DOI:
10.1039/C6QM00176A
(Research Article)
Mater. Chem. Front., 2017,
1, 654-659
A supramolecular hydrogel with identical cross-linking point density but distinctive rheological properties†
Received
16th August 2016
, Accepted 15th September 2016
First published on 26th September 2016
Abstract
Preparation of hydrogels with solely variable mechanical properties is greatly desired in the study of cell-matrix interactions in situ as well as cellular behaviors. Here, we fabricated a supramolecular polypeptide–DNA hydrogel, via the DNA self-assembly strategy, which possesses identical cross-linking point density but distinctive rheological properties. Keeping the cross-linking density constant, the rheological properties of the hydrogel can be adjusted by tailoring the stability of the DNA cross-linkers via easy programming of the DNA sequences. Through elongating and shortening the length of the DNA linker or inserting mismatch sites, the hydrogel can be strengthened, softened, or even fluidized, respectively. In addition, the mechanical properties can also be modulated by a pH-triggered conformation transition of the polypeptide backbone. Combining rapid physiological gelation and reversible thermal responsiveness, these supramolecular polypeptide–DNA hydrogels with solely tuned mechanical strength will promote studies on mechanical induced cell differentiation in situ, and have great potential in tissue engineering.
Introduction
Hydrogels are important soft scaffold materials in tissue engineering and regenerative medicine, acting as natural extracellular matrix (ECM) mimetics to provide biophysical support and biochemical guidance for encapsulated cells.1–4 Specifically, the mechanical properties of hydrogels play critical roles in modulating cell–matrix interactions as well as regulating biological processes including cell proliferation, spread and differentiation.5–10 For example, H. M. Blau et al. showed that the elasticity of the 2D PEG-hydrogel substrate can regulate muscle stem cell (MuSC) fate in vitro where soft substrates enhance the survival of MuSCs but prevent differentiation;11 in contrast to 2D platforms, J. L. West et al. fabricated 3D biomechanical patterning of hydrogels by photolithography for guiding HT-1080 fibrosarcoma cell migration.12 Meanwhile, various environmental stimuli like light,13,14 redox,15–16 heat17etc. are employed to trigger the cross-linking or de-cross-linking of the hydrogel network for controlling the mechanical properties of the hydrogel. In general, all above mentioned strategies are based on alteration of the cross-linking point density, which also change the network topology as well as the pore size distribution, and influence the molecular diffusion in a 3D environment.18–20 It could be argued that these subsequent changes might significantly affect the results too, thus the preparation of a hydrogel with solely variable mechanical properties is greatly desired for studying cellular behavior in situ, as well as in biomaterial science and tissue engineering. Recently, DNA has received continuous attention as a promising building block to create attractive nano-devices,21–25 nano-materials,26–30 as well as bulk materials like DNA hydrogels.31–41 Benefiting from the unique properties of DNA, such as precise recognition, a well-defined structure, a 50 nm persistence length of duplex,42,43 the supramolecular DNA hydrogels possess interesting properties such as swelling–shrinking behavior, shear-thinning properties, and a self-healing ability, etc.44–46 Furthermore, it is noteworthy that the stability of the duplex is closely related to the chain length, composition and especially the analog of the bases.47,48 With the same base composition, by simply shifting positions of two different bases in one chain of the duplex, the stabilities of these two duplexes could be enormously different.
Herein, we demonstrate that the rheological properties of a supramolecular DNA hydrogel could be solely changed without altering its network topology and cross-linking point density. By treating the lastly formed cross-linking duplexes as ‘cross-linking points’ in a typical network, the mechanical properties of the hydrogel are basically decided by the stability of the DNA cross-linkers. Based on the rational design of the DNA sequences, the value of the storage modulus (G′) of a DNA hydrogel could vary from several Pascal to several thousand Pascal whilst keeping the hydrogel cross-linking point density and the topological network unchanged. We believe that this material will become the footstone for methods that can enable strictly scientific studies on mechanical strength induced cell differentiation, and subsequently help in understanding embryonic development and tissue regeneration.
Experimental
Materials
All oligonucleotides (see ESI,† Table S1) were synthesized using an ABI 394 DNA synthesizer and purified using HPLC. All other chemicals were bought from Sigma-Aldrich and used directly without further purification unless noted.
Synthesis of the polypeptide–DNA conjugate
In a typical reaction, poly(L-glutamic acid-co-γ-propargyl-L-glutamate), namely p(LGA-co-PLG) (22.1 mg) was dissolved in water (100 μL) as a stock solution. 25 μL of this solution was mixed with 25 μL of CuSO4 solution (1 M), 150 μL THTA (200 mM), 1000 μL of azido-DNA solution (2.5 mM), and 100 μL sodium ascorbate (1 M). After incubating at 50 °C overnight with shaking, the solution was separated using a Millipore Microcon YM-30 filter to remove the unreacted DNA and other small molecules. The amount of DNA (m1) grafted on the polypeptide backbone was determined by measuring the DNA absorbance at 260 nm using a UV-Vis spectrophotometer, and the amount of polypeptide–DNA conjugate (m2) was determined by the weight of the polypeptide–DNA conjugate in the dry state; therefore the amount of polypeptide was m3 = m2 − m1. The final product was diluted with a corresponding buffer at 15 wt% as a stock solution.
Hydrogel preparation
A stock solution of two components was prepared: polypeptide–DNA (15 wt%, 13.8 mM with respect to DNA) and a DNA linker (5 mM). Calculated volumes of the two components were added in 1.5 mL EP tubes in PBS buffer (100 mM, pH 7.4) containing 150 mM NaCl and mixed quickly. For the preparation of hydrogels with different pH values, the protocol was the same as described above except that the buffers used were phosphate buffers (100 mM) with pH 5.0 or pH 8.0 containing 150 mM NaCl.
Rheological tests
Rheological tests were carried out on an AR-G2 rheometer (TA Instruments) equipped with a temperature controller. Four types of rheological experiments were performed in 8 mm parallel-plate geometry using 40 μL of hydrogels (resulting in a gap size of 0.15 mm): (i) in order to find the linear viscoelastic region of the time sweeps, an oscillatory strain sweep (0.01–100%) was conducted at 25 °C; (ii) frequency sweep tests were carried out between 0.05 and 100 rad s−1 at 25 °C at a fixed strain of 1%; (iii) Time-scan tests were done at a fixed frequency and strain of 1 Hz and 1%, respectively, at 25 °C for 3 min; (iv) temperature-ramp tests were performed at a fixed frequency and strain of 1 Hz and 1%, and the changes of G′ and G′′ were measured from 25 °C to 60 °C at a rate of 2 °C min−1.
Circular dichroism spectroscopy
The circular dichroism (CD) spectra were collected on an Applied Photophysics Chirascan spectropolarimeter which was used in the range of 180–350 nm at room temperature in a quartz cell of 1 mm optical length with a scanning speed of 60 nm min−1. Ellipticity ([θ] in deg cm2 dmol−1) = (millidegrees × mean residue weight)/(path length in millimeters × concentration of polypeptide in mg mL−1).
Results and discussion
Preparation and characterization of the polypeptide–DNA hydrogel
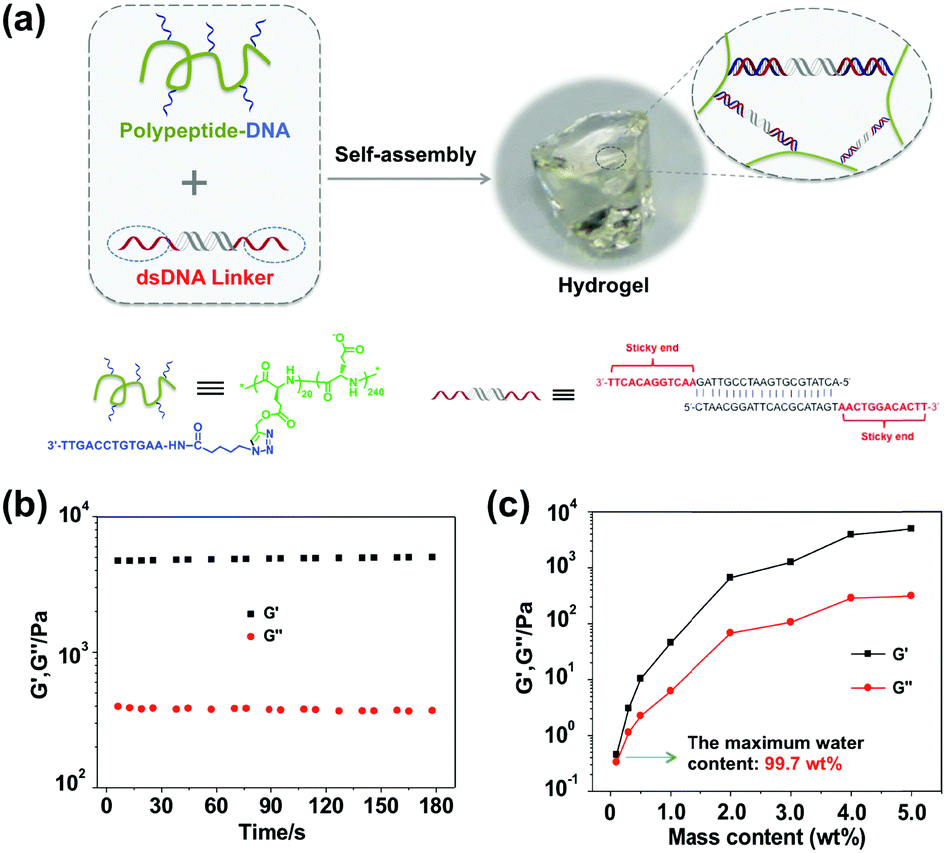
Fig. 1a illustrates the mechanism of our hydrogel formation via mixing two components: component 1 “polypeptide–DNA” is a poly(L-glutamic acid) derived polypeptide functionalized with 5–6 single stranded DNAs (ssDNAs), which was synthesized via a Cu+-catalyzed “click reaction” between azido-DNA and poly(L-glutamic acid240-co-γ-propargyl-L-glutamate20) (p(LGA240-co-PLG20), Mw 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 060, PDI 1.4) following an established method (Fig. S1, ESI†).49 Component 2 “DNA linker” is a double stranded DNA (dsDNA) with two identical “sticky ends” (Fig. S2, ESI†). The sequences of the “sticky ends” are fully or partially complementary to that of ssDNA grafted onto the polypeptide backbone (for the exact sequences, see Table S1, ESI†). When the two components are mixed at a 1:1 molar ratio of “sticky ends” with a total mass content of 5 wt% in 100 mM PBS buffer (pH 7.4 with 150 mM NaCl), an optically transparent hydrogel is formed within seconds. As shown in Fig. 1b, when the oscillatory shear rheological test was carried out at a fixed strain of 1% and a frequency of 1 Hz at 25 °C, we can see that G′ (storage modulus, 4845.5 ± 95.6 Pa) was significantly higher than G′′ (loss modulus, 376.1 ± 8.1 Pa). This result further verified that the hydrogel was indeed formed as expected. As usual, we systematically investigated the hydrogel mechanical strength with different mass contents and found that the higher the mass content, the denser the cross-linking point, and the stronger the mechanical strength. It is noteworthy that the maximum water content of our hydrogel can be as high as 99.7 wt% (Fig. 1c), demonstrating the excellent water storage capacity of the hydrogel.
060, PDI 1.4) following an established method (Fig. S1, ESI†).49 Component 2 “DNA linker” is a double stranded DNA (dsDNA) with two identical “sticky ends” (Fig. S2, ESI†). The sequences of the “sticky ends” are fully or partially complementary to that of ssDNA grafted onto the polypeptide backbone (for the exact sequences, see Table S1, ESI†). When the two components are mixed at a 1:1 molar ratio of “sticky ends” with a total mass content of 5 wt% in 100 mM PBS buffer (pH 7.4 with 150 mM NaCl), an optically transparent hydrogel is formed within seconds. As shown in Fig. 1b, when the oscillatory shear rheological test was carried out at a fixed strain of 1% and a frequency of 1 Hz at 25 °C, we can see that G′ (storage modulus, 4845.5 ± 95.6 Pa) was significantly higher than G′′ (loss modulus, 376.1 ± 8.1 Pa). This result further verified that the hydrogel was indeed formed as expected. As usual, we systematically investigated the hydrogel mechanical strength with different mass contents and found that the higher the mass content, the denser the cross-linking point, and the stronger the mechanical strength. It is noteworthy that the maximum water content of our hydrogel can be as high as 99.7 wt% (Fig. 1c), demonstrating the excellent water storage capacity of the hydrogel.
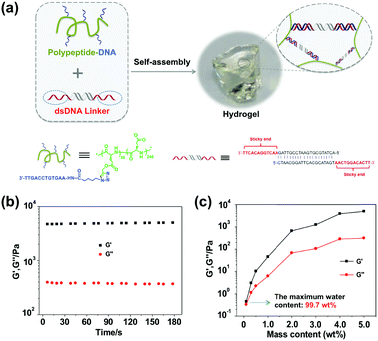 |
| | Fig. 1 Supramolecular hydrogel preparation and mechanical characterization. (a) The hydrogel is formed via DNA hybridization by mixing two complementary components, i.e. polypeptide–DNA and the dsDNA linker. (b) Rheological properties of the hydrogel (5 wt%) were characterized at a fixed strain of 1% and a frequency of 1 Hz at 25 °C. (c) The mechanical properties of hydrogels with different mass contents were tested at a fixed strain of 1% and a frequency of 1 Hz at 25 °C. And the maximum water content is as high as 99.7 wt% (the weight of salt is ignored here). | |
Tuning mechanical properties via adjusting the cross-linker stability
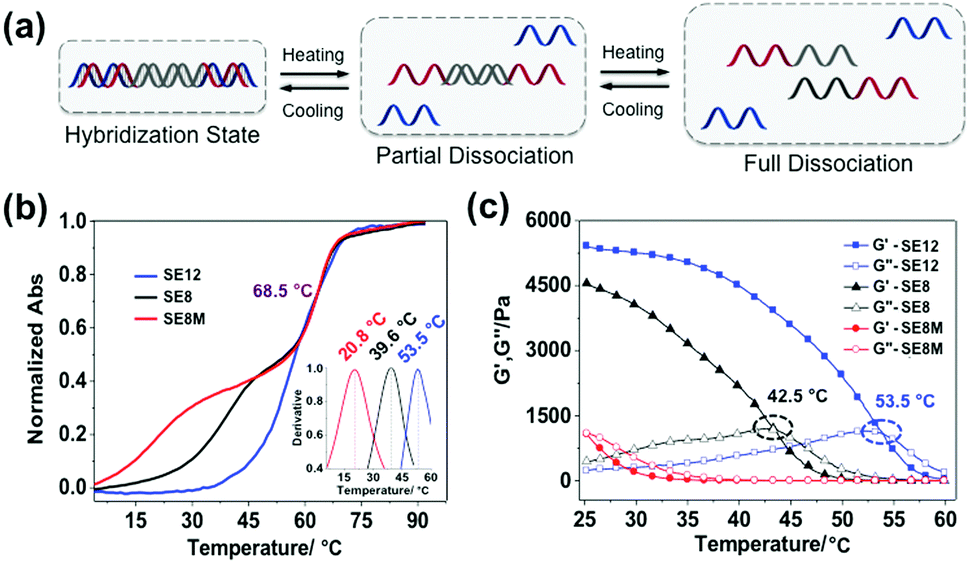
The rheological properties of these polypeptide–DNA hydrogels could be adjusted by tuning the DNA cross-linker stability while keeping the cross-linking density the same. To verify our strategy, a linear dsDNA linker was designed to possess a 20 base-pair (20 bp) middle double-stranded region, which is longer than the length of “sticky ends” ensuring its higher stability. By rational design, we selected three typical “sticky ends”: 12-base long ssDNA, which is fully complementary to the sequences grafted on the polypeptide (named SE12, for a detailed sequence see Table S1, ESI†); 8-base long ssDNA which is fully complementary to the grafted DNA from the 3′ end (named SE8); and SE8M which is a single-base mismatch sequence at the 4th base from the 5′ end to SE8. As illustrated in Fig. 2a, upon heating, the assembled DNA structures will collapse from the weakest link. Here we refer to the “sticky end” region formed with SE12, SE8 or SE8M; then the 20 bp region will also disassociate at higher temperatures. These designs are verified by monitoring their melting profiles from 4 °C to 90 °C using UV-Vis spectroscopy at 260 nm. From Fig. 2b, we can find that all these three assemblies have two distinct melting regions, which indicate two separate dissociation processes. According to different sequences, the lower melting points (Tms) of SE12, SE8 and SE8M are measured as 53.5 °C, 39.6 °C and 20.8 °C respectively, but they all have a common higher Tm at 68.5 °C. These results illustrate that, by changing the sequence analog, the stability of the DNA linker can be obviously different and suitable for the comparison of the mechanical properties.
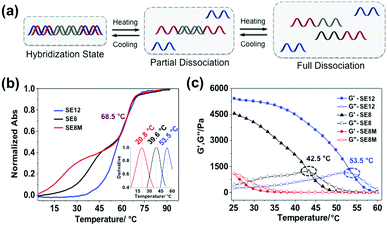 |
| | Fig. 2 Controlling the thermal stability of the DNA duplex via programming of the DNA sequences. (a) Schematic representation of the stepwise de-hybridization process triggered by heating. (b) Melting points of the DNA linker with different lengths of “sticky ends” were tested using UV-thermal spectra from 4 °C to 90 °C and calculated by the first derivative of each melting cure from 4 °C to 60 °C (inset curve). (c) The gel-to-sol transition process was characterized via a temperature sweep from 25 °C to 60 °C with a fixed frequency of 1 Hz, a strain of 1% and a temperature increasing speed of 2 °C min−1. | |
Then these three DNA linkers were applied to the protocol described before to prepare 5 wt% hydrogels and their thermal/mechanical properties were studied using a rheological temperature sweep test. As shown in Fig. 2c, the G′ of the hydrogels containing the SE12 linker continuously decreased whilst G′′ slowly increased along with increasing temperature, and they crossed at 53.5 °C, which indicates the transition from the hydrogel to the sol state. It is interesting to find that this transition temperature matches the lower Tm of SE12 very well. We can propose that the dissociation behavior of the ‘weakest link’ in the network is the deciding factor for their mechanical properties, because similar behavior has been seen with the SE8 system. For the SE8M system, there is no obvious transition point that has been observed and the G′ was not higher than G′′ over the entire temperature scan from 25 °C to 60 °C, indicating the system is in the sol state as the Tm of the SE8M duplex domain is only 20.8 °C. These results demonstrated that the thermal behavior of the mechanical properties of the hydrogel is strongly related to the strength of the cross-linker if its cross-linking point density and network topology are fixed.
In another way, under a given environmental temperature, the influence of cross-linker stability on the mechanical properties of the hydrogel can be observed directly. As shown in Fig. 3a, at 25 °C, the SE8M system (5 wt%) turns out to be in a sol state which collapsed on the substrate, while the SE8 system (5 wt%) is in a soft gel state and the SE12 system keeps a stiff gel state, which could maintain its original shape when put on the 2D substrate. As summarized in Fig. 3b, the SE8M system (5 wt%) has a G′ (168.5 ± 4.6 Pa) that was significantly lower than G′′ (482.2 ± 7.6 Pa), further indicating that it is in the sol state; in the meantime, the SE8 system has a G′ at 3952.4 ± 209.4 Pa and G′′ at 737.8 ± 3.9 Pa, which is a standard gel; and unsurprisingly, the SE12 system shows a higher G′ at 4929.7 ± 117.7 Pa and G′′ at 310.4 ± 9.4 Pa. In these three systems, there is no difference except the stability of the cross-linkers, thus we believe that the stability of the cross-linkers could definitely decide the mechanical strength of the hydrogel formed or even prevent the formation of the network. It is also noteworthy that the G′′ of SE12 is lower than that of SE8, which makes the tan δ (the value of G′′/G′) of SE12 significantly lower than that of SE8, which means it is much more stiff. This trend illustrates that the stability of the cross-linkers also plays an important role in deciding the stiffness of the material.50 As the stability of a DNA duplex could be continuously tuned by its composition and analog, we can propose that this strategy will enable the preparation of hydrogels with finely tuned mechanical strength from tens to thousands Pascal as well as stiffness, which will benefit the understanding of mechanical influences on cell differentiation.
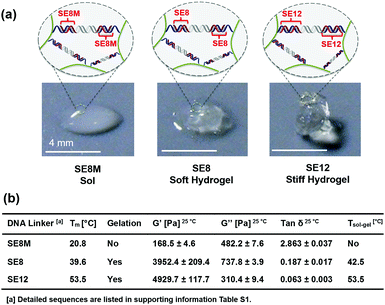 |
| | Fig. 3 (a) Tuning the mechanical properties of the hydrogels by simply increasing or decreasing the length of the “sticky ends” or inserting a mismatch without changing the cross-linking point density and the network topology. (b) The stability of DNA linkers with different lengths of “sticky ends” is listed and the resultant distinctive hydrogel modulus and sol–gel transition temperature are shown. | |
Tuning mechanical properties via pH-control
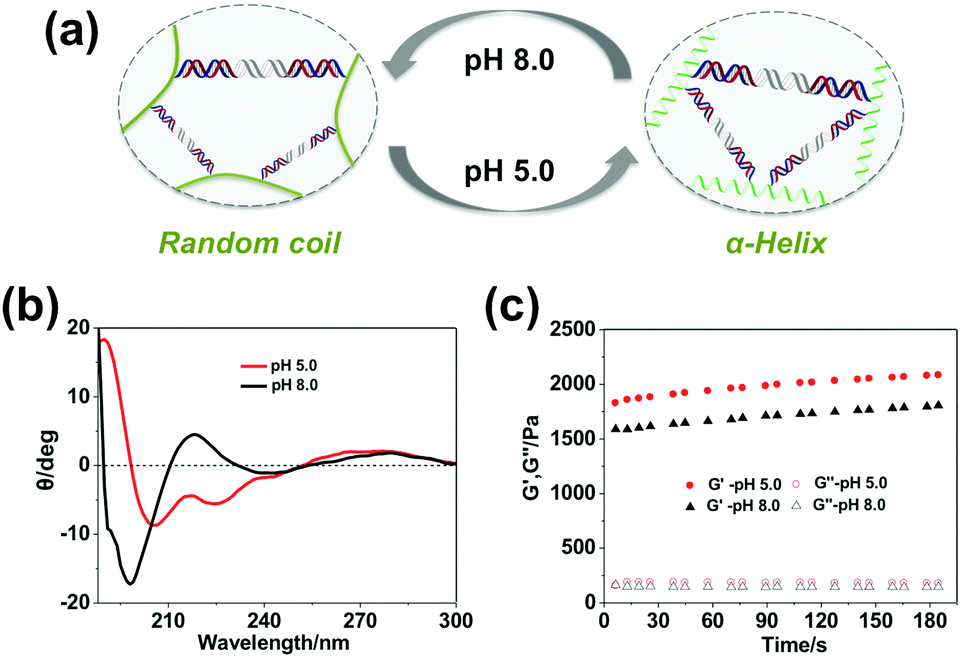
Additionally, besides tailoring the thermal stability of the DNA cross-linkers, altering the conformation of the polypeptide backbone can also change the mechanical properties of the hydrogel. It was found that the secondary structure of the polypeptide backbone could undergo reversible transitions between α-helix and random coil triggered by environmental pH (Fig. 4a), which was verified through circular dichroism (CD) characterization. As shown in Fig. 4b, the polypeptide backbone exhibited a predominantly α-helical conformation (negative bands: 222 nm and 208 nm) at pH 5.0 because of protonation of the carboxyl group, whereas random-coil conformation (positive band: 220 nm; negative band: 198 nm) predominated due to deprotonation of the carboxyl group at pH 8.0. As shown in Fig. 4c, when the pH value changed from 5.0 to 8.0, the storage modulus (G′) of 3 wt% hydrogels containing the SE12 linker changes from 2032.7 ± 98.8 Pa to 1755.5 ± 93.4 Pa, indicating that the hydrogel became much more stiff under the acidic environment. It has been demonstrated that the stability of DNA duplexes will decrease with the pH value;51 this phenomenon can only be attributed to the conformational changes of the polypeptide backbone, where the α-helix at pH 5.0 is more rigid and shorter than the random-coil at pH 8.0. These results demonstrate that the polypeptide–DNA hydrogel networks could have more responsiveness upon the rational design of their polypeptide backbone.
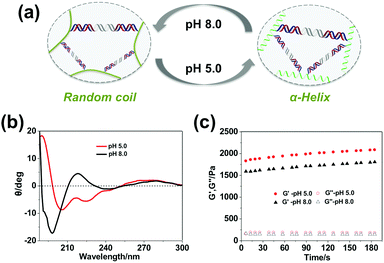 |
| | Fig. 4 Tuning the mechanical properties of the hydrogels by changing the environmental pH. (a) The secondary structure of the polypeptide backbone is sensitive to pH: at pH 5.0, it shows an α-helix; at pH 8.0, it shows a random coil. (b) CD characterization of the conformations of polypeptide–DNA at different pH. (c) The mechanical properties of the hydrogels (3 wt%) at different pH values, which was characterized at a fixed strain of 1% and a frequency of 1 Hz at 25 °C. | |
Thermal responsiveness
Due to the intrinsic non-covalent properties of DNA recognition, our supramolecular hydrogel should undergo predictable, reversible gel–sol transition via hybridization or de-hybridization controlled by temperature changes. Based on the thermal stability study of the DNA cross-linkers, using here the SE12 system as an example, 25 °C and 60 °C were chosen as the two fixed temperatures to investigate the sol–gel transition behaviors. As shown in Fig. 5a, the G′ of the hydrogel (5 wt%) was significantly higher than the G′′ at 25 °C which is a typical feature of a hydrogel; however, the G′ became much lower than G′′ when the temperature changed from 25 °C to 60 °C, indicating that the hydrogel network was broken, resulting in a sol-state. This sol-state could return to the original gel-state when the temperature returned to 25 °C. Furthermore, this gel-to-sol switching process is highly reversible and can be cycled many times, demonstrating that the hydrogels possess varying mechanical properties during different temperatures. It should be noted that our hydrogel can also be mechanically tuned by other methods such as varying the relative molar ratio of the two components. In our study, the DNA linker (bp12) and polypeptide–DNA were mixed together with different molar ratios of “sticky ends” at 0.6:1, 0.8:1, 1.0:1 and 1.2:1, respectively, with a fixed total mass content of 5 wt%. Theoretically, the mechanical properties of the hydrogels should be the highest when the “sticky ends” from the DNA linker and polypeptide–DNA are in the ratio of 1:1, i.e., the networks are fully cross-linked. In our experiment, as shown in Fig. 5b, the hydrogel at the ratio of 0.8:1 and 1.0:1 exhibited the highest mechanical strength, and beyond this range, when the system had either polypeptide–DNA or the DNA-linker in excess, the hydrogel exhibited a lower mechanical strength. These results further verified that the mechanical properties of the polypeptide–DNA hydrogel can also be fine-tuned by varying the temperature or molar ratio of two components.
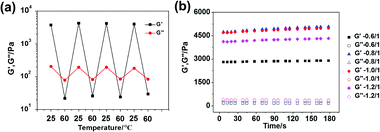 |
| | Fig. 5 (a) A thermal-triggered sol–gel transition process between 25 °C and 60 °C was performed at a fixed frequency (1 Hz) and strain (1%) for the SE12 system. (b) The mechanical properties can be tuned by varying the molar ratio of the two components with a fixed mass content of 5 wt% for the SE12 system. | |
Conclusions
In summary, we prepared a supramolecular hydrogel cross-linked by the formation of DNA duplexes. By modulating the stability of the DNA cross-linking points through rational design sequences, the mechanical strength of the hydrogels can be tuned from several Pascal to several thousand Pascal while keeping the cross-linking density and the network topology unchanged. In addition, the pH-triggered transformation of the polypeptide backbone can also influence the rheological properties of the hydrogel. Together with physiological gelation, thermal responsiveness, excellent applicability in three-dimensional ink-jet cell printing technology and demonstrated biocompatibility,49 we believe that these hydrogels with solely tuned mechanical strength will promote studies on mechanical induced cell differentiation in situ, and have great potential in tissue engineering.
Acknowledgements
The authors thank the financial support by the National Basic Research Program of China (973 program, 2013CB932803), the National Natural Science Foundation of China (No. 21534007, 91427302, 21421064), the NSFC-DFG joint project TRR61 and Beijing Municipal Science and Technology Commission. Dr P. Chen acknowledges the National Natural Science Foundation of China (51403073).
Notes and references
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869 CrossRef CAS PubMed.
- D. Seliktar, Science, 2012, 336, 1124 CrossRef CAS PubMed.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352 CrossRef CAS PubMed.
- M. C. Cushing and K. S. Anseth, Science, 2007, 316, 1133 CrossRef CAS PubMed.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677 CrossRef CAS PubMed.
- W. L. Murphy, T. C. McDevitt and A. J. Engler, Nat. Mater., 2014, 13, 547 CrossRef CAS PubMed.
- R. H. Pritchard, Y. Y. Huang and E. M. Terentjev, Soft Matter, 2014, 10, 1864 RSC.
- V. Vogel and M. Sheetz, Nat. Rev. Mol. Cell Biol., 2006, 7, 265 CrossRef CAS PubMed.
- P. M. Gilbert, K. L. Havenstrite, K. E. Magnusson, A. Sacco, N. A. Leonardi, P. Kraft, N. K. Nguyen, S. Thrun, M. P. Lutolf and H. M. Blau, Science, 2010, 329, 1079 CrossRef PubMed.
- M. S. Hahn, J. S. Miller and J. L. West, Adv. Mater., 2006, 18, 2679 CrossRef CAS.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59 CrossRef CAS PubMed.
- M. Guvendiren and J. A. Burdick, Nat. Commun., 2012, 3, 792 CrossRef PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- Y. Zhang, B. Zhang, Y. Kuang, Y. Gao, J. Shi, X. X. Zhang and B. Xu, J. Am. Chem. Soc., 2013, 135, 5008 CrossRef CAS PubMed.
- M. J. Glassman, J. Chan and B. D. Olsen, Adv. Funct. Mater., 2013, 23, 1182 CrossRef CAS PubMed.
- M. Gottlieb, C. W. Macosko, G. S. Benjamin, K. O. Meyers and E. W. Merrill, Macromolecules, 1981, 14, 1039 CrossRef CAS.
- S. K. Patel, S. Malone, C. Cohen, J. R. Gillmor and R. H. Colby, Macromolecules, 1992, 25, 5241 CrossRef CAS.
-
A. M. Grillet, N. B. Wyatt, L. M. Gloe, Polymer gel rheology and adhesion, 2012, InTech, 59.
- D. Liu, E. Cheng and Z. Yang, NPG Asia Mater., 2011, 3, 109 CrossRef.
- Y. Dong, Z. Yang and D. Liu, Acc. Chem. Res., 2014, 47, 1853 CrossRef CAS PubMed.
- J. Bath and A. J. Turberfield, Nat. Nanotechnol., 2007, 2, 275 CrossRef CAS PubMed.
- X. Liu, C. Lu and I. Willner, Acc. Chem. Res., 2014, 47, 1673 CrossRef CAS PubMed.
- H. Liang, X. Zhang, Y. Lv, L. Gong, R. Wang, X. Zhu, R. Yang and W. Tan, Acc. Chem. Res., 2014, 47, 1891 CrossRef CAS PubMed.
- N. C. Seeman, Nature, 2003, 421, 427 CrossRef PubMed.
- Z. Yang, H. Liu and D. Liu, NPG Asia Mater., 2015, 7, e161 CrossRef CAS.
- M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 840 CrossRef CAS PubMed.
- A. Samanta, Z. Deng, Y. Liu and H. Yan, Nano Res., 2013, 6, 853 CrossRef CAS.
- M. Kwak and A. Herrmann, Chem. Soc. Rev., 2011, 40, 5745 RSC.
- D. Yang, M. R. Hartman, T. L. Derrien, S. Hamada, D. An, K. G. Yancey, R. Cheng, M. Ma and D. Luo, Acc. Chem. Res., 2014, 47, 1902 CrossRef CAS PubMed.
- S. H. Um, J. B. Lee, N. Park, S. Y. Kwon, C. C. Umbach and D. Luo, Nat. Mater., 2006, 5, 797 CrossRef CAS PubMed.
- E. Cheng, Y. Xing, P. Chen, Y. Yang, Y. Sun, D. Zhou, L. Xu, Q. Fan and D. Liu, Angew. Chem., Int. Ed., 2009, 121, 7796 CrossRef.
- E. Cheng, Y. Li, Z. Yang, Z. Deng and D. Liu, Chem. Commun., 2011, 47, 5545 RSC.
- Y. Xing, E. Cheng, Y. Yang, P. Chen, T. Zhang, Y. Sun, Z. Yang and D. Liu, Adv. Mater., 2011, 23, 1117 CrossRef CAS PubMed.
- J. Jin, Y. Xing, Y. Xi, X. Liu, T. Zhou, X. Ma, Z. Yang, S. Wang and D. Liu, Adv. Mater., 2013, 25, 4714 CrossRef CAS PubMed.
- W. Guo, R. Orbach, I. Mironi-Harpaz, D. Seliktar and I. Willner, Small, 2013, 9, 3748 CrossRef CAS PubMed.
- Z. Zhu, C. Wu, H. Liu, Y. Zou, X. Zhang, H. Kang, C. J. Yang and W. Tan, Angew. Chem., Int. Ed., 2010, 49, 1052 CrossRef CAS PubMed.
- N. Dave, M. Y. Chan, P.-J. J. Huang, B. D. Smith and J. Liu, J. Am. Chem. Soc., 2010, 132, 12668 CrossRef CAS PubMed.
- B. Wei, I. Cheng, K. Q. Luo and Y. Mi, Angew. Chem., Int. Ed., 2008, 47, 331 CrossRef CAS PubMed.
- H. Jiang, V. Pan, S. Vivek, E. R. Weeks and Y. Ke, ChemBioChem, 2016, 17, 1156 CrossRef CAS PubMed.
- J. E. Godfrey and H. Eisenberg, Biophys. Chem., 1976, 5, 301 CrossRef CAS PubMed.
- G. S. Manning, Biopolymers, 1981, 20, 1751 CrossRef CAS.
- L. Peng, M. You, Q. Yuan, C. Wu, D. Han, Y. Chen, Z. Zhong, J. Xue and W. Tan, J. Am. Chem. Soc., 2012, 134, 12302 CrossRef CAS PubMed.
- C. Li, M. Rowland, Y. Shao, T. Cao, C. Chen, H. Jia, X. Zhou, Z. Yang, O. Scherman and D. Liu, Adv. Mater., 2015, 27, 3298 CrossRef CAS PubMed.
- C. Li, P. Chen, Y. Shao, X. Zhou, Y. Wu, Z. Yang, Z. Li, T. Weil and D. Liu, Small, 2015, 11, 1138 CrossRef CAS PubMed.
- Y. K. Cheng and B. M. Pettitt, Prog. Biophys. Mol. Biol., 1992, 58, 225 CrossRef CAS PubMed.
- F. X. Jiang, B. Yurke, R. S. Schloss, B. L. Firestein and N. A. Langrana, Biomaterials, 2010, 31, 1199 CrossRef CAS PubMed.
- C. Li, A. Faulkner-Jones, A. R. Dun, J. Jin, P. Chen, Y. Xing, Z. Yang, Z. Li, W. Shu, D. Liu and R. R. Duncan, Angew. Chem., Int. Ed., 2015, 54, 3957 CrossRef CAS PubMed.
- W. C. Yount, D. M. Loveless and S. L. Craig, Angew. Chem., Int. Ed., 2005, 44, 2746 CrossRef CAS PubMed.
- X. Zhou, C. Li, Y. Shao, C. Chen, Z. Yang and D. Liu, Chem. Commun., 2016, 52, 10668 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qm00176a |
|
| This journal is © the Partner Organisations 2017 |

 a,
Zhongqiang
Yang
a,
Zhibo
Li
c and
Dongsheng
Liu
*a
a,
Zhongqiang
Yang
a,
Zhibo
Li
c and
Dongsheng
Liu
*a