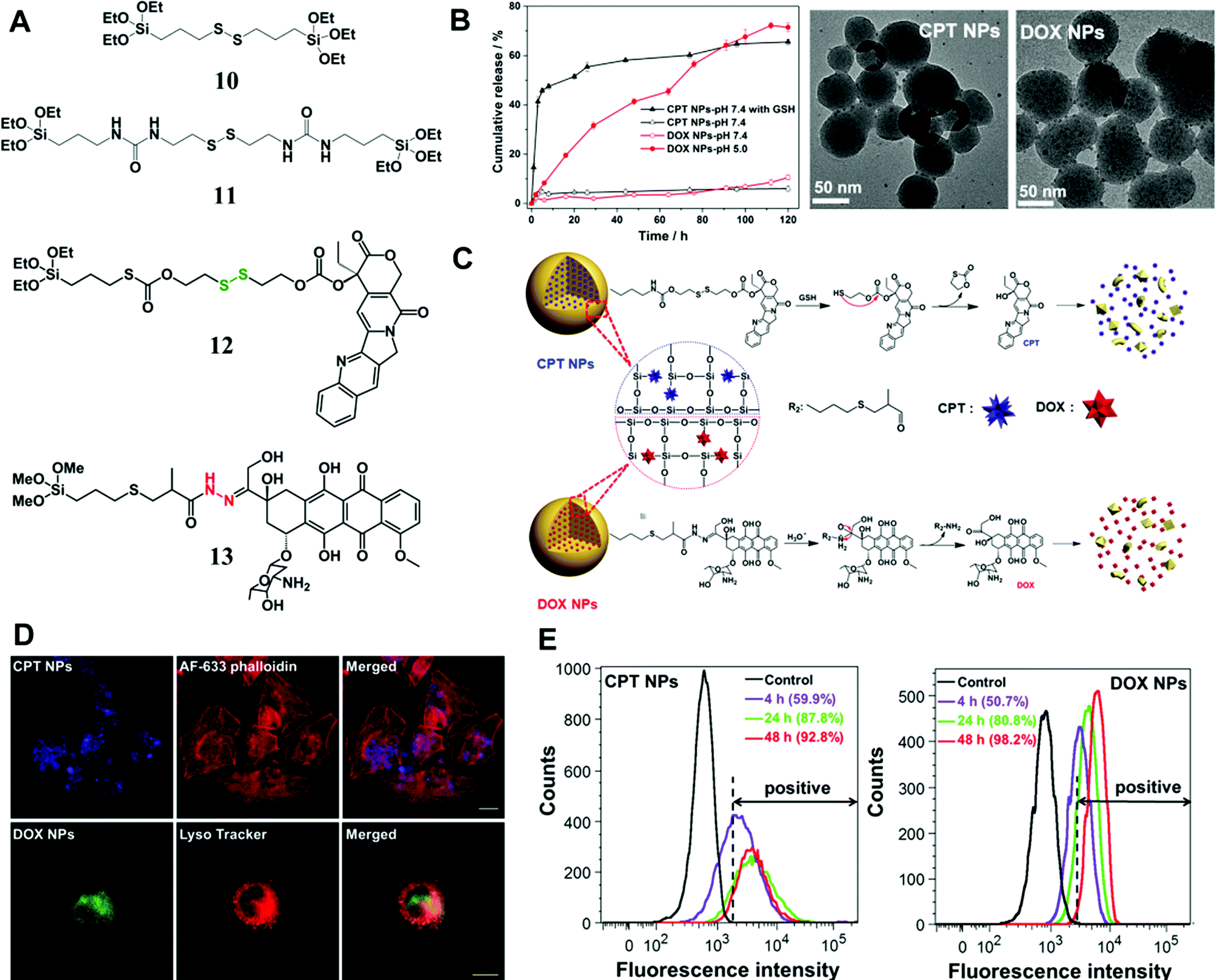 | ||
| Fig. 7 (A) The chemical structure of stimuli-responsive silane bridged with a disulfide bond and two types of prodrug monomers (GSH-responsive CPT (12) and pH-responsive DOX (13) prodrug monomers); (B) in vitro CPT or DOX release from the prodrug NPs at 37 °C in PBS (pH 7.4) with/without 10 mM GSH and under different pH conditions, respectively; representative TEM images of CPT or DOX prodrug NPs after immersion in PBS (pH 7.4) containing 10 mM GSH or in PBS (pH 5.0) for 72 h, respectively. (C) Proposed degradation mechanism and stimuli-triggered drug release from the prodrug NPs. (D) CLSM images of CPT prodrug NPs in HeLa cells after 20 h of incubation; the lysosome co-localization analysis of DOX prodrug NPs was obtained after 5 h of incubation in HeLa cells, and the fluorescence of Lyso Tracker and DOX prodrug NPs were pseudo-labeled red and green, respectively. Scale bar: 20 μm. (E) Flow cytometric analysis of (a) CPT prodrug NPs and (b) DOX prodrug NPs internalized in HeLa cells at different incubation times. The unlabeled cells were tested as controls. Reprinted with permission from ref. 97. Copyright 2015 Royal Society of Chemistry. | ||
Later, Xu et al. conjugated two commercial drug molecules (camptothecin (CPT) and DOX) with triethoxysilane groups through a covalent GSH-responsive disulfide bond and pH-responsive hydrazone bond, respectively (Fig. 7),97 which significantly reduced drug leaching from NPs compared with those by physical encapsulation. The prodrug monomers were introduced into silica NPs through condensation with a tetraalkoxysilane process, resulting in NPs with controlled sizes from 50 to 200 nm. The in vitro release profile (Fig. 7B) demonstrated excellent stability of the silica prodrug NPs under physiological conditions, whereas the drug release was sensitive to stimuli caused by the cleavage of disulfide and hydrazone bonds (Fig. 7C). Cell experiments revealed the intracellular localization (Fig. 7D and E) of the prodrug CPT or DOX NPs in tumor cells and particularly the co-localization of DOX NPs with lysosomes. More importantly, simultaneous application of these two prodrug NPs showed a synergistic effect for eradication of tumor cells, suggesting a promising combinatorial therapeutic platform based on nonporous silica NPs.
Although nonporous silica NP-based drug delivery platforms have recently attracted more attention and shown promising potential, there are some major challenges in their application for nanomedicine. First, the drug-loading efficiency needs to be improved without compromising the stability of the cargo–prodrug silica nanocomplex. Cheng and co-workers85 significantly improved the drug-loading efficiency, whereas the stability of prodrug NPs under physiological conditions was still far from satisfactory. Xu et al.97 designed silica prodrug NPs with excellent stability under physiological conditions, whereas the drug-loading efficiency was lower than expected. Second, most of the existing stimuli-responsive prodrug silica NPs are based on the stimulus of an enzyme, GSH or low pH. The targeting specificity of the prodrugs to tumoral tissues could be further improved if they are functionalized with specific targeting ligands, such as folic acid, antibodies, peptides or aptamers.
4. Applications in bioimaging and diagnosis
Precise and early diagnosis is crucial in cancer therapy, which can not only reduce the risk of fatality but also avoid excessive or inadequate therapy.106–112 Other than drug molecules, organic fluorophores or inorganic NPs (e.g., QDs, magnetic NPs) can also be introduced within or onto the surface of silica NPs for in vivo and in vitro imaging and diagnosis.113–132 It is known that most of the existing organic molecules or fluorophores, especially those for near infrared (NIR) imaging, have poor solubility or stability that limits their practical applications for diagnosis. An effective way to address this issue is to incorporate the imaging agents into inorganic nanoparticles (e.g., silica, QDs), which can improve the photophysical and photochemical properties of the organic fluorophores or other biomolecules.114,124–127 Owing to the inherent advantages of silica NPs in terms of controllable size and stability, imaging platforms based on silica NPs have potential for diagnostic and imaging applications.There are two common strategies to prepare silica NP-based imaging platforms: physical and chemical. Wang et al. designed a class of NIR imaging agent with a large Stokes shift (Fig. 8A) by introducing two NIR fluorescent molecules into silica NPs. The resulting NPs showed significantly reduced background fluorescence and crosstalk between the excitation light and emitting signals that was attributed to the small Stokes shift of traditional NIR dyes. Alternatively, organic or inorganic fluorophores can be modified onto the surface of silica carriers. Recently, Lee et al. reported a type of ultrasensitive and biocompatible silica NPs with highly bright CdSe@CdS@ZnS QDs conjugated onto the surface for bioimaging.116 The surface conjugation did not show any effect on the quantum yield of QDs and thus significantly enhanced the fluorescence signals in vivo.
 | ||
| Fig. 8 (A) Chemical structure of 14, 15 and 16 fluorescence molecules; (B) schematic illustration of LSS-NFSiNPs; (C) LSS-NFSiNPs were injected intravenously into nude mice to obtain real-time FRET imaging of the abdomen in vivo. Reprinted with permission from ref. 117. Copyright 2012 American Chemical Society. | ||
Bezdetnaya et al. also reported a silica NP-based platform for NIR imaging of the lymphatic system using a silane (16 in Fig. 8A), which had a stable core–shell structure and tunable surface charge. This platform was able to accurately map lymph nodes for the diagnosis of potential metastatic and draining nodes during surgical procedures. Furthermore, these NPs could be efficiently excreted through the hepatobiliary route, showing no obvious toxicity in vivo (mouse model) up to 3 months after injection. Different from conventional incorporation of NIR dyes in a silica matrix, Chen et al. developed a class of novel NIR fluorescent silica NPs with a large Stokes shift (LSS-NFSiNPs) based on the principle of fluorescence resonance energy transfer (FRET) (Fig. 8B and C).117 Two highly soluble organic dyes, methylene blue (MB) and tris(2,2-bipyridyl)-dichlororuthenium(II) hexahydrate (RuBpy), were used as the model donor–receptor pair. These silica NPs showed strong fluorescence and a Stokes shift greater than 200 nm, which could facilitate real-time and deep-tissue imaging in vivo.
Other molecular probes, such as antibodies or aptamers, can be functionalized on imaging platforms to realize precise bioimaging and highly specific molecular diagnosis. Wu et al. developed silica NPs coated with antibody-conjugated QDs by employing dual-signal amplification immunosensing for identification of low-abundance circulating tumor cells (CTCs) in peripheral blood (Fig. 9).122 These silica NP tracers were able to identify two disease-specific biomarkers on Hep3B liver cancer cells and achieved a limit of detection down to 10 cells per mL. Recently Ban et al. designed silica NPs (Dual-SiNP) modified with dual aptamers (MUC1 and HER2), which was highly sensitive and selective for the detection of four distinct types of breast cancer cells.67 The obtained NPs with controllable size and low cytotoxicity can be used in the diagnosis and prognosis of breast cancer metastasis.
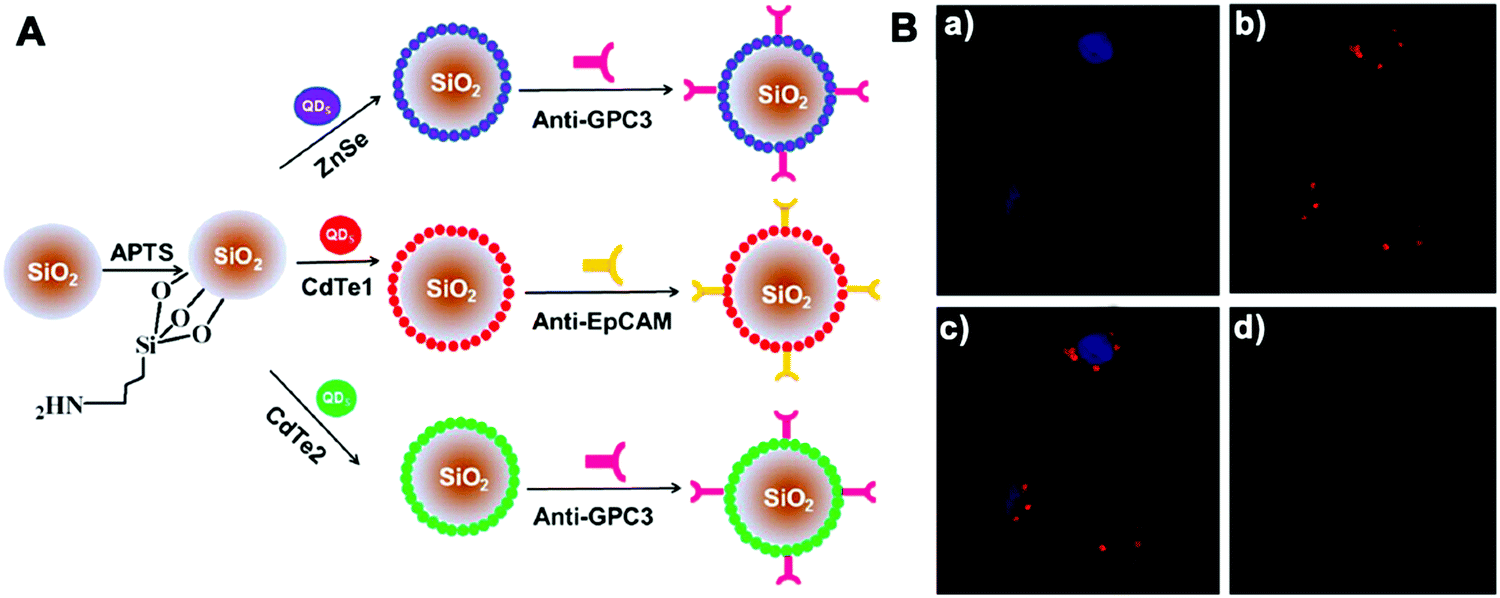 | ||
| Fig. 9 (A) Schematic illustration of the structure of three QD-coated silica NPs; (B) CLSM images of MCF-7 cells incubated with NPs. The fluorescence of (a) Hoechst 33342 and (b) Si/Cd1/anti-EpCAM NPs was pseudo-labeled blue and red, (c) a merged image of (a) and (b). (d) Negative control. Copyright of 2013 American Chemical Society. Reprinted with permission from ref. 122. Copyright 2013 American Chemical Society. | ||
5. Applications as bio-adhesives
Tissue bio-adhesive play an important part in hemostasis and wound therapy. To date, various natural and artificial materials, including fibrin glue, protein and synthetic adhesives, have been developed for conventional hemostats and wound closure methods.133–137 Among these reported bio-adhesive platforms, polymer-based tissue adhesives have adjustable chemical structure and controllable material properties (mechanical, degradation and adhesion).138–140 However, conventional polymeric glues usually require heating, chemical reactions and an appropriate pH condition during the synthetic process, and may also induce toxic effects. It is desirable to develop functional bio-adhesive materials that are biocompatible, nontoxic, sterilisable, cost-effective and can also be easily manufactured.To improve bio-adhesive efficiency, Leibler et al. recently proposed a strategy of gluing swollen polymer networks together using nonporous silica NPs (Fig. 10), taking advantage of the strong affinity between NPs and the polymeric network chains.141 Due to the different chemical nature of the particle solutions, such as TM-50 silica solution, the surface chemistry allowed adsorption of NPs on both gels of polymer networks (Fig. 10A and B). For demonstration in biological tissues, the TM-50 silica solution was first spread on the surface of two slices of calf liver. After simple pressing using a finger for ≈30 s, the two tissues could adhere together readily. The strong affinity at the tissue interface was due to the ideal match between the size of NPs and the polymeric network mesh size on the tissue surface, where the NPs act as “connectors” between polymer chains. Under external stress, the polymer chains adsorbed onto NPs were able to reorganize, which allowed large deformation and energy dissipation and thus created a strong bond without affecting the rigidity of the assembly. The same research team further proposed a nano-bridging method to assist wound closure using silica NPs,142 which could form abundant connectors at the wound edges. After silica NP solution was spread onto the surface of wounded tissue, the wound edges were brought into contact by gentle pressing. Finally, the silica NPs adsorbed on the surface of tissue components at the site of injury formed numerous connectors that linked the wound edges. This method was able to effectively repair the skin injury on the thin abdominal skin and thick dorsal skin of a Wistar rat. In addition, fast hemostasis after hepatectomy could be achieved using silica NPs. No pathological inflammation or necrosis was found at the site of treatment 3 days after hepatectomy. This simple and rapid nano-bridging method can be used for urgent control of bleeding, tissue repair or other clinical applications.
 | ||
| Fig. 10 (A) The concept of “gluing” swollen polymer networks together using silica NPs. (B) The irreversible adsorption on the surface of the NPs. (C) SEM characterization of the TM-50 silica NPs adsorbed on a S0.1 gel surface after multiple soaking and washing for several days in water (left); lap-joint geometry. Video extensometers marked by white dots were used to measure the displacement (right). (D) Two slices of calf liver glued by spreading TM-50 silica NP solution on the interface and pressing (left); the corresponding normalized force–displacement curves of the joint liver slices (right). Reprinted with permission from ref. 141. Copyright 2013 Nature Publishing Group. | ||
Although silica NPs have demonstrated good potential as tissue bio-adhesive materials, most existing methods rely on the native properties of silica NPs.143 On the other hand, there are many strategies to further improve silica NP-based adhesives using biocompatible polymers, such as PEG or other synthetic polymers, to functionalize silica NPs. Moreover, silica NP-based bio-adhesives could be loaded with prodrugs or biological factors to enhance the therapeutic effect and, therefore, significantly promote tissue regeneration during wound healing.
6. Conclusions and perspectives
As an emerging and rapidly advancing application of nanotechnology, nanomedicine has shown powerful potential as a universal theranostic platform for the healthcare industry. In the past few decades, there have been numerous and increasing numbers of novel systems utilizing various nanoscale agents for drug delivery, phototherapy, bioimaging, biosensing, tissue regeneration and many other critical biomedical applications reported each year. The size of the nanotheranostic agents is comparable with biomolecules and subcellular organelles, providing an accurately engineered platform for interfacing with complex and heterogeneous biological systems. The functionalities of the nanomaterial substrates can be tailored by different physical and chemical methods and customized for specific applications, leading to a vehicle capable of delivery of therapeutic agents, contrast agents, or other analytical nanodevices.However, several key challenges exist and have hindered the broad medical applications of nanomedicine in practice. One of the major concerns is the potential toxicity or undesirable side effects of various nanotheranostic agents to healthy cells and tissues. The pharmaceutical developers usually need to spend dauntingly expensive resources and time within many rounds of strict pre-clinical and clinical trials for a new nanomedicine to clear the mandatory administrative authorization. The nanotoxicity could be due to several reasons. One of them is related to the substrate materials that construct the matrix of the nanocarrier. Another is attributed to the nonspecific toxicity of the loaded theranostic agents. Silica-based NPs represent an important class of nanomaterials and have demonstrated unique advantages in drug delivery, bioimaging, bio-adhesives and other biomedical applications. Compared with many inorganic nanomaterials, silica-based NPs exhibit lower toxicity and excellent biocompatibility, and their degradation products (orthosilicic acid) can be efficiently excreted through the renal system. Nonporous silica NPs carry theranostic agents mainly by encapsulation or covalent conjugation, which ensures high stability of the carrier–cargo complex and minimizes cargo leakage as well as the induced nonspecific toxicity to healthy tissues.
Other features of nonporous silica NPs as a highly advantageous theranostic platform include tunable size control and flexible approaches for functionalization. Following the well-defined synthetic route based on the Stöber method, the size of the NPs can be precisely tuned down to 20 nm with superior monodispersity and morphological control, which covers the regular size range for various biomedical applications. To enhance the interaction with biological systems for desirable theranostic efficacy, functional ligands can be grafted within or onto the surface of NPs to achieve disease-specific cellular internalization and biodistribution, stimuli-responsive cargo release, or to improve the stability, biocompatibility and degradation rate of the carrier.
As described in this overview, there have been rapid advances in the design and synthesis of nonporous silica NPs as versatile theranostic platforms, many of which are subjected to further pre-clinical explorations. It is expected that new advances will be made towards integrating nonporous silica NPs with other systems, such as polymeric and small molecular materials, as well as the strategic synthesis and scale-up production of more “intelligent” and multimodal theranostic nanoagents. It is very encouraging to witness that the FDA has recently approved a type of nonporous silica-based NPs called Cornell Dots (C-dots), for clinical applications in cancer diagnostics and therapeutics.144,145 Further research effort in this area could help design novel silica-based nanomaterials to better understand and control their interaction with complex biological systems.
Acknowledgements
This work was supported by start-up grants (SWU115058, SWU115059), Fundamental Research Funds for Central Universities from Southwest University (XDJK2017D006, XDJK2016A010), Basic and Frontier Research Project of Chongqing (cstc2016jcyjA0078) and the National Natural Science Foundation of China (31671037).Notes and references
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- V. P. Chauhan and R. K. Jain, Nat. Mater., 2013, 12, 958–962 CrossRef CAS PubMed.
- A. N. Zelikin, C. Ehrhardt and A. M. Healy, Nat. Chem., 2016, 8, 997–1007 CrossRef CAS PubMed.
- E. K.-H. Chow and D. Ho, Sci. Transl. Med., 2013, 5, 216rv214 Search PubMed.
- M. W. Tibbitt, J. E. Dahlman and R. Langer, J. Am. Chem. Soc., 2016, 138, 704–717 CrossRef CAS PubMed.
- W. Chen, R. Sheng, P. D. Baade, S. Zhang, H. Zeng, F. Bray, A. Jamal, X. Q. Yu and J. He, Ca-Cancer J. Clin., 2016, 66, 115–132 CrossRef PubMed.
- K. Riemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef PubMed.
- P. T. Wong and S. K. Choi, Chem. Rev., 2015, 115, 3388–3432 CrossRef CAS PubMed.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
- H. Wei, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2013, 38, 503–535 CrossRef CAS.
- J. Liu, W. Huang, Y. Pang and D. Yan, Chem. Soc. Rev., 2015, 44, 3942–3953 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- D. Ling, N. Lee and T. Hyeon, Acc. Chem. Res., 2015, 48, 1276–1285 CrossRef CAS PubMed.
- Y. Liu, C. Lou, H. Yang, M. Shi and H. Miyoshi, Curr. Cancer Drug Targets, 2011, 11, 156–163 CrossRef CAS PubMed.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2013, 26, 435–451 CrossRef.
- M. S. Bradbury, E. Phillips, P. H. Montero, S. M. Cheal, H. Stambuk, J. C. Durack, C. T. Sofocleous, R. J. Meester, U. Wiesner and S. Patel, Integr. Biol., 2013, 5, 74–86 RSC.
- L. Tang and J. Cheng, Nano Today, 2013, 8, 290–312 CrossRef CAS PubMed.
- W. Q. Lim, S. Z. F. Phua, H. V. Xu, S. Sreejith and Y. Zhao, Nanoscale, 2016, 8, 12510–12519 RSC.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- Z. Xu, D. Wang, M. Guan, X. Liu, Y. Yang, D. Wei, C. Zhao and H. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 3424–3431 CAS.
- J.-H. Park, L. Gu, G. Von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331–336 CrossRef CAS PubMed.
- H. Ow, D. R. Larson, M. Srivastava, B. A. Baird, W. W. Webb and U. Wiesner, Nano Lett., 2005, 5, 113–117 CrossRef CAS PubMed.
- S. Kaur, C. Prasad, B. Balakrishnan and R. Banerjee, Biomater. Sci., 2015, 3, 955–987 RSC.
- H. Mekaru, J. Lu and F. Tamanoi, Adv. Drug Delivery Rev., 2015, 95, 40–49 CrossRef CAS PubMed.
- D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- E. J. Anglin, L. Cheng, W. R. Freeman and M. J. Sailor, Adv. Drug Delivery Rev., 2008, 60, 1266–1277 CrossRef CAS PubMed.
- S. Banerjee and A. Datta, Langmuir, 2009, 26, 1172–1176 CrossRef PubMed.
- I.-Y. Kim, E. Joachim, H. Choi and K. Kim, J. Nanomed. Nanotechnol., 2015, 11, 1407–1416 CrossRef CAS PubMed.
- H. Wang, L. Tang, Y. Liu, I. T. Dobrucki, L. W. Dobrucki, L. Yin and J. Cheng, Theranostics, 2016, 6, 1467–1476 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- U. Prabhakar, H. Maeda, R. K. Jain, E. M. Sevick-Muraca, W. Zamboni, O. C. Farokhzad, S. T. Barry, A. Gabizon, P. Grodzinski and D. C. Blakey, Cancer Res., 2013, 73, 2412–2417 CrossRef CAS PubMed.
- H. Meng, M. Xue, T. Xia, Z. Ji, D. Y. Tarn, J. I. Zink and A. E. Nel, ACS Nano, 2011, 5, 4131–4144 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC.
- D. K. Yi, S. T. Selvan, S. S. Lee, G. C. Papaefthymiou, D. Kundaliya and J. Y. Ying, J. Am. Chem. Soc., 2005, 127, 4990–4991 CrossRef CAS PubMed.
- L. Tang, X. Yang, Q. Yin, K. Cai, H. Wang, I. Chaudhury, C. Yao, Q. Zhou, M. Kwon and J. A. Hartman, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15344–15349 CrossRef CAS PubMed.
- S.-W. Ha, C. E. Camalier, G. R. Beck Jr and J.-K. Lee, Chem. Commun., 2009, 2881–2883 RSC.
- G. Alberto, G. Caputo, G. Viscardi, S. Coluccia and G. Martra, Chem. Mater., 2012, 24, 2792–2801 CrossRef CAS.
- A. Bumb, S. K. Sarkar, N. Billington, M. W. Brechbiel and K. C. Neuman, J. Am. Chem. Soc., 2013, 135, 7815–7818 CrossRef CAS PubMed.
- K. Ma, D. Zhang, Y. Cong and U. Wiesner, Chem. Mater., 2016, 28, 1537–1545 CrossRef CAS.
- T. Ribeiro, S. Raja, A. S. Rodrigues, F. Fernandes, J. P. S. Farinha and C. Baleizão, RSC Adv., 2013, 3, 9171–9174 RSC.
- B. Chen, M. Zuberi, R. B. Borgens and Y. Cho, J. Biol. Eng., 2012, 6, 1–8 CrossRef PubMed.
- D. Kudela, S. A. Smith, A. May-Masnou, G. B. Braun, A. Pallaoro, C. K. Nguyen, T. T. Chuong, S. Nownes, R. Allen and N. R. Parker, Angew. Chem., Int. Ed., 2015, 54, 4018–4022 CrossRef CAS PubMed.
- Z. Xu, K. Zhang, X. Liu and H. Zhang, RSC Adv., 2013, 3, 17700–17702 RSC.
- V. Marzaioli, J. A. Aguilar-Pimentel, I. Weichenmeier, G. Luxenhofer, M. Wiemann, R. Landsiedel, W. Wohlleben, S. Eiden, M. Mempel and H. Behrendt, Int. J. Nanomed., 2014, 9, 2815 Search PubMed.
- W. Scarano, P. Souza and M. H. Stenzet, Biomater. Sci., 2015, 3, 163–174 RSC.
- B. Kim, G. Han, B. J. Toley, C. K. Kim, V. M. Rotello and N. S. Forbes, Nanotechnology, 2010, 5, 465–472 CAS.
- R. P. Bagwe, L. R. Hilliard and W. Tan, Langmuir, 2006, 22, 4357–4362 CrossRef CAS PubMed.
- Z. G. Estephan, J. A. Jaber and J. B. Schlenoff, Langmuir, 2010, 26, 16884–16889 CrossRef CAS PubMed.
- S. Legrand, A. Catheline, L. Kind, E. C. Constable, C. E. House-croft, L. Landmann, P. Banse, U. Pielesa and A. Wirth-Heller, New J. Chem., 2008, 32, 588–593 RSC.
- X. X. He, H. L. Nie, K. M. Wang, W. H. Tan, X. Wu and P. F. Zhang, Anal. Chem., 2008, 80, 9597–9603 CrossRef CAS PubMed.
- S. Bonacchi, D. Genovese, R. Juris, M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Angew. Chem., Int. Ed., 2011, 50, 4056–4066 CrossRef CAS PubMed.
- M. Nakamura and K. Ishimura, Langmuir, 2008, 24, 12228–12234 CrossRef CAS PubMed.
- M. Ninell P, H. Gregory B, W. Wei, F. Carmen M, N. Prakash D and R. Scott T, Nanoscale, 2013, 5, 6372–6380 RSC.
- N. Reinhardt, L. Adumeau, O. Lambert, S. Ravaine and S. Mornet, J. Phys. Chem. B, 2015, 119, 6401–6411 CrossRef CAS PubMed.
- B.-H. Jun, D. W. Hwang, H. S. Jung, J. Jang, H. Kim, H. Kang, T. Kang, S. Kyeong, H. Lee, D. H. Jeong, K. W. Kang, H. Youn, D. S. Lee and Y.-S. Lee, Adv. Funct. Mater., 2012, 22, 1843–1849 CrossRef CAS.
- A. J. Moro, J. Schmidt, T. Doussineau, A. Lapresta-Fernandez, W. Joachim and G. J. Mohr, Chem. Commun., 2011, 47, 6066–6068 RSC.
- A. Bumb, S. K. Sarkar, N. Billington, M. W. Brechbiel and K. C. Neuman, J. Am. Chem. Soc., 2013, 135, 7815–7818 CrossRef CAS PubMed.
- I. de Feijter, L. Albertazzi, A. R. A. Palmans and I. K. Voets, Langmuir, 2015, 31, 57–64 CrossRef CAS PubMed.
- L. Yang, X. B. Zhang, M. Ye, J. H. Jiang, R. H. Yang, T. Fu, Y. Chen, K. M. Wang and W. H. Tan, Adv. Drug Delivery Rev., 2011, 63, 1361–1370 CrossRef CAS PubMed.
- Y. F. Guo, H. H. Liu, D. D. Tang, C. X. Li and Y. L. Zhao, Polym. Chem., 2015, 6, 2647–2658 RSC.
- G. W. Jia, Z. Q. Cao, H. Xue, Y. S. Xu and S. Y. Jiang, Langmuir, 2009, 25, 3196–3199 CrossRef CAS PubMed.
- Y. F. Wu, P. Xue, Y. J. Kang and K. M. Hui, Anal. Chem., 2013, 85, 3166–3173 CrossRef CAS PubMed.
- H. Jo, J. Her and C. Ban, Biosens. Bioelectron., 2015, 71, 129–136 CrossRef CAS PubMed.
- M. Li, J. W. Y. Lam, F. Mahtab, S. J. Chen, W. J. Zhang, Y. N. Hong, J. Xiong, Q. C. Zheng and B. Z. Tang, J. Mater. Chem. B, 2013, 1, 676–684 RSC.
- M. T. Hurley, Z. F. Wang, A. Mahle, D. Rabin, Q. Liu, D. S. English, M. R. Zacharia, D. Stein and P. DeShong, Adv. Funct. Mater., 2013, 23, 3335–3343 CrossRef CAS.
- Y. Chen, H. Chen and J. Shi, Adv. Mater., 2013, 23, 3144–3176 CrossRef PubMed.
- H. Su, J. M. Koo and H. Cui, J. Controlled Release, 2015, 219, 383–395 CrossRef CAS PubMed.
- J. Tom, K. Ohno and S. Perrier, Polym. Chem., 2016, 7, 6075–6083 RSC.
- A. R. Jennings, S. M. Budy, C. J. Thrasher and S. T. Iacono, Chem. Mater., 2016, 8, 16212–16220 CAS.
- C. H. J. Choi, J. E. Zuckerman, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6656–6661 CrossRef CAS PubMed.
- X. X. He, H. L. Nie, K. M. Wang, W. H. Tan, X. Wu and P. F. Zhang, Anal. Chem., 2008, 80, 9597–9603 CrossRef CAS PubMed.
- R. Kumar, I. Roy, T. Y. Ohulchanskky, L. A. Vathy, E. J. Bergey, M. Sajjad and P. N. Prasad, ACS Nano, 2010, 4, 699–708 CrossRef CAS PubMed.
- R. K. Singh, K. D. Patel, K. W. Leong and H.-W. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 10309–10337 CAS.
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 CrossRef CAS PubMed.
- D. M. Yang, P. A. Ma, Z. Y. Hou, Z. Y. Cheng, C. X. Li and J. Lin, Chem. Soc. Rev., 2015, 44, 1416–1448 RSC.
- D. B. Pacardo, F. S. Ligler and Z. Gu, Nanoscale, 2015, 7, 3381–3391 RSC.
- Y. Wang, M. S. Shim, N. S. Levinson, H.-W. Sung and Y. Xia, Adv. Funct. Mater., 2014, 24, 4206–4220 CrossRef CAS PubMed.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243–4268 RSC.
- M. Karimi, I. Chaudhury, C. Jianjun, M. Safari, R. Sadeghi, M. Habibi-Rezaei and J. Kokini, J. Mol. Catal. B: Enzym., 2014, 104, 48–55 CrossRef CAS.
- L. Tang, N. P. Gabrielson, F. M. Uckun, T. M. Fan and J. Cheng, Mol. Pharmaceutics, 2013, 10, 883–892 CrossRef CAS PubMed.
- L. Tang, T. M. Fan, L. B. Borst and J. Cheng, ACS Nano, 2012, 6, 3954–3966 CrossRef CAS PubMed.
- L. Tang, X. Yang, L. W. Dobrucki, I. Chaudhury, Q. Yin, C. Yao, S. Lezmi, W. G. Helferich, T. M. Fan and J. Cheng, Angew. Chem., Int. Ed., 2012, 124, 12893–12898 CrossRef.
- A. Mignot, C. Truillet, F. Lux, L. Sancey, C. Louis, F. Denat, F. Boschetti, L. Bocher, A. Gloter, O. Stephan, R. Antoine, P. Dugourd, D. Luneau, G. Novitchi, L. C. Figueiredo, P. C. de Morais, L. Bonneviot, B. Albela, F. Ribot, L. Van Lokeren, I. Dechamps-Olivier, F. Chuburu, G. Lemercier, C. Villiers, P. N. Marche, G. Le Duc, S. Roux, O. Tillement and P. Perriat, Chem. – Eur. J., 2013, 19, 6122–6136 CrossRef CAS PubMed.
- G. Ding, D. Li, Y. Liu, M. Guo, Y. Duan, J. Li and Y. Cao, J. Nanopart. Res., 2014, 16, 1–10 CrossRef CAS.
- A. Das, P. Mukherjee, S. Singla, P. Guturu, M. Frost, D. Mukhopadhyay, V. H. Shah and C. R. Patra, Nanotechnology, 2010, 21, 305102 CrossRef PubMed.
- P. Botella, I. Abasolo, Y. Fernández, C. Muniesa, S. Miranda, M. Quesada, J. Ruiz, S. Schwartz Jr. and A. Corma, J. Controlled Release, 2011, 156, 246–257 CrossRef CAS PubMed.
- H. N. Li, Y. W. Mu, S. S. Qian, J. S. Lu, Y. K. Wan, G. D. Fua and S. Q. Liu, Analyst, 2015, 140, 567–573 RSC.
- R. Gangwar, G. B. Tomar, V. Dhumale, S. Zinjarde, R. B. Sharma and S. Datar, J. Agric. Food Chem., 2013, 61, 9632–9637 CAS.
- C.-H. Lai, T.-C. Chang, Y.-J. Chuang, D.-L. Tzou and C.-C. Lin, Bioconjugate Chem., 2013, 24, 1698–1709 CrossRef CAS PubMed.
- P. Zhang and J. Kong, Talanta, 2015, 134, 501–507 CrossRef CAS PubMed.
- S. V. Bhosale, Chem. – Eur. J., 2014, 20, 5253–5257 CrossRef CAS PubMed.
- S. Zhang, Z. Q. Chu, C. Yin, C. Y. Zhang, G. Lin and Q. Li, J. Am. Chem. Soc., 2013, 135, 5709–5716 CrossRef CAS PubMed.
- Z. G. Xu, S. Y. Liu, Y. J. Kang and M. F. Wang, Nanoscale, 2015, 7, 5859–5868 RSC.
- Y. Gao, Y. Q. Wang, A. Fu, W. Shi, D. Yeo, K. Q. Luo, H. Ow and C. J. Xu, J. Mater. Chem. B, 2015, 3, 1245–1253 RSC.
- M. Benezra, E. Phillips, M. Overholtzer, P. B. Zanzonico, E. Tuominen, U. Wiesner and M. S. Bradbury, Small, 2015, 11, 1721–1732 CrossRef CAS PubMed.
- H. Wei, R. X. Zhuo and X. Z. Zhang, Prog. Polym. Sci., 2013, 38, 503–535 CrossRef CAS.
- S. F. Medeiros, A. M. Santos, H. Fessi and A. Elaissari, Int. J. Pharm., 2011, 403, 139–161 CrossRef CAS PubMed.
- F. J. Xu, F. B. Su, S. B. Deng and W. T. Yang, Macromolecules, 2010, 43, 2630–2633 CrossRef CAS.
- C. H. Lu and I. Willner, Angew. Chem., Int. Ed., 2015, 54, 12212–12235 CrossRef CAS PubMed.
- S. V. Bhosale, Chem. – Eur. J., 2014, 20, 5253–5257 CrossRef CAS PubMed.
- D. D. Wang, Z. G. Xu, Z. J. Chen, X. Y. Liu, C. L. Hou, X. Y. Zhang and H. X. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 12600–12608 CAS.
- H. Su, J. M. Koo and H. G. Cui, J. Controlled Release, 2015, 219, 383–395 CrossRef CAS PubMed.
- R. C. Qian, L. Ding, L. W. Yan, M. F. Lin and H. X. Ju, J. Am. Chem. Soc., 2014, 136, 8205–8208 CrossRef CAS PubMed.
- J. W. Tian, L. Ding, H. X. Ju, Y. C. Yang, X. L. Li, Z. Shen, Z. Zhu, J. S. Yu and C. Y. J. Yang, Angew. Chem., Int. Ed., 2014, 53, 9544–9549 CrossRef CAS PubMed.
- T. Hayashi, Y. Yasueda, T. Tamura, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2015, 137, 5372–5380 CrossRef CAS PubMed.
- T. Yoshii, K. Mizusawa, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2014, 136, 16635–16642 CrossRef CAS PubMed.
- D. J. Ye, A. J. Shuhendler, L. N. Cui, L. Tong, S. S. Tee, G. Tikhomirov, D. W. Felsher and J. H. Rao, Nat. Chem., 2014, 6, 519–526 CrossRef CAS PubMed.
- M. H. Lee, Z. G. Yang, C. W. Lim, Y. H. Lee, D. B. Sun, C. H. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed.
- K. M. Wang, X. X. He, X. H. Yang and H. Shi, Acc. Chem. Res., 2013, 46, 1367–1376 CrossRef CAS PubMed.
- W. Arap, R. Pasqualini, M. Montalti, L. Petrizza, L. Prodi, E. Rampazzo, N. Zaccheroni and S. Marchiò, Curr. Med. Chem., 2013, 20, 2195–2211 CrossRef CAS PubMed.
- T. Aubert, F. Cabello-Hurtado, M. A. Esnault, C. Neaime, D. Lebret-Chauvel, S. Jeanne, P. Pellen, C. Roiland, L. L. Polles, N. Saito, K. Kimoto, H. Haneda, N. Ohashi, F. Grasset and S. Cordier, J. Phys. Chem. C, 2013, 117, 20154–20163 CAS.
- B. H. Jun, D. W. Hwang, H. S. Jung, J. Jang, H. Kim, H. Kang, T. Kang, S. Kyeong, H. Lee, D. H. Jeong, K. W. Kang, H. Youn, D. S. Lee and Y. S. Lee, Adv. Funct. Mater., 2012, 22, 1843–1849 CrossRef CAS.
- X. X. He, Y. S. Wang, K. M. Wang, M. Chen and S. Y. Chen, Anal. Chem., 2012, 84, 9056–9064 CAS.
- S. Moom, M. Kurian and D. Losic, Biomater. Sci., 2014, 2, 10–34 RSC.
- R. Joshi, V. Feldmann, W. Koestner, C. Detje, S. Gottschalk, H. A. Mayer, M. G. Sauer and J. Engelmann, Biol. Chem., 2012, 394, 125–135 Search PubMed.
- V. Skripachevaa, A. Mustafinaa, N. Davydovb, V. Burilovb, A. Konovalova, S. Solovevaa and I. Antipin, Mater. Chem. Phys., 2012, 132, 488–493 CrossRef.
- M. Soster, R. Juris, S. Bonacchi, D. Genovese, M. Montalti, E. Rampazzo, N. Zaccheroni, P. Garagnani, F. Bussolino, L. Prodi and S. Marchiò, Int. J. Nanomed., 2012, 7, 4797–4807 CAS.
- Y. F. Wu, P. Xue, Y. J. Kang and K. M. Hui, Anal. Chem., 2013, 85, 3166–3173 CrossRef CAS PubMed.
- E. Lipani, S. Laurent, M. Surin, L. V. Elst, P. Leclère and R. N. Muller, Langmuir, 2013, 29, 3419–3427 CrossRef CAS PubMed.
- X. L. Guével, B. Hötzer, G. Jung and M. Schneider, J. Mater. Chem., 2011, 21, 2974–2981 RSC.
- S. L. C. Pinho, H. Faneca, C. F. G. C. Geraldes, M. H. Delville, L. D. Carlos and J. Rocha, Biomaterials, 2012, 33, 925–935 CAS.
- L. Accomasso, E. C. Rocchietti, S. Raimondo, F. Catalano, G. Alberto, A. Giannitti, V. Minieri, V. Turinetto, L. Orlando, S. Saviozzi, G. Caputo, S. Geuna, G. Martra and C. Giachino, Small, 2012, 8, 3192–3200 CrossRef CAS PubMed.
- N. Wartenberg, P. Fries, O. Raccurt, A. Guillermo, D. Imbert and M. Mazzanti, Chem. – Eur. J., 2013, 19, 6980–6983 CrossRef CAS PubMed.
- G. Lapadula, A. Bourdolle, F. Allouche, M. P. Conley, I. D. Rosal, L. Maron, W. W. Lukens, Y. Guyot, C. Andraud, S. Brasselet, C. Copéret, O. Maury and R. A. Andersen, Chem. Mater., 2014, 26, 1062–1073 CrossRef CAS.
- M. Helle, E. Rampazzo, M. Monchanin, F. Marchal, F. Guillemin, S. Bonacchi, F. Salis, L. Prodi and L. Bezdetnaya, ACS Nano, 2013, 7, 8645–8657 CrossRef CAS PubMed.
- X. Q. Zhang, X. Y. Zhang, L. Tao, Z. Chi, J. Xu and Y. Wei, J. Mater. Chem. B, 2014, 2, 4398–4414 RSC.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243–4268 RSC.
- C. Caltagirone, A. Bettoschi, A. Garau and R. Montis, Chem. Soc. Rev., 2015, 44, 4645–4671 RSC.
- J. Kim, S. Pramanick, D. Lee, H. Park and W. Kim, Biomater. Sci., 2015, 3, 1002–1007 RSC.
- A. N. Gent, Langmuir, 1996, 12, 4492–4496 CrossRef CAS.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS PubMed.
- J. C. Wheat and J. S. Wolf Jr., Urol. Clin. N. Am., 2009, 36, 265–275 CrossRef PubMed.
- W. D. Spotnitz and S. Burks, Transfusion, 2008, 48, 1502–1516 CrossRef PubMed.
- B. Bharti, J. Meissner and G. H. Findenegg, Langmuir, 2011, 27, 9823–9833 CrossRef CAS PubMed.
- M. Mehdizadeh and J. Yang, Macromol. Biosci., 2013, 13, 271–288 CrossRef CAS PubMed.
- S. J. Frost, D. Mawad, J. Hook and A. Luto, Adv. Healthcare Mater., 2016, 5, 401–414 CrossRef CAS PubMed.
- S. Rose, A. Prevoteau, P. Elzière, D. Hourdet, A. Marcellan and L. Leibler, Nature, 2014, 505, 382–385 CrossRef CAS PubMed.
- A. Meddahi-Pellé, A. Legrand, A. Marcellan, L. Louedec, D. Letourneur and L. Leibler, Angew. Chem., Int. Ed., 2014, 53, 6369–6373 CrossRef PubMed.
- D. Pedrazzoli and A. Pegoretti, Compos. Sci. Technol., 2013, 76, 77–83 CrossRef CAS.
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 87–108 CrossRef PubMed.
- R. Friedman, JNCI, J. Natl. Cancer Inst., 2011, 103, 1428–1429 CrossRef PubMed.
| This journal is © the Partner Organisations 2017 |