
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and synthesis of squaramide-based MOFs as efficient MOF-supported hydrogen-bonding organocatalysts†
Xiaoping
Zhang‡
ab,
Zhenjie
Zhang‡
a,
Jake
Boissonnault
a and
Seth M.
Cohen
*a
aDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, USA. E-mail: scohen@ucsd.edu
bDepartment of Chemistry, Key Laboratory of Advanced Energy Materials Chemistry (MOE), Collaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, P. R. China
First published on 14th June 2016
Abstract
Herein, we utilize a new, squaramide-based ligand, combined with a postsynthetic exchange (PSE) synthetic approach to prepare a series of Cu(II)–squaramide MOFs that are active catalysts for the Friedel–Crafts reaction.
Among various organocatalysts, those based on the thiourea core are broadly utilized in the field of H-bond donor catalysis.1 Recently, bifunctional squaramide moieties have emerged as powerful hydrogen-bonding groups for Lewis acid catalysis.2–8 Squaramides are four-membered ring systems, which are derived from squaric acid and possess hydrogen-bond accepting and donating functionality through their carbonyl and N–H groups, respectively. The squaramide functionality possesses features such as ditopic binding, structural rigidity, high N–H acidity, and ease of preparation.9 Squaramide compounds have been shown to be competent for biomimetic transport,5,10,11 molecular recognition,12 ion sensing,13,14 and organocatalysis.15,16 The high propensity for hydrogen-bonding is driven via a concomitant increase in aromaticity on the squaramide ring.17 Although the increased acidity of squaramides can be useful in hydrogen-bond donating organocatalysis, the strong hydrogen-bonding also drives self-association/aggregation of squaramides that impedes catalysis.18 To prevent self-association and enhance the catalytic performance of squaramides, one strategy is to immobilize these groups within porous materials such as metal–organic frameworks (MOFs).19,20
MOFs are a class of porous, crystalline materials composed of inorganic nodes and organic linkers. They have uniform 3-dimensional structures of high surface area, large pore sizes, low density, and have potential applications in gas sorption,21,22 molecular recognition,23 proton conductivity,24 and organocatalysis.25–29 Along with their modular synthesis and tunable porosity, MOFs constitute attractive candidates as platforms for heterogeneous catalysis.
To incorporate squaramide catalysts into MOFs, a feasible design approach is to employ squaramide compounds as ligand struts for MOFs. Hupp, Farha, and Mirkin developed a mixed-ligand MOF catalyst, UiO-67-Squar/bpdc,20 which was constructed using a combination of unfunctionalized and squaramide-appended biphenyl dicarboxylate (bpdc) ligands. In this system, the squaramide group was attached as a ‘dangling’ substituent off of the bpdc ligand framework. The percentage of squaramide ligand incorporation was varied from 0–100%. The authors reported that MOFs with 100% squaramide ligand incorporation showed low catalytic activity, similar to those MOFs with 100% unfunctionalized bpdc ligand (e.g. no catalytic sites). MOFs with 50% squaramide ligand exhibited the highest catalytic activity, which was attributed to an optimal balance of catalytic sites and sufficient retention of MOF porosity. Herein, an alternative approach presented for squaramide-based MOF catalysts is described. A new squaramide ligand with four carboxylate ligating groups (5,5′-(3,4-dioxocylcobut-1-ene-1,2-diyl)bis(azanediyl)diisophthalic acid, H4dbda, Fig. 1) has been designed and synthesized. Using H4dbda, we developed a stable Cu-MOF, [Cu2(dbda)(CH3OH)2] (Cu(dbda)), through a postsynthetic exchange (PSE) reaction from its Zn(II) analogue, [Zn2(dbda)(H2O)2] (Zn(dbda)). The Friedel–Crafts reaction, a prototypical reaction for hydrogen-bonding organocatalyst,30–34 was selected to evaluate the catalytic performance of these MOFs (Fig. 1). Cu(dbda) demonstrated high catalytic efficiency for the Friedel–Crafts reaction between indole and substituted β-nitrostyrenes while Zn(dbda) was not a catalyst due to its poor stability. The Cu(dbda) system allows for 100% incorporation of the H4dbda squaramide ligand without a significant loss of MOF porosity.
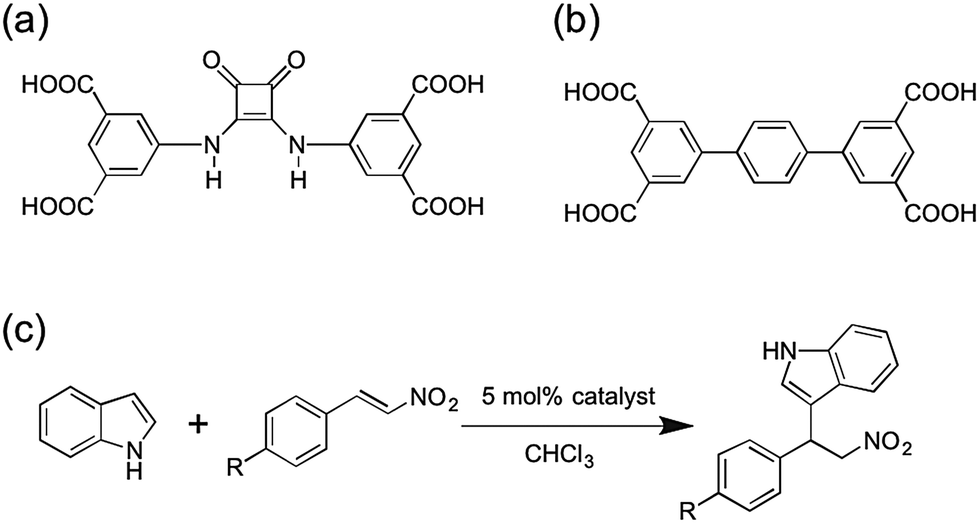 | ||
| Fig. 1 (a) Structure of the H4dbda ligand. (b) Structure of H4tptc ligand. (c) Friedel–Crafts reaction between indole and substituted β-nitrostyrenes. | ||
The H4dbda ligand was synthesized using a modified literature procedure.35 Both 1H NMR and MS data confirmed the composition of the ligand (Fig. S1 and S2, ESI†). Combining H4dbda and Zn(NO3)2 in a mixture of DMF and EtOH at 80 °C for 24 h afforded pale-yellow crystals of Zn(dbda). Single-crystal X-ray diffraction (XRD) structure determination revealed that Zn(dbda) crystallized in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group with unit cell parameters a = b = 18.8687(9) Å, c = 38.3555(17) Å, α = β = 90°, γ = 120° (Table S1, ESI†). As shown in Fig. 2, Zn(dbda) is built by [Zn2(COO)4] paddlewheel secondary building units (SBUs). The 1,3-benzenedicarboxylate (1,3-bdc) moieties in dbda4− are linked by [Zn2(COO)4] SBUs to form a 2-dimensional Kagomé lattice that are connected via the squaramide core to form an overall 3-dimensional framework with nbo topology (Fig. S3 and S4, ESI†). There are large channels with approximate dimensions of 13 × 5 Å along the crystallographic c-axis. N2 sorption of Zn(dbda) showed essentially no uptake (Fig. 3), which is attributed to a loss of Zn(dbda) crystallinity upon thermal activation (60 °C, 10 h) as evidenced by powder X-ray diffraction (PXRD, Fig. S5, ESI†). Zn(dbda) was found to be stable in common organic solvents such as CHCl3 for 24 h at room temperature, but not stable in water (Fig. S6 and S7, ESI†). Despite these stability limitations, the catalytic performance of Zn(dbda) in the Friedel–Crafts reaction was examined. Indole (0.15 mmol) and β-nitrostyrene (0.1 mmol) were chosen as test substrates, and the reaction was performed at 50 °C in CHCl3 with 5 mol% loading of the Zn(dbda) catalyst (based on an empirical formula of Zn4(dbda)2(H2O)4); however, no products were observed after 24 h (Fig. S8, ESI†). PXRD of Zn(dbda) after 24 h revealed that the MOF had lost crystallinity under the reaction conditions (Fig. S9, ESI†).
m space group with unit cell parameters a = b = 18.8687(9) Å, c = 38.3555(17) Å, α = β = 90°, γ = 120° (Table S1, ESI†). As shown in Fig. 2, Zn(dbda) is built by [Zn2(COO)4] paddlewheel secondary building units (SBUs). The 1,3-benzenedicarboxylate (1,3-bdc) moieties in dbda4− are linked by [Zn2(COO)4] SBUs to form a 2-dimensional Kagomé lattice that are connected via the squaramide core to form an overall 3-dimensional framework with nbo topology (Fig. S3 and S4, ESI†). There are large channels with approximate dimensions of 13 × 5 Å along the crystallographic c-axis. N2 sorption of Zn(dbda) showed essentially no uptake (Fig. 3), which is attributed to a loss of Zn(dbda) crystallinity upon thermal activation (60 °C, 10 h) as evidenced by powder X-ray diffraction (PXRD, Fig. S5, ESI†). Zn(dbda) was found to be stable in common organic solvents such as CHCl3 for 24 h at room temperature, but not stable in water (Fig. S6 and S7, ESI†). Despite these stability limitations, the catalytic performance of Zn(dbda) in the Friedel–Crafts reaction was examined. Indole (0.15 mmol) and β-nitrostyrene (0.1 mmol) were chosen as test substrates, and the reaction was performed at 50 °C in CHCl3 with 5 mol% loading of the Zn(dbda) catalyst (based on an empirical formula of Zn4(dbda)2(H2O)4); however, no products were observed after 24 h (Fig. S8, ESI†). PXRD of Zn(dbda) after 24 h revealed that the MOF had lost crystallinity under the reaction conditions (Fig. S9, ESI†).
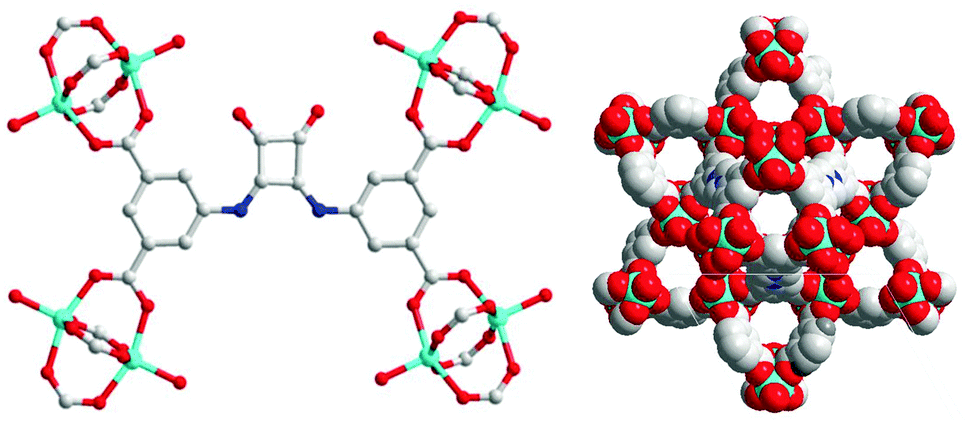 | ||
| Fig. 2 Coordination environment of Zn(dbda) (left). Packing of Zn(dbda) along the crystallographic c-axis (right). Color code: O, red; N, blue; Zn, cyan; C, gray. | ||
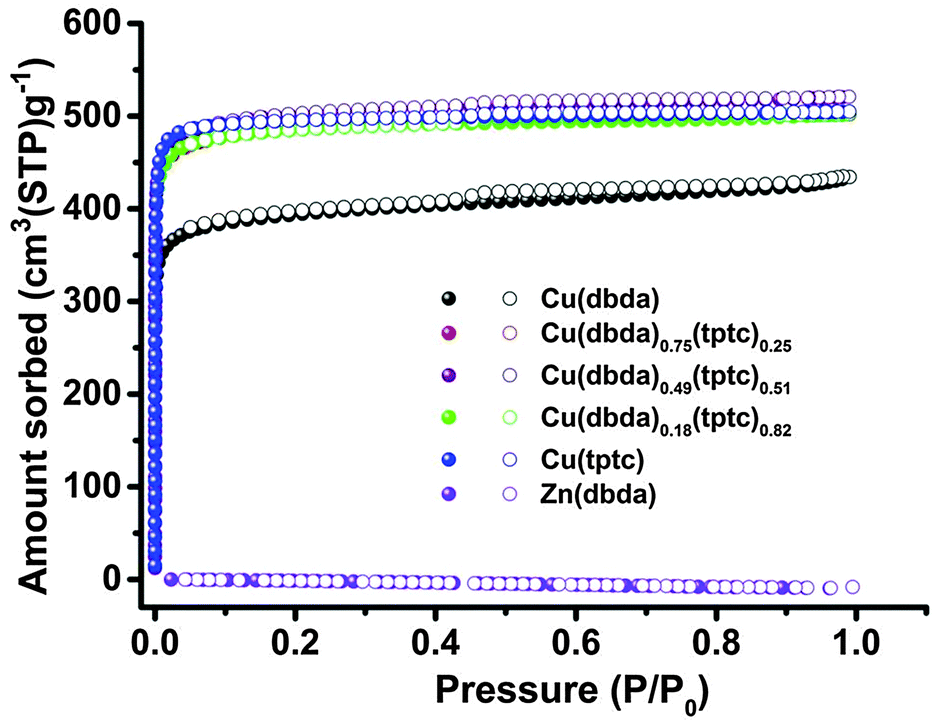 | ||
| Fig. 3 N2 gas sorption of Cu(dbda)x(tptc)1−x (x = 1, 0.75, 0.49, 0.18, 0). Adsorption and desorption branches are shown with filled and empty symbols, respectively. | ||
Literature reports suggest that [Zn2(COO)4] SBUs may possess low chemical stability.36 However, there are reports of Zn(II) SBUs that can undergo PSE with Cu(II) cations and the resulting Cu(II)-based SBUs showing greater chemical stability.37 PSE of Zn(dbda) with Cu(NO3)2 produced a new compound Cu(dbda) (Fig. S10, ESI†); attempts to prepare Cu(dbda) directly from H4dbda and various Cu salts were unsuccessful under a variety of reaction conditions. Both EDX (∼99% Cu) and ICP-OES (∼99% Cu) showed that the Zn(II) ions were essentially completely replaced by Cu(II) by this PSE process (Fig. S11 and Table S2, ESI†). XRD of Cu(dbda) revealed that Cu(dbda) exhibited the same structure as Zn(dbda), with only slight differences in cell parameters: a = b = 18.2623(6) Å, c = 39.6543(13) Å. Cu(dbda) possesses a slightly smaller unit cell (V = 11453.3(7) Å3) than Zn(dbda) (V = 11826.1(10) Å3) (Table S3, ESI†) due to the shorter Cu–O bonds. For Zn(dbda), the average distance of Zn–O (carboxylate) bonds is 2.036 Å, while for Cu(dbda), the average Cu–O (carboxylate) distance is 1.947 Å. Moreover, the metal–metal distance in the paddlewheel SBUs decreased from 2.999 Å (Zn–Zn) to 2.629 Å (Cu–Cu). The permanent porosity of Cu(dbda) was measured by N2 absorption at 77 K, which showed a typical type I isotherm,38 giving a BET surface area of 1516 ± 66 m2 g−1 and a total pore volume of 0.662 cm3 g−1 (Fig. 3). The PXRD pattern of Cu(dbda) remains intact upon immersion in organic solvents for 90 h, suggesting improved stability consistent with the gas sorption data; however, Cu(dbda) was unstable in water (Fig. S12 and S13, ESI†).
The model reaction between indole (0.15 mmol) and β-nitrostyrene (0.10 mmol, Fig. 1) was examined with 5 mol% loading of Cu(dbda) (based on an empirical formula of Cu4(dbda)2(CH3OH)4) as a catalyst. Under identical conditions to those reported for UiO-67-Squar/bpdc (CD2Cl2, 25 °C, 24 h), Cu(dbda) gave a yield of only 32%, compared to 78% for UiO-67-Squar/bpdc.20 Therefore, reaction conditions were optimized for Cu(dbda) including the use of different solvents (toluene, CH3CN, CHCl3) and temperatures (25, 35, and 50 °C) (Table S4 and Fig. S14, ESI†). It was found that CHCl3 gave the best results, and activity could be increased with increasing temperature from ∼60% (25 °C), to ∼78% (35 °C), to >99% (50 °C). Control reactions either without catalyst or with free H4dbda ligand (not as part of a MOF) were also performed under the same optimized reaction conditions. Without catalyst, no product was observed. The catalytic activity of free H4dbda was much lower than the MOF, giving a yield of only 33% after 24 h (Fig. S15, ESI†). The improved activity of Cu(dbda) suggests that the MOF prevents squaramide self-aggregation, which likely causes the sluggish activity of the free H4dbda ligand. A time-dependent study was performed from 30 min to 24 h (Fig. S16, ESI†). The conversion of β-nitrostyrene was ∼60% (turnover frequency, TOF = 3.0 × 10−3 s−1) after 1 h, with 95% conversion after 8 h (TOF = 6.6 × 10−4 s−1), and complete consumption of the starting material after 24 h.
An advantage of heterogeneous catalysts is reusability, which was also investigated for Cu(dbda). Only a small decrease in activity (from >99% to ∼96%) was observed after 5 runs (Fig. S17, ESI†). Characterization of Cu(dbda) after the 5th run showed the catalyst retained its crystallinity, with the PXRD pattern of the recycled Cu(dbda) in good agreement with the calculated patterns (Fig. S18, ESI†). A test was performed to confirm the heterogeneity of the catalysts (and rule out soluble species) by removing the MOF catalyst by filtration after 30 min (at which time the yield was ∼22%). The filtrate was then re-analyzed after a total of 24 h showing no formation of new product and indicating that the catalyst is heterogeneous and there is no leaching of a catalytic species into solution (Fig. S16, ESI†).20 The substrate scope for various substituted-nitrostyrene derivatives was also examined to assess the utility of Cu(dbda) (Fig. S19, ESI†). Good to excellent yields, ranging from ∼61% for 4-nitro-β-nitrostyrene (poor solubility in CHCl3) to 97% for 4-chloro-β-nitrostyrene, were obtained (Table 1). Overall, these results show that Cu(dbda) is an efficient, recyclable, heterogeneous catalyst for the Friedel–Crafts reaction.
| Entry | Ra | Catalyst | Temp. (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| Conditions: indole (0.15 mmol), β-nitrostyrenes (0.10 mmol), and catalyst (5 mol%) in CHCl3 (1 mL). Yields monitored by 1H NMR. See ESI for details.a Catalyst loading was 10 mol%.b Catalyst loading was 20 mol%.c Conditions: indole (0.02 mmol), β-nitrostyrene (0.014 mmol), and catalyst (10 mol%) in toluene (0.7 mL), see ref. 20 for details. | |||||
| 1 | H | None | 50 | 24 | 0 |
| 2 | H | H4dbdaa | 50 | 24 | 32 |
| 3 | H | Cu(NO3)2b | 50 | 24 | 28 |
| 4 | H | Zn(dbda) | 50 | 24 | 0 |
| 5 | H | Cu(dbda) | 25 | 24 | 60 |
| 6 | H | Cu(dbda) | 35 | 24 | 78 |
| 7 | H | Cu(dbda) | 50 | 1 | 60 |
| 8 | H | Cu(dbda) | 50 | 8 | 95 |
| 9 | H | Cu(dbda) | 50 | 24 | >99 |
| 10 | Cl | Cu(dbda) | 50 | 24 | 97 |
| 11 | F | Cu(dbda) | 50 | 24 | 91 |
| 12 | Br | Cu(dbda) | 50 | 24 | 92 |
| 13 | CH3 | Cu(dbda) | 50 | 24 | 90 |
| 14 | OCH3 | Cu(dbda) | 50 | 24 | 74 |
| 15 | NO2 | Cu(dbda) | 50 | 24 | 61 |
| 16 | H | Cu(dbda)0.75(tptc)0.25 | 50 | 24 | 99 |
| 17 | H | Cu(dbda)0.49(tptc)0.51 | 50 | 24 | 95 |
| 18 | H | UiO-67-Squar/bpdcc | 50 | 24 | 95 |
| 19 | H | UiO-67-Urea/bpdcc | 50 | 24 | 79 |
| 20 | H | Cu(dbda)0.18(tptc)0.82 | 50 | 24 | 88 |
| 21 | H | Cu(tptc) | 50 | 24 | 38 |
To further investigate the catalytic activity of Cu(dbda), a series of isostructural MOF catalysts, [Cu2(dbda)x(tptc)1−x(CH3OH)2] (Cu(dbda)x(tptc)1−x, x = 0.75, 0.49, 0.18, 0; H4tptc = p-terphenyl-3,5,3′′,5′′-tetracarboxylic acid, Fig. 1), were prepared via a mixed-ligand strategy; the single-crystal structure of Cu(tptc) was previously reported in 2006.39 The structures of the two tetracarboxylic ligands (H4dbda and H4tptc) are very similar, both using square-planar 4-connected nodes with the distance between 4 and 4′ carbon atoms in H4dbda of 11.57 Å versus 11.38 Å in H4tptc. The synthesis of Cu(dbda)x(tptc)1−x (x = 0.75, 0.49, 0.18) was achieved via a PSE approach using Zn(dbda)x(tptc)1−x MOFs as precursors. Both EDX and ICP-OES results (Table S2 and Fig. S20–S23, ESI†) showed the metal content of Cu(II) in all of the Cu(dbda)x(tptc)1−x (x = 0.75, 0.49, 0.18, 0) was >92%, thus indicating near complete PSE. The PXRD patterns of the mixed-ligand MOFs Cu(dbda)x(tptc)1−x were all in good agreement with calculated patterns (Fig. S24, ESI†). The ratio of dbda4− and tptc4− in Cu(dbda)x(tptc)1−x (x = 0.75, 0.49, 0.18) determined by 1H NMR (Fig. S25, ESI†) were very close to the expected values (0.75, 0.50, and 0.25 based on the ratio of starting materials). In addition, a direct solvothermal synthesis was examined for the preparation of Cu(dbda)x(tptc)1−x.39 However, the loading of dbda4− through solvothermal methods was much lower than the ratio used in the synthesis, with initial ratios of dbda4− of 0.50 and 0.25 giving incorporation of only x = 0.31 and 0.12, respectively. Moreover, Cu(dbda) could not be obtained using solvothermal methods (Fig. S26 and S27, ESI†). The permanent porosity of Cu(dbda)x(tptc)1−x were also evaluated by N2 sorption isotherms at 77 K, giving the expected type-I isotherms (Fig. 3). The BET surface areas of Cu(dbda)x(tptc)1−x (x = 0.75, 0.49, 0.18) were 1884 ± 61 m2 g−1, 1946 ± 124 m2 g−1, and 1887 ± 45 m2 g−1, respectively, with total pore volumes of Cu(dbda)x(tptc)1−x (x = 0.75, 0.49, 0.18) of 0.775 cm3 g−1, 0.803 cm3 g−1, and 0.775 cm3 g−1, respectively. These results show that the porosity of the Cu(dbda)x(tptc)1−x fall in the expected range between those of the single-ligand MOFs, Cu(dbda) (1516 m2 g−1) and Cu(tptc) (1942 m2 g−1).
Cu(dbda)x(tptc)1−x was employed as catalyst for the Friedel–Crafts reaction between indole (0.15 mmol) and β-nitrostyrene (0.1 mmol), carried out at 50 °C in CHCl3 for 24 h (5 mol% loading, based on the empirical formula Cu4(dbda)2x(tptc)2−2x(H2O)4, x = 0.75, 0.49, 0.18). Using Cu(tptc) (5 mol% loading, no squaramide sites) as a catalyst gave a low yield of ∼39% (Fig. S28, ESI†); the residual activity may arise from the Lewis acid Cu(II) sites, as Cu(II)-complexes have been reported to show catalytic activity for the Friedel–Crafts reaction.40 Consistent with this observation, Cu(NO3)2, gave a 28% conversion under the same reaction conditions. In contrast, for Cu(dbda)x(tptc)1−x (x = 1, 0.75, 0.49, 0.18, 0), activity increased with increasing dbda4− content: 39% (Cu(tptc)), 88% (Cu(dbda)0.18(tptc)0.82), 95% (Cu(dbda)0.49(tptc)0.51), 99% (Cu(dbda)0.75(tptc)0.25), and >99% (Cu(dbda)), respectively. These results verify that the squaramide group is the governing catalytic functional group in these MOFs. For a more direct comparison to the prior reports,20 a 2.45 mol% loading of catalyst Cu(dbda)0.49(tptc)0.51 achieved the same catalytic performance as the reported UiO-67-Squar/bpdc at 10 mol% loading (95% yield at 50 °C for 24 h).20 The result further verified the high activity of the dbda4−-based MOF.
Taken together, Cu(dbda) has features that compliment and distinguish it from the previously reported UiO-67-Squar/bpdc catalyst.20Cu(dbda) has a different MOF structure type from UiO-67-Squar/bpdc, wherein the catalytic group is part of the ligand ‘backbone’ rather than a dangling component. Depending on catalyst design, one can envision scenarios where one or the other functional group arrangement might be preferable. Also, in contrast to UiO-67-Squar/bpdc, Cu(dbda) allows for 100% functional ligand incorporation without loss of surface area. Indeed, using H4tptc as a complimentary, unfunctionalized ligand the Cu(dbda) system is not limited with respect to the amount of squaramide ligand that can be included while maintaining a high degree of porosity and activity. Both Cu(dbda)x(tptc)1−x and UiO-67-Squar/bpdc catalysts show good activity under similar conditions, and hence each helps advance the ability of utilizing squaramides in MOF-based catalytic systems.
In summary, we prepared a squaramide tetracarboxylate ligand, and developed catalytic squaramide MOFs prepared via metal PSE from Zn(II) precursor MOFs. The Cu(II) MOFs were more stable and hence showed good catalytic performance when compared to the unstable parent Zn(II) MOF, in a Friedel–Crafts reaction of indole with β-nitrostyrenes. A series of isostructural MOFs Cu(dbda)x(tptc)1−x (x = 0.75, 0.49, 0.18, 0) showed that the catalytic performance of these MOFs increased with increasing amounts of the squaramide ligand, demonstrating that squaramide MOFs are promising MOF-supported heterogeneous organocatalysts. Our ongoing progress with squaramide MOFs are focusing on their applications in molecular recognition and ion sensing.
This work was financially supported by a grant from the Division of Chemistry of the National Science Foundation (CHE-1359906) and China Scholarship Council.
Notes and references
- S. J. Connon, Chem. – Eur. J., 2006, 12, 5418–5427 CrossRef PubMed.
- J. Aleman, A. Parra, H. Jiang and K. A. Jorgensen, Chem. – Eur. J., 2011, 17, 6890–6899 CrossRef CAS PubMed.
- R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330–2346 RSC.
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416–14417 CrossRef CAS PubMed.
- D. Roca-Lopez, U. Uria, E. Reyes, L. Carrillo, K. A. Jorgensen, J. L. Vicario and P. Merino, Chem. – Eur. J., 2016, 22, 884–889 CrossRef CAS PubMed.
- Y. Wang, J. Pan, R. Jiang, Y. Wang and Z. Zhou, Adv. Synth. Catal., 2016, 358, 195–200 CrossRef CAS.
- H. Zhang, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2014, 136, 16485–16488 CrossRef CAS PubMed.
- C. H. Cheon and H. Yamamoto, Tetrahedron Lett., 2009, 50, 3555–3558 CrossRef CAS.
- P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Adv. Synth. Catal., 2015, 357, 253–281 CrossRef CAS.
- Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153–156 CrossRef CAS PubMed.
- H. Jiang, M. W. Paixao, D. Monge and K. A. Jorgensen, J. Am. Chem. Soc., 2010, 132, 2775–2783 CrossRef CAS PubMed.
- B. Soberats, L. Martinez, E. Sanna, A. Sampedro, C. Rotger and A. Costa, Chem. – Eur. J., 2012, 18, 7533–7542 CrossRef CAS PubMed.
- X. Wu, N. Busschaert, N. J. Wells, Y. B. Jiang and P. A. Gale, J. Am. Chem. Soc., 2015, 137, 1476–1484 CrossRef CAS PubMed.
- C. Gaeta, C. Talotta, P. Della Sala, L. Margarucci, A. Casapullo and P. Neri, J. Org. Chem., 2014, 79, 3704–3708 CrossRef CAS PubMed.
- B. Shan, Y. Liu, R. Shi, S. Jin, L. Li, S. Chen and Q. Shu, RSC Adv., 2015, 5, 96665–96669 RSC.
- A. Rostami, C. J. Wei, G. Guerin and M. S. Taylor, Angew. Chem., Int. Ed., 2011, 50, 2059–2062 CrossRef CAS PubMed.
- D. Quiñonero, R. Prohens, C. Garau, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2002, 351, 115–120 CrossRef.
- A. Portell, R. Barbas, D. Braga, M. Polito, C. Puigjaner and R. Prohens, CrystEngComm, 2009, 11, 52–54 RSC.
- J. V. Alegre-Requena, E. Marqués-López, R. P. Herrera and D. D. Díaz, CrystEngComm, 2016, 18, 3985–3995 RSC.
- C. M. McGuirk, M. J. Katz, C. L. Stern, A. A. Sarjeant, J. T. Hupp, O. K. Farha and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 919–925 CrossRef CAS PubMed.
- L. Du, Z. Lu, K. Zheng, J. Wang, X. Zheng, Y. Pan, X. You and J. Bai, J. Am. Chem. Soc., 2013, 135, 562–565 CrossRef CAS PubMed.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Nature, 2015, 527, 357–361 CrossRef CAS PubMed.
- B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed.
- N. T. Nguyen, H. Furukawa, F. Gandara, C. A. Trickett, H. M. Jeong, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 15394–15397 CrossRef CAS PubMed.
- M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- T. Sawano, N. C. Thacker, Z. Lin, A. R. McIsaac and W. Lin, J. Am. Chem. Soc., 2015, 137, 12241–12248 CrossRef CAS PubMed.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- N. T. S. Phan, K. K. A. Le and T. D. Phan, Appl. Catal., A, 2010, 382, 246–253 CrossRef CAS.
- K.-I. Shimizu, K. Niimi and A. Satsuma, Appl. Catal., A, 2008, 349, 1–5 CrossRef CAS.
- T. B. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903–2915 CrossRef CAS PubMed.
- G. Dessole, R. P. Herrera and A. Ricci, Synlett, 2004, 2374–2378 CAS.
- E. A. Hall, L. R. Redfern, M. H. Wang and K. A. Scheidt, ACS Catal., 2016, 6, 3248–3252 CrossRef CAS.
- A. Rostami, A. Colin, X. Y. Li, M. G. Chudzinski, A. J. Lough and M. S. Taylor, J. Org. Chem., 2010, 75, 3983–3992 CrossRef CAS PubMed.
- M. Bosch, M. Zhang and H.-C. Zhou, Adv. Chem., 2014, 2014, 1–8 CrossRef.
- Z. Wei, W. Lu, H.-L. Jiang and H.-C. Zhou, Inorg. Chem., 2013, 52, 1164–1166 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–609 CrossRef CAS.
- X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schroder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CrossRef CAS PubMed.
- W. Li, Catal. Lett., 2014, 144, 943–948 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, FTIR, TGA, PXRD, 1H NMR, and crystallographic data. CCDC 1472314 and 1472315. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc03190k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |