
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fragment-oriented synthesis: β-elaboration of cyclic amine fragments using enecarbamates as platform intermediates†
Alexandre F.
Trindade
a,
Emily L.
Faulkner
 a,
Andrew G.
Leach
b,
Adam
Nelson
*ac and
Stephen P.
Marsden
*a
a,
Andrew G.
Leach
b,
Adam
Nelson
*ac and
Stephen P.
Marsden
*a
aSchool of Chemistry, University of Leeds, Leeds LS2 9JT, UK. E-mail: s.p.marsden@leeds.ac.uk; a.s.nelson@leeds.ac.uk
bDepartment of Pharmacy, University of Manchester, Oxford Road, Manchester M13 9PL, UK
cAstbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, UK
First published on 26th June 2020
Abstract
A strategy for the β-sp3 functionalisation of cyclic amines is described. Regioselective conversion of protected amines to enecarbamates is achieved through electrochemical oxidation; these intermediates can be derivatised by functionalised alkyl halides under photoredox catalysis. The potential of the methods is highlighted by direct growth of a DCP2B-binding fragment.
Fragment-based ligand discovery is well established as a powerful approach for the discovery of new drugs, with four molecules developed in this way in clinical usage and many more in advanced trials.1 Fragments are typically smaller molecules than those used in high-throughput screening, meaning they explore chemical space more efficiently and, while the absolute potency of hit molecules is generally low, they bind their targets with high ligand efficiency (binding energy per non-hydrogen atom). A key step in the process is to ‘grow’ fragment hits from potentially any position without disrupting existing favourable binding interactions, in order to improve potency whilst preserving high ligand efficiency.2
Medicinal chemistry is dominated by a limited reaction toolkit that is heavily focused on heteroatom functionalisation.3 Given the strict molecular weight constraints applied to fragments, there are usually limited opportunities to grow them by elaborating functional groups that are not engaged in binding interactions.4 Fragment growth thus frequently requires time- and resource-intensive de novo synthesis, and can become a bottleneck in discovery.5 This is illustrated in the discovery of 1 (Fig. 1, panel A), a sub-micromolar lead compound against X-linked Inhibitor of Apoptosis Protein.6 Analysis of the binding pose of the low potency fragment hit 2 by protein crystallography identified elaboration of the 2- and 5-positions of the piperazine ring as key opportunities. The preparation of the lead compound 1 necessitated multi-step de novo synthesis of the disubstituted piperidine core.
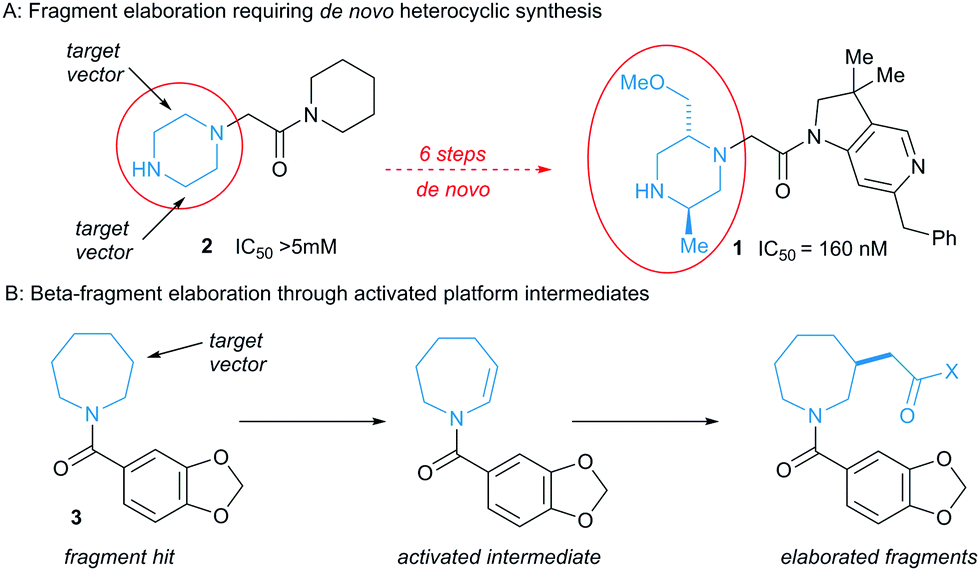 | ||
| Fig. 1 De novo synthesis versus direct fragment elaboration. | ||
A more attractive alternative would be to directly elaborate the hit fragment or a closely-related ‘stock’ precursor. In this context, C–H functionalisation chemistry presents an attractive opportunity to decorate otherwise inert atoms, and the many methods developed for direct functionalisation of sp2 C–H bonds7 complements the wide availability of sp2-rich fragments. Given their ubiquity in drug molecules and other bioactives,8 saturated cyclic amines are attractive sub-structures within sp3-rich fragments. Many methods have been developed for direct functionalization of saturated cyclic amines α-to nitrogen9 and indeed a recent report describes α-amino radical generation/Minisci heteroarylation as a means of selective α-functionalisation of protected cyclic amines in the context of fragment growth.10 By contrast, however, methods for the β- and γ-functionalisation of cyclic amines are much less common,11 meaning fragment growth along these vectors would likely fall back upon scaffold re-synthesis.
We envisaged that oxidation of protected cyclic amines to enamides or enecarbamates would afford a platform for the elaboration of fragment hits such as 3 at the β-position with a variety of functional groups, circumventing the need for de novo synthesis (Fig. 1, panel B). The utility of this process would rest both upon the efficient generation of the unsaturated amine derivatives, and the ability to introduce medicinally-relevant functionality to the β-carbon. We describe here the successful development of this approach.
Carbamate-protected amines would be useful precursors to fragments in which the nitrogen is either undecorated or, through N-functionalisation, a point of diversity, and so we sought methods to convert such species into enecarbamates. Employing the anodic oxidation first described by Shono,12N-Cbz piperidine 4a gave excellent conversion into methyl aminoacetal 5a (isolable in 90% yield). Attempted elimination failed using Brønsted acid12b,13 but the combination of TMSOTf and Hunig's base gave clean conversion to enecarbamate 6a (unoptimised 65% yield).14 Gratifyingly, the steps could be telescoped, giving 6a in 90% yield after purification. The protocol was extended to other carbamates (Scheme 1).
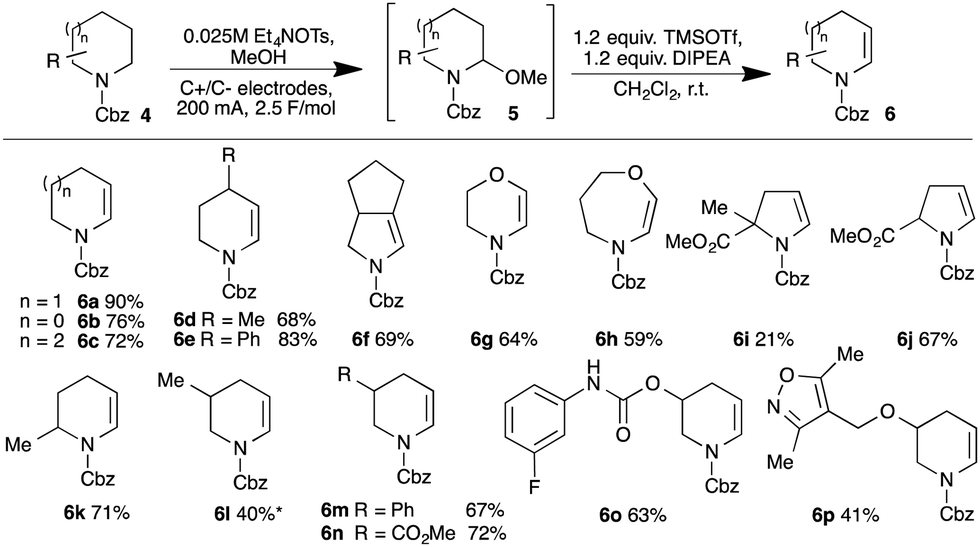 | ||
Scheme 1 Formation of enecarbamates by electrochemical oxidation/elimination. * 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of alkene regioisomers. :1 mixture of alkene regioisomers. | ||
The method proved robust across a broad range of substrates, encompassing various ring sizes (e.g.6a–c), the presence of β-heteroatoms (6g,h) and a range of appended substituents including arenes (6e,m), esters (6i,j,n), secondary carbamates (6o) and heteroarenes (6p). The oxidation of unsymmetrical substrates introduces the challenge of regiocontrol. With 2-substituted carbamates, literature precedent supports the predominant formation of the less-substituted 2,n-aminoacetals (although the origin of this selectivity remains unclear)15 and we did indeed observe formation of the less-substituted enecarbamates 6j and 6k.
The prospects for regiocontrol from more distal substituents seemed less encouraging, but we were delighted to find reactions of 3-substituted piperidines were often also highly selective. Of the five substrates investigated, four gave single regioisomers of the isolated enecarbamates 6m–p; the reaction of N-Cbz-3-methylpiperidine was less selective, with the less-substituted regioisomer 6l dominating in an inseparable 2:1 mixture. The origin of this regioselectivity was probed through DFT calculations (M06-2X/6-31+G** level of theory; see ESI†). The proposed mechanism involves formation of an (alkoxycarbonyl)amino radical cation, which undergoes proton loss to generate α-amino radicals; subsequent one-electron oxidation gives the corresponding iminium ions (Fig. 2). Radical A, leading to the observed favoured iminium ion, is higher in energy than radical B in both the gas and solution phase. Conversely, the observed iminium ion A is lower in energy than iminium B in the solution phase (moderate preference for B in the gas phase), but invoking iminium stability as the origin of the selectivity requires equilibration of the two isomers. We favour instead a kinetic explanation: modelling the transition states for solvent-mediated deprotonation of the (alkoxycarbonyl)amino radical cation revealed path A was favoured in both the gas (98:2) and solution (92:8) phase. The corresponding calculations with the 3-methyl variant showed a modest preference for path B (observed ratio within the intrinsic error of the calculations), supporting the lower observed selectivity. The origin of the selectivity is therefore likely steric.
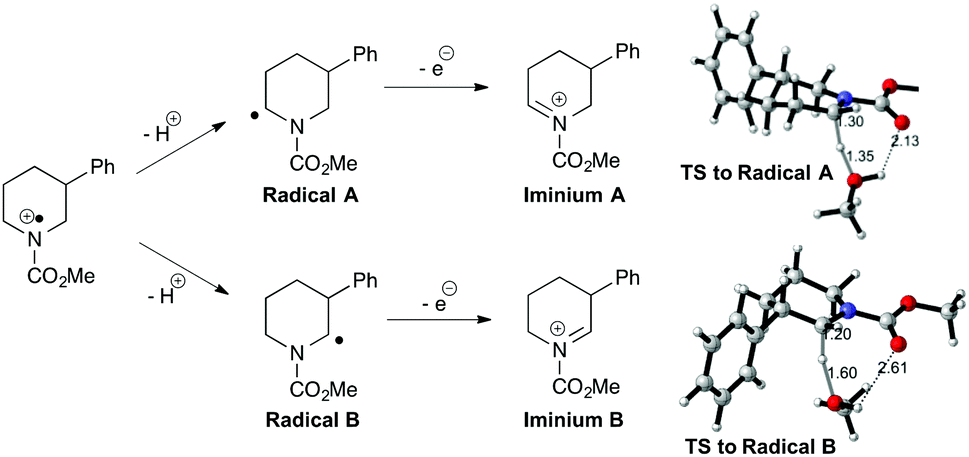 | ||
| Fig. 2 Regioselectivity in electrochemical carbamate oxidation. | ||
We now turned our attention to methods for elaboration of the enecarbamates to facilitate fragment growth along the valuable vector from the β-carbon atom. Specifically, we targeted introduction of functionalised alkyl groups capable of making additional beneficial interactions with a target protein by coupling of α-functionalised alkyl radicals with the enecarbamates as acceptor alkenes. While the coupling of enamides and enecarbamates has been reported with bromomalonates and bromoketoesters,16 reactions of the more useful monoactivated electrophiles (e.g. bromoacetates) have only been reported with highly nucleophilic enamines17,18 and their coupling with secondary enamides or enecarbamates has not been demonstrated to date.19 We therefore investigated the coupling of ethyl bromoacetate with enecarbamate 6a and pleasingly, following limited optimisation (ESI†), we identified Ir(ppy)3 as an effective photocatalyst for the reaction. In methanol as solvent, the initial product of the reaction was aminoacetal 7a; subsequent treatment of this crude material with triethylsilane in the presence of BF3 etherate gave substituted Cbz-piperidine 8a in 92% isolated yield (Scheme 2). In addition, the reaction also worked with N-acetyl and N-benzoyl enamides (ESI†), demonstrating the feasibility of direct elaboration of amide-containing fragments.
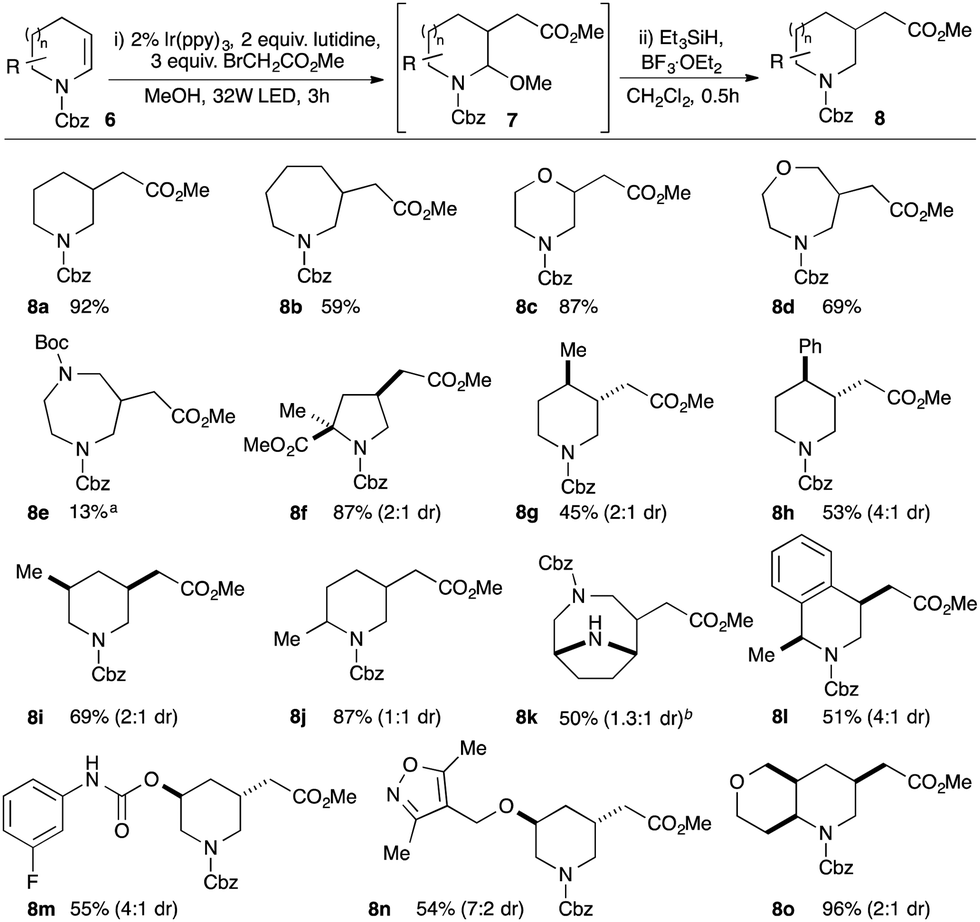 | ||
| Scheme 2 Substrate scope of enecarbamates in photoredox-mediated β-carboxyalkylation; ayield after N-Boc reprotection; bN-Boc protection removed in reductive step. | ||
We next investigated substrate scope and found that enecarbamates derived from piperidines, azepanes, morpholines, oxazepanes and diazepanes are all suitable partners in the reaction (e.g.8a–8e). However, Cbz-dihydropyrrole did not return the desired product, which we attributed to oxidative decomposition via Cbz-pyrrole. Consistent with this, blocking oxidation through α,α-disubstitution allowed generation of substituted pyrrolidine 8f. Substitution on the aliphatic backbone of the enecarbamates was tolerated, allowing access to 2,5-, 3,5- and 3,4-disubstituted piperidine products such as 8g–j: even more notably, potentially sensitive and medicinally relevant heteroatom-containing substrates also performed well (e.g.8m,n). Addition to more complex bicyclic substrates was also successful, exemplified in the construction of both bridged (8k) and fused (8o) products. Pleasingly, alternative monoactivated alkyl electrophiles could also be utilised, allowing for introduction of side-chains bearing ketone, nitrile and sulfonyl substituents (see ESI†). Details of mechanistic studies can be found in the ESI.†
Amides are present in ca. 25% of drug candidates and amide bond formation is amongst the most widely used reactions in drug discovery3b and development.20 The direct introduction of amidoalkyl groups to fragments would therefore be valuable but, despite extensive efforts, we were unable to achieve efficient coupling of cyclic enecarbamates with α-bromoacetamides (maximum 20% yield), possibly reflecting a more difficult reduction to give α-acetamidyl radicals.21 We therefore considered an alternative approach wherein coupling of a suitably activated ester electrophile would give an intermediate that could undergo substitution with an amine to deliver the amide in a telescoped process. As a further benefit, this approach would enable the synthesis of arrays of elaborated fragments from a single common activated ester intermediate. Pleasingly, reaction of N-hydroxysuccinimidyl bromoacetate 9 with enecarbamate 6a using 2 mol% Ir(ppy)3 in acetonitrile gave clean conversion into the substituted enecarbamate 10a in 77% NMR yield (Scheme 3). Simple addition of two equivalents of an amine and stirring led to the formation of the amidomethyl-substituted enecarbamates 11 in good yield over the two-step, one-pot sequence. The reactions worked well with a broad range of primary and secondary aliphatic amines (panels a and b), including hindered α-branched variants (e.g.11a,j,k,o). Medicinally-relevant substituents such as oxetanes (11k), morpholines (11n), (protected) piperazines (11m) and azine heterocycles (11c) were also tolerated, although reactions of (hetero)aromatic amines were unsuccessful even under forcing conditions. We also demonstrated that the crude enecarbamates could be directly reduced by exposure to Et3SiH and TFA, giving the saturated derivatives 12a–f in good yield (panel c).
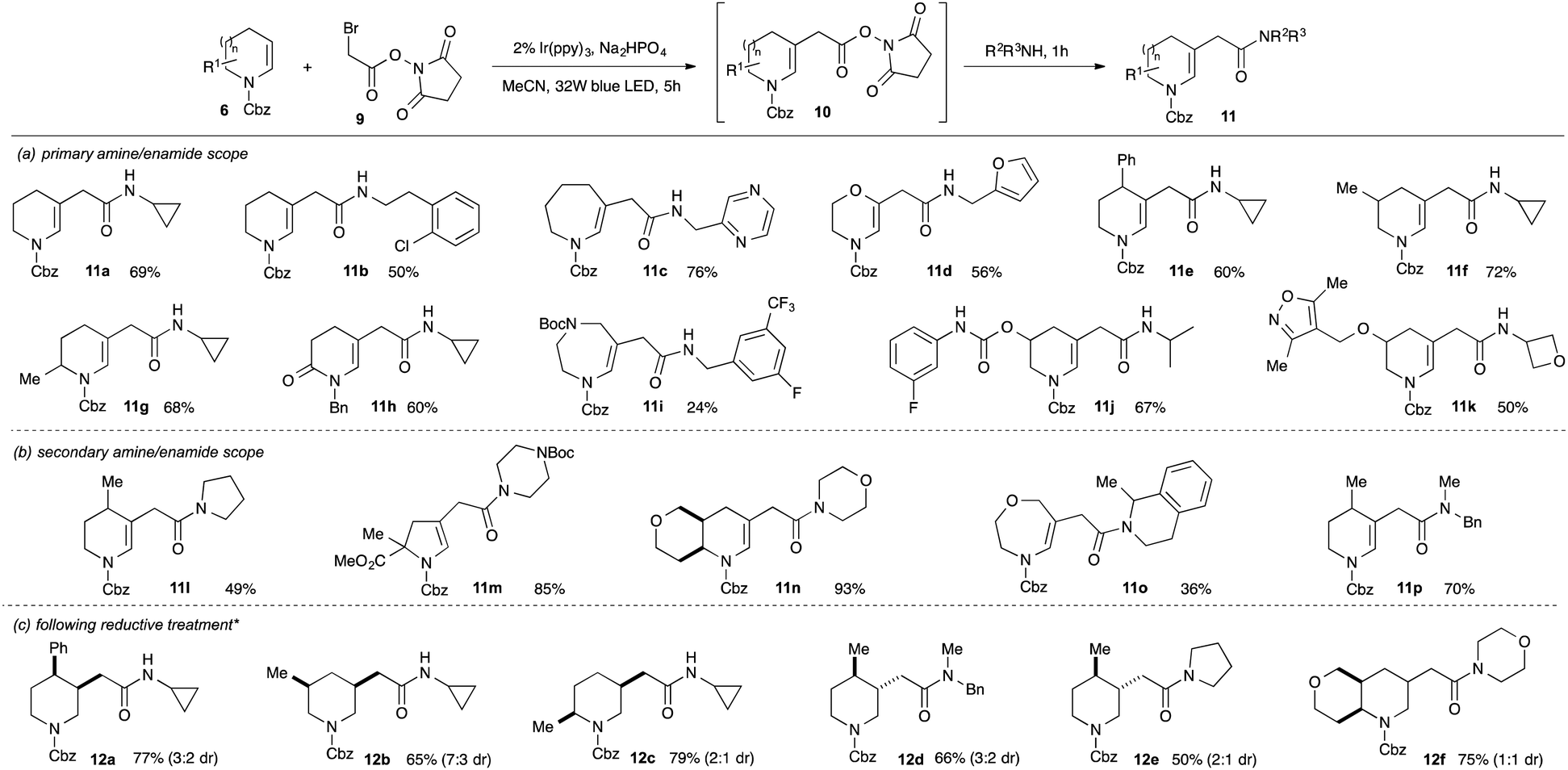 | ||
| Scheme 3 Synthesis of acetamidomethyl-substituted enamides by coupling of enecarbamates with activated esters. * Panel c only: the crude reaction mixture was treated with triethylsilane and TFA in dichloromethane. | ||
Finally we wished to demonstrate the application of the β-alkylation chemistry to the growth of a fragment hit. The piperonyl azepane 3 (Fig. 1) has been found via high-throughput X-ray crystallography to be a fragment hit for the m7GpppN-mRNA hydrolase DCP2B.22 The carbon atoms β-to the nitrogen have been identified as potential vectors for fragment growth,22 yet 3-carboxalkyl-substituted azepanes are not catalogue commodities,23 meaning de novo synthesis would be inevitable to exploit this vector. Instead, we were able to demonstrate introduction of a carboxymethyl group to enamide 13 by photoredox-mediated alkylation and reduction, giving ester 14 in 59% yield (Scheme 4). Alternatively, amidoalkyl groups could be introduced using the protocol via the intermediate N-hydroxysuccinimidyl ester, giving e.g. amide 15 in 36% yield. We also used both approaches to effect late-stage functionalization of the anti-depressant paroxetine (ESI†).
 | ||
| Scheme 4 Direct fragment growth and late-stage functionalisation of paroxetine using enecarbamate alkylation: (i) 2.2 mol% Ir(ppy)3, 3 equiv. BrCH2CO2Me, 2,6-lutidine, MeOH, 32 W blue LED, 3 h; (ii) Et3SiH, BF3·OEt2, CH2Cl2; (iii) 2.2 mol% Ir(ppy)3, 3 equiv. 9, Na2HPO4, MeCN, 32 W blue LED, 5 h, then cC3H5NH2, 1 h; (iv) Et3SiH, TFA, CH2Cl2. | ||
In summary, we have described methods for the challenging β-functionalisation of cyclic amines through regioselective conversion to enecarbamates and the subsequent installation of medicinally-relevant carboxyalkyl functionalities. We have further shown that the method can be used for regiospecific growth of fragment hits and in late-stage functionalization of complex products. The development of further methods for the direct elaboration of enecarbamates with medicinally-relevant functionality, as well as applications in fragment-based discovery projects, are ongoing.
We thank EPSRC (EP/N025652/1, EP/P016618/1) and EPSRC/Astex (studentship to ELF) for funding. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. MSCA-IF-2017-795189. Professor Frank von Delft (Diamond Light Source), and Drs Rachel Grainger and David Norton (Astex) are thanked for insightful comments. We acknowledge help from Research IT and use of the Computational Shared Facility (University of Manchester).
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug. Discov., 2016, 15, 605–619 CrossRef CAS.
- A. L. Hopkins, G. M. Keseru, P. D. Leeson, D. C. Rees and C. H. Reynolds, Nat. Rev. Drug. Discov., 2014, 13, 105–121 CrossRef CAS PubMed.
- (a) J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Nat. Rev. Drug. Discov., 2018, 17, 709–727 CrossRef PubMed; (b) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- P. N. Mortenson, D. A. Erlanson, I. J. P. de Esch, W. Jahnke and C. N. Johnson, J. Med. Chem., 2019, 62, 3857–3872 CrossRef CAS PubMed.
- C. W. Murray and D. C. Rees, Angew. Chem., Int. Ed., 2016, 55, 488–492 CrossRef CAS PubMed.
- G. Chessari, I. M. Buck, J. E. Day, P. J. Day, A. Iqbal, C. N. Johnson, E. J. Lewis, V. Martins, D. Miller, M. Reader, D. C. Rees, S. J. Rich, E. Tamanini, M. Vitorino, G. A. Ward, P. A. Williams, G. Williams, N. E. Wilsher and A. J. Woolford, J. Med. Chem., 2015, 58, 6574–6588 CrossRef CAS PubMed.
- For representative reviews, see: (a) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed; (b) R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, 17–117 CrossRef CAS; (c) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed.
- (a) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- For some recent examples, see: (a) A. J. J. Lennox, S. L. Goes, M. P. Webster, H. F. Koolman, S. W. Djuric and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 11227–11231 CrossRef CAS PubMed; (b) J. B. McManus, N. P. R. Onuska and D. A. Nicewicz, J. Am. Chem. Soc., 2018, 140, 9056–9060 CrossRef CAS PubMed; (c) W. Chen, L. Ma, A. Paul and D. Seidel, Nat. Chem., 2018, 10, 165–169 CrossRef CAS PubMed; (d) M. H. Shaw, V. M. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304–1308 CrossRef CAS PubMed; (e) D. T. Ahneman and A. G. Doyle, Chem. Sci., 2016, 7, 7002–7006 RSC.
- R. Grainger, T. D. Heightman, S. V. Ley, F. Lima and C. N. Johnson, Chem. Sci., 2019, 10, 2264–2271 RSC.
- See e.g.: (a) W. Chen, A. Paul, K. A. Abboud and D. Seidel, Nature Chem., 2020, 12, 545–550 CrossRef CAS PubMed; (b) R. Oeschger, B. Su, I. Yu, C. Ehinger, E. Romero, S. He and J. F. Hartwig, Science, 2020, 368, 736–741 CrossRef CAS PubMed; (c) J. Zhang, S. Park and S. Chang, J. Am. Chem. Soc., 2018, 140, 13209–13213 CrossRef CAS PubMed; (d) A. Millet, P. Larini, E. Clot and O. Baudoin, Chem. Sci., 2013, 4, 2241–2247 RSC; (e) N. Takasu, K. Oisaki and M. Kanai, Org. Lett., 2013, 15, 1918–1921 CrossRef CAS PubMed; (f) B. Sundararaju, M. Achard, G. V. M. Sharma and C. Bruneau, J. Am. Chem. Soc., 2011, 133, 10340–10343 CrossRef CAS PubMed.
- (a) T. Shono, H. Hamaguchi and Y. Matsumara, J. Am. Chem. Soc., 1975, 97, 4264–4268 CrossRef CAS; (b) T. Shono, Y. Matusmara, M. Ogaki and O. Onomura, Chem. Lett., 1987, 1447–1450 CrossRef CAS.
- S. Torii, T. Inokuchi, F. Akahosi and M. Kubota, Synthesis, 1987, 242–245 CrossRef CAS.
- T. Bach and H. Brummerhop, J. Prakt. Chem., 1999, 341, 312–315 CrossRef CAS.
- (a) A. M. Jones and C. E. Banks, Beilstein J. Org. Chem., 2014, 10, 3056–3072 CrossRef PubMed; (b) S. S. Libendi, Y. Demizu, Y. Matsumura and O. Onomura, Tetrahedron, 2008, 64, 3935–3942 CrossRef CAS.
- (a) T. Courant and G. Masson, Chem. – Eur. J., 2012, 18, 423–427 CrossRef CAS PubMed; (b) O. K. Koleoso, M. R. J. Elsegood, S. J. Teat and M. C. Kimber, Org. Lett., 2018, 20, 1003–1006 CrossRef CAS PubMed; (c) J.-H. Choi and C.-M. Park, Adv. Synth. Cat., 2018, 360, 3553–3562 CrossRef CAS.
- Isolated enamines: B. Hu, H. Chen, Y. Liu, W. Dong, K. Ren, X. Xie, H. Xu and Z. Zhang, Chem. Commun., 2014, 50, 13547–13550 RSC.
- In situ generated enamines: (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed; (b) Y. Zhu, L. Zhang and S. Luo, J. Am. Chem. Soc., 2014, 136, 14642–14645 CrossRef CAS PubMed; (c) A. Gualandi, M. Marchini, L. Mengozzi, M. Natali, M. Lucarini, P. Ceroni and P. G. Cozzi, ACS Catal., 2015, 5, 5927–5931 CrossRef CAS.
- For isolated examples, see: (a) primary enamides: J. Wu, M. Lang and J. Wang, Org. Lett., 2017, 19, 5653–5656 CrossRef CAS PubMed; (b) enephosphamides: A. Franchino, A. Rinaldi and D. J. Dixon, RSC Adv., 2017, 7, 43655–43659 RSC.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- T. Q. Chen and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2019, 58, 14584–14588 CrossRef CAS PubMed.
- Accessible at fragalysis.diamond.ac.uk.
- A search of commercially available compounds at reaxys.e-Molecules.com (18/11/2019) reveals a single compound (2-(1-Boc-azepane-3-yl)-acetic acid) available from a sole supplier by synthesis on demand.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and spectral characterisation. See DOI: 10.1039/d0cc03934a |
| This journal is © The Royal Society of Chemistry 2020 |