
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pseudomorphic transformation of amorphous calcium carbonate films follows spherulitic growth mechanisms and can give rise to crystal lattice tilting†
Joe
Harris
a,
I.
Mey
b,
M.
Hajir
c,
M.
Mondeshki
c and
Stephan E.
Wolf
*a
aDepartment of Materials Science and Engineering, Chair of Glass and Ceramics (WW3), Friedrich-Alexander-University Erlangen-Nürnberg. Martensstrasse 5, 91058 Erlangen, Germany. E-mail: stephan.e.wolf@fau.de; Fax: +49 9131 85 28311; Tel: +49 9131 85 27565
bInstitute of Organic and Biomolecular Chemistry, Georg-August-University of Göttingen, Tammannstrasse 2, 37077 Göttingen, Germany
cInstitute of Inorganic and Analytical Chemistry, Johannes Gutenberg-University of Mainz, Duesbergweg 10-14, Germany
First published on 30th April 2015
Abstract
Amorphous calcium carbonate films synthesized by the polymer-induced liquid-precursor (PILP) process convert into crystallographically complex calcite spherulites. Tuning the experimental parameters allows for the generation of crystal lattice tilting similar to that found in calcareous biominerals. This contribution evidences the role of spherulitic growth mechanisms in pseudomorphic transformations of calcium carbonate.
Calcareous biominerals, such as sea urchin spines and mollusk shells, are biogenically formed composite ceramics which show superb adaptation for given tasks. The 542 million years of evolution since the Cambrian explosion has rendered biominerals an immense source of inspiration for both material design and for the creation of ceramic materials at ambient temperature.1 The key step during genesis of these biogenic ceramics is the controlled solid-amorphous to crystalline transformation from a transient precursor to a highly co-oriented mosaic crystal.2–4 This phase transformation is pseudomorphic, i.e. it proceeds with conservation of the macro- and microscopic morphology of the mineral body which eventually leads to non-equilibrium shaped crystalline forms. For future mimesis of these intricate ceramics, the “percolation of crystallinity” through an initially fully amorphous mineral body has to be thoroughly understood. So far, it is generally accepted that biominerals initially form by aggregation of individual and amorphous nanoparticles.3,5–8 In a subsequent state, crystallinity propagates9 through the now fully aggregated10,11 and anhydrous,12 space-filling13/accreted10,11 amorphous mineral body. This eventually generates a mosaic crystal with an insignificant angle spread, often referred to as a mesocrystal.‡4,9,14,15 Ultrastructural analysis of different, mature calcareous biominerals revealed that they, in their fully crystalline state, still embody a nanogranular fine structure; a clear remnant of formation via aggregation and accretion processes of initially amorphous particles which eventually give rise to the conchoidal fracture behaviour of biominerals.6,14–16 In extension to the recent work of Seto et al.,14 Wolf et al. recently proposed that the apparent single crystallinity often observed in biominerals can be seen as an epitaxial nucleation process of the fundamental nanogranular building blocks, i.e. an already crystalline nanogranule bequeaths its crystal orientation to an adjacent and still amorphous granule by isoepitaxial nucleation.15,16 Metaphorically speaking, crystallinity jumps to neighbouring granules and, by this, percolates through the entire mineral body eventually resulting in the formation of an apparently single-crystalline body. This speculative model, which still awaits further corroboration, is capable of explaining the emergence of single-crystallinity and also more peculiar observations recently made on biominerals, such as the preservation of intracrystalline amorphicity in biominerals as evidenced, for instance, by the Gilbert group by the use of XANES-PEEM mapping techniques.9,12,16–18 In this special case, the revised model predicts that individual (patches of) nanogranules do not undergo phase transformation as they are either separated from the surrounding crystallinity by an impenetrable organic coating which ensheathes the granules or because their chemical composition does not meet the requirements, e.g. they contain too much water, foreign ions like magnesium, polymeric impurities, or improper stoichiometry.16,19 Employing XANES-PEEM on an extensive set of nacroprismatic mollusks shells, Gilbert and co-workers recently found the first example of a structure–property relationship in biogenic calcitic prisms.18 They observed a gradual tilting of the crystal lattice orientation in individual prisms of the Akoya pearl oyster Pinctada fucata and of the Black-Lip pearl oyster Pinctada margaritifera. In these species, the calcite crystal lattice orientation bends gradually over distances in the order of up to 100 μm leading to an increased angle spread of crystal orientations of 10° to 20°. On the macroscale, the shells of P. fucata and P. margaritifera feature a notable increase in hardness which correlates with the observed crystal orientation angle spread. Gilbert and co-workers inferred that increased crystal lattice tilting represents a significant evolutionary advantage as it provides a higher hardness and wear resistance and, therefore, better protection against predators and the environment.
In this communication, we evidence that crystal lattice tilting in calcite can be generated in vitro by pseudomorphic phase transformation of a nanogranular amorphous calcium carbonate precursor. We point out that this pseudomorphic phase transformation process involves spherulitic growth mechanisms which give rise to the observed crystal lattice tilting.
In order to easily study the solid-amorphous to crystalline transformation of amorphous calcium carbonate to calcite, two-dimensional thin films of amorphous calcium carbonate were selected as a model system, which made additional sample preparation for characterization of the crystalline state of the film obsolete. The polymer-induced liquid-precursor (PILP) process was employed as it allows for facile generation of thin amorphous films of calcium carbonate at the air–water interface.20–22 The PILP process employs polyelectrolytes such as poly-aspartate or poly(acrylic acid) to suppress classical nucleation and crystal growth and instead promotes the formation of an initial liquid-condensed mineral precursor phase.19 This so-called PILP phase subsequently solidifies by dehydration processes and the final amorphous-to-crystalline transformation then proceeds in a pseudomorphic fashion, aided by water. This solidification process is akin to the processes which occur during biogenic mineralization of calcium carbonate. Moreover, the nanoscopic organization of mineral entities synthesized via the PILP process bear a virtually indistinguishable similarity to that exhibited by calcareous biominerals: both are composed of individual nanogranules of similar size (up to 100 nm in diameter) which form a space-filling and fully accreted mineral body. Even the locus of the occluded/intracrystalline organic material seems to be consistent.6,15,16,23 Amorphous mineral bodies generated by the PILP process are thus optimal specimens for studying the transformation of a solid amorphous material to a solid crystalline body, a process which is rendered hard to follow in situ in biominerals.
The experiments were carried out employing the slow-diffusion technique, i.e. by incubation of a calcium chloride solution (10 mM, adjusted typically to pH = 5) in the vapour of decomposing ammonium carbonate. The reaction was performed in the presence of small amounts of poly (acrylic acid) (PAA) sodium salt (100 μg ml−1) which sets the reaction course to the PILP process. The development of the film was followed in situ with the aid of a digital microscope camera and the properties of the mineral film were extensively characterized ex situ by polarized light microscopy, powder X-ray diffraction (PXRD), Raman and FTIR spectroscopy, atomic force microscopy (AFM), coupled thermogravimetric analysis and differential scanning calorimetry (TGA/DSC) and solid-state nuclear magnetic resonance (SS-NMR).
In situ digital microscopy revealed that a thin mineral film formed at the air–water interface within 16 hours of crystallization. The non-birefringent mineral film was extremely brittle and fractured upon drying when sampled. When the reaction time was extended further, the mineral film thickened which manifested in decrease in film fragility. After 40 hours of reaction time, a thick film could be removed from the interface which, upon drying, partially crystalized as evidenced by the formation of small domains exhibiting the typical Maltese cross pattern of crystalline birefringent calcium carbonate polymorphs (Fig. S1†). Further extension of reaction time to >42 h resulted in formation of crystalline patches within the amorphous film at the air–water interface which showed birefringence under polarised optical microscopy (Fig. S2†). Both in situ digital microscopy and ex situ polarized light microscopy clearly showed that the crystalline patches form within the amorphous film. This is clearly evidenced by flakes in which both amorphous and crystalline domains are present (Fig. S3†). Raman microscopy evidenced that the birefringent domains consisted of crystalline calcite (Raman absorbance bands at 1091, 716, 276 and 146 cm−1 corresponding to the ν1, ν4 and lattice modes, see Fig. S4†) whereas the non-birefringent regions are composed of amorphous calcium carbonate (broad Raman peaks at 1082, and 712 cm−1 corresponding to the ν1 and ν4 modes of CO32− respectively, see Fig. S5†). From 72 hours of reaction time onwards, the amorphous domains dissolved slowly, leaving spatially independent, white coloured patches floating on the air–water interface after 96 h, with diameters spanning in excess of 2 mm and a constant thickness of 3 μm cross the breadth of each flake. Ex situ PXRD phase analysis (Fig. S6†), Raman microscopy analysis (Raman absorbance bands at 1085, 711, 278 and 153 cm−1 arising from the ν1, ν4 and lattice modes of calcite respectively, see Fig. S7†) and FTIR spectroscopy (IR bands at 1400, 872 and 712 cm−1 corresponding to the ν3, ν2 and ν4 CO32− bands of calcite, see Fig. S8†) showed concordantly that the crystallites were composed of calcite.
Close inspection by polarised light microscopy revealed the crystalline patches to be circularly shaped, flower-like calcite flakes (Fig. 1); some of which show a conspicuous similarity to ice flowers (Fig. 1B). The morphology clearly marks these radially polycrystalline aggregates as two-dimensional calcite spherulites.§We found two clearly distinguished types of spherulitic morphologies whose ratio/rate of formation depended on reaction parameters such as polymer concentration or initial reaction solution pH. The first spherulite morphology, type A, is remarkably uniform and characterized by radially-symmetric features with straight fibres (Fig. 1A); it is classically representative of a coarse (i.e. straying fibres are distinguishable) and closed (i.e. space-filling) spherulite.24 The evident Maltese extinction cross indicates clearly that the growth direction of the fibres is parallel to the principal axis of the optical uniaxial indicatrix of calcite (Fig. S9†), i.e. the c-axis. These fully radially-symmetric spherulites are prevalent when the initial pH of the reaction solution was elevated (pH 7 or 9, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 by surface area at pH = 7). The second spherulite variant, type B (Fig. 1B), dominated at lower starting pH (25:1 by surface area at pH = 5) and bears extraordinary crystallographic features. In stark contrast to the spherulites of the first type, the closed, coarse spherulites of type B lacked radial symmetry, prominently shown by their rough and non-circular edge. Polarized light microscopy revealed separate domains of varying contrast which radiated out from the centre of the spherulite (Fig. 1B). These domains do not feature straight delineations but run in curved paths resulting in unique, interwoven and tilted non-symmetrical structures (Fig. 1C); such structures were not observed for the spherulites of the first type. The curvature of these domains is non-uniform and cross over between straight and tilted delineations was observed occasionally. Light intensity line profiles obtained on polarised optical microscopy images of curved domains of the type B spherulites, show alteration in intensity along the domain (i.e. from the centre of the flakes to the edge, see Fig. S10†). This change in intensity is indicative of an alteration in the orientation of the calcite c-axis, and demonstrates the lattice tilting of the calcite crystallites.
:3 by surface area at pH = 7). The second spherulite variant, type B (Fig. 1B), dominated at lower starting pH (25:1 by surface area at pH = 5) and bears extraordinary crystallographic features. In stark contrast to the spherulites of the first type, the closed, coarse spherulites of type B lacked radial symmetry, prominently shown by their rough and non-circular edge. Polarized light microscopy revealed separate domains of varying contrast which radiated out from the centre of the spherulite (Fig. 1B). These domains do not feature straight delineations but run in curved paths resulting in unique, interwoven and tilted non-symmetrical structures (Fig. 1C); such structures were not observed for the spherulites of the first type. The curvature of these domains is non-uniform and cross over between straight and tilted delineations was observed occasionally. Light intensity line profiles obtained on polarised optical microscopy images of curved domains of the type B spherulites, show alteration in intensity along the domain (i.e. from the centre of the flakes to the edge, see Fig. S10†). This change in intensity is indicative of an alteration in the orientation of the calcite c-axis, and demonstrates the lattice tilting of the calcite crystallites.
 | ||
| Fig. 1 Micrographs of calcite spherulites obtained after 4 days at a starting pH of 5 obtained by polarized light microscopy under fully crossed polarizers. Constant thickness of the calcite spherulites was checked by AFM and SEM analysis of cross-sections of spherulites embedded in epoxy resin; therefore the varying contrast levels arise due to changing crystal orientation and not due to thickness variations. (A) A radially-symmetric two-dimensional calcite spherulite, a typical representative of spherulite type A, featuring clear Maltese cross extinction which evidences that the growth direction of each fibre is parallel to the c-axis, the principal axis of the optical uniaxial indicatrix of calcite. Close inspection reveals that subfibres nucleating on parent fibres also follow this growth direction. (B) A generic example of spherulites belonging to type B which are characterized by complex crystallographic features and intraspherulitic domain formation. The delineations of the subdomains are not straight but curved which cause the observed breaking of the radial symmetry. Closer analysis reveals that this morphology is accompanied by a tilting of the crystal lattice orientation. (C) A prominent example of strongly interwoven, interpenetrating type B spherulites. In all subfigures, arrows are placed as a guide for the eye to highlight selected straight resp. curved morphological features. | ||
In absence of the polymeric additive or when PAA was replaced by the non-charged polymer poly(vinylpyrrolidone) (PVP), no film formation was observed; instead vaterite and calcite crystal were formed at the air–water interface at variable ratios (Fig. S11 and S12†). Alteration of the initial PAA content of 100 μg ml−1 affected the morphology of the final crystals. At lower PAA concentrations (50 μg ml−1), precipitates with non-equilibrium morphologies characteristic of the PILP process were mainly observed in addition to a small amount of coarse closed spherulites without bent fibres (type A). At elevated polymer concentration (200 μg ml−1), spherulites were the prevalent crystal morphology found at the air–water interface, with coarse closed spherulites the most dominant in addition to a small amount of coarse closed spherulites with regions of fibre bending. At high PAA concentrations (1000 μg ml−1) only closed, coarse spherulites were observed. Addition of magnesium ions, as often used in PILP processes as an additive promoting formation and stabilization of ACC,25–30 prevented the formation of spherulites at the air–water interface: after 96 hours a thick amorphous film was generated which remained amorphous even upon drying.
TGA and DSC analysis of the spherulites indicated the presence of tightly bound water by a mass decrease of 0.62% weight/weight at temperatures above 120 °C prior to the onset of PAA decomposition (300 °C, as determined from TGA/DSC of pure PAA, see Fig. S13†). This suggested the presence of a hydrated calcium carbonate species, probably in the form of hydrated ACC as no evidence of the hydrated crystalline phases ikaite or calcium hexahydrate was found by PXRD. However, DSC analysis could not corroborate the presence of an amorphous phase, probably due to the low amount in which it was present within the spherulites. The mass decrease of 1.8% from 300 °C to the onset of calcite decomposition gave an approximate value of PAA incorporation within the spherulites, a value noteworthy as it is similar to that found for intracrystalline organics in biominerals.31–34
We thus used a set of different 13C solid state NMR spectroscopic techniques to probe for an additional amorphous mineral fraction beside the crystalline calcite phase and to evidence the presence of organic inclusions in the spherulites. The fully relaxed 13C single pulse (SP) NMR spectrum recorded with a recycle delay of 2200 s (Fig. 2) depicts a single resonance signal with a chemical shift of 168.4 ppm and full width at half height (FWHH) of 41 Hz. The FWHH, a sensitive measure for the local order,16,35 shows clearly that the crystalline bulk phase of the spherulites was highly ordered calcite. Reducing the recycle delay lead to saturation of the slowly relaxing crystalline signal contribution (i.e.13CO32− in a calcitic structure) allowed for the observation of faster relaxing components which experience a higher degree of local disorder, e.g. amorphous mineral components or disordered regions due to intra- or pericrystalline organic inclusions. More strictly speaking, the relaxation time decreases because in the disordered regions protons are inherently present which allow for dipolar relaxation by coupling with 13C nuclei in their vicinity. ACC is often monohydrated which gives rise to increased dipolar relaxation. At a recycle delay of 20 s, the 13C SP spectrum (Fig. 2A) shows the resonance calcite signal shifted to 168.3 ppm and broadened (FWHH = 48 Hz) indicating the presence of a second mineral phase with increased disorder with a near-range order similar to calcite, i.e. amorphous calcium carbonate in the proto-calcite form.36
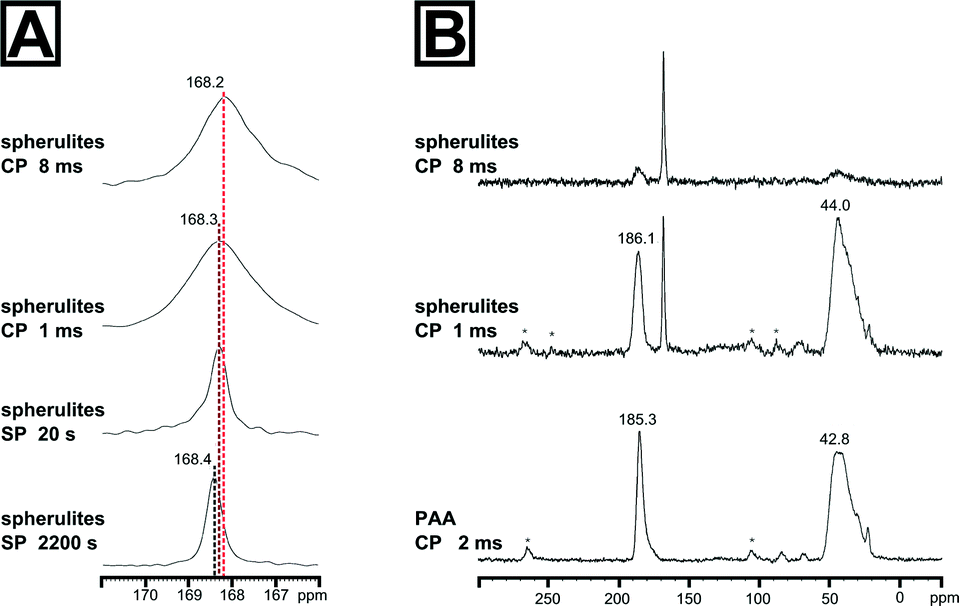 | ||
| Fig. 2 13C solid state NMR spectra of the calcite spherulites evidencing the presence of a second amorphous phase and the incorporation of the polymeric additive PAA into the spherulites at grain boundaries. (A) Carbonyl region of the 13C solid state NMR spectra of the calcite spherulites, recorded as single pulse (SP) experiments with 2200 s or 20 s recycle delay, and 1H-13C cross-polarization transfer (CP), recorded with 3 s recycle delay at 1 ms and 8 ms contact time. (B) 13C CP NMR spectra of the spherulites and of the pure PAA polymer standard recorded with 2 ms cross-polarization time. The signal at 185.3 ppm with a full width at half height (FWHH) of 457 Hz of the PAA spectrum corresponds to the 13C resonance of the carbonyl group, the broad signal at 42.8 ppm with a FWHH of 1578 Hz is related with the overlapping CH and CH2 signals of the vinyl backbone of the polymer. Additionally, two broad spinning sidebands, spaced by the spinning frequency, are observed at ca. 105 and 265 ppm which arise from the significant chemical shift anisotropy of the carbonyl group. | ||
We further probed the organization of the amorphous fraction by heteronuclear 1H-13C dipole–dipole coupling in a cross-polarization (CP) experiment as a function of the contact time. Such polarization transfers are active in coupled proton-carbon pairs if the internuclear distance is in the range of up to 5 Å. Only carbons with neighbouring protons will appear as resonance signals in such spectra, i.e.13C sites in the vicinity to the polymer additive will be significantly enhanced, e.g. at the interface between the mineral and the organic inclusions. The 13C CP NMR spectrum of the spherulites recorded with a cross-polarization time of 1 ms is shown in Fig. 2B. The maximum of the calcite resonance detected at 168.3 ppm is slightly shifted to higher field. However, its FWHH increased more than fourfold to about 170 Hz compared to the FWHH of the bulk crystalline phase probed in the SP experiments with long relaxation time. This dramatic increase of the FWHH for the calcite signal points to an interaction between PAA and the mineral surface, i.e. the polymer additive introduced disorder at the mineral interface. On the basis of the reference 13C CP spectrum of PAA recorded with a contact time of 2 ms, the remainder of the signals can be assigned to the polymeric additive incorporated in the spherulites. Comparing the 13C CP NMR spectrum recorded with 1 ms cross-polarization time, the signal of the polymeric carbonyl groups is shifted by 0.8 ppm to lower field and shows a significant signal broadening to 617 Hz in comparison to the CP NMR spectrum of the pure polymer. This finding nicely correlates with a broadening of the amorphous calcium carbonate signal indicating CaCO3 – PAA interaction. Interestingly, the broadening of the signals of both interacting components at the mineral/inclusion interface, i.e. the proto-calcite phase and the PAA carbonyl groups, is quite comparable (130 Hz for proto-calcite and 160 Hz for PAA). The slight discrepancy of about 30 Hz is probably related to the inherently disordered nature of the polymer. The signal of the polymer backbone in the 13C spectrum of the synthetic sample resonates at slightly lower field (44.0 ppm) and remains practically unchanged (1578 Hz) compared to the pure polymer. However, the top of the vinyl peak changed visibly when PAA was incorporated in the spherulites. One may speculate that the decrease of the distribution of the resonance frequencies at the maximum of the backbone peak indicates a conformational change as a result of the mineral/polymer interaction. Turning to the 13C CP NMR spectrum recorded with 8 ms contact time, polymer signals are weakened and confirm that the polymeric additive remains at the interface region and only a small amount is entrapped within the crystal. In comparison to the spectrum recorded with 1 ms contact time, the calcite signal maximum shifts to 168.2 ppm and the FWHH lowered slightly to 143 Hz.
In order to further probe the nanoscopic organization of the calcareous films, we undertook atomic force microscopy on the spherulitic flakes. Images were recorded in bimodal dual AC imaging mode, in which cantilever probes were excited at the fundamental and third resonant frequency resulting in two amplitude and two phase images.37 The higher eigenmodes provide enhanced contrast for material properties both in the phase and amplitude channels which give more detailed mapping especially for small and sharp structures. We sampled multiple sites on both sides of the spherulites, i.e. the spherulite's surface which faces the mother solution and the one facing air; we additionally characterized cross-sections of embedded spherulites in epoxy resin and the film in its still amorphous stage. In all cases, we found a similar colloidal texture, the hallmark of PILP products (Fig. 3A, Fig. S14†). This clearly evidenced that the pseudomorphic transformation preserved the granular features at the nanoscale level. Both surfaces of the spherulites fully resemble the cross-section and consisted entirely of accreted, fully space-filling nanogranules of up to 80 nm in diameter as estimated by grain size analysis. They form a compact material; no interstices or intergranular cavities were observed. The phase micrograph (Fig. 3B) shows a clear contrast between the singular granules which might indicate that the granules are formed from two individual components; a majority component, from which the granules are composed, and a second minor component of different mechanical stiffness which apparently resides at the granules' surface/rim. Since the surface of the spherulites is intrinsically rough, this observation has to be taken with care because the phase shift can also arise due to complex interactions of the probe with the grooves of the surface.
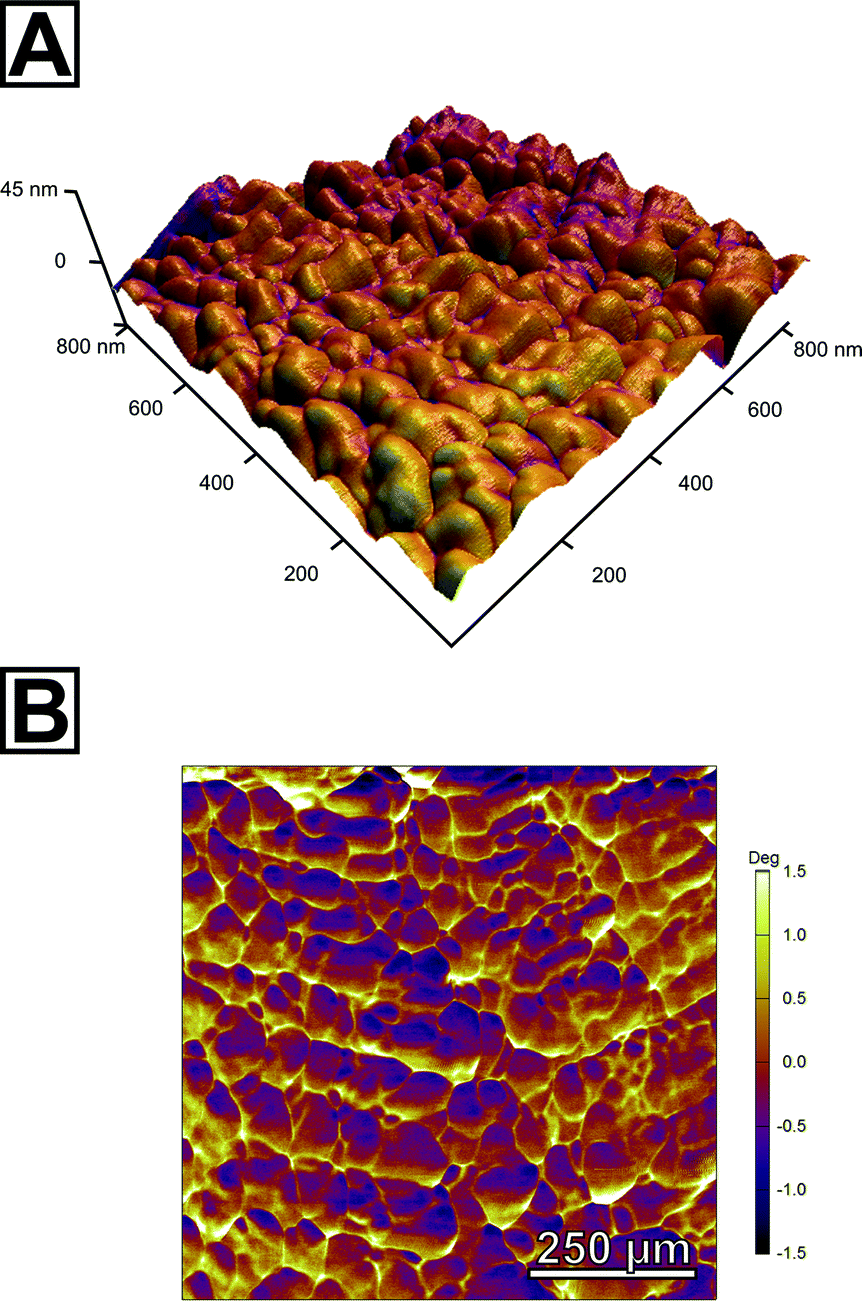 | ||
| Fig. 3 Atomic force micrographs of the calcite spherulites exhibiting their nanoscopic organization. (A) Topographical height image textured with the amplitude of the third resonant frequency shows densely accreted granules of calcium carbonate with an approximate diameter of 80 nm forming a space-filling mineral body. (B) The phase image of the overtone suggests a second component of different mechanical stiffness located/enriched at intergranular regions; this might be the intracrystalline fraction of the polymeric additives as evidenced by SS-NMR. | ||
Conclusions
Calcium carbonate spherulites formed in solution have been known for more than century; the earliest reports date back to the works of Pieter Harting from 1827, followed by works of T. S. Hunt, E. von Lengyel, E. Häckel and others.26,38–44 In this work, we show for the first time that calcite spherulites with bent crystal lattice orientation can form via a pseudomorphic phase transformation from a transient amorphous precursor.Our work strongly supports the admonition of Andreassen et al.,41–44 that spherulitic growth mechanisms play an important role in the calcium carbonate system in general and a central role in pseudomorphic phase transformations of calcium carbonate, e.g. under PILP conditions. Reviewing the literature, spherulitic crystal growth in calcium carbonate thin films is frequently reported.20,25,27,45–60 Predominantly these film have been prepared on substrates,20,25,27,45,47–50,53–60 and only in rare cases were the films prepared at the air–water interface.46,51,52 However, roughly one half of these publications omit to denote the observed crystal morphology correctly as spherulites.25,54–60 This demonstrates that the central role of spherulitic growth during the pseudomorphic transformation of calcium carbonate, especially under PILP conditions, has not gained general awareness.
The key finding of this contribution is that, under certain experimental conditions, calcium carbonate spherulites can feature bent morphologies and crystal lattice tilting, i.e. the gradual tilting of the crystal lattice orientation. None of the publications discussed above report this feature. As a matter of fact, spherulites in general are well known to exhibit this feature and it is commonly attributed to noncrystallographic branching mechanisms, which can be favoured by the presence of inclusions or impurities which slow down the phase transformation process.61,62 In our system, the emergence of crystal lattice tilting is favoured by a lower initial pH of the reaction solution. This change of the starting pH yields a slightly different pH profile of the precipitation reaction (Fig. S15†). We believe that this change in the reaction profile leads to the formation of an amorphous film with a composition slightly deviating from that formed at elevated initial pH, be it by a slightly increased amount of polymer entrapped within the film, increased amount of water or a different stoichiometry (e.g. increased amount of bicarbonate). This change of composition may then slow down the phase transformation process favouring the formation of the observed crystal lattice tilting.
We believe that the tendency of the investigated system to undergo spherulitic growth is an ultimate outcome of its nanogranular structure in which a significant amount of additive is incorporated, probably at intergranular sites. Such impurities residing at grain boundaries are known to give rise to noncrystallographic branching and therefore to stimulate spherulite formation.62 It is important to note that the employed system shares this trait with calcareous biominerals.6,16,19 We therefore expect that spherulitic growth mechanisms take place in the phase transformation processes of calcareous biominerals. Moreover, our observation that crystal lattice tilting can be generated by a pseudomorphic transformation of a transient amorphous precursor suggests clearly that the crystal lattice tilting observed in calcite prisms of P. fucata and P. margaritifera finds origin in spherulitic growth mechanisms. However, the ultimate mechanism, which gives rise to the observed crystal lattice tilting both in vitro and in vivo, still awaits elucidation. The subject of future research should lie in whether the observed crystal lattice tilting, both in vitro and in vivo, is induced by small angle twinning, e.g. controlled by the intergranular organic matrix, or follows noncrystallographic branching mechanisms.
In summary, we have shown that spherulitic growth mechanisms and non-crystallographic branching can play a crucial role in the pseudomorphic transformation of amorphous calcium carbonate eventually leading to crystal lattice tilting. As seen in Nature, such crystal lattice tilting may pave the way for novel toughening mechanisms in a new class of bio-inspired ceramic materials and spherulitic growth mechanisms represent one route for creating such bent crystallinity at will.
Acknowledgements
We gratefully acknowledge financial support by an Emmy Noether research grant issued by the German Research Foundation (DFG, no. WO1712/3-1). We thank the Cluster of Excellence “Engineering of Advanced Materials – Hierarchical Structure Formation for Functional Devices” (Research Area E1 – Lightweight Materials) for partial support. Last but not least, we thank Emily Asenath-Smith, Cornell University, for insightful and fruitful discussions.Notes and references
- E. Bäuerlein, P. Behrens and M. Epple, Handbook of Biomineralization, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2007 Search PubMed.
- L. B. Gower, Chem. Rev., 2008, 108, 4551–4627 CrossRef CAS PubMed.
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959–970 CrossRef CAS PubMed.
- H. Cölfen and M. Antonietti, Mesocrystals and Nonclassical Crystallization, Wiley-VCH Verlag GmbH, 2008 Search PubMed.
- A. Baronnet, J.-P. Cuif, Y. Dauphin, B. Farre and J. Nouet, Mineral. Mag., 2008, 72, 617–626 CrossRef CAS PubMed.
- Y. Dauphin, Mineral. Mag., 2008, 72, 243–246 CrossRef CAS PubMed.
- D. E. Jacob, R. Wirth, A. Soldati, U. Wehrmeister and A. Schreiber, J. Struct. Biol., 2011, 173, 241–249 CrossRef CAS PubMed.
- F. Nudelman, H. H. Chen, H. A. Goldberg, S. Weiner and L. Addadi, Faraday Discuss., 2007, 136, 139 RSC.
- C. E. Killian, R. Metzler, Y. U. T. Gong, I. C. Olson, J. Aizenberg, Y. Politi, F. H. Wilt, A. Scholl, A. Young, A. Doran, M. Kunz, N. Tamura, S. N. Coppersmith and P. U. P. A. Gilbert, J. Am. Chem. Soc., 2009, 131, 18404–18409 CrossRef CAS PubMed.
- A. Gal, K. Kahil, N. Vidavsky, R. T. DeVol, P. U. P. A. Gilbert, P. Fratzl, S. Weiner and L. Addadi, Adv. Funct. Mater., 2014, 24, 5420–5426 CrossRef CAS PubMed.
- A. Gal, W. Habraken, D. Gur, P. Fratzl, S. Weiner and L. Addadi, Angew. Chem., 2013, 125, 4967–4970 CrossRef PubMed.
- Y. U. T. Gong, C. E. Killian, I. C. Olson, N. P. Appathurai, A. L. Amasino, M. C. Martin, L. J. Holt, F. H. Wilt and P. U. P. A. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 2012, 1–6 Search PubMed.
- L. Yang, C. E. Killian, M. Kunz, N. Tamura and P. U. P. A. Gilbert, Nanoscale, 2011, 3, 603–609 RSC.
- J. Seto, Y. Ma, S. A. Davis, F. C. Meldrum, A. Gourrier, Y.-Y. Kim, U. Schilde, M. Sztucki, M. Burghammer, S. Maltsev, C. Jäger and H. Cölfen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3699–3704 CrossRef CAS PubMed.
- S. E. Wolf, I. Lieberwirth, F. Natalio, J.-F. Bardeau, N. Delorme, F. Emmerling, R. Barrea, M. Kappl and F. Marin, Faraday Discuss., 2012, 159, 433 RSC.
- S. E. Wolf, C. Böhm, J. Harris, M. Hajir, M. Mondeshki and F. Marin, Key Engineering Materials, special volume Biomineralix – European COST Action TD0903, 2015, in print Search PubMed.
- I. C. Olson, A. Z. Blonsky, N. Tamura, M. Kunz, B. Pokroy, C. P. Romao, M. A. White and P. U. P. A. Gilbert, J. Struct. Biol., 2013, 184, 454–463 CrossRef CAS PubMed.
- I. C. Olson, R. Metzler, N. Tamura, M. Kunz, C. E. Killian and P. U. P. A. Gilbert, J. Struct. Biol., 2013, 183, 180–190 CrossRef CAS PubMed.
- S. E. Wolf and L. B. Gower, in New Perspectives on Mineral Nucleation and Growth, ed. M. Kellermeier, A. van Driessche, L. Benning and D. Gebauer, Springer, 2015, in print Search PubMed.
- L. B. Gower and D. J. Odom, J. Cryst. Growth, 2000, 210, 719–734 CrossRef CAS.
- E. DiMasi, V. M. Patel, M. Sivakumar, M. J. Olszta, Y. P. Yang and L. B. Gower, Langmuir, 2002, 18, 8902–8909 CrossRef CAS.
- Y. Jiang, H. Gong, M. Grzywa, D. Volkmer, L. B. Gower and H. Cölfen, Adv. Funct. Mater., 2013, 23, 1547–1555 CrossRef CAS PubMed.
- D. E. Jacob, A. Soldati, R. Wirth, J. Huth, U. Wehrmeister and W. Hofmeister, Geochim. Cosmochim. Acta, 2008, 72, 5401–5415 CrossRef CAS PubMed.
- H. D. Keith and F. J. Padden, J. Appl. Phys., 1963, 34, 2409–2421 CrossRef CAS PubMed.
- B. Cantaert, A. Verch, Y.-Y. Kim, H. Ludwig, V. N. Paunov, R. Kröger and F. C. Meldrum, Chem. Mater., 2013, 25, 4994–5003 CrossRef CAS PubMed.
- S. Valiyaveettil, P. K. Ajikumar, L. G. L. G. Wong, G. Subramanyam and R. Lakshminarayanan, Cryst. Growth Des., 2005, 5, 1129–1134 Search PubMed.
- Y.-Y. Kim, E. P. Douglas and L. B. Gower, Langmuir, 2007, 23, 4862–4870 CrossRef CAS PubMed.
- F. F. Amos, D. M. Sharbaugh, D. R. Talham, L. B. Gower, M. Fricke and D. Volkmer, Langmuir, 2007, 23, 1988–1994 CrossRef CAS PubMed.
- H. Gong, M. Pluntke, O. Marti, P. Walther, L. B. Gower, H. Cölfen and D. Volkmer, Colloids Surf., A, 2010, 354, 279–283 CrossRef CAS PubMed.
- A. S. Schenk, H. Zope, Y.-Y. Kim, A. Kros, N. A. J. M. Sommerdijk and F. C. Meldrum, Faraday Discuss., 2012, 159, 327 RSC.
- F. Marin and G. Luquet, Unusually Acidic Proteins in Biomineralization, in Handbook of Biomineralization: Biological Aspects and Structure Formation, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2007 Search PubMed.
- F. Marin, R. Amons, N. Guichard, M. Stigter, A. Hecker, G. Luquet, P. Layrolle, G. Alcaraz, C. Riondet and P. Westbroek, J. Biol. Chem., 2005, 280, 33895–33908 CrossRef CAS PubMed.
- F. Marin, B. Marie, S. Ben Hamada, P. Silva, N. Le Roy, S. E. Wolf, C. Montagnani, C. Joubert and D. Piquemal, in Recent Advances in Pearl Research, ed. S. Watabe, K. Maeyama and H. Nagasawa, Terrapub, 2013, pp. 149–166 Search PubMed.
- B. Marie, I. Zanella-Cléon, M. Corneillat, M. Becchi, G. Alcaraz, L. Plasseraud, G. Luquet, F. Marin and I. Zanella-cle, FEBS J., 2011, 278, 1–14 CrossRef PubMed.
- H. Nebel, M. Neumann, C. Mayer and M. Epple, Inorg. Chem., 2008, 47, 7874–7879 CrossRef CAS PubMed.
- D. Gebauer, P. N. Gunawidjaja, J. Y. P. Ko, Z. Bacsik, B. Aziz, L. Liu, Y. Hu, L. Bergström, C.-W. Tai, T.-K. Sham, M. Edén and N. Hedin, Angew. Chem., 2010, 122, 9073–9075 CrossRef PubMed.
- R. Proksch, Appl. Phys. Lett., 2006, 89 Search PubMed.
- E. von Lengyel, Z. Kristallogr., 1936, 97, 67–87 Search PubMed.
- T. S. Hunt, Am. J. Sci., 1866, 42(124), 49–67 CrossRef.
- E. Häckel, Kristallseelen. Studien über das anorganische Leben, Alfred Kroner Verlag, Leipzig, 1917 Search PubMed.
- J.-P. Andreassen, J. Cryst. Growth, 2005, 274, 256–264 CrossRef CAS PubMed.
- J.-P. Andreassen, E. M. Flaten, R. Beck and A. E. Lewis, Chem. Eng. Res. Des., 2010, 88, 1163–1168 CrossRef CAS PubMed.
- J.-P. Andreassen, R. Beck and M. Nergaard, Faraday Discuss., 2012, 159, 247 RSC.
- R. Beck and J.-P. Andreassen, Cryst. Growth Des., 2010, 10, 2934–2947 CAS.
- X. Xu, J. T. Han and K. Cho, Chem. Mater., 2004, 16, 1740–1746 CrossRef CAS.
- X. Xu, J. T. Han, D. H. Kim and K. Cho, J. Phys. Chem. B, 2006, 110, 2764–2770 CrossRef CAS PubMed.
- N. Hosoda and T. Kato, Cellulose, 2001, 688–693 CAS.
- J. T. Han, X. Xu, D. H. Kim and K. Cho, Chem. Mater., 2005, 17, 136–141 CrossRef CAS.
- J. T. Han, X. Xu, D. H. Kim and K. Cho, Adv. Funct. Mater., 2005, 15, 475–480 CrossRef CAS PubMed.
- X. Cheng, P. L. Varona, M. J. Olszta and L. B. Gower, J. Cryst. Growth, 2007, 307, 395–404 CrossRef CAS PubMed.
- J. Pecher, P. Guenoun and C. Chevallard, Cryst. Growth Des., 2009, 9, 1306–1311 CAS.
- C. Li, G. Hong, H. Yu and L. Qi, Chem. Mater., 2010, 22, 3206–3211 CrossRef CAS.
- F. Zhu, T. Nishimura, H. Eimura and T. Kato, CrystEngComm, 2014, 16, 1496–1501 RSC.
- N. Hosoda, A. Sugawara and T. Kato, Macromolecules, 2003, 36, 6449–6452 CrossRef CAS.
- D. C. Popescu, E. N. M. Van Leeuwen, N. Rossi, S. J. Holder, J. A. Jansen and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2006, 45, 1762–1767 CrossRef CAS PubMed.
- N. A. J. M. Sommerdijk, E. N. M. van Leeuwen, M. R. J. Vos and J. A. Jansen, CrystEngComm, 2007, 9, 1209 RSC.
- J. Pretula, K. Kaluzynski, B. Wisniewski, R. Szymanski, T. Loontjens and S. Penczek, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 830–843 CrossRef CAS PubMed.
- T. Sakamoto, A. Oichi, T. Nishimura, A. Sugawara and T. Kato, Polym. J., 2009, 41, 522–523 CrossRef CAS.
- B. Cantaert, Y.-Y. Kim, H. Ludwig, F. Nudelman, N. A. J. M. Sommerdijk and F. C. Meldrum, Adv. Funct. Mater., 2012, 22, 907–915 CrossRef CAS PubMed.
- A. S. Schenk, B. Cantaert, Y. Kim, Y. Li, E. S. Read, M. Semsarilar, S. P. Armes and F. C. Meldrum, Chem. Mater., 2014, 26, 2703–2711 CrossRef CAS.
- A. G. Shtukenberg, Y. O. Punin, A. Gujral and B. Kahr, Angew. Chem., Int. Ed., 2014, 53, 672–699 CrossRef CAS PubMed.
- A. G. Shtukenberg, Y. O. Punin, E. Gunn and B. Kahr, Chem. Rev., 2012, 112, 1805–1838 CrossRef CAS PubMed.
- C. G. Darwin, Philos. Mag., 1922, 43, 800–820 CrossRef CAS PubMed.
- A. Blatter and C. Ortiz, J. Cryst. Growth, 1994, 139, 120–128 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, polarized light micrographs of transient states of film formation; Raman, IR and PXRD analyses; line intensity profiles of type A and B spherulites; TGA/DSC profiles of spherulites; control crystallization experiments and AFM micrographs of the amorphous film. See DOI: 10.1039/c5ce00441a |
| ‡ Please note that the term mesocrystal does not and should not embrace any connotation about a specific formation pathway. In this paper, we use the term mesocrystal to describe mosaic crystals whose internal structure and morphology is suggestive of their particle-mediated mode of growth; for definition of a mosaic crystal please see the pertinent work of Darwin dating back to the 1920's.63 |
| § Spherulites, defined as “with an outer spherical envelope”, have to be clearly distinguished from dendritic (skeletal) crystals: spherulites do not have a single crystallographic orientation. The radiating domains form from the central nucleus by noncrystallographic branching, i.e. the crystallographic orientation of the daughter crystals and the parent fibre/nucleus is not preserved. Flat, two-dimensional spherulites may be correctly denominated as cylindrulites.64 The term spherulite observed in the literature is often misused; for an excellent review on this, please see Shtukenberg et al.62 |
| This journal is © The Royal Society of Chemistry 2015 |