
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iodine-enabled organoelectrocatalysis: enantioselective cross dehydrogenative coupling of sulfides and aldehydes†
Zhen
Wang
,
Marcel
Gausmann
,
Jan-Hendrik
Dickoff
and
Mathias
Christmann
 *
*
Freie Universität Berlin Institute of Chemistry and Biochemistry, Takustr. 3, 14195 Berlin, Germany. E-mail: mathias.christmann@fu-berlin.de
First published on 19th February 2024
Abstract
A method for the direct asymmetric α-sulfenylation of aldehydes with sulfides was developed. By merging electrochemistry and asymmetric organocatalysis, we obtained α-sulfenylated aldehydes with good to excellent enantioselectivities. Mechanistic investigations indicated a pivotal role of iodine as a catalytic mediator, not only facilitating redox transformations but also possibly contributing to the formation of sulfenyl iodide (RSI) intermediates derived from electrochemically generated disulfides. Our metal-free protocol offers a sustainable and efficient route to access a wide array of α-sulfenylated aldehydes. Remarkably, these transformations take place at room temperature, obviating the need for additional stoichiometric oxidants, thus exemplifying an environmentally friendly and practical synthetic strategy.
Introduction
Enantiomerically enriched α-sulfenylated aldehydes are key intermediates for preparing important chiral building blocks,1 such as β-hydroxysulfides existing widely in natural products and pharmaceuticals,2 chiral secondary alcohols and 1,2-disubstituted epoxides3 (Fig. 1A). Furthermore, thioethers can be converted to sulfoxides and sulfones, which are prevalent in active pharmaceutical ingredients (APIs) and natural products4 (Fig. 1B). Consequently, many efficient methods have been devised to construct the α-sulfenylated aldehyde motif.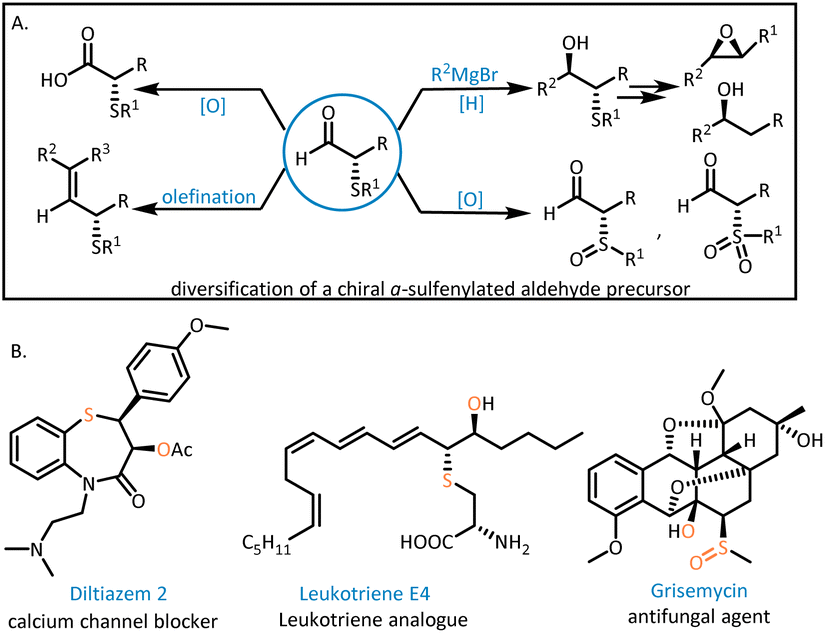 | ||
| Fig. 1 Diversification of chiral α-sulfenylated aldehydes (A) and derivatives of chiral α-sulfenylated aldehydes in natural products and pharmaceuticals (B). | ||
In the most common approach, enamines react with sulfur-based electrophiles to give chiral α-sulfenylated aldehydes.5 Most sulfenylation reactions feature reagents with a weak N–S bond as a “S+” equivalent. As a consequence, large nitrogen-based leaving groups are formed as by-products thus reducing the reaction's atom economy6 (Scheme 1a). A complementary approach has been reported by the Jørgensen group utilizing an oxidative umpolung strategy via α-substituted O-bound quinol ethers (Scheme 1b).7 This method is very effective for α-branched aldehydes but also requires DDQ or other stoichiometric oxidants. The direct dehydrogenative C–H/S–H cross-coupling is the most attractive and environmentally benign approach to construct chiral α-sulfenylated aldehydes8 (Scheme 1c). Unfortunately, a rapid and effective enantioselective method using unmodified thiols is less developed.
 | ||
| Scheme 1 Methods for the synthesis of enantiomerically enriched α-sulfenylated aldehydes. | ||
Recently, merging organocatalysis and electrocatalysis has helped to realize dehydrogenative cross-couplings in asymmetric synthesis.9 With the tunability of the redox potential, traditional oxidants or reductants can be avoided. For instance, Jørgensen demonstrated the possibility of a direct intermolecular α-arylation of aldehydes using electron-rich aromatic compounds wherein a transformation is impossible by Friedel–Crafts reactions.10 Luo used an asymmetric enamine addition to an anode-generated benzyne intermediate to construct α-arylated and α-cyclohexenylated cyclic β-ketocarbonyls.11 Mei reported an unusual asymmetric Shono-type oxidation with acyclic amines by means of anodic oxidation and organocatalysis.12 In related studies, Meggers,13 Lin,14 and Guo15 have demonstrated the effectiveness of employing transition-metal catalysts towards this goal.
Despite these significant advancements, the direct synthesis of chiral α-sulfenylated aldehydes using thiols remains challenging. First, nucleophilic thiols are not reactive toward nucleophilic enol or enamine intermediates. To address this limitation, it is required to identify a suitable mediator effecting a polarity reversal at sulfur. Second, thiols immediately form disulfides at the anode according to a literature report,16 and the amino catalyst can possibly react with disulfides.17 In addition, thiol radicals could be generated in the reaction system. The amino catalyst and enamine intermediate can form radical intermediates in an oxidative environment that may lead to undesired side reactions.18 Finally, the α-sulfenylated product could be further oxidized to the sulfoxides and sulfones.
Inspired by iodine-catalyzed electrooxidative cross-coupling reactions19 and our continuing efforts in asymmetric organic synthesis,20 we envisioned merging organo- and electrocatalysis. With the anode oxidation, we propose that iodine as a mediator assists the electrophilic sulfenylation by converting sulfides/disulfides into an electrophilic sulfur species 7 (Fig. 5). The hydrogen iodide byproduct of the reaction is electrochemically converted into hydrogen with concomitant regeneration of iodine. Furthermore, the redox mediator iodine can decrease the oxidation potential of the reaction, avoiding the overoxidation of the sulfenylated product. Herein, we report our study on the direct enantioselective α-sulfenylation of aldehydes between thiols and aldehydes via organoelectrocatalysis with catalytic amount of iodine (Scheme 1c). Compared with traditional multi-step methods,3,5 the outstanding features of our protocol for the preparation of α-sulfenylated aldehydes include mild conditions, no additional stoichiometric oxidants, and high atom economy, which all together are in line with the 12 principles of green chemistry.21
Results and discussion
Reaction optimization
We commenced our studies by using hydrocinnamaldehyde (1a) and 4-methyl thiophenol (2a) as substrates, I2 and secondary amine (3a) as catalysts, n-Bu4NPF6 (2.0 equiv.) as the electrolyte and 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) MeCN/H2O as the solvent in an undivided cell equipped with two platinum plate electrodes to screen the optimized reaction conditions (Table 1). Several commonly used chiral amine catalysts were tested first. To our delight, the desired product 4a was obtained in 20% yield with 80% ee value using (2S,5R)-2-(tert-butyl)-3,5-dimethylimidazolidin-4-one (3a) as the catalyst (entry 1). In contrast, L-proline 3b provided the product as a racemate (entry 2). The primary anime 3e did not afford any desired product (entry 5). Increasing the constant current to 5 mA afforded the product with high yield and enantioselectivity (entry 6). Further increase of the current lowered the yield and the enantiomeric excess (entry 7). As solvent, 300:1 (v/v) MeCN/H2O was found to give the optimal yields and highest ee values. H2O was an essential factor for good ee value but further increasing the amount of H2O had a deleterious effect on the yield (entries 8–10). This phenomenon may be attributed to the fact that an appropriate amount of H2O facilitates the hydrolysis of iminium intermediates while excess H2O leads to catalyst deactivation.22
:1 (v/v) MeCN/H2O as the solvent in an undivided cell equipped with two platinum plate electrodes to screen the optimized reaction conditions (Table 1). Several commonly used chiral amine catalysts were tested first. To our delight, the desired product 4a was obtained in 20% yield with 80% ee value using (2S,5R)-2-(tert-butyl)-3,5-dimethylimidazolidin-4-one (3a) as the catalyst (entry 1). In contrast, L-proline 3b provided the product as a racemate (entry 2). The primary anime 3e did not afford any desired product (entry 5). Increasing the constant current to 5 mA afforded the product with high yield and enantioselectivity (entry 6). Further increase of the current lowered the yield and the enantiomeric excess (entry 7). As solvent, 300:1 (v/v) MeCN/H2O was found to give the optimal yields and highest ee values. H2O was an essential factor for good ee value but further increasing the amount of H2O had a deleterious effect on the yield (entries 8–10). This phenomenon may be attributed to the fact that an appropriate amount of H2O facilitates the hydrolysis of iminium intermediates while excess H2O leads to catalyst deactivation.22
|
|
|||||
|---|---|---|---|---|---|
| Entry | 3 | Electric current | Solvent | Yieldb (%) | eec,d (%) |
| a General conditions: 1a (0.90 mmol), 2a (0.30 mmol), 3 (30 mol%), I2 (10 mol%), n-Bu4NPF6 (0.6 mmol), MeCN (3 mL) and H2O (10 μL) in an undivided cell with two platinum electrodes (each 1.0 × 1.0 cm2) electrolysis at room temperature for 6 h. b The yield of the aldehyde product was determined by 1H NMR analysis with CH2Br2 as an internal standard. c The aldehyde product was reduced by NaBH4 to the corresponding alcohol and then isolated yield (two steps and one pot) and ee value was calculated. d Enantioselectivities were determined by chiral HPLC analysis. n.d. = not detected. e Without iodine. | |||||
| 1 | 3a | 2 mA | 300:1 MeCN/H2O |
20 | 81 |
| 2 | 3b | 2 mA | 300:1 MeCN/H2O |
40 | 0 |
| 3 | 3c | 2 mA | 300:1 MeCN/H2O |
31 | 0 |
| 4 | 3d | 2 mA | 300:1 MeCN/H2O |
34 | 44 |
| 5 | 3e | 2 mA | 300:1 MeCN/H2O |
0 | — |
| 6 | 3a | 5 mA | 300:1 MeCN/H2O |
84(80)c | 85 |
| 7 | 3a | 6 mA | 300:1 MeCN/H2O |
52 | 74 |
| 8 | 3a | 5 mA | MeCN | 86 | 37 |
| 9 | 3a | 5 mA | 400:1 MeCN/H2O |
84 | 75 |
| 10 | 3a | 5 mA | 200:1 MeCN/H2O |
40 | 89 |
| 11 | 3a | 5 mA | Toluene | 0 | 0 |
| 12 | 3a | 5 mA | DMF | Trace | — |
| 13e | 3a | 5 mA | MeCN | n.d. | — |
| 14 | — | 5 mA | MeCN | n.d. | — |
| 15 | 3a | — | MeCN | n.d. | — |

After extensive efforts in screening reaction parameters, an optimized system employing Pt as the electrodes with 5 mA constant current at room temperature and using 3a (30 mol%) as organocatalyst, I2 (10 mol%) as mediator, three equivalents of aldehyde 1a in a 0.1 M mixture of MeCN and H2O (300:1 (v/v) MeCN/H2O) containing n-Bu4NPF6 (2.0 equiv., 0.2 M) as electrolyte for 6 h, was able to give 5a with 80% isolated yield and 85% ee. Control experiments showed that the reaction did not proceed in the absence of I2, organocatalysts or electric current (entries 13–15). These results imply that these conditions are crucial for a successful transformation.
Substrate scope
With the optimized conditions in hand, we investigated the reaction scope with respect to the thiol substrates by using the hydrocinnamaldehyde as the aldehyde first (Table 2). Thiophenols bearing electron-neutral substituents, such as 4-methyl, 3,4-dimethyl and hydrogen (2a, 2e and 2f) showed good yields (65–80%) with high to excellent enantioselectivity (86–91% ee). A slight decrease in yield was observed when the steric bulk in para position of the thiophenol increased (2b and 2c) with little influence on the enantioselectivity. Notably, 4-hydroxyl thiophenol failed to afford the corresponding product under the standard reaction conditions, but 4-methoxy thiophenol gave the product 5d with 78% yield and 91% ee. Furthermore, thiophenols bearing electro-withdrawing substituents (2g–2j) proceeded smoothly to afford the α-sulfenylated products in moderate to good yields (48–84%) and enantioselectivities (61–87% ee). Aliphatic thiols could be employed as substrates (2k and 2l) to furnish the products under this mild condition albeit with lower yields. These substrates are not commonly employed in electrochemistry because of their instability.23 Interestingly, the corresponding products can be obtained in excellent enantioselectivity (>99% and 95% ee). Moreover, the substrate scope can be extended to the hetaryl thiol. 2-Thiophenothiol (2m) could react smoothly to furnish the desired product in 76% yield with 89% ee.| a Reactions were performed on a 0.30 mmol scale under the standard conditions. Unless noted otherwise, the α-sulfenylated aldehyde products were reduced to the corresponding alcohol by NaBH4 and shown are their total isolated yields for the two steps. The ee values were determined by chiral HPLC analysis. b The yields and ee values were determined by the aldehyde form. |
|---|

|
Next, we explored the asymmetric α-sulfenylation reaction with different aldehydes (Table 2). Different substitutions on the phenyl ring of phenylpropanals are explored firstly. Electron-donating group (1n, –OMe) and halogen (1o and 1p) lead to a slight decrease on the yield. (35–45%) and ee value (31–76%). The reaction was also found to be amenable to aldehyde with different chain lengths, which gave the desired products in good yields and enantioselectivities. For example, two unbranched aldehydes afforded the corresponding α-sulfenylated products 5q and 5r in 67% and 53% yields with 71% and 84% ee, respectively. Moreover, cyclohexyl- (1s–t) and phenyl-substituted aldehydes (1u) can also react smoothly, affording products 5s–5u in 64–85% yields and 71–85% ee. The introduction of alkene, alkyne groups were also well tolerated under the standard conditions, in 65% and 60% yields with moderate enantioselectivity (53% and 29% ee, respectively). Nitrogen heterocycles are reactive substrates often used in electrochemistry. They are prone to Shono-type oxidations followed by nucleophilic attack. Gratifyingly, we observed that the N-Boc-protected piperidine aldehyde could be used as a substrate to afford 4x in moderate yield and 71% ee. Finally, a derivative of 5s (Fig. 2) was synthesized and its absolute configuration was determined by a single-crystal X-ray diffraction study of 6a. The absolute configurations of other products were determined by depicted in analogy.
 | ||
| Fig. 2 Absolute configuration of 6a. | ||
To test the scalability of this protocol, a synthesis of 5a on a 3.0 mmol-scale was carried out, smoothly affording the desired product 5a in 65% yield with 80% ee. Furthermore, considering the practicability, we conducted the reaction on gram-scale (9.0 mmol) using graphite electrodes instead of platinum electrodes, which afforded 2.16 g of 5a (93% yield and 57% ee) (see the ESI† for detailed information). We further carried out simple nucleophilic addition to directly obtain β-hydroxysulfide 6b with 69% yield and 94:6 dr value (relative configuration tentatively was assigned in analogy to reference).24 Olefination with triethylphosphonoacetate afforded (E)-ester 6c with minor racemization (65%, 83% ee) (Fig. 3).
 | ||
| Fig. 3 Derivation of α-sulfenylated aldehydes. aDetermined by chiral HPLC analysis. bThe ratio of E/Z was determined by 1H NMR spectroscopy. | ||
Mechanistic studies
Next, a series of control experiments were conducted to support the proposed reaction pathway depicted in Fig. 5. First, a cyclic voltammetry experiment shows a significant decrease in the highest oxidation potential of the reaction mixture with the addition of I2 to the reaction mixture (Fig. 4A, from 1.44 V to 1.09 V vs. Fc+/Fc), which implies that I2 can decrease the reaction oxidation potential via accelerating the electron transfer at the electrode/electrolyte interface. Furthermore, an oxidation peak of product 4a in acetonitrile was observed at 1.31 V vs. Fc+/Fc, where other substrates are preferred to be oxidized under the current of 5 mA (see the ESI, Fig. S2 and S4†), explaining why the sulfenylated product will not suffer overoxidation to the sulfoxide or sulfone directly under the standard conditions. In addition, according to our monitoring of the reaction process with 1H NMR, the anodic oxidation will quickly oxidize the thiol in 1.5 h to the corresponding disulfide (see ESI Fig. S6† for the details), which is accordance with literature reports.16 Hence, we replaced the thiophenol with disulfide and subjected it to the standard reaction conditions. As expected, the sulfenylated product 4a was formed though with a slightly decreased yield of 63% (eqn (1)). Moreover, to rule out the attack of enamine intermediate to disulfide straightly, the reaction was carried out without iodine in the presence of disulfide under standard conditions. As expected, no desired product formed (eqn (2)). In addition, according to our hypothesis, the sulfenylated iodide 7 is a key intermediate. The proposed reactive intermediate was generated in situ by reaction of disulfide of 2ii′ and I2. After they were stirred for 10 minutes, aldehyde and catalyst 3a were then added to the above mixture. The reaction was stirred overnight in the absence of electric current. After the reduction workup with NaBH4, we could separate the corresponding alcohol product in 8% yield while the expected yield is 10% with 0.1 equiv. I2 participating the reaction (eqn (3)). To further validate the presence of the key intermediate in our reaction, we synthesized intermediate 7i using a direct approach (see ESI† for the details). Subsequently, we exposed the intermediate to a mixture of aldehyde and catalyst, and the corresponding alcohol product could be finally separated with 55% yield and 52% ee (eqn (4)). In addition, we replaced the electrochemical oxidation by using TBHP (tert-butyl hydroperoxide) as the oxidant with other conditions unchanged. The sulfenylated product was formed in 31% yield and α,β-unsaturated product 9 was also isolated with 20% yield, which was probably formed by the elimination of the corresponding sulfoxide product (eqn (5)). Furthermore, when TEMPO was used as the radical trapping reagent using thiophenol as the substrate under the standard conditions, the product was completely oxidized to then furnish the α,β-unsaturated product 9 with 35% yield finally (eqn (6)).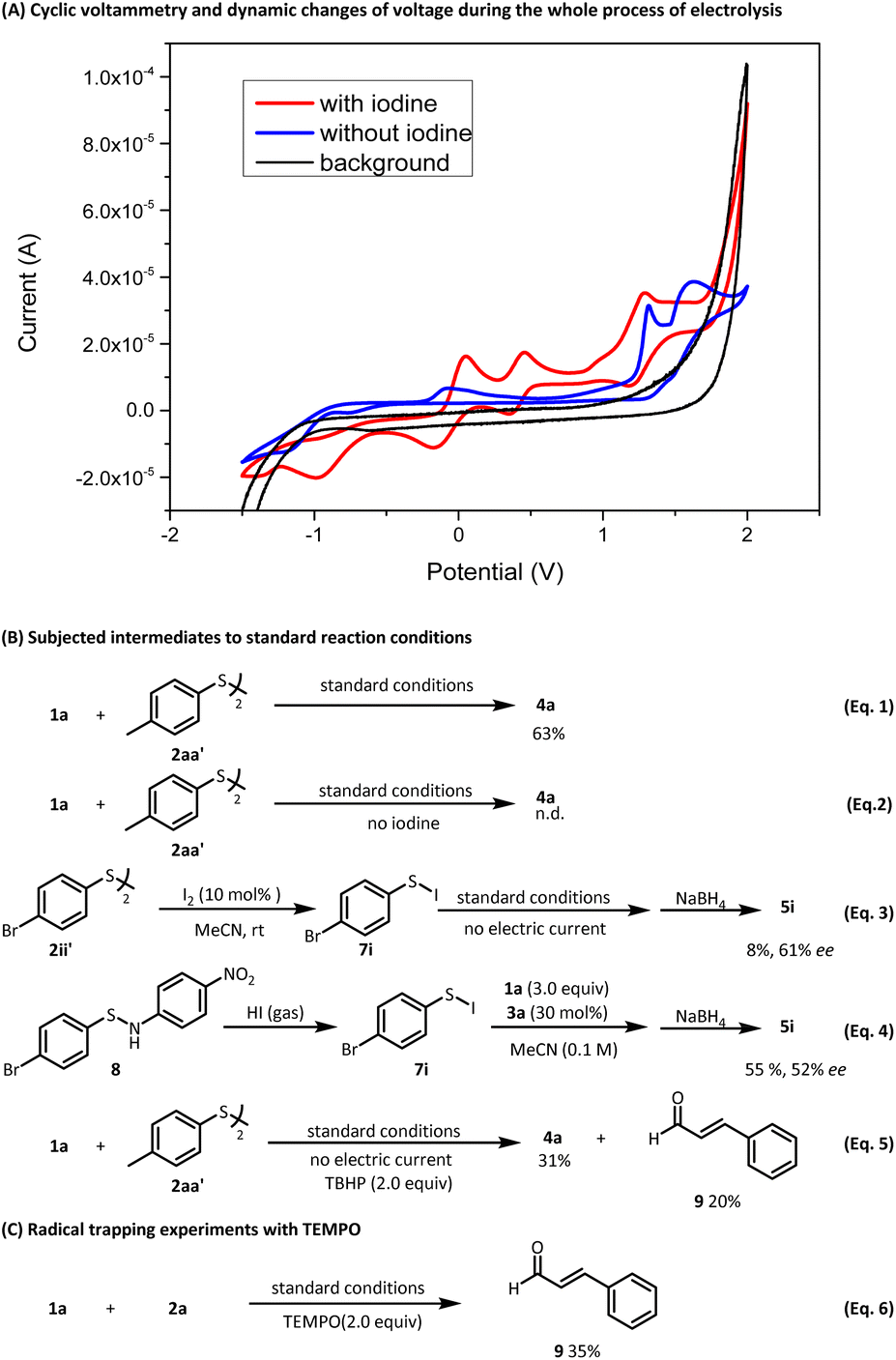 | ||
| Fig. 4 (A) Cyclic voltammograms (CV): using Pt wire as anode and cathode, ferrocene as the internal reference electrode, 0.1 M n-Bu4NPF6 with scanning rate 100 mV s−1. All the potential values were adjusted relative to Fc/Fc+. Yields of 4a were determined by 1H NMR analysis with CH2Br2 as an internal standard. The isolated yields of 5i and 9 were given and the enantioselectivities were determined by chiral HPLC analysis. (B) Subjected intermediates to standard reaction conditions. (C) Radical trapping experiments with TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy). | ||
 | ||
| Fig. 5 Proposed mechanism for the iodine-mediated α-sulfenylation of aldehydes. | ||
Based on these above experiments and previous literature,25 we proposed a novel reaction pathway (Fig. 5). Initially, condensation of aldehyde with amine catalyst 3a produces enamine intermediate I. Concomitantly, the thiol is converted to the corresponding disulfide via anodic oxidation. Reaction of I2 with the disulfide is then proposed to generate sulfenyl iodide (R'S-I) 7 as the reactive electrophile. The iminium intermediate II is then produced by the nucleophilic attack of I to 7. Iminium hydrolysis affords the α-sulfenylated adduct 4 with regeneration of organocatalyst 3a. The iodide anion which will be oxidized to iodine under the anodic oxidation conditions.
Conclusions
In conclusion, we have developed a direct and highly enantioselective method to construct the α-sulfenylated aldehydes from aldehydes and thiophenols by merging anodic oxidation and organocatalysis. This mild and metal-free protocol operates simply in one step and does not require additional stoichiometric oxidants. Mechanistic investigations indicate that the presence of catalytic amounts of iodine enables the formation of the key RSI intermediate in situ and take effect as a redox mediator to avoid the sulfenylated product to be overoxidized. This atom-economic method is currently being extended in our laboratory to other asymmetric α-functionalization reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. Wang thanks the China Scholarship Council for doctoral scholarship. We thank members from research group of Malischewski and Eigler for suggestions on measuring cyclic voltammetry. C. Groneberg, A. Peuker and G. Drendel (Freie Universität Berlin) are acknowledged for analytical support.References
- B. M. Trost, Chem. Rev., 1978, 78, 363–382 CrossRef CAS.
- F. Zhao, K. Lauder, S. Liu, J. D. Finnigan, S. B. R. Charnock, S. J. Charnock and D. Castagnolo, Angew. Chem., Int. Ed., 2022, 61, e202202363 CrossRef CAS PubMed.
- F. Rota, L. Benhamou and T. D. Sheppard, Synlett, 2016, 27, 33–36 CAS.
- (a) T. G. Back, K. N. Clary and D. Gao, Chem. Rev., 2010, 110, 4498–4553 CrossRef CAS PubMed; (b) C. Zhao, K. P. Rakesh, L. Ravidar, W.-Y. Fang and H.-L. Qin, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS PubMed.
- (a) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794–797 CrossRef CAS PubMed; (b) A. Armstrong, L. Challinor and J. H. Moir, Angew. Chem., Int. Ed., 2007, 46, 5369–5372 CrossRef CAS PubMed.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- J. Blom, G. J. Reyes-Rodríguez, H. N. Tobiesen, J. N. Lamhauge, M. V. Iversen, C. L. Barløse, N. Hammer, M. Rusbjerg and K. A. Jørgensen, Angew. Chem., Int. Ed., 2019, 58, 17856–17862 CrossRef CAS PubMed.
- Q. Jiang, J. Luo and X. Zhao, Green Chem., 2024 10.1039/D3GC04154A.
- (a) X. Chang, Q. Zhang and C. Guo, Angew. Chem., Int. Ed., 2020, 59, 12612–12622 CrossRef CAS PubMed; (b) Q. Lin, L. Li and S. Luo, Chem. – Eur. J., 2019, 25, 10033–10044 CrossRef CAS PubMed; (c) M. S. Ghosh, V. S. Shinde and M. Rueping, Beilstein J. Org. Chem., 2019, 15, 2710–2746 CrossRef CAS PubMed.
- K. L. Jensen, P. T. Franke, L. T. Nielsen, K. Daasbjerg and K. A. Jørgensen, Angew. Chem., Int. Ed., 2010, 49, 129–133 CrossRef CAS PubMed.
- L. Li, Y. Li, N. Fu, L. Zhang and S. Luo, Angew. Chem., Int. Ed., 2020, 59, 14347–14351 CrossRef CAS PubMed.
- Z.-H. Wang, P.-S. Gao, X. Wang, J.-Q. Gao, X.-T. Xu, Z. He, C. Ma and T.-S. Mei, J. Am. Chem. Soc., 2021, 143, 15599–15605 CrossRef CAS PubMed.
- P. Xiong, M. Hemming, S. I. Ivlev and E. Meggers, J. Am. Chem. Soc., 2022, 144, 6964–6971 CrossRef CAS PubMed.
- L. Song, N. Fu, B. G. Ernst, W. H. Lee, M. O. Frederick, R. A. DiStasio and S. Lin, Nat. Chem., 2020, 12, 747–754 CrossRef CAS PubMed.
- Q. Zhang, X. Chang, L. Peng and C. Guo, Angew. Chem., Int. Ed., 2019, 58, 6999–7003 CrossRef CAS PubMed.
- G. Laudadio, E. Barmpoutsis, C. Schotten, L. Struik, S. Govaerts, D. L. Browne and T. Noël, J. Am. Chem. Soc., 2019, 141, 5664–5668 CrossRef CAS PubMed.
- I. V. e. Koval’, Russ. Chem. Rev., 1994, 63, 735–750 CrossRef.
- T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS.
- K. Liu, C. Song and A. Lei, Org. Biomol. Chem., 2018, 16, 2375–2387 RSC.
- (a) S. Ponath, M. Menger, L. Grothues, M. Weber, D. Lentz, C. Strohmann and M. Christmann, Angew. Chem., Int. Ed., 2018, 57, 11683–11687 CrossRef CAS PubMed; (b) C. Apel, S. S. Hartmann, D. Lentz and M. Christmann, Angew. Chem., Int. Ed., 2019, 58, 5075–5079 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green chemistry: theory and practice, Oxford university press, 2000 Search PubMed.
- G. Hutchinson, C. Alamillo-Ferrer and J. Burés, J. Am. Chem. Soc., 2021, 143, 6805–6809 CrossRef CAS PubMed.
- P. Wang, S. Tang, P. Huang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009–3013 CrossRef CAS PubMed.
- T. Sheppard, F. Rota and L. Benhamou, Synlett, 2015, 27, 33–36 CrossRef.
- H.-A. Du, R.-Y. Tang, C.-L. Deng, Y. Liu, J.-H. Li and X.-G. Zhang, Adv. Synth. Catal., 2011, 353, 2739–2748 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2263955. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc03828a |
| This journal is © The Royal Society of Chemistry 2024 |