Selectively sensing first-row transition metal ions through fluorescence enhancement
Sanchari Pal
,
Nabanita Chatterjee
and
Parimal K. Bharadwaj
*
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208016, India. E-mail: pkb@iitk.ac.in
First published on 29th May 2014
Abstract
Transition metal ions, especially the first-row ones are ubiquitous in nature and they perform many biological functions. However, high accumulation of these ions can be quite detrimental to health. Their spatial distribution with high fidelity inside as well as outside cells is, therefore, of paramount importance. In addition to biological sensing, fluorescence enhancement by transition metal ions can be potentially useful in other areas of science. However, most of the transition metal ions are paramagnetic and they very effectively quench fluorescence. Surmounting this problem of fluorescence quenching, a number of systems have been reported which are the topic of the present review.
 Sanchari Pal | Sanchari Pal was born in West Bengal, India. She received her M.Sc. (Gold medalist) from Jadavpur University, in 2010. She is currently pursuing her Ph.D. at the Department of Chemistry, IIT Kanpur under the supervision of Prof. P. K. Bharadwaj. Her research interests focus on Fluorescence signaling, Cryptands and MOFs. |
 Nabanita Chatterjee | Nabanita Chatterjee was born in West Bengal, India. She received her M.Sc. from University of Mysore, in 2010. She is currently pursuing her Ph.D. at the Department of Chemistry, IIT Kanpur under the supervision of Prof. P. K. Bharadwaj. Her research interests focus on Fluorescence signaling and MOFs. |
 Parimal K. Bharadwaj | Parimal K. Bharadwaj received his Ph.D. (1979) from the Indian Institute of Technology Kharagpur. After spending a year as a UNESCO fellow (1980) at the Tokyo Institute of Technology, he moved to Rutgers University in NJ where he carried out research in Bioinorganic Chemistry (1980–1985) first as a Postdoctoral Fellow and then as an Assistant Professor (non-tenure track). Thereafter, he spent two years (1986–1987) at the University of California at Davis working on radiopharmaceuticals. He returned to India as an Assistant Professor of Chemistry at the Indian Institute of Technology Kanpur where he became a full Professor in 1995. His current research is focused on fluorescence signaling, FRET, using cryptands as platforms for photoinduced energy and charge separation and on the design and synthesis of functional metal-organic framework materials. |
1. Introduction
Many transition metal ions are ubiquitous in nature and they perform1 several biological functions while at the same time, excess accumulation of these ions in biosystems can lead to diseases2 and ultimately to fatal consequences. Their spatial as well as temporal distribution inside and outside the cells is, therefore, of paramount importance to understand physiological conditions. Fluorescence spectroscopy3 is a sensitive tool to detect and measure concentration of an analyte with high fidelity. The basic design of the signaling system is, however, consists of a signaling (fluorophore) and a guest-binding (receptor) moieties that may be integrated or covalently attached through a spacer (Scheme 1). Out of these two designs, the one with a spacer is preferable as either the fluorophore or the receptor or simultaneously both units can be varied at will. An ideal chemosensor will be the one that can recognize a particular metal ion in presence of many other competing ones. It should be designed with new recognition principles or fine tuning the existing ones, both of which are formidable challenges to chemists. The right donor atoms should be placed at strategic positions to match the coordination preferences of the metal ion. Biologically relevant paramagnetic transition metal ions quench emission quite effectively and in many cases, emission quenching has been adopted4 as a signaling mode to detect the presence of an analyte. However, for practical as well as sensitivity reasons, fluorescence enhancement and not quenching should be the preferred mechanism in fabricating a sensor. In simple terms, blocking of the fluorescence quenching by a paramagnetic transition metal ion requires that metal ion–receptor (M–R) interaction should be greater than the metal ion–fluorophore (M–F) communication. This idea can be incorporated in designing the overall architecture of the signaling system. Choice of the fluorophore should be made based on its excitation/emission characteristics, photochemical as well as thermal stability, magnitude of the Stokes shift, solubility properties and so on. In cases where a spacer is used, the nature of the later is also important5 in the signal transduction process. Although most of the emission techniques and uses revolve around small organic molecules as receptors, recent years have witnessed a large number of supramolecular systems with interesting photochemical as well as photophysical properties. In addition to the sensory roles, it can also be used for information processing6 that opens up a whole new window of opportunities. Finally, accessing a signal via remote control with waveguides and fiber-optics technology has led to widespread use of the fluorescence techniques in chemistry related sensing and switching applications. In the present review, only first-row transition metal ions are covered without Zn2+ ion as several excellent review articles7 on Zn2+ ion sensing are available. Generally, literature from 2005 onwards has been covered. Of course, in some cases chemosensors reported prior to 2005 are included especially for those metal ions for which not many examples are known. However, the literature coverage is not exhaustive and at the outset, we apologize if we have not covered any important work in the present review. There are several reviews available in the literature8 on transition metal ion chemosensors using fluorescence.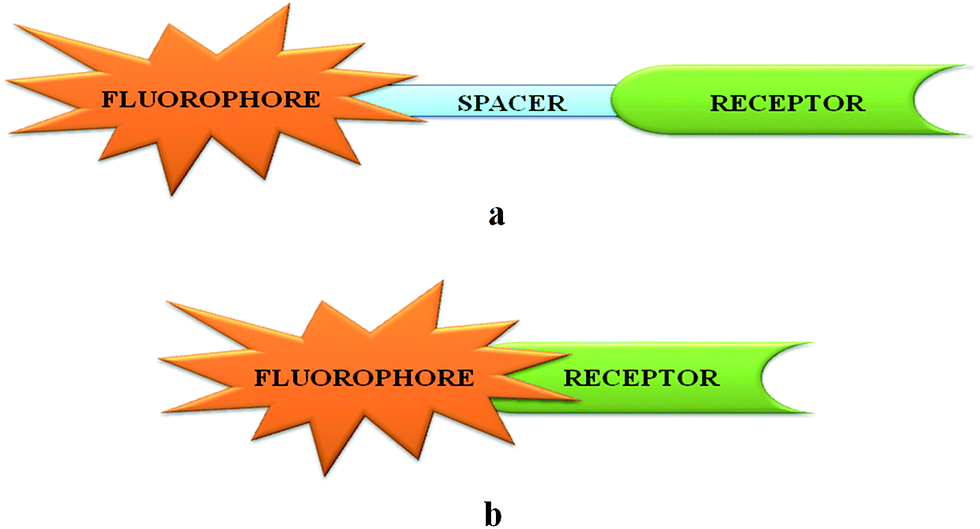 | ||
| Scheme 1 Two common designs used in fluorescence signaling unit (a) through spacer signaling unit and (b) integrated signaling unit. | ||
2. Sensing of metal ions
2.1. Scandium chemosensors
Pure organic stereoisomers are much sought after synthetic compounds in pharmaceutical, agrochemical, and other industries. These aspects have spurred research in asymmetric synthesis and the development of analytical techniques for determining their stereochemical purity. Fluorescence enhancement upon binding of Sc3+ with stereoisomers has been used as a high throughput technique for determining the purity of stereoisomers. Thus, chemosensor 1,8-bis(3-tert-butyl-9-acridyl)naphthalene N,N′-dioxide (1), has been synthesized from 3-tert-butylaniline and 2-chlorobenzoic acid.9 Free 1 does not show any significant emission upon excitation. However, with Sc3+ ion, it forms a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex showing high chelation-enhanced fluorescence enhancement (CHEF) while other competing metal ions like Cu2+, Zn2+, Yb3+, Sn2+ and In3+ do not exhibit such enhancement. Compound 1 thus serves as a selective fluorescence-on chemosensor for the Sc3+ ion. Stereoselective displacement of Sc3+ from its complex with concomitant loss of fluorescence output can be used to determine the presence of a particular enantiomer.
:1 complex showing high chelation-enhanced fluorescence enhancement (CHEF) while other competing metal ions like Cu2+, Zn2+, Yb3+, Sn2+ and In3+ do not exhibit such enhancement. Compound 1 thus serves as a selective fluorescence-on chemosensor for the Sc3+ ion. Stereoselective displacement of Sc3+ from its complex with concomitant loss of fluorescence output can be used to determine the presence of a particular enantiomer.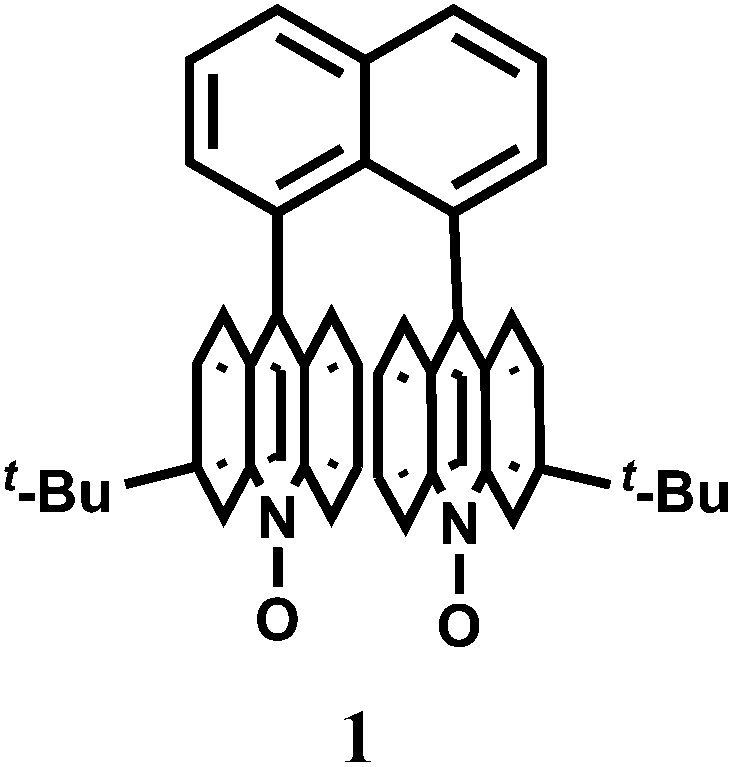
The compound 2,3,5,6-tetrakis(2-pyridyl)pyrazine (TPPZ) (2) itself shows negligible fluorescence but in presence of 1 equivalent of Sc3+ ion in MeCN affords a strong fluorescence due to the formation of a 1:1 complex10 (Scheme 2) albeit with a low association constant of (1.4 ± 0.1) × 102. Since Zn2+ ion strongly interferes, it must be absent for sensing of Sc3+ ion.
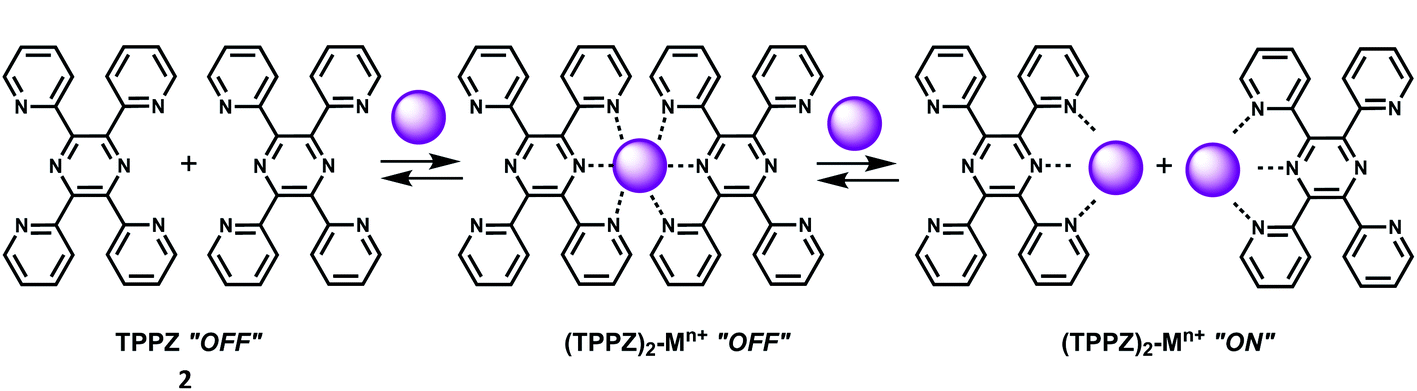 | ||
| Scheme 2 Binding mode of 2 with metal ion. | ||
2.2. Vanadium chemosensors
Vanadium is an essential trace element due to its significant roles in various enzyme systems.11 Vanadate ion (VO43−) is a structural analogue of phosphate; it is also a transition state analogue of phosphate in the penta-coordinated form. So, vanadate interacts with a broad range of phosphate-metabolizing enzymes.12 Besides, vanadate is also found as the cofactor of vanadium haloperoxidases (V-HPOs),13 which are important enzymes that catalyze halogenation of natural compounds.14 In aqueous medium, however, vanadate ion undergoes complicated hydrolysis and oligomerization reactions that can make it a difficult one to trace. Epidemiological evidences suggest that vanadium may also play a beneficial role in the prevention of heart-disease.15 However, it is quite toxic and at high concentration level, vanadium is a potentially dangerous chemical pollutant that can play havoc with the agricultural system. It is thus quite important to have a method available for quantitative detection of the metal ion in aqueous medium.The rhodamine-derived chemosensor 3 has been synthesized by reacting rhodamine hydrazide with 5-chlorosalicylaldehyde in ethanol16 to provide a N2O donor set. Metal free 3 does not show any emission when excited at 530 nm in aqueous MeOH (1:1, v/v, pH 7.2) as the spirolactam ring remains intact. However, in presence of 10−9 M VO2+ at pH 7–8, a 40-fold enhancement of emission is observed at 583 nm due to spiro-bond cleavage (Scheme 3). On the other hand, Cu2+ affords only 3.6 fold enhancement. Other competing metal ions (Bi3+, Tb3+, Pb2+, Cr3+, Al3+, Eu3+, Ru3+, Sm3+, Sn4+, Ni2+, Co2+, Yb3+, Hg2+, Ga3+, Zr4+, Er3+, Ho3+, Fe3+, Zn2+, Mg2+, Mn2+, La3+, Nd3+) including heavy metal ions do not give any emission. The VO2+ ion which has a preference for O donors, forms a square pyramidal complex with the chemosensor. ESI-MS data supports the formation of a 1:1 complex although no stability constant value for the complexation is available.
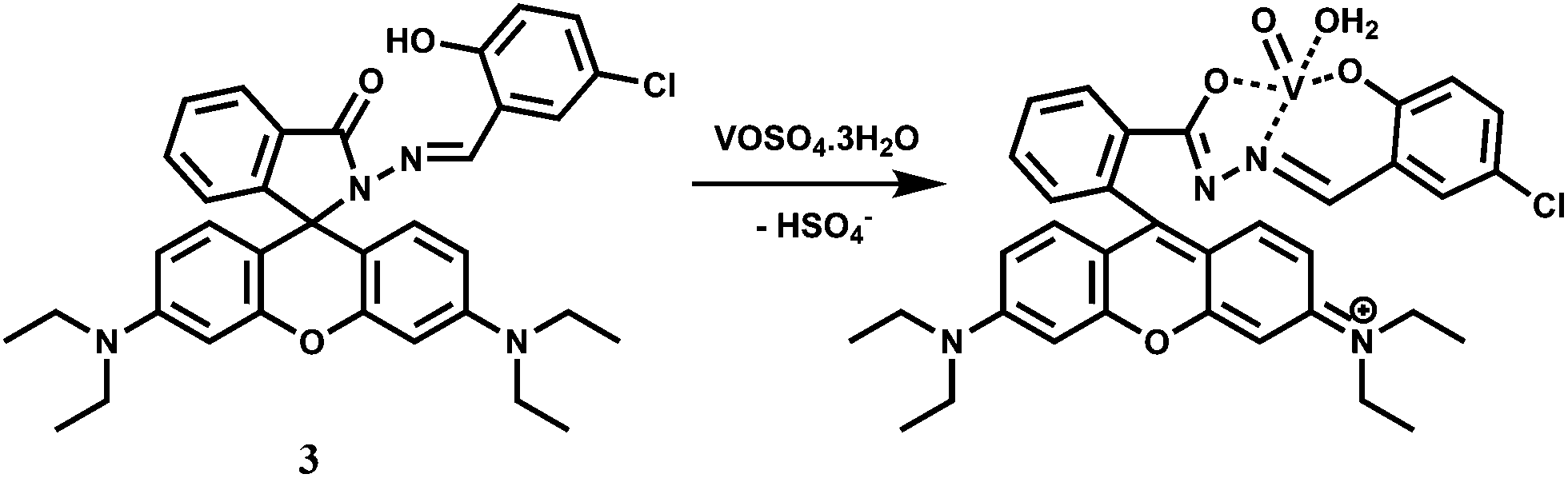 | ||
| Scheme 3 Proposed sensing mechanism of 3 to VO2+. | ||
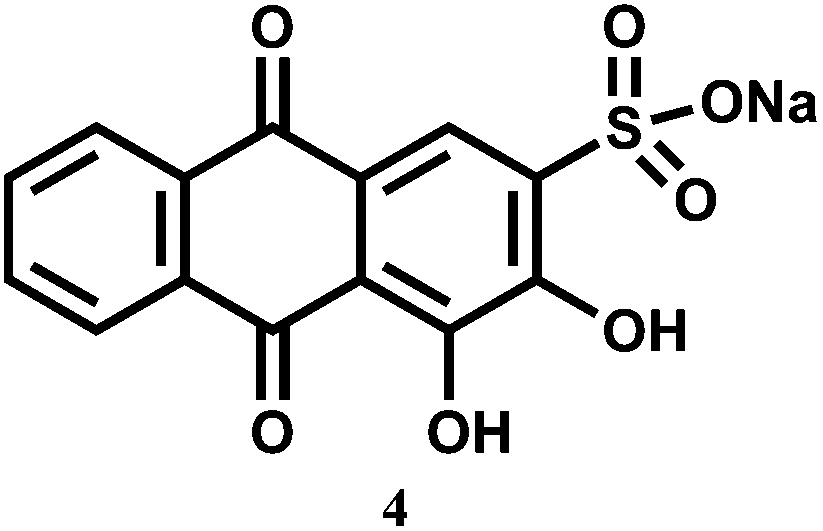
A flow method has been developed for the determination of pentavalent V in different media. In this method, continuous fluorescence measurement is done on the complex formed between V5+ and 1,2-dihydroxyanthraquinone-3-sulfonate (4) sorbed on anionic resin beads.17 Although no association constant data are available, it is found that the dye shows increased sensitivity by about 4 × 103 times higher compared to the same system without immobilizing beads. Besides, immobilization of the reagent in the solid support18 makes possible the production of a more rigid environment which closes deactivation pathway. Both these factors allow nM amounts of V5+ to be detected in different kinds of samples, viz., water from various sources, urine and serum, as well as mussel tissues.
The receptor 5 has been designed for sensing vanadates and its oligomers.19 The pyrene groups in 5 with their strong stacking tendency, can make the receptor pre-organized20 and this can open up to accommodate dimer and trimers of vanadate. The chemosensor is found to bind preferentially the vanadate VO43− ion in DMSO (Scheme 4). The fluorescence spectrum of 5 in DMSO exhibits pyrene monomer emission bands at 390, 410 and 437 nm as well as a small peak at 607 nm. Upon addition of VO43−, the monomer emissions along with the emission at 607 nm are quenched while a peak at 508 nm, assigned to the emission of the pyrene excimer21 and a shoulder at 564 nm appear, confirming stacking of the pyrene units. With the addition of 7 equivalents of VO43−, the intensity of the major peak at 513 nm, with a 5 nm red-shift, increased significantly with a 30-fold enhancement. After about 8 h, the peak at 513 nm disappears and a new peak at 564 nm appears showing a 50-fold enhancement due to binding of the oligomers that are in equilibrium with the monomer. This sensor, therefore, represents a design to tackle complicated hydrolysis and oligomerization of the vanadate ion in aqueous medium.
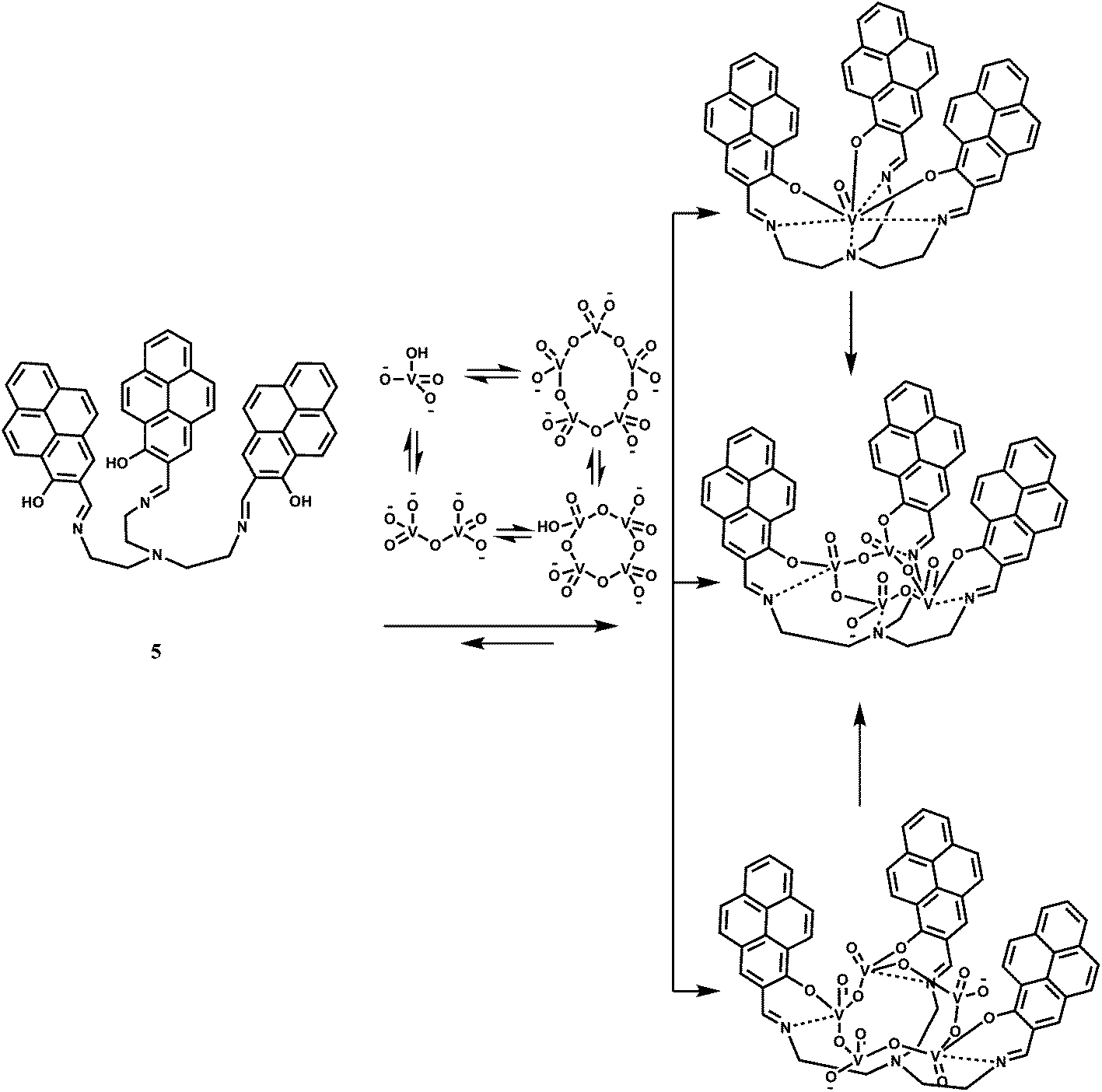 | ||
| Scheme 4 Proposed binding mechanism of 5 with different vanadate species. | ||
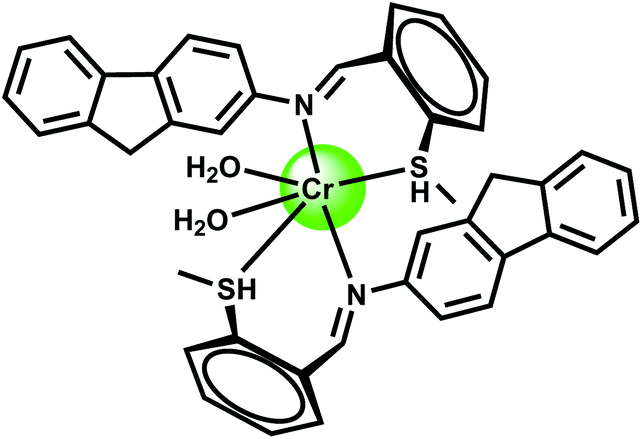
2.3. Chromium chemosensors
Cr3+ is an essential trace element in human nutrition. It exerts great impacts on the metabolism of carbohydrates, fats, proteins and nucleic acids by activating certain enzymes and stabilizing proteins and nucleic acids.22 Insufficient dietary intake of Cr3+ leads to increased risk factors associated with diabetes and cardiovascular diseases, including elevated levels of circulating insulin, glucose, triglycerides and total cholesterol and impaired immune function.23 As a contaminant, chromium is found mostly in Cr(VI) form and its bacterial reduction to Cr3+ is considered as one of the promising strategies for bioremediation. However, exposure to high levels of Cr3+ can negatively affect cellular structures. The environmental protection agency (USA) has set the maximum permissible level of total chromium at 100 ppb. Fluorescence turn-on reagents for monitoring intracellular Cr3+ concentration remain underdeveloped especially in aqueous media. In earlier reports on Cr3+ sensors, several divalent and trivalent metal ions like Cu2+, Zn2+, Cd2+, Ni2+ or Al3+ were found24 to interfere with the detection processes. Amongst different designs, spirolactam ring opening in presence of the metal ion has been a popular method of detection of the Cr3+ ion.25The chemosensor 6 is based on rhodamine B where 8-hydroxyquinoline moiety has been attached along with a ferrocene substituent.26 This is found to be highly selective for the Cr3+ ion in presence of a host of other biologically relevant metal ions. The association constant for Cr3+ is estimated to be 7.5 × 103 M−1 in the aqueous ethanol (1:1, v/v, pH 7.4) solution. On excitation at 530 nm in 50% aqueous ethanol, metal-free 6 shows very low fluorescence at 580 nm since the spirocyclic form of rhodamine prevail. Upon addition of Cr3+ ion in the pH range of 5–10 in aqueous ethanol, a 15-fold increase in fluorescence is observed within 1 min, with a peak at 587 nm due to opening of the spirolactam ring. Under similar experimental conditions, only Hg2+ exhibits slight enhancement. However, other metal ions do not show any discernible change compared to the metal-free chemosensor. The most likely donor atoms for Cr3+ are the conjugated moiety including carbonyl O, imino N, and quinoline N and O atoms (Scheme 5). Due to its instantaneous response and solubility in aqueous ethanol, 6 is used in cell imaging studies. When the HeLa cells are stained with a 20 μM solution in ethanol–PBS buffer (1:99, v/v) for 30 min at 25 °C a very faint intracellular fluorescence is observed. Addition of Cr3+ at 37 °C, affords a significant fluorescence in the intracellular domain in 2.5 h. Bright-field measurements after the Cr3+ treatment indicates that the cells remain viable throughout the period. Moreover, differential pulse voltammetric studies in ethanol gives a significant shift of the ferrocenium/ferrocene potential in absence/presence of Cr3+ ion indicating that 6 can be used as a redox chemosensor of Cr3+.
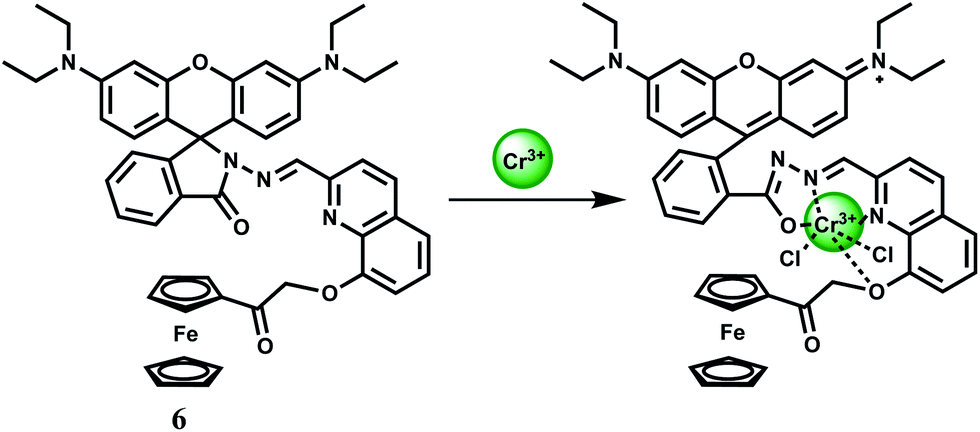 | ||
| Scheme 5 Probable complexation mechanism of 6 with Cr3+. | ||
The rhodamine 6G based chemosensor 7 has been designed incorporating a diethylene triamine moiety as the binding site for Cr3+ ion at pH 7.2 in aqueous medium.27 The association constant (Ka) with Cr3+ is found to be 4.16 × 104 M−1. This sensor gives about 60-fold enhancement upon addition of 5 equivalents of Cr3+ due to spiro-ring opening (Scheme 6) while other ions including Cd2+, Co2+, Cu2+, Ni2+, Zn2+, Mg2+, Ba2+, Pb2+, Na+ and K+ afford no distinct response. Only Fe3+ also forms a 1:1 complex giving a stability constant of 4.73 × 103 M−1 and shows a 17-fold enhancement upon addition of 5 equivalents of Fe3+ ion. Iron being much more biologically abundant than chromium, this chemosensor can only be useful in absence of Fe3+ ion which seriously limits its utility. This also suggests that cell imaging studies for temporal distribution of Cr3+ cannot be made with 7.
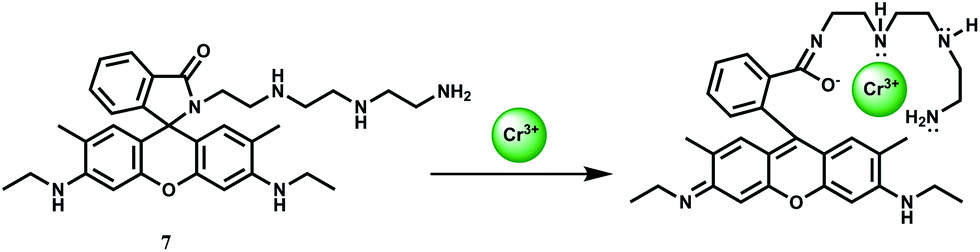 | ||
| Scheme 6 Proposed mechanism for fluorescence enhancement of 7 upon the addition of Cr3+. | ||
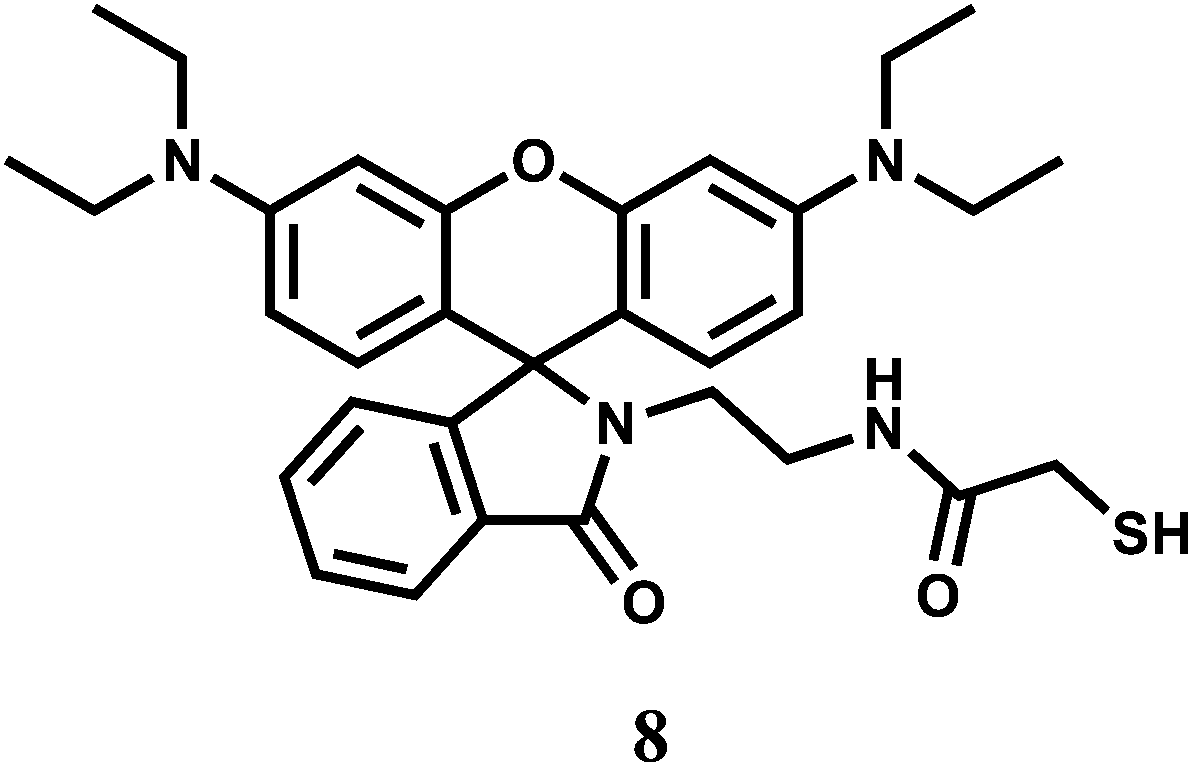
The rhodamine B derived chemosensor 8 forms a 2:1 complex with Cr3+ and exhibits appreciable response due to spiro-bond opening in the range, 0–10 μM with a detection limit of 1 ppm. The system is functional within a wide range of pH (4.0–8.0) in 20% aqueous methanolic medium.28 Other competing metal ions that include alkali/alkaline earth, first row transition and heavy metal ions, do not give any significant response. The response towards Cr3+ is found to be independent of the nature of anion used. Although response time for enhancement is found to be sluggish (∼10 min), no interference from competing metal ions makes it an excellent sensor for the Cr3+ ion. Its usability in aqueous alcohol allows for imaging of living MGC803 cell lines demonstrating its potential applications in biological systems.
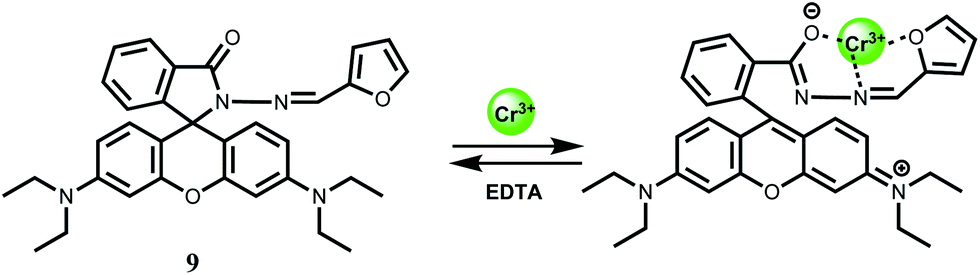
For sensing of Cr3+ ion in aqueous medium with low detection limit, the rhodamine B derivative 9 has been synthesized.29 This chemosensor gives Cr3+ ion induced spiro-bond opened turn-on fluorescence in aqueous ethanol within a wide pH (5–9) range. The detection limit is found to be quite low at 0.023 μM. Besides, the Cr3+ ion can be removed using EDTA making the system reversible. However, the metal–chemosensor stoichiometry of 1:1 proposed by the authors is not consistent with the strong preference of hexacoordintion for the Cr3+ ion (Scheme 7). No association constant value for complexation is available. However, since other competing metal ions do not show any significant emission, this system has been used for intracellular imaging in live cells and determination of the ion in tap water and river water.
 | ||
| Scheme 7 Proposed mechanism for fluorescence enhancement of 8 upon the addition of Cr3+. | ||
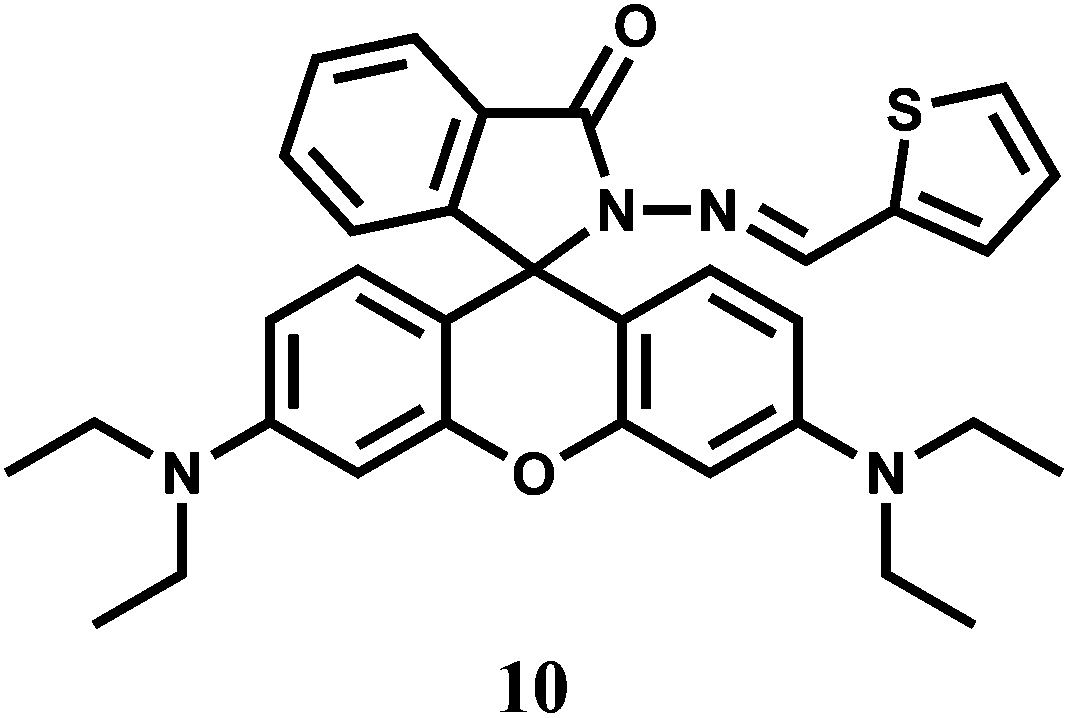
The chemosensor 10 is structurally quite similar to 9 except that a thiophene moiety has been used in place of a furan to bind a metal ion through the carbonyl O, inamine N and thiophene S donors.30 The sensor forms a 1:1 complex with Cr3+ giving a moderately high association constant of 2.0 × 104 M−1. Complexing with Cr3+ triggers the formation of the highly fluorescent ring-opened form which is pink in colour. It affords an emission centering at 583 nm with about 1200-fold enhancement. However, Hg2+ also gives significant emission at 576 nm with this fluorescent probe.
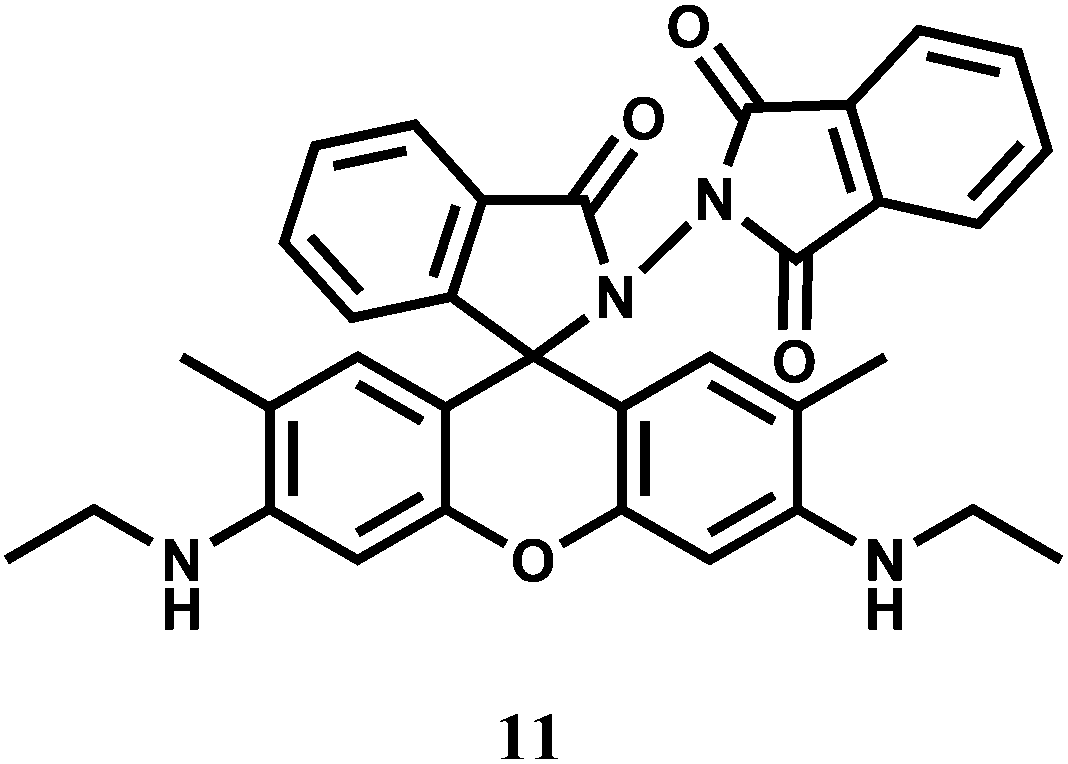
Rhodamine 6G derived chemosensors 11 and 12 are found to bind specifically to Hg2+ or Cr3+ in presence of large excess of other competing ions with associated changes in their optical and fluorescence spectral properties.31 For 11, the detection limit is lower than the permissible [Cr3+] or [Hg2+] in drinking water as per standard U.S. EPA norms. The chemosensor 12 can be used as a ratiometric sensor for detection of Cr3+ and Hg2+ based on the Förster resonance energy transfer (FRET) process involving the donor naphthalimide and the acceptor Cr3+/Hg2+-bound xanthene fragment (Scheme 8). Studies reveal that 12 can be used for recognition and sensing of Hg2+/Cr3+. Further, confocal microscopic studies confirm that 12 can also be used as an imaging probe for detection of these ions in A431 cells.
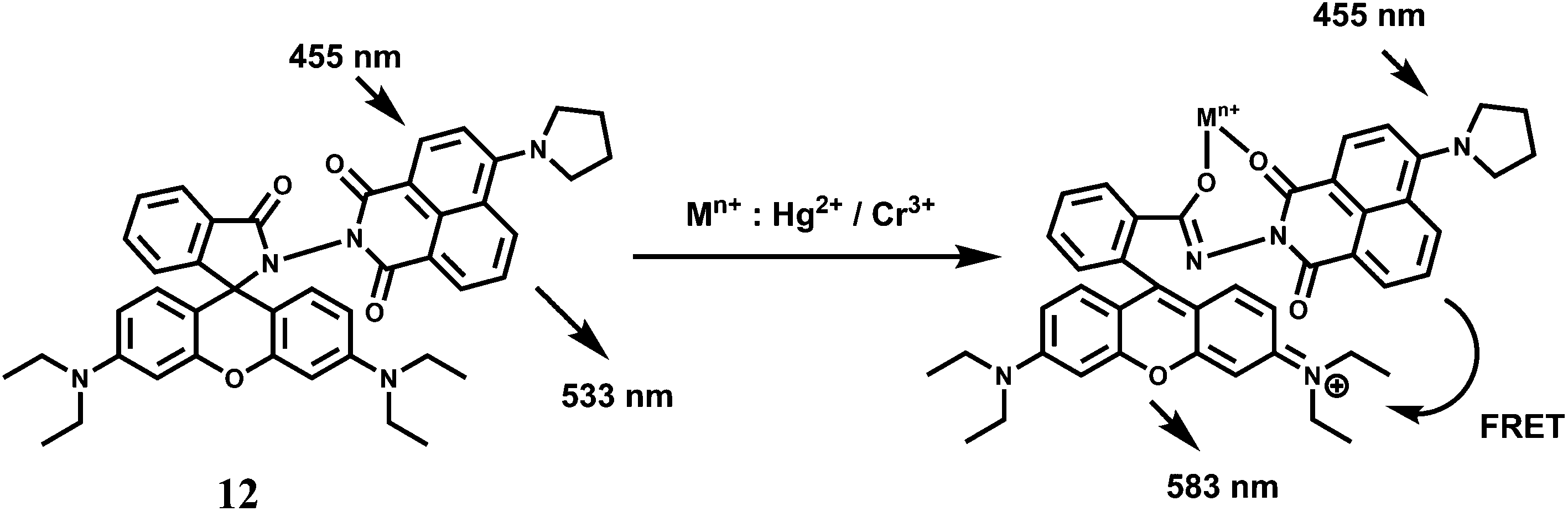 | ||
| Scheme 8 Probable metal ion binding mode of 12. | ||
On the basis of FRET from 1,8-naphthalimide to rhodamine, the dyad 13 has been synthesized as a Cr3+ selective fluorescent probe for monitoring Cr3+ ion concentration in living cells with ratiometric fluorescence methods.32 When excited at 405 nm in aqueous ethanol (1:2 v/v), a yellow fluorescence centered at 536 nm is observed attributable to an intramolecular charge transfer (ICT) band involving the 1,8-naphthalimide chromophore. Addition of 10 equivalents of Cr3+ ion causes the appearance of a strong absorption band centered at 568 nm changing the colour of the solution to red-orange from yellow. The Cr3+-complex on excitation at 515 nm, shows a 15-fold enhancement of fluorescence at 590 nm due to spirolactam ring opening (Scheme 9). Further, when excited at 405 nm, the ICT band intensity at 536 nm is reduced drastically with a concomitant increase of the intensity at 594 nm due to an efficient FRET from the naphthalimide to the ring-opened spirolactam. This fluorescent probe exhibits excellent selectivity over other metal ions. Competition experiments for Cr3+ mixed with excess of competing metal ions does not show any change in the emission band intensity, although Mn2+, Ag+ and Al3+ ions induces slight quenching or increasing of the ratio, F594/F536. Confocal microscopic studies confirm that 13 can be used for monitoring intracellular Cr3+ levels in living cells with general fluorescence and FRET methods. Such systems will be of help in studying the bioactivity of Cr3+ in biological systems.
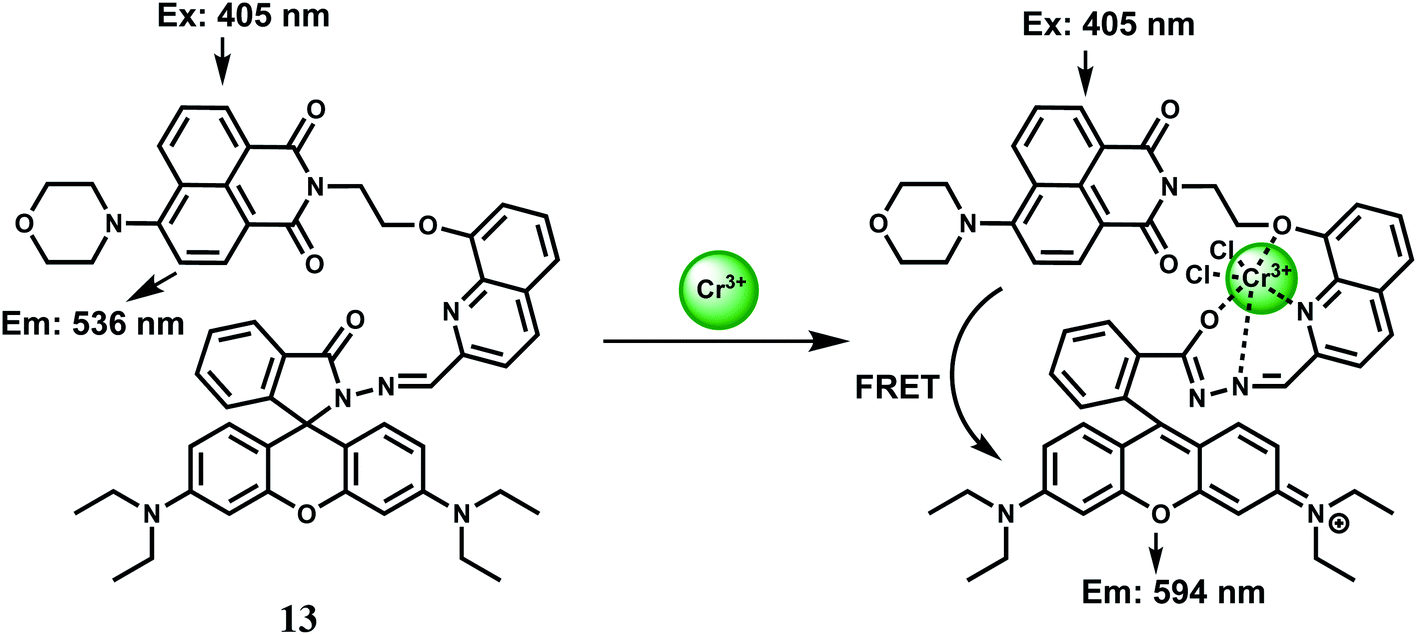 | ||
| Scheme 9 Probable sensing mechanism of 13. | ||
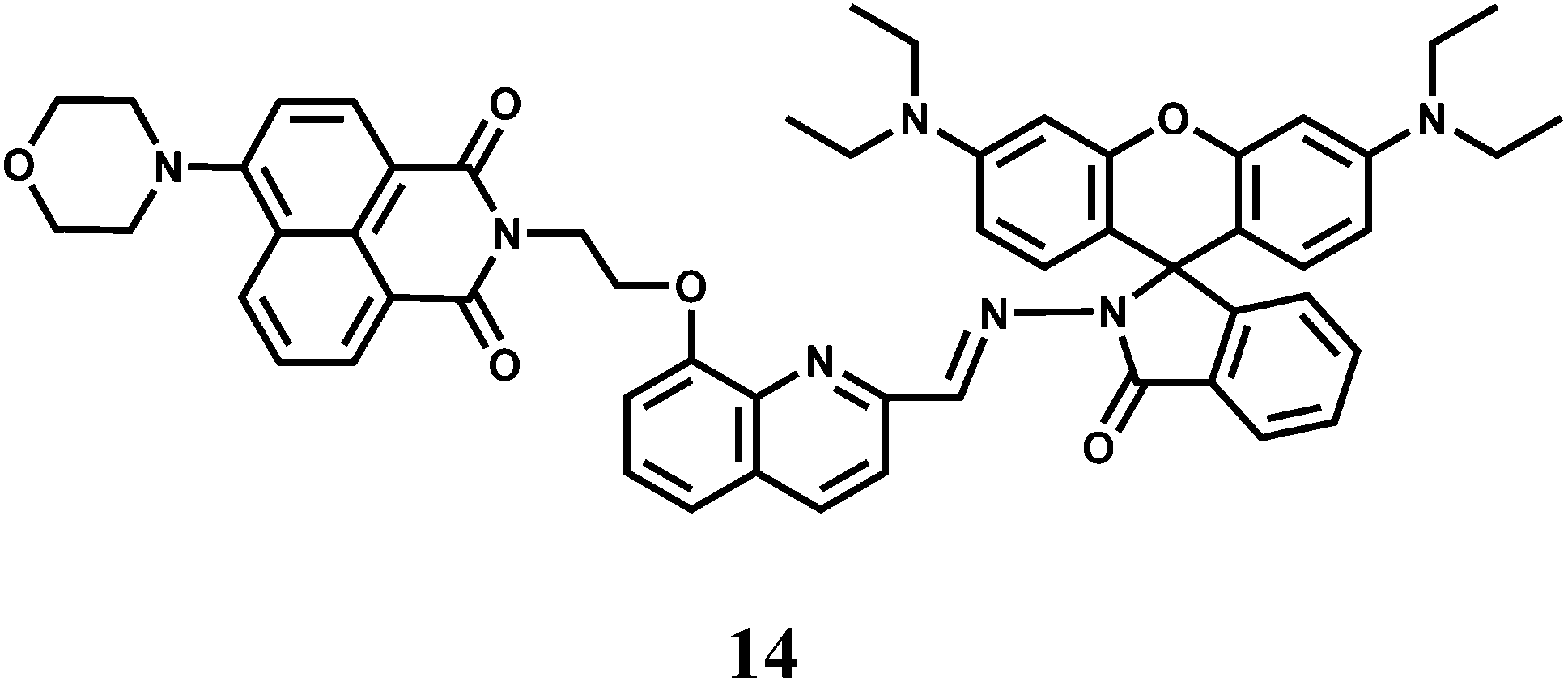
The absorption maximum of a rhodamine derivative normally appears around 550 nm and typically gives a 25 nm Stokes shift of the emission band. This can limit its application as an effective chemosensor in many biological applications.33,34 To circumvent this, the chemosensor 14 capable of showing two-photon excited fluorescence (TPEF) upon Cr3+ binding has been synthesized.35 In presence of Cr3+ ion, 14 changes to the open-ring form giving remarkable fluorescence enhancement both in one and two-photon excitations. The broad excitation wavelength, on/off fluorescence and high selectivity to Cr3+ (KS = 9.4 × 103 M−1) enable 14 to be a powerful Cr3+ cation sensor with potential applications in biological systems.
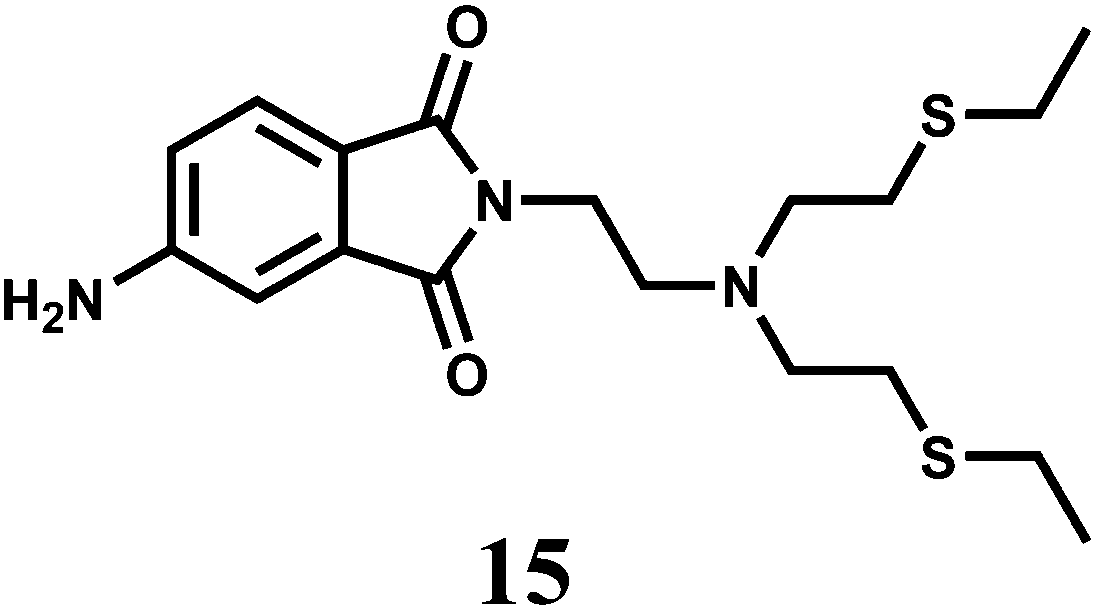
The photoinduced electron transfer (PET) chemosensor 15 represents one of the few fluorescence chemosensor for Cr3+ where a rhodamine is not used.36 The signaling and the receptor moieties are covalently attached. In THF, it exhibits 17-fold chelation enhanced fluorescence (CHEF) in presence of Cr3+. The receptor is found to be highly selective for Cr3+ ion against the background of environmentally and biologically relevant metal ions forming a 1:1 complex with a high association constant of 11.3 × 104 M−1.
The highly selective luminescent chemosensor 16 in aqueous solution has been assembled by a low-selectivity luminogenic receptor with Cu2+ as a metal quencher.37 Three tetranitrogen chelating sites are integrated into the multichannel receptor with a tris(1,10-phenanthroline)ruthenium(II) luminophore at the core. This receptor exhibits chelating affinity towards several transition metal cations, among which Cu2+ efficiently quenches the emission. Addition of Cr3+ into the Cu2+-complex of 16 results in a metal-exchange reaction with concomitant recovery of fluorescence. Other transition metal ions do not take part in the metal-exchange reaction. The quencher displacement sensing strategy in this design can be a simple but efficient approach for OFF–ON luminescent sensing of metal cations that inherently lack selective ligands. This strategy of fluorescence sensing is, however, not that useful for biological imaging studies as many side reactions might interfere with the determination of a particular ion.

Two PET chemosensors 17 and 18, incorporating dansyl dye as a fluorophore and various carboxhydrazones as receptors, have been reported.38 Sensor 17 contains a pyridine-carboxhydrazone tridentate coordination site, forming a 2:1 complex with Cr3+ (Kas = 6.07 ± 0.10 × 107 M−1). Metal-free 17 gives a weak dansyl based emission at 545 nm when excited at 350 nm in DMF–water (9:1, v/v). Upon addition of Cr3+ it affords a 5-fold enhancement in the pH range, 5–9. No emission is observed in presence of alkali, alkaline earth or competing first-row transition metal ions. Likewise, 18 offers a 4-fold enhancement of dansyl based emission selectively in presence of Cr3+ under similar experimental conditions (Scheme 10). The metal ion forms a 1:1 complex with 18 giving an association constant value of Kas = 9.59 ± 0.03 × 103 M−1.
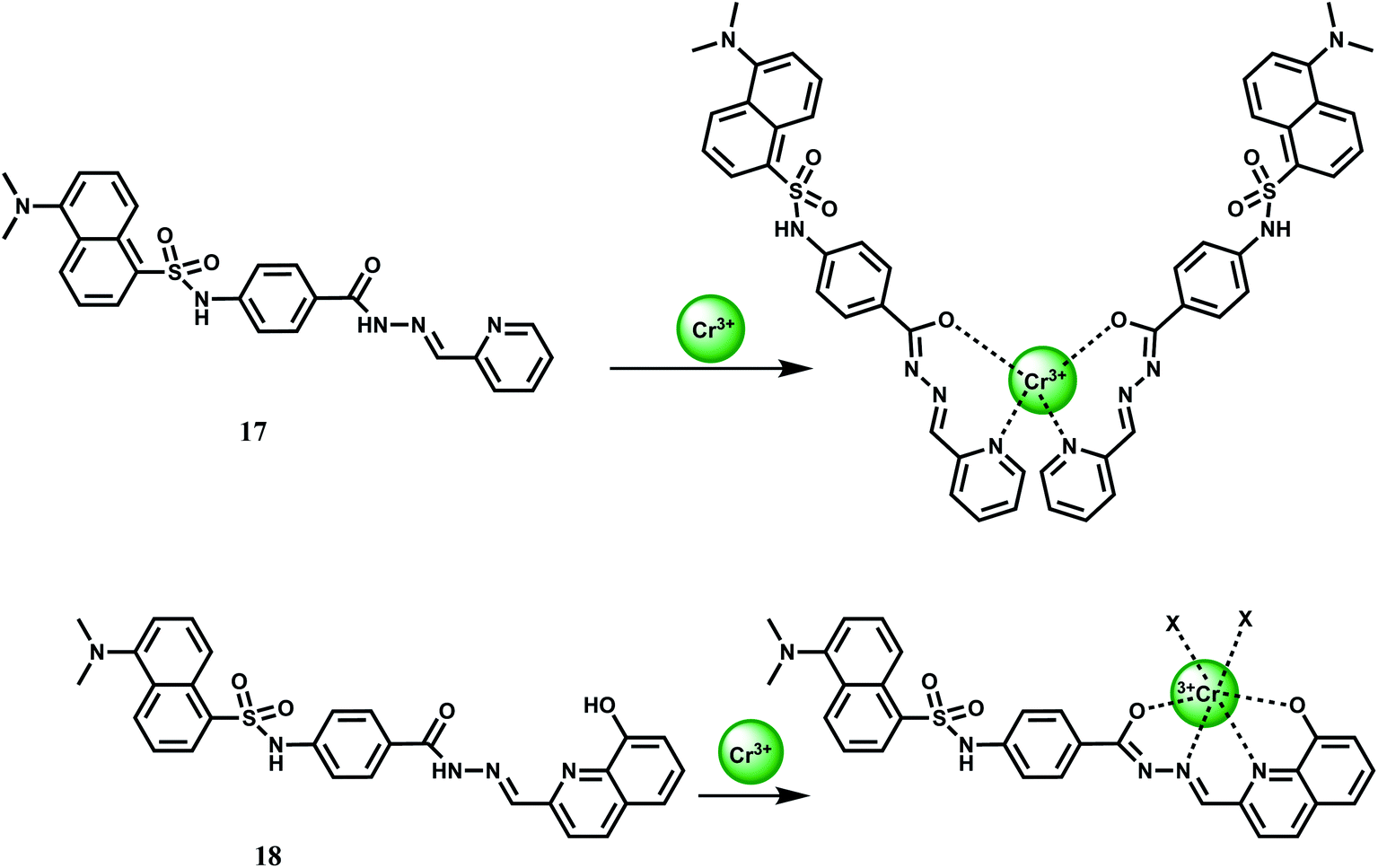 | ||
| Scheme 10 Possible binding mode of 17 and 18 with Cr3+ (X is the coordinating anion or solvent). | ||
Biaryl borondipyrromethene (BODIPY) fluorophore has high photostability with high fluorescence quantum yields. Besides, it is excitable with visible light, has narrow absorption and emission bands and is amenable to easy structural modification that can lead to spectral shifts in the absorption and emission bands to longer wavelengths. In 19, a BODIPY group is covalently connected to a receptor incorporating NO donors. In acetonitrile, metal-free 19 gives almost no fluorescence when excited at 560 nm due to efficient PET from the amino N to the BODIPY.39
Upon addition of Cr3+ ion, initially it forms a 2:1 complex (Scheme 11) giving weak fluorescence because the uncoordinated amine nitrogens can take part in the PET process to the photoexcited BODIPY moieties. With gradual increase of Cr3+ ion concentration, it forms a 2:2 complex (Scheme 11) engaging all the N lone-pairs that blocks PET to the BODIPY group. As a result, a strong BODIPY based emission with nearly 2800-fold enhancement factor is observed around 640 nm. The enhancement is observed within 10 s indicating that 19 can detect Cr3+ very rapidly. Other competing metal ions do not interfere with Cr3+ sensing. However, in presence of more than 1% water in MeCN, Fe3+, Cu2+ and Hg2+ strongly quenches fluorescence and thus seriously limits the use of 19 as a probe. Besides, the detailed coordination mode of Cr3+ with 19 requires further studies.
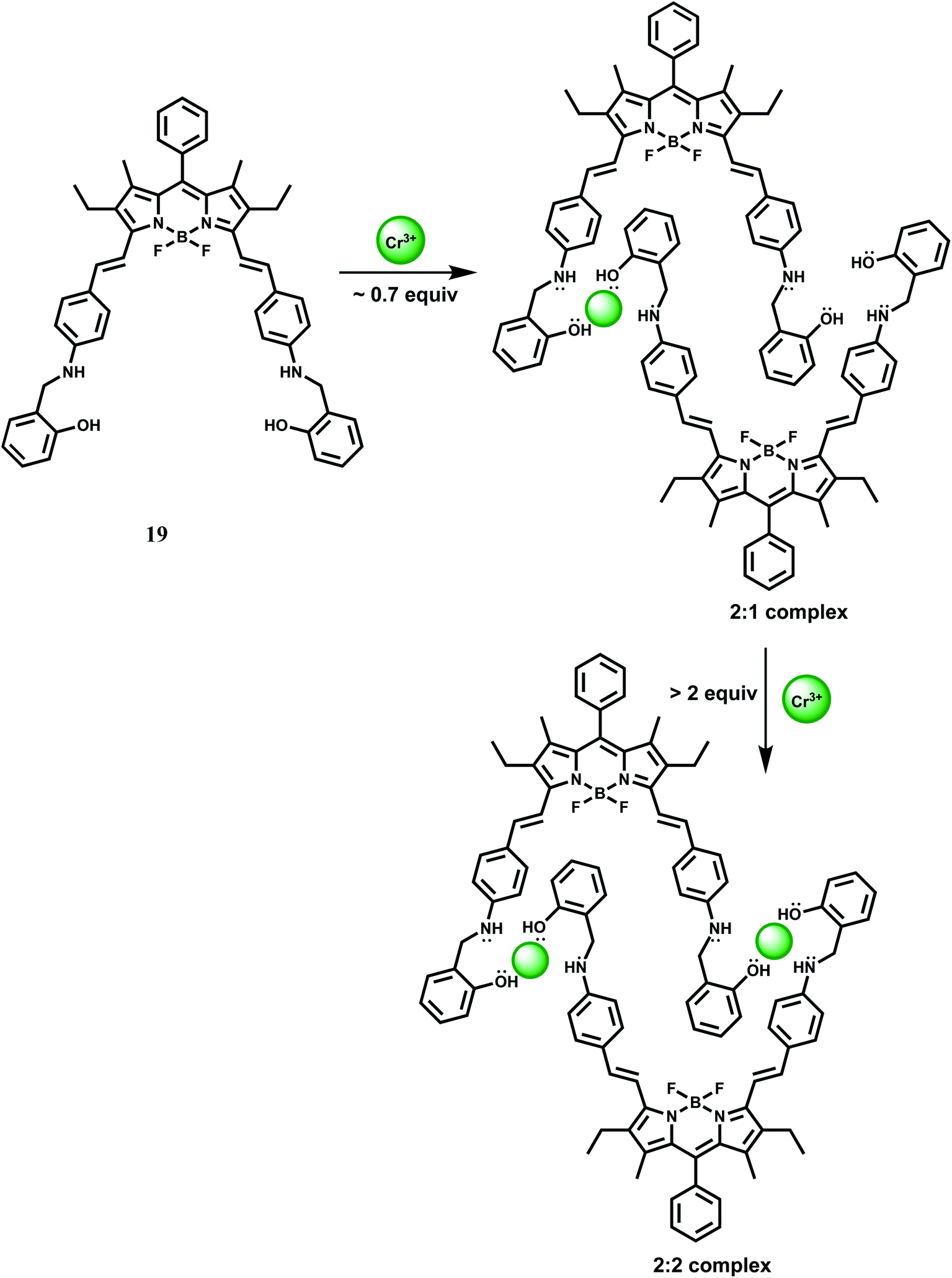 | ||
| Scheme 11 Possible sequence for coordination between 19 and Cr3+. | ||
For ratiometric fluorescence sensing of Cr3+ in aqueous medium, a rhodamine derivative (RG1) and a coumarin derivative (CG1) have been used.40 Together, they constitute the chemosensor 20 as shown in Scheme 12. Upon addition of Cr3+ to a buffered solution (pH = 6) containing equimolar amounts of CG1 and RG1, the metal ion binds to both the components as shown (Scheme 12). When the coumarin derivative is excited at 450 nm, the metal ion acts as a conduit for efficient FRET from the coumarin moiety to the ring-opened rhodamine moiety.41 Fluorescence measurements also show an excellent selectivity toward Cr3+ over metal ions such as alkali, alkaline earth and other first-row transition metal ions. Even the presence of heavy metal ions cannot bring any obvious fluorescence change. Confocal fluorescence microscopic investigation on HeLa cells are found to be capable of detecting subcellular concentration of Cr3+ ion.
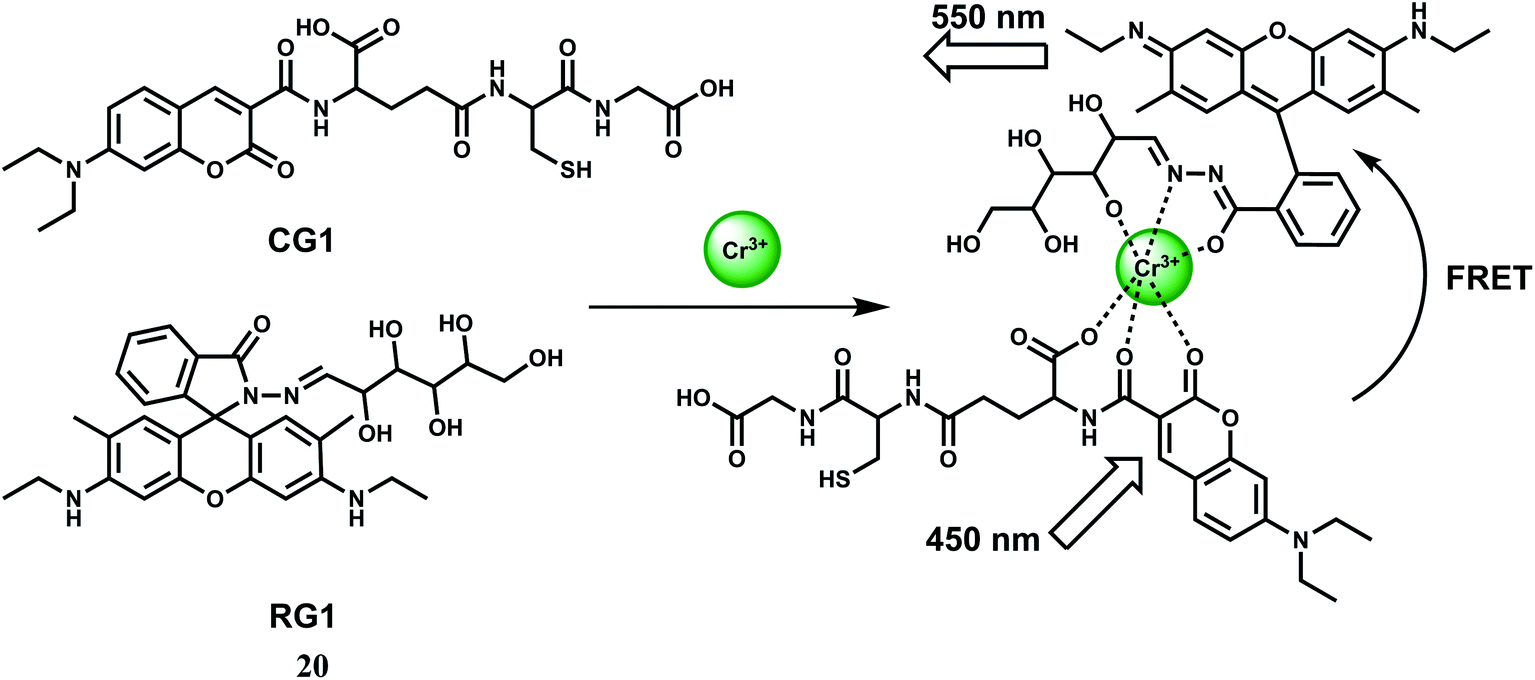 | ||
| Scheme 12 Possible sensing mechanism of 20. | ||
The fluorescent probes 21(a–d) having N, O/S/Se donors show donor specific ‘turn-on’ recognition property42 towards Cr3+, Fe2+ and Cu2+. Here, the fluorescence signals are controlled by the conformational change of the ligand framework upon binding with the metal ion (Scheme 13). Theoretical studies at the B3LYP/6-31G* level supports the mode of binding. However, they suffer from poor selectivity and only provide the starting point for the development of chemosensors with better selectivity and sensitivity.
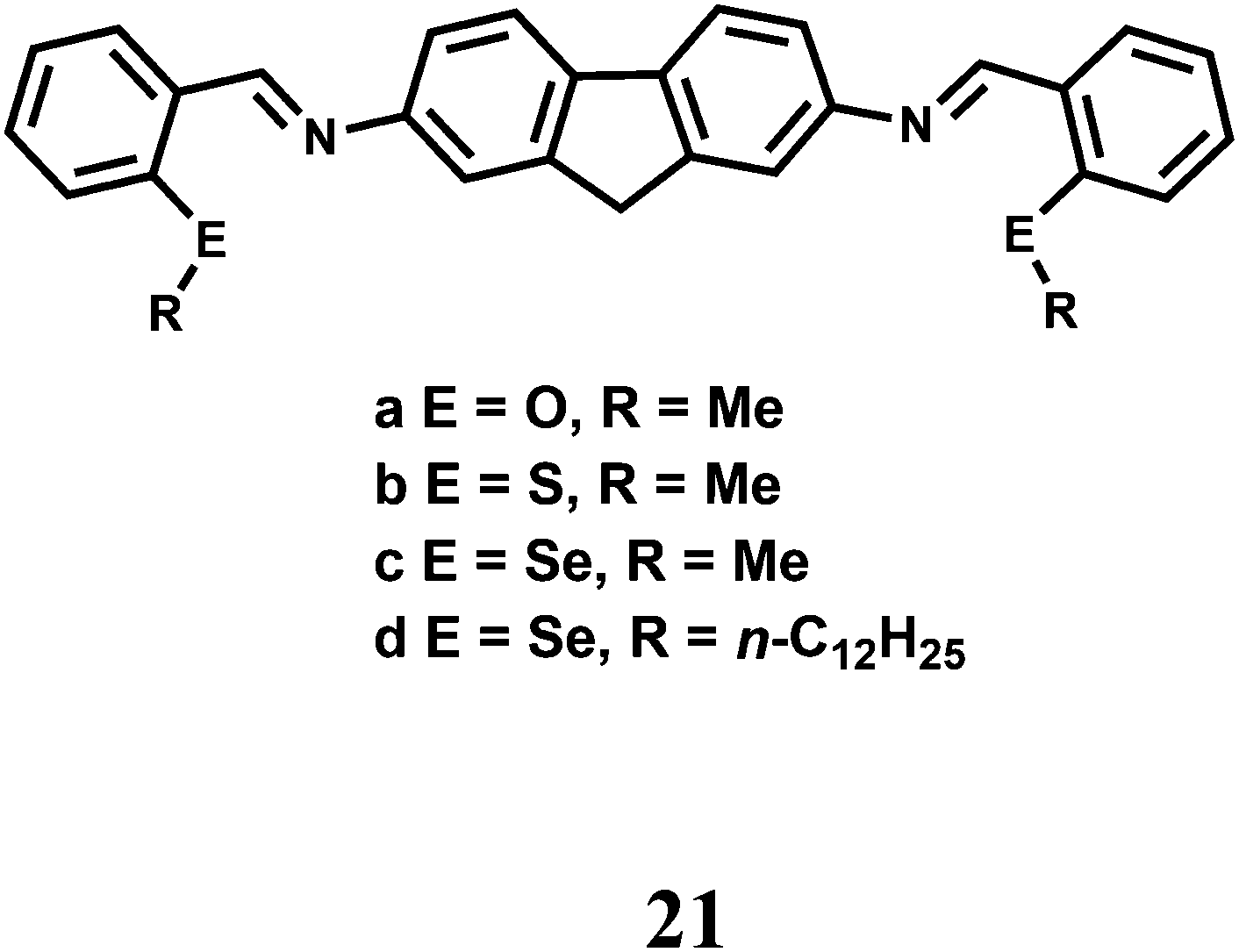
 | ||
| Scheme 13 The binding mode of Cr3+ ion with the fluorescent probe 21. | ||
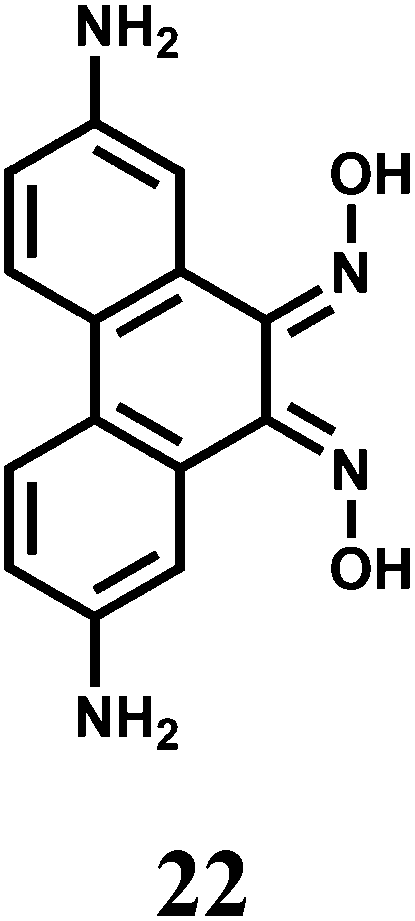
The bis-dioxime chemosensor 22 upon excitation at 417 nm gives an emission at 515 nm that shows about 60% CHEF enhancement in presence of Cr3+ while quenching in presence of Fe3+.43 Therefore, in absence of Fe3+, this sensor can be used for detecting Cr3+ ion.
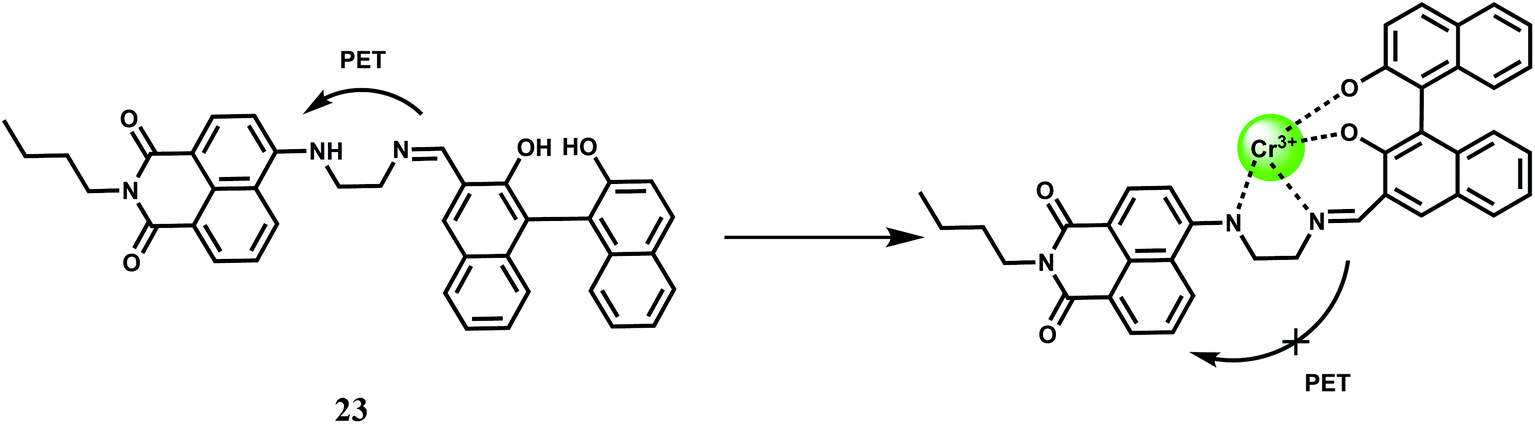
The chemosensor 23 with the naphthalimide based fluorophore connected to a BINOL framework shows low emission in THF–H2O (85/15, v/v) due to an efficient PET from the receptor.44 It forms a 1:1 complex (Scheme 14) with Cr3+ ion with a moderately high association constant of 2.4 × 104 M−1 in THF–H2O (85/15, v/v). Once a Cr3+ ion binds at the receptor it gives a 600-fold emission enhancement with the band centering at 498 nm. Other competing metal ions gives slight quenching although in the competition experiments they do not interfere. The detection limit is calculated to be 0.20 μM and response time is found to be about 10 s making it a fast responsive and highly sensitive probe for Cr3+.
 | ||
| Scheme 14 The binding mode of Cr3+ ion with the fluorescent probe 23. | ||
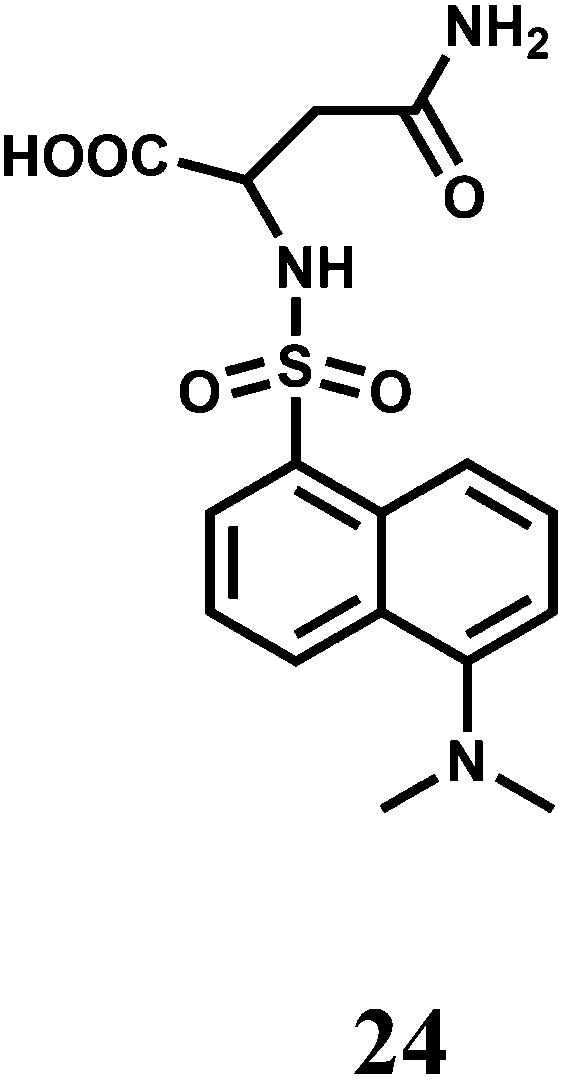
A dansyl fluorophore is covalently attached to asparagine to have the fluorescent probe 24.45 The probe shows a fluorescence “turn-on” and can detect Cr3+ at trace levels (5.2 ppb) in aqueous solution under acidic pH (5.9). When a Cr3+ ion binds at the sulphonamide group, the ICT from the sulfonamide to the dansyl is inhibited giving a 1.6-fold fluorescence enhancement. However, first row transition metal ions and few heavy metal ions also exhibit enhancement to a lesser degree and hence 24 should be regarded as a signaling system with poor chemoselectivity.
A spirobenzopyran derivative 25 with an amide moiety as the binding site for Cr3+ has been designed for use in MeCN.46 The spiro-bond breaking in presence of Cr3+ (Scheme 15) gives 25-fold enhancement of fluorescence at 675 nm. The metal ion forms a 1:1 complex with the association constant (Ka) of 7.5 × 104 M−1. This is one of the few probes that gives emission in the NIR region although it is workable in organic media and cannot be used in imaging studies. Efforts should be made to modify the system at sites away from the receptor for enhanced water solubility.
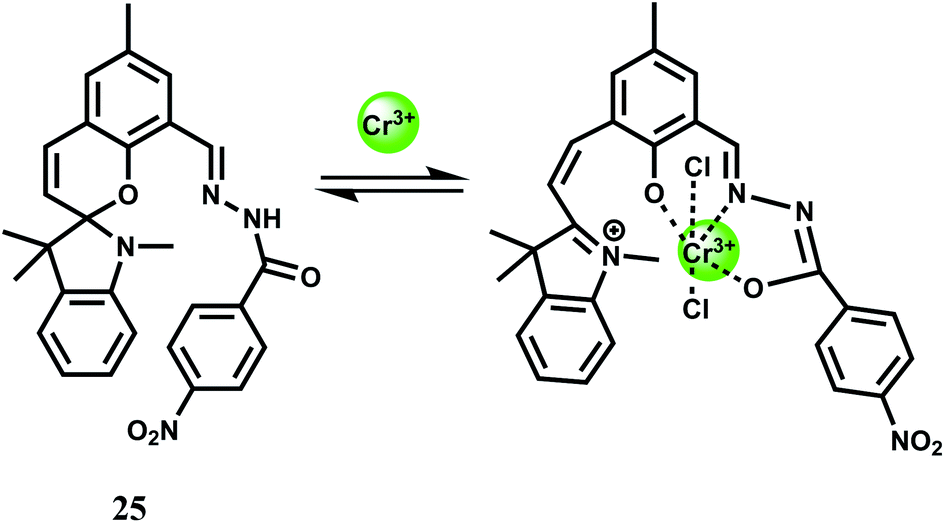 | ||
| Scheme 15 Possible sensing mechanism of 25. | ||
A fairly strong interaction between Cr3+ and the PET probe 26 is found in aqueous MeCN at pH 7.4 with 1:1 stoichiometry and a binding constant value of 8 × 104 M−1.47 The metal-free fluorescent probe does not emit significantly when excited at 409 nm. Upon addition of Cr3+, a 50-fold enhancement at 550 nm is observed due to snapping of the PET (Scheme 16). Competition experiments with other biologically relevant metal ions show that none of the metal ions interfere. Cell viability tests for the fluorescent probe is encouraging but use of organic solvent limits its use in live cell imaging studies.
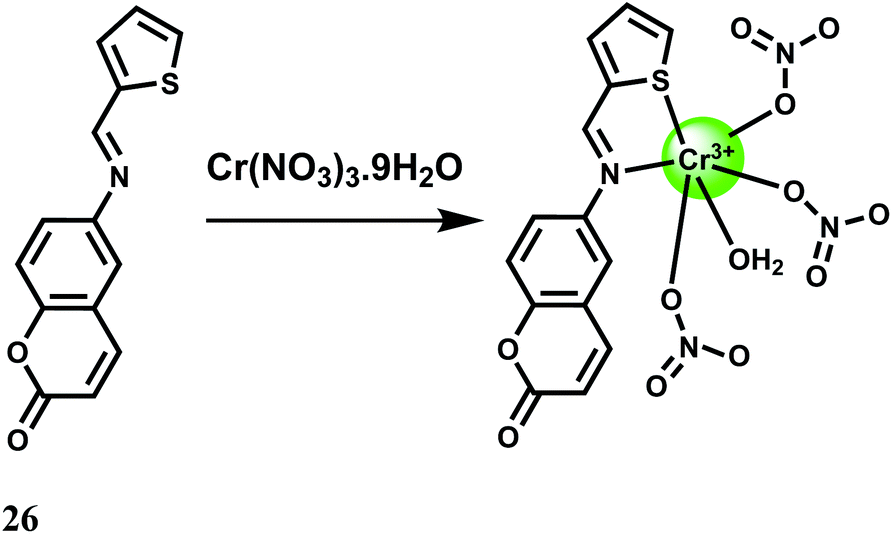 | ||
| Scheme 16 Possible sensing mechanism of 26. | ||
2.4. Manganese chemosensors
Manganese is an essential metal in all forms of life.48 The manganese enzymes that have attracted considerable attention are the O2 evolving complex of photosystem II and superoxide dismutase of prokaryotes and of the organelles of eukaryotes. This metal is also used widely as a versatile tool for biological studies. For example, high-spin Mn2+ is an excellent MRI relaxation agent that has been used in clinical diagnosis and is of widespread interest as a tool in neurobiological research.49 Chronic overexposure to manganese can result in movement disorders, mental disturbances and other brain-related toxicities.50 Fluorescent probes for detection and quantification of Mn2+ in biological systems will be of tremendous importance. However, development of an effective fluorescent probe for Mn2+ faces several challenges such as its paramagnetic nature, selectivity over abundant cellular metal ions especially Ca2+, aqueous solubility for cell imaging and so on. Overcoming these hurdles, presently there are few Mn2+ specific fluorescence signaling systems available. One such system (27) is a conjugate of 1,2-bis(2-pyren-1-ylmethylamino-ethoxy)ethane (NPEY) and graphene nanosheets (GNs) via π–π stacking that can be synthesized by simply mixing the diluted aqueous solutions of both components.51 The conjugate itself does not show any significant emission due to an efficient PET from NPEY to GN. Upon addition of Mn2+ ion that binds to the NPEY blocking the PET to give enhancement of fluorescence. The NPEY–GN combination (27) not only shows good selectivity toward Mn2+ with the detection limit as low as 4.6 × 10−5 M, but also shows “turn-on” response both in vitro and in living cells (Scheme 17). These sensing capabilities of NPEY–GNs in living cells make it a robust candidate for many biological fields, such as intracellular tracking, intracellular imaging, etc.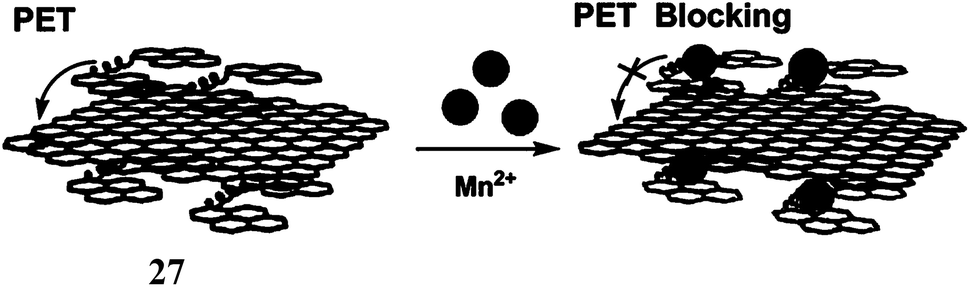 | ||
| Scheme 17 Schematic demonstration of fluorescence “Turn-On” mechanism for Mn2+ detection by 27. | ||
The receptor (O,O′-bis(2-aminophenyl)ethyleneglycol-N,N,N′,N′-tetraacetic acid) popularly known as BAPTA, is an excellent receptor for Ca2+ ion and several Ca2+ sensors are built around this moiety. Chemosensors 28 and 29 have been designed to differentiate binding affinities of Mn2+ over Ca2+ by partially replacing hard carboxylate oxygens of BAPTA by softer pyridines.52 Both Mn2+ and Ca2+ are classified as “hard” metals and, therefore, prefer to bind hard donors. However, Mn2+ appears to be more tolerant toward softer donors compared to Ca2+.53,54
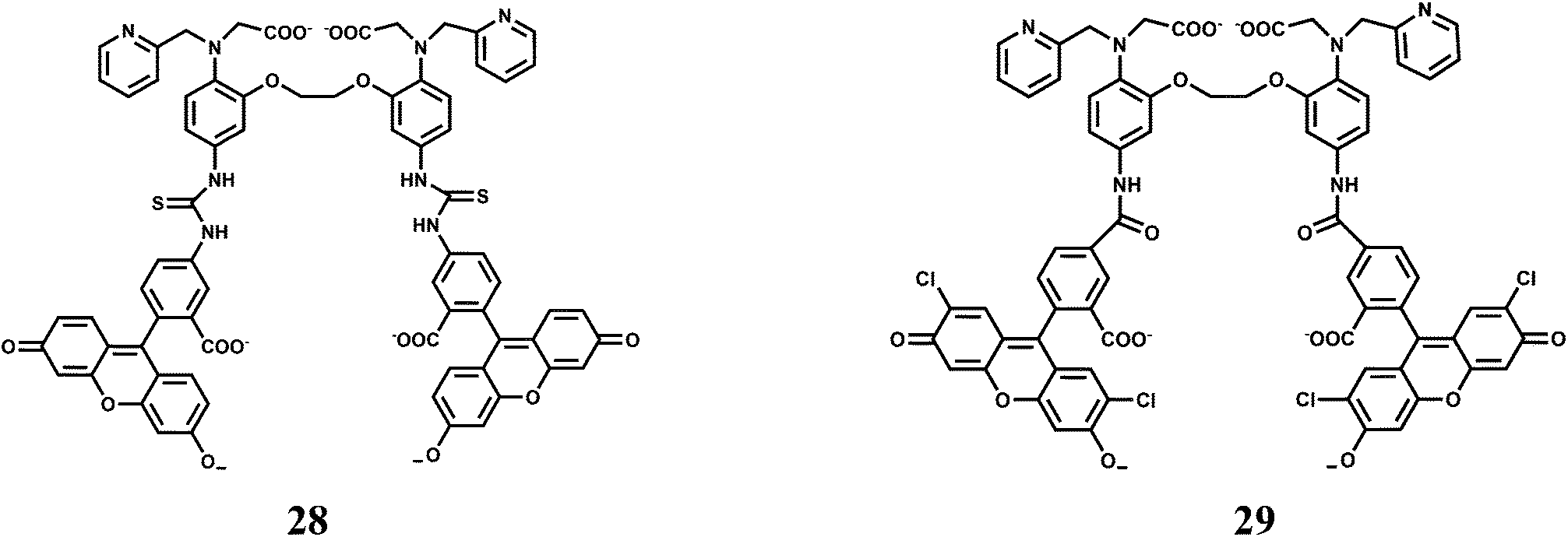
The association constant ratio (logKMn/logKCa) is found to be 2.368 for 28 while it is 2.402 for 29. When excited at 493 nm, metal-free 28 shows a low intensity emission centering at 519 nm in aqueous medium. Upon addition of Mn2+, a 4-fold enhancement is observed until saturation after 1 equivalent. On the other hand, metal-free 29 can be excited at 505 nm to obtain an emission maximum at 530 nm. It also affords almost a 4-fold enhancement in presence of 1 equivalent of Mn2+ ion. Both the probes have been used for intracellular Mn2+ with HeLa cell lines. However, it is to be noted that biological concentration of Ca2+ is about thousand times that of Mn2+ and hence cell imaging studies for Mn2+ will not be suitable especially when it is present in trace quantities.
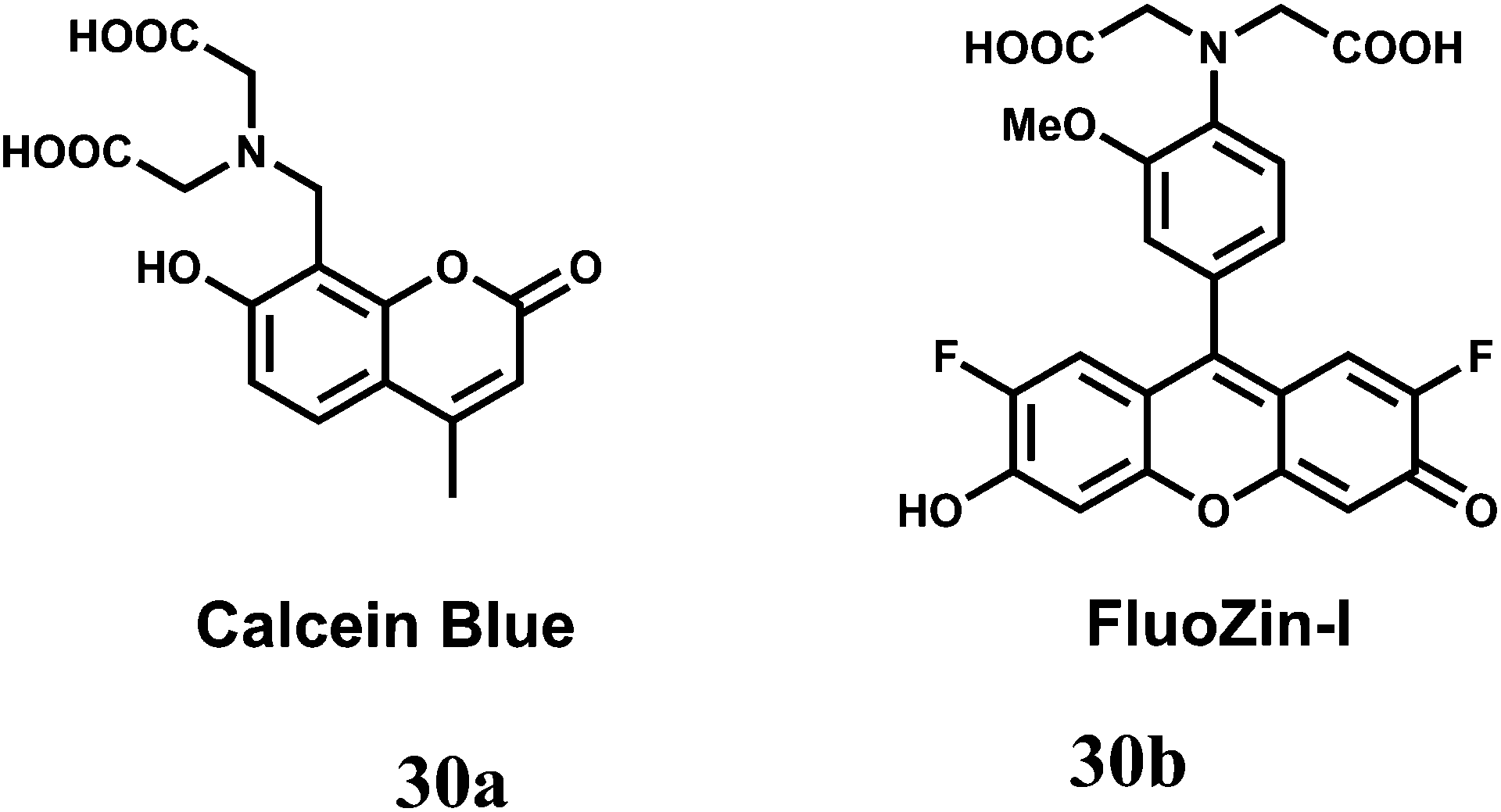
The two-metal two-dye displacement assay has been adopted for detection of Mn2+ ion in 30(a,b) where two commercially available dyes, viz., calcein blue (CB) and fluozin-1 (Fz1) have been used.55 It works due to differential binding abilities of the two dyes. Initially, Cd2+ is chelated by the stronger ligand CB and the resulting complex is highly fluorescent. The other ligand, Fz1 remain uncomplexed and gives a weak fluorescence. Upon addition of Mn2+ to the mixture, the Cd2+ is displaced from CB by Mn2+ resulting in quenching of fluorescence (Scheme 18). However, Cd2+ presently available in the solution, forms a complex with Fz1 accompanied by a strong fluorescence. The Cd2+-complex of CB gives a green fluorescence with the emission band at 493 nm upon excitation at 350 nm. On the other hand, Cd2+-complex of Fz1 gives an emission with maximum at 518 nm. Thus, it allows indirect detection of Mn2+ and also cell imaging studies.
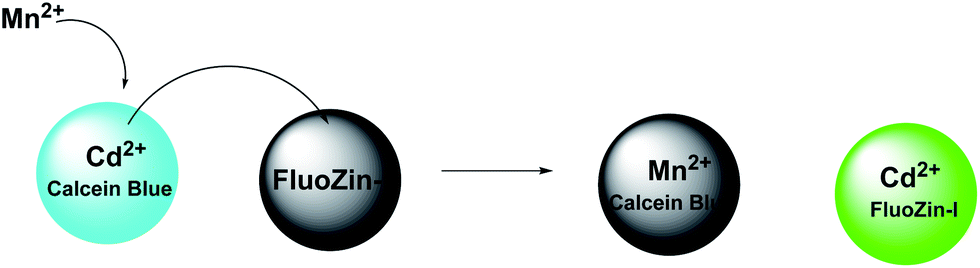 | ||
| Scheme 18 Possible metal ion binding mode of 30(a,b). | ||
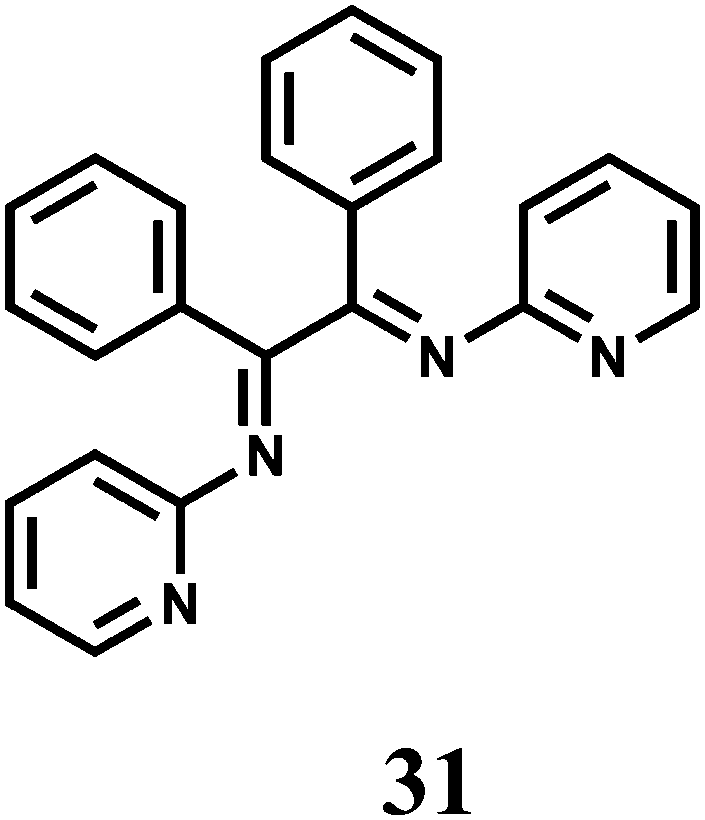
The PET chemosensor 31 forms a 1:1 complex with Mn2+ with logβ ≈ 3.0.56 On excitation at 310 nm in aqueous MeCN (1:1, v/v, pH 4.0) a 4-fold enhancement of fluorescence peaking at 360 nm is observed upon addition of Mn2+ ion due snapping of PET. However, alkaline earth metal ions also show enhancement albeit to lesser degrees.
2.5. Iron chemosensors
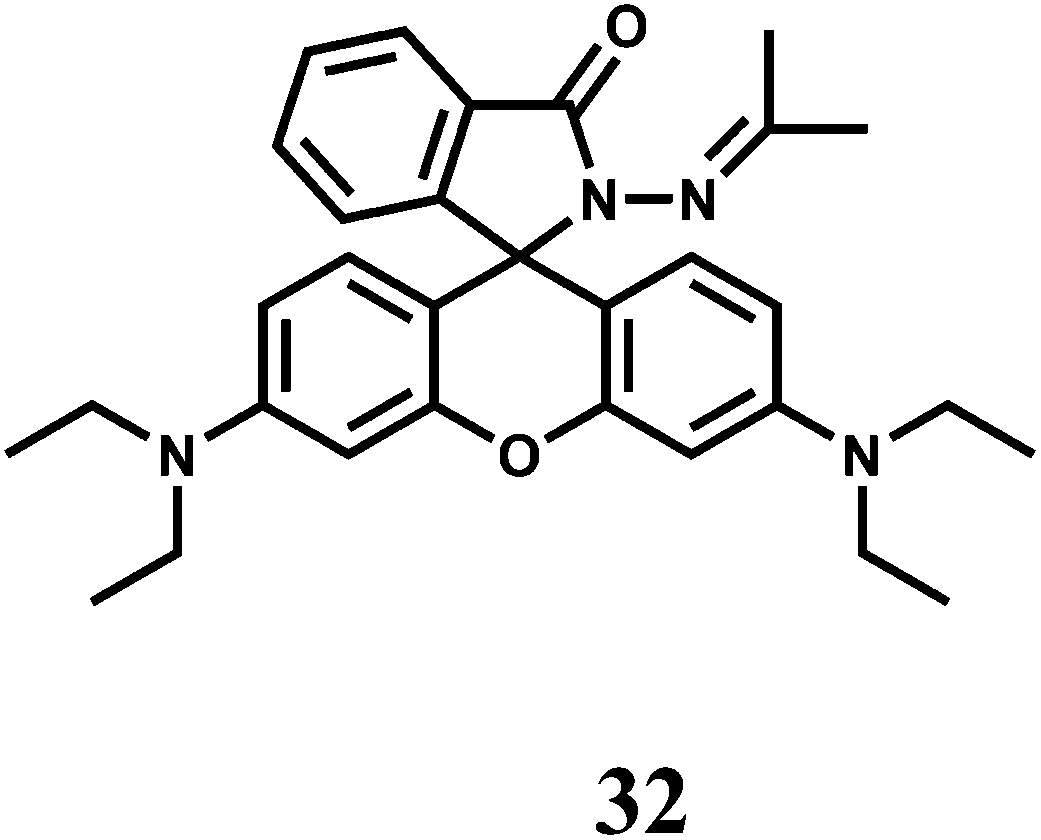
Iron is an essential element for many biological processes. But it is also detrimental to health as it causes the generation of highly destructive oxygen species. Accumulated data on iron metabolism suggest that alterations in the intracellular pool of ‘redox-active’ iron (also called ‘chelatable iron’) can contribute substantially to a variety of injurious processes such as ischemia-reperfusion injury, ethanol toxicity and oxidative injury after nutrient deprivation57 and so on. On the other hand, chelatable iron is increasingly found to be decisively involved in several intracellular signalling pathways.58 Most studies have been focused on detecting the cytosolic pool of chelatable, redox-active iron which is generally regarded as the cellular chelatable iron pool.59 However, a ‘transit’ pool of chelatable iron must also be assumed to exist within the mitochondria during haem synthesis. Iron is incorporated into protoporphyrin IX within the mitochondrial matrix. In addition, there is now increasing list of evidences that suggest a definite role of mitochondrial chelatable iron in the pathogenesis of various human diseases.60 It follows, therefore, that quantitative knowledge of spatial distribution of different forms of iron is of extreme importance.Like many first-row transition metal ions, fabrication of iron sensors has been dependent heavily on the spirolactam ring opening in presence of the metal ion. One or two rhodamine derivatives have been used in sensing the metal ion in 2+ and 3+ oxidation states. Thus, chemosensor 32 gives a spirolactam ring-opening fluorescence enhancement at 583 nm by a factor of 112 in aqueous MeCN in presence of Fe3+ while an enhancement of 18-fold is observed with Cu2+.61 Other biologically relevant alkali, alkaline earth and transition and heavy metal ions show very weak response. With Fe3+, 32 forms a 1:1 complex with the stability constant value of 2.3 × 104 M−1. This fluorescent probe has been used for live cell imaging studies although authors are silent about the solvent system used in imaging studies and possible interference from Cu2+ ion. It is important that cell imaging as well as cell viability studies are made in pure aqueous or aqueous ethanolic medium.
The rhodamine B hydroxamate fluorescent probe 33 having a biomimetic hydroxamate binding unit is prepared in two steps from rhodamine B.62 It binds Fe3+ in 1:1 stoichiometry in MeOH–MeCN (1:1, v/v) mixed solvent with the association constant of 2.3 × 104 M−1. Upon addition of Fe3+ to the probe solution, a 112-fold enhancement is observed due to spirolactam ring opening (Scheme 19). Other biologically relevant metal ions do not show any significant response or fluorescence response of Fe3+ is not affected by the presence of any other metal ion. The reversible binding of Fe3+ with 33 is demonstrated by observing immediate disappearance of fluorescence (and colour) upon the addition of excess of EDTA into the solution of 33 and Fe3+.
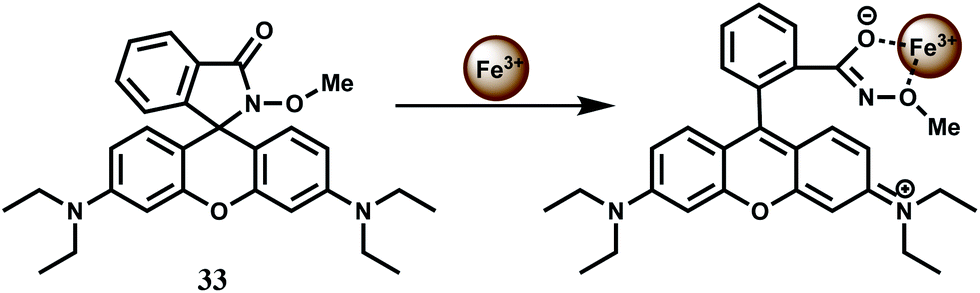 | ||
| Scheme 19 Possible binding mode of chemosensor 33. | ||
The imine linkage in the rhodamine 6G based chemosensor 34 is broken in presence of Fe3+ ion in aqueous medium and in the process, the spirolactam ring is also broken (Scheme 20).63 As a result, a strong emission band at 551 nm is observed. This Fe3+-catalyzed hydrolysis reaction of 34 is not influenced by excess amounts of other biologically active metal ions such as K+, Na+, Fe2+, Ca2+, Mg2+ or Mn2+ ion. Besides, heavy and transition-metals that are potential pollutants do not interfere in the binding of Fe3+ and subsequent emission enhancement. The high selectivity of this chemosensor for Fe3+ has been used to determine excess, free Fe3+ ions deposited into live cells using a ferric citrate-overloaded cell line. The greater concentration of fluorescent cells and the stronger intensities are found to be much more evident for the iron-overloaded cells compared to the normal untreated cells. Thus, this chemosensor can be used for determination of free Fe3+ in live liver cells without any detrimental effect on the Fe3+-based enzymes.
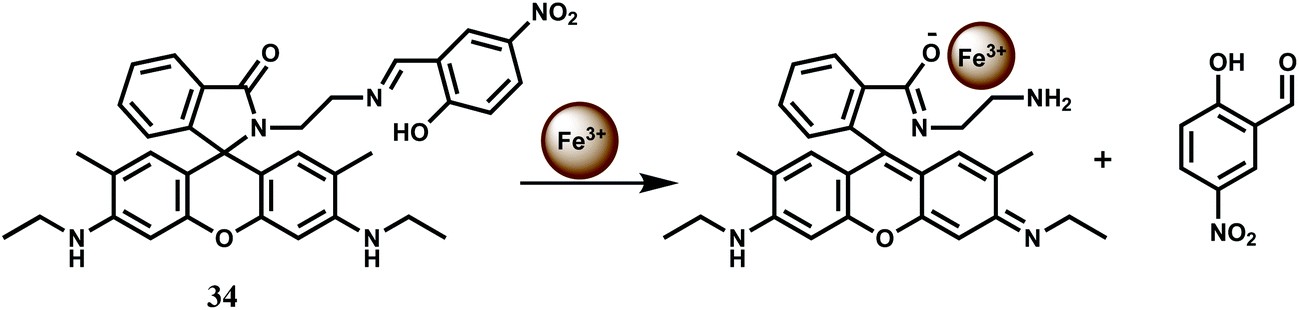 | ||
| Scheme 20 Possible mechanism of Fe3+-induced Schiff base hydrolysis and the spirolactam ring opening of 34. | ||
The rhodamine 6G–phenylurea conjugate 35 gives a strong fluorescence in the presence of Fe3+ in H2O–CH3CN (1:1, v/v) due to spirolactam ring opening.64 In presence of acetate ion, the metal ion is extracted with the formation of spirolactam bond once again thus offering a reversible fluorescence (Scheme 21). In response to acetate ions, the system not only provides remarkable fluorescence quenching but also exhibits a clear colour change from pink to colourless for naked eye observation. The background anions show small or no interference with the detection of acetate ions. This is the first chemosensor based on metal ion complexes that can selectively detect acetate ions over fluoride and dihydrophosphate ions in an aqueous environment. Such systems are expected to find a lot of practical applications.
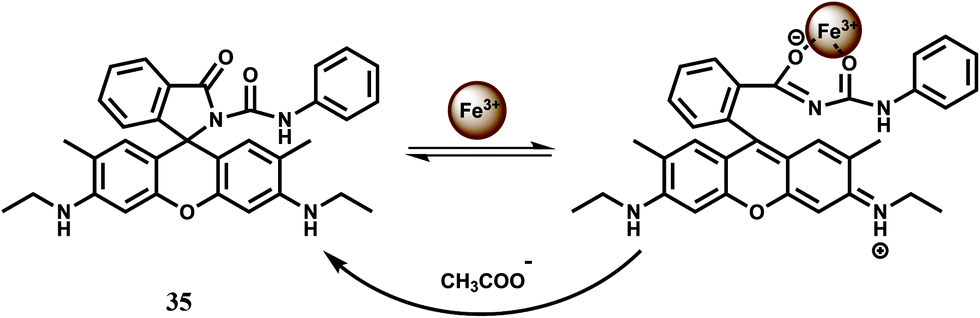 | ||
| Scheme 21 Possible binding mode of chemosensor 35. | ||
The chemosensor 36 containing quinoline moiety as the binding site for Fe3+ ion has been synthesized.65 Upon addition of Fe3+ in aqueous ethanol medium, the spirolactam bond is broken (Scheme 22) affording an approximately 190-fold enhancement of fluorescence with a peak at 559 nm following excitation at 505 nm. Fe3+ forms a 1:1 complex with 36 as shown in Scheme 22 with a high association constant of 1.1 × 106 M−1. While other transition, alkali and alkaline earth or heavy metal ions do not interfere, Cr3+ and Al3+ give mild enhancement of fluorescence. However, none of these metal ions are essential and hence the probe can be used in imaging studies. Structural studies of 36 indicate that the phthalimide side chain plays an important role in the ion selectivity due to its steric bulk. Upon addition of excess EDTA, Fe3+ is removed changing the solution to colourless with 98% quenching of fluorescence. Thus 36 can be classified as a reversible chemosensor for Fe3+ ion. Bioimaging studies of EJ cells with 36 shows that it can act as a Fe3+ sensor in living cells.
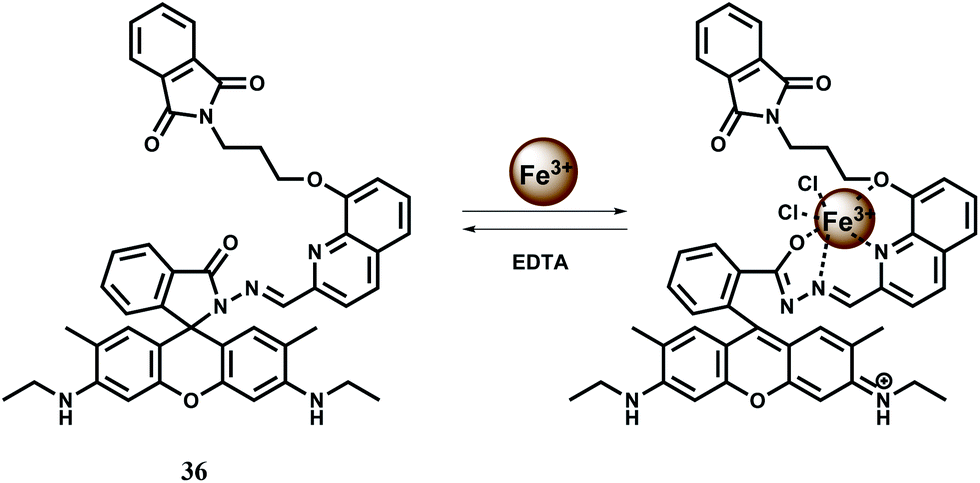 | ||
| Scheme 22 Possible binding mode of chemosensor 36. | ||
The rhodamine 6G derived chemosensor 37 has been synthesized that provides a definite binding site for Fe3+ (Scheme 23).66 The chemosensor forms a 1:1 complex with Fe3+ and the association constant is moderately high at 1.04 × 104 M−1. The free 37 gives only a weak emission at 550 nm when excited at 505 nm. A 23-fold enhanced green-yellow fluorescence is observed with the peak at 550 nm upon addition of Fe3+ in aqueous MeCN (1:1) at pH = 7.2. No interference from other competing ions is observed. The detection limit is found to be in the 10−8 M range. Gradual addition of Na2–EDTA to the Fe3+-complex causes decrease in fluorescence intensity and upon addition of 10 equivalents of EDTA2− ion, Fe3+ is totally removed with complete quenching of the fluorescence making it a reversible chemosensor for Fe3+ ion. However, its lack of solubility in aqueous ethanolic medium does not allow its use in live cell imaging studies.
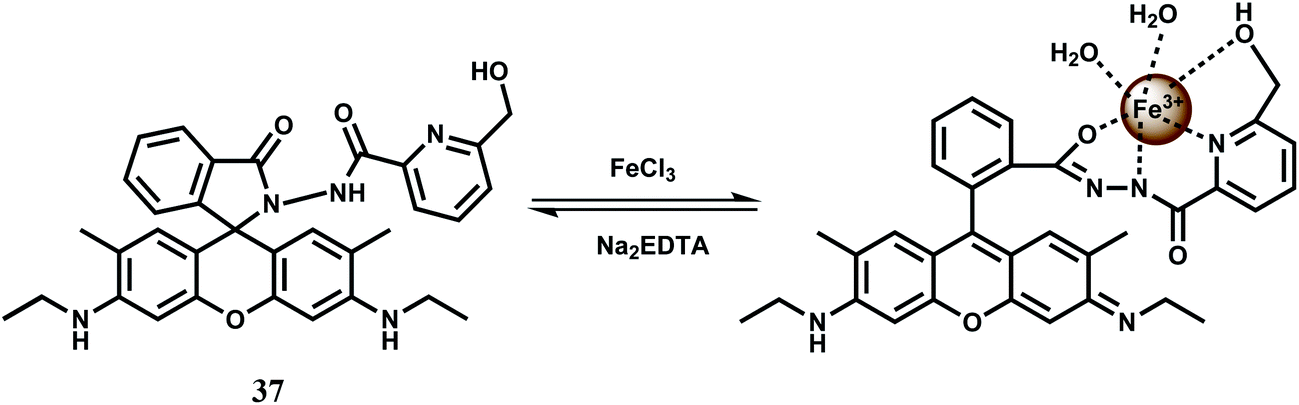 | ||
| Scheme 23 Possible binding mode of chemosensor 37. | ||
For increasing water solubility, rhodamine B hydrazide derived chemosensor 38 has been synthesized. This binds Fe3+ with 1:1 stoichiometry (Scheme 24) with the association constant Ka showing 7.52 × 104 M−1.67 The fluorescent probe gives spiro-bond opened 10-fold enhancement of fluorescence in presence of Fe3+ in water containing less than 1% organic co-solvent. Other biologically relevant metal ions exerted negligible effect even when present in excess. However, Cr3+ also gives enhancement to a lesser extent although that may not be a problem as compared to chromium, iron is present in large amount in biosystems. This probe is also found to be cell permeable and suitable for monitoring intracellular Fe3+ with low cytotoxicity in living cells by confocal microscopy.
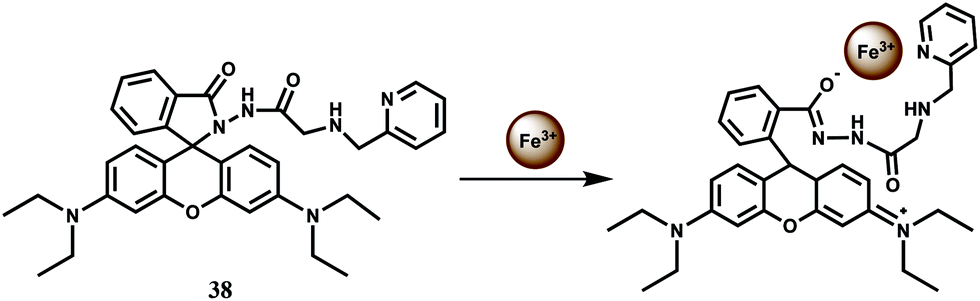 | ||
| Scheme 24 Possible sensing mechanism of 38. | ||
To improve selectivity towards Fe3+, a different coordination moiety has been added to rhodamine B to have 39.68 Indeed, this probe offers a spiro-ring opened (Scheme 25) 23-fold enhancement with Fe3+ at 586 nm when excited at 530 nm in aqueous buffer solution. No other competing metal ions show any enhancement. The dissociation constant (Kd) of Fe3+ complex is found to be 1.87 × 10−5 M. The fluorescent probe is found to be safe for long hours at low concentration; it is cell permeable and suitable for the real-time imaging of Fe3+ in living MGC803 cells.
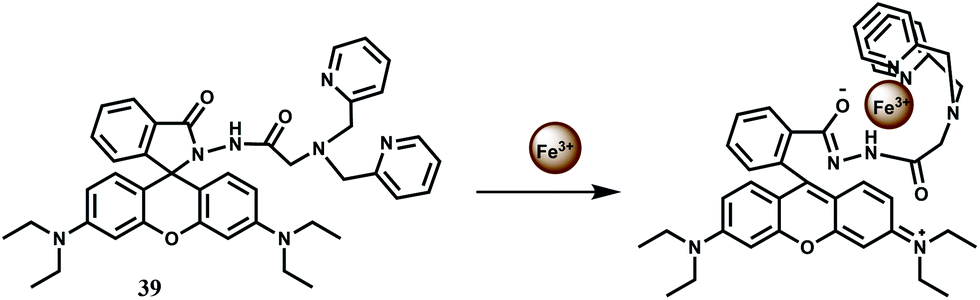 | ||
| Scheme 25 Possible metal ion binding mode of 39. | ||
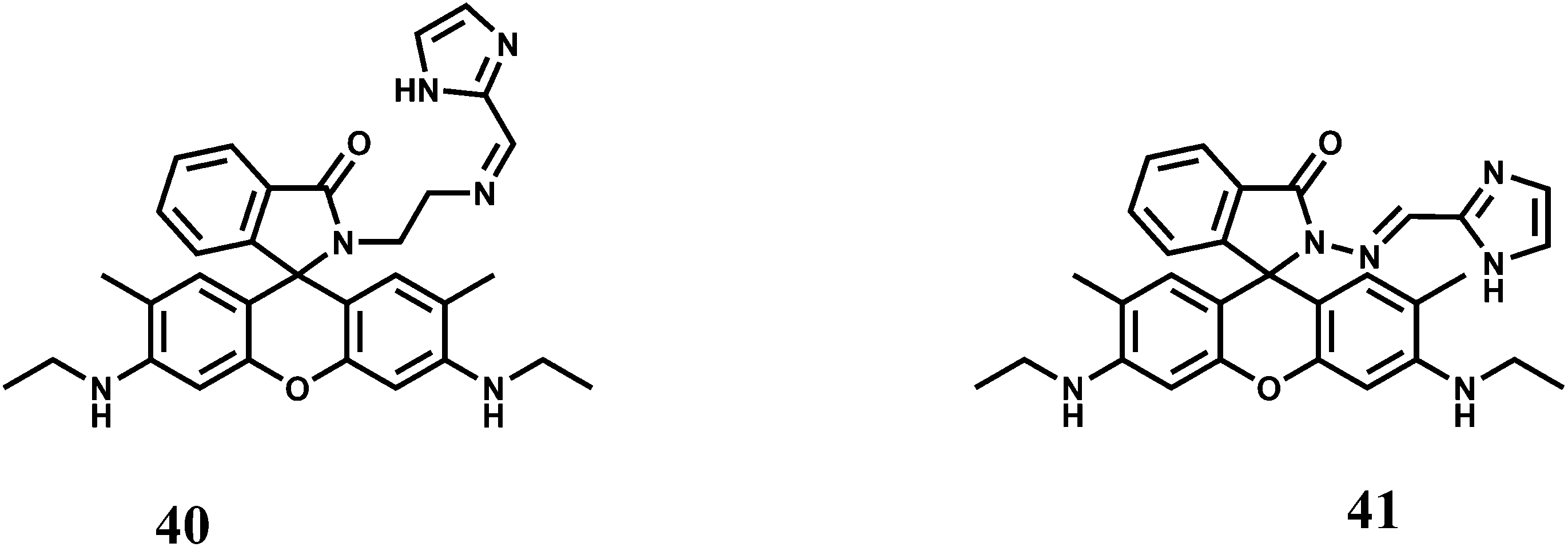
The rhodamine 6G derived chemosensors 40 and 41 are found to show excellent selectivity towards Fe3+ with the binding constant values of 6.74 × 104 M−1 and 3.946 × 104 M−1 respectively with 40 and 41 in aqueous DMSO at pH 7.4.69 Upon excitation at 480 nm both the probes show substantial spiro-ring opened enhancement at 553 nm in presence of Fe3+. None of the other coexisting metal or heavy metal ions shows any enhancement. Live cell imaging experiments with each probe demonstrated its value of practical applications in biological systems.
Although several rhodamine based chemosensors for Fe3+ are available as mentioned above, in most cases either a pure organic solvent or an organic co-solvent will be required to guarantee a high affinity of Fe3+ towards a chemosensor. However, in some cases, it is required that the probe be highly soluble in water for detecting Fe3+ ions in natural water samples. For increased solubility in water the N-stabilized rhodamine hydrazide fluorescent probe 42 has been synthesized. The N-stablization of rhodamine increases its rigidity resulting in larger Stokes shift and higher intensity of fluorescence.70 This chemosensor itself does not show any fluorescence but in presence of Fe3+ in aqueous medium (pH 5–8) gives about 164-fold enhancement in less than a minute (Scheme 26).71 Under similar conditions other biologically relevant metal ions do not show any fluorescence except Cr3+ ion which also gives substantial enhancement. This probe forms a 1:1 complex with Fe3+ and although no association constant data are available, this binding is found to be reversible. Upon addition of 20 equiv. of K3PO4 to the 42–Fe3+ complex in water, the fluorescence is completely quenched. Further addition of 20 equiv. of Fe3+ restores the fluorescence to its original value. This chemosensor is found to be useful in imaging MGC803 cells for monitoring Fe3+ ion.
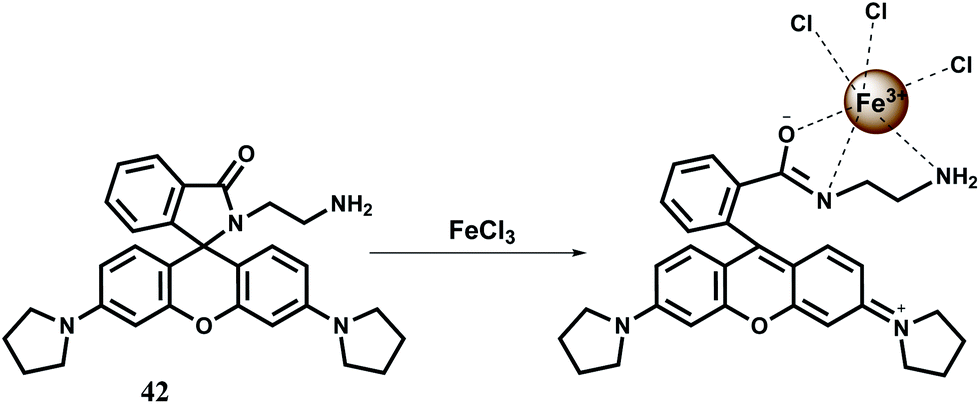 | ||
| Scheme 26 Possible sensing mechanism of 42. | ||
The fluorescent probe 43 has two fluorophores: a dansyl unit as a donor and a rhodamine 101 as an acceptor for FRET based signaling.72 In FRET based sensing, the pseudo-Stokes shifts are larger than the Stokes shift of either fluorophore. This avoids self-quenching as well as fluorescence detection errors due to backscattering effects from the excited source.73 The probe forms a complex of 1:1 stoichiometry with Fe3+ showing a moderately high association constant (Ka) of 1.74 × 104 M−1. The Fe3+ induced spirolactam ring-opening of the rhodamine moiety transforms it as a FRET acceptor (Scheme 27). When the dansyl group is excited at 380 nm, the FRET acceptor emits at 605 nm. The probe displayed a linear response to Fe3+ in the range of 5.5–25 μM with a detection limit of 0.64 μM. Other competing metal ions do not give any fluorescence and their presence do not change the fluorescence profile of the Fe3+ complex. Thus, this novel rhodamine–dansyl conjugate can selectively sense Fe3+ ion in presence of a host of other ions.
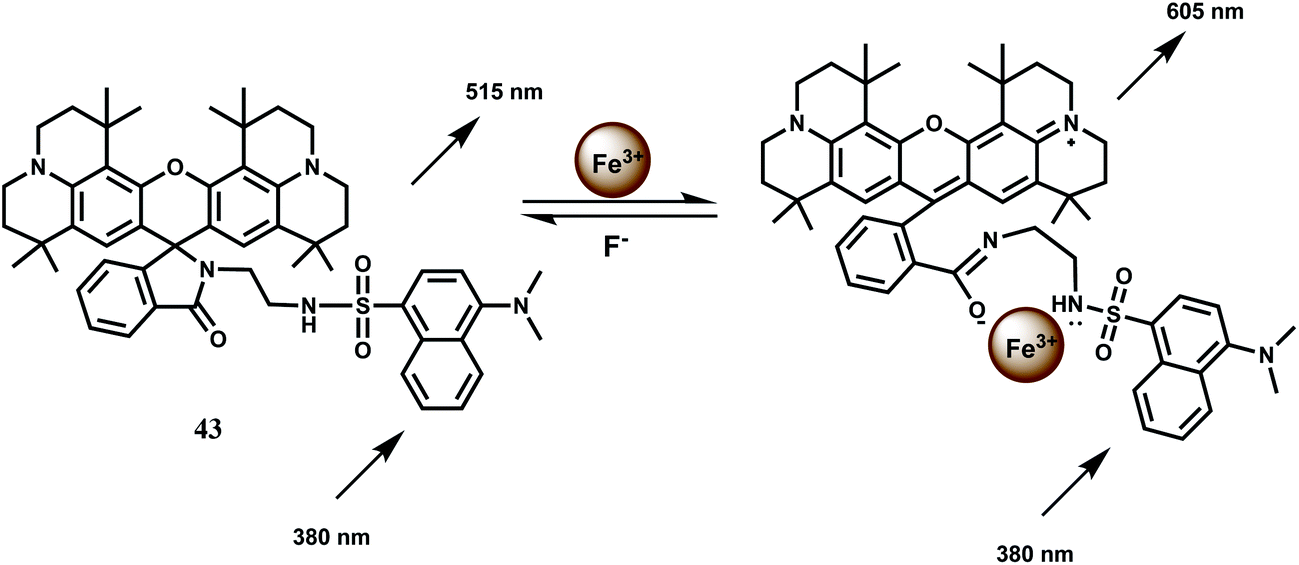 | ||
| Scheme 27 Possible sensing mechanism of 43. | ||
Another FRET based chemosensor on the rhodamine platform for Fe3+ is provided by the system 44 has been shown below. A quinoline fluorescent group is covalently linked74 to a rhodamine moiety. Metal free 44 exhibits a weak emission at 482 nm which changes upon addition of Fe3+ ion, due to an efficient FRET from the conjugated quinoline to rhodamine. Thus, the excitation interference of the new rhodamine based probe is eliminated as a result of efficient FRET (Scheme 28). Binding of the metal ion also causes opening of the spiro-bond of the rhodamine moiety resulting in an enhancement by a factor of 186 times indicating that the new sensor can be used as a highly sensitive Fe3+ probe especially when other competing metal ions taken in excess, do not interfere with the Fe3+ determination. Its aqueous solubility makes it an useful probe for live cell imaging studies.
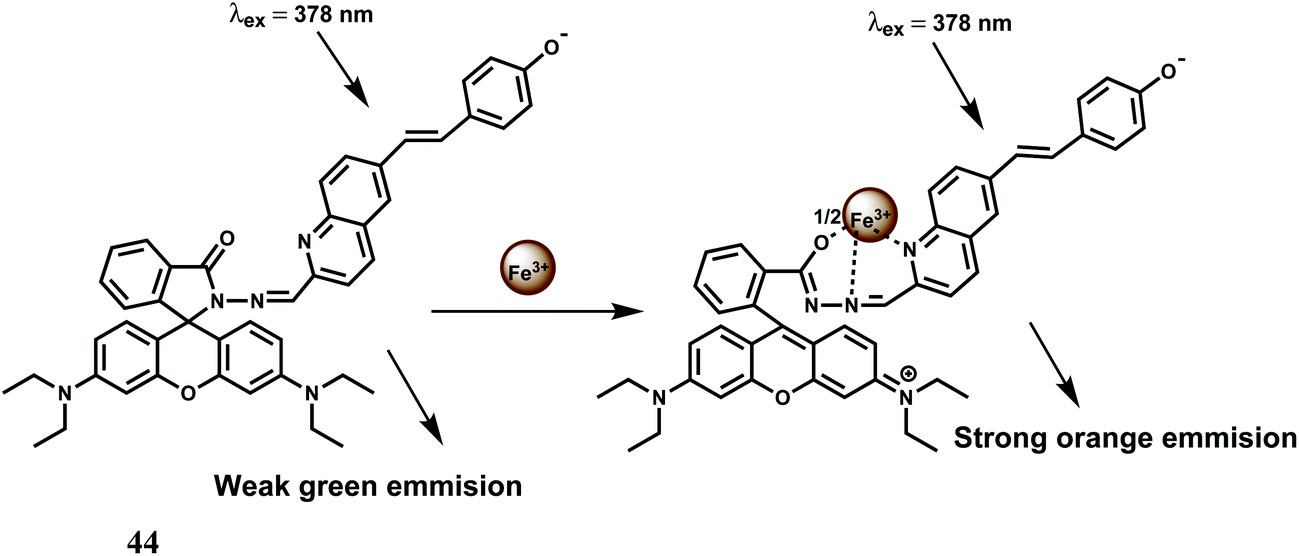 | ||
| Scheme 28 Possible mechanism of Fe3+ induced FRET. | ||
A rhodamine B and a coumarin are connected to have the ratiometric fluorescent probe 45 for Fe3+ with high sensitivity and selectivity.75 Upon addition of Fe3+ it binds to both the moiety in aqueous medium with the appearance of spiro-ring opened fluorescence peak at 580 nm and PET suppressed fluorescence from the coumarin moiety at 460 nm (Scheme 29). The association constant for Fe3+ (Ka) is found to be 1.172 × 104 M−1 which is quite significant. But a host of other competing metal ions also give enhancement at 460 nm belong to the coumarin moiety. Thus, the design principle is not at all clear in this case.
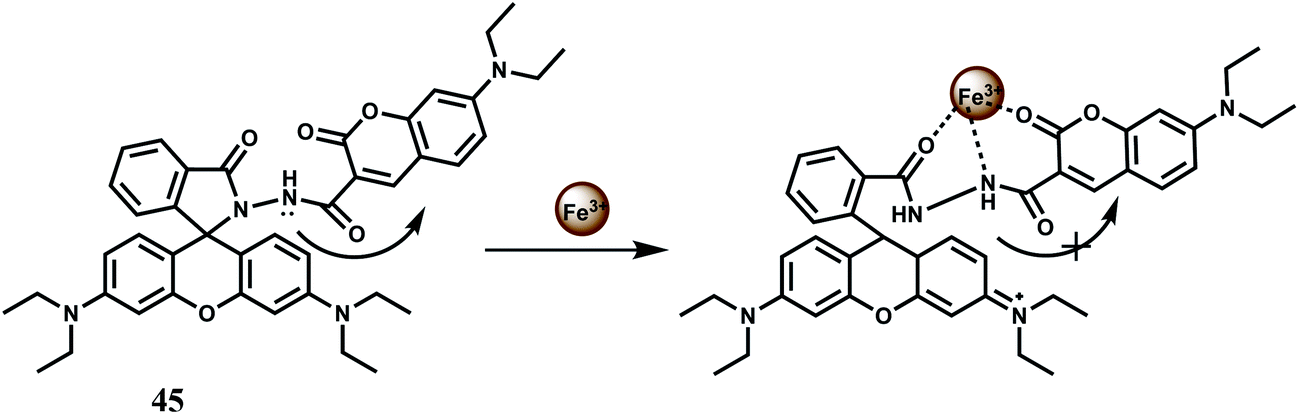 | ||
| Scheme 29 Possible sensing mechanism of 45. | ||
In a novel approach, NP conjugate of Fe3O4–Rh6G has been synthesized as the chemosensor 46 and found to be sensitive and selective in detecting Fe3+ ion.76 It forms a 1:1 complex with Fe3+ with high stability constant (5.0 × 106 M−1). A ∼60-fold enhancement due to spirolactam ring opening is observed for Fe3+ with the detection limit reaching as low as 2 ppb (Scheme 30). Amongst other metal ions, only Cr3+ shows some enhancement. Since the system can work in aqueous medium, confocal microscopic studies have been made on HeLa cells. The chemosensor 46 is more sensitive compared to the system where no NP is used due to its low solubility.
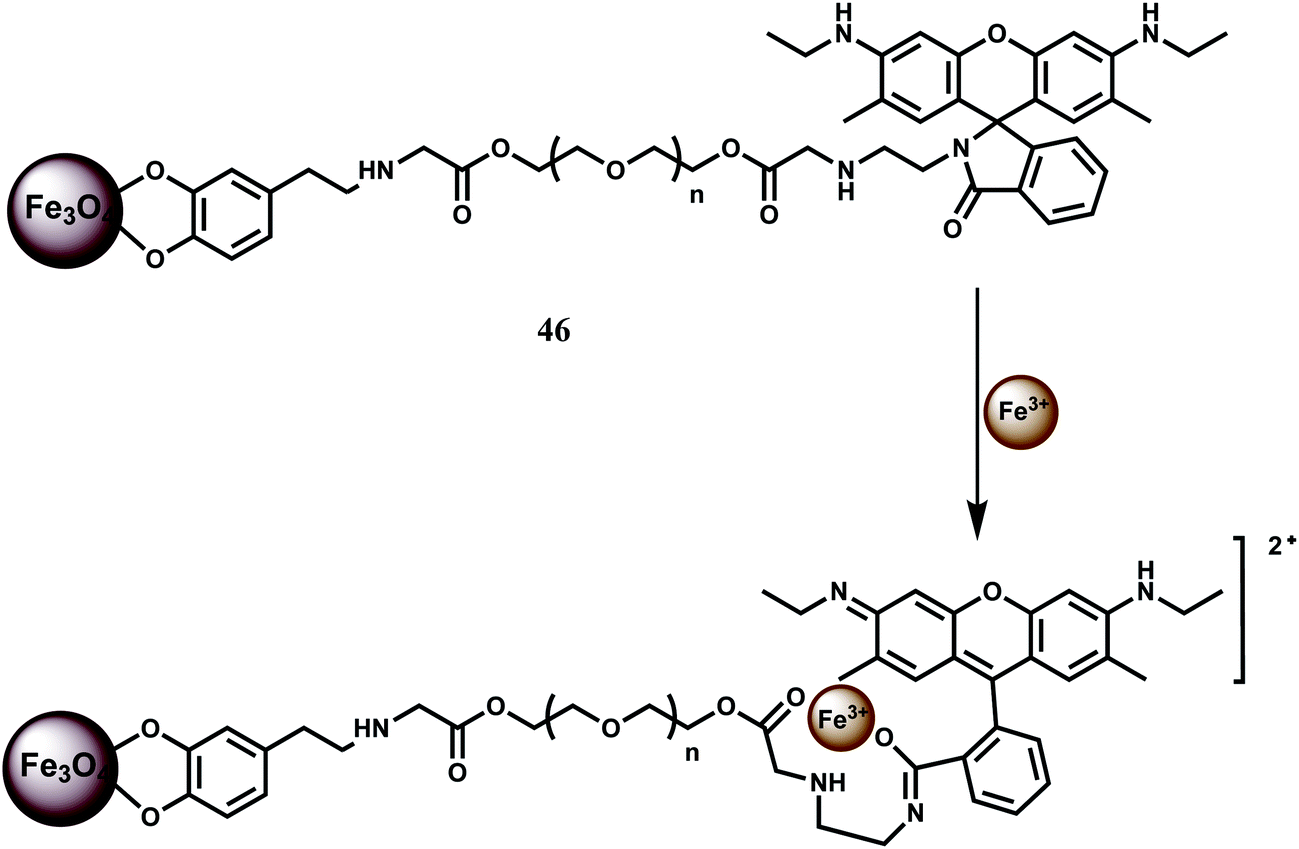 | ||
| Scheme 30 Binding of Fe3+ to the chemosensor 46. | ||
In addition to mono-rhodamine derived systems, a number of bis-rhodamine derivatives have been designed for detection of iron in different oxidation states. One such bis-rhodamine based chemosensor 47 is easily synthesized from rhodamine B and diethylenetriamine.77 The free sensor is colourless in absolute ethanol or in water in the pH range, 5.0–9.0 and does not show any significant emission suggesting that the spirolactam bonds are intact. Upon addition of Fe3+, colour of the solution changes immediately to purple both in ethanol and water giving an orange fluorescence while other competing metal ions do not show any change. However, in ethanol medium, Cr3+, Fe2+ and Cu2+ show emission to a small extent. Addition of Fe3+ causes both the spirocyclic rings to the open form with a ∼110 and ∼40-fold enhancement at 580 nm in ethanol and water respectively when excited at 510 nm (Scheme 31).
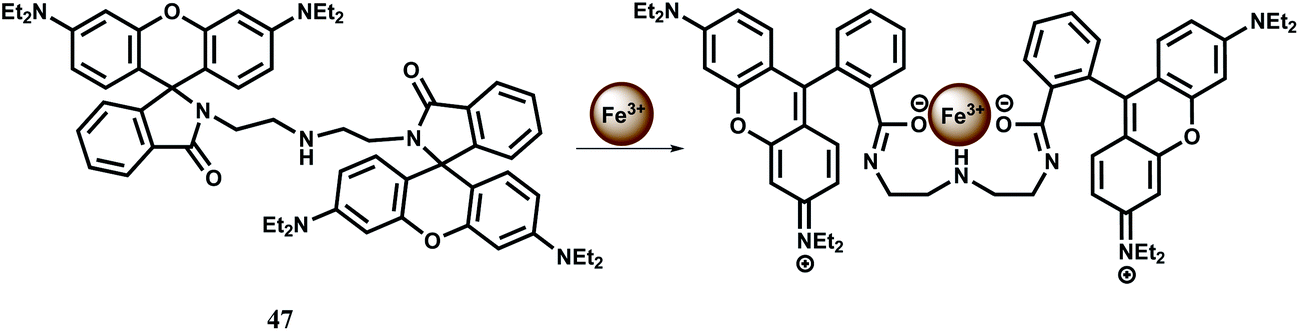 | ||
| Scheme 31 Binding of Fe3+ to the chemosensor 47. | ||
The bis-rhodamine-based dual sensor 48 upon complexation with Fe3+ triggers the formation of a highly fluorescent ring-opened form.78 This sensing mechanism is reversible, as indicated by disappearance of the emission with the addition of excess EDTA. However, Cu2+ interferes and Fe3+ cannot be detected in presence of Cu2+ ion (Scheme 32).
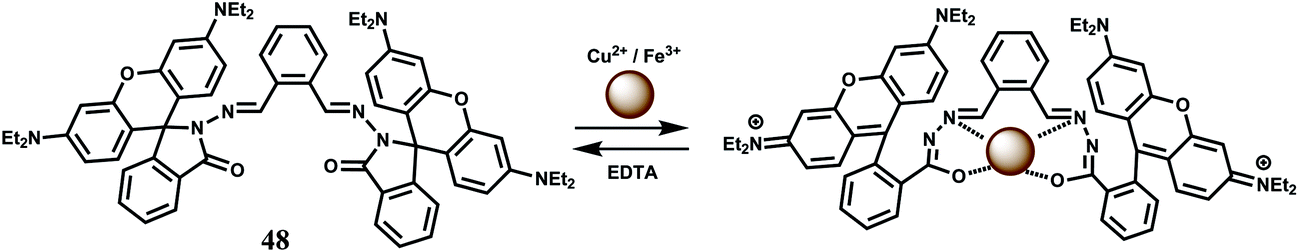 | ||
| Scheme 32 Reversible binding of Fe3+/Cu2+ to the chemosensor 48. | ||
Another bis(rhodamine)-based chemosensor 49 exhibits high selectivity for Fe3+ over other transition metal ions in 50% aqueous ethanol.79 The spirocyclic rings are opened and a 115-fold enhancement of fluorescence at 575 nm is observed. However, Cr3+ also gives enhancement of emission albeit to a lesser degree.
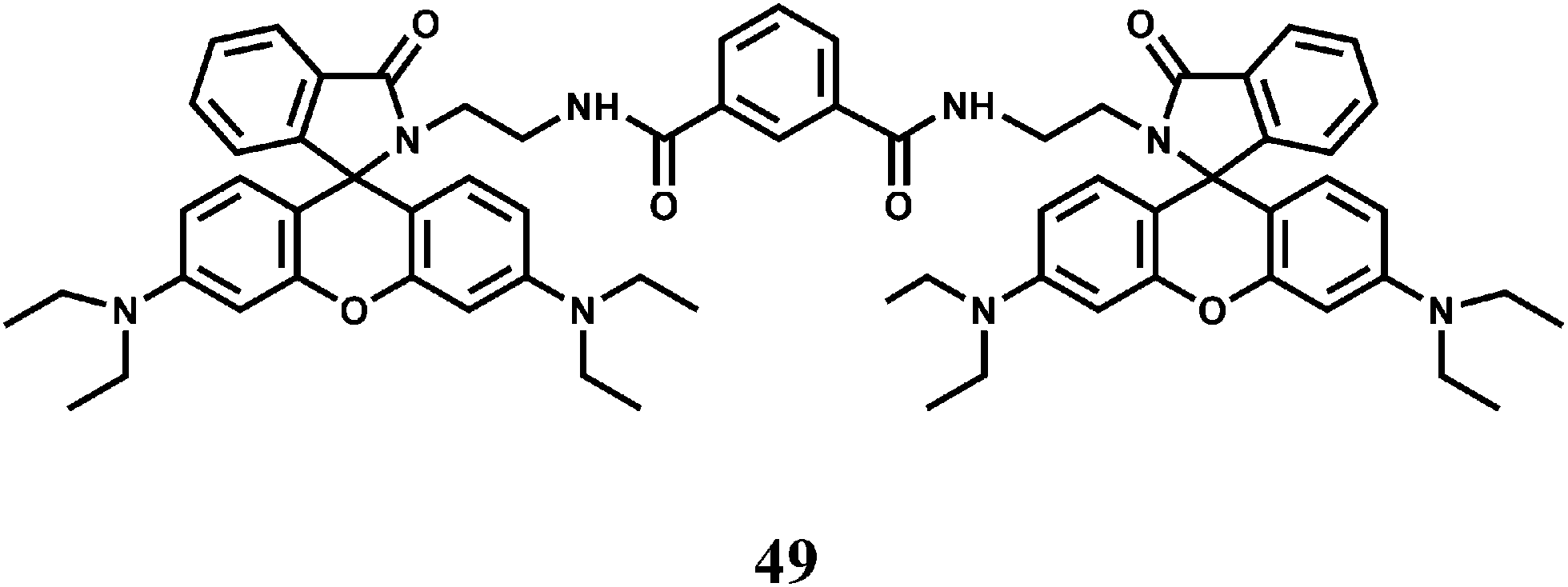
When two rhodamine moieties are connected through a disulfide linkage to have chemosensor 50, it gives about 60-fold enhancement in presence of Fe3+ in aqueous ethanol within a wide pH range.80 However, as is found for most of the rhodamine derivatives, Cr3+ ion also affords enhancement to a less amount. The association constant (Ka) with Fe3+ is found to be 7.4 × 104 M−1. Since cytotoxic measurements are found to be negative, the probe has been used for live cell imaging studies.
In an elegant approach, a 12-membered dithia-aza-oxa macrocycle is attached to two different fluorophores through para-substituted benzene to have the chemosensors 51 and 52.81 In this design, the fluorophores are well-separated from the receptor to minimize metal-fluorophore interaction and possible quenching. The response of Fe3+ in presence of potentially interfering metal ions are studied in solvents of different polarity. In organic solvents, fluorescence of metal-free sensors are quenched due to ICT/PET when excited. Addition of Fe3+ leads to recovery of the fluorescence without pronounced spectral shifts. Also, metal-free 52 shows dual emissions in water and can be employed for the selective ratiometric signaling of Fe3+ in buffered aqueous solution.
The fluorophore 53 has two di-(2-picolyl)-amine (DPA) groups as the metal binding sites attached to a perylene tetracarboxylic diimide fluorophore via benzene as the spacer.82 Perylene tetracarboxylic diimides have excellent thermal and photo-stability, high luminescence efficiency and novel optoelectronic properties.83 The receptor and the fluorophore moieties being separated through the rigid spacer, gives low M–F interactions when a metal ion is bound at the receptor. Upon addition of Fe3+, PET from the dipicolyl moiety to the fluorophore is blocked leading to recovery of fluorescence. An enhancement factor of 138 in presence of Fe3+ is observed although metal ions such as Co2+, Zn2+ and Cu2+ also give emission to a small extent. The Zn2+ ion although diamagnetic shows low enhancement and hence it appears that binding strength of the metal ion at the receptor decides the extent of enhancement.
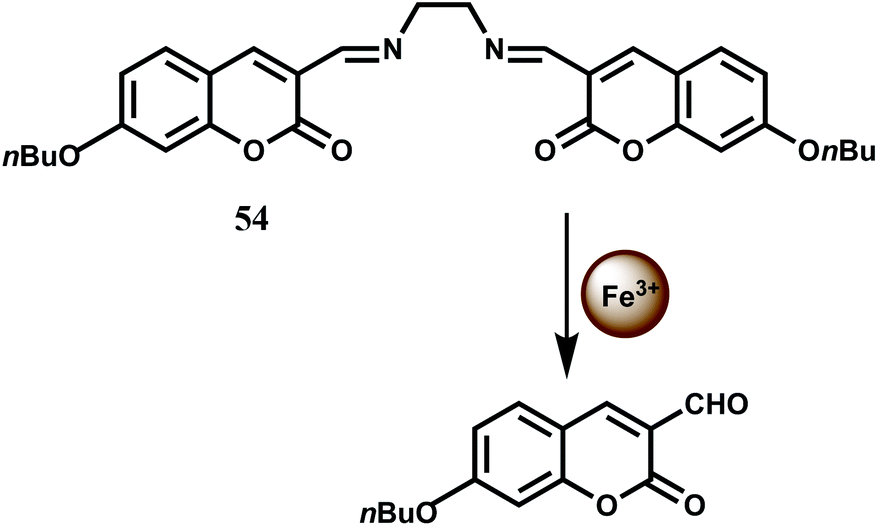
The bis(coumarinyl) Schiff base 54 does not show any significant emission in MeOH–H2O (98:2, v/v) medium.84 This low-fluorescence character of free 54 is attributed to a photoinduced charge transfer (PCT) process from the electron donating C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N moiety to the coumarin ring leading to quenching of the excited state of coumarin. Upon addition of Fe3+ to 54 which catalyzes the hydrolysis of the imine linkage that releases strongly fluorescent coumarin derivative offering about a 140-fold enhancement in fluorescence (Scheme 33). Other first-row transition metal ions do not catalyze the hydrolysis and hence does not interfere.
N moiety to the coumarin ring leading to quenching of the excited state of coumarin. Upon addition of Fe3+ to 54 which catalyzes the hydrolysis of the imine linkage that releases strongly fluorescent coumarin derivative offering about a 140-fold enhancement in fluorescence (Scheme 33). Other first-row transition metal ions do not catalyze the hydrolysis and hence does not interfere.
 | ||
| Scheme 33 Probable Fe3+ induced sensing mechanism of 54. | ||
It is known that Fe3+ has a strong binding affinity with imidazole N atom.85 The association constant Ka of 55 for Fe3+ ion is found to be (2.9 ± 0.2) × 103 M−1 with the metal ion engaging four N donors equatorially while the axial sites are occupied by counter anions as proven by different spectroscopic techniques. When excited at 288 nm in MeCN–H2O (95:5, v/v) the metal-free 55 gives an emission at 412 nm.86 Addition of Fe3+ to the solution causes the emission at 412 nm to decrease in intensity with the appearance of a new band at 475 nm. Thus, this chemosensor can be used as a ratiometric probe for Fe3+. Other competing metal ions do not afford any ratiometric response neither they interfere with the response obtained with Fe3+ ion.
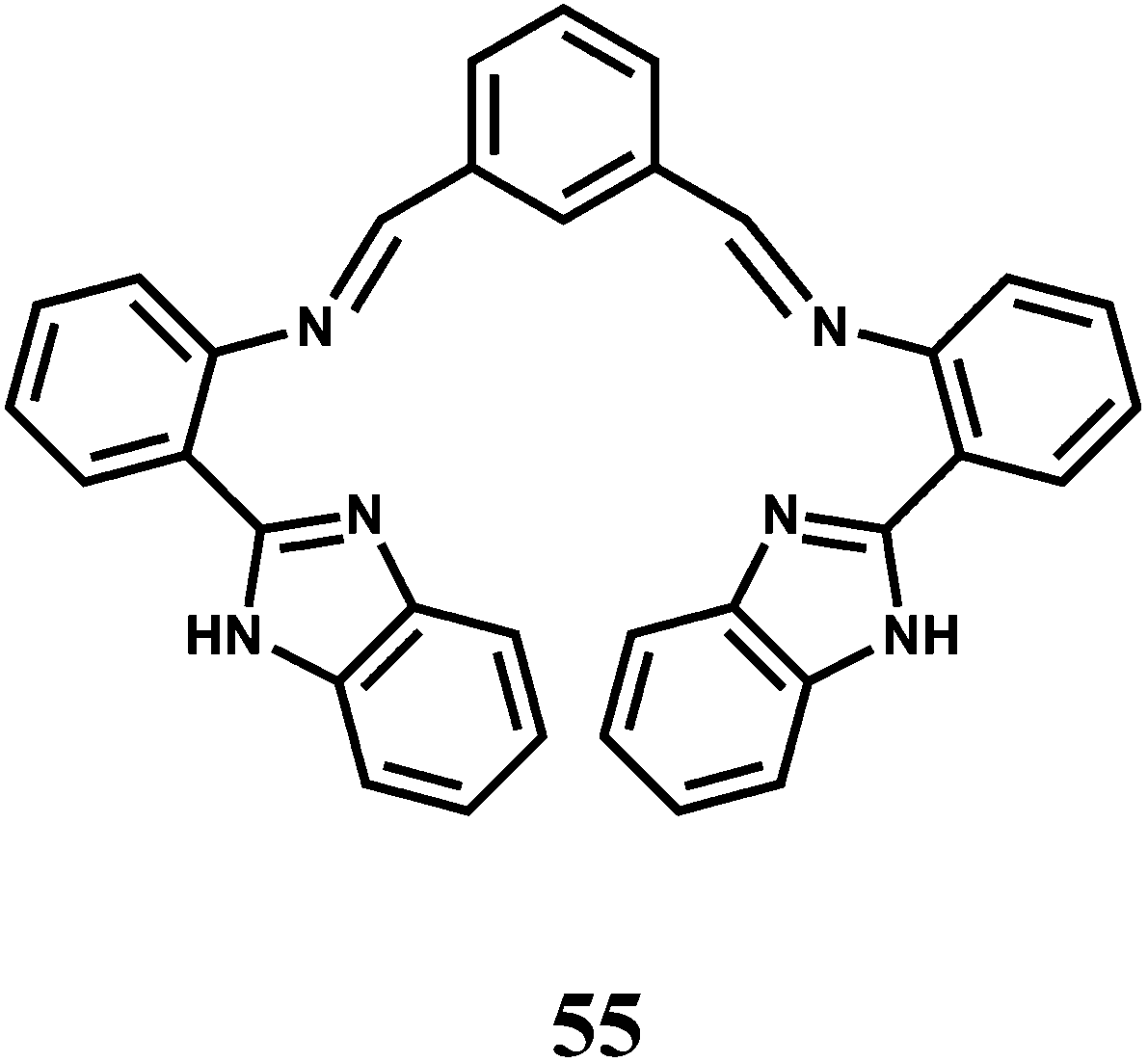
The PET chemosensor 56 has a squarate hydroxamate and a coumarin signaling units linked through a spacer. The squarate dianion itself does not bind first- and second-row transition-metal ions in a chelating fashion because the bite angle imposed by the cyclobutene framework is too large to sustain such coordination. That is why, the squarate hydroxamate has been used which is known to bind Fe3+ effectively.87 The amine linking the squaramide moiety and the fluorophore is likely to cause a PET-type quenching of the chromophore. The linker moiety is designed to be rigid to prevent a bound metal to have any significant communication with the fluorophore and to lessen the effects of the paramagnetic quenching.88
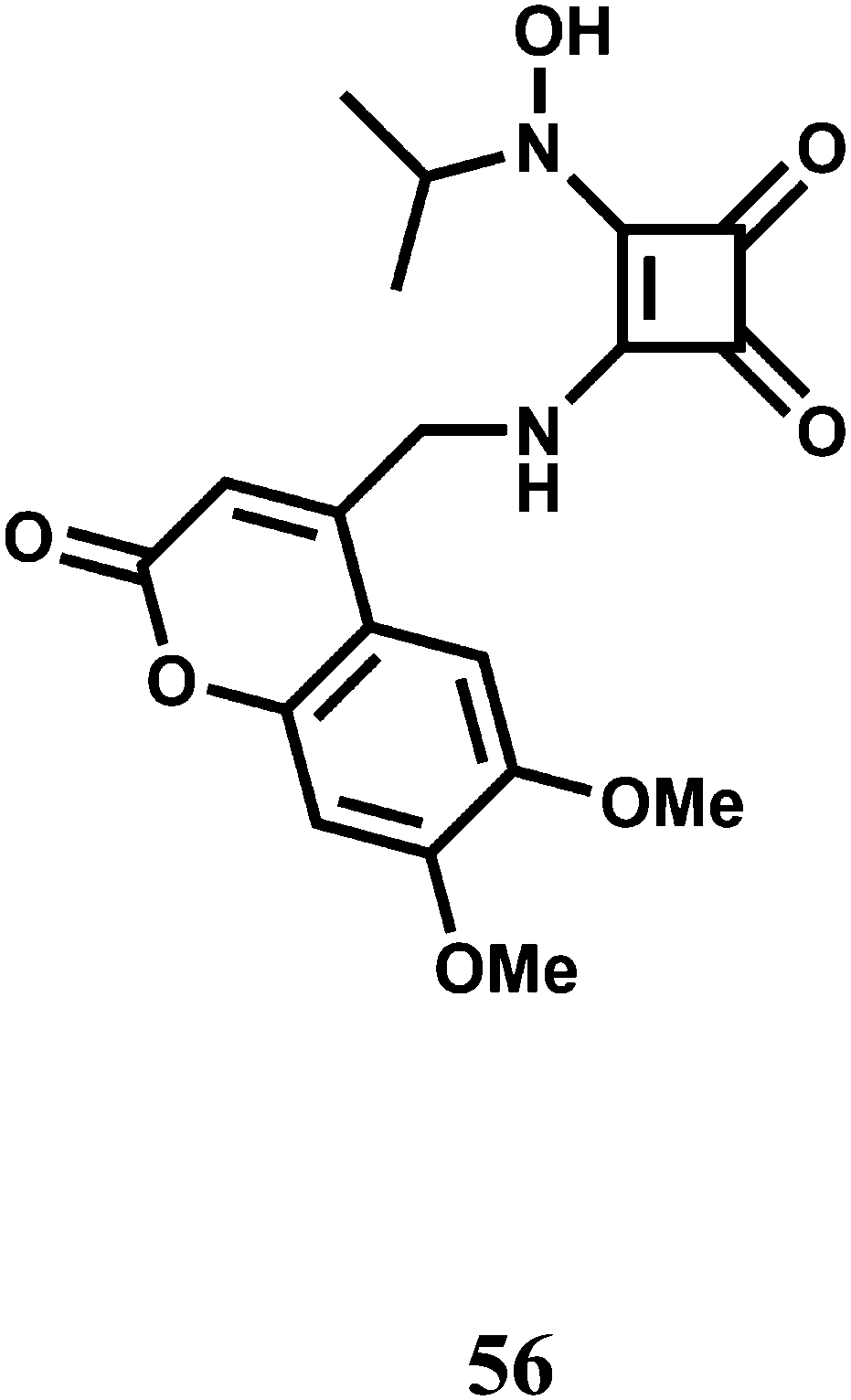
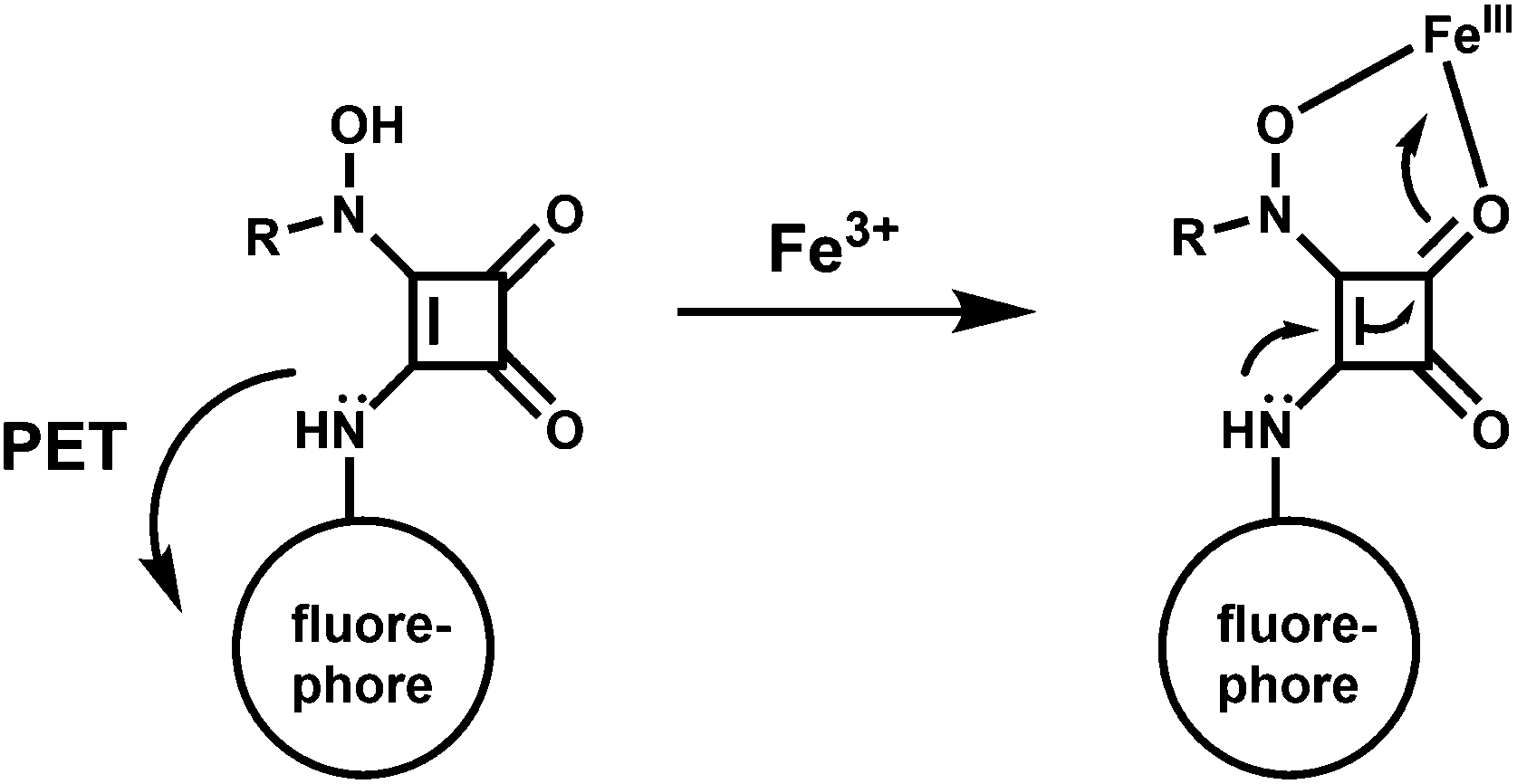
The metal-free sensor shows low emission suggestive of an efficient PET quenching of the coumarin chromophore. Upon addition of Fe3+ to an aqueous buffered (pH, 4.4) solution of 56 a 9-fold enhancement of fluorescence is observed due to blocking of the PET (Scheme 34). At a 150 μM concentration of 56 in the presence of 1 equiv. of Fe3+ at ambient temperature, equilibration of the emission intensity is reached only after 15 min suggesting its slow response. However, the signal does not degrade over the course of 48 h. Although no stability constant value is given, the fluorescence signal does not diminish even in presence of 1000-fold excess of EDTA.
 | ||
| Scheme 34 Possible binding mode of chemosensor 56. | ||
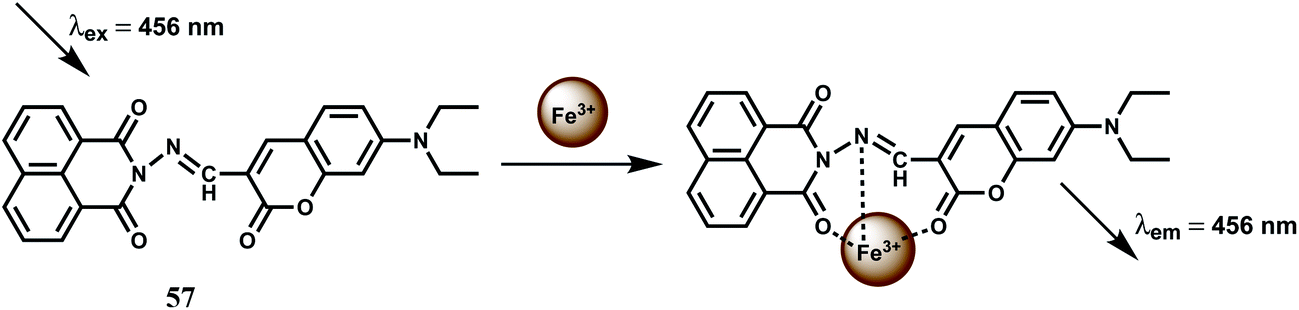
The chemosensor 57 has been built with two fluorophores viz., a coumarin and a naphthalimide for sensing Fe3+ (Scheme 35).89 The metal free sensor shows very weak emission when excited at 456 nm. In presence of Fe3+ ion, however, it gives an enhancement at 504 nm with high selectivity over other biologically relevant metal ions in aqueous THF solution. The association constant Ka value is, however, found to be moderate at 2.589 ± 0.206 × 103 M−1. Utility of using two fluorophores is not clear from the studies and also possible interference from Cr3+ ion has not been probed for this chemosensor.
 | ||
| Scheme 35 Possible sensing mechanism of 57. | ||
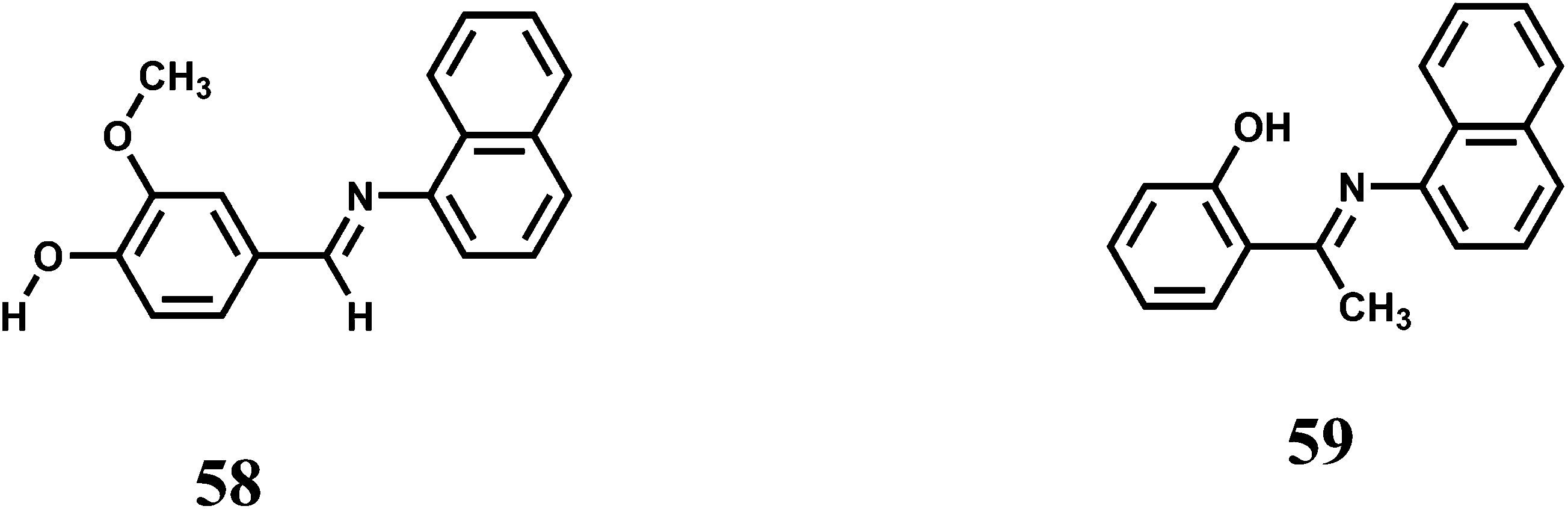
Two 1-naphthylamine derived ICT probes 58 and 59 are found to act as fluorescence turn-on sensors selectively for Fe3+ ion in methanol.90 Other competing metal ions show quenching of fluorescence like the metal-free probes. However, it is necessary to study the efficiency of enhancement when other metal ions are added in presence of Fe3+ ion for its effective use.
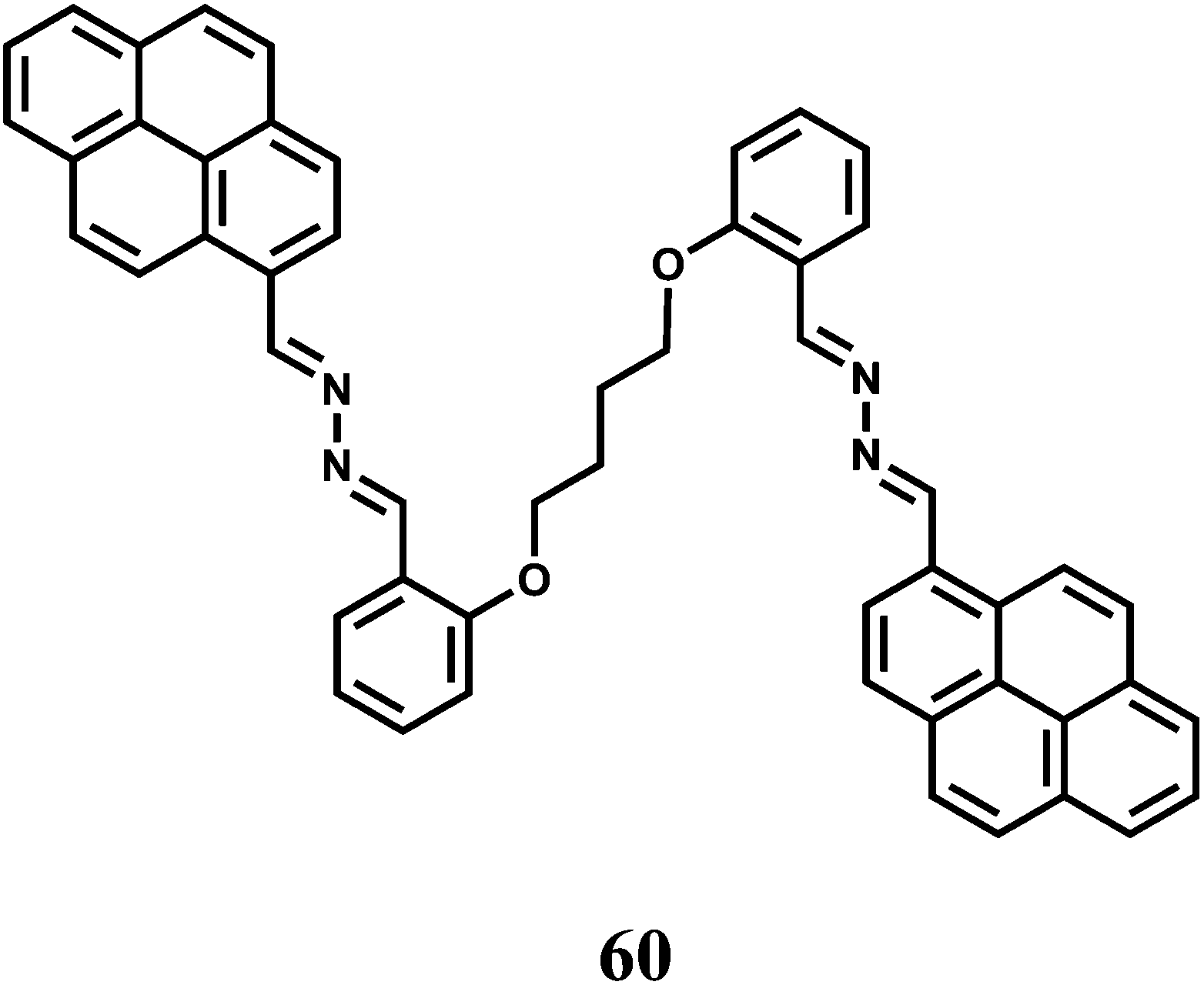
Two pyrene moieties are present at the two ends of a linear chain incorporating O and N donors to have the fluorescent probe 60.91 In the metal-free state, the two pyrene groups are far apart for any possible excimer emission. Besides, no monomer emission is observed due to an efficient PET from the imine N to the excited pyrene. Selectively in presence of Fe3+ ion, the probe folds around the metal ion that brings the two pyrene moieties close to one another to give an excimer band at 507 nm when excited at 330 nm. However, no monomer emission is mentioned arising out of the blocked of PET upon Fe3+ binding. The association constant of the Fe3+ complex is found to be moderately high at 1.27 × 104 M−1.
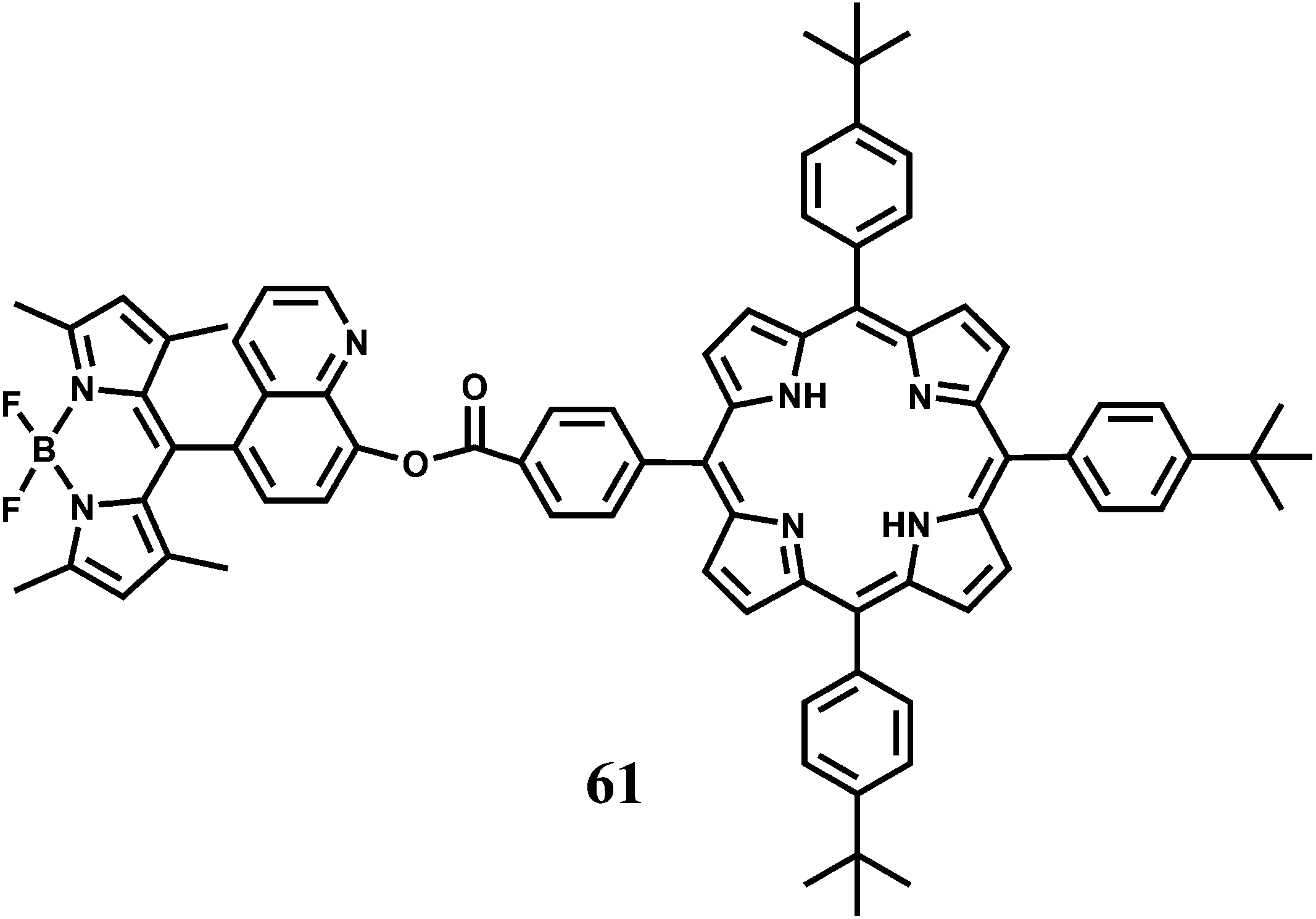
A BODIPY covalently linked with 8-hydroxyquinoline has been attached to a benzoate linked porphyrin to have the dyad 61.92 The 8-hydroxyquinoline benzoate moiety not only facilitates the energy transfer process but also provides binding sites for Hg2+ and Fe2+ ions. On irradiation at 470 nm where the BODIPY unit absorbs most of the light, 61 gives not only the BODIPY emission at 516 nm but also the porphyrin emission at 650 nm. Addition of Hg2+ leads to a significant decrease in the BODIPY emission (almost 90% in comparison with that of the metal-free 61) and a slight increase in the porphyrin emission decreasing the F516/F650 ratio from 16.4 to 1.24. In presence of Fe2+, however, an opposite change in the corresponding emission intensities are observed. The emission from BODIPY is remarkably enhanced (83.62% relative to that of metal-free 61) and porphyrin emission, simultaneously diminished slightly, with the F516/F650 ratio increasing from 16.4 to 26.67. It is believed that binding of either metal ion at the 8-hydroxyquinoline site leads to twisting of the dyad bringing the two fluorophores closer. This facilitates FRET from BODIPY to porphyrin. Presence of Fe2+ induces partial restraint in the FRET due to non-radiative quenching in the excited states associated with the unpaired electrons in Fe2+. This system represents a rare example of Fe2+ detection.
2.6. Cobalt chemosensors
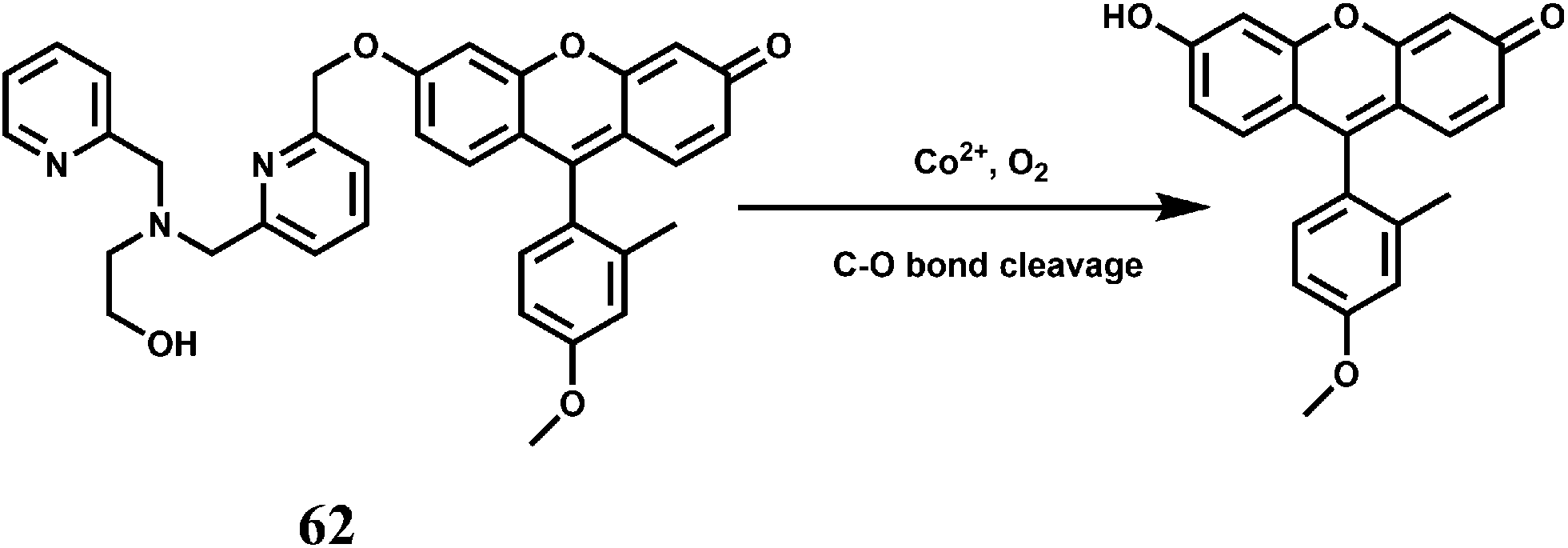
Cobalt is an essential trace element in human body and found in cobalamin and also in a few metalloproteins.93 Apart from its biological roles, exposure to high levels of cobalt can lead to health hazards like decreased cardiac output, cardiac and thyroid enlargements, heart disease, elevated red blood cells accompanied by increased cells in the bone marrow, increased blood volume, vasodilation, flushing and so on.94 Monitoring its concentration in human is, therefore, of crucial importance. However, there is hardly any fluorescence signaling systems available for Co2+ that can be utilized for tracking this transition metal in living cells or other biological specimens. Two principal challenges of Co2+ detection in biosystems are its paramagnetic nature and its biologically relevant divalent metal ion competitors like Ni2+, Cu2+ and Zn2+ that typically bind more tightly to common ligands compared to Co2+ owing to Irving–Williams series considerations.95 In spite of these difficulties, there are several chemosensors available for detecting the Co2+ ion.The chemosensor 62 has been synthesized by connecting a Tokyo Green (TG) dye derivative with a tetradentate N3O chelator ligand through a cleavable ether linkage.96 In presence of O2, the Co2+ ion can break the C–O bond releasing TG free which does not contain a bound, quenching paramagnetic metal center (Scheme 36). This transforms a weakly fluorescent probe into a highly fluorescent dye, that gives about 18-fold fluorescence enhancement in aqueous medium. Cellular concentrations of other competing and biologically relevant s- and d-block metal ions do not elicit any significant fluorescence enhancement. This sensor can also detect changes in exchangeable Co2+ pools in living cells as found in case of lung carcinoma A549 cell line.
 | ||
| Scheme 36 Possible sensing mechanism of chemosensor 62. | ||
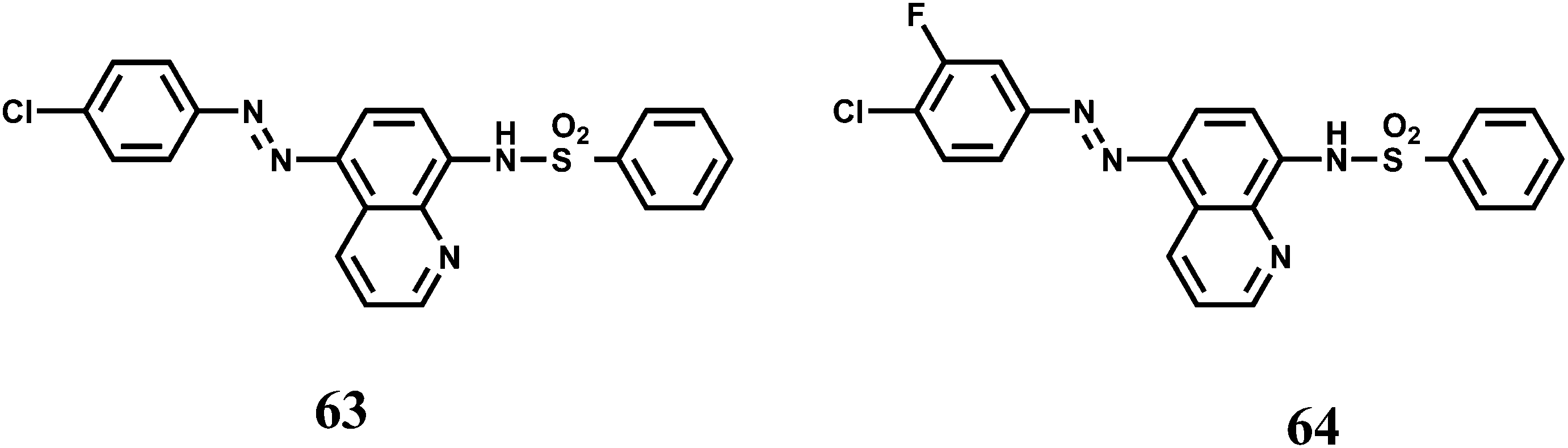
Two 8-sulfonamidoquinoline derivatives 63 and 64 were designed for the determination of Co2+ in presence of H2O2.97 It has been observed that under basic medium it forms a chelate with the sulphonamide group resulting in intense emission in the ultraviolet region where a range of metal ions, including Cr3+, Cu2+ and Ni2+ do not interfere with the exception of Fe3+. This makes these probes useful for the determination of trace amount of Co2+ in food and hair samples. While speciation of the complex is not clear, it is likely that Co2+ is oxidized to the kinetically inert Co3+ complex. Interestingly, no emission is observed in absence of H2O2.
Cyanide anions are extremely poisonous as they inhibit the process of cellular respiration in mammals by interacting strongly with a heme unit in the active site of cytochrome a3.98 Also, the high level of intracellular Ca2+ concentration is caused by the uptake of CN− and triggers a cascade of enzymatic reactions to increase the level of reactive oxygen species that finally inhibits the antioxidant defense system.99 It is thus crucially important to have a sensor to monitor the CN− concentration in biosystems. The coumarinylsalen fluorescence chemosensor 65 binds a Co2+ ion at the salen moiety as shown in Scheme 37.100 While the metal-free ligand shows emission, when it is bound to Co2+ no emission is observed due to an efficient PET from the coumarin moieties to the metal center. Upon addition of CN− anion which occupies the two axial sites (association constant, K1 ≥ 107 M−1 and K2 = 4.0 × 105 M−1) the LUMO of the cobalt complex is raised above the HOMO of the fluorophore. This blocks the donor excited PET to give a strong fluorescence recovery.
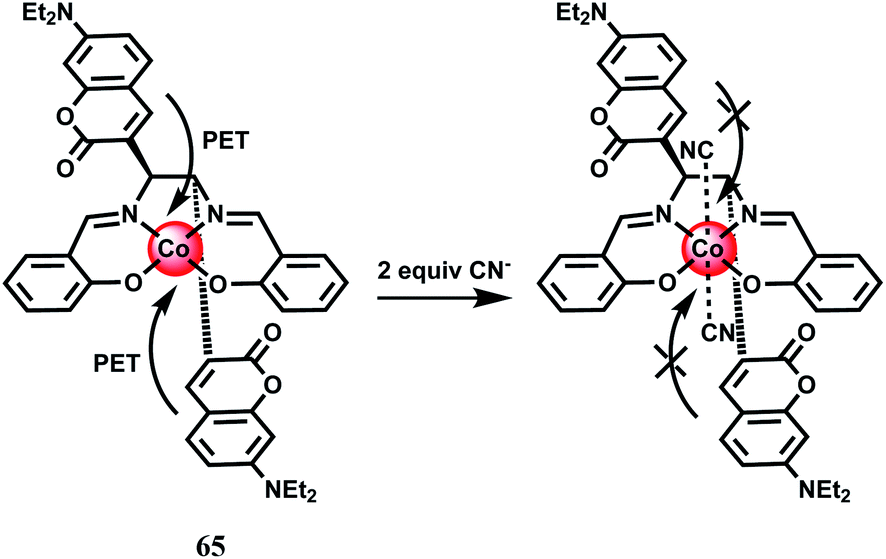 | ||
| Scheme 37 Complexation of the cyanide ion with 65·Co. | ||
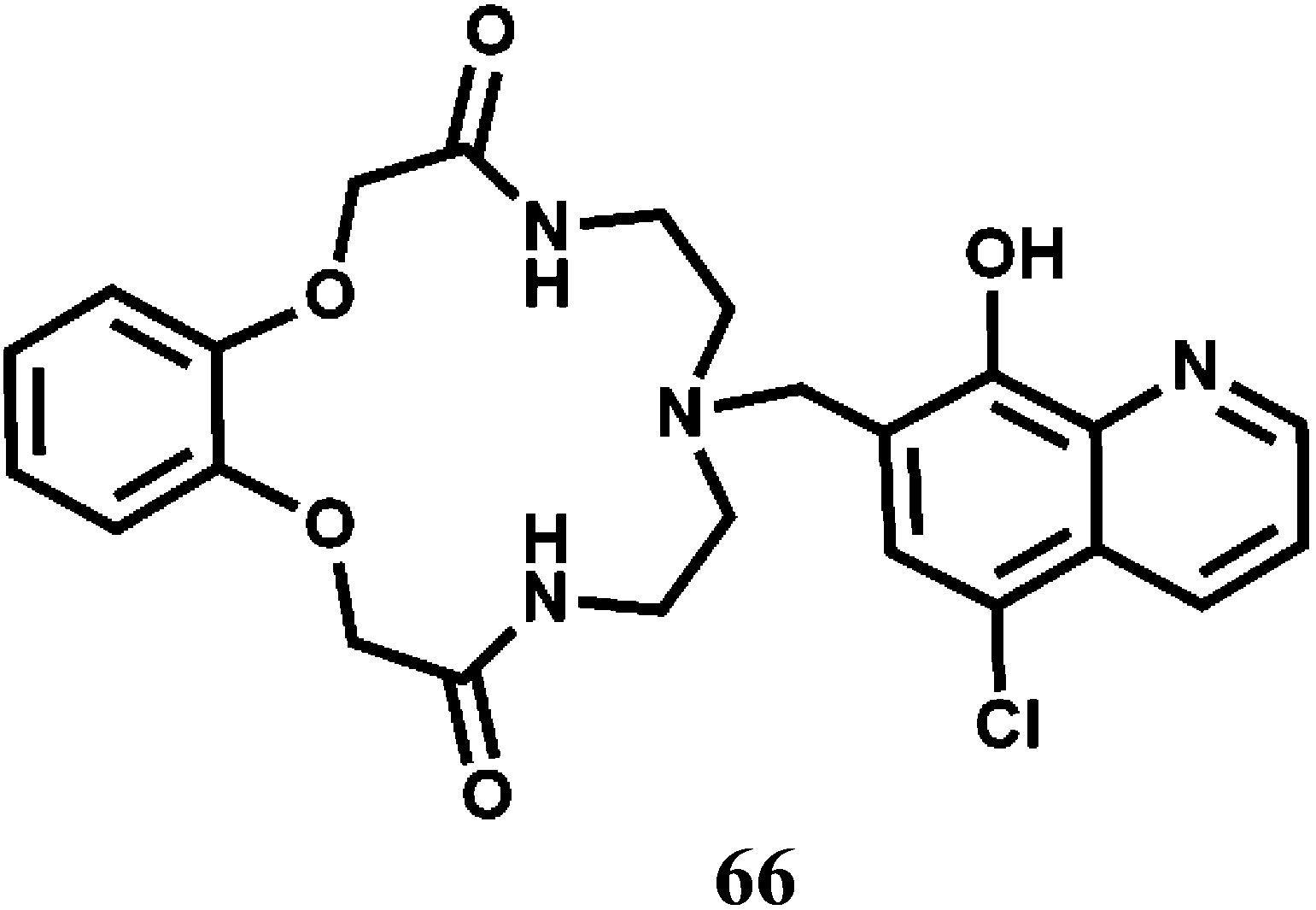
An efficient and selective fluorescent optode membrane based on the chemosensor 66 has been fabricated in the plasticized PVC membrane containing sodium tetraphenylborate as a liphophilic anionic additive.101 The metal-free sensor upon excitation at 345 nm gives a strong emission at 520 nm at pH 5.0. Upon addition of Co2+ ion, this emission is quenched with the appearance of two new bands in the region, 385–420 nm. Quenching of the 520 nm band and appearance of the 420 nm band ratio can be used for quantitative estimation of Co2+ up to the concentration range of 5.0 × 10−7 to 2.0 × 10−2 M with a relatively fast response time of less than 5 min. The proposed fluorescence optode has been successfully applied to find out cobalt content of vitamin B12 ampoule, cobalt cake, cobalt alloy and tap water samples.
The chemosensor 67 based on ICT mechanism has been reported.102 The free probe on excitation at 404 nm gives a weak, structureless emission band at 428 nm in buffered MeOH–H2O (1:1 v/v) at pH 7.4. Upon addition of Co2+, the metal ion binds at the site shown that results in the colour modulation as well as increases the ICT transition by a 37-fold enhancement in the fluorescence output. The selectivity of Co2+ arises as it fits well in the pseudo-cavity with N2O donor set (Scheme 38). Other biologically relevant metal ions give negligible enhancement. A slight modification of the receptor can make this system highly sensitive and selective for Co2+ ion.
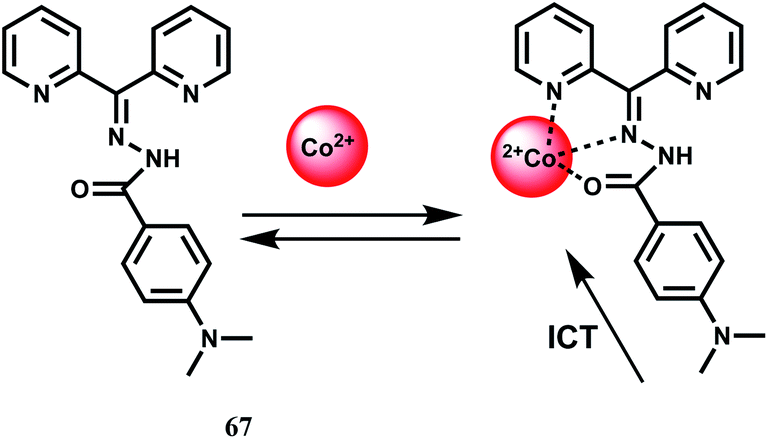 | ||
| Scheme 38 Possible Co2+ binding mode of 67. | ||
Chemosensor 68 based on 9,10-anthraquinone has been introduced to show that binding of Co2+ changes the colour from yellow to pale green.103 It also affords a 100-fold CHEF with green fluorescence in 20% aqueous methanol at pH 7 with the Co2+ ion. However, metal ions like Cu2+ and Ni2+ interfere by completely quenching the fluorescence (in presence of Cu2+) or significantly reduce the emission (in presence of Ni2+). This fluorescent probe is, therefore, not suitable for use in biological samples.
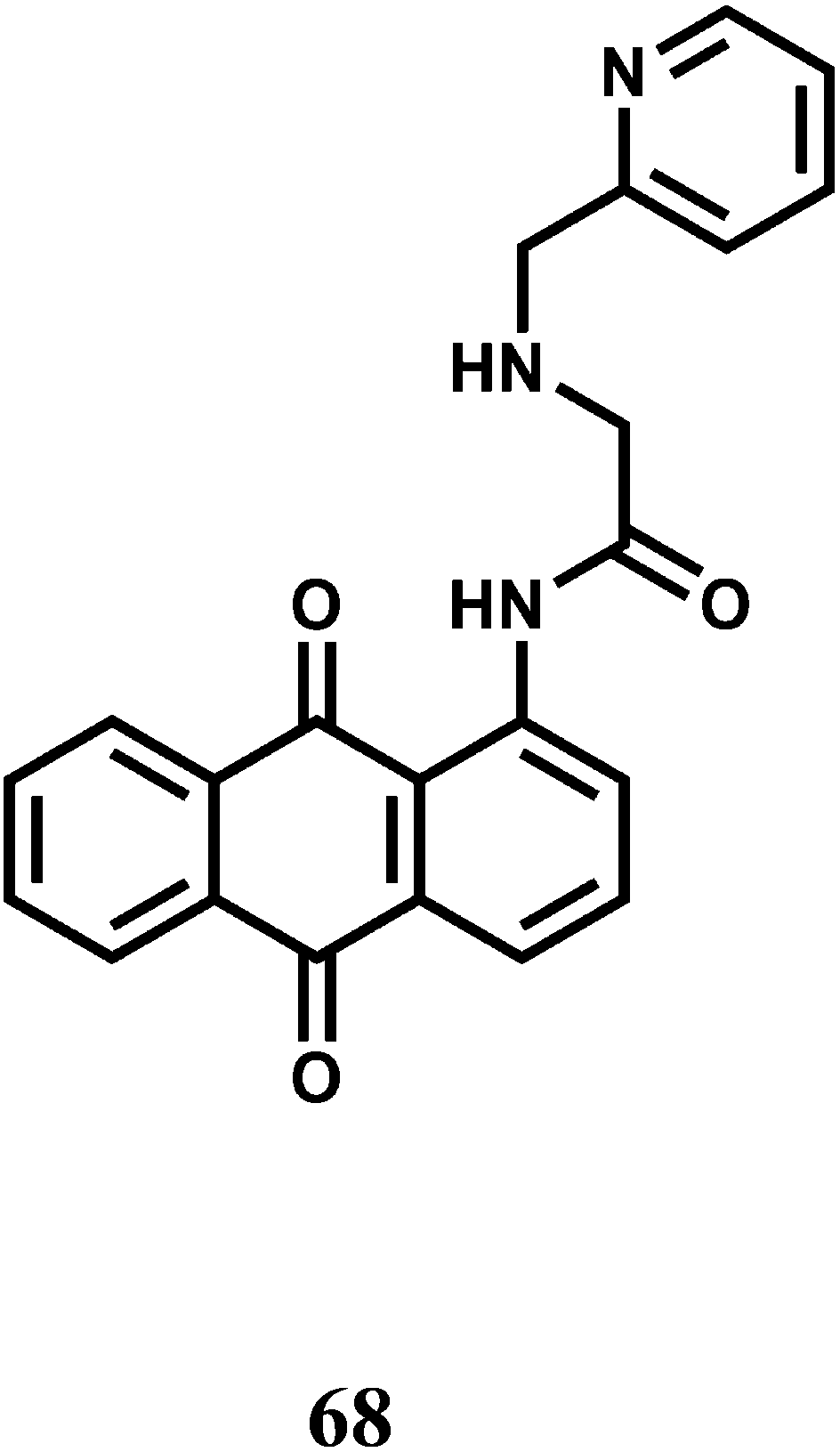
The fluorescent probe 69 with a benzyl ether linker on excitation at 350 nm emits at 380 nm in aqueous solution at pH 7.2.104 Addition of Co2+ to the solution leads to quenching of this emission band and appearance of a new band at 460 nm. The rapid fluorescence quenching of the 380 nm band is due to binding of the paramagnetic Co2+. However, Co2+ mediated cleavage of the benzyl ether bond releases highly fluorescent 2-(2′-hydroxyphenyl)benzothiazole that gives emission at 460 nm without any metal bonded to it.
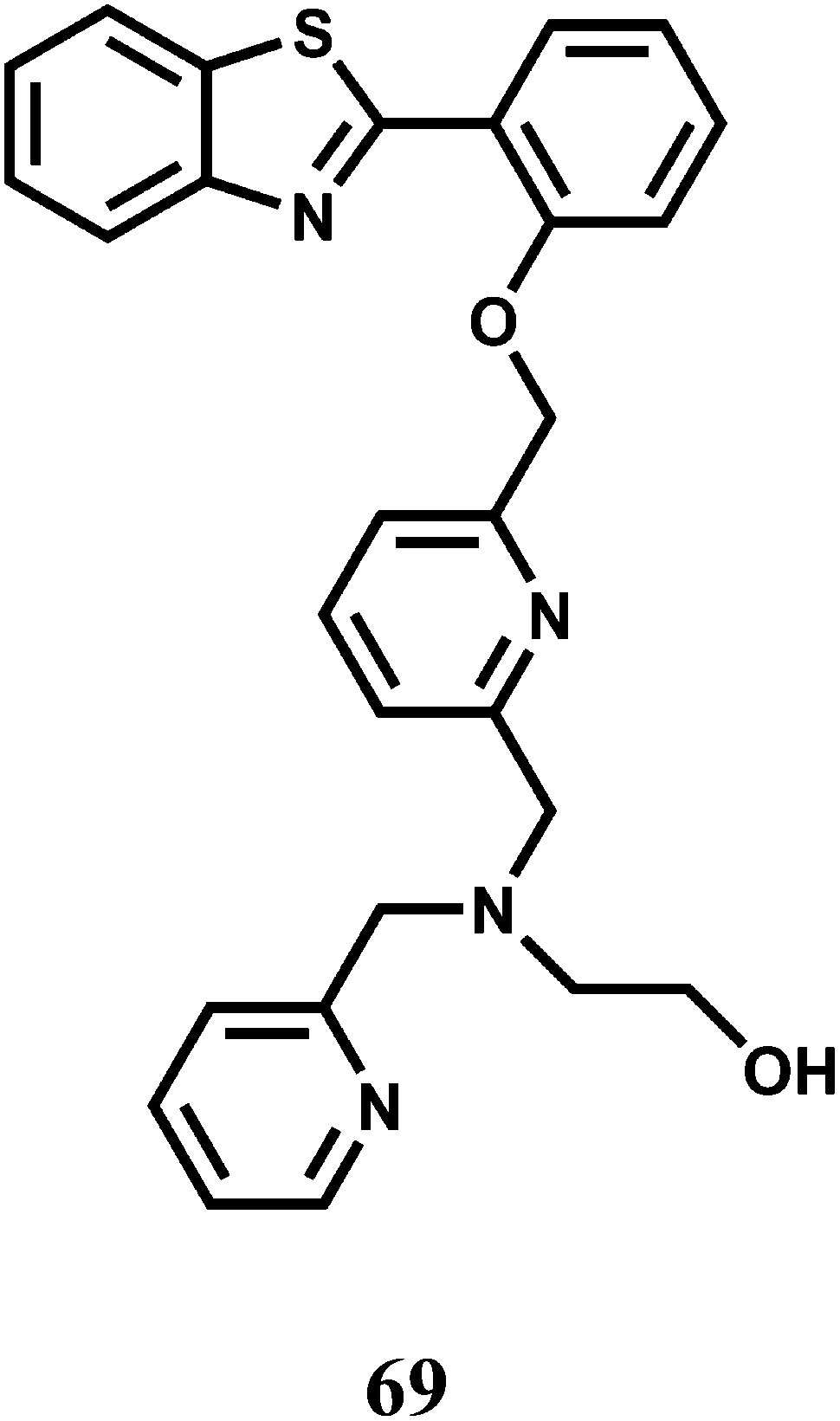
The coumarin–quinoline hybrid 70 affords turn-on fluorescence in presence of paramagnetic Co2+ and Ni2+ ions at 392 nm when excited at 338 nm in methanolic water (9:1, v/v).105 Heavy metal ions like Ag+ also elicit slight fluorescence showing its poor chemoselective nature. The fluorescence appears to be due to binding a metal ion at the site providing NO2 coordination that stops the deactivation pathway.
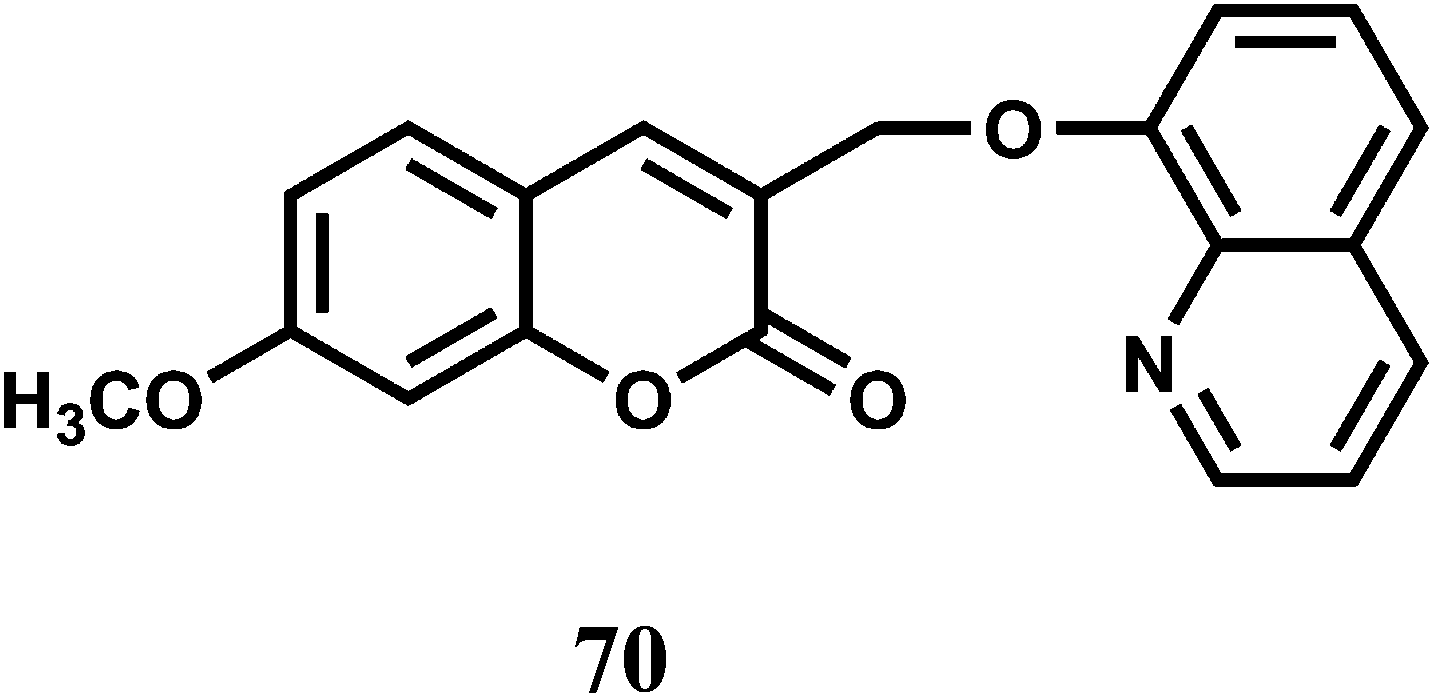
2.7. Nickel chemosensors
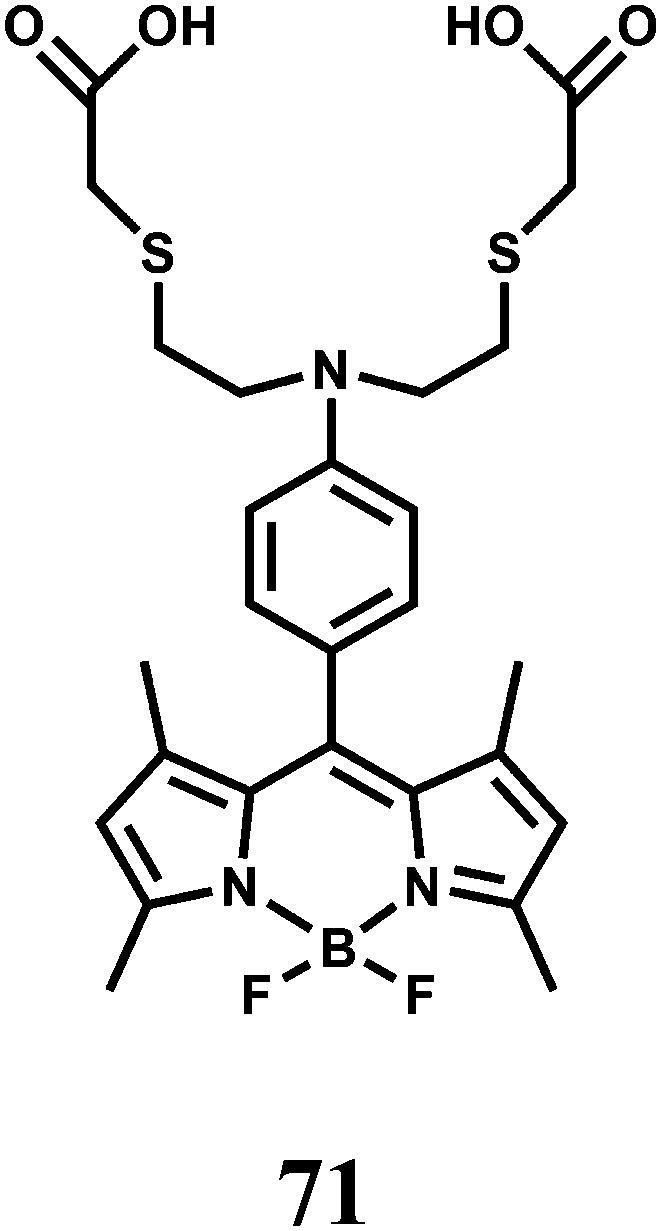
Nickel is an essential element in plants and many other biosystems. It is implicated in various enzyme activities106 that include ureases, hydrogenases, superoxide dismutases, acetyl-coenzyme A synthases, carbon monoxide dehydrogenases, methyl-coenzyme M reductases and frequently used in catalytic processes. However, as an industrial pollutant, nickel is a toxic element that can cause lung injury, allergy and carcinogenesis. It is, therefore, of importance to have sensors for nickel. A major obstacle is to design receptors selective for Ni2+ ion over other biologically relevant metal ions.In chemosensor 71, a BODIPY unit is covalently connected to a mixed N/O/S receptor to coordinate the Ni2+ cation. The receptor forms a 1:1 complex with Ni2+ with an approximate dissociation constant of (195 ± 5) × 10−6 M.107 The probe gives a 25-fold fluorescence enhancement upon Ni2+ binding. This turn-on response is reversible as treatment of the Ni2+-loaded 71 with the metal ion chelator TPEN restores the fluorescence back to the free 71 level. Biologically relevant alkali, alkaline earth and transition metal ions except Cu2+ do not show any enhancement or interfere with the Ni2+ response. Cu2+ quenches the enhancement obtained with Ni2+ restricting its usefulness. Confocal microscopic studies show that this chemosensor can monitor changes in Ni2+ levels within living mammalian cells.
Rhodamine derived chemosensor108 72 show spiro-ring opened 200-fold fluorescence enhancement in presence of Co2+ and Ni2+ while other transition metal ions do not interfere in aqueous medium containing 2% DMSO. Both Co2+ and Ni2+ form 1:1 complexes with the association constants of 2.1 × 104 and 4.5 × 103 M−1, respectively. Binding to both ions is reversible, as the metal can be extracted with EDTA with concomitant reduction in the fluorescence to the metal-free state. Since Co2+ competes fairly well with Ni2+, the detection of Ni2+ can be done only if Co2+ is not present.
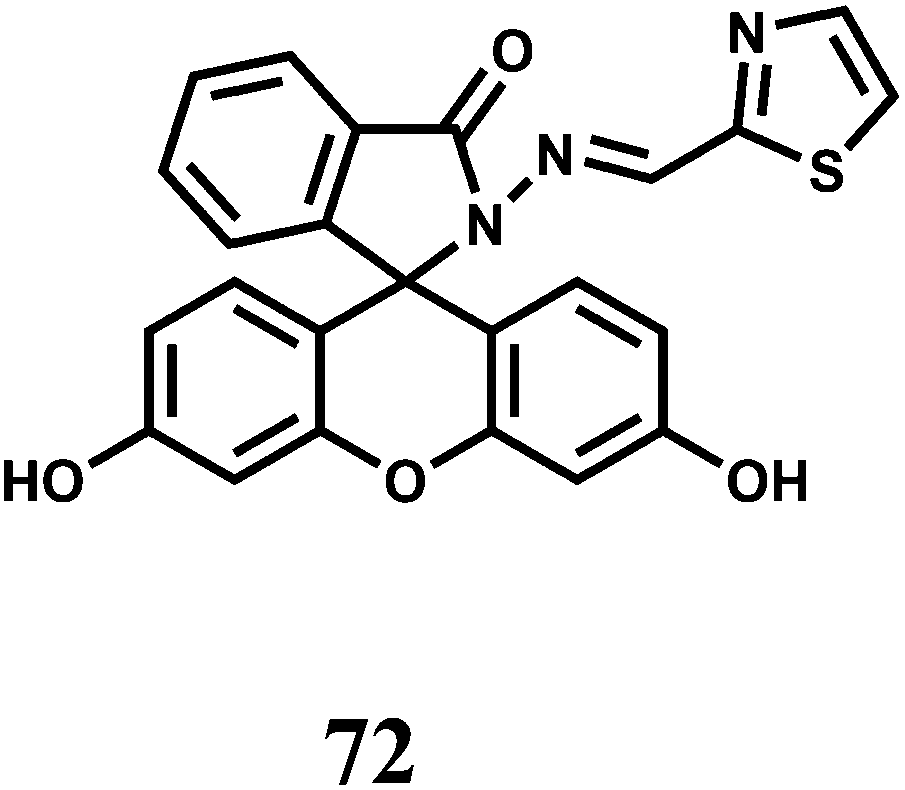
The PET chemosensor 73 designed with coumarin attached to a piperazine moiety works in alkaline pH.109 It affords a 7-fold enhancement in presence of large excess of Ni2+ ion. However, Zn2+ ion also gives a 10-fold enhancement and therefore, limits its utility.
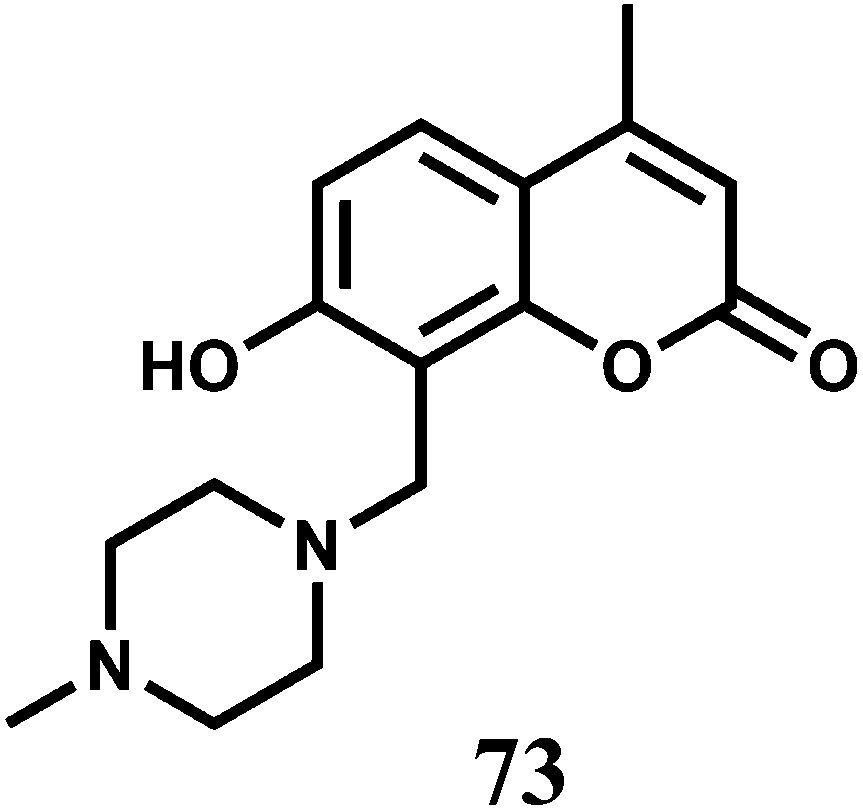
The novel Zn2+ Schiff base complex 74 bearing acacen2− (dianion of N,N′-ethylenebis(acetylacetonylideneaminato)) moieties have been constructed.110 The crystal structure of its dinickel complex shows that the two Ni2+ ions occupy the two metallacycles. In DMF, 74 gives an emission at 361 upon excitation at 293 nm. When Ni2+ is added, the structure becomes more rigid blocking its deactivation pathway and hence, a significant increase in the emission quantum yield is observed.
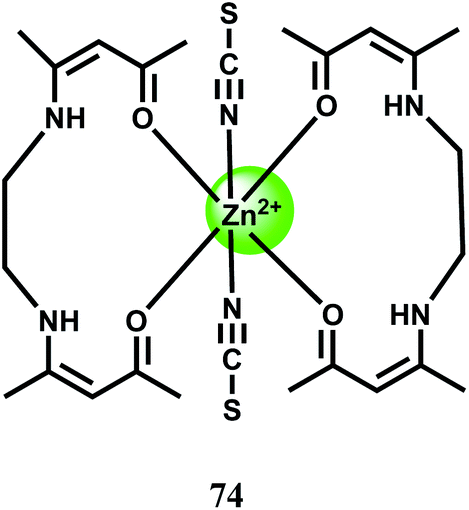
The malonohydrazide based PET fluorescent probe 75 forms a 1:1 complex in DMSO–water (1:1, v/v) with Ni2+ ion with the association constant of Ka = 3.3 × 104 M−1.111 The free probe gives an emission peak at 629 nm on excitation at 317 nm which increases significantly and selectively in presence of Ni2+ ion (Scheme 39). The Ni2+ binding at the site is quite selective and strong due to good fitting of the metal ion in the binding pocket. The cause of fluorescence enhancement is not mentioned by the authors but it is more likely to be due to increased rigidity of the system that blocks the deactivation pathway.
 | ||
| Scheme 39 Possible metal ion binding mode of 75. | ||
2.8. Copper chemosensors
Copper is the third-most abundant transition metal in the human body. Copper containing proteins are involved in a wide variety of biochemical functions112 including copper transport (ceruloplasmin), copper storage (metallothionein), protective roles (Cu–Zn superoxide dismutase), terminal oxidase for O2 respiration (cytochrome c oxidase) and so on. Copper ions are mainly metabolized by the liver. Copper in the cytoplasm predominantly binds to metallothionein, and the excess copper is excreted into the bile mainly through a lysosome-to-bile pathway.113 The level of copper in lysosomes is closely related to a number of neurodegenerative diseases, that include Wilson's disease,114 Menkes disease115 and Alzheimer's disease.116 Copper commonly accumulates in the lysosomes of patients suffering from these diseases. It is therefore, interesting to know the spatial distribution of copper in the biosystems. It follows, therefore, those sensors that can signal the presence of copper ion selectively is of high demand. However, some of the shortcomings for practical applications such as cross-sensitivities toward other metal cations and low fluorescence quantum yields are also to be kept in mind. Also, aqueous medium should be preferred for biological studies. Definitely, development of new Cu2+-selective turn-on fluorescent probes is of utmost importance and necessity.In the PET chemosensor 76117 a N2O donor receptor has been connected via Schiff base condensation to an electron-withdrawing naphthalimide derivative. It forms a highly stable 1:1 complex with Cu2+ (Ka = 1.1 × 1010 M−1) in acetonitrile–water (70:30, v/v). Free 75 shows a weak emission at 519 nm due to an efficient PET from the receptor to the fluorophore (Scheme 40). Binding of Cu2+ stops the PET giving a 5-fold fluorescence enhancement. The detection limit is calculated to be 1.5 × 10−7 M while changes in fluorescence is observed within 10 s. Other biologically relevant metal ions show negligible enhancement or do not interfere with the Cu2+-induced emission.
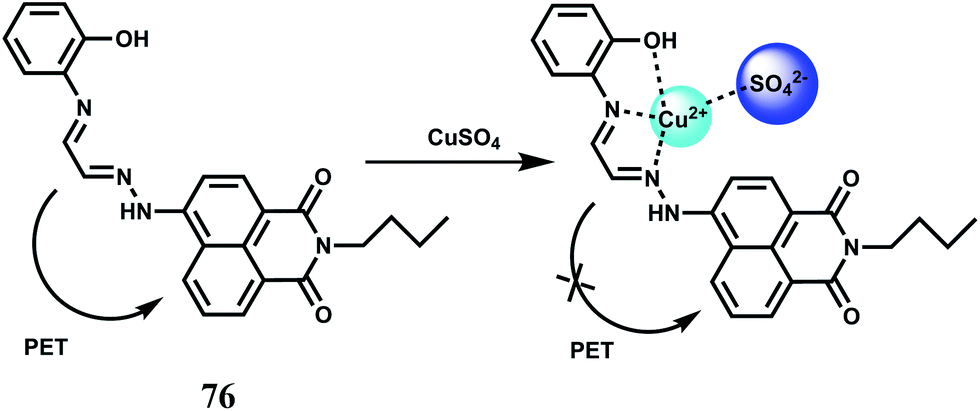 | ||
| Scheme 40 Proposed binding mode of 76 with Cu2+. | ||
The PET chemosensor 77 incorporated with two thioether and three amine units has been designed to detect Cu2+ ion in CH3CN–H2O (4:1) at pH 7.118 Besides, the larger cavity size of the receptor has lowered the association constant for the 1:1 complex with Cu2+ to logK= 4.1 ± 0.1. Ni(II), Cd(II), Zn(II), Ag(I), and Hg(II) (1000 μM) do not interfere in the fluorescence studies. Significant participation of amine nitrogen in binding with Cu2+ ion is responsible for the enhancement of fluorescence due to suppression of the PET.
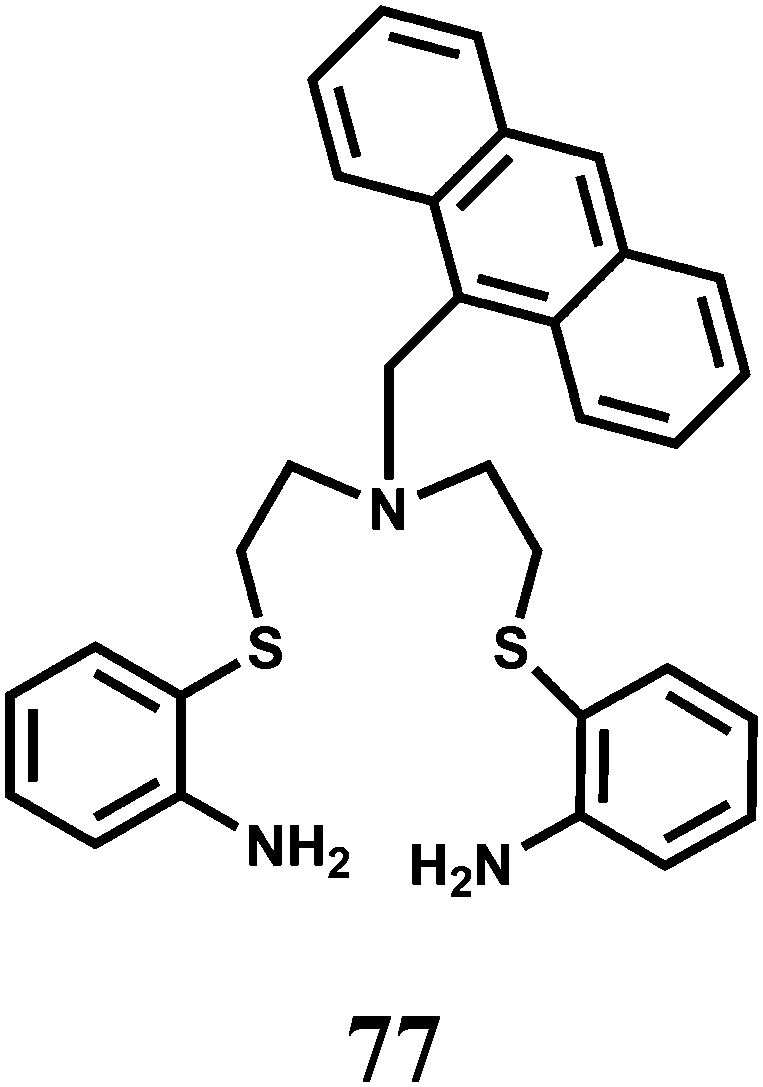
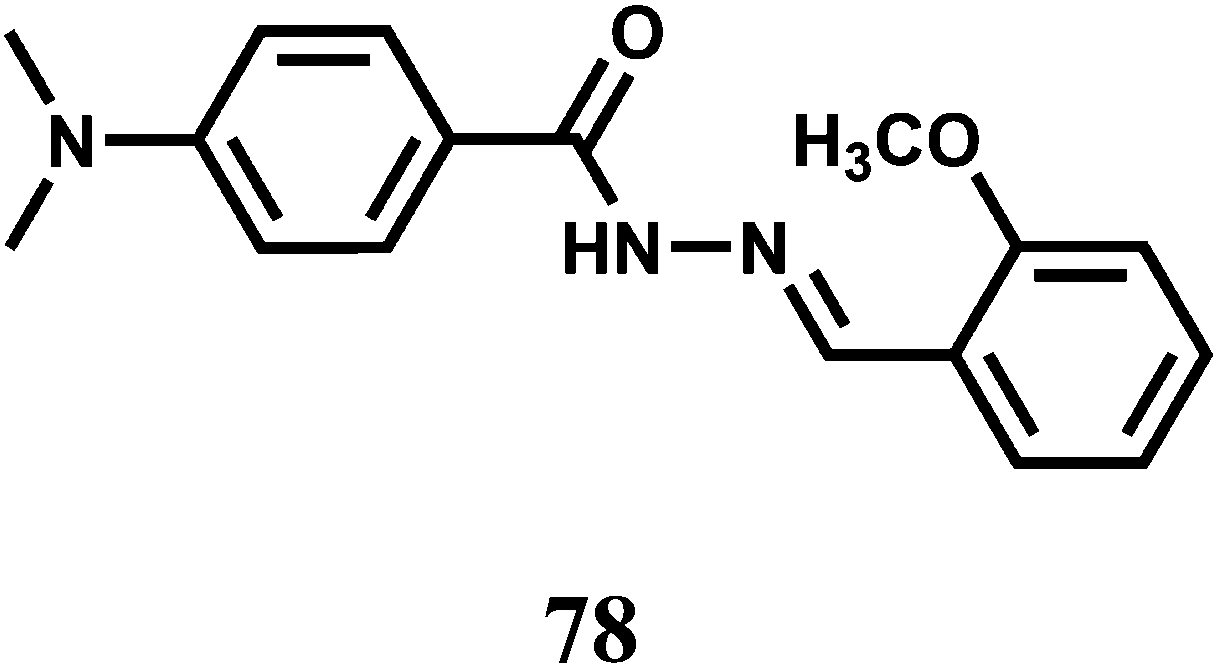
In addition to the PET chemosensors mentioned above, intramolecular charge transfer emission has also been used as a mechanism for Cu2+ detection. The chemosensor 78 has been synthesized by connecting 2-methoxybenzaldehyde hydrazone with 4-(N,N-dimethylamino)benzamide.119 The methoxy group rather than a phenolic group has been used to nullify excited-state intramolecular proton transfer (ESIPT) that would have opened another channel of deactivation. The metal ions Cu2+, Zn2+, Hg2+ and Pb2+ bind to the chemosensor with 1:1 stoichiometry to similar extent in the ground state as the binding constant of comparable value (order, 104 M−1) is obtained with each metal ion. In MeCN, 77 emits dual fluorescence, an excited state CT pertaining to the 4-(N,N-dimethylamino)benzamide group at 525 nm and a LE emission at 350 nm. A host of biologically relevant as well as heavy metal ions are used for probing the response of the CT band. These metal ions either quench the fluorescence or have no effect. Only Cu2+ exhibits a prominent 30-fold enhancement of the CT band with a substantial blue-shift from 525 to 460 nm. The fluorescence enhancement factor decreased with increasing water content in MeCN, but can still be significant even when the water content is 90% by volume. The low fluorescence quantum yield of 77 in absence of Cu2+ can be attributed to radiationless transitions from the nπ* state. Upon addition of Cu2+ the metal ion coordinates with the lone pair of the carbonyl O raising the energy level of the nπ* state such that the ππ* state becomes the lowest excited state, leading to a substantial increase in the fluorescence quantum yield. Besides, Cu2+ binding can also lead to a conformation restriction which can block a channel of the deactivation pathway.
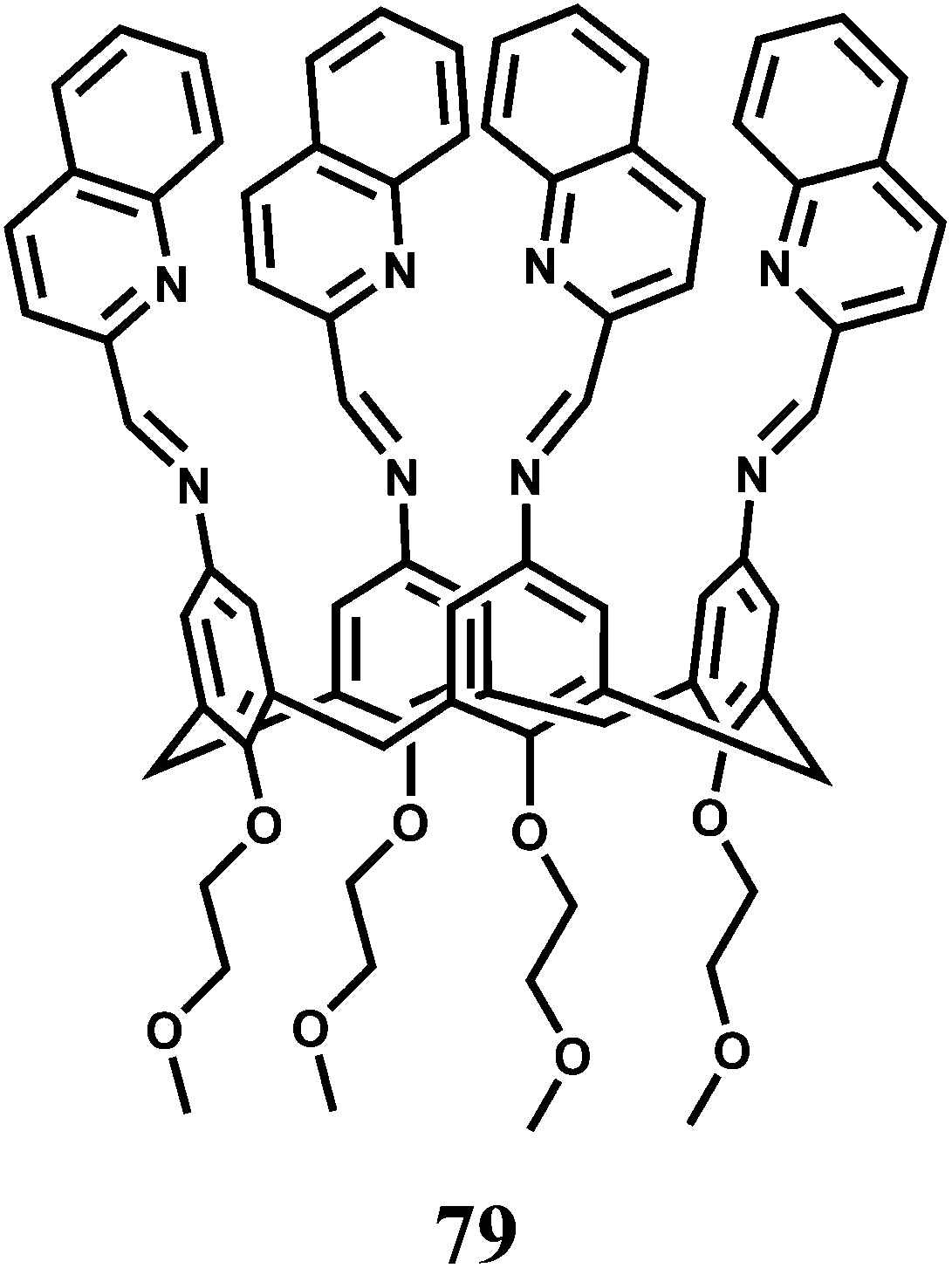
The PET chemosensor 79 has been fabricated on a calix[4]arene platform by modifying its upper rim with four iminoquinoline subunits.120 Metal free 79 shows a very low fluorescence quantum yield in MeCN due to an efficient PET from the imine N atoms. A Cu2+ ion coordinates the four imine N atoms with high association constant (Ka) of 3.67 × 107 M−1 to block the PET accompanied by about 1200-fold enhancement of fluorescence. While other transition metal ions do not bind at this site, alkali/alkaline metal ions are expected to bind at the lower rim to the ethereal O atoms without affecting fluorescence. While this is a simple design, its lack of solubility in water restricts its usefulness.
Like in the cases of other transition metal sensors, a number of turn-on sensors for Cu2+ has been designed based on rhodamine derivatives. The probe 80 has been synthesized by condensing two rhodamine B hydrazides with terephthalaldehyde. In aqueous medium, free 80 shows very weak emission in aqueous medium. Addition of Cu2+ ion gives a remarkably high fluorescence at 575 nm due to opening of spirolactam bond while other metal ions do not exhibit any significant emission (Scheme 41).121 Spectroscopic data suggest a 1:1 stoichiometry although no association constant value is provided.
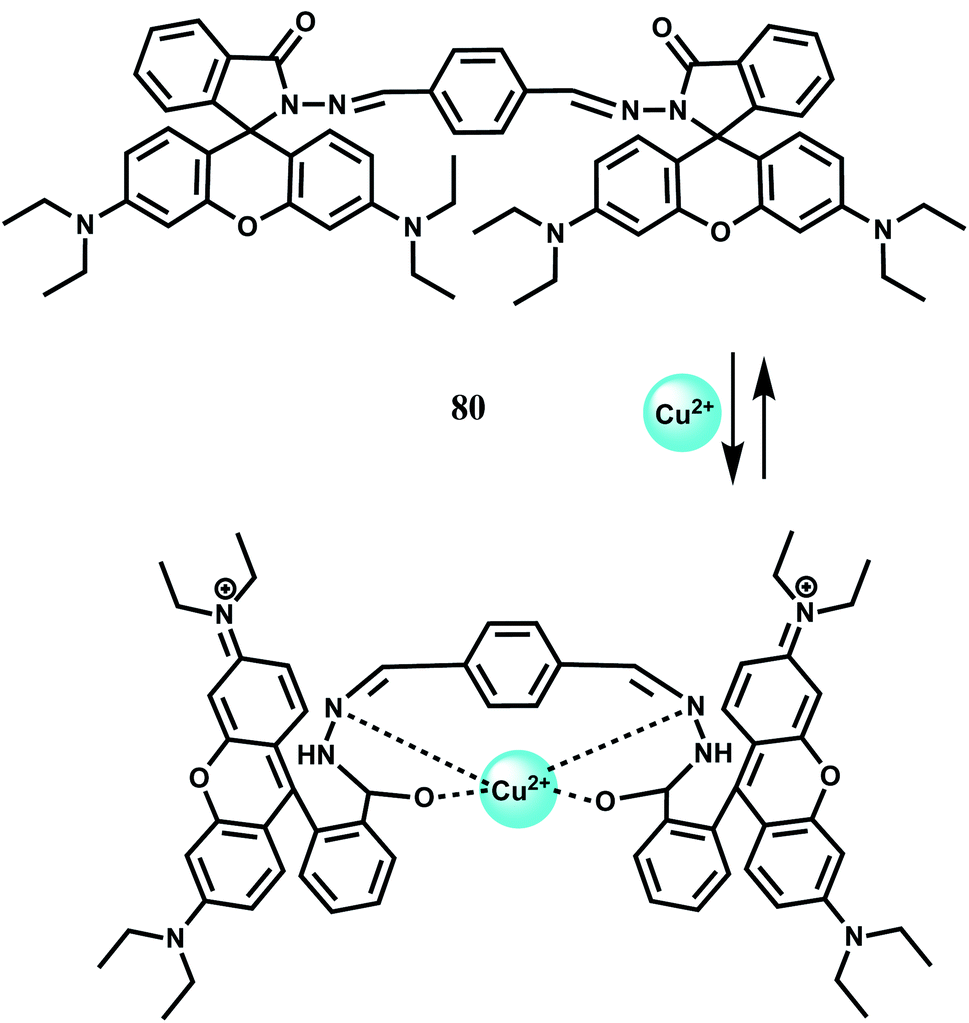 | ||
| Scheme 41 Proposed binding mode of 80 with Cu2+. | ||
In chemosensor 81, a Cu2+ ion is anchored at the site shown in Scheme 42 leading to opening of the spirolactam as well as hydrolytic cleavage of the imine bonds giving high fluorescence.122 Since it works in aqueous medium, it affords imaging studies of different cell lines with confocal microscopy. However, the hydrolysis step is sluggish and takes at least 1 h and so this probe cannot be used for measuring temporal distribution of Cu2+ ion in biological systems.
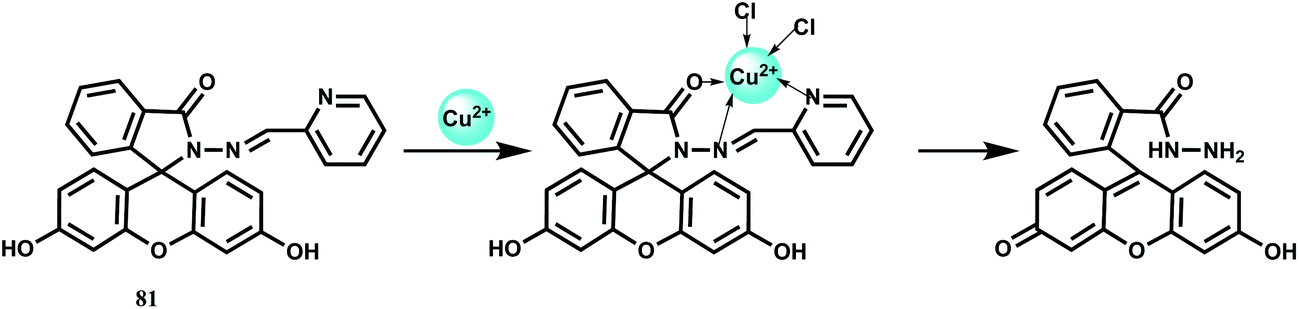 | ||
| Scheme 42 Proposed binding mode of 81 with Cu2+. | ||
The chemosensor 82 is possibly the first example of a boronic acid-linked chemosensor for Cu2+ ion (Scheme 43).123 The metal ion is believed to occupy the site as shown giving an association constant value of 2.8 × 103 M−1. When the boronic acid group is replaced by hydrogen, the Cu2+ ion does not elicit spiro-ring opened fluorescence enhancement. Thus, boronic acid moiety is important in binding a Cu2+ ion at the site. Free 81 forms a colourless solution in 50% aqueous MeCN showing very low emission. Upon addition of Cu2+ the colour changes to pink accompanied by spirolactam ring-opened fluorescence enhancement at 585 nm. Other biologically relevant metal ions do not give any fluorescence. Also, metal ion competition experiments show that binding of Cu2+ to the chemosensor is unaffected by other metal ions even when used in large excess. When an excess of cyclen is added to the solution containing the Cu2+ complex of 82, the fluorescence disappear and the original structure of the chemosensor is regenerated. Thus, in this case, Cu2+-induced spirolactam ring opening is a reversible process. Confocal microscopic experiments show that 81 can pass through cell membranes and displays a strong fluorescence response to intracellular copper ions in different mammalian cell lines.
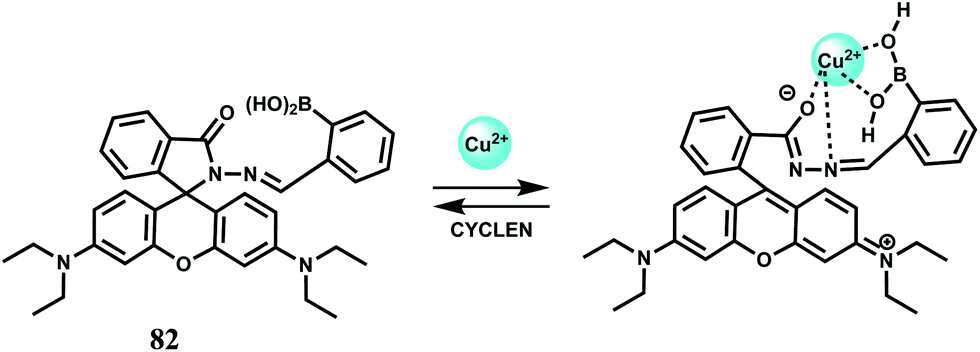 | ||
| Scheme 43 Proposed binding mode of 82 with Cu2+. | ||
The rhodamine B based chemosensor 83 with a six-membered reactive ring was developed by inserting a nitrogen atom in the known fluorescent probe rhodamine B spiro thiohydrazide.124 This sensor is shown to be an efficient “turn-on” fluorescent chemodosimeter for Cu2+ in a neutral aqueous medium due to opening of the spiro ring (Scheme 44). Other biologically relevant or heavy metal ions do not induce ring-opened fluorescence enhancement. Titration experiments reveal that the fluorescence intensity at 597 nm increases linearly with increasing concentrations of Cu2+ between 0.10 and 10.0 μM and further smoothly increase until a maximum is reached up to 20.0 μM Cu2+, where an approximately 400-fold enhancement is observed.
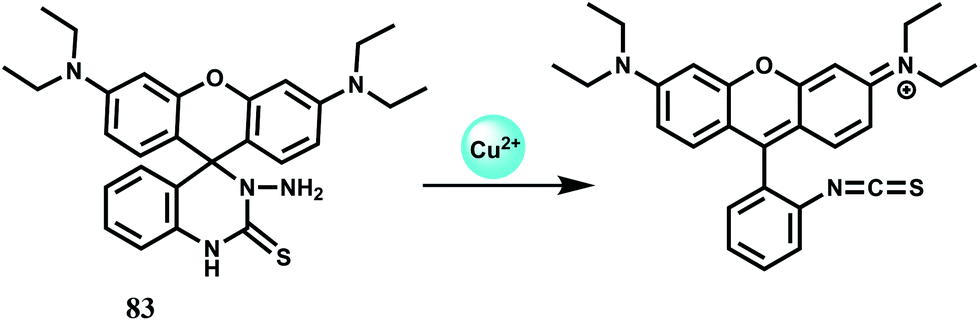 | ||
| Scheme 44 Proposed sensing mechanism of Cu2+ by 83. | ||
The chemosensor 84 has also been found to selectively bind Cu2+ ion with the association constant estimated to be 8.9 × 103 M−1 in aqueous MeCN (1:1, v/v). Upon binding of Cu2+, strong spiro-ring opened fluorescence enhancement is observed (Scheme 45)125 while other competing metal ions do not bring any discernible fluorescence changes. Also, Cu2+ induced fluorescence of 84 is reversible as addition of excess EDTA completely quenches the fluorescence. Experiments with living cells indicate that the chemosensor is cell viable and can detect intracellular Cu2+ ion.
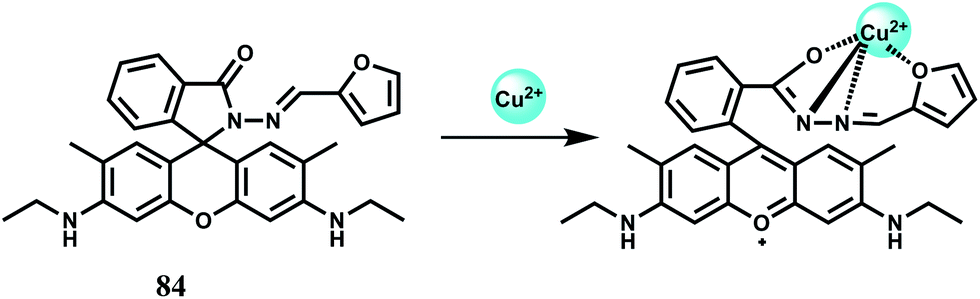 | ||
| Scheme 45 Proposed binding mode of 84 with Cu2+. | ||
Chemosensors 85 and 86 can be easily synthesized using click chemistry.126 These compounds are non-fluorescent but with increase in pH in presence of Cu2+ lead to Fenton reaction promoting ring-opening of triazole linked fluorescein lactone (Scheme 46) that leads to strong emission with the detection limit down to 200 nM. Selectivity of this protocol has been evaluated by screening various biologically relevant metal ions that shows none of these metal ions can elicit fluorescence enhancement.
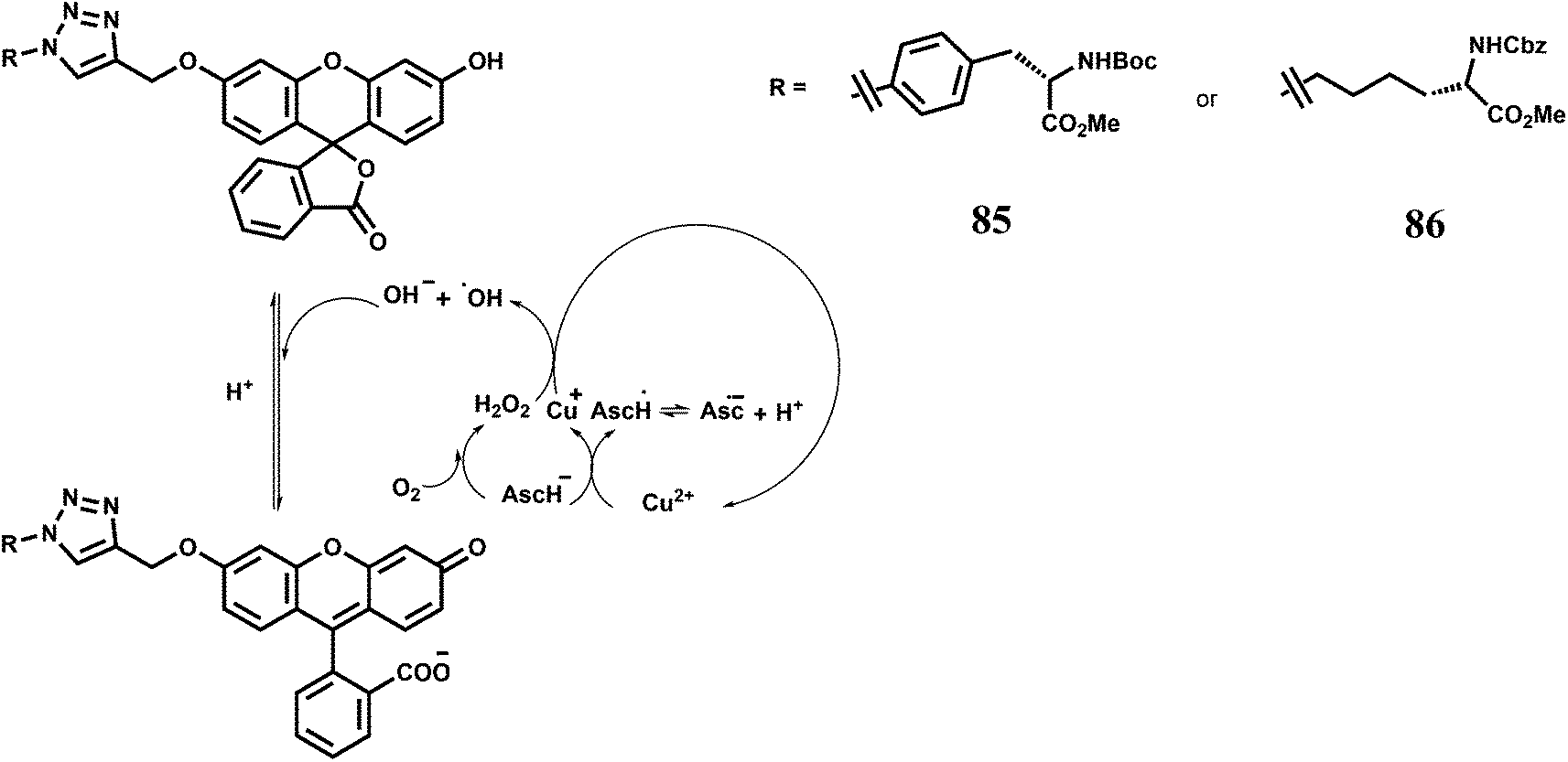 | ||
| Scheme 46 Proposed mechanism for detection of Cu2+ by 85 and 86 based on the Fenton reaction. | ||
The indole derivative of rhodamine 87 specifically binds to Cu2+ ion with an association constant of 1.71 × 104 M−1 in presence of large excess of other competing ions.127 The metal-free dye emits at 490 nm when excited at 340 nm in aqueous MeCN due to the indole moiety. Binding of the Cu2+ ion breaks the spiro-bond of the rhodamine moiety (Scheme 47). At this stage, excitation at 340 nm results in FRET from the donor indole to the Cu2+ bound xanthene acceptor accompanied by a strong emission at 587 nm with concomitant reduction of the emission due to the indole moiety. The detection limit of 87 for Cu2+ ion is found to be 3.6 ppb, which is much lower than the U.S. EPA maximum allowable limit for Cu2+ ion (1.3 ppm) in drinking water. Besides, fluorescence microscopic studies confirmed that the chemosensor can be used as an imaging probe to study the uptake of Cu2+ in HeLa cells.
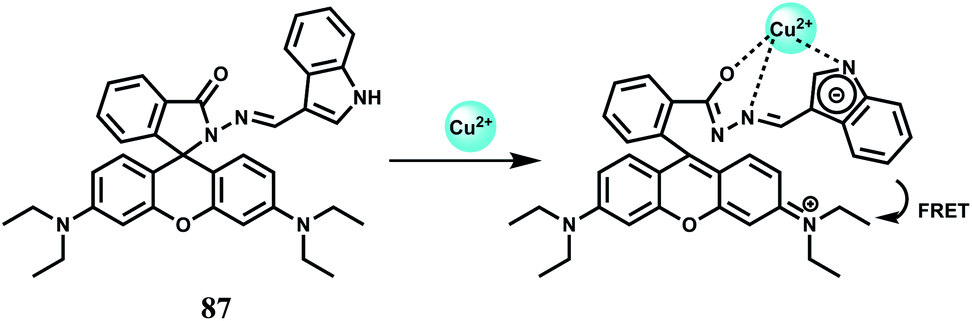 | ||
| Scheme 47 Proposed binding mode of 87 with Cu2+. | ||
The rhodamine B semicarbazide 88 show high spiro-bond opened fluorescence (Scheme 48) in aqueous methanol (1:1, v/v), exhibiting a fast response time (in seconds) and a detection limit of 1.6 × 10−7 mol L−1 at neutral pH.128 The quick response time allows determination of Cu2+ in waste water samples and also monitoring of intracellular Cu2+ levels in living cells using confocal fluorescence spectroscopy. The high selectivity of the chemosensor for Cu2+ ion might be due to close matching between the Cu2+ radius and the size of the binding pocket. However, no association constant value is available.
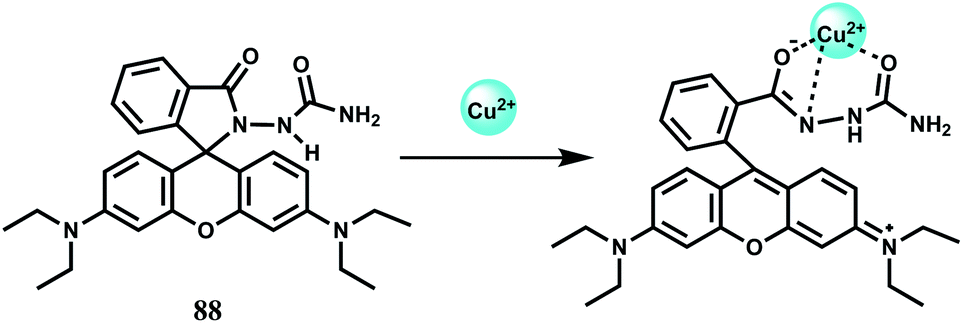 | ||
| Scheme 48 Possible sensing mechanism of 88. | ||
The strategy of metal induced bond breaking has been adopted for the coumarin-334 derived chemosensor 89.129 This compound is non-emissive due to an efficient PET from the imine N atom. Addition of Cu2+ catalyzes breaking of the imine linkage to free the coumarin-334. Since the paramagnetic Cu2+ is not bound to the coumarin, it offers a strong fluorescence (Scheme 49). The detection limit for Cu2+ is found to be 8.7 × 10−8 M which is within the EPA (US) acceptable limit in drinking water. Another important property of the chemodosimeter is its high selectivity for Cu2+ over other metal ions. The competing metal ions do not catalyze hydrolysis of the imine bond.
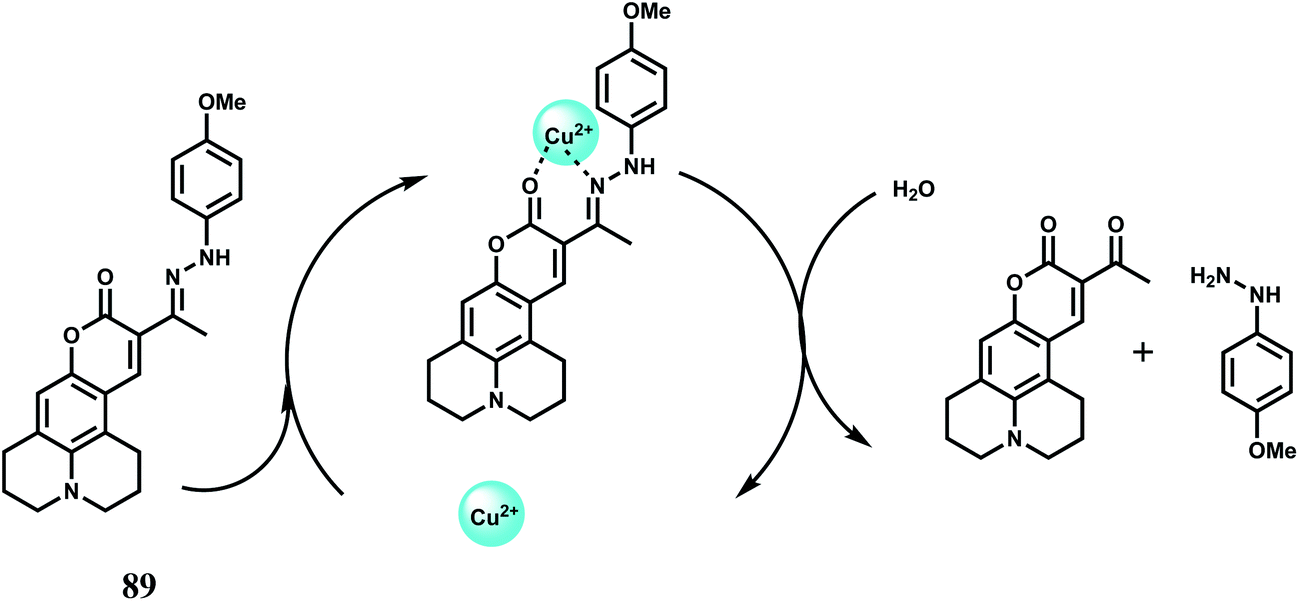 | ||
| Scheme 49 Cu2+-induced catalytic sensing cycle of 89. | ||
BODIPY is an important fluorophore and has been used for several metal ions because of its visible excitation and emission characteristics. The chemosensor 90 based on BODIPY has been designed to have a receptor specific for Cu2+ ion.130 To one arm of the fluorophore is also attached a p-diacetylene benzene moiety to extend conjugation of the system for emission at longer wavelength. Excitation at 620 nm in CH3CN in presence of Cu2+ ion affords an emission with maximum at 651 nm.
Two more BODIPY derived chemosensors 91 and 92 are reported.131 Compound 91 gives large CHEF enhancement in presence of Pb2+ or Cu2+ among the metal ions examined. The association constants of 91 with Pb2+ and Cu2+ are found to be 8.8 × 103 and 5.5 × 104 M−1 respectively. On the other hand, 92 exhibits absolute chemoselectivity for Cu2+ ion over other competing ions including Pb2+ and gives high fluorescence in presence of Cu2+ ion through hydrolysis of the acetyl groups (Scheme 50).
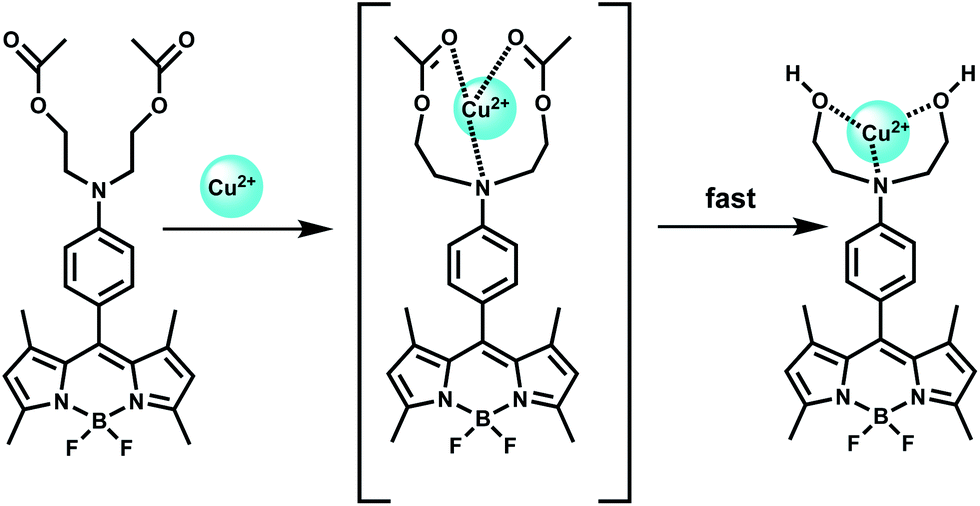 | ||
| Scheme 50 Proposed sensing mechanism of chemosensor 92. | ||
When the receptor contains several soft thioether donors as in 93, the BODIPY based system exhibits excellent selectivity towards the soft Cu+ ion132 giving about 10-fold turn-on response and shows excellent selectivity for Cu+ over biologically relevant metal ions, including Cu2+. Confocal microscopic studies indicate that this sensor can be used for detecting Cu+ levels within living cells.
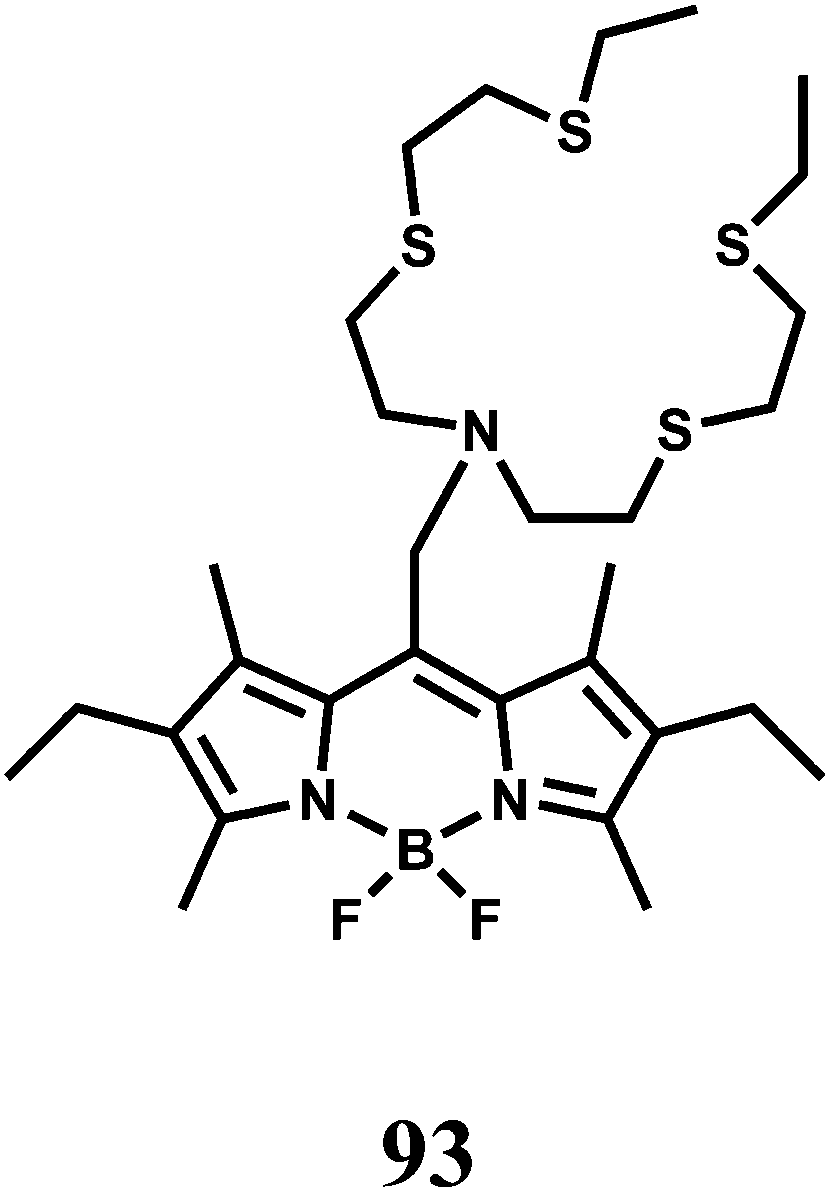
A slightly different derivative of a BODIPY reporter with the same thioether-based receptor as above has been constructed to obtain 94 for selective detection of Cu+ ion over other biologically relevant metal ions, including Cu2+ and Zn2+.133 Live-cell confocal microscopy experiments show that 94 is membrane-permeable and can sense changes in the levels of labile Cu+ pools within living cells by ratiometric imaging, including expansion of endogenous stores of exchangeable intracellular Cu+ triggered by ascorbate stimulation in kidney and brain cells. Such knowledge of spatial distribution of Cu+ is important to monitor physiology of this important metal.
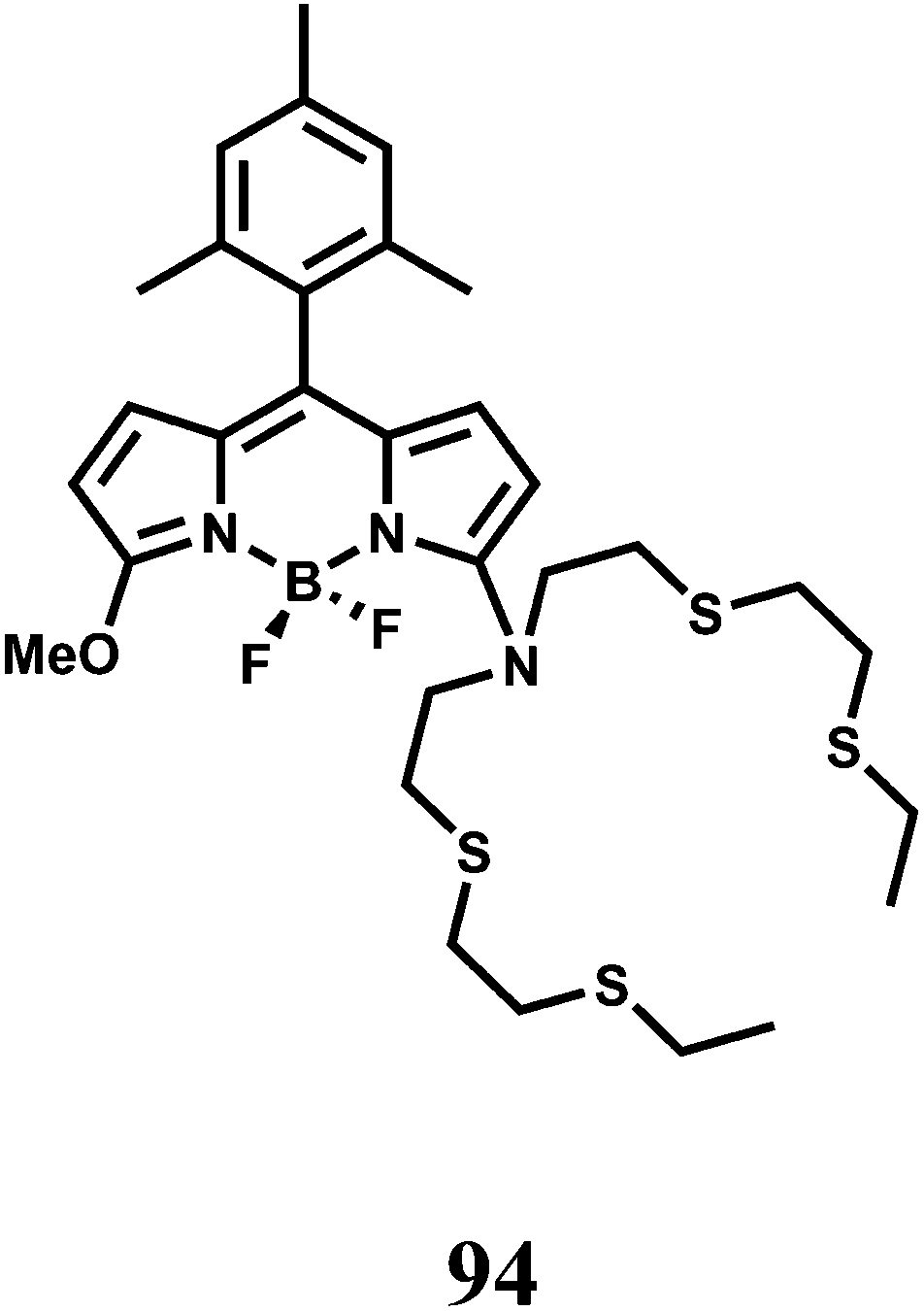
The fluorescent probe 95 has been designed as a new type of targetable fluorescent chemosensor for imaging exchangeable mitochondrial copper pools in living cells.134 The sensor has two distinctive sites: a Cu+-responsive fluorophore and a mitochondrial-targeting triphenylphosphonium moiety for localizing the probe to this organelle. The fluorescence intensity of 95 increases 10-fold with a slight blue shift of the emission maximum to 558 nm with the addition of 1 equiv. of Cu+. The observed dissociation constant (Kd) value for Cu+ complex is found to be 7.2 × 10−12 M. The fluorescence response of the Cu+ bound 95 is not affected by the presence of physiologically relevant concentrations of Ca2+, Mg2+, and Zn2+. Besides, other bioavailable divalent transition metal ions do not induce a change in the emission intensity of 95 or do not interfere with the Cu+ response. This PET sensor is found to be very useful in detecting labile Cu+ in the mitochondria of living cells.
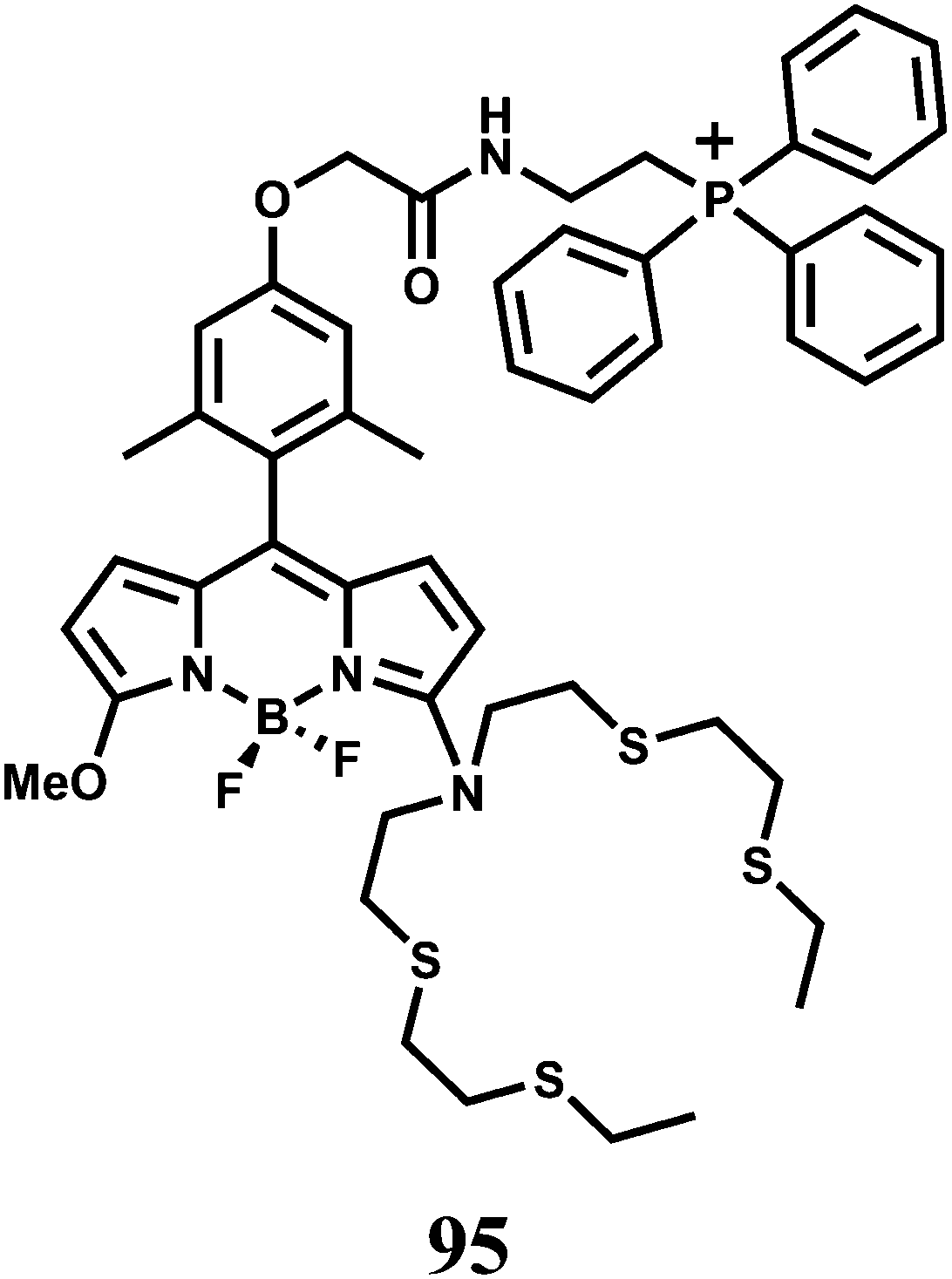
Two-photon fluorescence is a preferable technique for imaging live cells. Two-photon microscopy has a number of advantages over one photon microscopy including increased penetration depth (4500 mm), localized excitation and reduced photodamage and photobleaching effects, thereby allowing tissue imaging for a longer period of time. Besides, no interference from natural amino acid fluorophores occurs which allows for sharp images. The chemosensor 96 has been designed with the same receptor as above for binding a Cu+ ion inside. The Cu+ complex of 96 affords a TPA (two-photon absorption) value of 67 GM at 750 nm.135
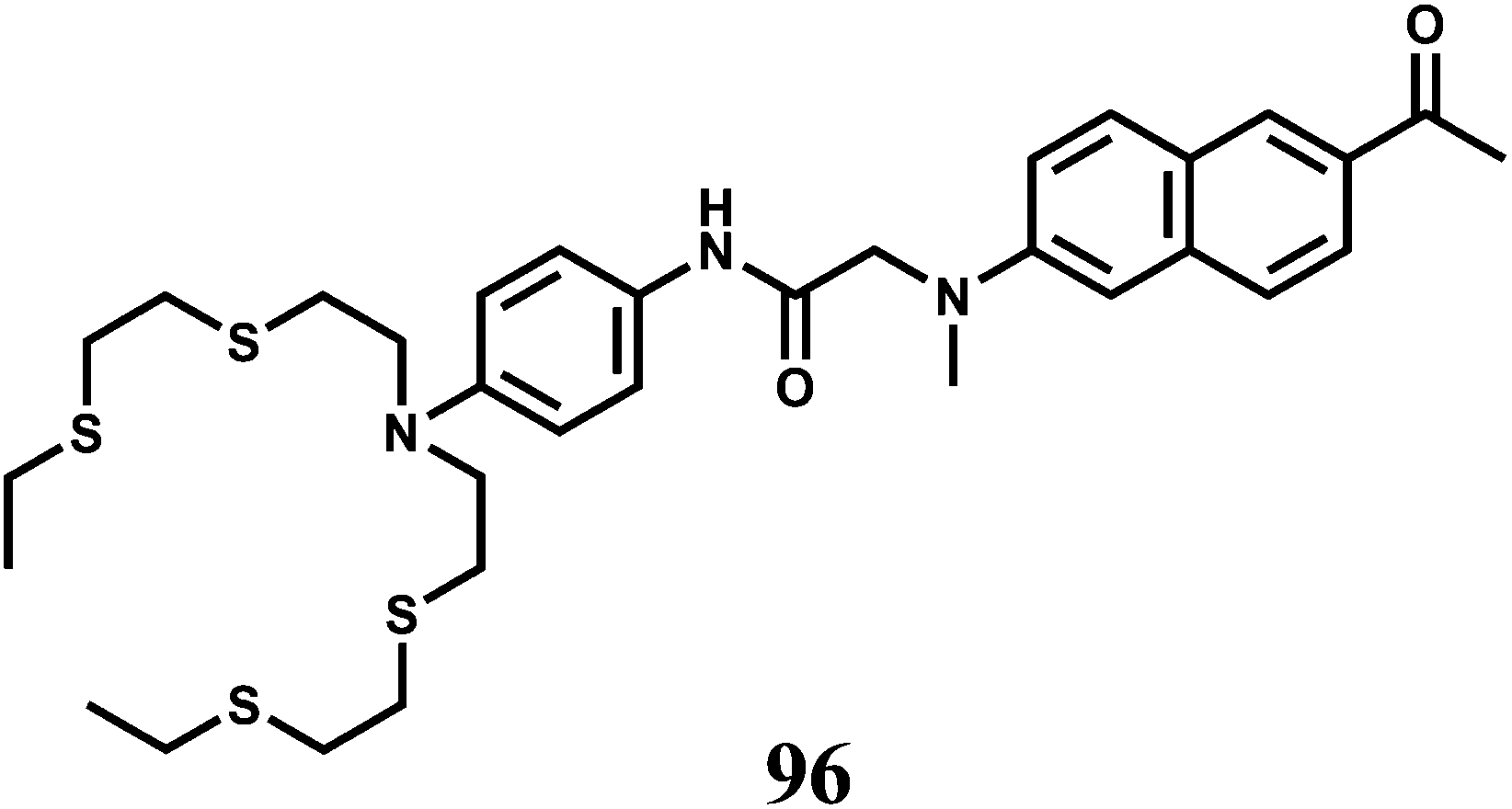
The chemosensor 97 is based on coumarin fluorophore that displays high selectivity for Cu2+ in aqueous medium136 with high emission quantum yield due to blocking of PET from the imine N atom to the fluorophore with association constant Ka = 3.34 × 104 M−1. Its operation in aqueous medium allows for microscopic imaging in LLC-MK2 cells (in vitro) and several living organs (in vivo).
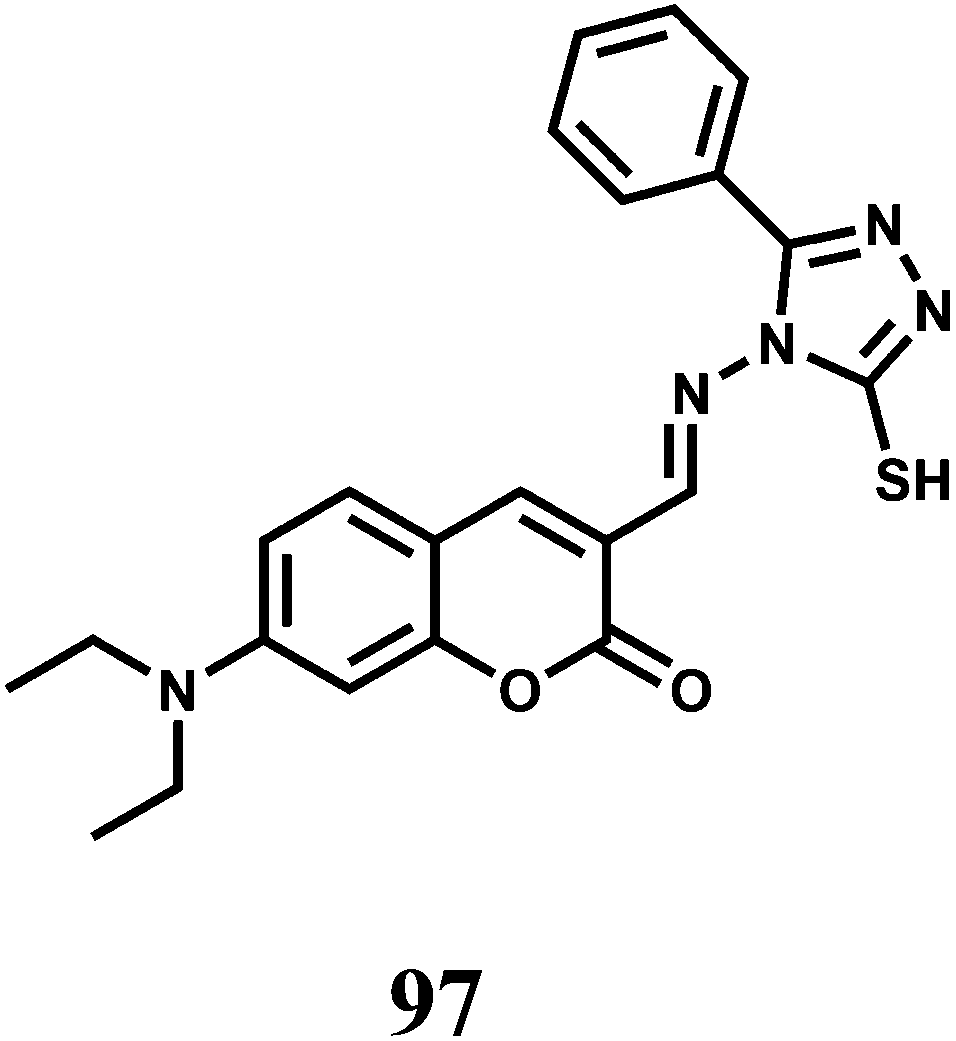
The weakly fluorescent thiosemicarbazone derivative 98 was found to be a selective optical and “turn-on” fluorescent chemodosimeter for Cu2+ ion in aqueous ethanolic medium.137 It is argued that Cu2+ selectivity results from an oxidative cyclization of 98 into the highly fluorescent rigid 4,5-dihydro-5,5-dimethyl-4-(naphthalen-5-yl)-1,2,4-triazole-3-thione (Scheme 51). The chemodosimetric reaction, in combination with selective metal ion-induced catalysis, can be a promising approach to develop selective detection methods for different metal ions.
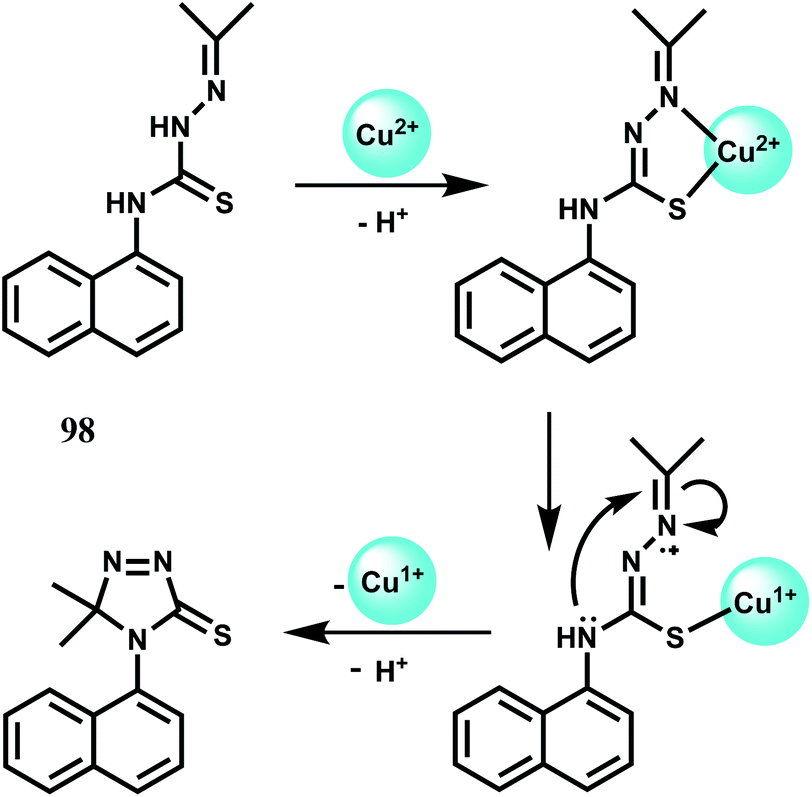 | ||
| Scheme 51 Proposed oxidative cyclization mechanism of 98 by Cu2+. | ||
A coumarin moiety is covalently attached on either side of benzyl dihydrazone to have the sensor 99.138 Upon excitation at 468 nm, the aqueous solution of the metal-free sensor gives an emission at 534 nm with very low quantum yield due to efficient PET from the hydrazone moiety to the coumarin fluorophore association constant (Ka) 3.67 × 106 M−1. Upon addition of Cu2+ it undergoes conformational change as shown (Scheme 52) and at the same time, blocks both the PET pathways to afford a strong fluorescence. This system functions in aqueous medium and therefore, is suitable for use in live cell imaging studies.
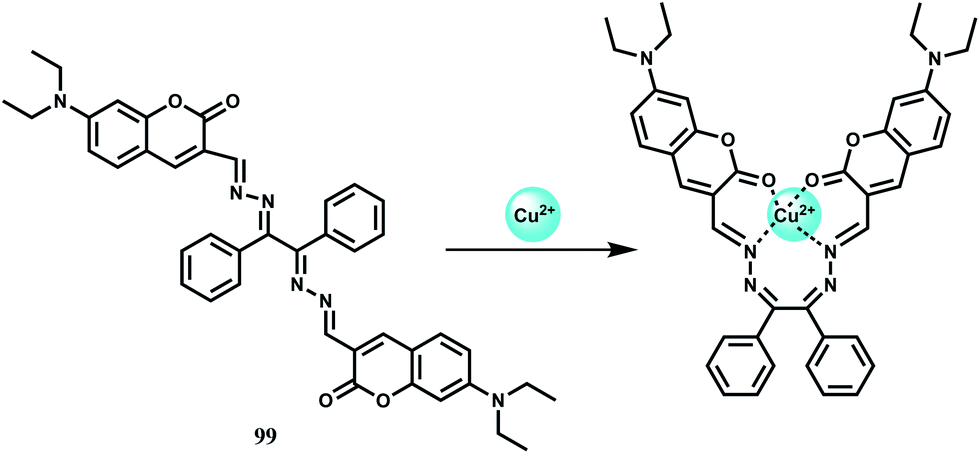 | ||
| Scheme 52 Proposed binding mode of 99 with Cu2+. | ||
In designing the PET sensor 100, a tricarbocyanine dye is used as the NIR fluorophore and 2,2′-azanediyl bis(N-hydroxyacetamide) as the Cu2+ receptor.139 The specificity of Cu2+ binding at the receptor is accomplished due to matching of the cavity of the binding site with radius of Cu2+ ion (Scheme 53). The metal-free system shows very weak fluorescence, however, in presence of Cu2+ ion, the PET is blocked giving high fluorescence at 800 nm in aqueous medium with a dissociation constant of 2.7 ± 0.3 μM in buffered aqueous medium. The emission maximum lies within the biological window and more importantly, aqueous medium can be used both of which make this chemosensor quite important.
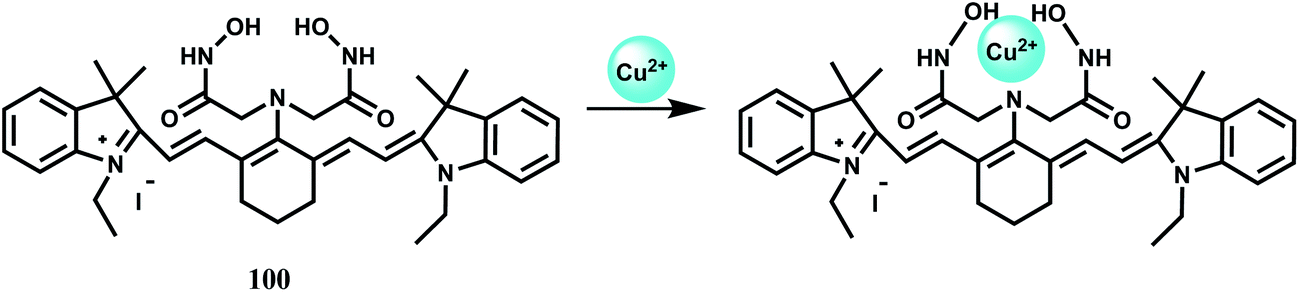 | ||
| Scheme 53 Proposed binding mode of 100 with Cu2+. | ||
The chemosensor 101 obtained by derivatizing the upper rim of a thiacalix[4]arene by a crown ether and the lower rim by two pyrene moieties.140 Upon addition of Cu2+ in aqueous organic medium, two Cu2+ ions are believed to occupy the sites as shown (Scheme 54) with the binding constant >1011. This makes the two pyrene moieties well-separated and offering only monomer emission due to blocking of PET. However, addition of Li+ ion which occupies the crown ether cavity leads to ejection of the Cu2+ ions. This brings the two pyrene moieties closer for π-stacking giving an excimer emission.
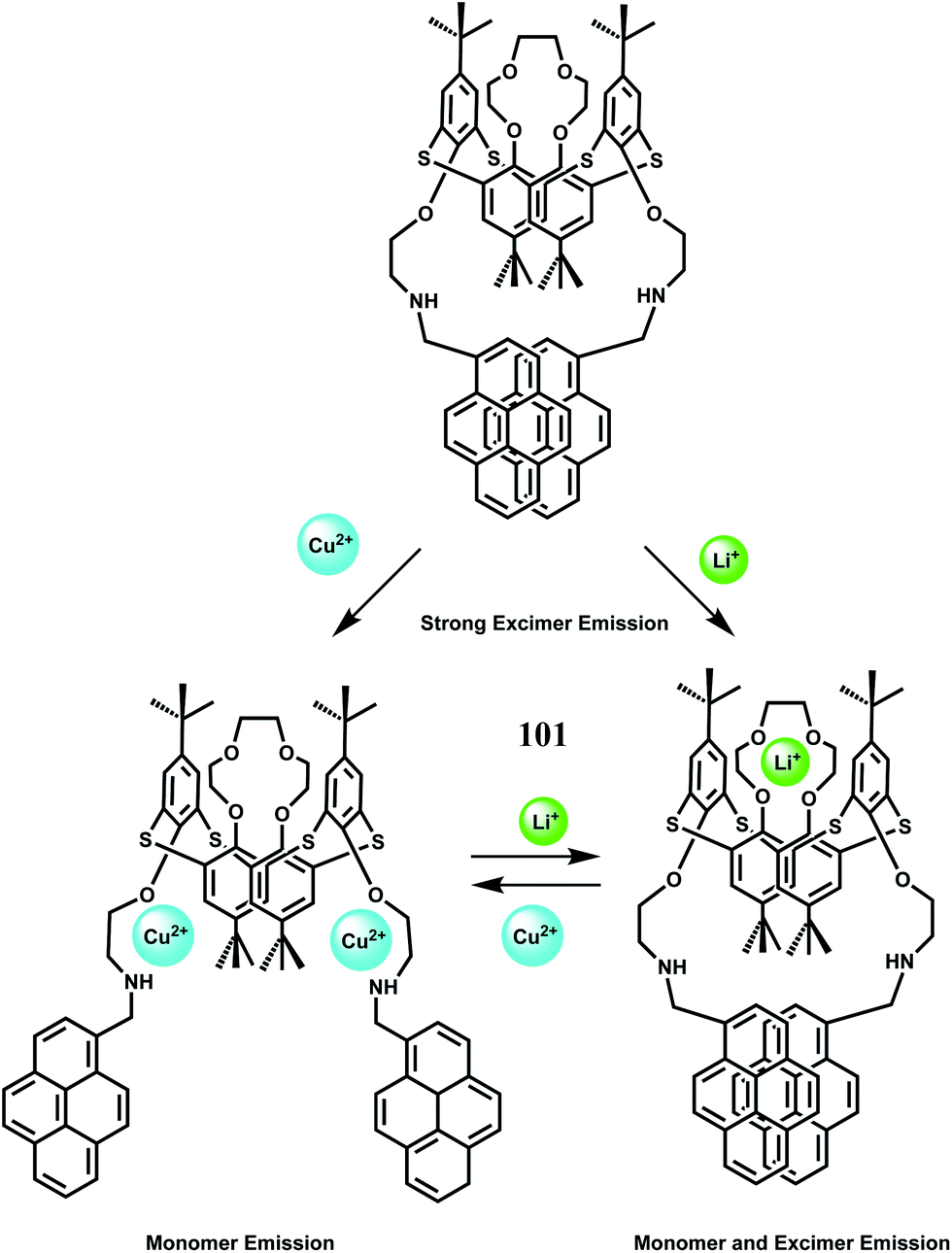 | ||
| Scheme 54 Proposed binding mode of 101. | ||
The electron-withdrawing fluorophore 7-nitrobenz-2-oxa-1,3-diazole (NBD) derived chemosensor 102 binds a Cu2+ ion at the pocket shown (Scheme 55).141 This mode of binding by the Cu2+ ion blocks the PET recovering fluorescence in aqueous MeCN.
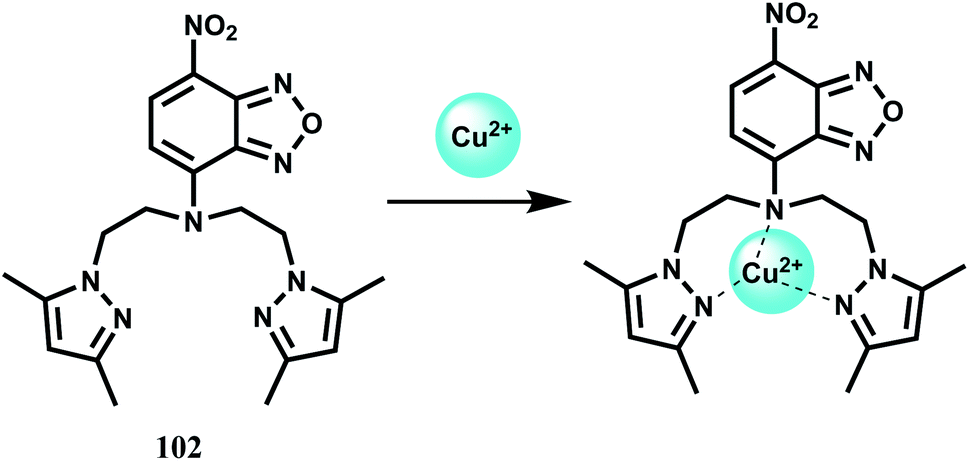 | ||
| Scheme 55 Proposed binding mode of 102. | ||
In an elegant approach, a sensor system that separates the sensing and the signaling events have been designed. Two different fluorogenic ligands 103 and 104 are capable of metal chelation.142 The system functions as illustrated below (Scheme 56).

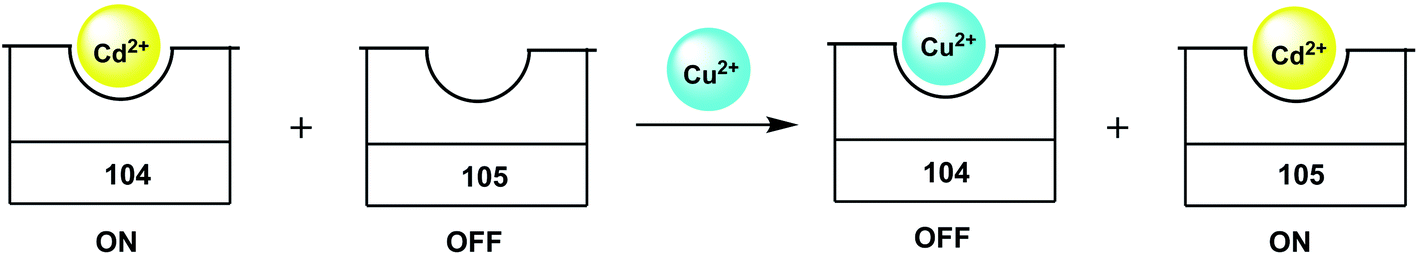 | ||
| Scheme 56 Schematic representation of a ratiometric Cu2+ sensor system. | ||
In presence of 1 equivalent of Cd2+, it preferably chelates with 103 giving strong emission centering at 435 nm due to stoppage of PET. When an equivalent amount of Cu2+ is added it displaces Cd2+ from 103 leading to fluorescence quenching from the paramagnetic ion. However, Cd2+ is now bonded to 104 giving strong emission at 518 nm again due to blocking of PET. The entire operation can be performed in aqueous media in HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer at pH 7.2. This way, detection of Cu2+ is indirect.
The chemosensor 105 acts as an ICT based ratiometric and colourimetric fluorescent chemosensor selective for Cu2+ ion (Scheme 57).143 A Cu2+ ion binds fairly strongly at the site shown with an association constant estimated to be 2.58 × 104 M−1. Upon addition of Cu2+, the metal free absorption band shifts from 328 nm to about 540 nm due to deprotonation as shown with the disappearance of the ICT band centering at 496 nm and the appearance of a new band at 620 nm.
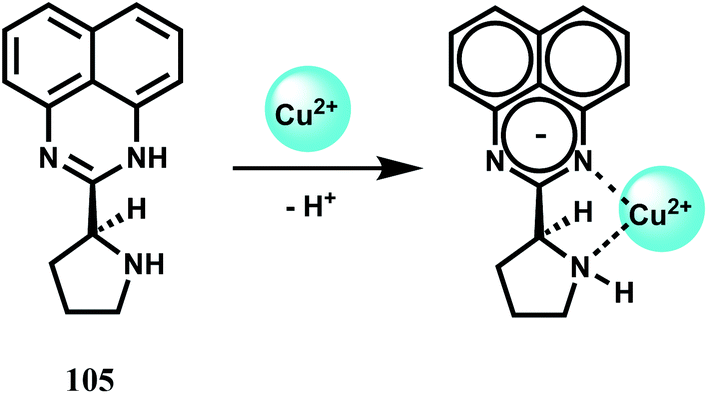 | ||
| Scheme 57 Proposed binding mode of 105 with Cu2+. | ||
The quinoline-indene derived PET chemosensor 106 was found to be sensitive towards Cu2+ ion in 1:1 aqueous MeCN giving a 5-fold enhancement of fluorescence (Scheme 58).144 The receptor cavity binds a Cu2+ ion with an association constant estimated to be (logK) 6.48 ± 0.48.
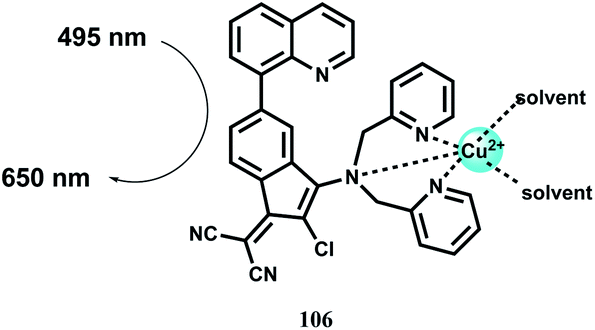 | ||
| Scheme 58 Proposed binding mode of 106 with Cu2+. | ||
Two 4,5-disubstituted-1,8-naphthalimide derivatives 107 and 108 have been synthesized as Cu2+ ion sensors.145 The free sensor 107 displays a broad emission centering at 534 nm in aqueous medium. Upon addition of Cu2+ this band reduces in intensity with the appearance of a new band centered at 478 nm due to complex formation with the probe. Compound 108 on the other hand, quenches emission of the free probe although colour of the solution changes sharply from yellow to pink.
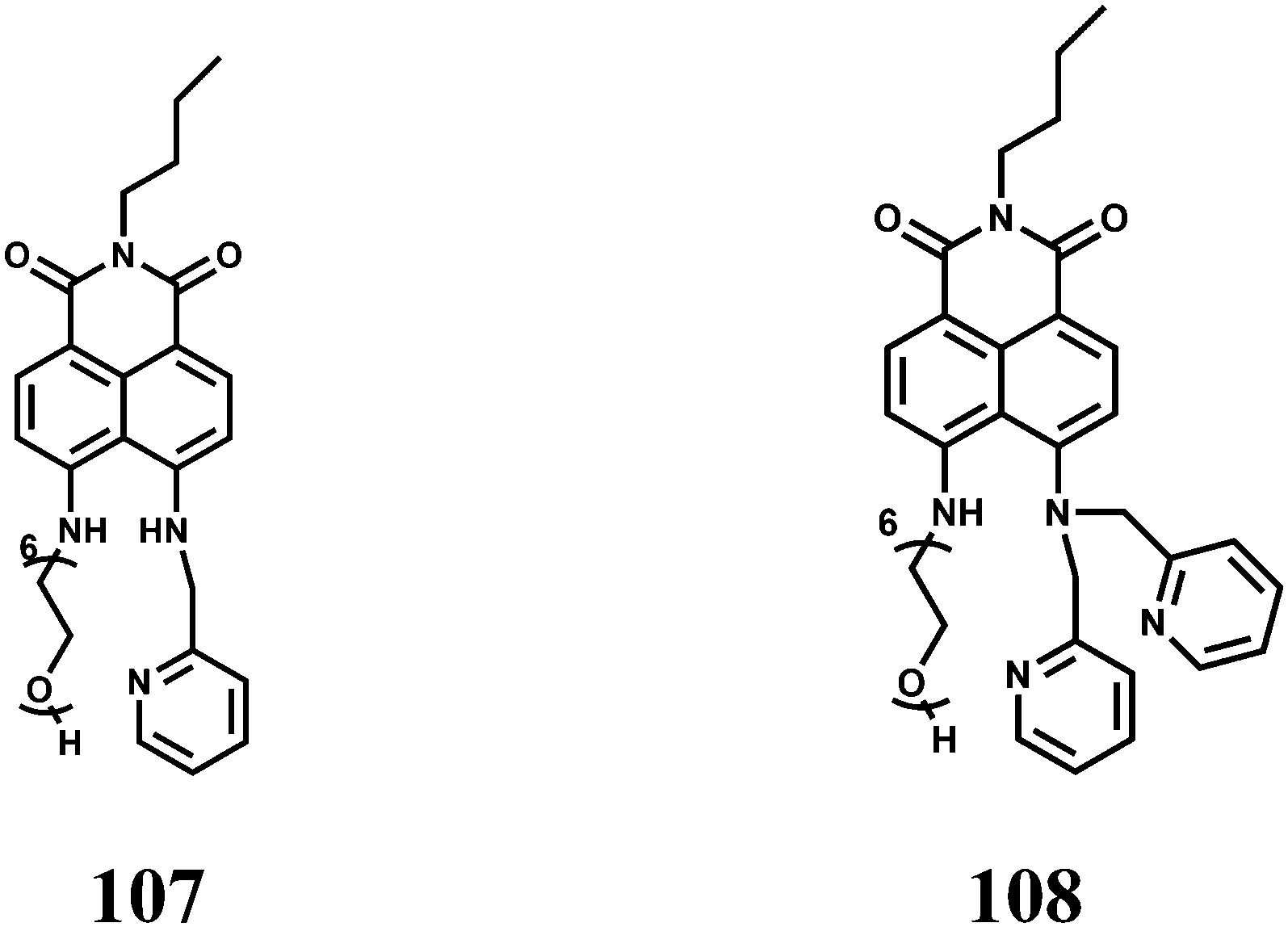
The compound 109 is non-emissive although in presence of Cu2+ ion,146 it undergoes oxidative cyclization (Scheme 59) to form a highly fluorescent benzotriazole. This cyclization reaction can be triggered by μM level concentration of Cu2+ in water at pH = 6–8, at room temperature. The cyclic product exhibits a green emission at 530 nm.
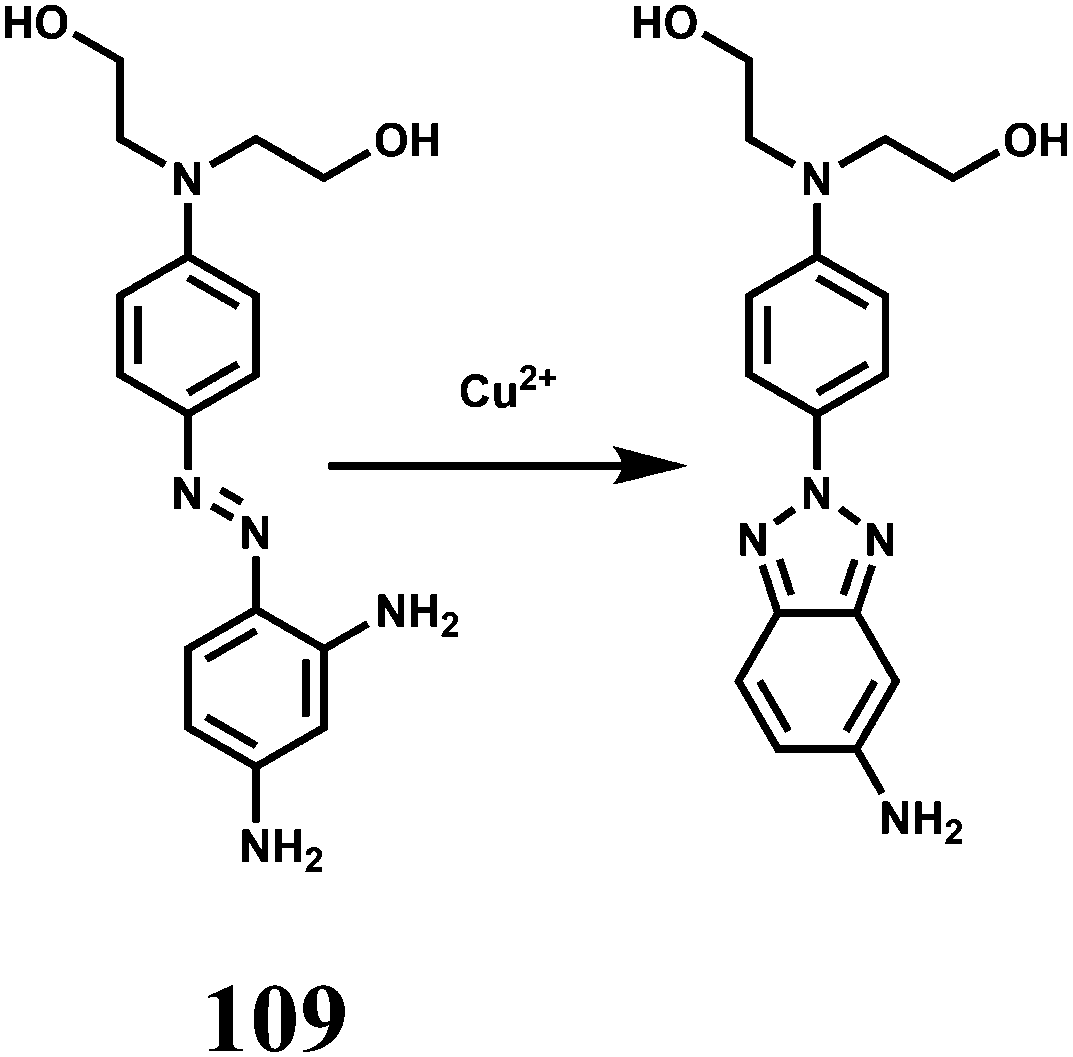 | ||
| Scheme 59 Cu2+ ion induced oxidative cyclization of 109. | ||
The receptor in the fluorescent probe 110 can accommodate a Cu2+ ion with a high association constant of 2.95 × 105 M−1. Once the metal ion occupies the site, the two naphthalimide moieties come closer to each other. Upon addition of 1 equiv. of Cu2+ ion, the two naphthalimide moieties come closer giving an excimer emission at 558 nm. Further addition of another equiv. of Cu2+, triggers a change in the conformation of the chemosensors where the two naphthalimide groups are move far apart from each other giving a naphthalimide monomer emission at 461 nm (Scheme 60).147
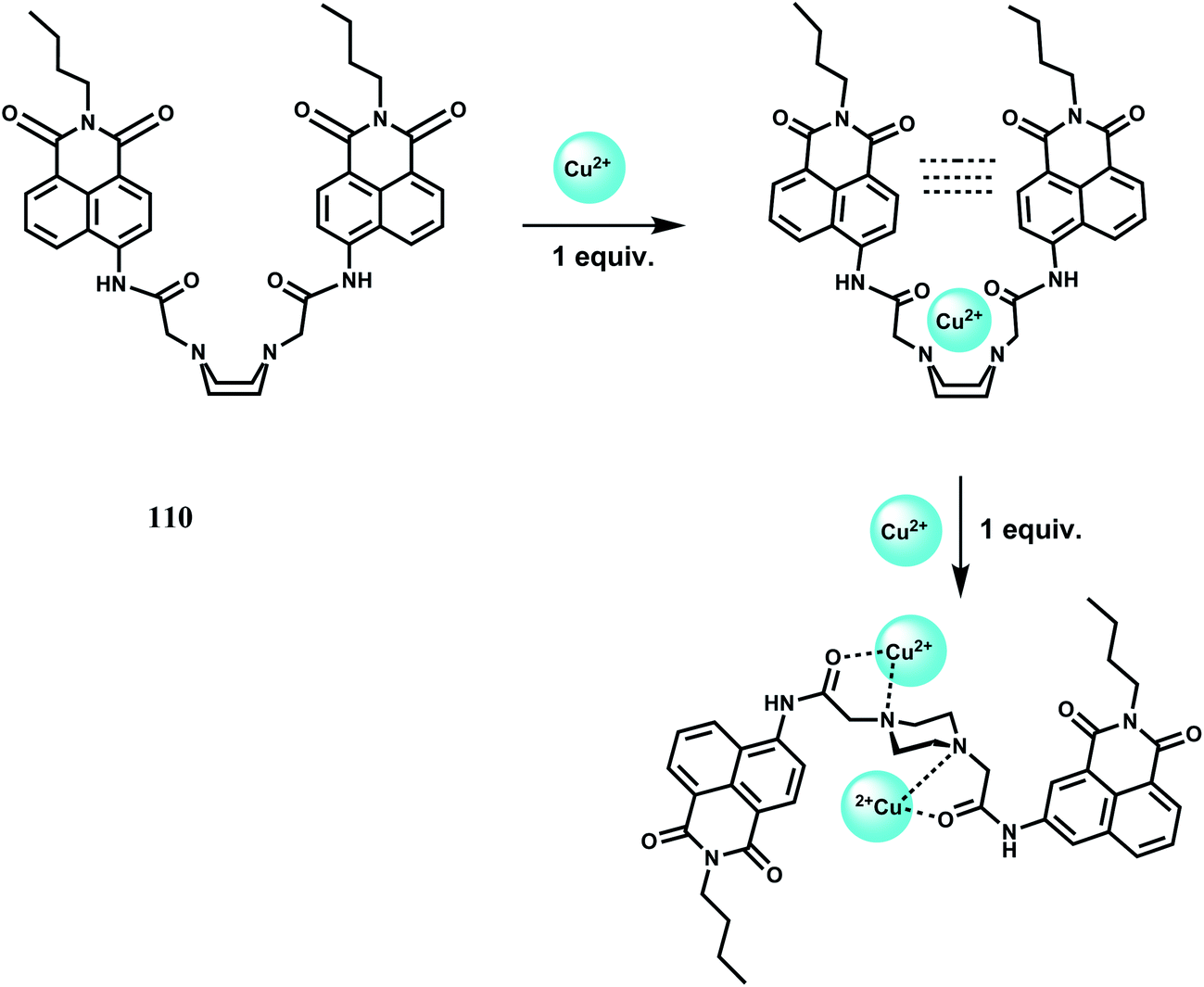 | ||
| Scheme 60 Proposed binding mode of 110 with Cu2+. | ||
The chemosensor 111 has been designed based on the strong tendency of the pyrene moiety to form intermolecular dimers.148 The free sensor exhibits pyrene monomer emission at 388 nm upon excitation in acetonitrile solution. Upon addition of Cu2+ ion, the monomer emission is partially quenched but a strong excimer emission at 460 nm is observed. It is argued that interaction of the quinolinylamide group with the Cu2+ ion induces an intermolecular π–π* interaction which is responsible for the fluorescence changes. With increasing the length between the pyrene and quinolinylamide moieties, excimer formation is hindered.
The thiophene based Schiff base 112 was found to be highly selective fluorescence turn-on PET chemosensor for Cu2+ in aqueous medium.149 It affords a high binding constant of 4.23 × 105 M−1 in aqueous medium with the detection limit of 0.418 nM. The sensor gives a 35-fold enhancement at 430 nm in presence of Cu2+ when excited at 370 nm. In presence of Cr3+ and Sn2+ the enhancement maximum is obtained at 465 nm while other metal ions do not interfere. This probe can be used for studying Cu2+ ion in the living cell by confocal microscopy.
The fluorescent probe 113 was easily synthesized via Schiff base condensation as a chemosensor for Cu2+.150 The free probe does not give any emission when excited at 400 nm in acetonitrile due to an efficient PET. Upon addition of Cu2+ it affords 120-fold fluorescence enhancement at 468 nm as the PET process is blocked. The N and S atoms provide a binding scaffold for recognition of Cu2+ (Scheme 61) with fairly high affinity (Ka = 4.35 × 104 M−1). Other competing metal ions do not interfere. The simplicity and elegance of the probe design should spur further research for water soluble signaling systems for biological applications.
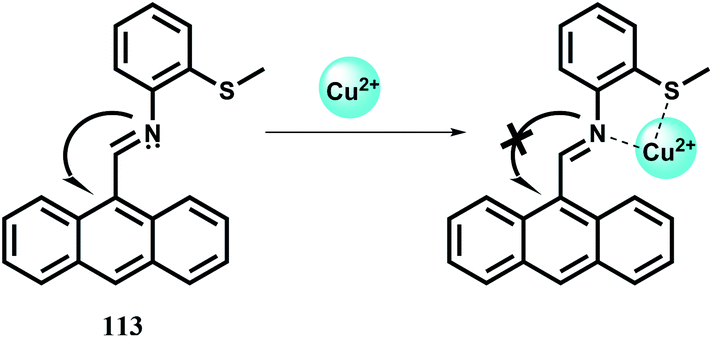 | ||
| Scheme 61 Possible sensing mechanism of 113. | ||
The coumarin derived fluorescent probe 114 is found to be highly selective for Cu+ ion.151 The benzylic ether bond of the probe can be cleaved selectively in presence of Cu+ under a physiological condition to release the coumarin derivative that is highly fluorescent (Scheme 62). Other competing metal ions do not give any fluorescence as they cannot cleave the benzylic ether bond.
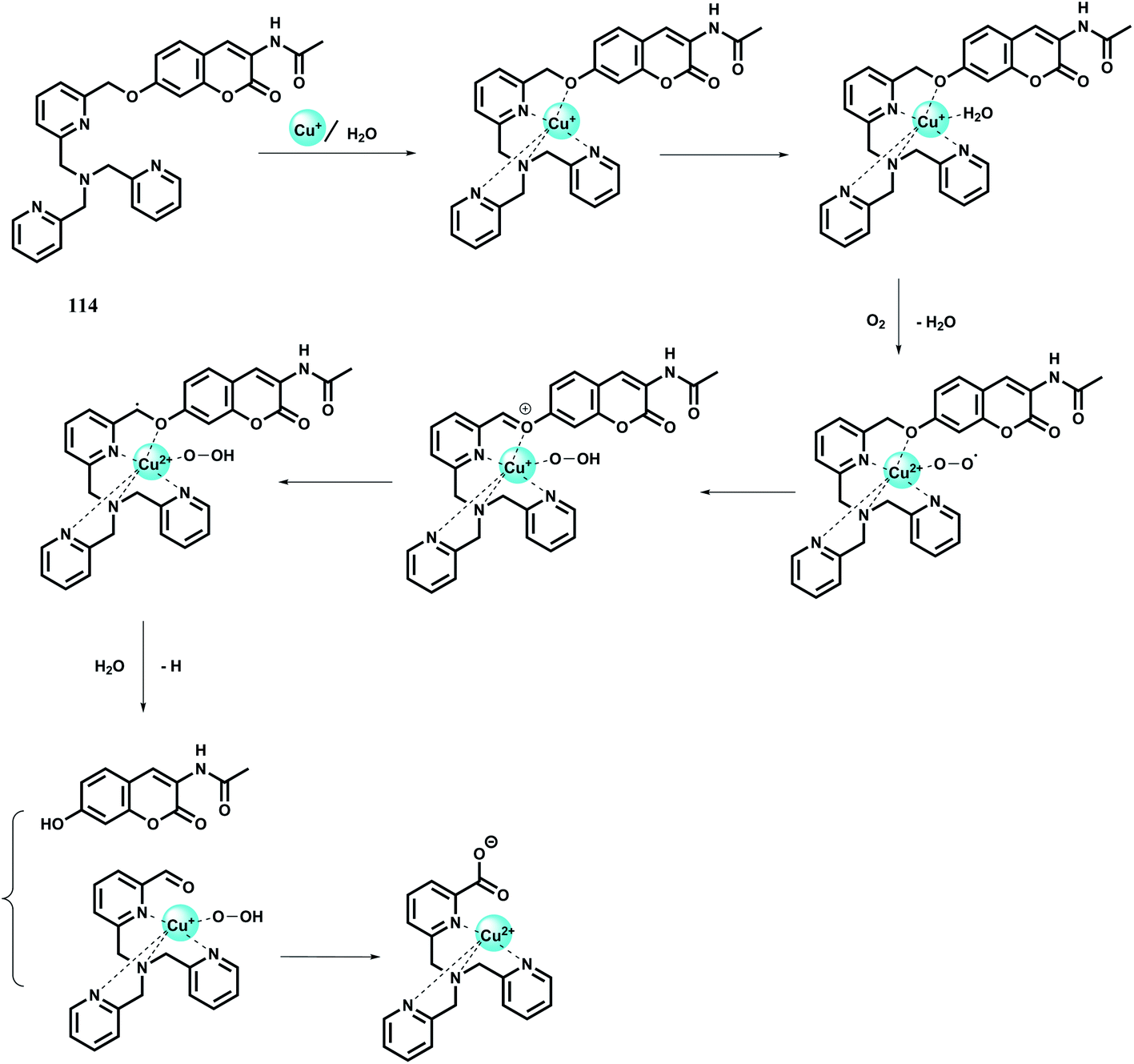 | ||
| Scheme 62 Possible sensing mechanism of 114. | ||
For monitoring real-time copper fluxes in living animals and thus offers an opportunity to examine copper physiology, the chemosensor 115 is designed.152 It consists of a near-infrared emitting cyanine dye with a sulfur-rich receptor to provide a selective and sensitive turn-on response to Cu+. This probe with negligible cytotoxicity is capable of monitoring fluctuations in exchangeable copper stores in living cells and mice under basal conditions, as well as in situations of copper overload or deficiency. This new generation sensor is definitely expected to spur research on sensors for monitoring fluctuations in exchangeable metal ions in biosystems.
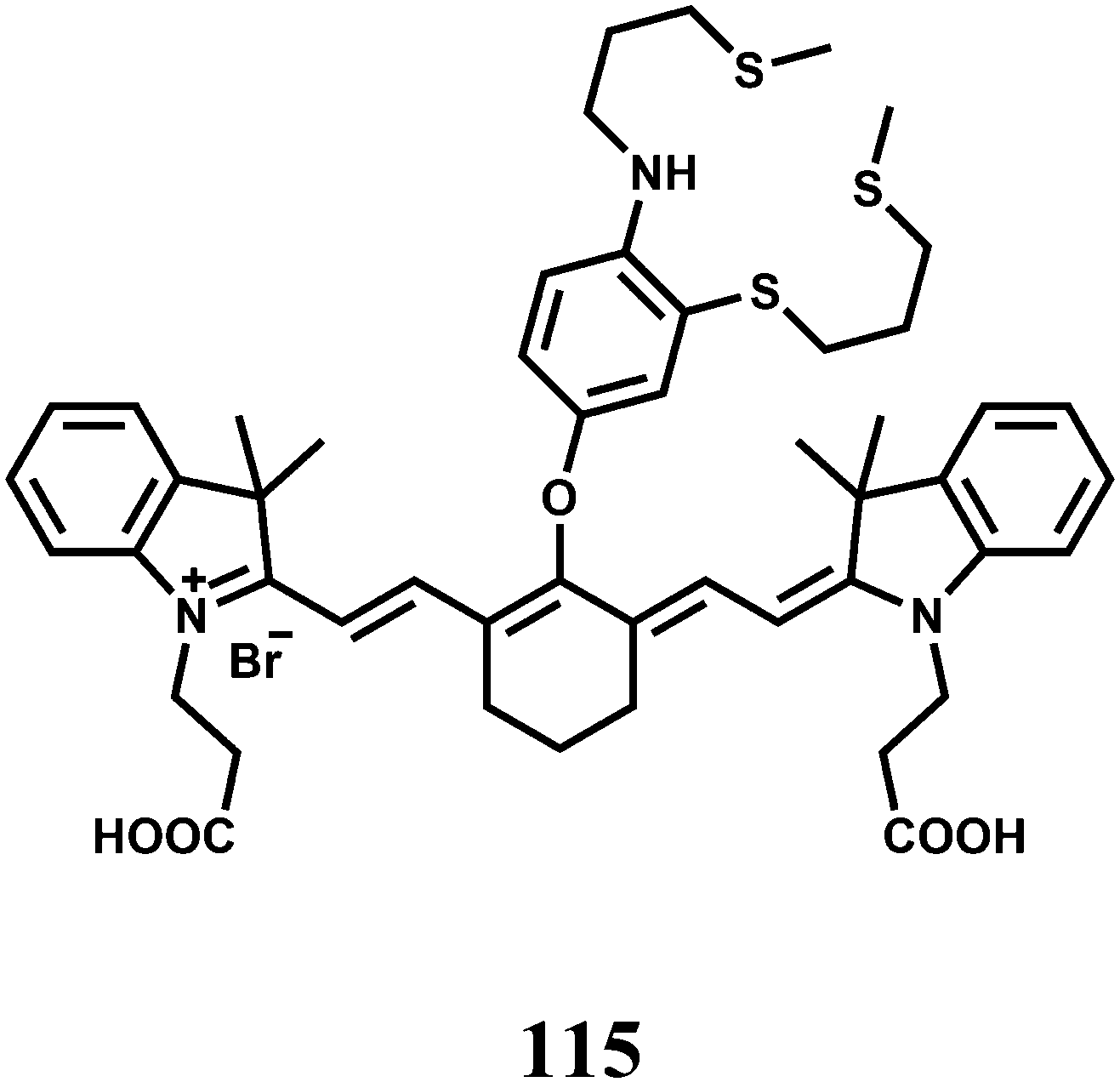
3. Conclusions
It is now clear that several design principles have been adopted for signalling the presence of first-row transition metal ions through turn-on fluorescence. The nature of the fluorophore assumes great significance. It is often found that PET/PCT based sensors often lead to quenching of fluorescence. Of all the designs, rhodamine derived sensors are particularly attractive as they are capable of overcoming fluorescence quenching by paramagnetic transition metal ions. Several key issues still remain in the fluorescence detection of biologically relevant first-row transition metal ions. Most important being aqueous solubility of the probe and a very quick response with high selectivity and sensitivity for temporal distribution with high fidelity of the metals in live cells. Of course, cell viability of the sensor is an important parameter that should always be kept in mind. For live cell imaging studies, systems that show two-photon fluorescence are of crucial importance as they can nullify the background effect of natural fluorophores. Finally, availability of variable oxidations states of transition metals have not been exploited in designing chemosensors. Thus, despite considerable advances, this subject still requires a lot of attention from chemists to purposely build fluorescence signalling probes.Acknowledgements
Financial support for this work is provided by the DST, New Delhi, India and DRDO, New Delhi, India. SP and NC wish to thank CSIR for the senior research fellowship. PKB wishes to thank all his students who made significant contributions in the area of fluorescence signalling and fluorescence based logic gates over the years.Notes and references
- (a) W. Kaim and B. Schwedeski, in Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life, John Wiley and Sons, New York, 1994 Search PubMed; (b) Perspectives on Bioinorganic Chemistry, ed. R. W. Hay, J. R. Dilworth and K. B. Nolan, JAI Press, London, 1991 Search PubMed; (c) Metal Ions in Biological Systems, ed. H. Sigel, Marcel Dekker, New York, 1973 Search PubMed; (d) R. Wever and K. Kustin, Adv. Inorg. Chem., 1990, 53, 81 CrossRef.
- (a) P. S. Dobbin and R. C. Hider, Chem. Br., 1990, 26, 565 CAS; (b) W. Mertz, Science, 1981, 213, 1332 CAS; (c) A. Butler and C. J. Carrano, Coord. Chem. Rev., 1991, 109, 61 CrossRef CAS.
- (a) J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006 Search PubMed; (b) B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3 CrossRef CAS.
- (a) L. Fabbrizzi, M. Licchelli, P. Pallavicini, L. Parodi and A. Taglietti, in Transition Metals in Supramolecular Chemistry, ed. J. P. Sauvage, John Wiley & Sons Ltd., New York, 1999, vol. 5 Search PubMed; (b) L. Fabbrizzi and A. Poggi, Chem. Soc. Rev., 1995, 24, 197 RSC.
- G. Das, P. K. Bharadwaj, M. Basu-Roy and S. Ghosh, Chem. Phys., 2002, 277, 145 CrossRef CAS.
- (a) A. P. de Silva, H. Q. N. Gunaratne and C. P. McCoy, Nature, 1993, 42, 364 Search PubMed; (b) A. P. de Silva, N. D. McClenaghan and C. P. McCoy, in Electron Transfer in Chemistry, ed. V. balzani, Wiley-VCH, Weinheim, 2001, vol. 5 Search PubMed; (c) A. P. de Silva and S. Uchiyama, Top. Curr. Chem., 2011, 300, 1 CrossRef.
- (a) Z. Xu, J. Yoon and D. R. Spring, Chem. Soc. Rev., 2010, 39, 1996 RSC; (b) E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517 CrossRef CAS PubMed; (c) E. Tomat and S. J. Lippard, Curr. Opin. Chem. Biol., 2010, 14, 225 CrossRef CAS PubMed; (d) E. M. Nolan and S. J. Lippard, Acc. Chem. Res., 2009, 42, 193 CrossRef CAS PubMed; (e) K. Kikuchi, K. Komatsu and T. Nagano, Curr. Opin. Chem. Biol., 2004, 8, 182 CrossRef CAS PubMed; (f) P. Jiang and Z. Guo, Coord. Chem. Rev., 2004, 248, 205 CrossRef CAS PubMed.
- (a) Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568 RSC; (b) H. N. Kim, Z. Guo, W. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79 RSC; (c) J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483 RSC; (d) D. T. Quang and J. S. Kim, Chem. Rev., 2010, 110, 6280 CrossRef CAS PubMed; (e) Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS PubMed; (f) M. Formica, V. Fusi, L. Giorgi and M. Micheloni, Coord. Chem. Rev., 2010, 256, 170 CrossRef PubMed; (g) A. P. de Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385 RSC; (h) X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590 CrossRef CAS PubMed; (i) K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564 CrossRef CAS PubMed; (j) P. K. Bharadwaj, Prog. Inorg. Chem., 2003, 51, 251 CAS.
- (a) S. Liu, J. P. C. Pestano and C. Wolf, J. Org. Chem., 2008, 73, 4267 CrossRef CAS PubMed; (b) D. P. Iwaniuk, K. Yearick-Spangler and C. Wolf, J. Org. Chem., 2012, 77, 5203 CrossRef CAS PubMed.
- J. Yuasa and S. Fukuzumi, J. Am. Chem. Soc., 2006, 128, 15976 CrossRef CAS PubMed.
- K. H. Thompson, J. H. McNeill and C. Orvig, Chem. Rev., 1999, 99, 2561 CrossRef CAS PubMed.
- (a) D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, Chem. Rev, 2004, 104, 849 CrossRef CAS PubMed; (b) R. L. VanEtten, P. P. Waymack and D. M. Rehkop, J. Am. Chem. Soc., 1974, 96, 6782 CrossRef CAS; (c) W. Plass, Angew. Chem., Int. Ed., 1999, 38, 909 CrossRef CAS; (d) D. R. Davies and W. G. J. Hol, FEBS Lett., 2004, 577, 315 CrossRef CAS PubMed.
- H. Vilter, Phytochemistry, 1984, 23, 1387 CrossRef CAS.
- A. Butler and J. N. Carter-Franklin, Nat. Prod. Rep., 2004, 21, 180 RSC.
- M. Mracova, D. Jirova, H. Janci and J. Lener, Sci. Total Environ., 1993, Part 1, 16 Search PubMed.
- F.-J. Huo, J. Su, Y.-Q. Sun, C.-X. Yin, H.-B. Tong and Z.-X. Nie, Dyes Pigm., 2010, 86, 50 CrossRef CAS PubMed.
- M. J. R. Rama, A. R. Medina and A. M. Díaz, Talanta, 2005, 66, 1333 CrossRef PubMed.
- A. M. García Campaña, F. Alés Barrero and M. Román Ceba, Anal. Sci., 1996, 12, 647 CrossRef.
- Y. Zhou, J. Y. Jung, H. R. Jeon, Y. Kim, S.-J. Kim and J. Yoon, Org. Lett., 2011, 13, 2742 CrossRef CAS PubMed.
- X. Zhang and W.-D. Woggon, J. Am. Chem. Soc., 2005, 127, 14138 CrossRef CAS PubMed.
- (a) Z. Xu, N. J. Singh, J. Lim, J. Pan, H. N. Kim, S. Park, K. S. Kim and J. Yoon, J. Am. Chem. Soc., 2009, 131, 15528 CrossRef CAS PubMed; (b) F. Wang, R. Nandhakumar, J. H. Moon, K. M. Kim, J. Y. Lee and J. Yoon, Inorg. Chem., 2011, 50, 2240 CrossRef CAS PubMed.
- (a) W. Mertz and K. Schwarz, Arch. Biochem. Biophys., 1955, 58, 504 CrossRef CAS; (b) H. Arakawa, J. Biol. Chem., 2000, 275, 10150 CrossRef CAS PubMed; (c) R. Anderson and R. A. Chromium, Trace Elements in Human and Animal Nutrition, Academic Press, New York, 1987 Search PubMed.
- J. B. Vincent, Nutr. Rev., 2000, 58, 67 CrossRef CAS PubMed.
- (a) C. Núñez, R. Bastida, A. Macías, E. Bértolo, L. Fernandes, J. L. Capelo and C. Lodeiro, Tetrahedron, 2009, 65, 6179 CrossRef PubMed; (b) M. J. E. Resendiz, J. C. Noveron, H. Disteldorf, S. Fischer and P. J. Stang, Org. Lett., 2004, 6, 651 CrossRef CAS PubMed.
- (a) M. Sarkar, S. Banthia and A. Samanta, Tetrahedron Lett., 2006, 47, 7575 CrossRef CAS PubMed; (b) J. Mao, L. N. Wang, W. Dou, X. L. Tang, Y. Yan and W. S. Liu, Org. Lett., 2007, 9, 3188 CrossRef PubMed.
- K. Huang, H. Yang, Z. Zhou, M. Yu, F. Li, X. Gao, T. Yi and C. Huang, Org. Lett., 2008, 10, 2557 CrossRef CAS PubMed.
- J. Mao, L. Wang, W. Dou, X. Tang, Y. Yan and W. Liu, Org. Lett., 2007, 9, 4567 CrossRef CAS PubMed.
- M. Li, D. Zhang, Y. Liu, P. Ding, Y. Ye and Y. Zhao, J. Fluoresc., 2014, 24, 119 CrossRef CAS PubMed.
- Y. Zhou, J. Zhang, L. Zhang, Q. Zhang, T. Ma and J. Niu, Dyes Pigm., 2013, 148, 97 Search PubMed.
- A. J. Weerasinghe, C. Schmiesing and E. Sinn, Tetrahedron Lett., 2009, 50, 6407 CrossRef CAS PubMed.
- P. Mahato, S. Saha, E. Suresh, R. D. Liddo, P. P. Parnigotto, M. T. Conconi, M. K. Kesharwani, B. Ganguly and A. Das, Inorg. Chem., 2012, 51, 1769 CrossRef CAS PubMed.
- Z. Zhou, M. Yu, H. Yang, K. Huang, F. Li, T. Yi and C. Huang, Chem. Commun., 2008, 3387 RSC.
- R. H. Lipson, Y. J. Shi, and D. Lacey, in An Introduction to Laser Spectroscopy, ed. D. L. Andrews and A. A. Demidov, Kluwer Academic/Plenum Publishers, New York, 2nd edn, 2002, pp. 257–309 Search PubMed.
- Z. Xiaolin, X. Yi and Q. Xuhong, Angew. Chem., Int. Ed., 2008, 47, 8025 CrossRef PubMed.
- Y. Wan, Q. Guo, X. Wang and A. Xia, Anal. Chim. Acta, 2010, 665, 215 CrossRef CAS PubMed.
- M. Sarkar, S. Banthia and A. Samanta, Tetrahedron Lett., 2006, 47, 7575 CrossRef CAS PubMed.
- J.-N. Wang, Q. Qi, L. Zhang and S.-H. Li, Inorg. Chem., 2012, 51, 13103 CrossRef CAS PubMed.
- H. Wu, P. Zhou, J. Wang, L. Zhao and C. Duan, New J. Chem., 2009, 33, 653 RSC.
- D. Wang, Y. Shiraishi and T. Hirai, Tetrahedron Lett., 2010, 51, 2545 CrossRef CAS PubMed.
- X. Hu, X. Zhang, G. He, C. He and C. Duan, Tettrahedron, 2011, 67, 1091 CAS.
- A. E. Albers, V. S. Okreglak and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 9640 CrossRef CAS PubMed.
- S. Panda, P. B. Pati and S. S. Zade, Chem. Commun., 2011, 47, 4174 RSC.
- V. Bravo, S. Gil, A. M. Costero, M. N. Kneeteman, U. Llaosa, P. M. E. Mancini, L. E. Ochando and M. Parra, Tetrahedron, 2012, 68, 4882 CrossRef CAS PubMed.
- S. Wu, K. Zhang, Y. Wang, D. Mao, X. Liu, J. Yu and L. Wang, Tetrahedron Lett., 2014, 55, 351 CrossRef CAS PubMed.
- L.-J. Ma, W. Cao, J. Liu, M. Zhang and L. Yang, Sens. Actuators, B, 2013, 181, 782 CrossRef CAS PubMed.
- S. Goswami, A. K. Das, A. K. Maity, A. Manna, K. Aich, S. Maity, P. Sahab and T. K. Mandal, Dalton Trans., 2014, 43, 231 RSC.
- S. Guha, S. Lohar, A. Banerjee, A. Sahana, I. Hauli, S. K. Mukherjee, J. S. Matalobos and D. Das, Talanta, 2012, 91, 18 CrossRef CAS PubMed.
- J. Emsley, in Nature's Building Blocks: An A-Z Guide to the Elements, Oxford University Press, Oxford, 2001, pp. 249–253 Search PubMed.
- J. H. Lee and A. P. Koretsky, Curr. Pharm. Biotechnol., 2004, 5, 529 CAS.
- J. A. Roth, Biol. Res., 2006, 39, 45 CAS.
- Xi. Mao, H. Su, D. Tian, H. Li and R. Yang, ACS Appl. Mater. Interfaces, 2013, 5, 592 CAS.
- J. Liang and J. W. Canary, Angew. Chem., Int. Ed., 2010, 49, 7710 CrossRef CAS PubMed.
- Z. D. Dai, K. N. Khosla and J. W. Canary, Supramol. Chem., 2009, 21, 296 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581 CrossRef CAS.
- F. Gruppi, J. Liang, B. B. Bartelle, M. Royzen, D. H. Turnbullb and J. W. Canary, Chem. Commun., 2012, 48, 10778 RSC.
- K. Dutta, R. C. Deka and D. K. Das, J. Fluoresc., 2013, 23, 1173 CrossRef CAS PubMed.
- (a) R. Baliga, N. Ueda and S. V. Shah, Biochem. J., 1993, 291, 901 CAS; (b) G. Balla, G. M. Vercellotti, J. W. Eaton and H. S. Jacob, J. Lab. Clin. Med., 1990, 116, 546 CAS; (c) K. Ŏllinger and K. Roberg, J. Biol. Chem., 1997, 272, 23707 CrossRef PubMed; (d) O. Sergent, I. Morel, P. Cogrel, M. Chevanne, N. Pasdeloup, P. Brissot, G. Lescoat, P. Cillard and J. Cillard, Biol. Trace Elem. Res., 1995, 47, 185 CrossRef CAS PubMed; (e) A. Voogd, W. Sluiter, H. G. van Eijk and J. F. Koster, J. Clin. Invest., 1992, 90, 2050 CrossRef CAS PubMed.
- J. Fandrey, S. Frede, W. Ehleben, T. Porwol, H. Acker and W. Jelkmann, Kidney Int., 1997, 51, 492 CrossRef CAS.
- (a) W. Breuer, S. Epsztejn and Z. I. Cabantchik, J. Biol. Chem., 1995, 270, 24209 CrossRef CAS PubMed; (b) S. Epsztejn, O. Kakhlon, H. Glickstein, W. Breuer and Z. I. Cabantchik, Anal. Biochem., 1997, 248, 31 CrossRef CAS; (c) S. P. Young and P. Aisen, in The Liver: Biology and Pathobiology, ed. I. M. Arias, J. L. Boyler and N. Fausto, Raven Press Ltd., New York, 3rd edn, 1994, pp. 597–617 Search PubMed.
- (a) M. B. Delatycki, R. Williamson and S. M. Forrest, J. Med. Genet., 2000, 37, 1 CrossRef CAS; (b) H. Puccio and M. Koenig, Hum. Mol. Genet., 2000, 9, 887 CrossRef CAS PubMed; (c) M. F. Beal, Biochim. Biophys. Acta, 1998, 1366, 211 CrossRef CAS.
- M. Zhang, Y. Gao, M. Li, M. Yu, F. Li, L. Li, M. Zhu, J. Zhang, T. Yia and C. Huang, Tetrahedron Lett., 2007, 48, 3709 CrossRef CAS PubMed.
- S. Bae and J. Tae, Tetrahedron Lett., 2007, 48, 5389 CrossRef CAS PubMed.
- M. H. Lee, T. V. Giap, S. H. Kim, Y. H. Lee, C. Kang and J. S. Kim, Chem. Commun., 2010, 46, 1407 RSC.
- Z.-Q. Hu, X.-M. Wang, Y.-C. Feng, L. Ding, M. Li and C.-S. Lin, Chem. Commun., 2011, 47, 1622 RSC.
- L. Huang, F. Hou, J. Cheng, P. Xi, F. Chen, D. Baib and Z. Zeng, Org. Biomol. Chem., 2012, 10, 9634 CAS.
- S. Goswami, S. Das, K. Aich, D. Sarkar, T. K. Mondal, C. K. Quah and H. K. Fun, Dalton Trans., 2013, 42, 15113 RSC.
- S. Ji, X. Meng, W. Ye, Y. Feng, H. Sheng, Y. Cai, J. Liu, X. Zhu and Q. Guo, Dalton Trans., 2014, 43, 1583 RSC.
- H. Sheng, X. Meng, W. Ye, Y. Feng, H. Sheng, X. Wang and Q. Guo, Sens. Actuators, B, 2014, 195, 534 CrossRef CAS PubMed.
- G. Sivaraman, V. Sathiyaraja and D. Chellappa, J. Lumin., 2014, 145, 480 CrossRef CAS PubMed.
- J. Arden-Jacob, K.-H. Drexhage, R. Herrmann and H.-P. Josel, Pentacyclic compounds and their use as absorption or fluorescent dyes, US Pat., 5 750 409, 1998.
- J. Wang, D. Zhang, Y. Liu, P. Ding, C. Wang, Y. Ye and Y. Zhao, Sens. Actuators, B, 2014, 191, 344 CrossRef CAS PubMed.
- P. Xie, F. Guo, R. Xia, Y. Wang, D. Yao, G. Yang and L. Xie, J. Lumin., 2014, 145, 849 CrossRef CAS PubMed.
- L. Tolosa, K. Nowaczyk and J. Lakowicz, in An Introduction to Laser Spectroscopy, ed. D. L. Andrews and A. A. Demidov, Kluwer Academic/Plenum Publishers, New York, 2nd edn, 2002 Search PubMed.
- S. Wang, X. Meng and M. Zhu, Tetrahedron Lett., 2011, 52, 2840 CrossRef CAS PubMed.
- F. Ge, H. Ye, H. Zhang and B.-X. Zhao, Dyes Pigm., 2013, 99, 661 CrossRef CAS PubMed.
- B. Wang, J. Hai, Z. Liu, Q. Wang, Z. Yang and S. Sun, Angew. Chem., Int. Ed., 2010, 49, 4576 CrossRef CAS PubMed.
- Y. Xiang and A. Tong, Org. Lett., 2006, 8, 1549 CrossRef CAS PubMed.
- A. J. Weerasinghe, F. A. Abebe and E. Sinn, Tetrahedron Lett., 2011, 52, 5648 CrossRef CAS PubMed.
- X. Chen, H. Hong, R. Han, D. Zhang, Y. Ye and Y.-F. Zhao, J. Fluoresc., 2012, 22, 789 CrossRef CAS PubMed.
- C.-Y. Li, C.-X. Zou, Y.-F. Li, J.-L. Tang and C. Weng, Dyes Pigm., 2014, 104, 110 CrossRef CAS PubMed.
- J. L. Bricks, A. Kovalchuk, C. Trieflinger, M. Nofz, M. Büschel, A. I. Tolmachev, J. Daub and K. Rurack, J. Am. Chem. Soc., 2005, 127, 13522 CrossRef CAS PubMed.
- H. Wang, D. Wang, Q. Wang, X. Li and C. A. Schalley, Org. Biomol. Chem., 2010, 8, 1017 CAS.
- (a) M. R. Wasielewski, J. Org. Chem., 2006, 71, 5051 CrossRef CAS PubMed; (b) J. A. A. W. Elemans, R. van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251 CrossRef CAS; (c) H. Langhals, Helv. Chim. Acta, 2005, 88, 1309 CrossRef CAS; (d) F. Würthner, Chem. Commun., 2004, 1564 RSC.
- W. Lin, L. Yuan, J. Feng and X. Cao, Eur. J. Org. Chem., 2008, 2689 CrossRef CAS.
- (a) A. S. Chauvin, Y. M. Frapart, J. Vaissermann, B. Donnadieu, J. P. Tuchagues, J. C. Chottard and Y. Li, Inorg. Chem., 2003, 42, 1895 CrossRef CAS PubMed; (b) H. Ohta, Y. Sunatsuki, M. Kojima, Y. Ikuta, Y. Goto, N. Matsumoto, S. Iijima, H. Akashi, S. Kaizaki, F. Dahan and J.-P. Tuchagues, Inorg. Chem., 2004, 43, 4154 CrossRef PubMed; (c) L. L. Bénisvy, S. Halut, B. Donnadieu, J.-P. Tuchagues, J.-C. Chottard and Y. Li, Inorg. Chem., 2006, 45, 2403 CrossRef PubMed.
- H. J. Jung, N. Singh and D. O. Jang, Tetrahedron Lett., 2008, 49, 2960 CrossRef CAS PubMed.
- N. C. Lim, M. D. Morton, H. A. Jenkins and C. Brückner, J. Org. Chem., 2003, 68, 9233 CrossRef CAS PubMed.
- N. C. Lim, S. V. Pavlova and C. Brückner, Inorg. Chem., 2009, 48, 1173 CrossRef CAS PubMed.
- Z. Li, Y. Zhou, K. Yin, Z. Yu, Y. Li and J. Ren, Dyes Pigm., 2014, 105, 7 CrossRef CAS PubMed.
- K. Ghosh, S. Rathi and R. Kushwaha, Tetrahedron Lett., 2013, 54, 6460 CrossRef CAS PubMed.
- P. K. Chung, S.-R. Liu, H.-F. Wang and S.-P. Wu, J. Fluoresc., 2013, 23, 1139 CrossRef CAS PubMed.
- Y. Chen, L. Wan, X. Yu, W. Li, Y. Bian and J. Jiang, Org. Lett., 2011, 13, 5774 CrossRef CAS PubMed.
- (a) C. Little, S. E. Aakre, M. G. Rumsby and K. Gwarsha, Biochem. J., 1982, 207, 117 CAS; (b) M. Dennis and P. E. Kolattukudy, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5306 CrossRef CAS; (c) W. Maret and B. L. Vallee, Methods Enzymol., 1993, 226, 52 CAS; (d) K. W. Walker and R. A. Bradshaw, Protein Sci., 1998, 7, 2684 CrossRef CAS PubMed.
- (a) A. Leonard and R. Lauwerys, Mutat. Res., 1990, 239, 17 CrossRef CAS; (b) A. I. Seldén, C. Norberg, C. Karlson-Stiber and E. Hellström-Lindberg, Environ. Toxicol. Pharmacol., 2007, 23, 129 CrossRef PubMed.
- H. Irving and R. J. P. Williams, J. Chem. Soc., 1953, 3192 RSC.
- H. Y. Au-Yeung, E. J. New and C. J. Chang, Chem. Commun., 2012, 48, 5268 RSC.
- Q. Maa, Q.-E. Cao, Y. Zhao, S. Wu, Z. Hu and Q. Xu, Food Chem., 2000, 71, 123 CrossRef.
- (a) O. Warburg, Hoppe-Seyler's Z. Physiol. Chem., 1911, 76, 331 CrossRef PubMed; (b) D. Kellin, Proc. R. Soc. London, Ser. B, 1929, 104, 206 CrossRef; (c) B. Vennesland, E. E. Comm, C. J. Knownles, J. Westly and F. Wissing, Cyanide in Biology, Academic Press, London, 1981, pp. 487–494 Search PubMed.
- (a) J. D. Johnson, T. L. Meisenheimer and G. E. Isom, Toxicol. Appl. Pharmacol., 1986, 84, 464 CrossRef CAS; (b) B. K. Ardelt, J. L. Borowitz and G. E. Isom, Toxicology, 1989, 56, 146 CrossRef.
- J. H. Lee, A. R. Jeong, I.-S. Shin, H.-J. Kim and J.-I. Hong, Org. Lett., 2010, 12, 764 CrossRef CAS PubMed.
- M. Shamsipur, M. Sadeghi, K. Alizadeh, H. Sharghi and R. Khalifeh, Anal. Chim. Acta, 2008, 57, 630 Search PubMed.
- S. H. Mashraqui, M. Chandiramani, R. Betkar and K. Poonia, Tetrahedron Lett., 2010, 51, 1306 CrossRef CAS PubMed.
- S.-P. Wu, K.-J. Du and Y.-M. Sung, Dalton Trans., 2010, 39, 4363 RSC.
- D. Maity, V. Kumar and T. Govindaraju, Org. Lett., 2012, 14, 6008 CrossRef CAS PubMed.
- W. Lin, L. Yuan, Z. Cao, J. Feng and Y. Feng, Dyes Pigm., 2009, 83, 14 CrossRef CAS PubMed.
- H. Küpper and P. M. H. Kroneck, in Nickel and Its Surprising Impact in Nature, ed. A. Sigel, H. Sigel and R. K. O. Sigel, John Wiley & Sons Ltd., U. K., 2007, vol. 2, pp. 31–62 Search PubMed.
- S. C. Dodani, Q. He and C. J. Chang, J. Am. Chem. Soc., 2009, 131, 18020 CrossRef CAS PubMed.
- F. A. Abebe, C. S. Eribal, G. Ramakrishna and E. Sinn, Tetrahedron Lett., 2011, 52, 5554 CrossRef CAS PubMed.
- N. Chattopadhyay, A. Mallick and S. Sengupta, J. Photochem. Photobiol., A, 2006, 177, 55 CrossRef CAS PubMed.
- G.-B. Li, H.-C. Fang, Y.-P. Cai, Z.-Y. Zhou, P. K. Thallapally and J. Tian, Inorg. Chem., 2010, 49, 7241 CrossRef CAS PubMed.
- U. Fegade, J. Marek, R. Patil, S. Attarde and A. Kuwar, J. Lumin., 2014, 146, 234 CrossRef CAS PubMed.
- Copper Proteins, ed. T. G. Spiro, John Wiley, New York, 1981 Search PubMed.
- R. S. Britton, Semin. Liver Dis., 1996, 16, 3 CrossRef CAS PubMed.
- (a) S. C. Leary, P. A. Cobine, B. A. Kaufman, G. H. Guercin, A. Mattman, J. Palaty, G. Lockitch, D. R. Winge, P. Rustin, R. Horvath and E. A. Shoubridge, Cell Metab., 2007, 5, 9 CrossRef CAS PubMed; (b) S. Lutsenko, A. Gupta, J. L. Burkhead and V. Zuzel, Arch. Biochem. Biophys., 2008, 476, 22 CrossRef CAS PubMed.
- (a) S. G. Kaler, Nat. Rev. Neurol., 2011, 7, 15 CrossRef CAS PubMed; (b) I. Bertini and A. Rosato, Cell. Mol. Life Sci., 2008, 65, 89 CrossRef CAS PubMed.
- (a) K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205 CrossRef CAS PubMed; (b) E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995 CrossRef CAS PubMed; (c) D. R. Brown and H. Kozlowski, Dalton Trans., 2004, 1907 RSC.
- Z. Chen, L. Wang, G. Zou, J. Tang, X. Cai, M. Teng and L. Chen, Spectrochim. Acta, Part A, 2013, 105, 57 CrossRef CAS PubMed.
- S. Kaur and S. Kumar, Chem. Commun., 2002, 2840 RSC.
- Z.-C. Wen, R. Yang, H. Hea and Y.-B. Jiang, Chem. Commun., 2006, 106 RSC.
- G.-K. Li, Z.-X. Xu, C.-F. Chen and Z.-T. Huang, Chem. Commun., 2008, 1774 RSC.
- Y. Zhou, J. Zhang, H. Zhou, Q. Zhang, T. Ma and J. Niu, J. Lumin., 2012, 132, 1837 CrossRef CAS PubMed.
- F.-J. Huo, C.-X. Yin, Y.-T. Yang, J. Su, J.-B. Chao and D.-S. Liu, Anal. Chem., 2012, 84, 2219 CrossRef CAS PubMed.
- K. M. K. Swamy, S.-K. Ko, S. K. Kwon, H. N. Lee, C. Mao, J.-M. Kim, K.-H. Lee, J. Kim, I. Shin and J. Yoon, Chem. Commun., 2008, 5915 RSC.
- C. Wu, Q.-N. Bian, B.-G. Zhang, X. Cai, S.-D. Zhang, H. Zheng, S.-Y. Yang and Y.-B. Jiang, Org. Lett., 2012, 14, 4198 CrossRef CAS PubMed.
- L. Huang, X. Wang, G. Xie, P. Xi, Z. Li, M. Xu, Y. Wu, D. Bai and Z. Zeng, Dalton Trans., 2010, 39, 7894 RSC.
- Y.-B. Ruan, C. Li, J. Tangb and J. Xie, Chem. Commun., 2010, 46, 9220 RSC.
- C. Kar, M. D. Adhikari, A. Ramesh and G. Das, Inorg. Chem., 2013, 52, 743 CrossRef CAS PubMed.
- M. Saleem and K.-H. Lee, J. Lumin., 2014, 145, 843 CrossRef CAS PubMed.
- M. H. Kim, H. H. Jang, S. Yi, S.-K. Chang and M. S. Han, Chem. Commun., 2009, 4838 RSC.
- S. Yin, V. Leen, S. V. Snick, N. Boensa and W. Dehaen, Chem. Commun., 2010, 46, 6329 RSC.
- X. Qi, E. J. Jun, L. Xu, S.-J. Kim, J. S. J. Hong, Y. J. Yoon and J. Yoon, J. Org. Chem., 2006, 71, 2881 CrossRef CAS PubMed.
- L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 10 CrossRef CAS PubMed.
- D. W. Domaille, L. Zeng and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 1194 CrossRef CAS PubMed.
- S. C. Dodani, S. C. Leary, P. A. Cobine, D. R. Winge and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 8606 CrossRef CAS PubMed.
- C. S. Lim, J. H. Han, C. W. Kim, M. Y. Kang, D. W. Kang and B. R. Cho, Chem. Commun., 2011, 47, 7146 RSC.
- K. C. Ko, J.-S. Wu, H. J. Kim, P. S. Kwon, J. W. Kim, R. A. Bartsch, J. Y. Lee and J. S. Kim, Chem. Commun., 2011, 47, 3165 RSC.
- A. Basu and G. Das, Dalton Trans., 2011, 40, 2837 RSC.
- G. He, X. Zhao, X. Zhang, H. Fan, S. Wu, H. Li, C. He and C. Duan, New J. Chem., 2010, 34, 1055 RSC.
- P. Li, X. Duan, Z. Chen, Y. Liu, T. Xie, L. Fang, X. Li, M. Yin and B. Tang, Chem. Commun., 2011, 47, 7755 RSC.
- M. Kumar, N. Kumar and V. Bhalla, Dalton Trans., 2012, 41, 10189 RSC.
- T. Mistri, R. Alam, M. Dolai, S. K. Mandal, A. R. Khuda-Bukhsh and M. Ali, Org. Biomol. Chem., 2013, 11, 1563 CAS.
- M. Royzen, Z. Dai and J. W. Canary, J. Am. Chem. Soc., 2005, 127, 1612 CrossRef CAS PubMed.
- S. Goswami, D. Sen and N. K. Das, Org. Lett., 2010, 12, 856 CrossRef CAS PubMed.
- E. Ballesteros, D. Moreno, T. Gómez, T. Rodríguez, J. Rojo, M. Gracía-Valverde and T. Torroba, Org. Lett., 2009, 11, 1269 CrossRef CAS PubMed.
- Z. Xu, J. Pan, D. R. Spring, J. Cui and J. Yoon, Tetrahedron, 2010, 66, 1678 CrossRef CAS PubMed.
- J. Jo, H. Y. Lee, W. Liu, A. Olasz, C.-H. Chen and D. Lee, J. Am. Chem. Soc., 2012, 134, 16000 CrossRef CAS PubMed.
- Z. Xu, J. Yoon and D. R. Spring, Chem. Commun., 2010, 46, 2563 RSC.
- H. S. Jung, M. Park, D. Y. Han, E. Kim, C. Lee, S. Ham and J. S. Kim, Org. Lett., 2009, 11, 3378 CrossRef CAS PubMed.
- S. Suganya, S. Velmathi and D. MubarakAli, Dyes Pigm., 2014, 114, 116 CrossRef PubMed.
- L. Yang, Q. Song, K. Damit-Og and H. Cao, Sens. Actuators, B, 2013, 176, 181 CrossRef CAS PubMed.
- K.-K. Yu, K. Li, J.-T. Hou and X.-Q. Yu, Tetrahedron Lett., 2013, 54, 5771 CrossRef CAS PubMed.
- T. Hirayama, G. C. Van de Bittner, L. W. Gray, S. Lutsenko and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2228 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |