Study on effective extraction of chicken feather keratins and their films for controlling drug release†
Xiao-Chun
Yin
,
Fang-Ying
Li
,
Yu-Feng
He
,
Yan
Wang
and
Rong-Min
Wang
*
Key Laboratory of Eco-Environment-Related Polymer Materials of Ministry of Education, Key Laboratory of Polymer Materials of Gansu Province, Institute of Polymer, Northwest Normal University, Lanzhou 730070, China. E-mail: wangrm@nwnu.edu.cn; Tel: +86-931-7970358
First published on 26th February 2013
Abstract
We report the fabrication of keratin films with porosity using keratin, for controlled drug delivery systems. Feathers are currently an important waste product from the poultry industry, however abandoned down products cause an environmentally difficult disposal problem as they are not efficiently extracted and applied. In this paper, feather keratin was extracted using a high-efficiency method. The yield of pure keratin was up to 90%. Its molecular weight was 20 kDa with low dispersity. The films, based on the extracted keratin, were prepared using a simple method, and showed good mechanical properties. Furthermore, the feather keratin films were used to load and release drugs. The resultant feather keratin biopolymer films were pH-responsive and showed controllable drug-release behavior. In summary, we achieved a simple method to reutilize disused feathers. It could be an attractive candidate for applications in the biomedical field.
Introduction
Over the past decade, the use of biodegradable polymers for the administration of pharmaceuticals and biomedical agents has increased dramatically. The most important biomedical applications of biodegradable polymers are in the areas of controlled drug delivery systems (DDSs).1 In controlled DDSs, an active therapeutic is incorporated into a polymeric network structure in such a way that the drug is released from the material in a predefined manner.2 A variety of synthetic and natural polymers have been applied as drug carriers.3 In general, synthetic polymers offer greater advantages over natural ones in that they can be tailored to give a wider range of properties. However, toxicity is more likely to be associated with these synthetic polymers. Therefore, a safer carrier becomes necessary. Natural polymers may generally be considered safer than synthetic polymers. Therefore, natural polymers have certain advantages as drug delivery carriers.Several proteins, including collagen, albumin, gelatin, fibroin and keratin, have been investigated in the development of naturally-derived biomaterials.4 Of these proteins, keratin-based materials show promise for revolutionizing the biomaterial world5 because of their biocompatibility, biodegradability,6 mechanical durability, and natural abundance. Over the past century, research has focused on the extraction, purification and characterization of keratin from hair, wool fibers, etc. It has led to the development of keratin based biomaterials, which can be applied in controlled DDSs. Feathers, the most abundant keratinous biomass worldwide, are a valuable and renewable protein source.7 However, feathers are currently an important waste product in the poultry industry, causing an environmentally difficult disposal problem as they are not efficiently extracted and applied. Therefore, from both an economic and environmental point of view, it is quite desirable to develop an effective and profitable process to use these kinds of resources.8 Feathers are mainly composed of the structural protein, keratin. Keratin is not a single substance. In fact, it is composed of a complex mixture of proteins. Keratin is insoluble in many common solvents, such as dilute acids, alkalines, water, and organic solvents.9
There are several procedures available to dissolve feather keratin. For example, acid and alkali hydrolysis,10 the reduction of disulfide bonds with alkaline sodium sulfide solutions, a combination of enzymatic and chemical treatment,11 the use of ammonium copper hydroxide,12 and the oxidation of disulfide bonds with performic acid.13 These dissolution methods are in addition to peptide bond scission. A common method to extract keratin involves the use of reducing agents because the native form is hard to obtain, due to its highly cross-linked state by disulfide bonds. The use of the Shindai method, which omits the use of detergent, provides a sufficient amount of extracted proteins for analysis and can avoid protein hydrolysis.14 Reducing agents, such as dithiothreitol and 2-mercaptoethanol, work by cleaving keratin’s disulfide bonds and thus increasing it’s solubility.9
The ability to fabricate a variety of drug delivery constructs with different morphologies such as films, gels, foams, microparticles and scaffolds contributes to a broad application of the biomaterial.15 It is possible to develop drug delivery carriers based on keratin. The aim of this study was to obtain a simple and effective method for extracting keratin from chicken feathers, to prepare keratin films based on the extracted feather keratin and investigate the controllable drug release properties of keratin films. Here, rhodamine B was chosen as a kind of water-soluble model drug molecule, which measured the drug delivery function of the keratin carriers.
Materials and methods
Chicken feathers were collected at a local poultry farm in Gansu Province, western China. All the chemicals used in the experiments were of analytical grade and used without further purification.Extraction of keratin from feathers
Feather keratin was extracted under reducing conditions with the so-called Shindai method.19 The process of extraction is described in the ESI,† and included three steps: ethanol pretreatment, hydrochloric acid pretreatment and 2-mercaptoethanol deoxidization. The typical procedure was as follows: raw feathers were washed 3 times with water to remove sand and dust. Then, they were dried and cut into small filaments with lengths of 1–2 mm. 3 g of clean feathers were refluxed in EtOH–H2O for 2 h. Then they were pretreated with HCl solution for 2 h to remove grease. The ethanol and HCl were filtered off and the clean pretreated feathers were stored at room temperature.The pretreated feathers were immersed in 150 mL of a solution containing urea (0.33 mol L−1), SDS (0.05 mol L−1), 2-mercaptoethanol (0.095 mol L−1) and Tris (0.016 mol L−1) at pH 8.0. The mixture was stirred at 70 °C for 2 h under a N2 atmosphere. After being filtered, a keratin solution was obtained. The solution was acidified, and the keratin was precipitated with ethanol. Subsequently, the keratin sedimentation was washed five times with distilled water and lyophilized.
Reactions were performed in a flask containing 150 mL reduced keratin solution, the solution was agitated at 70 °C. Then, 5 mL of chloroacetic acid solution (0.14 g mL−1) was added dropwise in 5 min for regulating the pH value to 8–9. After reacting for 1 h, the reaction was stopped and acidified. The keratin was precipitated with HCl solution, washed with water and lyophilized, which afforded the modified keratin.
Preparation of feather keratin films
The aqueous dispersion of the reduced keratin was mixed with a certain amount of glycerol. The mixture was stirred for 1 h at room temperature. These film-forming dispersions were poured into polystyrene petri dishes (10 cm diameter) and dried in a ventilated oven at 50 °C for 24 h. The films were then removed from the casting surface (petri dish) and stored for analysis. The thickness of the films was controlled by varying the volume of the keratin dispersion poured into the petri dish. Film thickness was measured with a micrometer with a sensitivity of 0.001 mm. Five to ten thickness measurements were carried out for each film, from which an average was obtained.FT-IR spectroscopy
The FT-IR spectra of the feather keratin were measured with a model FTS 3000 FT-IR Nexus spectrometer from DIGILAB equipped with a ZnSe ATR cell (model Smart Performer). The analysis was performed in triplicate.Circular dichroism (CD)
CD spectra were measured using a model J-810 CD spectrometer from JASCO with a 1 mm path length at room temperature. The wavelength was 180 nm < λ < 300 nm. Solutions were highly diluted by the tripartite water. Then, it was supplied for CD measurement.Elemental analysis and amino acid analysis
Elemental analysis (C, H, N, and S) was performed on the freeze-dried samples of the feather keratin using an elementary analyzer (Vario EL from Elementar, Germany). The amino acid composition of the feather keratin was determined using a Hitachi 835-50 Amino Acid Analyzer. Free amino acids were analyzed by HPLC according to the AccQ-Tag method (Waters). The eluate was detected at 254 nm. The column of keratin solution samples was 10 mL using tetrahydrofuran as solvent. Samples were analyzed in duplicate (error: <2%). The quantitative amino acid composition, expressed as mol% for each amino acid, was determined by external standard calibration (Amino Acid Standard H, Pierce).MALDI-TOF MS
The molar mass range of the extracted feather keratin was measured by a matrix-assisted laser desorption ionization time-of-flight mass spectrometer (MALDI-TOF MS) (Biflex III from Bruker Daltonics) equipped with a 337 nm nitrogen laser. The matrix was trans-3,5-dimethoxy-4-hydroxycinnamic acid.Scanning electron microscopy (SEM)
The surface morphology of the keratin film was observed with scanning electron microscopy (JSM-6701F, Japan). The specimen was washed in ethanol, dried, coated with gold under vacuum and observed.Thermogravimetry measurement
All the samples (10.0 mg) used had been dried in a vacuum at 70 °C for 12 h. The thermal degradation test was conducted on a TG/DTA (Pyris Diamond, PerkinElmer) at a heating rate of 10 °C min−1 under nitrogen. The change in the weight differential difference with temperature was recorded.Mechanical properties
Stress–strain curves were determined by uniaxial measurement using an INSTRON 5500 mechanical tester (Instron, USA). For that, the films were cut into rectangular pieces (10 × 20 mm2) and fixed on the grips of the device with a gap of 20 mm. The films were tractioned at a speed of 20 mm min−1. The ultimate strength, the ultimate elongation and Young's modulus were calculated from five independent measurements.Preparation of drug-loaded keratin films and controllable release properties
25 mg of rhodamine B (RB) was stirred in 5 mL of distilled water at room temperature until a homogeneous solution was obtained. 0.5 g of glycerol was added to the 30 mL of the aqueous dispersion containing the reduced keratin (1 g keratin/30 mL dispersion), which was then mixed with the drug solution. The film-forming dispersion was prepared under constant magnetic stirring at room temperature and mixed for 1 h. The films were then poured into polystyrene petri dishes (10 cm diameter) and dried in a ventilated oven at 50 °C for 24 h. After this, the drug-loaded keratin films were removed from the casting surface (petri dish) and stored. The drug-loaded keratin films were immersed in 300 mL water. At appropriate time intervals, the amount of drug released was determined by UV-vis spectra (in aqueous solution measured at 553.5 nm).Results and discussion
Extraction of keratin from chicken feathers
Keratin, present in feathers, is insoluble in polar solvents like water, weak acids and bases, as well as in apolar solvents while it is active because cystine can be reduced, oxidized and hydrolyzed.16 The addition of reducing agent for scissoring cystine decreases the strength of the fibers. Therefore, it is possible to dissolve keratin using solvents by scissoring –S–S– bonds and adding hydrogen bonds.17 Here, a simple and powerful procedure was designed which involved two steps of pretreatment and using 2-mercaptoethanol to scissor the disulfide bonds under moderately alkaline conditions, and urea to break the hydrogen bonds. Although it has been reported that 2-mercaptoethanol and urea have been used in reducing keratins for wool keratins,18 many procedures have involved lots of reacted agents.19 In order to calculate the yield of the extracted keratin, the feathers were cleaned, dried and weighed before pretreatment.Due to the presence of numerous disulfide bonds and hydrogen bonds, excess reducing agent is necessary. In this study, 2-mercaptoethanol was used for the scission of disulfide bonds. The amount of 2-mercaptoethanol used was lower than that of Schrooyen's method,18 which used white body feathers from broilers. Sodium dodecyl sulfate (SDS), a typical surfactant, was used to protect the reduced keratin as it formed complexes with the keratin, and prevented extensive aggregation of the protein after dissolution. The yield of pure keratin obtained was 93%. This exceeded the results obtained by Schrooyen who obtained a yield of about 75%, based on the starting weight of the feathers.
In general, some treatment conditions, such as temperature, time, pH value and dosage of the reagents, had a significant influence on the yield and the molecular weight of the extracted keratin. Therefore, these factors were investigated.
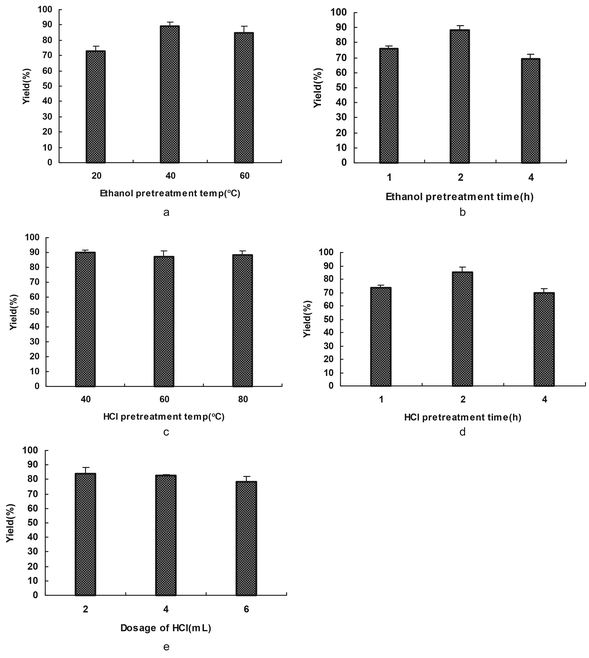 | ||
| Fig. 1 The influence of pretreatment on solubilization. (a) and (c) show the effects of ethanol pretreatment temperature and HCl pretreatment temperature on the keratin yield. (b) and (d) show the effects of time in ethanol and HCl pretreatment on the keratin yield. (e) shows the effect of the dosage of HCl on the keratin yield. | ||
As reduced keratin can be re-oxidized easily in air, a gel was rapidly formed as a result of the cross-linking of the polyfunctional keratin chains. Here, SDS, was used as a highly effective surfactant that lowers the surface tension of aqueous solutions. SDS is a negatively charged surfactant; this new negative charge is greater than the original charge of that protein, so the charge of the protein can be ignored. The electrostatic repulsion that is created by the binding of SDS causes proteins to unfold. Thus, SDS was used to protect the reduced keratin as it formed complexes with the keratin and prevented extensive protein aggregation after the keratin was reduced. We thought that this procedure could also be carried out by chemical modification with ingredients which contain carboxyl compounds.
In summary, the optimal processes and conditions for extracting up to 90% of the keratin from feathers were as follows: (I) EtOH pretreatment: 40 °C, 2 h. (II) HCl pretreatment: 1 mL of HCl for 1 g of feathers at 40 °C for 2 h. (III) 2-Mercaptoethanol deoxidization: 2 g of feathers, 0.5 mL of 2-mercaptoethanol, feather![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SDS = 1:0.8(g/g), feather:urea = 1:1(g/g), 70 °C, 2 h under a nitrogen atmosphere.
:SDS = 1:0.8(g/g), feather:urea = 1:1(g/g), 70 °C, 2 h under a nitrogen atmosphere.
Characterization of extracted feather keratin
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, and occurred in the range 1700–1600 cm−1. Amide II, which falls in the 1540–1520 cm−1 range, was related to N–H bending and the C–H stretching vibration. Amide III occurred in the range 1220–1300 cm−1, and resulted from in phase combination of C–N stretching and the CO bending vibration. The peaks at 990 cm−1 and 580 cm−1 were the characteristic absorptions of the C–S and S–S bonds.22,23 In addition, the positions of these bands indicated the conformations of the protein material:24,25 1650 cm−1 and 1630 cm−1 for amide I, 1540 cm−1 and 1520 cm−1 for amide II, and 1230 cm−1 and 1270 cm−1 for amide III.26
O stretching, and occurred in the range 1700–1600 cm−1. Amide II, which falls in the 1540–1520 cm−1 range, was related to N–H bending and the C–H stretching vibration. Amide III occurred in the range 1220–1300 cm−1, and resulted from in phase combination of C–N stretching and the CO bending vibration. The peaks at 990 cm−1 and 580 cm−1 were the characteristic absorptions of the C–S and S–S bonds.22,23 In addition, the positions of these bands indicated the conformations of the protein material:24,25 1650 cm−1 and 1630 cm−1 for amide I, 1540 cm−1 and 1520 cm−1 for amide II, and 1230 cm−1 and 1270 cm−1 for amide III.26
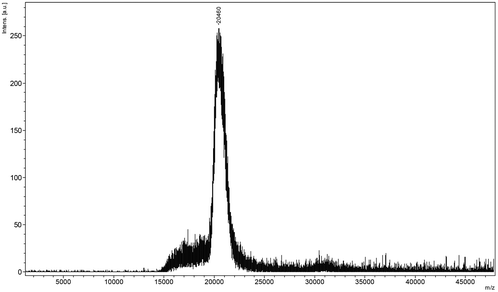 | ||
| Fig. 2 The MALDI-TOF chart of the extracted keratin. | ||
| a The amount was corrected with the results of elemental analysis. | ||||||
|---|---|---|---|---|---|---|
| Amino acid | Ala | Cysa | Pro | Ser | Glu | Leu |
| Content in keratin (mol%) | 29.54 | 14.42 | 10.12 | 6.98 | 5.58 | 5.07 |
| Amino acid | Gly | Val | Asp | Arg | Thr | Ile |
| Content in keratin (mol%) | 4.75 | 4.26 | 3.97 | 3.7 | 3.62 | 3.29 |
| Amino acid | Phe | Tyr | Lys | His | Met | |
| Content in keratin (mol%) | 2.55 | 1.16 | 0.66 | 0.19 | 0.13 | |
As mentioned above, we found that dry keratin could not be redissolved in water. The reason is pure keratin can be re-oxidized easily in air, causing the cross-linking of the keratin chains by reforming S–S bonds. The application of dry keratin is therefore limited. While, carboxyl compound, glycine or chloroacetic acid was also used to modify the dissolved feather keratin, and water-soluble keratin derivatives were obtained. Dry water-soluble keratin was obtained by deposition and lyophilization. The dry keratin could also be redissolved in water. This meant that water-soluble keratin was obtained which should increase the possible application. Compared with the research of Schrooyen,18 who modified the feather keratin, the modifiers we used were cheap and non-toxic.
The average sulfur content of feather keratin was 4.0% (w/w), as determined by elemental analysis. These sulfur groups originated from different amino acids, such as cystine disulfide bonds (–S–S–), cysteine free thiol groups (–SH), or methionine. It was reported that the amount of free thiol groups was less than 5% (w/w) of the total cysteine in feathers.28 The content of methionine in feathers is low.29 It is assumed that the 4.0% (w/w) of sulfur measured was mainly due to disulfide linkages. Therefore, one feather keratin chain contained approximately 13 disulfide groups. This meant that there were 26 cysteine residues in one keratin chain if all the disulfide bonds were broken. In this study, to scissor the disulfide bonds, a 22-fold molar excess of 2-mercaptoethanol with respect to the disulfide bonds was used. This was less than the amount used by Schrooyen (100-fold molar excess).18
Keratin is a major structural fibrous protein and provides an outer covering. It contains scores of cysteine residues (14–15%) which can build intermolecular cross-links by disulfide bonding. The hydrophobic amino acid residues (60%) of keratin chains also play a very important role in keratin configuration (see Fig. 3a and 3b). The molecular weight of the extracted keratin was 20 kDa, based on the results of MALDI-TOF MS. An average molecular weight of 96.32 for each amino acid residue of the extracted keratin was calculated. There were about 202 amino acids in the extracted keratin. This MALDI-TOF MS data agrees with the published data that feather keratin consists of 96 amino acid residues.30 Based on the results of FT-IR and CD, the secondary structure of the feather keratin mainly consisted of the β-sheet conformation. During the extraction procedure, the β-sheet conformation was partly destroyed, and the keratin chains had a random coil conformation.
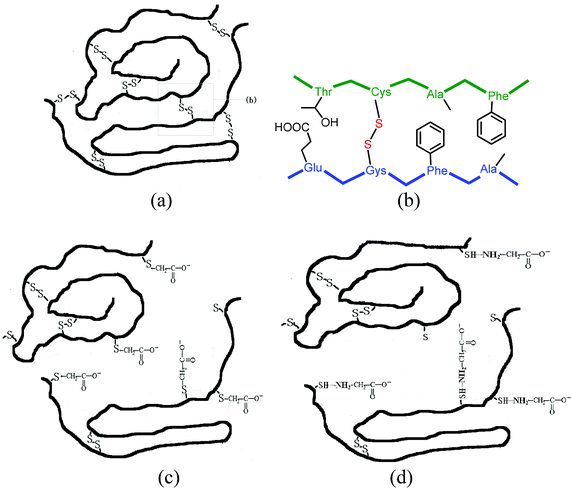 | ||
| Fig. 3 (a) and (b) show the cross-links of disulfide bonds in feather keratin. (c) and (d) show the suggested structures of reversible water-soluble keratin. | ||
The reduced keratin could be re-oxidized easily in air, which caused the solubility to decrease. In this study, SDS, a typical surfactant, was used to protect the reduced keratin as it formed complexes with the keratin. We found that the extracted keratin re-aggregated due to cross-linking of the polyfunctional keratin chains, when the mixture was dialyzed extensively against distilled water. The solid soluble extracted keratin became insoluble if stored for more than 3 days. In order to prevent aggregation of the extracted keratins, carboxy compounds, such as glycine and chloroacetic acid, were used to modify the extracted keratin. It was found that the extracted keratin did not re-aggregate when carbonyl compounds were introduced to the cysteine residues of the keratin. This meant that completely soluble keratin was obtained. Based on the results of MALDI-TOF MS, the molecular weight of chloroacetic acid-modified feather keratin was 20.735 kDa, which was higher than that of feather keratin. In the IR spectra of chloroacetic acid-modified feather keratin, the characteristic absorption peaks at 1643 cm−1 (CO) and 1527 cm−1 (N–H) were shifted compared to feather keratin. At the same time, the absorption intensity of CO was increased. That meant that the ingredients were inserted into the keratin chain. The mode of combining the extracted keratin with the ingredients is suggested in Fig. 3c and 3d. The obtained reversible water-soluble derivatives may be applied in the areas of controlled drug delivery systems.
Characterization of keratin films
| Glycerol conc (g gly per g FK) | Ultimate strength (MPa) | Elongation (%) | Young's modulus (MPa) |
|---|---|---|---|
| 0.1 | — | — | — |
| 0.2 | — | — | — |
| 0.3 | 7.56 | 111.37 | 27.61 |
| 0.4 | 6.64 | 121.52 | 14.41 |
| 0.5 | 4.56 | 103.29 | 10.88 |
 | ||
| Fig. 4 The micrographs of the chicken feather keratin film (a) and keratin powder (b). | ||
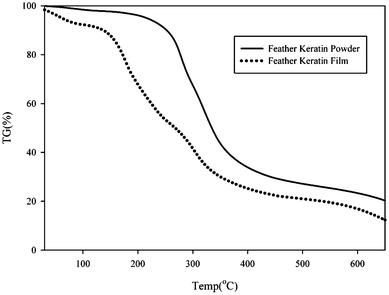 | ||
| Fig. 5 The TG curve of the feather keratin powder and feather keratin film. | ||
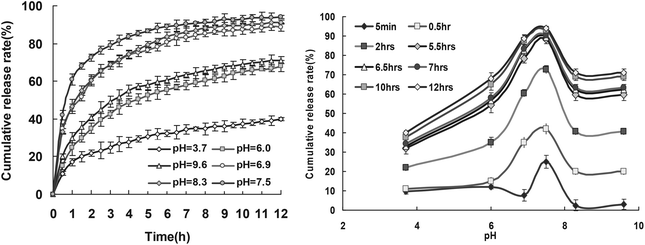 | ||
| Fig. 6 Release profiles of RB from drug loaded keratin films at 20 °C. | ||
Conclusion
In summary, keratin films with porosity possessed excellent controlled release properties. Firstly, the high pure keratin had been extracted and feather keratin was stable and homogeneous. The yield of keratin from the feathers was 93%, the molecular weight of the feather keratin was 20 kDa with low dispersity, and included 14–15% cysteine residues. Secondly, the films based on the extracted keratin were easily prepared. They showed good mechanical properties. Furthermore, the feather keratin films were used to load drugs. The pH-sensitivity and biocompatible skeleton will surely make the resultant film an attractive candidate for application in the biomedical field. We achieved a simple method to reutilize disused feathers. Meanwhile, a new kind of raw material which could be used as cosmetic, iatrical and officinal materials, and biodegradable materials was obtained.Acknowledgements
The authors thank Dr Shaoxiang Xiong, Professor of the Institute of Chemistry, Chinese Academy of Sciences, Beijing, China, for MALDI-TOF MS analysis. We also thank Professor Ming Lin of the Health Science Center in Peking University for their help in the amino acid analysis. The project was supported by the NSFC (no. 21263024, 21244003), PCSIRT (IRT1177), the Gansu Sci & Techn Support Project (1011GKCA017), and Funda Res Funds Gansu Univ (2010-176).References
- Y. Lu and S. C. Chen, Adv. Drug Delivery Rev., 2004, 56, 1621 CrossRef CAS.
- V. Jogani, K. Jinturkar, T. Vyas and A. Misra, Recent Pat. Drug Delivery Formulation, 2008, 2, 25 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3 CrossRef CAS.
- Q. Lu, S. J. Zhang, K. Hu, Q. L. Feng, C. B. Cao and F. Z. Cui, Biomaterials, 2007, 28, 2306 CrossRef CAS.
- A. J. Poole, J. S. Church and M. G. Huson, Biomacromolecules, 2009, 10, 1 CrossRef CAS.
- S. Balaji, R. Kumar, R. Sripriya, P. Kakkar, D. Vijaya Ramesh, P. Neela Kanta Reddy and P. K. Sehgal, Mater. Sci. Eng., C, 2012, 32, 975 CrossRef CAS.
- A. A. Onifade, N. A. Al-Sane, A. A. Al-Musallam and S. Al-Zarban, Bioresour. Technol., 1998, 66, 1 CrossRef CAS.
- Y.-X. Wang and X.-J. Cao, Process Biochem., 2012, 47, 896 CrossRef CAS.
- H. H. Bragulla and D. G. Homberger, J. Anat., 2009, 214, 516 CrossRef CAS.
- Y. Nagai and T. Nishikawa, Agric. Biol. Chem., 1970, 34, 16 CrossRef.
- P. G. Dalev, Bioresour. Technol., 1994, 48, 265 CrossRef CAS.
- Y. Nagai and T. Nishikawa, Agric. Biol. Chem., 1970, 34, 575 CrossRef CAS.
- B. S. Harrap and E. F. Woods, Biochem. J., 1964, 92, 81 Search PubMed.
- A. Nakamura, M. Arimoto, K. Takeuchi and T. Fujii, Biol. Pharm. Bull., 2002, 25, 569 CAS.
- E. Wenk, H. P. Merkle and L. Meinel, J. Controlled Release, 2011, 150, 128 CrossRef CAS.
- R. Endo, K. Kamei, I. Iida and Y. Kawahara, J. Archaeol. Sci., 2008, 35, 1240 CrossRef.
- P. M. M. Schrooyen, P. J. Dijkstra, R. C. Oberthur, A. Bantjes and J. Feijen, J. Agric. Food Chem., 2000, 48, 4326 CrossRef CAS.
- P. M. M. Schrooyen, P. J. Dijkstra, R. C. Oberthur, A. Bantjes and J. Feijen, J. Colloid Interf. Sci., 2001, 240, 30 CrossRef CAS.
- G. R. P. Moore, S. M. Martelli, C. Gandolfo, P. J. A. Sobral and J. B. Laurindo, Food Hydrocolloids, 2006, 20, 975 CrossRef CAS.
- K. H. Dahl and J. S. McKinley-McKee, Bioorg. Chem., 1981, 10, 329 CrossRef CAS.
- N. H. Leon, Textile Progress, 1975, 7, 1 CrossRef.
- A. Vasconcelos, G. Freddi and A. Cavaco-Paulo, Biomacromolecules, 2008, 9, 1299 CrossRef CAS.
- E. Wojciechowska, A. Wlochowicz and A. Weselucha-Birczynska, J. Mol. Struct., 1999, 511–512, 307 CrossRef CAS.
- N. V. Bhat and S. M. Ahirrao, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 1273 CrossRef CAS.
- S. W. Ha, A. E. Tonelli and S. M. Hudson, Biomacromolecules, 2005, 6, 1722 CrossRef CAS.
- M. Tsukada, Y. Gotoh, M. Nagura, N. Minoura, N. Kasai and G. Freddi, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 961 CrossRef CAS.
- N. Greenfield and G. D. Fasman, Biochemistry, 1969, 8, 4108 CrossRef CAS.
- K. Arai, T. Hirata, S. Nishimura, M. Hirano and S. Naito, J. Appl. Polym. Sci., 1993, 47, 1973 CrossRef CAS.
- K. Akahane, S. Murozono and K. Murayama, J. Biochem., 1977, 81, 11 CAS.
- K. M. Arai, R. Takahashi, Y. Yokote and K. Akahane, Eur. J. Biochem., 1983, 132, 501 CrossRef CAS.
- T. Tanabe, N. Okitsu, A. Tachibana and K. Yamauchi, Biomaterials, 2002, 23, 817 CrossRef CAS.
- P. M. M. Schrooyen, P. J. Dijkstra, R. C. Oberthur, A. Bantjes and J. Feijen, J. Agric. Food Chem., 2001, 49, 221 CrossRef CAS.
- S. Reichl, M. Borrelli and G. Geerling, Biomaterials, 2011, 32, 3375 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm00158j |
| This journal is © The Royal Society of Chemistry 2013 |