
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recombinatorial approach for the formation of surface-functionalised alkaline-stable lignin nanoparticles and adhesives†
Federica
Ferruti
 ab,
Ievgen
Pylypchuk
a,
Luca
Zoia
*c,
Heiko
Lange
c,
Marco
Orlandi
c,
Adrian
Moreno
a and
Mika H.
Sipponen
*a
ab,
Ievgen
Pylypchuk
a,
Luca
Zoia
*c,
Heiko
Lange
c,
Marco
Orlandi
c,
Adrian
Moreno
a and
Mika H.
Sipponen
*a
aDepartment of Materials and Environmental Chemistry, Stockholm University, Svante Arrhenius väg 16 C, 106 91, Stockholm, Sweden. E-mail: mika.sipponen@mmk.su.se
bCorimav Pirelli, Department of Material Science, University of Milano-Bicocca, Via R. Cozzi 55, 20125, Milan, Italy
cDepartment of Earth and Environmental Sciences, University of Milano-Bicocca, Piazza della Scienza 1, 20126, Milan, Italy. E-mail: luca.zoia@unimib.it
First published on 15th December 2022
Abstract
Lignin nanoparticles (LNPs) are considered as intriguing green, renewable alternatives to fossil-based nanomaterials. However, the predisposition of LNPs to dissolve under alkaline conditions makes covalent surface functionalisation in the dispersion state difficult and limits applications demanding morphological stability under challenging pH conditions. Mechanistic studies suggest that during the formation of LNPs by nanoprecipitation the higher molecular weight fractions of lignin likely start precipitating first, while the low molecular weight fractions tend to deposit later and thus locate on the outer shell. Capitalising this aggregation pattern, the present work presents a strategy to prepare surface-functionalised LNPs that can find applications as adhesives and alkaline stable LNPs. The entire process is based on a single-step solvent fractionation of lignin using either ethanol or ethyl acetate, subsequent functionalisation of selected fractions with epichlorohydrin, and recombination according to the original mass proportions in line with the so-called zero waste principle. Aqueous colloidal dispersions of lignins were synthesised by nanoprecipitation of epoxidised low molecular weight (MW) fractions combined with the corresponding unmodified high MW ones, and vice versa. Upon thermal treatment, LNPs containing the epoxidised insoluble fraction underwent intraparticle crosslinking, proving dimensional stability at pH 12. Conversely, LNPs including epoxidised solvent-soluble fractions resulted in interparticle crosslinking upon heating, which confirmed the surface localisation of such low MW fractions. The latter system was exploited to develop green LNP-based adhesives for aminated glass with lap shear strength outperforming prior adhesive systems based on lignin particles.
Introduction
Lignin is one of the major constituents of wood alongside cellulose and hemicellulose, and it is one of the most abundant biomass components. It is thus a promising, green and renewable alternative to fossil-based technology for the production of chemicals and materials for diverse applications including areas of material sciences and medicine. The structural variability and complexity of lignins depend on botanical source and isolation process, this so far hampering the conversion of lignin into higher value-added products, and dooming this biomass to combustion for energy recovery. This is particularly true for softwood kraft lignin (SKL), derived from the kraft pulping process during which wood is exposed to elevated pH values and temperatures of over 160 °C for an extended time, i.e., 2–3 h. Such harsh conditions alter the native structure of lignin by depolymerisation and subsequent repolymerising lignin-based fragments; these processes lead to an even wider variety of inter-unit bondings than already present in planta, and a broad distribution of molecular weights which further limits the use of kraft lignin in many value-added applications.1For the rational design of functional materials, it is pivotal to overcome the structural heterogeneity of SKL by separating different lignin constituents with different characteristic physicochemical features, such as to arrive at fractions with more predictable and tuneable behaviour. This separation can be attained by fractionation which reportedly leads to the isolation of fractions of narrow dispersity with a simultaneous modulation of functional groups including phenols, aliphatic alcohols and carboxylic acids. Fractionation can be carried out by different methods including solvent- and membrane-mediated processes.2–8 Among those strategies, the most commonly adopted procedure is solvent fractionation which allows for the isolation of lignin cuts whose composition and physicochemical features can be tuned by selecting the polarity and H-bond capacity of the solvent.9,10 However, at the same time it is important to attempt to utilize a maximum amount of the original lignin in a material-efficient way.
One of the emerging approaches to valorise lignin includes the synthesis of lignin nanoparticles (LNPs), also referred to as colloidal lignin particles (CLPs), which seem promising as drug nanocarriers,11 surfactants in Pickering emulsions, UV-blockers, biocides, reinforcing fillers, nano-glues, etc.12,13 The most popular procedure for the preparation of LNPs includes solvent polarity shift by pouring a lignin solution in a mixture of water-miscible organic solvent and water into a non-solvent, i.e., water, or vice versa, leading to the formation of spherical nanoparticles due to minimisation of surface energy.14 It was demonstrated that upon addition of the antisolvent, particles form by molecular weight-dependent precipitation, with high molecular weight (MW) fractions in the core and low MW cuts on the surface of nanoparticles.11,15,16
Owing to the small size and rather compact internal morphology of LNPs,17 the surface-exposed functional groups largely define their reactivity. In many instances, the hydroxyl and carboxylic acid groups do not provide sufficient reactivity and further chemical activation is required.18 The surface chemistry of LNPs is, however, dependent on the localisation of low MW fractions on the outer shell of the particles, which hampers surface-specific modification of the particles due to dissolution and aggregation in alkaline pH and organic solvents. Functionalisation of LNPs can be attained by chemical modification of lignin prior to nanoparticle formation; however, this approach lacks surface specificity. Known methods for surface functionalisation of LNPs are based on the physical absorption of cationic polymers, oligomers and proteins on the surface of LNPs in the dispersion state.19–22 Surface-specific functionalisation involving formation of covalent bonds can be carried out on inherently stable, non-crosslinked nanoparticles of pre-functionalised lignin23 and internally cross-linked LNPs.24,25 Literature25 suggests that lignin–epoxy hybrid particles are capable to undergo internal or interparticle curing reactions as function of the mass fractions of the epoxy crosslinker. However, these prior works do not consider the effects of molecular weight distribution and reactivity of functionalised lignin on its solubility and ability to form nanoparticles with localised reactive groups.
Here we propose a covalent surface-specific functionalisation of LNPs which takes advantage on the mechanism of molecular weight-driven nanoprecipitation11 and consists in the solvent fractionation of SKL for the isolation of low and high MW fractions, selective functionalisation of the low MW component and recombination with the corresponding high MW portion for the formation of LNPs in order to valorise the entire starting material. Furthermore, by exploiting the mechanism of nanoprecipitation, we show that it is possible to prepare alkaline stable, internally cross-linked LNPs by solvent fractionation of SKL, functionalisation of high MW components, recombination with the corresponding low MW fractions and thermal curing. Moreover, we demonstrated as a proof of concept that the surface-functionalised LNPs could be used as waterborne adhesives for aminated glass.
Materials and methods
Materials
All experiments were based on BIOPIVA™ 100 pine Kraft lignin (SKL) (UPM, Finland). All chemicals and solvents were purchased from Sigma Aldrich, Merck, Fischer, Fluka or VWR and were used as received.Lignin fractionation
Single-step solvent fractionation of lignin was reported elsewhere.26 Dried SKL (10 g) was dissolved in 100 mL of solvent, i.e., either ethanol or ethyl acetate, under magnetic stirring at room temperature for 3 h. After that, the insoluble residue was filtered off on a Büchner filter through paper filters (Whatman, pore size 0.7 μm) and dried at 45 °C for one night. The filtered solution was concentrated by evaporation under reduced pressure.Etherification of lignin fractions
Etherification of lignin fractions followed a procedure already reported in literature.27 In a 250 mL round-bottom flask equipped with a condenser, lignin (500 mg) was dissolved in a mixture of acetone and water (50% v/v, 75 mL) in the presence of NaOH, using 3 eq. with respect to the number of OH-groups of the lignin fraction, and epichlorohydrin, 20 eq. with respect to the number of OH-groups of the lignin fraction. The mixture was kept under vigorous stirring at 50 °C for 5 h. Water (50 mL) was added, and pH was lowered to 3.5 with 0.1 m HCl. The precipitated product was isolated by centrifugation, washed twice with excess deionised water and freeze-dried in order to recover the product as a brown powder.Preparation of LNPs
LNPs were prepared by nanoprecipitation as reported in literature.25SKL (360 mg) was dissolved in 12 g of acetone/water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w) under magnetic stirring at room temperature for 2 h. Solid residues were filtered off through paper filters (Whatman, pore size 0.7 μm). Deionised water (36 g, solution:water = 1:3, w/w) was rapidly poured into the SKL solution under vigorous magnetic stirring and LNPs instantly formed. Acetone was removed by evaporation under reduced pressure. The larger aggregates were filtered off using paper filters (Whatman, pore size 0.7 μm) and subsequently though glass fibre filters (Whatman). The final dispersion concentration was 4.3 g L−1. In the case of NPs obtained by mingling fractions, epoxidised and unmodified fractions were recombined according to their mass ratio in the original SKL.
:1 w/w) under magnetic stirring at room temperature for 2 h. Solid residues were filtered off through paper filters (Whatman, pore size 0.7 μm). Deionised water (36 g, solution:water = 1:3, w/w) was rapidly poured into the SKL solution under vigorous magnetic stirring and LNPs instantly formed. Acetone was removed by evaporation under reduced pressure. The larger aggregates were filtered off using paper filters (Whatman, pore size 0.7 μm) and subsequently though glass fibre filters (Whatman). The final dispersion concentration was 4.3 g L−1. In the case of NPs obtained by mingling fractions, epoxidised and unmodified fractions were recombined according to their mass ratio in the original SKL.
Thermal curing of LNPs
LNPs in the dispersed state were submitted to thermal curing in a sealed vial at 105 °C for 16 h.Sample preparation for adhesive tests
Glass slides were functionalised with an amino group slightly modifying a procedure already reported in literature.28 Microscope glass slides (26 mm × 76 mm, VWR) were rinsed with distilled water, sonicated in acetone for 5 minutes and iPrOH for additional 5 minutes. Then, they were air dried. 10 mL of 3-aminopropyltriethoxysilane (APTES) was dropwise added to 200 mL of iPrOH/water 95:5 (v/v) kept at room temperature under vigorous stirring in beaker. When a whitish suspension was formed, glass slides were immersed in the suspension and soaked overnight. Glass slides were then rinsed, sonicated for 5 minutes and rinsed again with iPrOH before being air-dried. LNP dispersion (4.3 g L−1, 50 μL) was deposited on a 26 × 26 mm section (geometry requirements stated in ASTM D5868 for single-lap shear testing29) of aminated glass. Another aminated glass was placed on top and the system was cured at 105 °C for 16 h.
Gel permeation chromatography (GPC)
GPC analyses were performed on samples acetylated according to the procedure reported in literature30 and run by using an Agilent 1100 liquid chromatography system equipped with a VWD G1314 UV detector set at 280 nm. The GP-column apparatus comprised Agilent PL gel 5 μm, (500 Å) and Agilent PL gel 5 μm (104 Å) column. Tetrahydrofuran was used as sample solvent and eluent at a flow rate of 1 mL min−1. Polymer standards of polystyrene (Mp range 162–3114000 Da) from Polymer Laboratories were employed for calibration. Number-average molecular weight (Mn), weight-average molecular weight (Mw), and the peak molecular weight (Mp) of lignin samples were evaluated. Moreover, the dispersity index (PDI), defined as the ratio Mw/Mn, was calculated. Calculated molecular masses were rounded to full hundreds.Phosphorus nuclear magnetic resonance spectroscopy (31P NMR)
A quantitative determination of the hydroxyl groups of lignin fractions was obtained by 31P NMR using a Bruker Avance NMR spectrometer operating at 500.130 MHz. Samples were prepared according to the following procedure.31 Dried lignin fractions (ca. 20 mg) were dissolved in a mixture of pyridine-deuterated chloroform (1.6:1 v/v mL, 700 μL) containing 1 mg mL−1 of chromium(III) acetylacetonate (Cr(acac)3). A solution of endo-N-hydroxy-5-norbornene-2,3-dicarboximide (100 μL, 121.5 mm, pyridine/CDCl3 4.5:0.5 v/v) and 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (100 μL) were added as internal standard and derivatising agent, respectively. Reported values are mean values of two replicates with percentage errors generally <5% relative to the mean.
Fourier transform infrared (FT-IR) spectroscopy
ATR FT-IR/spectra were recorded on a Varian 610-IR FT-IR spectrometer in the range of 450–4000 cm−1.Dynamic light scattering (DLS)
The hydrodynamic diameter of LNPs in aqueous suspension was measured by a Zetasizer Nano ZS90 instrument (Malvern Instruments Ltd., U.K.). The absorption and refractive index (RI) of LNPs were set at 0.9 and 1.4, respectively. The viscosity and RI of the dispersant, i.e., water if not stated otherwise, were set at 0.8872 cP and 1.33, respectively. Three replicates of 11 scans each were run for every sample. The values reported are mean values of analyses run on three different samples.Scanning electron microscopy (SEM)
Air-dried LNPs and disrupted glass joints were visualised by a scanning electron microscope JSM-7000F (JEOL, Japan) without any coating of the sample.Mechanical property testing of adhesives
Uniaxial shear mechanical tests on glass joints were performed using an Instron 5960 universal testing machine (Instron, USA) with a 100 N load cell at a strain rate of 1 mm min−1 at 50% relative humidity at 25 °C.32 The analyses were run on samples having a geometry meeting the requirements stated in ASTM D5868.29 The reported results are one repetition test for each sample. Additional shear mechanical tests were performed by applying a load of increasing weight to the glass joint with a grip (ESI, Fig. S9†) and results are reported as an average of three repetitions for each sample.Environmental impact assessment
E-factor33 and solvent demand were calculated for the production process of LNP-based adhesives using the contribution of each step to the overall E-factor and estimating solvent demand from literature-reported procedures.34Results and discussion
Structural characterisation of lignin fractions and etherification with epichlorohydrin
The reactivity of LNPs largely depends on the functional groups exposed on the surface; tuning it by selectively modifying external functionalities would significantly facilitate the design of materials with specific properties. In parallel, the stabilisation of LNPsvia intraparticle crosslinking would help overcome the restrictions due to their tendency to dissolve in alkaline and organic media. Aiming at these goals, lignin was submitted to a single step solvent fractionation in order to isolate high and low MW cuts which were optionally separately etherified with epichlorohydrin (Scheme 1) and recombined according to their original mass proportions, as outlined in Scheme 2. | ||
| Scheme 1 Single-step solvent fractionation of SKL followed by etherification of the fractions with epichlorohydrin. | ||
 | ||
| Scheme 2 Synthesis of LNPs by nanoprecipitation (a) and schematic depiction of LNP structure and crosslinking before and after thermal treatment (b). | ||
Firstly, SKL was treated with green organic solvents of different polarity, namely ethanol (EtOH) and ethyl acetate (EtOAc), in order to isolate narrowly disperse, high and low molecular weight fractions by a single-step solvent fractionation. The solubilisation yield, molecular weight (MW) and dispersity index (PDI) of soluble lignin fractions increased with increase in the hydrogen bonding capacity and polarity of the solvent (Fig. 1).10 More specifically, fractionation with ethyl acetate allowed for the isolation of a more homogeneous soluble fraction with a narrower MW distribution compared to the extraction with ethanol. Independent of the solvent, when compared to the insoluble fractions, the soluble cuts showed lower molecular weights and noticeably higher number of phenolic end groups which are in agreement with the presence of smaller polymeric chains.1 The number of carboxylic acids decreased while the content in aliphatic alcohols increased with increasing MW of the fraction (Table 1). This general tendency of SKL to fractionate in polar organic solvents has previously been observed by several groups.26,35,36
 | ||
| Fig. 1 GPC analysis of different fractions of SKL from ethanol (a) and ethyl acetate (b) treatment, and quantitative 31P NMR spectroscopy experiments (c). | ||
| SKL | SolEtOH | InsEtOH | SolEtOAc | InsEtOAc | |
|---|---|---|---|---|---|
| Properties | |||||
| Extraction yield/% m m−1 | 100 | 56 | 44 | 36 | 64 |
| Molecular weight characteristics | |||||
| M n g−1 mol−1 | 2100 | 1100 | 3700 | 700 | 4000 |
| M W g−1 mol−1 | 6700 | 3300 | 9000 | 1300 | 12400 |
| PDI | 3.3 | 3.0 | 2.4 | 1.8 | 3.2 |
| Hydroxyl group content/mmol g −1 | |||||
| Aliphatic-OH | 1.86 | 1.36 | 2.31 | 0.80 | 2.41 |
| Condensed phenolic-OH | 1.53 | 1.93 | 1.74 | 1.63 | 1.64 |
| G- and H- phenolic-OH | 1.98 | 2.80 | 1.77 | 2.54 | 1.72 |
| Total phenolic-OH | 3.87 | 4.73 | 3.51 | 4.17 | 3.35 |
| Carboxylic acids | 0.35 | 0.46 | 0.18 | 0.25 | 0.29 |
SKL and their fractions were then etherified with epichlorohydrin (Scheme 1) following a literature procedure.1 Yields and ATR FT-IR characterisations of the epoxidised samples are reported in ESI (Fig. S1–4†). FT-IR/ATR spectra showed the structural characteristics of fractions, including the presence of aromatic rings, aliphatic alcohols, phenols and carboxylic acids. The corresponding epoxidised derivatives show a decrement in intensity of the band at 1370 cm−1 associated to O–H stretching in phenols and the simultaneous appearance of a band at 920 cm−1 typical of epoxy groups.
Dimensional and morphological characterisation of LNPs before and after thermal curing
LNPs were synthesized following a well-established procedure25 which includes the dissolution of lignin into a 3:1 w/w mixture of acetone and water, followed by the addition of excess water as poor solvent and removal of the organic solvent by evaporation under reduced pressure to obtain a stable aqueous colloidal dispersion. Upon addition of water, lignin macromolecules rearranged into spherical nanoparticles to minimise the contact area with the poor solvent.37 In order to valorise the whole starting material, LNPs were prepared by merging epoxidised soluble fractions with the corresponding insoluble fraction and, vice versa, epoxidised insoluble fractions were recombined with the related soluble portion (Scheme 2). LNPs in the dispersed state were subsequently submitted to thermal curing in a sealed vial at 105 °C for 16 h.25
The standard LNPs and the particles prepared by recombining the epoxidised soluble (GlySol) and unmodified insoluble (Ins) lignin fractions were all colloidally stable at room temperature (Fig. 2a). Upon thermal treatment at 105 °C for 16 h, regular LNPs remained essentially unchanged with respect to particle size. In contrast, the particles that contained epoxidised lignin showed increased particle sizes, which is likely due to the thermally initiated ring-opening reactions causing inter-particle cross-linking. The most obvious increase in in the average hydrodynamic diameter was observed with LNPs generated on the basis of fully epoxidised SKL, followed by the particles containing epoxidised low molecular weight fractions. In all of these cases the colloidal dispersions became visibly unstable (Fig. 2b). This phenomenon was ascribed to the crosslinking achieved by the nucleophilic opening of the epoxy groups, which was confirmed by the disappearance of the bands related to epoxy groups in the ATR FT-IR spectra (ESI, Fig. S5–8†). In contrast, the particles containing epoxidised high MW fractions underwent markedly lower extent of inter-particle reactions and thus remained dispersed with average hydrodynamic diameter less than 400 nm (Fig. 2b).
 | ||
| Fig. 2 Analysis of hydrodynamic diameter by DLS before (a) and after (b) thermal curing. SEM images of air-dried LNPs (c) and thermally cured LNPs (d). | ||
The observed variation in the dimensions of the LNPs led us to conclude that the epoxidised soluble fractions were located at the particle surfaces and that thermal curing induced intraparticle crosslinking, as depicted in Scheme 2b. When compared to LNPs derived from ethanol fractionation, the ones obtained upon ethyl acetate fractionation, i.e., GlySolEtOAc+ InsEtOAcLNPs, experienced a higher degree of interparticle crosslinking, as reflected by the extent of the dimensional growth upon thermal treatment. The observed higher degree of crosslinking in the case of lignin fractions from ethyl acetate was ascribed to the high selectivity of this solvent in solubilising a low molecular weight, narrowly polydisperse lignin fraction38,39 which, after functionalisation with epoxy groups, specifically locates on the outer shells of LNPs, as expected from the molecular weight-dependent precipitation mechanism described in the literature, assuming a rather unchanged solubility profile for functionalised lignin fractions under tested conditions.11 On the other hand, ethanol fractionation resulted in a soluble portion that exhibited a broader molecular weight distribution. After functionalisation, hence, in GlySolEtOH+ InsEtOHLNP systems, the epoxidised ethanol-soluble fraction can be speculated to be comparably unspecifically distributed partly in the outer shell and partly in the core of LNPs, leading to a less evident interparticle crosslinking and dimensional increase.
SEM images evidence that LNPs exhibited a spherical morphology which was preserved even after the thermal curing (Fig. 2c-d). From SEM images of thermally cured GlySolEtOAc+ InsEtOAcLNPs, a superficial crosslinking can be inferred by observing the three-dimensional structure and piling up of LNPs, which is not visible in the images of the same LNPs before thermal curing, allowing for exclusion of simple aggregation or ripening effects that may lead to a similar observation.
LNPs comprised of epoxidised insoluble and unmodified soluble fractions, i.e., Sol + GlyIns LNPs, displayed a peculiar behaviour upon thermal treatment. In the case of ethyl acetate fractionation giving rise to SolEtOAc+ GlyInsEtOAcLNPs, no marked change in the average hydrodynamic diameter was detected in LNPs after curing, as for LNPs from unmodified SKL. This observation indicates that the crosslinking, corroborated by the disappearance of the diagnostic band at 920 cm−1 associated to epoxy groups in their FT-IR/ATR spectra (ESI, Fig. S5–8†), was confined to the internal parts of the particles. Such internal stabilisation induced a high dimensional stability in these LNPs even under alkaline conditions, i.e., at pH 12 (Fig. 3). Conversely, in the case of ethanol fractionation allowing for the generation of SolEtOH+ GlyInsEtOHLNPs, a limited increase in hydrodynamic dimension was observed by DLS. This behaviour finds an explanation from the poor selectivity encountered in the ethanol fractionation which allowed for isolation of two fractions with only slightly different molecular weights, which causes a less specific locating of the fractions on the outer shell or in the core of LNPs during the MW-driven particle formation process. Therefore, epoxidised ethanol-insoluble fractions could expose part of the reactive groups on the surface, leading to a modest interparticle crosslinking and dimensional increase.
 | ||
| Fig. 3 Analysis of hydrodynamic diameter of LNPs by DLS at pH 4.5 (a) and at pH 12 before (b) and after (c) thermal curing at 105 °C. | ||
LNPs are known to increase their size due to polyelectrolyte swelling induced by ionisation of functional groups and disruption of intramolecular hydrogen bonding under alkaline conditions.40 This swelling ultimately leads to disassembly and dissolution of the particles. LNPs from SKL thermally treated at 105 °C for 16 h are no exception in this respect (Fig. 3a-b). Conversely, thermally cured LNPs deriving from epoxidised insoluble and unmodified soluble fractions, i.e.Sol + GlyIns LNPs, exhibited a high dimensional stability under alkaline conditions (pH 12), if compared to the corresponding uncured particles under the same conditions (Fig. 3b-c). The increased dimensional stability under alkaline conditions paves the way to the covalent surface functionalisation of LNPs.
Covalently surface-functionalised LNPs as competitive adhesives for aminated glass
The ability to restrict functionalization of lignin to low or high MW fractions according to the desired properties complies with green chemistry principles in form of minimization of waste and unnecessary derivatizations. Moreover, LNPs displaying a covalently linked functionalisation only on the surface, and thus a very responsible use of derivatising chemicals, could find a wide variety of applications such as green tailored adsorbents, green flocculants, or green sensors.12 Also bio-based adhesives are acquiring more and more attention as a renewable alternative to common fossil-based technologies. In the latter context, lignin is being studied as a glue for both soft and hard materials, i.e., wood, hydrogels, etc. on the one hand, and cement, ceramics, etc. on the other hand. A recent study41 highlighted the importance of confined evaporation-induced self-assembly of colloidal lignin and non-covalent interactions between glue and substrate to achieve anisotropic adhesives with a good performance. As a proof of concept, the potential use of the above-described functional LNPs as waterborne adhesives for aminated glass (Scheme 3) was evaluated. To this aim, common microscope glass slides were first washed and then treated with (3-aminopropyl)triethoxysilane (APTES) in order to cover the surface of the glass with amino groups that provide, upon thermal curing, a site for covalent binding to LNPs exhibiting an epoxidised surface. The latter were added in the form of a colloidal dispersion in water, following literature-reported procedures41 for a straightforward comparison. Among all the LNPs described in the previous section, GlySolEtOAc+ InsEtOAcLNPs gave the most promising inter-particle crosslinking behaviour, confirming the desired localisation of epoxy groups at the surface of LNPs. The location of the epoxy groups at the surface renders these particles also capable of covalently binding to the surface of the aminated glass slides. GlySolEtOAc+ InsEtOAcLNPs were thus selected for in plane (shear) adhesive tests and compared to standard SKL NPs.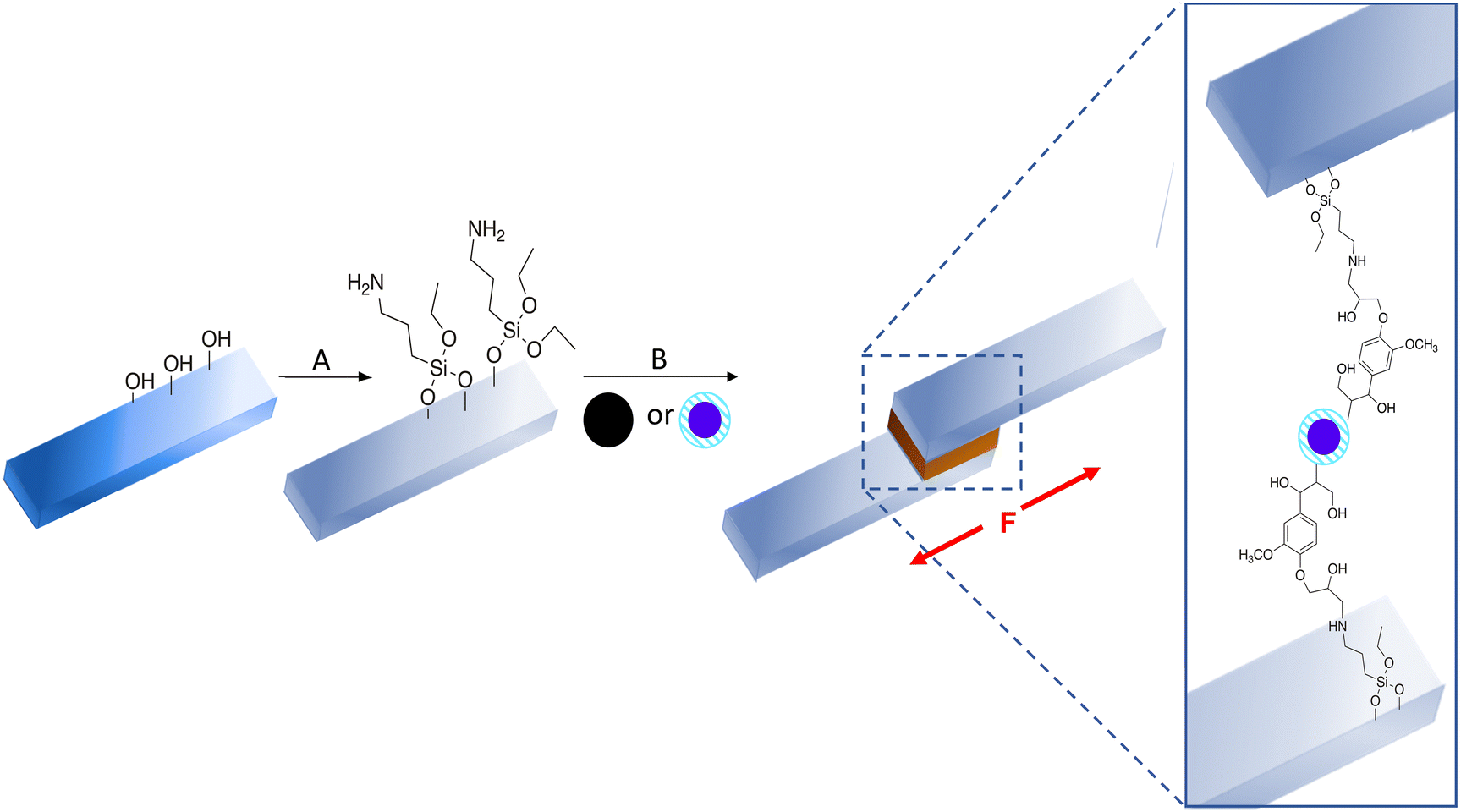 | ||
| Scheme 3 Surface functionalisation of glass slides and application of LNP-based adhesives. (A): Amination of glass slides. (i) (3-aminopropyl)triethoxysilane, iPrOH/H2O, 95:5 (v/v), 25 °C, overnight; (ii) iPrOH/H2O washing. (B): (i) Deposition of 0.43% (w/w) LNP aqueous dispersions (black: SKL, blue: GlySolEtOAc+ InsEtOAcLNPs) and junction of aminated glass slides; (ii) Thermal treatment, 105 °C, 16 h. | ||
Glass joints macroscopically showed an even distribution of LNPs (Fig. 4) without any accumulation of material on the rims of the joint, unlike what has been reported for organosolv lignin NPs on untreated glass.41 After the rupture of the joints during the shear tests, SEM images evidenced that the macroscopic structure comprised multiple layers of LNPs piling up, highlighting the importance of particle–particle interactions. In addition, both of the glass slides resulted to be covered with LNPs after rupture, showing that a strong particle–substrate interaction was achieved, in force exceeding intraparticle linking and inner-particle intermolecular forces. Noteworthy, GlySolEtOAc+ InsEtOAcLNPs showed two-fold better adhesive properties than SKL NPs, with ultimate shear strengths of 5.1 ± 0.8 MPa and 2.4 ± 0.2 MPa, respectively, on aminated glass, as expected due to the surface chemistry, which allowed for a covalent binding of surface-epoxidised LNPs on the support upon thermal treatment (Fig. 4).
 | ||
| Fig. 4 Images of LNPs glass joints and schematic depiction of LNPs used as adhesives (a). Stress to strain curves of glass joints using different LNP aqueous dispersions (black: SKL NPs, light blue: GlySolEtOAc+ InsEtOAcLNPs) obtained by the tensile testing machine (b). Ultimate tensile strength evaluated in the lab by applying a load to the jointed glass slides (c). Scanning electron microscopy images of joints ruptured after tensile test for adhesives based on SKL NPs (d) and GlySolEtOAc+ InsEtOAcLNPs (e). | ||
When comparing the shear strength of adhesives (Table 2) obtained by LNP dispersions of similar concentration, SKL NPs on aminated glass slides proved to be nearly 20 times more efficient than LNPs based on organosolv lignin on pristine glass.41 This behaviour can be ascribed to higher interaction energies between lignin and amines than lignin and hydroxyl groups exposed on pristine glass surfaces. While SKL NPs reached the lower limit of strength required from urea-formaldehyde adhesives according to the ASTM D4690 standard,47GlySolEtOAc+ InsEtOAcLNPs exhibited even better adhesive properties on aminated glass, this feature being ascribable to the covalent binding between epoxy groups exposed on the outer layers of such LNPs and amino groups disposed on the surface of the treated glass substrate (Scheme 3 and Fig. 4). A rather low surface concentration of GlySolEtOAc+ InsEtOAcLNPs was sufficient to outperform formulations of lignin with epoxide-based crosslinkers originally intended as wood adhesives, which needed a nearly thousandfold higher resin loading at the interface to guarantee a shear strength of nearly 2.2 MPa.42 The same interparticle crosslinked LNPs demonstrated improvement also when compared to the previously reported results for intraparticle-crosslinked hybrid bisphenol A/diglycidyl ether-modified lignin nanoparticles developed as wood adhesives which reached an ultimate shear strength of 4.0 ± 0.8 MPa with a relatively high concentration of NPs on the junction surface of 0.1 kg m−2.25 In a recent work,43 a novel wood adhesive was prepared by crosslinking water-soluble epoxidised lignin with LNPs, a maximum ultimate shear strength of 13 MPa, being more than twice the value obtained for GlySolEtOAc+ InsEtOAcLNPs, was achieved by applying an interface loading which was a thousand times higher than that shown in the present study. When considering other bio-based adhesives for glass, LNPs on aminated glass showed similar performances as chitin-amyloid mixtures44 and chitin nanocrystals45 on pristine glass. Standard SKL NPs displayed an ultimate shear strength of the same magnitude as cellulose nanocrystals (CNCs). GlySolEtOAc+ InsEtOAcLNPs replicated the results reported for CNCs, but, most noteworthy, in a more material-efficient manner, i.e., with an interface loading of 3.2 × 10−4 kg m−2 for GlySolEtOAc+ InsEtOAcLNPs compared to 2.2 × 10−2 kg m−2 for CNCs.46 Finally, we note that the weight fraction of the epoxy derivative in GlySolEtOAc+ InsEtOAcLNPs is only 10.3 wt% which compares favourably with 30 wt% of bisphenol A diglycidyl ether in colloidal lignin particles in a previous study.25
| Material | Support | Interface loading/kg m−2 | Shear strength/MPa | Ref. |
|---|---|---|---|---|
| GlySol EtOAc + Ins EtOAc LNPs | Aminated glass | 3.2 × 10−4 | 5.1 ± 0.8 | This work |
| SKL NPs | Aminated glass | 3.2 × 10−4 | 2.4 ± 0.2 | This work |
| Organosolv LNPs | Pristine glass | 3.2 × 10−4 | 0.11 ± 0.04 | 41 |
| Chitin nanocrystals | Pristine glass | 8.0 × 10−4 | 5.3 ± n.a. | 45 |
| Cellulose nanocrystals | Pristine glass | 2.2 × 10−2 | 4.7 ± 0.7 | 46 |
| Chitin-amyloid mixtures | Pristine glass | 2.6 × 10−4 | 1.4 ± 0.4 | 44 |
| Crosslinked water-soluble epoxidised lignin/LNPs | Wood | 3 × 10−1 | 13 ± n.a. | 43 |
| Bisphenol A diglycidyl ether-modified LNPs | Wood | 1 × 10−1 | 4.0 ± 0.8 | 25 |
| Lignin with epoxide-based crosslinkers | Wood | 4 × 10−1 | 2.2 ± n.a. | 42 |
Environmental impact assessment
The environmental impact of GlySolEtOAc+ InsEtOAcLNPs was compared to the one of LNPs from epoxidised SKL, i.e., GlySKL NPs, as the reference material which would display a superficial functionalisation and allow for similar interparticle crosslinking behaviour, and thus expected adhesive performances, upon thermal curing at 105 °C for 16 h. In particular, the E-factor33 was taken into account as a simple metric. Additionally, the solvent demand was calculated as an independent indicator and excluded from E-factor analysis since solvents and non-volatile wastes require distinctive processing. These two metrics can obviously only provide a partial estimation of the environmental impact of the process without considering toxicity issues and energy demand.The analysis of the contribution of each step to the overall values of E-factor and solvent demand (Fig. 5) was performed according to the equations described in literature.34 This approach evidenced the impact of fractionation, epoxidation and LNP formation on the overall E-factor and solvent demand of the entire process. While considering GlySolEtOAc+ InsEtOAcLNPs, fractionation led to a quantitative recovery of the starting material. This holistic use of the starting material allows for its neutrality in the E-factor estimation performed by excluding the solvents. The solvent employed in the generation of the material under analysis, i.e., EtOAc, was considered seperately only for the calculation of solvent demand. The splitting of the indicators into the single contributions of each step renders visible the remarkable impact of epoxidation, which involved excess epichlorohydrin and high dilution for the reaction to proceed. By epoxidizing just the soluble fraction, the quantity of material to be functionalized dropped from 100% to 36% for the EtOAc fractionation, thus reducing both E-factor and solvent demand associated to this step to one third of their original value. The solvent demand associated to fractionation, which was required for GlySolEtOAc+ InsEtOAcLNPs but not for GlySKL NPs, was more than compensated by the diminution of solvent consumption connected to epoxidation. In conclusion, the production of GlySolEtOAc+ InsEtOAcLNPs resulted to have a better environmental impact in terms of E-factor and solvent demand compared to the synthesis of GlySKL NPs.
 | ||
| Fig. 5 Contributions of fractionation, epoxidation and LNP formation steps to the overall E-factor calculated excluding solvents (a) and solvent demand (b) for the formation of fully epoxidised SKL NPs and GlySolEtOAc+ InsEtOAcLNPs. | ||
Conclusions
Solvent fractionation allowed for the refinement of softwood kraft lignin (SKL) into well-characterised fractions with specific MW and narrow dispersity and, therefore, for the development of materials with tailored properties. Solvent polarity and H-bonding capacity are crucial parameters affecting yield and selectivity of fractionation. Lignin nanoparticles (LNPs) prepared by mixing an epoxidised high MW fraction and the corresponding low MW fraction proved dimensionally stable upon thermal treatment and under alkaline conditions after thermal curing, confirming thus indirectly the hypothesis that epoxidised high MW fractions constituted the core of these LNPs. It was also possible to prepare LNPs with a surface-specific covalent functionalisation after fractionation, derivatising the low MW lignin fraction and recombining it with the corresponding high MW fraction. The lower environmental impact of the recombinatorial approach was shown in terms of reduced E-factor and solvent demand. This material-efficient technology allowed to valorise all of the starting material and could be used to develop performance-competitive adhesives for aminated glass surfaces, as such paving way for a new class of lignin-based functional materials.Author contributions
FF: conceptualization, investigation, formal analysis, visualization, writing – original draft; IP: methodology, project administration, writing – review & editing; MO: funding acquisition; HL: methodology, writing – review & editing; LZ: funding acquisition, project administration, writing – review & editing; AM: methodology, writing – review & editing; MHS: funding acquisition, project administration, methodology, supervision, writing – review & editing.Conflicts of interest
The authors declare we have no conflicts of interest to disclose.Acknowledgements
FF, LZ, HL and MO acknowledge financial support of Corimav Pirelli. FF is additionally grateful for the support by the Erasmus+ programme of the European Union. MHS acknowledges Vetenskapsrådet for financial support(grant number 2020-03752).References
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- C. Cui, R. Sun and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2014, 2, 959–968 CrossRef CAS.
- R. Mörck, H. Yoshida and K. P. Kringstad, Holzforschung, 1986, 40, 51–60 CrossRef.
- R. W. Thring, M. N. Vanderlaan and S. L. Griffin, J. Wood Chem. Technol., 1996, 16, 139–154 CrossRef CAS.
- A. Toledano, L. Serrano, A. Garcia, I. Mondragon and J. Labidi, Chem. Eng. J., 2010, 157, 93–99 CrossRef CAS.
- M. Helander, H. Theliander, M. Lawoko, G. Henriksson, L. Zhang and M. E. Lindström, BioResources, 2013, 8, 2270–2282 CrossRef.
- A. Keyoumu, R. Sjödahl, G. Henriksson, M. Ek, G. Gellerstedt and M. E. Lindström, Ind. Crops Prod., 2004, 20, 143–150 CrossRef CAS.
- O. Sevastyanova, M. Helander, S. Chowdhury, H. Lange, H. Wedin, L. Zhang, M. Ek, J. F. Kadla, C. Crestini and M. E. Lindström, J. Appl. Polym. Sci., 2014, 131, 40799–40810 CrossRef.
- M. Gigli and C. Crestini, Green Chem., 2020, 22, 4722–4746 RSC.
- V. Ponnuchamy, O. Gordobil, R. H. Diaz, A. Sandak and J. Sandak, Int. J. Biol. Macromol., 2021, 168, 792–805 CrossRef CAS PubMed.
- M. H. Sipponen, H. Lange, M. Ago and C. Crestini, ACS Sustainable Chem. Eng., 2018, 6, 9342–9351 CrossRef CAS.
- A. Moreno and M. H. Sipponen, Mater. Horiz., 2020, 7, 2237–2257 RSC.
- S. Beisl, A. Miltner and A. Friedl, Int. J. Mol. Sci., 2017, 18, 1244–1268 CrossRef CAS PubMed.
- F. Xiong, Y. Han, G. Li, T. Qin, S. Wang and F. Chu, Ind. Crops Prod., 2016, 83, 663–669 CrossRef CAS.
- I. V. Pylypchuk, P. A. Lindén, M. E. Lindström and O. Sevastyanova, ACS Sustainable Chem. Eng., 2020, 8, 13805–13812 CrossRef CAS.
- I. V. Pylypchuk, A. Riazanova, M. E. Lindström and O. Sevastyanova, Green Chem., 2021, 23, 3061–3072 RSC.
- T. Zou, N. Nonappa, M. Khavani, M. Vuorte, P. Penttilä, A. Zitting, J. J. Valle-Delgado, A. M. Elert, D. Silbernagl, M. Balakshin, M. Sammalkorpi and M. Österberg, J. Phys. Chem. B, 2021, 125, 12315–12328 CrossRef CAS.
- A. Moreno, J. Liu, M. Morsali and M. H. Sipponen, in Micro and Nanolignin in Aqueous Dispersions and Polymers, ed. D. Puglia, C. Santulli and F. Sarasini, Elsevier, 2022, 385–431 Search PubMed.
- T. Zou, M. H. Sipponen and M. Österberg, Front. Chem., 2019, 7, 1–12 CrossRef.
- M. H. Sipponen, M. Smyth, T. Leskinen, L.-S. Johansson and M. Österberg, Green Chem., 2017, 19, 5831–5840 RSC.
- M. Lievonen, J. J. Valle-Delgado, M.-L. Mattinen, E.-L. Hult, K. Lintinen, M. A. Kostiainen, A. Paananen, G. R. Szilvay, H. Setälä and M. Österberg, Green Chem., 2016, 18, 1416–1422 RSC.
- T. Leskinen, J. Witos, J. J. Valle-Delgado, K. Lintinen, M. Kostiainen, S. K. Wiedmer, M. Österberg and M.-L. Mattinen, Biomacromolecules, 2017, 18, 2767–2776 CrossRef CAS PubMed.
- A. Moreno, J. Liu, R. Gueret, S. E. Hadi, L. Bergström, A. Slabon and M. H. Sipponen, Angew. Chem., Int. Ed., 2021, 60, 20897–20905 CrossRef CAS PubMed.
- M. Morsali, A. Moreno, A. Loukovitou, I. Pylypchuk and M. Sipponen, Biomacromolecules, 2022, 23(11), 4597–4606 CrossRef CAS PubMed.
- T. Zou, M. H. Sipponen, A. Henn and M. Österberg, ACS Nano, 2021, 15, 4811–4823 CrossRef CAS PubMed.
- A. Duval, F. Vilaplana, C. Crestini and M. Lawoko, Holzforschung, 2016, 70, 11–20 CAS.
- C. Gioia, M. Colonna, A. Tagami, L. Medina, O. Sevastyanova, L. A. Berglund and M. Lawoko, Biomacromolecules, 2020, 21, 1920–1928 CrossRef CAS PubMed.
- N. Majoul, S. Aouida and B. Bessaïs, Appl. Surf. Sci., 2015, 331, 388–391 CrossRef CAS.
- ASTM, Stand. Pract. Lap Shear Adhes. Fibre-Reinf. Plast. FRP Bond., 2001.
- L. Zoia, A. Salanti, P. Frigerio and M. Orlandi, BioResources, 2014, 9, 6540–6561 CrossRef.
- A. Salanti, L. Zoia and M. Orlandi, Green Chem., 2016, 18, 4063–4072 RSC.
- I. Ribca, M. E. Jawerth, C. J. Brett, M. Lawoko, M. Schwartzkopf, A. Chumakov, S. V. Roth and M. Johansson, ACS Sustainable Chem. Eng., 2021, 9, 1692–1702 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- J. Andraos, Green Process. Synth., 2019, 8, 324–336 CAS.
- V. Passoni, C. Scarica, M. Levi, S. Turri and G. Griffini, ACS Sustainable Chem. Eng., 2016, 4, 2232–2242 CrossRef CAS.
- A.-S. Jääskeläinen, T. Liitiä, A. Mikkelson and T. Tamminen, Ind. Crops Prod., 2017, 103, 51–58 CrossRef.
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Green Chem., 2020, 22, 2712–2733 RSC.
- A. Salanti, M. Orlandi and L. Zoia, ACS Sustainable Chem. Eng., 2020, 8, 8279–8287 CrossRef CAS.
- A. Salanti, M. Orlandi, H. Lange, F. Ferruti and L. Zoia, ACS Sustainable Chem. Eng., 2022, 10, 11680–11691 CrossRef CAS.
- M. B. Agustin, P. A. Penttilä, M. Lahtinen and K. S. Mikkonen, ACS Sustainable Chem. Eng., 2019, 7, 19925–19934 CrossRef CAS.
- S. Beisl, J. Adamcyk, A. Friedl and H. Ejima, Colloid Interface Sci. Commun., 2020, 38, 100306–100311 CrossRef CAS.
- R. Jingxian Li, J. Gutierrez, Y.-L. Chung, C. W. Frank, S. L. Billington and E. S. Sattely, Green Chem., 2018, 20, 1459–1466 RSC.
- K. A. Henn, S. Forssell, A. Pietiläinen, N. Forsman, I. Smal, P. Nousiainen, R. P. B. Ashok, P. Oinas and M. Österberg, Green Chem., 2022, 24, 6487–6500 RSC.
- L. G. Greca, K. J. D. France, J. Majoinen, N. Kummer, O. I. V. Luotonen, S. Campioni, O. J. Rojas, G. Nyström and B. L. Tardy, J. Mater. Chem. A, 2021, 9, 19741–19753 RSC.
- H. Liu, Y. Feng, X. Cao, B. Luo and M. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 11356–11368 CrossRef CAS PubMed.
- B. L. Tardy, J. J. Richardson, L. G. Greca, J. Guo, H. Ejima and O. J. Rojas, Adv. Mater., 2020, 32, 1906886–1906893 CrossRef CAS PubMed.
- ASTM, ASTM D4690, Standard Specification for Urea-Formaldehyde Resin Adhesives, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03406a The Excel spreadsheet containing the calculations of E-factors is available from authors upon reasonable request. |
| This journal is © The Royal Society of Chemistry 2023 |