
A series of novel mercury(I) selenites and tellurites containing SOJT Mo6+ cations†
Xue-Li
Cao
ab,
Fang
Kong
a,
Chun-Li
Hu
a and
Jiang-Gao
Mao
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China
bUniversity of the Chinese Academy of Sciences, Beijing 100039, P. R. China. E-mail: mjg@fjirsm.ac.cn; Fax: (+86)591-83714946; Tel: (+86)591-83704836
First published on 15th October 2014
Abstract
Using Hg2Cl2, MoO3, and TeO2 (or SeO2) as starting materials, four new mixed-metal tellurites and selenites have been obtained in the unexplored mercury(I)–Mo6+–Se4+/Te4+–O system, namely, Hg2MoSeO6 (1), α-Hg2MoTeO6 (2), β-Hg2MoTeO6 (3), and Hg2Mo2TeO9 (4). They represent the first mercury(I) tellurites and selenites containing octahedrally coordinated d0 transition metal (TM) cations. All four compounds are centrosymmetric. They show three types of 3D structures based on Hg22+ dumbbells, MoO6 octahedra, QO32− (Q = Se, Te) or TeO44− anions. Compounds 1–3 feature similar 1D anionic chains of [MoO3(QO3)]2− (Q = Se or Te), in which the 1D chains of corner-sharing MoO6 octahedra are further decorated by bidentate bridging selenite or tellurite anions. Compounds 1 and 2 are isostructural and their 3D network structures are based on 2D mercury tellurite or selenite layers in the ac plane pillared by MoO6 octahedra, whereas the 3D network of β-Hg2MoTeO6 (3) is composed of 1D mercury tellurite chains along the a axis and 1D chains of [MoO3(TeO3)]2− anions along the b axis. Compound 4 features an unusual 3D network structure composed of novel [Mo2O5(TeO4)]2− double layers interconnected by Hg22+ cations. Within the [Mo2O5(TeO4)]2− double layer, the cyclo-Mo4O20 tetramers are further interconnected via corner sharing into a 2D molybdenum oxide double layer with TeO4 groups capping the walls of the thus-formed eight-membered rings (8-MRs). Thermal stabilities and optical properties as well as theoretical calculations based on density functional theory (DFT) methods were also performed.
Introduction
Metal selenites and tellurites can form a diversity of unusual structures due to the presence of the stereochemically active lone-pair in the Se4+ and Te4+ cations, which means they can serve as structure-directing agents.1 In addition, Te(IV) can be coordinated by three, four or five oxygen atoms to form TeOX (X = 3, 4, 5) polyhedra, which are further condensed into various clusters, 1D chains, 2D layers and 3D networks, whereas only Se2O52− can be formed for the selenite anion.2 Furthermore, the combination of a QO32− (Q = Se, Te) group and an octahedrally-coordinated d0 TM cation such as Ti4+, Nb5+, Mo6+, W6+, Ta5+ and V5+, both of which are susceptible to second-order Jahn–Teller (SOJT) distortion, favors the formation of noncentrosymmetric (NCS) or polar structures with possible second-harmonic generation (SHG).2b,3–9 For example, BaMo2TeO9 displays a very strong SHG response of 600 × α-SiO2, and its large crystals have also been obtained successfully.1c,9So far, counter cations used to balance the negative charges of d0 TM selenites and tellurites include alkali,10a,b alkaline earth,1c,9 rare earth,10c transition metal10d and post-transition metal main group elements.10e No compounds have been reported in similar systems with Hg+/Hg2+ as balanced cations, though a few ternary mercury(I) or mercury(II) selenites and tellurites have been structurally characterized,11–13 including HgSeO3,12a Hg3(HSeO3)2(SeO3)2,12b HgSeO3·0.5H2O,12c α-, β- and γ-Hg2SeO3,12d HgTeO3,13a Hg2Te2O7,13b and Hg2TeO3.13c
In this paper, we will focus our research attention on the unexplored mercury(I)–Mo6+–Se4+/Te4+–O system due to the fact that the combination of the Hg+ cations, which are unique in bonding character with two types of SOJT building units, may result in a variety of new compounds with novel structures and interesting physical properties. Our research in this aspect has led to the isolation of four new compounds, namely, Hg2Mo2TeO9 (1), α-Hg2MoTeO6 (2), β-Hg2MoTeO6 (3), and Hg2MoSeO6 (4). Herein, we report their syntheses and crystal structures as well as electronic structure calculations.
Experimental section
Materials and instruments
Hg2Cl2 (99+%, AR), MoO3 (99.5+%, AR), SeO2 (99+%, AR), TeO2 (99+%, AR), and BaSO4 (99.9+%, AR) were used as received. All of the chemicals were supplied by the Shanghai Reagent Factory. The X-ray powder diffraction patterns were collected in continuous mode on a Rigaku MiniFlex II diffractometer at room temperature (Cu-Kα radiation, flat plate geometry). Data were collected in the 2θ range of 5–70° with a step size of 0.02°. Microprobe elemental analyses for the Hg, Mo, Te and Se elements were carried out on a field-emission scanning electron microscope (FESEM, JSM6700F) equipped with an energy dispersive X-ray spectroscope (EDS, Oxford INCA). Infrared spectra were collected on a Magna 750 FT-IR spectrometer as KBr pellets in the spectral range of 4000–400 cm−1 with a resolution of 2 cm−1 at room temperature. The UV-Vis diffuse reflectance spectra were measured with a PE Lambda 900 UV-Vis spectrophotometer in the range of 190–2500 nm at room temperature. A BaSO4 plate was used as a standard (100% reflectance) for the baseline correction. The absorption spectrum was converted from the reflectance spectra using the Kubelka–Munk function: α/S = (1 − R)2/2R = K/S,14 where α, R, S and K represent absorption coefficient, reflectance, scattering coefficient and absorption, respectively. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed with a NETZCH STA449C unit at a heating rate of 10 °C min−1 under a N2 atmosphere.Syntheses
The four compounds were hydrothermally synthesized by reaction of a mixture of Hg2Cl2, MoO3 and TeO2 (or SeO2) in 6 mL of deionized water. The loaded compositions are as follows: Hg2Cl2 (141.6 mg, 0.3 mmol), MoO3 (43.2 mg, 0.3 mmol), SeO2 (88.8 mg, 0.8 mmol) and H2O (6 mL) for Hg2MoSeO6 (1); Hg2Cl2 (141.6 mg, 0.3 mmol), MoO3 (57.6 mg, 0.4 mmol), TeO2 (127.7 mg, 0.8 mmol), and H2O (6 mL) for α-Hg2MoTeO6 (2); Hg2Cl2 (141.6 mg, 0.3 mmol), MoO3 (57.6 mg, 0.4 mmol), TeO2 (159.6 mg, 1.0 mmol), and H2O (6 mL) for β-Hg2MoTeO6 (3); Hg2Cl2 (141.6 mg, 0.3 mmol), MoO3 (86.4 mg, 0.6 mmol), TeO2 (79.8 mg, 0.5 mmol) and H2O (6 mL) for Hg2Mo2TeO9 (4). The mixtures were sealed in an autoclave equipped with a Teflon liner (23 mL) and heated at 210 °C (for 1) or 220 °C (for 2, 3 and 4) for 4 days, followed by slow cooling to room temperature at a rate of 2 °C h−1. Yellow prism-shaped crystals of 1 were obtained as a single phase in a yield of about 92% based on Mo, which has been confirmed by X-ray diffraction (XRD) studies (ESI, Fig. S1a†). Yellow block-shaped single crystals of 3 and red flake-shaped crystals of 4 were collected in ca. 52% and 45% yields (based on Mo), respectively, as confirmed by X-ray diffraction (XRD) studies (ESI, Fig. S1b and S1c†). Though many attempts have been made to synthesize a pure phase of α-Hg2MoTeO6 (2) by changing the molar ratios of the starting materials and the reaction temperatures, products always contain compounds 3 and 4 as impurities, hence α-Hg2MoTeO6 was not subject to optical and thermal measurements. For compounds 2, 3 and 4, the main impurities are rodlike crystals of MoTe2O7. Results of EDS elemental analyses on the single crystals of 1–4 gave molar ratios of Hg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo:Q (Q = Se or Te) of 2.02:1.12:1, 2.12:1.15:1, 2.23:1.17:1, and 2.04:2.00:1, respectively, which are in good agreement with those determined from single-crystal X-ray structural studies.
:Mo:Q (Q = Se or Te) of 2.02:1.12:1, 2.12:1.15:1, 2.23:1.17:1, and 2.04:2.00:1, respectively, which are in good agreement with those determined from single-crystal X-ray structural studies.
X-Ray crystallography
A yellow brick crystal of Hg2MoSeO6 (0.43 × 0.24 × 0.17 mm3), a yellow needle-like crystal of α-Hg2MoTeO6 (0.23 × 0.09 × 0.08 mm3), and a yellow brick crystal of β-Hg2MoTeO6 (0.36 × 0.33 × 0.27 mm3), as well as a red plate-shaped crystal of Hg2Mo2TeO9 (0.26 × 0.22 × 0.05 mm3) were used for single-crystal X-ray data collections. Diffraction data were measured on a Mercury CCD diffractometer (for Hg2MoSeO6, β-Hg2MoTeO6, and Hg2Mo2TeO9) and a Saturn724 CCD diffractometer (for α-Hg2MoTeO6), with Mo-Kα radiation (λ = 0.71073 Å) at room temperature. All four data sets were corrected for Lorentz and polarization factors as well as absorption by the multi-scan method.15a All four structures were solved by direct methods and refined by full-matrix least-squares fitting on F2 using SHELXL-97.15b All four structures were checked for possible missing symmetry elements with PLATON,15c but none were found. All of the atoms were refined with anisotropic thermal parameters. Crystallographic data, structural refinements and important bond distances are summarized in Tables 1 and 2. More details of the crystallographic studies are given as ESI.† Further details of the crystal structure studies can be obtained from the FIZ Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (Fax: (49)7247808666; E-mail: crysdata@fiz-karlsruhe.de), on quoting the depository numbers CSD 428420–428423.| a R 1 = ∑||Fo| − |Fc||/∑|Fo|, wR2 = {∑w[(Fo)2 − (Fc)2]2/∑w[(Fo)2]2}1/2. | ||||
|---|---|---|---|---|
| Formula | Hg2MoSeO6 | α-Hg2MoTeO6 | β-Hg2MoTeO6 | Hg2Mo2TeO9 |
| fw | 672.08 | 720.72 | 720.72 | 864.66 |
| Crystal system | Orthorhombic | Orthorhombic | Orthorhombic | Monoclinic |
| Space group | Pbcm | Pbcm | Pbca | P2(1)/c |
| a/Å | 6.4578(7) | 6.429(4) | 11.592(8) | 9.5533(9) |
| b/Å | 13.3556(19) | 13.452(9) | 7.343(5) | 7.6391(7) |
| c/Å | 7.2072(10) | 7.277(5) | 14.763(10) | 11.3555(10) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 90 | 90 | 90 | 92.675(6) |
| γ (°) | 90 | 90 | 90 | 90 |
| V/Å3 | 621.61(14) | 629.3(7) | 1256.7(14) | 827.81(13) |
| Z | 4 | 4 | 8 | 4 |
| D c (g cm−3) | 7.182 | 7.606 | 7.618 | 6.938 |
| μ(Mo-Ka) (mm−1) | 57.081 | 55.139 | 55.226 | 43.417 |
| GOF on F2 | 1.113 | 1.248 | 1.174 | 1.116 |
| R 1, wR2 [I > 2σ(I)]a | 0.0261, 0.0554 | 0.0242, 0.0470 | 0.0284, 0.0496 | 0.0371, 0.0837 |
| R 1, wR2 (all data) | 0.0277, 0.0563 | 0.0256, 0.0475 | 0.0301, 0.0504 | 0.0394, 0.0850 |
| Hg2MoSeO6 | |||
| Hg(1)–O(1) | 2.182(8) | Mo(1)–O(4) | 1.971(3) |
| Hg(1)–Hg(2) | 2.528(7) | Mo(1)–O(4) | 1.971(3) |
| Hg(2)–O(1) | 2.343(8) | Mo(1)–O(2) | 2.214(6) |
| Hg(2)–O(4) | 2.477(7) | Mo(1)–O(2) | 2.214(6) |
| Hg(2)–O(2) | 2.551(5) | Se(1)–O(2) | 1.699(6) |
| Hg(2)–O(2) | 2.551(5) | Se(1)–O(2) | 1.699(6) |
| Mo(1)–O(3) | 1.717(6) | Se(1)–O(1) | 1.720(7) |
| Mo(1)–O(3) | 1.717(6) | ||
| α-Hg2MoTeO6 | |||
| Hg(1)–O(1) | 2.155(7) | Mo(1)–O(4) | 1.957(3) |
| Hg(1)–Hg(2) | 2.532(1) | Mo(1)–O(4) | 1.957(3) |
| Hg(2)–O(1) | 2.286(7) | Mo(1)–O(2) | 2.185(5) |
| Hg(2)–O(2) | 2.523(6) | Mo(1)–O(2) | 2.185(5) |
| Hg(2)–O(2) | 2.523(6) | Te(1)–O(2) | 1.868(5) |
| Hg(2)–O(4) | 2.577(7) | Te(1)–O(2) | 1.868(5) |
| Mo(1)–O(3) | 1.732(5) | Te(1)–O(1) | 1.895(7) |
| Mo(1)–O(3) | 1.732(5) | ||
| β-Hg2MoTeO6 | |||
| Hg(1)–O(2) | 2.152(6) | Mo(1)–O(3) | 2.088(6) |
| Hg(1)–Hg(2) | 2.519(1) | Mo(1)–O(6) | 2.115(6) |
| Hg(2)–O(1) | 2.123(6) | Mo(1)–O(2) | 2.305(6) |
| Hg(2)–O(4) | 2.597(6) | Te(1)–O(1) | 1.863(6) |
| Mo(1)–O(5) | 1.714(6) | Te(1)–O(3) | 1.881(6) |
| Mo(1)–O(4) | 1.769(6) | Te(1)–O(2) | 1.922(6) |
| Mo(1)–O(6) | 1.804(6) | ||
| Hg2Mo2TeO9 | |||
| Hg(1)–O(8) | 2.198(7) | Mo(2)–O(9) | 1.699(8) |
| Hg(1)–O(1) | 2.419(7) | Mo(2)–O(8) | 1.745(7) |
| Hg(1)–Hg(2) | 2.5324(6) | Mo(2)–O(5) | 1.902(7) |
| Hg(2)–O(4) | 2.159(7) | Mo(2)–O(1) | 1.937(6) |
| Mo(1)–O(6) | 1.700(8) | Mo(2)–(3) | 2.273(7) |
| Mo(1)–O(7) | 1.705(7) | Mo(2)–O(2) | 2.274(7) |
| Mo(1)–O(5) | 1.930(7) | Te(1)–O(4) | 1.886(7) |
| Mo(1)–O(3) | 2.011(7) | Te(1)–O(2) | 1.907(6) |
| Mo(1)–O(2) | 2.141(6) | Te(1)–O(3) | 2.080(7) |
| Mo(1)–O(4) | 2.380(7) | Te(1)–O(1) | 2.113(7) |
Computational descriptions
Single-crystal structural data of the four compounds were used for theoretical calculations. All calculations (including band structures and density of states (DOS)) were carried out using the total-energy code of CASTEP.16 The exchange and correlation effects were calculated using Perdew–Burke–Ernzerhof (PBE) in the generalized gradient approximation (GGA).17 The interactions between the ionic cores and the electrons were described by the norm-conserving pseudopotential.18 The following valence-electron configurations were considered in the computation: Hg-5d106s2, Mo-4d55s1, Te-5s25p4, Se-4s24p4, and O-2s22p4. The numbers of plane waves included in the basis sets were determined by a cutoff energy of 750 eV for all four compounds. The numerical integration of the Brillouin zone was performed using Monkhorst–Pack k-point samplings of 4 × 2 × 3, 4 × 2 × 3, 2 × 3 × 2, and 3 × 3 × 2, respectively. The other parameters and convergent criteria were the default values of the CASTEP code.Results and discussion
Syntheses
Four new mercury(I) molybdenum(VI) mixed-metal tellurites and selenites, namely, Hg2MoSeO6 (1), α-Hg2MoTeO6 (2), β-Hg2MoTeO6 (3), and Hg2Mo2TeO9 (4), have been successfully synthesized by hydrothermal reactions. Compounds 2, 3, and 4 were prepared by slightly altering the ratios of the same reaction materials. Using the same starting materials Hg2Cl2, MoO3, and TeO2, the molar ratios of Hg:Mo:Te were 3:2:4, 3:2:5, and 3:3:2.5 for α-Hg2MoTeO6, β-Hg2MoTeO6, and Hg2Mo2TeO9, respectively. Obviously, the Mo:Te ratio plays an important role in the chemical composition of the final products. A relatively large Mo:Te ratio favors the formation of a molybdenum oxide layer composed of corner-sharing MoO6 octahedra as in Hg2Mo2TeO9 (4), whereas for the other three compounds with Mo:Q molar ratios of 1:1, the MoO6 octahedra form a 1D chain via corner sharing.
Structures of Hg2MoSeO6 and α-Hg2MoTeO6
Hg2MoSeO6 and α-Hg2MoTeO6 are isostructural and crystallize in the centrosymmetric space group Pbcm (no. 57). In this case, only the structural details of α-Hg2MoTeO6 will be described. The structure of α-Hg2MoTeO6 features a new 3D network based on 1D chains of [MoO3(TeO3)]2− along the c axis, which are interconnected by dumbbell Hg22+ cations (Fig. 1). Its asymmetric unit contains two Hg+, one Mo6+, and one Te(IV) atom. Both the Mo6+ and Te4+ cations are in asymmetric coordination environments attributed to SOJT effect. The Mo6+ cation is in a distorted octahedral geometry composed of two tellurite oxygen atoms from two tellurite groups and four oxo anions. The Mo–O bond distances are two long [2.185(5) Å], two normal [1.957(3) Å], and two short [1.732(5) Å], hence the MoO6 octahedron is distorted toward an edge (local C2 direction). The magnitude of the out-of-center distortion (Δd) was calculated to be 0.92.2b The Te4+ cation is in a ψ-TeO3 trigonal pyramidal environment with the pyramidal site occupied by the lone pair of electrons. The Te–O bond lengths range from 1.868(5) to 1.895(7) Å. Neighbouring MoO6 octahedra are interconnected via corner sharing (O4) into a 1D chain along the c axis, and Te(1)O3 groups are grafted on the 1D chain in a bidentate bridging fashion, leading to the formation of the 1D [MoO3(TeO3)]2− chain along the c axis (see Fig. 1a).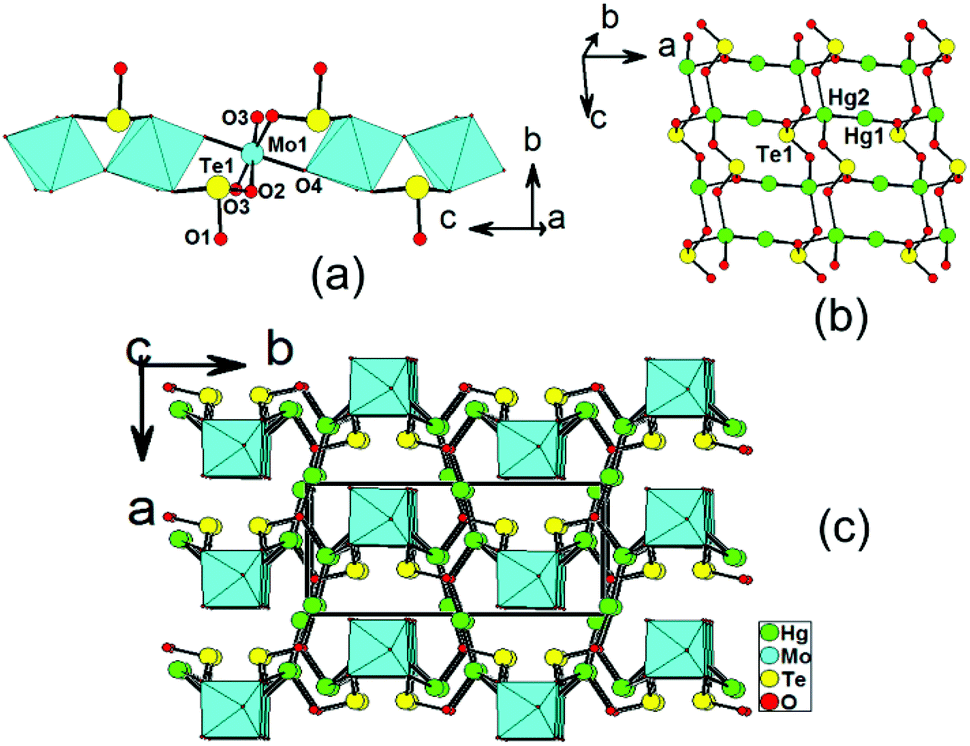 | ||
| Fig. 1 1D [MoO3(TeO3)]2− down the c axis (a), a Hg2TeO3 layer in the ac plane (b) and a view of the structure of α-Hg2MoTeO6 down the c axis (c). Hg, Mo, Te and O atoms are drawn as green, cyan, yellow and red circles, respectively. The MoO6 octahedra are shaded in cyan. | ||
As we all know, Hg+ is present in a coupled fashion as Hg22+, with a Hg(1)–Hg(2) bond length of 2.5322(17) Å, which is close to those reported in other mercury(I) oxide compounds.11,12 In α-Hg2MoTeO6, the Hg(1) atom is bonded to one unidentate TeO3 group, whereas the Hg(2) atom is connected by three unidentate TeO3 groups and one oxo anion (ESI, Fig. S2a†). The Hg–O bond lengths are in the range of 2.155(7)–2.577(7) Å, which are close to those reported in other mercury(I) tellurites.13 Each Te(1)O3 group is hexadentate and bridges two Mo6+ cations, one Hg(1) and three Hg(2) atoms (ESI, Fig. S2b†). Bond valence calculations on α-Hg2MoTeO6 gave values of 5.90 and 3.93 for the Mo and Te atoms, respectively, indicating that they are in oxidation states of 6+ and 4+, respectively.20
It is interesting to note that neighbouring dumbbell Hg22+ cations are linked by bridging TeO3 groups, leading to a 2D Hg2TeO3 layer parallel to the ac plane (Fig. 1b). The above 1D molybdenum tellurite chains and 2D mercury(I) tellurite layers are further interconnected by Hg–O–Mo and Hg–O–Te bridges into a pillared layered 3D network (Fig. 1c).
The overall structure of Hg2MoSeO6 is the same as that of α-Hg2MoTeO6, the main difference is the TeO3 groups being replaced by SeO3 groups. The magnitude of the out-of-center distortion (Δd) for the MoO6 octahedron in Hg2MoSeO6 was calculated to be 1.01, which is slightly larger than that in α-Hg2MoTeO6. Furthermore, the b and c axial lengths of Hg2MoSeO6 are slightly smaller than those of α-Hg2MoTeO6 due to the smaller size of Se4+ compared with Te4+.
Structure of β-Hg2MoTeO6
β-Hg2MoTeO6 crystallizes in the centrosymmetric space group Pbca (no. 61). Its structure can be described as a different 3D network composed of 1D chains of [MoO3(TeO3)]2− along the b axis, interconnected by dumbbell Hg22+ cations (Fig. 2). The network can also be viewed to be formed by the interconnection of 1D [MoO3(TeO3)]2− chains along the b axis and 1D mercury(I) tellurite chains along the a axis (Fig. 2a and 2b). Its asymmetric unit contains two Hg+, one Mo6+, and one Te4+ cation. Both the Mo6+ and Te4+ cations are in asymmetric coordination environments, attributed to SOJT effects. The Mo6+ cation is in a distorted octahedral geometry composed of two tellurite oxygen atoms and four oxo anions. The Mo–O bond distances are three long [2.088(6)–2.305(6) Å] and three short [1.714(6) Å–1.804(6) Å], hence the MoO6 octahedron is distorted toward a face (local C3 direction). The magnitude of the out-of-center distortion (Δd) was calculated to be 1.29,2b which is slightly larger than that for MoO6 in α-Hg2MoTeO6. The unique Te4+ cation is in a ψ-TeO3 trigonal pyramidal environment with the pyramidal site occupied by the lone pair of electrons. The Te–O bond lengths range from 1.863(6) to 1.922(6) Å. Neighbouring MoO6 octahedra are interconnected via corner sharing (O6) into a 1D chain along the b axis, which is further decorated by the TeO3 groups in a bidentate bridging fashion, so as to form a 1D [MoO3(TeO3)]2− anionic chain (Fig. 2a). This chain is slightly different from that in α-Hg2MoTeO6, the main differences being the different axes that the chains are extended along and the different distortion directions associated with the MoO6 octahedra.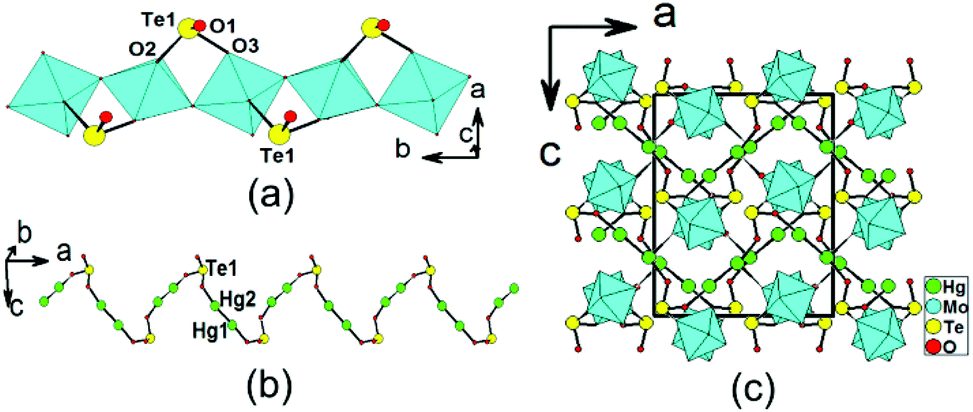 | ||
| Fig. 2 A 1D [MoO3(TeO3)]2− chain along the b axis (a), a 1D Hg2TeO3 chain along the a axis (b), and a view of the structure of β-Hg2MoTeO6 down the b axis (c). Hg, Te and O atoms are drawn as green, yellow and red circles, respectively. The MoO6 octahedra are shaded in cyan. | ||
As with α-Hg2MoTeO6, Hg+ also exists as dumbbell Hg22+. The Hg(1) atom is bonded to one unidentate TeO3 group, whereas the Hg(2) atom is connected by one unidentate TeO3 group, and one oxo anion (ESI, Fig. S3a†). The Hg–O bond lengths are in the range of 2.123(6)–2.597(6) Å. The Te(1)O3 connects with two Mo6+ and two Hg+ cations (ESI, Fig. S3b†). Bond valence calculations on β-Hg2MoTeO6 gave values of 5.98 and 3.82 for Mo and Te atoms, respectively, indicating that they are in oxidation states of 6+ and 4+, respectively.20
It is interesting to note that the interconnection of the Hg22+ ions by bridging TeO3 groups leads to a zigzag 1D Hg2TeO3 chain parallel to the a axis (Fig. 2b). The above two types of 1D chains are further interconnected by Hg–O–Mo and Hg–O–Te bridges into a complicated 3D network (Fig. 2c).
The main differences between the α- and β-forms of Hg2MoTeO6 lie in the different axes along which the 1D [MoO3(TeO3)]2− anionic chains are extended and the different substructures for mercury(I) tellurite (layered in α-Hg2MoTeO6 and 1D in β-Hg2MoTeO6).
Structure of Hg2Mo2TeO9
Hg2Mo2TeO9 shows a novel 3D framework structure in which 2D [Mo2O5(TeO4)]2− double layers are bridged by Hg22+ dumbbells (Fig. 3). There are two Hg+, two Mo6+, and one Te(IV) atom in the asymmetric unit. Both the Mo6+ and Te4+ cations are in asymmetric coordination environments attributed to SOJT effect. Both the Mo(1) and Mo(2) atoms are in distorted octahedral geometries composed of six oxygen atoms.19e The Mo–O bond distances are two long [2.141(6)–2.380(7) Å], two short [1.699(8) Å–1.745(7) Å], and two normal [1.902(7)–1.937(6) Å], hence both MoO6 octahedra are distorted toward an edge (local C2 direction). The magnitudes of the out-of-center distortions (Δd) were calculated to be 1.26 and 1.18 for Mo(1)O6 and Mo(2)O6, respectively,2b,12 which are comparable to those of β-Hg2MoTeO6 and A4Mo5O15(SeO3)2(H2O)2 (A = K, Rb).19e Different from those in the other three phases, the Te4+ cation in Hg2Mo2TeO9 is in a ψ-TeO4 square pyramidal environment with the pyramidal site occupied by the lone pair of electrons (the seesaw geometry). The Te–O bond lengths range from 1.886(7) to 2.113(7) Å (Table 2). Two pairs of Mo(1)O6 and Mo(2)O6 octahedra are corner-sharing to form a cyclic [Mo4O20]16− unit (Fig. 3a). These clusters are further interconnected via corner sharing into a novel 2D [Mo2O9]6− layer with eight-membered rings (8-MRs) (Fig. 3a). TOPOS analysis reveals that the above 2D layer is a 4-connected 2D network with the Schläfli symbol of {44; 62}.21 TeO4 groups are capped on the walls of the 8-MRs of the [Mo2O9]6− layer, forming a unique [Mo2O5(TeO4)]2− double layer in the bc plane (Fig. 3b).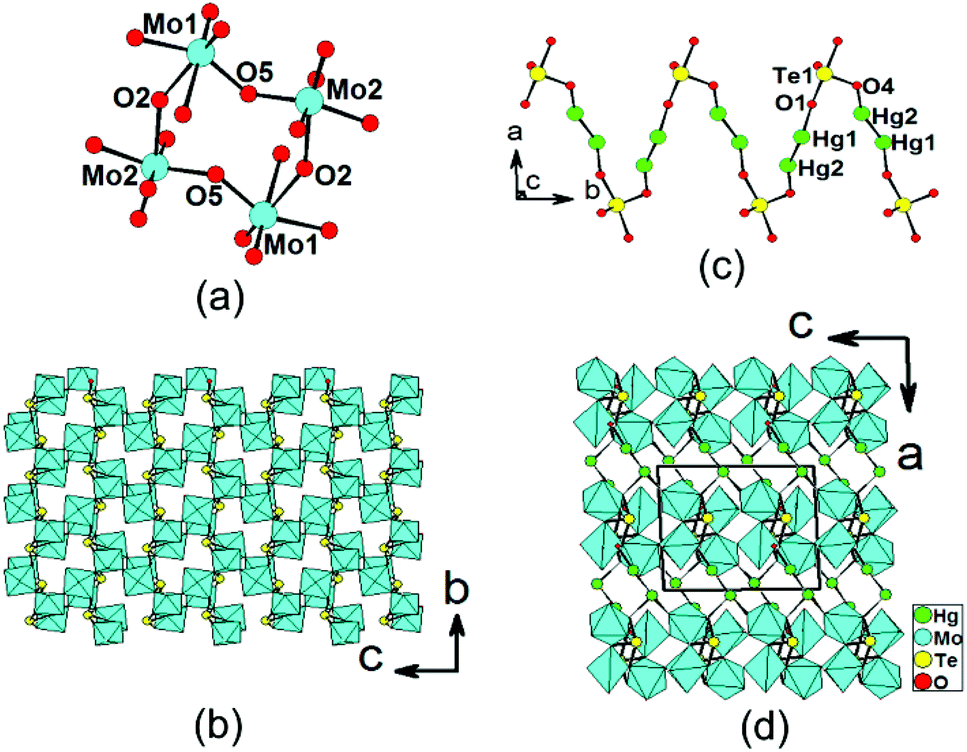 | ||
| Fig. 3 A cyclic [Mo4O20] cluster unit (a), a 2D [Mo2O5(TeO4)]2− layer in the bc plane (b), a 1D Hg2TeO4 chain along the b axis (c), and a view of the structure of Hg2Mo2TeO9 down the b axis (d). Hg, Mo, Te and O atoms are drawn as green, cyan, yellow and red circles, respectively. The MoO6 octahedra are shaded in cyan. | ||
The Hg(1) atom is coordinated by one unidentate TeO4 group and one oxo anion, whereas the Hg(2) atom is bonded to one unidentate TeO4 group (ESI, Fig. S4a†). The Hg–O bond lengths are in the range of 2.159(7)–2.419(7) Å. The interconnection of the Hg22+ dumbbells by TeO4 groups results in a zigzag chain of [Hg2TeO4]2− along the b axis (Fig. 3c). The above 2D [Mo2O5(TeO4)]2− double layers are interconnected by mercury(I) ions via Hg–O–Te bridges into a complicated 3D framework (Fig. 3d). Overall, each Te(1)O4 group is hexadentate, bridging two Hg+ and four Mo6+ cations (Fig. S4b†). Bond valence calculations on Hg2Mo2TeO9 gave values of 5.98 and 3.93 for the Mo and Te atoms, respectively, indicating that they are in oxidation states of 6+ and 4+, respectively.20
It is interesting to compare the [Mo2O5(TeO4)]2− double layer in Hg2Mo2TeO9 with those reported in monoclinic and orthorhombic forms of BaTeMo2O9.1c,22 In the monoclinic form of BaMo2TeO9 in a polar space group P21,1c dimeric Mo2O11 units are corner-sharing with each other to form a Mo–O double layer with six-membered rings capped by TeO3 groups. In the orthorhombic form of BaMo2TeO9 in a polar space group Pca21,22 dimeric Mo2O11 units are corner-sharing with each other to form Mo–O double chains, which are further bridged by pairs of TeO3 groups to form a double layer with Mo6Te2 8-MRs. The monoclinic form displays strong SHG response of 600 × α-SiO2, whereas the orthorhombic form only exhibits a weak SHG response of 0.2 × KDP. Due to the much larger size of the Hg22+ cation, the interlayer distance of about 9.55 Å for Hg2Mo2TeO9 is significantly larger than those of both forms of BaTeMo2O9 (both close to 8.8 Å).
Regrettably, although two building units favouring the formation of NCS or polar structures were employed, no NCS or polar structures have been obtained owing to the fact that the asymmetric groups are arranged in an antiparallel manner. Furthermore, the sizes of the constituent cations, hydrogen-bonding and flexible coordination may affect the materials’ symmetries, according to previous reports. However, to design new functional asymmetric materials rationally is still a formidable mission.2f,4f,23
TGA and DSC
Results of the TGA and DSC analyses indicate that Hg2MoSeO6, β-Hg2MoTeO6, and Hg2Mo2TeO9 are all stable up to 325 °C (Fig. S5†). Hg2MoSeO6 displays two main steps of weight loss in the temperature range of 337–710 °C and exhibits two endothermic peaks at 417 and 695 °C, during which 1 mol of SeO2 and 2 mol of Hg per formula unit are released.12d The observed weight loss of 76.1% for Hg2MoSeO6 is close to the calculated one (76.2%). In the temperature range of 325–664 °C, β-Hg2MoTeO6 shows three steps of weight loss whereas Hg2Mo2TeO9 reveals two steps of weight loss. The observed weight losses of 55.3% and 46.5% respectively for β-Hg2MoTeO6 and Hg2Mo2TeO9 correspond to the release of 2 mol Hg per formula unit, and are very close to their calculated values (55.7% and 46.4%).12d The final residues were not characterized due to their melting with the TGA buckets (made of Al2O3) under high temperatures.Optical properties
The IR spectra of Hg2MoSeO6, β-Hg2MoTeO6, and Hg2Mo2TeO9 have been measured in the wavelength range of 4000–400 cm−1 at room temperature. All three compounds are transparent in the range of 4000–1000 cm−1. The bands associated with the Te(Se)–O, Mo–O and O–Mo–O vibrations appeared in the wave number range of 400–1000 cm−1 (Fig. S6, Table S1†). The Mo–O stretching vibrations were observed at 799–926 cm−1, whereas Te(Se)–O stretching vibrations appeared at 651–772 cm−1. The absorption bands occurring below 651 cm−1 can be assigned to Mo–O–Te(Se) bending vibrations. These assignments are in good agreement with the literature.24–27 The IR spectra and assignments are shown in the ESI.†UV absorption spectra of Hg2MoSeO6, β-Hg2MoTeO6, and Hg2Mo2TeO9 show no absorption in the range of 760–2500 nm (0.76–2.5 μm) (Fig. S7†). Hence, all three compounds are transparent in the range of 0.76–2.5 μm, corresponding to the visible, near-IR and middle-IR regions. The UV-Vis diffuse reflectance spectrum measurements indicate that the band gaps are approximately 2.71 eV, 2.72 eV and 2.25 eV for Hg2MoSeO6, β-Hg2MoTeO6, and Hg2Mo2TeO9, respectively, hence all three compounds are wide band gap semiconductors.
Theoretical calculations
Theoretical calculations based on DFT methods were made.16 The calculated band structures of Hg2MoSeO6, α-Hg2MoTeO6, β-Hg2MoTeO6, and Hg2Mo2TeO9 along high symmetry points of the first Brillouin zone are plotted in Fig. 4. The state energies (eV) of the lowest conduction bands (L-CB) and the highest valence bands (H-VB) of the four compounds are listed in the ESI, Table S2.† The calculated band structures (Fig. 4, Table S2†) indicate that all four compounds are indirect band gap semiconductors with band gaps of 2.42, 2.34, 2.46, and 2.00 eV for Hg2MoSeO6, α-Hg2MoTeO6, β-Hg2MoTeO6, and Hg2Mo2TeO9, respectively, which are slightly smaller than their experimental values (2.71, 2.72, and 2.25 eV, respectively, for Hg2MoSeO6, β-Hg2MoTeO6, and Hg2Mo2TeO9). This is not surprising as it is well-known that the GGA does not accurately describe the eigenvalues of the electronic states, which usually causes quantitative underestimation of band gaps for semiconductors and insulators.28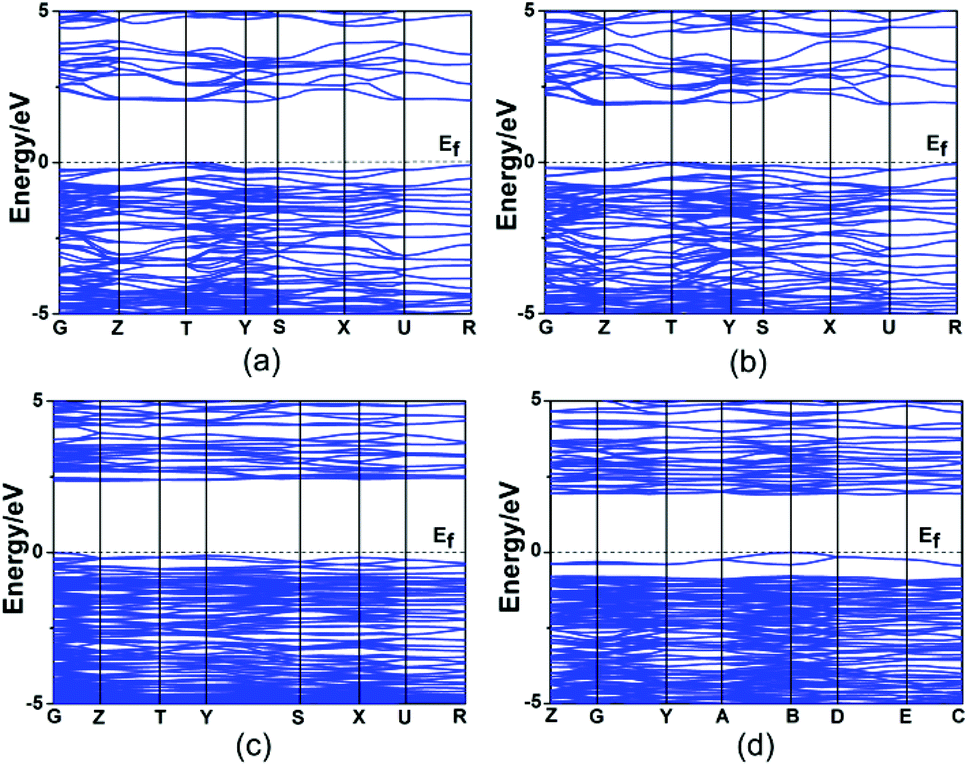 | ||
| Fig. 4 The scissor-added band structures of Hg2MoSeO6 (a), α-Hg2MoTeO6 (b), β-Hg2MoTeO6 (c) and Hg2Mo2TeO9 (d). | ||
The bands can be assigned according to the total and partial densities of states (DOS) as plotted in Fig. 5. DOS plots of the four compounds are very similar, hence Hg2Mo2TeO9 is used as a representative. For Hg2Mo2TeO9 (Fig. 5c), the bottom-most VBs from −19.8 to −16.3 eV originate from the O 2s mixing with a small amount of the Te 5s, Te 5p and Mo 4d states. The bands between −10.0 and −8.8 eV are composed of the Te 5s and O 2p states. We will focus on the VB and the CB in the vicinity of the Fermi level (between −8.0 and −7.8 eV), which counts for most of the bonding characteristic in a compound. In the region of −8.0–0 eV, the Hg 5d state overlaps fully with O 2p, and in the region of −8.0–7.8 eV, the Te 5p and Mo 4d states overlap fully with O 2p, indicative of the Hg–O, Mo–O and Te–O bonding interactions.
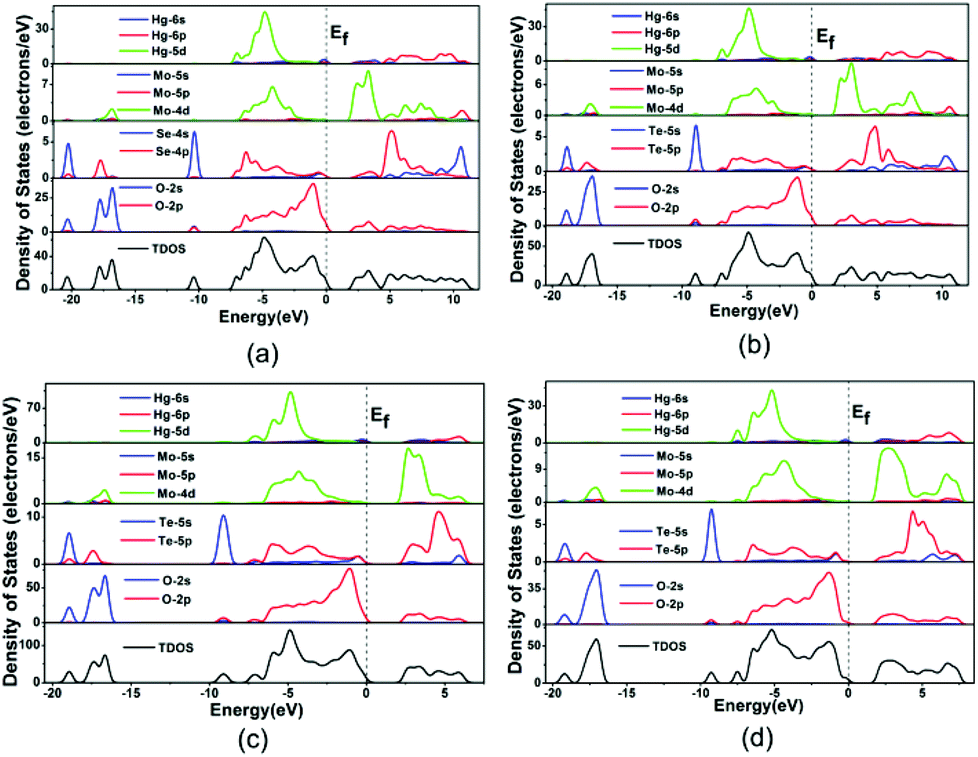 | ||
| Fig. 5 The total and partial density of states of Hg2MoSeO6 (a), α-Hg2MoTeO6 (b), β-Hg2MoTeO6 (c) and Hg2Mo2TeO9 (d). | ||
Population analyses give more information about quantitative bond analysis. The calculated bond orders of Hg–Hg, Hg–O, Mo–O, and Te(Se)–O bonds are 0.33–0.71, 0.02–0.35, 0.14–1.05, and 0.35–0.53 e, respectively, which indicate that the Hg–O bonds have more ionic character whereas the Hg–Hg, Mo–O and Te(Se)–O bonds are much more covalent in nature.
Conclusions
In summary, we have successfully synthesized four new mercury(I) molybdenum(VI) selenites and tellurites, namely, Hg2MoSeO6 (1), α-Hg2MoTeO6 (2), β-Hg2MoTeO6 (3), and Hg2Mo2TeO9 (4). They represent the first mercury(I) selenites and tellurites containing octahedrally coordinated d0 transition metal (TM) ions. Compounds 1–3 feature 1D anionic chains of [MoO3(QO3)]2− (Q = Se, Te) whereas Hg2Mo2TeO9 (4) features a novel [Mo2O5(TeO4)]2− anionic double layer. The results of our studies indicate that small changes in reaction conditions in the same system can lead to different chemical compositions and structures of the final products obtained. Our future research efforts will be focused on the introduction of other octahedrally-coordinated d0 transition metal cations such as V5+, Ti4+, W6+, etc. into the mercury selenite or tellurite systems.Acknowledgements
This work was supported by National Natural Science Foundation of China (grants no. 21231006, 21403232 and 91222108), NSF of Fujian Province (grant no. 2011J05037), and Key Laboratory of Optoelectronic Materials Chemistry and Physics.Notes and references
- (a) F. Kong, S. P. Huang, Z. M. Sun, J. G. Mao and W. D. Cheng, J. Am. Chem. Soc., 2006, 128, 7750 CrossRef CAS PubMed; (b) J.-G. Mao, H.-L. Jiang and F. Kong, Inorg. Chem., 2008, 47, 8498 CrossRef CAS PubMed; (c) H. S. Ra, K. M. Ok and P. S. Halasyamani, J. Am. Chem. Soc., 2003, 125, 7764 CrossRef CAS PubMed; (d) K. M. Ok and P. S. Halasyamani, Inorg. Chem., 2004, 43, 4248 CrossRef CAS PubMed; (e) R. T. Hart, K. M. Ok, P. S. Halasyamani and J. W. Zwanziger, Appl. Phys. Lett., 2004, 85, 938 CrossRef CAS PubMed; (f) J. Goodey, J. Broussard and P. S. Halasyamani, Chem. Mater., 2002, 14, 3174 CrossRef CAS.
- (a) K. M. Ok and P. S. Halasyamani, Chem. Mater., 2001, 13, 4278 CrossRef CAS; (b) P. S. Halasyamani, Chem. Mater., 2004, 16, 3586 CrossRef CAS; (c) N. Sau Doan and P. S. Halasyamani, Inorg. Chem., 2013, 52, 2637 CrossRef PubMed; (d) J. Yeon, S.-H. Kim, N. Sau Doan, H. Lee and P. S. Halasyamani, Inorg. Chem., 2012, 51, 609 CrossRef CAS PubMed; (e) S.-e. Bang, D. W. Lee and K. M. Ok, Inorg. Chem., 2014, 53, 4756 CrossRef CAS PubMed; (f) D. W. Lee and K. M. Ok, Inorg. Chem., 2013, 52, 5176 CrossRef CAS PubMed.
- (a) W.-L. Zhang, W.-D. Cheng, H. Zhang, L. Geng, C.-S. Lin and Z.-Z. He, J. Am. Chem. Soc., 2010, 132, 1508 CrossRef CAS PubMed; (b) S. Pan, J. P. Smit, B. Watkins, M. R. Marvel, C. L. Stern and K. R. Poeppelmeier, J. Am. Chem. Soc., 2006, 128, 11631 CrossRef CAS PubMed; (c) H. Jo, Y. H. Kim, D. W. Lee and K. M. Ok, Dalton Trans., 2014, 43, 11752 RSC; (d) C. Bai, C. Lei, S. Pan, Y. Wang, Z. Yang, S. Han, H. Yu, Y. Yang and F. Zhang, Solid State Sci., 2014, 33, 32 CrossRef CAS PubMed; (e) H. Wu, S. Pan, K. R. Poeppelmeier, H. Li, D. Jia, Z. Chen, X. Fan, Y. Yang, J. M. Rondinelli and H. Luo, J. Am. Chem. Soc., 2011, 133, 7786 CrossRef CAS PubMed.
- (a) T. Sivakurnar, H. Y. Chang, J. Baek and P. S. Halasyamani, Chem. Mater., 2007, 19, 4710 CrossRef; (b) H. Y. Chang, S.-H. Kim, K. M. Ok and P. S. Halasyamani, Chem. Mater., 2009, 21, 1654 CrossRef CAS; (c) N. Sau Doan, S.-H. Kim and P. S. Halasyamani, Inorg. Chem., 2011, 50, 5215 CrossRef PubMed; (d) J. Zhang, X. Tao, Y. Sun, Z. Zhang, C. Zhang, Z. Gao, H. Xia and S. Xia, Cryst. Growth Des., 2011, 11, 1863 CrossRef CAS; (e) J. Zhang, Z. Zhang, Y. Sun, C. Zhang, S. Zhang, Y. Liu and X. Tao, J. Mater. Chem., 2012, 22, 9921 RSC; (f) S.-J. Oh, D. W. Lee and K. M. Ok, Inorg. Chem., 2012, 51, 5393 CrossRef CAS PubMed.
- (a) C.-F. Sun, C.-L. Hu, X. Xu, J.-B. Ling, T. Hu, F. Kong, X.-F. Long and J.-G. Mao, J. Am. Chem. Soc., 2009, 131, 9486 CrossRef CAS PubMed; (b) B.-P. Yang, C.-L. Hu, X. Xu, C.-F. Sun, J.-H. Zhang and J.-G. Mao, Chem. Mater., 2010, 22, 1545 CrossRef CAS; (c) C.-F. Sun, C.-L. Hu, X. Xu, B.-P. Yang and J.-G. Mao, J. Am. Chem. Soc., 2011, 133, 5561 CrossRef CAS PubMed.
- (a) X.-A. Chen, L. Zhang, X.-A. Chang, H.-G. Zang and W.-Q. Xiao, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, I76 Search PubMed; (b) K. M. Ok and P. S. Halasyamani, Inorg. Chem., 2005, 44, 2263 CrossRef CAS PubMed; (c) C.-F. Sun, C.-L. Hu, F. Kong, B.-P. Yang and J.-G. Mao, Dalton Trans., 2010, 39, 1473 RSC; (d) C.-F. Sun, T. Hu, X. Xu and J.-G. Mao, Dalton Trans., 2010, 39, 7960 RSC.
- (a) T. C. Shehee, R. E. Sykora, M. K. Kang, P. S. Halasyamani and T. E. Albrecht-Schmitt, Inorg. Chem., 2003, 42, 457 CrossRef CAS; (b) X. A. Chen, X. A. Chang, H. G. Zang, Q. Wang and W. Q. Xiao, J. Alloys Compd., 2005, 396, 255 CrossRef CAS PubMed.
- (a) R. E. Sykora, D. M. Wells and T. E. Albrecht-Schmitt, Inorg. Chem., 2002, 41, 2304 CrossRef CAS PubMed; (b) R. E. Sykora, S. M. McDaniel, D. M. Wells and T. E. Albrecht-Schmitt, Inorg. Chem., 2002, 41, 5126 CrossRef CAS PubMed.
- W. Zhang, X. Tao, C. Zhang, Z. Gao, Y. Zhang, W. Yu, X. Cheng, X. Liu and M. Jiang, Cryst. Growth Des., 2008, 8, 304 CAS.
- (a) H. Y. Chang, S. W. Kim and P. S. Halasyamani, Chem. Mater., 2010, 22, 3241 CrossRef CAS; (b) H. Y. Chang, S. W. Kim and P. S. Halasyamani, Chem. Mater., 2010, 22, 3241 CrossRef CAS; (c) Y. L. Shen, H. L. Jiang, J. Xu, J. G. Mao and K. W. Cheah, Inorg. Chem., 2005, 44, 9314 CrossRef CAS PubMed; (d) H.-L. Jiang, F. Kong, Y. Fan and J.-G. Mao, Inorg. Chem., 2008, 47, 7430 CrossRef CAS PubMed; (e) X.-L. Cao, C.-L. Hu, X. Xu, F. Kong and J.-G. Mao, Chem. Commun., 2013, 49, 9965 RSC.
- Gmelins Handbuch der anorganischen Chemie, 34 Quecksilber B4, Verlag Chemie, Weinheim, 8th edn, 1969, p. 1094 Search PubMed.
- (a) M. Koskenlinna and J. Valkonen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1040 CrossRef; (b) M. Koskenlinna and J. Valkonen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 491 CrossRef; (c) M. Koskenlinna and J. Valkonen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 1070 CrossRef; (d) M. Weil, J. Solid State Chem., 2003, 172, 35 CrossRef CAS.
- (a) V. Kramer and G. Brandt, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 917 CrossRef; (b) M. Weil, Z. Kristallogr., 2003, 218, 691 CrossRef CAS; (c) J. D. Grice, Can.Mineral., 1989, 27, 133 CAS.
- W. M. Wendlandt and H. G. Hecht, Reflectance Spectroscopy, Interscience, New York, 1966 Search PubMed.
- (a) CrystalClear Version 1.3.5. Rigaku Corp., Woodlands, TX, 1999 Search PubMed; (b) G. M. Sheldrick, SHELXTL, Crystallographic Software Package, SHELXTL, Version 5.1, Bruker-AXS, Madison, WI, 1998 Search PubMed; (c) A. L. Spek, PLATON, Utrecht University, Utrecht, The Netherlands, 2001 Search PubMed.
- (a) V. Milman, B. Winkler, J. A. White, C. J. Pickard, M. C. Payne, E. V. Akhmatskaya and R. H. Nobes, Int. J. Quantum Chem., 2000, 77, 895 CrossRef CAS; (b) M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. S. Lin, A. Qteish, M. C. Payne and V. Heine, Phys. Rev. B: Condens. Matter, 1993, 47, 4174 CrossRef.
- (a) C. Robl and K. Haake, J. Chem. Soc., Chem. Commun., 1992, 1786 RSC; (b) G. Q. Huang, S. W. Zhang and M. C. Shao, Chem. J. Chin. Univ., 1995, 16, 670 CAS; (c) V. Balraj and K. Vidyasagar, Inorg. Chem., 1999, 38, 1394 CrossRef CAS; (d) A. Guesdon and B. Raveau, Chem. Mater., 2000, 12, 2239 CrossRef CAS; (e) F. Kong, C.-L. Hu, X. Xu, T.-H. Zhou and J.-G. Mao, Dalton Trans., 2012, 41, 5687 RSC.
- (a) N. E. Brese and M. Okeeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192 CrossRef; (b) I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- V. A. Blatov, IUCr CompComm Newsl., 2006, 7, 4 Search PubMed . http://www.topos.ssu.samara.ru.
- J. J. Zhang, Z. H. Zhang, W. G. Zhang, Q. X. Zheng, Y. X. Sun, C. Q. Zhang and X. T. Tao, Chem. Mater., 2011, 23, 3752 CrossRef CAS.
- (a) Y. H. Kim, D. W. Lee and K. M. Ok, Inorg. Chem., 2013, 52, 11450 CrossRef CAS PubMed; (b) R. E. Sykora, K. M. Ok, P. S. Halasyamani and T. E. Albrecht-Schmitt, J. Am. Chem. Soc., 2002, 124, 1951 CrossRef CAS PubMed; (c) J. Goodey, K. M. Ok, J. Broussard, C. Hofmann, F. V. Escobedo and P. S. Halasyamani, J. Solid State Chem., 2003, 175, 3 CrossRef CAS; (d) M.-H. Choi, S.-H. Kim, H. Y. Chang, P. S. Halasyamani and K. M. Ok, Inorg. Chem., 2009, 48, 8376 CrossRef CAS PubMed; (e) D. W. Lee, D.-b. Bak, S. B. Kim, J. Kim and K. M. Ok, Inorg. Chem., 2012, 51, 7844 CrossRef CAS PubMed.
- M. A. Frechero, O. V. Quinzani, R. S. Pettigrosso, M. Villar and R. A. Montani, J. Non-Cryst. Solids, 2007, 353, 2919 CrossRef CAS PubMed.
- W. T. A. Harrison, L. L. Dussack and A. J. Jacobson, J. Solid State Chem., 1996, 125, 234 CrossRef CAS.
- L. N. Kurilenko, N. V. Serebryakova, E. I. Saunin, V. V. Gromov and N. P. Sokolova, Russ. Chem. Bull., 1988, 37, 839 CrossRef.
- N. Sau Doan and P. S. Halasyamani, Inorg. Chem., 2012, 51, 9529 CrossRef PubMed.
- (a) R. W. Godby, M. Schluter and L. J. Sham, Phys. Rev. B: Condens. Matter, 1987, 36, 6497 CrossRef CAS; (b) H.-L. Jiang, F. Kong and J.-G. Mao, J. Solid State Chem., 2007, 180, 1764 CrossRef CAS PubMed; (c) C. M. I. Okoye, J. Phys.: Condens. Matter, 2003, 15, 5945 CrossRef CAS; (d) R. Terki, G. Bertrand and H. Aourag, Microelectron. Eng., 2005, 81, 514 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format, simulated and experimental XRD powder patterns, the calculated state energies of the L-CB and H-VB, IR spectra, UV spectra, TGA and DSC curves, coordination environments around the Hg and Te atoms for three compounds. See DOI: 10.1039/c4qi00132j |
| This journal is © the Partner Organisations 2014 |