
NAD+ modulation with nicotinamide mononucleotide coated 3D printed microneedle implants†
Masood
Ali
 ah,
Wenhao
Huang
a,
Yicheng
Huang
a,
Xiaoxin
Wu
bcd,
Sarika
Namjoshi
a,
Indira
Prasadam
d,
Heather A. E.
Benson
e,
Tushar
Kumeria
*fgh and
Yousuf
Mohammad
*ah
ah,
Wenhao
Huang
a,
Yicheng
Huang
a,
Xiaoxin
Wu
bcd,
Sarika
Namjoshi
a,
Indira
Prasadam
d,
Heather A. E.
Benson
e,
Tushar
Kumeria
*fgh and
Yousuf
Mohammad
*ah
aFrazer Institute, Faculty of Medicine, The University of Queensland, Brisbane, QLD 4102, Australia. E-mail: y.mohammed@uq.edu.au
bDepartment of Orthopaedic Surgery, The Second Xiangya Hospital, Central South University, Changsha 410011, China
cResearch Centre for Computer-aided Drug Discovery, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518005, China
dCentre for Biomedical Technologies, School of Mechanical, Medical and Process Engineering, Queensland University of Technology, QLD 4059, Australia
eCurtin Medical School, Curtin University, Bentley, WA 6102, Australia
fSchool of Materials Science and Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: t.kumeria@unsw.edu.au
gAustralian Centre for Nanomedicine, The University of New South Wales, Sydney, NSW 2052, Australia
hSchool of Pharmacy, The University of Queensland, Brisbane, QLD 4102, Australia
First published on 14th February 2025
Abstract
Nicotinamide adenine dinucleotide (NAD+) deficiency has been shown to cause pathogenesis of age-related functional decline and diseases. Investigational studies have demonstrated improvements in age-associated pathophysiology and disease conditions. However, invasive methods such as immunohistochemistry, metabolic assays, and polymerase chain reaction currently used to measure cell metabolism render cells unviable and unrecoverable for longitudinal studies and are incompatible with in vivo dynamic observations. We report a non-invasive optical technique to investigate the upregulation of nicotinamide adenine dinucleotide (NAD+) in keratinocytes (both in vitro and ex vivo) upon administration of nicotinamide mononucleotide (NMN) coated microneedle (μNDs) implants. Our technique exploits intrinsic autofluorescence of cells and tissues using multiphoton microscopy. Additionally, μND coating formulations to date have been evaluated using fluorescence microscopy to determine the coated amount, often an imprecise correlation between fluorescence intensity and the coated amount on the μND surface. We also show that rheomechanical attributes of the coating formulation (containing two different viscosity enhancers: sucrose and carboxy methyl cellulose) affect the flow mechanics of the coating formulation at micron scale, and thus the amount of drug coated on the μND surface. In vitro keratinocyte cells were investigated with four concentrations of NMN (50, 250, 500 and 1000 μg), and evaluated with time-dependent NMN (500 μg) treatment at 0, 5, 10, 30, 60, 360 and 1460 min. We demonstrate that intracellular keratinocyte fluorescence of the endogenous NADH shows a decreasing trend in both the average fluorescence lifetime (τm) and the free unbound NADH (τ1), with increasing dosage of NMN administration. A similar trend in the average fluorescence lifetime (τm) of endogenous NAD(P)H was also seen in mouse ear skin ex vivo skin upon administration of NMN. We show a promising, minimally invasive, alternative delivery system for the NAD+ precursor molecule that can enhance patient compliance and therapeutic outcomes.
1. Introduction
Nicotinamide adenine dinucleotide (NAD+) deficiency has been shown to cause pathogenesis of age-related functional decline and diseases. Surveillance studies have demonstrated that supplementing the nutraceutical NAD+ precursor molecule nicotinamide mononucleotide (NMN) has ameliorated age-associated pathophysiologies and disease conditions, such as restoring NAD+ biosynthesis in impaired pancreatic beta cells, improvement of metabolic disorders such as obesity and diabetes, alleviating neurological disorder such as Alzheimer's through NAD+ mediated pathways.1–6 Such studies suggest that the positive effects of NMN, and the therapeutic dose and mode of administration need to be further investigated. To date, NMN has been administered orally only, with a dose limit of 250–900 mg.7–10 The reason for the high dose is the first pass metabolism. This results in potential side effects and excessively high cost (e.g., a pack of 250 mg NMN costs $149 from Agemate). Therefore, there is a need to deliver an effective NMN dose through other means of delivery that can reduce the high dose burden of oral delivery, also providing a localised effect, and eventually lowering the cost of healthcare. Microneedle (μND) arrays have successfully managed to deliver various therapeutics using the transdermal route.11,12 While dissolving μNDs have been reported to be safe for repeated administration,13,14 it must remain in the skin for a long time in order to fully dissolve.15 Besides, the amount of drug delivered by dissolving μNDs depends on the size of the μND array (number of needles in an array, and the height of the needles), and the application time.11 To alleviate the administration frequency issue, extensive studies were conducted using μND implant arrays to minimise patch wear time.16–27 Moreover, sustained release of drugs from single wear μNDs is highly desirable to treat chronic diseases. Additionally, μND implant patches are cost effective, since the cost to make one implantable patch with the ability to deliver a drug dose over multiple days is equivalent to manufacturing multiple dissolving or solid μNDs to deliver that same amount of dose.28Typical visualisation methods to characterise drug delivery through μNDs include computed tomography (CT) scanning,29 optical coherence tomography (OCT),30 confocal laser scanning microscopy (CLSM),31,32 ultrasound scanning,30,33 matrix-assisted laser desorption/ionisation mass spectrometry imaging (MALDI-MSI),34 as well as Raman based techniques such as confocal Raman, Coherent anti-Stokes Raman scattering (CARS)35 and surface enhanced Raman spectroscopy (SERS),36 and recently multiphoton imaging tomography (MPT).31,37 While CT, OCT and ultrasound have been used non-invasively to visualise μNDs in the skin, they lack the chemical sensitivity and visualisation depth to image drug release within the skin epithelia. Furthermore, due to the limitation in the imaging area using OCT and ultrasound, only a small area of μND arrays can be quantified over time.35 Additionally, CLSM can potentially incur UVA-induced photodamage for near-infrared radiation wavelengths below 650 nm, limited depth penetration, scattering, photobleaching and photodamage.38,39 Although confocal Raman and MALDI-MSI34 can visualise drug in the skin, they are quite invasive, typically requiring prepared skin tissue slices. Advanced bi-modal imaging systems such as Coherent antistokes and surface enhanced Raman spectroscopy, respectively, can facilitate non-invasive, label-free chemically specific imaging in skin samples under near-physiological conditions with a high z-resolution investigating complete penetration of the μND in the skin;35 however, they still lack capturing endogenous signals from cellular components to investigate dynamic physiological changes in live cells and tissues. The range of endogenous fluorophores in skin provides both an opportunity and a challenge in the imaging of exogenously applied therapeutics.40 In this regard, MPT imaging techniques have shown great potential in the imaging of endogenous fluorophores such as NAD(P)H and FAD, which are established important metabolic cofactors of cellular redox (oxidation/reduction) reactions involved in ATP production and antioxidant defence.31,37,40,41 The capture of autofluorescence from the endogenous fluorophore molecules using MPT subsequently facilitates the visualisation of a detailed cellular morphological structure.40,41 Another advantage of MPT includes imaging with increasing depth of tissue penetration of up to 1350 nm where water strongly absorbs and attenuates light and radiation, respectively, providing the highest resolution of all in vivo tissue imaging techniques.40–42 To this extent, we have evaluated changes in endogenous skin NAD+ levels due to the presence of exogenous molecule NMN by exploiting the fluorescence lifetime imaging microscopy (FLIM) technique within MPT. In this technique, fluorophores, including endogenous fluorophores in the skin, are excited by a femtosecond pulsed laser, and the time taken for them to return to the ground state is measured as the fluorescence lifetime, typically measured in picoseconds to nanoseconds. Changes in fluorescence lifetimes are indicative of interactions with exogenous materials, cellular degeneration, or cellular toxicity resulting from exposure.42,43 In this study, we used the fluorescence lifetime properties of NAD(P) and NAD(P)H to monitor the NAD+ regulation in mouse ear skin ex vivo after treatment with NMN μND implant arrays. We show that MPT-FLIM of NMN μND implant treated ex vivo skin yields characteristic and reproducible data that can be used to monitor the increased amount of NAD+ synthesis from the upper layer of the viable epidermis. These observations may contribute to the development of a novel means to image and assess pharmacodynamic effects caused due to μND treated skin that is metabolically comparable to in vivo skin.
The inherent thermodynamic properties of μND coating formulation was first studied by Gill and Prausnitz in 2007, demonstrating that the amount of loaded drug is generally influenced by the coating formulation viscosity, surface tension, wettability and the number of dips.44 Since then, fluorescence microscopy assay has become a standard key qualitative performance method to evaluate the amount of coated drug, though it sometimes offers an imprecise correlation between fluorescence intensity and the actual drug amount on μND surfaces.45–48 Rheological measurements, on the other hand, provide a comprehensive understanding of the coating's performance characteristics by assessing its viscosity and elasticity,49 which cannot otherwise be captured by fluorescence microscopy alone. These attributes are critical for ensuring the even spread and adhesion of the coating under shear stress,50,51 maintaining consistency in flow mechanics,52 and preventing the coating from dripping or running off the μND surface.51 While fluorescence microscopy alone provides visual and spatial distribution data, rheology quantifies the material's response to stress, strain, and temperature changes, helping to identify optimal formulations with desired properties such as ease of application and uniform spreadability. Ideal non-Newtonian liquids exhibit a constant viscosity coefficient over a wide range of applied stresses, independent of shearing duration or history.53 However, coating formulations containing trace levels of viscosity enhancer and a surfactant, often display intermediate behaviour between that of a solid and a liquid.49,54 These materials have been found to exhibit recoverable, time-independent deformation/shear thinning under micro-stresses,55 followed by retention of original storage modulus (G′).56 The thixotropic phenomenon in coating formulations, in particular, shows a decrease in viscosity over time under constant shear stress, such as during the flow out of a micro-nozzle technology and/or dip coating techniques of materials, followed by recovery to their original viscosity once the stress is removed.49,54 The three interval thixotropy test (3ITT) is generally used in the food industry to generate rapid shear stress/shear rate deformation simulating effects of stirring during processing and handling steps of food57,58 and paint59–61 industries, respectively. 3ITT is different from the well-known and frequently used frequency sweep and creep/recovery tests62,63 as a quick “shear rate/shear stress” can be applied in the non-linear viscoelastic region (non-LVR) and deformation can be examined in both LVR and non-LVRs. This is a particularly important characteristic to investigate in coating formulations used in the dip coating of μNDs, as this determines the μ-flow mechanics of the coating formulation on the surface of the μNDs. We hypothesise during the dip coating of μNDs, the coating formulation undergoes three phases of molecular mechanical deformation: (a) pre-dipping phase, here the coating formulation doesn’t experience any shear, therefore, the shear conditions are at rest and well within the formulation's linear viscoelastic range (LVR) and constant G′ (storage modulus) and G′′ (loss modulus), (b) dipping phase, here the coating formulation experiences high-shear conditions which fall outside the LVR or non-linear viscoelastic region, causing the formulation's molecular bonds to break due to the dip coating process, and lastly (c) post dipping phase, here the coating formulation on the surface of μNDs tries to regain its original viscoelastic modulus G′ thus exhibiting pseudoplastic behaviour.64 By integrating rheological assessment with traditional microscopy techniques, a more robust evaluation of coating formulations can be achieved, leading to improved product performance and reliability.
Herein, we show that the thixotropic phenomenon of coating formulation containing viscosity enhancers such as sucrose and carboxy methyl cellulose (CMC), affects the amount of NMN coated onto the surface of μND implant tips. NMN coated μND implant arrays exhibited NMN release in vitro human skin for up to 3 days after single application, with 80% burst release within the first 24 h, followed by 20% release in the following 48 h. Finally, the NMN released from the NMN coated implant arrays increased NAD+ levels in mouse ear skin epidermis which was evaluated using MPT imaging modality. This proof-of-concept study may contribute to the development of a novel means to image and assess pharmacodynamic effects caused due to μND treated skin that is metabolically comparable to in vivo skin.
2. Methods and materials
2.1. Materials and equipment
All master μND arrays were printed from a UV-curable resin (eResin-PLA Pro Transparent, eSUN) using a desktop DLP 3D printer (Sonic Mini 4K, Phrozen, Taiwan). The 3D printing parameters were a layer thickness of 25 μm in the z-direction, with 3 s layer curing. The resin was modified by the addition of analytical grade methanol (Sigma). Pharmaceutical grade sucrose was purchased from Ajax Finechem (342.3 g mol−1, 0809450), pharmaceutical grade CMC was purchased from Sigma (high viscosity: 419273 and low viscosity: C5678), pharmaceutical grade Tween-20 was purchased from Sigma (P2287), and pharmaceutical grade NMN (99.9% purity) was purchased from Agemate (Australia). A Thermofisher spectrophotometer (Multiskan) was used for fluorescence detection and quantification. For all human skin in vitro studies, ethics approval was obtained. Full thickness healthy human donor skin was donated from female patients (within the age range 40–55 year) undergoing abdominoplasty at Brisbane (QLD) hospitals. Ethics approval was granted by The University of Queensland Human Research Ethics Unit, approval number: 03081 (HREC/16/QPAH/64). After collection, subcutaneous fat was removed with a scalpel blade, using an established protocol.65 The skin thickness was measured using digital Vernier calliper (Kincrome, Australia). Defatted skin was then frozen at −20 °C for further use.2.2. Fabrication of μND implants
The μND implant arrays were fabricated using a PDMS female mould using a two-step solvent casting method as previously described,66 with some modifications. PDMS female moulds were prepared from a 3D printed master67–71 with geometry consisting of a conical shaped tip (Fig. S1aiii (ESI†) 0.8 × 0.5 mm, Height × Base diameter), with a trapezium middle shaft (Fig. S1aii (ESI†) 0.4 × 1.4 mm, Height × Base diameter), 1.8 mm between needle tips (Fig. S1aiv, ESI†) and base height (Fig. S1ai, ESI†) respectively. This design allowed us to (i) make an on-demand master μND array, which was then used to make multiple moulds and (ii) optimise and use a fast-dissolving sugar–polymer complex as the middle shaft to dislodge the implants in the skin. Firstly, the tip implant portion of the μND was prepared, 4 μL of μND casting solution containing methacrylate resin (80%, v/v) was poured using a pipette into the mould cavities followed by centrifuging at 3200 rpm for 10 min. Any residual resin solution on the surface was scraped off using a glass coverslip. For visual confirmation in some cases, the two-step casting resin solution also contained 0.01% (w/v) Orange B dye (O8126, Sigma) to facilitate microscopy imaging. Then, the moulds were placed inside a UV-curing chamber (405 nm) to cure the tip implant for 8 min. Following that, 10 μL of a polymer sugar complex solution consisting of a mixture of hyaluronic acid (HA), polyvinyl pyrrolidone (PVP) and sucrose with a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3 (% w/v), was pipetted onto the mould surface, then centrifuged at 3200 rpm for 10 min to fill the middle portion of the PDMS female mould. The moulds were then left inside an incubator at 60 °C for 12 h. This was followed by the addition of the third layer of the resin polymer (500 μL) and curing the UV-chamber for 8 min. The μND implant arrays were peeled gently with forceps and stored in a desiccator at room temperature until further use.
:1:3 (% w/v), was pipetted onto the mould surface, then centrifuged at 3200 rpm for 10 min to fill the middle portion of the PDMS female mould. The moulds were then left inside an incubator at 60 °C for 12 h. This was followed by the addition of the third layer of the resin polymer (500 μL) and curing the UV-chamber for 8 min. The μND implant arrays were peeled gently with forceps and stored in a desiccator at room temperature until further use.
Zeiss Stem 2000-C stereo microscope with a stand-alone Axiocam ERc 5s microscope camera (Zeiss, Germany) was used to image μND arrays. The microscope was connected to the computer and run by an Axiovision 4 LE viewfinder app to capture images and make iterations such as interactive geometric measurements, scale magnification and scale units. Additionally, μND arrays were also visualised on a bench-top scanning electron microscope (SEM), by placing them on circular discs and scanning in high-vacuum mode using the ETD detector at 10−5 Torr and 15 kV (Joel, Neoscope). After initial visual inspection, parameter dimensions were approximated by performing an analysis on digital images of samples taken with the microscope.
2.3. Structural strength of μND implants
The structural strength of μND implant arrays was evaluated by measuring the compression and shear strength using the texture analyser (TA, CT3 Texture analyser, Brookfield), equipped with a 5 kg load force gauge, using a previously described method with some modification.66 Briefly, for the compression test, a single μND implant array was immobilised on the bottom plate of the TA instrument, and the cylindrical-shaped sensor probe was lowered onto the μNDs in the vertical direction at a speed of 0.05 mm s−1 (n = 3 arrays). When the probe sensor first detected the μND tips, the displacement was set to zero, starting the force measurement until the displacement reached 0.81 mm from the μND tips towards the patch backing. To test the shear strength of μND patches, a single μND patch was attached to the TA baseplate positioned ‘horizontally’ with needle tips facing sidewards using double-sided tape, and the cylindrical sensor probe, initially located above the μND patch, started moving vertically towards the μNDs at a speed of 0.05 mm s−1 (n = 3 arrays). When the probe sensor first detected the μND shaft (tips facing sidewards), the displacement was set to zero, starting the force measurement until the displacement (0.33 mm) reached a point to break the tip from the backing plate.In vitro μND detachment from the patch backing was studied using the time-course of μND detachment test following an established protocol.21 μND implant arrays (n = 3) were immobilised on the bottom surface of a Petri dish, and then phosphate-buffered saline (PBS, 37 °C) (P-3813, Sigma) was added. The PBS contained 138 mM NaCl and 2.7 mM KCl. The μND detachment time due to array-backing dissolution was monitored using a microscope (Zeiss, Germany) and the time was measured until all μNDs were detached from their backing. To investigate μND penetration into skin and detachment from the backing μND implant array, a skin insertion test was performed using full thickness human skin in vitro following an established protocol with slight modification.21 Briefly, μND implant arrays were applied on the skin using compression from the TA at 0.05 mm s−1 (hold for 5 min). Following that, the backing of the μND implant was removed gently. Brightfield and Fluorescence microscopy (Olympus IX73, USA) were used to image μND implant treated skin, with a DAPI filter to visualise the implant in the skin (an excitation/emission maxima of 350/465 nm).
2.4. Coating formulation rheological assessment
Two different viscosity enhancers were trialled to study the coating formulation's rheomechanical thixotropic properties using an established protocol with some modifications.57,72 Briefly, the rheological measurements of the coating formulations A–D (Table 1) were performed using a strain/stress-controlled rheometer (Anton Paar, MRC 302, Austria) equipped with a parallel plate (plate diameter 50 mm, angle 4°, gap size 0.5 mm) within the shear rate range of 0.01–100 s−1 at room temperature (21–24 °C). An amplitude sweep test was performed to determine the storage modulus (G′), and loss modulus (G′′) at a frequency of 1 Hz at room temperature (21–24 °C) collecting 25 points with a strain sweep of 0.1–1.0% to determine the Linear Viscoelastic Range (LVR). Afterwards, 400 μL of the sample was pipetted onto the lower plate, equilibrated to room temperature (21–24 °C) for 2.5 min before starting the run. Due to the presence of both a viscosity enhancer and a surfactant, the coating formulation started to solidify after 2.5 min. After determining the LVR, 3ITT was performed using a three-step operation: (a) rest phase: plateau curve obtained at constant shear rate of 0.1 s−1 for 6 s collecting 10 points, (b) shear phase: plateau curve obtained at constant shear rate of 100 s−1 for 6 s collecting 10 points, and lastly (c) retention phase: upward curve obtained constant shear rate of 0.1 s−1 for 6 s collecting 10 points. All measurements were carried out in triplicates. The aqueous coating formulations (w/v % unless otherwise specified) were prepared using MilliQ water accordingly (Table 1). The sampling size for each coating formulations A and B were n = 3.| Formulations | NMN (mg mL−1) | Viscosity enhancer (w/v, %) | Tween-20 (v/v %) |
|---|---|---|---|
| A | 50 | Sucrose (5 mg mL−1) | 0.01 |
| B | 50 | CMC (1% High Mw) | 0.01 |
2.5. Coating of μND implants
To ensure the uniformity of coating on dual layered μND arrays, coating tanks were prepared based on the dimensions of the tip of the μND only, such that only the implant tip was coated and not the middle shaft or the base, modifying the dimensions from our previous study.65 The coating tank was 3D printed with the aim of creating micro-tanks on a flat base that would facilitate the immersion of an individual μND tip implant only for the coating process. The dimension of the individual micro-tanks: micro-tank inner diameter approximately 500 μm and height 1.2 mm, with a 3 × 3 micro-tank array, with the distance between the micro-tanks set to 5 mm in CAD (Autodesk fusion) (Fig. S2, ESI†). The micro-tanks are hollow to facilitate the filling from the bottom of the base rather than the top, and secondly, any positive pressure or air bubbles created from the dipping of the μNDs will dissipate from the bottom opening of the base. Approximately 27 μL (3 μL × 9 micro-tanks) of the coating formulation was used to fill the entire array of micro-tanks. These geometrical dimensions ensure a perfect fit onto the respective coating tanks to avoid any coating both on the middle shaft and the base plate of the μND array. For the dip coating process, the number of dips were 3/5/7/9 and 5 min drying time in between dips at room temperature in a vacuum chamber was performed. The speed of dips was not controlled for the dip coating since the micro-tank is hypothesised to counteract the capillary pull49,73 of the coating formulation from coating the base of the μND. Following the coating process, the coated μND tips were washed with 500 μL PBS (pH 7.4), filtered through 0.5 μm filter, then injected into HPLC to determine the coated NMN amount. The dip coated μND tips were also visualised for coating efficiency using the SEM by placing them on circular discs and scanned in high-vacuum mode using the ETD detector at 10−5 Torr and 15 kV (Joel, Neoscope).2.6. Live cell imaging of keratinocytes: assessing NAD+ levels in keratinocyte cells in vitro using multiphoton tomography
To date, there are no literature studies interrogating the detection and regulation of NAD+ on keratinocytes in vitro after supplementing with NMN, using Multiphoton Tomography. Therefore, a systematic study was designed interrogating the regulation of NAD+in vitro post NMN administration following a 2-regimen study. Regimen 1 (dose-dependent): NMN treated cells for 6 h and 24 h at various amounts (0, 250, 500 and 1000 μg). Regimen 2 (time-dependent): The amount of NMN was constant at 500 μg, but the treatment time varied at 0, 5, 10, 30 min, and 1, 6, and 24 h.HaCat cell lines were used as they are the most commonly used keratinocyte cell lines.74 Cryopreservation, thawing and passaging (Passage 4) of HaCat cells were carried out using an established protocol.75 The frozen HaCat cells (in 5% DMSO) were thawed in a 37 °C incubator, then neutralised by adding full growth media (RPMI, supplemented with 10% FBS and 1% penicillin streptomycin) immediately, centrifuged at 300 rpm for 3 min prior to discarding the supernatant. The cells were then grown in a T75 flask in full growth media overnight prior to passaging. After 24 h, the cells (106 cells) were washed with sterile PBS and trypsinised (0.05%). The trypsinised cells were then transferred into a 15 mL falcon tube and centrifuged at 300 rpm for 3 min. Following that, the supernatant was discarded, and the cell pellet was dispersed in growth media and counted for viability using the trypan blue method. The cells (104 cells) were then grown on coverslips in a 6-well plate ensuring the majority of the cell density on the cover slip. The well plate was incubated at 37 °C with 5% CO2 overnight. After overnight incubation, the media was discarded and replaced with fresh media, followed by the treatment of the cells with various concentrations of NMN. Various NMN amounts (0, 250, 500 and 1000 μg) were dissolved in sterile water for injection and then filtered through a sterile 0.5 μm filter. After adding the required amount of NMN to the cells (n = 3 for each NMN amount), the well plate was placed in the incubator overnight. The following day, the coverslip (containing the NMN treated cells) was placed on a glass slide such that the cells would not detach. Two pieces of parafilm were placed on two sides of the glass side, and then the cover slip with the cells was placed on top of the parafilm such that there was a space (approximately 500 μm) in between the coverslip and the glass slide. Approximately 100 μL of the media was filled into that space with a pipette. The glass slide with the coverslip was then imaged using multiphoton microscopy. The imaging protocols were followed from a previously established protocol41,65 with slight modification. Briefly, the microscope laser power was at 12 mW to avoid photodamaging the cells, excitation wavelength of 740 nm (two photon), at an exposure of 47 s with an acquisition image size of 512 × 512 pixels (256 × 256 μm2), and a bandpass filter of 350–650 nm (BG39, Schott glass colour filter) was used to optically filter the emitted fluorescence light to capture live images of MPT, fluorescence lifetime imaging microscope (FLIM) detectors with band pass filters 350 to 450 nm (channel 1) and 450 to 515 nm (channel 2) to isolate NAD(P)H signals into separate channels. FLIM measurements were carried out by using a time-correlated single-photon counting SPC-830 detector system (Becker & Hickel, Berlin, Germany) integrated into the multiphoton microscopy. The acquired FLIM images were processed with SPCImage on the selected ROIs. The ROIs were selected focussing on a single cell or a cluster of cells only. The decay matrix was calculated with the following: binning 2 (increase or decrease the number of photons for the exponential decay curve fit, increasing the photon count reduces spatial resolution42), selected the ‘shift’ function (adjusts decay curve fit to the left/right) and unselected ‘Scatter’ (adjusts decay curve fit up or down), exported pixel intensity and pixel frequency of average fluorescent lifetime (τm), non-enzyme bound lifetime (τ1) and redox ratio (α1/α2).
2.7. Coated NMN μND implant in vitro permeation study
The in vitro NMN skin permeation from the coated μND implant arrayed treated skin was studied using the vertical Franz diffusion cell technique using a previously established protocol with slight modifications.76 Donated human abdomen skin portions were defatted and cut into appropriate size (2 × 2 cm2) to fit into the donor compartment of the Franz diffusion vertical system. Coated NMN μND implant arrays (n = 3 μND implant array, n = 2 biological replicate) were then applied onto the full thickness skins’ stratum corneum side using a texture analyser (0.5 mm s−1, 5 min hold). The μND implants were then dislodged and remained in the skin, this was followed by mounting the treated skin onto the receptor compartment of the Franz diffusion cell, then placing the donor compartment on top of the receptor compartment and securing it tight. The integrity of the skin membrane was evaluated by measuring the resistance between the donor and the receptor compartment respectively, using a multimeter (Digitor Q1563, DSE, Brisbane, Australia) equipped with Ag/AgCl electrodes. Skin which showed resistance lower than 20 kΩ cm2 was discarded. The receptor chamber was filled with 0.1 M PBS (pH 7.4, 0.05% NaN3, 0.5% Volpo). Each cell (donor and receptor chambers) temperature was thermostatically maintained at 37 ± 1 °C. The donor cell was un-occluded, except a small bob was placed atop the μND array to stop it from detaching from the skin. Sample collection points were taken every 15 min for the first hour, then at 2, 4, 6, 12, 24, 48 and 72 h, respectively. To determine the NMN content, Franz diffusion receptor chamber solution samples were filtered through 0.5 μm filter and injected into the HPLC-UV system.2.8. Statistical analysis
All data are expressed as average ± SD. Analysis of variance (ANOVA, one way or two way) was utilised to determine statistical significance. A p-value of <0.05 was considered statistically significant.3. Results
3.1. 3D printed master μND and female mould fabrication
Master μNDs were designed in such a way that its geometry consisted of a conical tip (height = 0.8 mm) with a trapezium middle shaft (height = 0.4 mm), the trapezium middle shaft had walls angled from the base, giving it a wider opening. The master μND arrays yielded an output conical height of 737.0 ± 40.41 μm and tip diameter of 57.94 ± 14.46 μm (Fig. S4a, ESI†). A tip diameter of less than 80 μm is one of the most important design factor considerations for a successful skin insertion.11 Additionally, the output μND base diameter was approximately 1403 ± 29.37 μm, a wider base diameter is preferable when working with shear sensitive therapeutics since a higher centrifugal force is not needed to fill the therapeutics into the female μND mould cavities.11 To confirm the geometric dimensional closeness of the female mould to the master μND array, PVP μND arrays were made from the female mould, yielding a μND height and tip diameter of 697.0 ± 38.51 μm and 40.71 ± 10.59 μm respectively (Fig. S4b, ESI†), demonstrating closeness in values. The lower dimensional characteristics of the PVP μND can be attributed to the drying of the polymer during the solvent casting method, a phenomenon quite commonly seen previously.20,693.1.1.1. Compression and shear load tests of μND implants. The μND implant array was designed to achieve facile μND tip detachment from the patch backing post skin insertion. This was achieved by two design criteria: the μND patch must exhibit (a) enough mechanical strength allowing successful skin penetration under compression and (b) structural weakness at the μND – backing interface for easy detachment upon patch removal from the skin under tension. The interfacial strength between the middle shaft portion and the μND tip and the μND backing was investigated. This was done by examining the shear and tensile forces (inset graph, Fig. 1a and b). In these scenarios, the solid μND exhibited strong compressive force of 28.81 ± 12.29 N per needle (Fig. 1a) and 5.76 ± 2.40 N per needle (Fig. 1b) under shear to cause buckling only. In contrast, μND implants showed consistent μND implant tip detachment at the μND – pedestal interface under both compression and shear force (Fig. 1a and b microscopy images). In addition, these forces were significantly lower compared to solid μNDs (p < 0.1 and p < 0.01), measuring 11.51 ± 3.964 N per needle under compression and 1.500 ± 0.638 N per needle under shear (Fig. 1a and b). A notable observation was that the μND implant array showed a drop in the force when the middle shaft detached from the base plate, at 0.69 mm displacement distance for compression test (Fig. 1a) and at 0.23 mm displacement distance for shear test (Fig. 1b). This is indicative of the metallic probe compressing or shearing the μND tip to a point where the interfacial μND middle portion attachment detaches from the tip and the backing, demonstrating the successful sufficient mechanical strength suitable to facilitate μND tip implant detachment upon removal from the skin under compression.
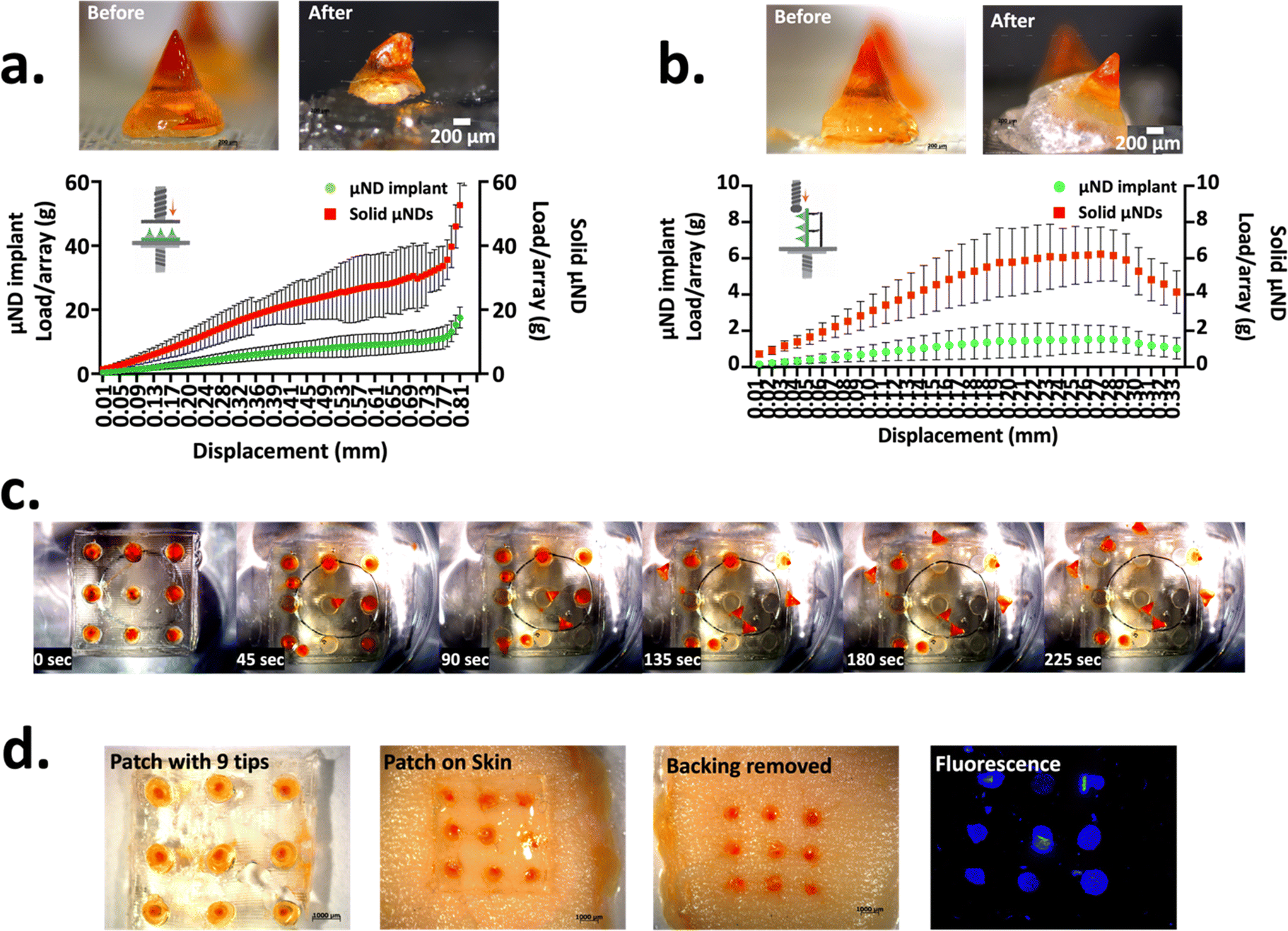 | ||
| Fig. 1 Characterisation of the mechanical strength of μND implant arrays. Representative compressive (a) and shear (b) force-displacement profile (under the microscopy images) of μND implants and solid μNDs, with inset: schematic illustration showing the compression stress (a) and shear stress (b) applied on the respective μNDs and bright-field microscopy images of the representative μND implant (one needle) before and after compressive stress (a) and shear stress (b). **p < 0.01 and *p < 0.1. All quantitative data are shown as Av. ± SD (n = 3, but data shown as 9 needles per array for the shear test only). (c) Time-lapse study of the μND implant tip showing detachment from solid backing through the dissolution of the middle shaft upon immersion in PBS (n = 3). Representative top-view images of μND patches in PBS (bright-field microscopy) over time (n = 3). To visualise MN detachment, an orange dye was included in the tip implant. (d) Insertion and detachment of the μND implant treated human skin in vitro. Representative bright-field and fluorescence microscopy images of μND implant treated skin after insertion and removal. Orange dye was used to visualize sites of implant penetration in the skin, which was fluorescently visualised using the DAPI filter with an excitation/emission maxima of 350/465 nm on the fluorescence microscope. | ||
3.1.1.2. μND implant detachment test (PBS and skin). To determine the middle shaft interface of the μND implants’ quick detachment through water absorption, μND implant arrays were dissolved in PBS, as well as assessed by insertion into skin. The μND implants were fixed inside a 10 cm dish followed by adding PBS to the dish to simulate the separation in the skin. The μND implant tip detachment time was measured by visual observation. Upon contacting with PBS, the μND implants middle shaft μNDs started detaching within 45 s, with complete needle tip dissociation within 225 s (Fig. 1c). This result suggests that adding a water soluble polymer–sugar complex middle shaft to the μND implants facilitated the detachment by faster dissolution of the μND backing.
The compressive and shear force measurements in Fig. 1a and b suggested that μND implants are mechanically strong under compression to be inserted into skin, but morphologically weak under tension to immediately detach following μND patch removal from the skin. To test this theory, the μND implants were pressed using a texture analyser against human skin in vitro (Fig. 1d). It was found that μND implant tips showed complete detachment from the backing 5 min post patch application (Fig. 1d). In addition, the applied patches had no remaining μNDs tips, meaning that they would not generate biohazardous sharps waste that can be easily and safely discarded without special handling in future clinical use. Taken together, the mechanical strength measurements (Fig. 1a and b) and μND detachment and skin insertion experiments (Fig. 1c and d), both indicate that the μND implants facilitate rapid tip detachment and implantation in the skin with no special instructions or skills required except for pressing the μNDs patch to the skin and then removing it after some time.
 | ||
| Fig. 2 (a) Rheological characterisation of two NMN coating formulations with sucrose (formulation-A) and CMC (formulation-B) as viscosity enhancers and Tween-20 as a surfactant. A rheometer was used to assess the formulations under shear stress (0.1–1 s−1) at room temperature (21–24 °C) (n = 3). Shear rate sweep tests revealed non-Newtonian pseudoplastic flow behavior with shear thinning at higher shear rates, simulating the flow mechanics of the coating formulation during μND dipping. Upon removal of shear, both formulations exhibited thixotropy, with elastic modulus retention. Formulation-B showed a higher viscosity (∼4 mPa s) than formulation-A (∼1 mPa s), resulting in delayed modulus recovery and reduced NMN coating adhesion. (b) Fluorescence microscopy images of μND implants coated with formulation-A and formulation-B (using fluorescein instead of NMN) demonstrating higher fluorescence intensity with formulation-A, indicating superior coating retention. (c) Light microscopy images of empty and dye filled 3D printed microtanks (i/iii) and 3D printed μNDs (ii/iv) (n = 3). (d) SEM images of coated μNDs (3/5/7/9 dips respectively) and the quantitative analysis of the average NMN coated amount (μg) as a function of the number of dips (n = 12, data shows Av ± SD and plotted in PRISM). | ||
To ensure that the coating formulation only coated on the μND implant tips and not the base, 3D printed micro-tanks (Fig. 2ci/iii) were fabricated with design dimensions of the micro-tank mouth facilitating the coating of the μND implant tip (Fig. 2cii) only. The micro-tank dimensions were designed with 500 μm in micro-tank inner diameter and 1.2 mm in height, such that when μND implant tips were dipped into the micro-tanks, the coating formulation didn’t reach the middle shaft portion of the μND implants (Fig. 2civ). The number of dips examined here were 3, 5, 7, and 9 at room temperature with coating formulation-A (NMN + sucrose + Tween-20) only. The average amount of coated NMN on the μND shafts increased with the number of dips (Fig. 2d). However, only 5 (90.22 ± 10.21 μg) and 7 (146.8 ± 17.30 μg) dips showed uniform coating which was mainly concentrated on the μND implant tips only (SEM images for 5/7 dips in Fig. 2d). On the other hand, although 9 dips achieved a larger coating amount (242.4 ± 36.79 μg), there was large standard deviation between subsequent coating events per μND implant array (interday precision). The standard deviation bars are shown for the specific experiments in the graphs in Fig. 2d, the average relative standard deviation (RSD) of the standard deviation bars for 5 and 7 dips were 11.31% and 11.78% respectively. Since the coating of μND arrays was performed manually, improved control of temperature and humidity in the coating environment, and automation of the coating process are postulated to further reduce the RSD to within acceptable limits.
3.1.3.1. NMN dose dependent study. Keratinocytes were treated with 0, 250, 500 and 1000 μg NMN and incubated overnight. The aim of the first study was to find out what effect did NMN exert on keratinocytes in a dose dependent manner, keeping time constant at 6 h and 24 h (time when the experiment was stopped, and the NAD+ in the cells were visualised). The cells were imaged using multiphoton microscopy, followed by the acquisition of the FLIM. Once the FLIM data were acquired, the images were entered into the TCSPC (time correlated single photon counting) software. The TCSPC software uses a bi-exponential decay profile to calculate the average fluorescence lifetime (τm), and free non-enzyme bound (τ1) NADH. When keratinocyte cells were treated with increasing NMN amounts in vitro, a significant reduction in the NAD+ fluorescence lifetime (τm and τ1) was observed (Fig. 3ci–ii). This trend is further observed in the FLIM images and decay matrix curves (Fig. 3a and b). A decay matrix curve provides an intuitive representation of the distribution of photons across various fluorescence lifetimes (number of photons vs. fluorescence lifetime). The peak in the decay matrix curves, for both the 6 and 24 h treatment (Fig. 3a and b), slowly shifts towards the lower fluorescence lifetime value (towards the blue gradient). The colour gradient in the decay matrix represents the fluorescence lifetime from low to high (blue to red). The FLIM images for the cells treated for 6 h (Fig. 3ai–iv) and 24 h (Fig. 3bi–iv) show a shift in colour from green-yellowish hue to green-blue hue. This qualitative observation demonstrates that, as the amount of NADH (converted from NMN) increases within a keratinocyte cell, the fluorescence lifetime decreases causing that shift in the decay-gram. After treating the cells for 6 h with NMN, a significant shift (p < 0.0001) in the average τm (Fig. 3ci) was seen from 1040 ± 180 ps (control) to 863 ± 161 ps (1000 μg NMN supplementation), while a similar shift in τ1 (Fig. 3cii, p < 0.001) was observed from 665 ± 123 ps (control) to 541 ± 168 ps (1000 μg NMN supplementation). The same trend was observed in the 24 h treatment, where the average τm (Fig. 3ci) shifted from 1071 ± 98 ps (control) to 803 ± 198 ps (1000 μg NMN supplementation), while the τ1 (Fig. 3cii) shifted from 1039 ± 63 ps (control, p < 0.0001) to 540 ± 90 ps (1000 μg NMN supplementation, p < 0.0001). These data demonstrate a linear increase in keratinocyte intracellular NAD+in vitro when treated with increasing amounts of NMN.
 | ||
| Fig. 3 Dose-dependent NAD+ regulation in the keratinocyte experiment. (a) Data showing FLIM images and decay matrix curves of NADH fluorescence within keratinocytes at two different times at 6 h (a) and 24 h (b), while treated with various amount of NMN at 0 μg (control i), 250 μg (ii), 500 μg (iii) and 1000 μg (iv). (c) Graphical presentation of (i) the average fluorescence lifetime (τm) and (ii) the free non-enzyme bound NADH (τ1) (n = 3). ****Statistical significance (p < 0.0001) was determined using the one-way ANOVA using multivariate Tukey test analysis. | ||
3.1.3.2. NMN time dependent study. The next study was to determine the τm and τ1 of NADH inside keratinocytes at 0, 5, 10, 30, 60 min, 6 and 24 h, while keeping the dosing regimen constant at 500 μg of NMN. Fig. 4 show the average τm and τ1 images taken at various time points. While the number of cells varies for each treatment (some images show more black areas with no cells), when processing the data, the areas containing the cells were selected as ROI for the bi-exponential decay calculations. A qualitative assessment of the FLIM acquired images (Fig. 4a) showed the pseudo-coloured keratinocytes change colour from green-yellow hue to green-blue hue. There was a notable shift in the average τm (Fig. 4bi) from 1060 ± 50 ps (0 min) to 835 ± 70 ps (24 h) (p < 0.0001), while the τ1 (Fig. 4bii) value shifts from 720 ± 80 ps (0 min) to 510 ± 60 ps (24 h) (p < 0.0001). This reduced fluorescence lifetime of photons rising from intracellular NADH is an indication of increased NMN to NAD+ conversion within the cells.
 | ||
| Fig. 4 Time-dependent NAD+ regulation in keratinocytes experiment. (a) FLIM images NADH fluorescence within keratinocytes at 0, 5, 10, 30, 60, 360 and 1460 min treated with 500 μg NMN. (b) Graphical presentation of (i) the average fluorescence lifetime (τm) and (ii) the free non-enzyme bound NADH (τ1). (c) Decay matrix curve of total photon inside a particular ROI showing (i) curve when 2-component vs. (iii) 3-component systems respectively. ****Statistical significance (p < 0.0001) was determined using the one-way ANOVA using multivariate Tukey test analysis. | ||
During the initial stages of data analysis, it was observed that the decay matrix curve yielded two distinct peaks (Fig. 4ci). The fluorescence lifetime τ1 of NADH was approximately 500 ps, however, another peak was also observed in the same decay matrix curve which was approximately 1000 ps. This additional peak only appeared when the TCSPC software tries to fit the photon lifetime value into the bi-exponential equation with a ‘2-exponential component’ system (synonymous to free and protein unbound: NADH and NADPH). When the decay matrix curve fit component was changed to 3-exponential component system, the additional peak has disappeared. In a vacuum system, an excited electron returns to ground state with the release of a photon, this photon will decay naturally due to a non-interacting environment.42,77 Biological systems, however, are considered highly-interactive environment with various random biological sinks (other +/− fluorophores molecules) that capture the photon and modifying its spontaneous decay process such that the decay rate of the photon itself is dependent on time, thus stretching the decay.42,77 Considering the complexity of the biological environment, it is not practical to calculate the individual decay times of other fluorophores, since these variations happen at molecular scale and therefore can’t be spatially resolved. In such cases, fitting to a 3-component system would imply an erroneous assumption of three discrete lifetimes.42,77,78 Although it may offer a better fit than the single-exponential or two-exponential models due to the additional fitting parameters, its use is not justified from a physical perspective. Thus, a higher order exponential decay model may enhance the ‘goodness of fit’ due to the additional fitting parameters rather than reflecting any underlying experimental decay dynamics.
 | ||
| Fig. 5 Illustration of skin puncture and NMN permeation profile due to NMN coated μND implants in vitro, and subsequent NAD+ synthesis in NMN-Coated μND implant treated Mouse Ear Skin ex vivo. (a) Graphical presentation of the NMN permeation profile from the NMN coated μND implants (5/7 dip coated μNDs implants only) treated skin showing: the Cumulative average NMN permeation amount profiles (μg cm−3 and %), and NMN amount in the skin and the patch post IVPT (n = 6 μND arrays, n = 2 biological replicate). (b) SEM image of μND implant treated human skin in vitro with inset showing magnified images of four implanted sites. (c) Graphical presentation of the NMN permeation profile from the NMN coated μND implants (7 dip coated μNDs implants only) treated mouse ear skin showing: the Cumulative average NMN permeation amount profiles (μg cm−3 and %), and NMN amount in the skin and the patch post IVPT (n = 6 μND arrays, n = 2 biological replicate). Individual permeation amount and percentages represented as Average ± SD. (c) NAD+ regulation in the NMN coated μND array treated mouse ear skin ex vivo (n = 2) in the Stratum Granulosum (SG) epidermal layer illustrated in the MPT-FLIM images of SG cells of epidermis at 30 μm in depth, control (untreated skin), μND implant untreated skin (−NMN), μND treated skin (+NMN). Images are pseudo-coloured by offset values to distinguish cellular NAD+ fluorescence levels (indicated by the colour bar below the four set of composite images, ranging from 0–2000 ps). (d) Graphical presentation of mean fluorescence lifetime (τm) and free unbound NADH (τ1) quantified from photons from the SG. Photon counts over time for NAD(P)H fluorescence decay in SG cells show a trend of decreased mean fluorescence lifetime (τm) in NMN-coated μND treated skin, suggesting enhanced NAD+ production and more enzyme-bound interactions compared to controls. Conversely, the free NAD(P)H lifetime (τ1) increases in NMN-treated samples, indicating a shift towards non-enzyme bound states. ****Statistical significance (p < 0.0001) was determined using the one-way ANOVA. | ||
The fluorescence of the endogenous fluorophores of the Stratum Granulosum (SG) allows visualisation of the detailed morphological cellular structure as well as changes caused by an external stimulant (NMN), noninvasively using the MPT.40 The photon counts over time for NAD(P)H of the SG layer, shown in Fig. 5c, can in turn be used to define fluorescence decay curve, mean fluorescence lifetimes (τm) and NAD(P)H- Free non-enzyme bound lifetime (τ1) (Fig. 5d). These two parameters are important to assess cellular metabolic function in a non-invasive manner. As seen in Fig. 5d, there was a decrease in the mean fluorescence lifetime τm in μND implant treated skin compared to control and μND implant minus NMN treated skin, respectively. The τm of untreated skin (control) and μND implant (minus NMN) treated skin remained at 1060 ± 79.95 ps and 1028 ± 118.0 ps across 6–72 h. The τm of NMN μND implant treated skin have, however, reduced 915 ± 59.00 ps over same period (Fig. 5d, τm) However, a non-significant increasing trend was observed in the free NAD(P)H non enzyme bound fluorescence lifetime (Control: 390 ± 134.0 ps, μND implant – NMN: 308 ± 21.56 ps and μND implant + NMN: 566 ± 95.58 ps).
4. Discussion
This study describes the design, development, and characterisation of μND implants for over three days, with a steady-state delivery of NAD+ precursor molecule NMN, which is otherwise generally administered orally as part of the existing drug product. NMN was coated onto the tip of the μND implants to be able to extend the release of NMN for more than one day. Intermediate-acting drug delivery systems such as coated μND patches, requires patches to be worn multiple times continuously. Additionally, general matrix drug delivery systems often achieve 1st order drug release, implant type delivery designs can enable zero-order release,80,81 which was shown in this study. Therefore, the design and conception of the μNDs implant array was motivated by the need to extend the release of NMN transdermally while reducing the frequency of periodic patching. Generally, μND implants, in the past, were designed to deliver drug which was embedded inside the μND tip that was made from a slow-dissolving polymer (PLLA, PLGA, etc.).81–84 This was not explored in this study, as the main aim was to investigate whether NMN coated μND implants would have the ability to diffuse NMN within human skin for more than one day from single wear.The 3D printed master μND arrays were designed with a specific geometry, including a conical tip and a trapezium middle shaft, intended to optimise insertion into the skin and facilitate therapeutic delivery loading. The achieved dimensions, particularly the tip diameter of 57.94 ± 14.46 μm, were critical since a tip diameter of less than 80 μm is essential for successful skin insertion. This demonstrated that the design process effectively considered and met the necessary criteria for functionality. At the same time, the output conical height and tip diameter of the master μND arrays showed relatively low variability (e.g., conical height 737.0 ± 40.41 μm), suggesting good consistency in the manufacturing process. However, it's important to consider the potential sources of this variability, such as slight differences in the moulding process or material inconsistencies. These variations could impact the performance and reliability of the μNDs, particularly in clinical applications. The wider base diameter of the μNDs (approximately 1403 ± 29.37 μm) is noted as advantageous for shear-sensitive therapeutics. This design choice reduces the need for high centrifugal forces to fill the female mould cavities, which could otherwise damage delicate therapeutic agents. The design appears well-suited for handling sensitive materials, potentially expanding the range of applicable therapeutics. The detailed understanding of the design and manufacturing process, including the factors influencing dimensional fidelity, is crucial for scaling up production and ensuring reproducibility in future applications.
The design of the μND implant array was strategically aimed at ensuring easy detachment of the μND tips from the patch backing post skin insertion. This was achieved through two critical design criteria: the patch needed to have sufficient mechanical strength to penetrate the skin under compression, and a deliberate structural weakness at the μND–backing interface to enable easy detachment upon patch removal under tension. The investigation into the interfacial strength between the μND middle shaft and the backing revealed distinct mechanical behaviours. Solid μNDs demonstrated robust compressive and shear forces, indicating a resistance to buckling, with forces recorded at 28.81 ± 12.29 N per needle and 5.76 ± 2.40 N per needle, respectively. In contrast, the μND implants showed consistent detachment of the tips at significantly lower forces of 11.51 ± 3.964 N per needle under compression and 1.500 ± 0.638 N per needle under shear. The critical observation of a force drop at specific displacement distances (0.69 mm for compression and 0.23 mm for shear) indicates the moment of detachment at the μND–pedestal interface, highlighting the effectiveness of the design in facilitating tip detachment. This detachment behaviour underscores the successful balance between achieving sufficient mechanical strength for skin penetration and ensuring easy tip release, critical for the intended application of the μND implant array. The study effectively demonstrated that the addition of a water-soluble polymer-sugar complex to the middle shaft of μND implants facilitates rapid detachment of the μND tips upon exposure to a hydrated environment, as evidenced by the complete needle tip dissociation within 225 seconds when immersed in PBS. This rapid detachment was also confirmed through in vitro skin insertion tests, where the μND tips detached entirely from the backing within 5 minutes post-application. These findings are supported by compressive and shear force measurements demonstrated in the literature,21,85 which indicate that while the μND implants possess sufficient mechanical strength for skin insertion, they are engineered to detach easily under tension, ensuring that the patch can be safely removed without leaving biohazardous sharps behind. The seamless detachment process highlights the practical potential of μND implants for clinical use, requiring minimal user intervention and ensuring safe disposal without the need for special handling. Additionally, based on our previous study utilising solid 3D-printed μNDs of 0.8 mm in shaft length—identical to the separating portion length of implantable μNDs—these solid μNDs demonstrated a skin penetration depth of approximately 100 μm.65 This depth effectively disrupts the stratum corneum and reaches into the viable epidermis, a critical factor for transdermal delivery of hydrophilic molecules such as NMN. Applying this knowledge to implantable μNDs in future in vivo experiments, it is likely that a similar penetration depth will be achieved, with potential slight variations due to factors like skin elasticity86 and hydration.87 The ability to disrupt both the stratum corneum and the upper viable epidermis supports efficient microchannel formation, offering a minimally invasive yet effective pathway for molecule transport across the skin barrier.
Although NMN loading within μNDs offers precise control over dosing and a sustained release profile,88–90 NMN coating presents several key advantages, particularly for applications requiring rapid therapeutic effects91,92 and simpler manufacturing processes.65 Coating NMN on μNDs enables a faster onset of action, as the NMN is immediately available on the surface for rapid release upon skin penetration, as shown in our previous work with solid coated μNDs.65 This burst release can be highly beneficial in cases where quick delivery is essential. Additionally, compared to two-layer rapidly separating μNDs and dissolving polymer μNDs, coated μNDs offer practical advantages, such as reliable performance in high humidity environments, broadening their potential applications.65,91 Moreover, coating methods such as dip-coating65 or spray-coating93 are generally less complex and more cost-effective to scale up compared to embedding NMN within the μND matrix. Despite potential concerns around stability and shorter duration of effect, the ability to quickly manufacture large quantities of NMN-coated μNDs makes this approach highly attractive for applications that prioritise speed, cost efficiency, and ease of production over extended, controlled release.
In exploring the rheological properties of formulations with two different viscosity enhancer, sucrose and CMC, shear stress tests using a rheometer revealed non-Newtonian flow behaviour, specifically shear thinning, under applied stress. This behaviour is consistent with the requirements for effective coating adhesion, as shear thinning facilitates the application of the coating while maintaining its integrity once applied, a phenomenon observed in paint and food industry. The increased viscosity of CMC based formulation led to slower recovery of the elastic modulus after shear stress removal, causing the formulation to flow excessively and drip off the μND shaft, thereby reducing the NMN coating's effectiveness. This observation aligns with findings in the literature where high-viscosity coatings often result in suboptimal retention on small-scale surfaces due to excessive flow post-application.94 The fluorescence intensity of the coatings further substantiated this, with sucrose based formulation demonstrating superior coating performance. This study emphasises the critical role of viscosity control and shear behaviour in the development of effective μND coatings, echoing the importance of rheological properties in ensuring consistent and reliable drug delivery.
Permeation studies in in vitro human skin were conducted using a controlled number of dip-coated μND implants (5 or 7). To ensure consistent NMN delivery, the study included multiple replicates, comprising biological replicates (n = 2) and technical triplicates (n = 3 μND arrays per biological replicate), to capture natural biological variability. Our study demonstrated the effectiveness of NMN-coated μND implants in penetrating the skin, rapidly releasing NMN, and achieving significant permeation (∼75% within 1 day, followed by ∼20% over the next 3 days). Minimal retention was observed in the array (∼1.7%), with moderate skin retention (3.7–4.2%). NMN diffusion was validated using a Franz diffusion cell, although minor discrepancies in NMN recovery indicate the need for further optimisation of the coating process. These findings highlight the potential of μND implants for transdermal drug delivery, while also identifying opportunities to enhance reproducibility and efficiency. The supplementation of NMN to keratinocytes in vitro has shown to increase the intercellular NAD(P)H levels. This increase in NAD(P)H levels was quantified using MPT, specifically using the FLIM modality. An important phenomenon that affects the endogenous NAD(P)H fluorescence lifetime τm is the dynamic fluorescence quenching or Förster Resonance Energy Transfer (FRET- a distance dependent nonradiative energy transfer process).95,96 As the intercellular NAD(P)H increases due to NMN supplementation, the intermolecular NAD(P)H distance reduces thus increasing the effect of FRET. Additionally, the intercellular free unbound NAD(P)H molecule itself have self-quenching properties due to the π–π (pie orbital bonds) interaction between the amide and the adenine rings,97 this further reduces the fluorescence lifetime. When the free unbound NAD(P)H binds to an intracellular protein, the π–π bond is disrupted thus increasing fluorescence lifetime of bound NADH (speculation and not assessed in this study). Nonetheless, this study confirmed the validation of NAD+ production in keratinocytes in vitro.
The observed decrease in τmin vitro suggests that a significant portion of the fluorophores are binding to enzymes, leading to quenching or energy transfer and resulting in a shorter average fluorescence lifetime (τm). This binding facilitates a faster non-radiative decay process, typically reflected in reduced τm.98,99 Conversely, the free fluorophores, reflected in τ1, exhibit increased lifetimes due to their presence in a less quenched or more favourable environment. These opposite trends in τm and τ1 indicate a heterogeneous population of fluorophores, where some are bound to enzymes in a quenched state while others remain free in a less quenched state.98,99 NMN delivery into the SG layer of the epidermis shifts this equilibrium, increasing the concentration of free NAD+, which raises τ1 while the enzyme-bound fraction contributes to the reduced τm due to enhanced quenching interactions.99 This phenomenon was also parallelled in mouse ear skin epidermis treated with a 7-dipped NMN-coated μND implant array, where ex vivo results mirrored the decrease in τm and increase in τ1. Notably, a difference in τ1 trends between in vitro and ex vivo systems highlight the influence of dynamic environments, with the simultaneous decrease in τm and τ1in vitro suggesting distinct molecular interactions or enzyme activities in a non-dynamic setting.99 To further elucidate these dynamics and confirm the fluorescence lifetime changes, complementary techniques such as time-resolved fluorescence anisotropy or Förster resonance energy transfer (FRET) studies are recommended.
Further technical advances are required to optimise the present μND implant fabrication process, specifically streamlining fabrication process, and minimising steps, improving scalability and automation. To manufacture distinct μND implants with sustained NMN release profile, the fabrication approach highlighted in this study may involve more steps compared with those that are used to create conventional μND systems for immediate and sustained releases. However, this process holds the potential for a greater throughput, is more scalable and is easier to automate due to the availability of low-cost desktop 3D printer.
5. Conclusion
This study developed μND implant arrays for sustained delivery of NMN in the skin. The incorporation of water-soluble polymer-sugar complex middle portion in the μND array retained sufficient mechanical strength during compression to allow μNDs to be inserted into the skin but weakened the μND interfacial interaction for easy detachment under shear. During the μND implant array removal, the μNDs were easily detached from the solid backing, which was achieved by mechanical weakening at the interface between the layers of the μNDs and accelerated dissolution of water-soluble middle portion material. The detached NMN coated μND implant tips showed sustained release of the NMN for times ranging from 1 to 3 days. Coating formulation rheomechanics showed the impact of viscosity enhancer with the flow mechanics and NMN coating efficiency on the μND surface. NMN administration resulted in a decrease in both the average fluorescence lifetime (τm) and the free unbound NADH fluorescence lifetime (τ1) in keratinocyte cells. Similar trends were observed in mouse ear skin ex vivo, indicating a consistent effect of NMN on endogenous NAD(P)H fluorescence lifetime. However, an opposite trend was observed for the free unbound NADH fluorescence lifetime (τ1) in the NMN treated mouse epidermal cells ex vivo, which can be attributed to complex dynamic cellular interactions and molecular quenching. Overall, this innovative implant drug delivery system demonstrated several significant aspects for its application of a NAD+ precursor molecule: (a) self-administration of drug-coated μNDs to the skin, (b) minimal patch wearing time, (c) generation of zero biohazardous sharps waste, and (d) sustained delivery of NMN. In conclusion, the μND implant arrays combined with sustained drug delivery of a NAD+ precursor molecule from the μNDs embedded in the skin may provide a simple, convenient and a promising option for drug delivery, thereby facilitating greater access and adherence to healthcare.Author contributions
Masood Ali wrote the original manuscript draft and performed editing, conducted the experiments, analysed data, developed methodology. Wenhao Huang conducted the experiments, analysed data and edited the manuscript. Yicheng Huang performed investigation and formal analysis. Xiaoxin Wu, Indira Prasadam, Sarika Namjoshi and Heather Benson edited the manuscript. Tushar Kumeria and Yousuf Mohammed conceptualised the idea, supervised and overlooked experiments, developed methodology, analysed data, provided support with manuscript editing and resources, submission, and management of the article.Data availability
The data that support the findings of this study are available from the corresponding author, [Dr Yousuf Mohammed], upon reasonable request. Data are not publicly available due to privacy and proprietary nature. However, detailed methodological information and a summary of the datasets can be accessed in the supplementary materials provided with this article.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was based on a part of Masood Ali's PhD thesis and Wenhao Huang's Master's thesis. The authors would like to express their appreciation and acknowledge the mouse ear skin (cadaver) donated by Professor Gabrielle Belz (The University of Queensland), from which the ex vivo imaging was possible. The authors acknowledge the facilities, and the scientific and technical assistance, of the Microscopy Australia Facility at the Centre for Microscopy and Microanalysis (CMM), The University of Queensland (UQ), and Dr Cameron Flegg at the Translational Research Institute (TRI) Microscopy core facility for his help in aiding with the imaging of skin. Xiaoxin Wu was supported by the National Natural Science Foundation of China (32300652).References
- P. W. Caton, J. Kieswich, M. M. Yaqoob, M. J. Holness and M. C. Sugden, Diabetologia, 2011, 54, 3083–3092 CrossRef.
- K. M. Ramsey, K. F. Mills, A. Satoh and S.-I. Imai, Aging Cell, 2008, 7, 78–88 CrossRef.
- J. R. Revollo, A. Körner, K. F. Mills, A. Satoh, T. Wang, A. Garten, B. Dasgupta, Y. Sasaki, C. Wolberger, R. R. Townsend, J. Milbrandt, W. Kiess and S.-I. Imai, Cell Metab., 2007, 6, 363–375 CrossRef PubMed.
- A. N. Long, K. Owens, A. E. Schlappal, T. Kristian, P. S. Fishman and R. A. Schuh, BMC Neurol., 2015, 15, 19 CrossRef.
- N. Klimova, A. Long and T. Kristian, J. Neurosci. Res., 2019, 97, 975–990 CrossRef CAS.
- J. Ji, D. Damschroder, D. Bessert, P. Lazcano, R. Wessells, C. A. Reynolds and M. L. Greenberg, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2022, 1867, 159094 CrossRef CAS.
- K. Tsubota, npj Aging Mech. Dis., 2016, 2, 16021 CrossRef.
- N. A. Youngson, G. M. Uddin, A. Das, C. Martinez, H. S. Connaughton, S. Whiting, J. Yu, D. A. Sinclair, R. J. Aitken and M. J. Morris, Reproduction, 2019, 158, 169–179 Search PubMed.
- L. Yi, A. B. Maier, R. Tao, Z. Lin, A. Vaidya, S. Pendse, S. Thasma, N. Andhalkar, G. Avhad and V. Kumbhar, GeroScience, 2023, 45, 29–43 CrossRef CAS.
- K. M. Pencina, S. Lavu, M. dos Santos, Y. M. Beleva, M. Cheng, D. Livingston and S. Bhasin, J. Gerontol., Ser. A, 2022, 78, 90–96 CrossRef.
- M. Ali, S. Namjoshi, H. A. E. Benson, Y. Mohammed and T. Kumeria, J. Controlled Release, 2022, 347, 561–589 CrossRef CAS PubMed.
- M. R. Prausnitz, Annu. Rev. Chem. Biomol. Eng., 2017, 8, 177–200 CrossRef CAS.
- R. Al-Kasasbeh, A. J. Brady, A. J. Courtenay, E. Larrañeta, M. T. C. McCrudden, D. O’Kane, S. Liggett and R. F. Donnelly, Drug Delivery Transl. Res., 2020, 10, 690–705 CrossRef CAS.
- E. M. Vicente-Perez, E. Larrañeta, M. T. C. McCrudden, A. Kissenpfennig, S. Hegarty, H. O. McCarthy and R. F. Donnelly, Eur. J. Pharm. Biopharm., 2017, 117, 400–407 CrossRef CAS PubMed.
- S. F. Lahiji, M. Dangol and H. Jung, Sci. Rep., 2015, 5, 7914 CrossRef CAS PubMed.
- Y. Lee, S. Park, S. I. Kim, K. Lee and W. Ryu, Adv. Mater. Technol., 2020, 5, 1901145 CrossRef.
- W. Li, R. N. Terry, J. Tang, M. R. Feng, S. P. Schwendeman and M. R. Prausnitz, Nat. Biomed. Eng., 2019, 3, 220–229 CrossRef PubMed.
- W.-J. Lee, M.-R. Han, J.-S. Kim and J.-H. Park, Expert Opin. Drug Delivery, 2019, 16, 199–206 CrossRef.
- C. Kuwentrai, J. Yu, L. Rong, B.-Z. Zhang, Y.-F. Hu, H.-R. Gong, Y. Dou, J. Deng, J.-D. Huang and C. Xu, Bioeng. Transl. Med., 2021, 6, e10202 CrossRef PubMed.
- A. Hou, G. Quan, B. Yang, C. Lu, M. Chen, D. Yang, L. Wang, H. Liu, X. Pan and C. Wu, Adv. Healthcare Mater., 2019, 8, 1900898 CrossRef.
- Y. Lee, W. Li, J. Tang, S. P. Schwendeman and M. R. Prausnitz, J. Controlled Release, 2021, 337, 676–685 CrossRef.
- L.-C. Su and M.-C. Chen, J. Mater. Chem. B, 2017, 5, 3355–3363 RSC.
- E. Dumkliang, P. Suriyaamporn, P. Patrojanasophon, T. Ngawhirunpat, T. Rojanarata, P. Opanasopit, S. Yoksan and B. Pamornpathomkul, J. Drug Delivery Sci. Technol., 2024, 92, 105377 CrossRef CAS.
- A. Aksit, D. N. Arteaga, M. Arriaga, X. Wang, H. Watanabe, K. E. Kasza, A. K. Lalwani and J. W. Kysar, Biomed. Microdevices, 2018, 20, 1–12 CrossRef CAS PubMed.
- G. Kang, M. Kim, H. Yang, J. Shin, J. Sim, H. Ahn, M. Jang, Y. Kim, H. S. Min and H. Jung, Adv. Funct. Mater., 2023, 33, 2210805 CrossRef CAS.
- Y. Kim, H. S. Min, J. Shin, J. Nam, G. Kang, J. Sim, H. Yang and H. Jung, Biomater. Res., 2022, 26, 53 CrossRef CAS PubMed.
- M. Sakamoto, Y. Hasegawa and M. Shikida, Microsyst. Technol., 2021, 27, 3907–3916 CrossRef.
- S. Sharma, A. Saeed, C. Johnson, N. Gadegaard and A. E. G. Cass, Sens. Biosens. Res., 2017, 13, 104–108 Search PubMed.
- E. Z. Loizidou, N. T. Inoue, J. Ashton-Barnett, D. A. Barrow and C. J. Allender, Eur. J. Pharm. Biopharm., 2016, 107, 1–6 CrossRef PubMed.
- P. Makvandi, M. Shabani, N. Rabiee, Q. K. Anjani, A. Maleki, E. N. Zare, A. H. B. Sabri, D. De Pasquale, M. Koskinopoulou, E. Sharifi, R. Sartorius, M. Seyedhamzeh, S. Bochani, I. Hirata, A. C. Paiva-Santos, L. S. Mattos, R. F. Donnelly and V. Mattoli, Adv. Mater., 2023, 2210034 CrossRef CAS.
- I. N. Haridass, J. C. J. Wei, Y. H. Mohammed, M. L. Crichton, C. D. Anderson, J. Henricson, W. Y. Sanchez, S. C. Meliga, J. E. Grice, H. A. E. Benson, M. A. F. Kendall and M. S. Roberts, J. Controlled Release, 2019, 306, 59–68 CrossRef CAS.
- Y. H. Mohammed, M. Yamada, L. L. Lin, J. E. Grice, M. S. Roberts, A. P. Raphael, H. A. E. Benson and T. W. Prow, PLoS One, 2014, 9, e101956 CrossRef PubMed.
- X. Luo, Q. Yu, L. Yang and Y. Cui, ACS Sens., 2023, 8(4), 1710–1722 CrossRef CAS.
- J. Nguyen, H. Lewis, A. Queja, A. N. Diep, G. Hochart and M. Ameri, J. Pharm. Sci., 2018, 107, 2192–2197 CrossRef.
- I. Iachina, A. H. Eriksson, M. Bertelsen, K. Petersson, J. Jansson, P. Kemp, K. M. Engell, J. R. Brewer and K. T. Nielsen, Eur. J. Pharm. Sci., 2023, 182, 106371 CrossRef.
- J. E. Park, N. Yonet-Tanyeri, E. Vander Ende, A.-I. Henry, B. E. Perez White, M. Mrksich and R. P. Van Duyne, Nano Lett., 2019, 19, 6862–6868 CrossRef.
- J. C. J. Wei, I. N. Haridass, M. L. Crichton, Y. H. Mohammed, S. C. Meliga, W. Y. Sanchez, J. E. Grice, H. A. E. Benson, M. S. Roberts and M. A. F. Kendall, Sci. Rep., 2018, 8, 17759 CrossRef PubMed.
- R. M. Williams, D. W. Piston and W. W. Webb, FASEB J., 1994, 8, 804–813 CrossRef PubMed.
- W. Sanchez, T. Prow, W. Sanchez, J. Grice and M. Roberts, J. Biomed. Opt., 2010, 15, 046008 CrossRef.
- Y. Mohammed, D. Barkauskas, A. Holmes, J. Grice and M. Roberts, J. Biomed. Opt., 2020, 25, 014509 Search PubMed.
- Y. H. Mohammed, I. N. Haridass, J. E. Grice, H. A. E. Benson and M. S. Roberts, J. Invest. Dermatol., 2020, 140, 1656–1659 CrossRef PubMed.
- K. König, A. P. Raphael, L. Lin, J. E. Grice, H. P. Soyer, H. G. Breunig, M. S. Roberts and T. W. Prow, Adv. Drug Delivery Rev., 2011, 63, 388–404 CrossRef.
- A. Zbinden, D. A. Carvajal Berrio, M. Urbanczyk, S. L. Layland, M. Bosch, S. Fliri, C.-E. Lu, A. Jeyagaran, P. Loskill, G. P. Duffy and K. Schenke-Layland, J. Biophotonics, 2020, 13, e202000375 CrossRef PubMed.
- H. S. Gill and M. R. Prausnitz, Pharm. Res., 2007, 24, 1369–1380 CrossRef.
- D. Jakka, A. V. Matadh, V. K. Shankar, H. N. Shivakumar and S. Narasimha Murthy, J. Pharm. Sci., 2022, 111, 2867–2878 CrossRef PubMed.
- D. Jung, N. S. Rejinold, J. E. Kwak, S. H. Park and Y. C. Kim, Colloids Surf., B, 2017, 159, 54–61 CrossRef PubMed.
- J. Chen, Y. Qiu, S. Zhang, G. Yang and Y. Gao, Drug Dev. Ind. Pharm., 2015, 41, 415–422 CrossRef.
- L. Liang, Y. Chen, B. L. Zhang, X. P. Zhang, J. L. Liu, C. B. Shen, Y. Cui and X. D. Guo, J. Drug Delivery Sci. Technol., 2020, 55, 101464 CrossRef.
- T. Kajiya, D. Sawai, K. Miyata, Y. Miyashita and H. Noda, Eur. Phys. J. E: Soft Matter Biol. Phys., 2022, 45, 76 CrossRef.
- R. J. A. Moakes, L. M. Grover and T. E. Robinson, Bioengineering, 2022, 10, 3 CrossRef PubMed.
- R. G. Larson and Y. Wei, J. Rheol., 2019, 63, 477–501 CrossRef.
- C. H. Lee, V. Moturi and Y. Lee, J. Controlled Release, 2009, 136, 88–98 CrossRef PubMed.
- O. Y. Mansour, M. Z. Sefain, M. M. Ibrahim and W. K. El-Zawawy, J. Appl. Polym. Sci., 2000, 77, 1666–1678 CrossRef.
- E. E. Peters, M. Ameri, X. Wang, Y.-F. Maa and P. E. Daddona, Pharm. Res., 2012, 29, 1618–1626 CrossRef.
- G. Mattei, L. Cacopardo and A. Ahluwalia, Materials, 2020, 13, 438 CrossRef.
- S. Cunha, M. Swedrowska, Y. Bellahnid, Z. Xu, J. M. Sousa Lobo, B. Forbes and A. C. Silva, Int. J. Pharm., 2022, 620, 121720 CrossRef.
- Y. Wei, Y. Lin, R. Xie, Y. Xu, J. Yao and J. Zhang, J. Food Eng., 2015, 166, 21–28 CrossRef.
- D.-q Li, M. Tohti, Y.-s Fu, Y. Zhang, Z.-w Xiong, J. Li and Y.-F. Guo, Int. J. Biol. Macromol., 2024, 264, 130453 CrossRef PubMed.
- V. L. Mucci, M. E. V. Hormaiztegui, J. I. Amalvy and M. I. Aranguren, J. Adhes. Sci. Technol., 2024, 38, 489–516 CrossRef.
- U. Kästner, Colloids Surf., A, 2001, 183, 805–821 CrossRef.
- J. H. Bieleman, Chimia, 2002, 56, 163 CrossRef.
- M. H. Elkomy, S. F. Elmenshawe, H. M. Eid and A. M. A. Ali, Drug Delivery, 2016, 23, 3294–3306 CrossRef.
- S. J. Yoon, J. Bak and B. Yoo, Int. J. Biol. Macromol., 2024, 264, 130600 CrossRef.
- M. Ali, Y. Yang, A. Abdoh and Y. Mohammed, RSC Appl. Interfaces, 2024, 1108–1128, 10.1039/D4LF00177J.
- M. Ali, S. Namjoshi, K. Phan, X. Wu, I. Prasadam, H. A. E. Benson, T. Kumeria and Y. Mohammed, ACS Biomater. Sci. Eng., 2024, 10(11), 7235–7255 CrossRef CAS PubMed.
- R. F. Donnelly, R. Majithiya, T. R. Singh, D. I. Morrow, M. J. Garland, Y. K. Demir, K. Migalska, E. Ryan, D. Gillen, C. J. Scott and A. D. Woolfson, Pharm. Res., 2011, 28, 41–57 CrossRef.
- D. Baykara, T. Bedir, E. Ilhan, M. E. Mutlu, O. Gunduz, R. Narayan and C. B. Ustundag, Front. Bioeng. Biotechnol., 2023, 11, 1157541 CrossRef PubMed.
- S. H. Lim, J. Y. Ng and L. Kang, Biofabrication, 2017, 9, 015010 CrossRef.
- K. J. Krieger, N. Bertollo, M. Dangol, J. T. Sheridan, M. M. Lowery and E. D. O’Cearbhaill, Microsyst. Nanoeng., 2019, 5, 42 CrossRef PubMed.
- S. Choo, S. Jin and J. Jung, Pharmaceutics, 2022, 14, 766 CrossRef.
- B. Venzac, S. Deng, Z. Mahmoud, A. Lenferink, A. Costa, F. Bray, C. Otto, C. Rolando and S. Le Gac, Anal. Chem., 2021, 93, 7180–7187 CrossRef.
- M. T. Yilmaz and C. Vatansever, Food Hydrocolloids, 2016, 61, 458–468 CrossRef CAS.
- M.-H. Hsiao, H.-F. Ye, T.-J. Liu and J. Wang, Adv. Chem. Eng. Sci., 2019, 09, 204–222 CrossRef CAS.
- M. D. Seo, T. J. Kang, C. H. Lee, A. Y. Lee and M. Noh, Biomol. Ther., 2012, 20, 171–176 CrossRef CAS.
- V. G. Wilson, Methods Mol. Biol., 2014, 1195, 33–41 CrossRef.
- C. M. R. R. Nastiti, T. Ponto, Y. Mohammed, M. S. Roberts and H. A. E. Benson, Pharmaceutics, 2020, 12, 108 CrossRef CAS PubMed.
- K. C. B. Lee, J. Siegel, S. E. D. Webb, S. Lévêque-Fort, M. J. Cole, R. Jones, K. Dowling, M. J. Lever and P. M. W. French, Biophys. J., 2001, 81, 1265–1274 CrossRef.
- C. Dysli, S. Wolf, M. Y. Berezin, L. Sauer, M. Hammer and M. S. Zinkernagel, Prog. Retinal Eye Res., 2017, 60, 120–143 CrossRef.
- H. Nadeeshani, J. Li, T. Ying, B. Zhang and J. Lu, J. Adv. Res., 2022, 37, 267–278 CrossRef PubMed.
- W.-W. Yang and E. Pierstorff, J. Lab. Autom., 2012, 17, 50–58 CrossRef PubMed.
- W. Li, J. Y. Chen, R. N. Terry, J. Tang, A. Romanyuk, S. P. Schwendeman and M. R. Prausnitz, J. Controlled Release, 2022, 347, 489–499 CrossRef PubMed.
- J. Wang, Y. Ye, J. Yu, A. R. Kahkoska, X. Zhang, C. Wang, W. Sun, R. D. Corder, Z. Chen and S. A. Khan, ACS Nano, 2018, 12, 2466–2473 CrossRef PubMed.
- P. Yang, C. Lu, W. Qin, M. Chen, G. Quan, H. Liu, L. Wang, X. Bai, X. Pan and C. Wu, Acta Biomater., 2020, 104, 147–157 CrossRef PubMed.
- K. T. Tran, T. D. Gavitt, N. J. Farrell, E. J. Curry, A. B. Mara, A. Patel, L. Brown, S. Kilpatrick, R. Piotrowska and N. Mishra, Nat. Biomed. Eng., 2021, 5, 998–1007 CrossRef.
- N. El-Sayed, L. Vaut and M. Schneider, Eur. J. Pharm. Biopharm., 2020, 154, 166–174 CrossRef PubMed.
- M. Ali, S. Namjoshi, H. A. E. Benson, T. Kumeria and Y. Mohammed, Nano TransMed, 2022, 1, e9130002 CrossRef.
- S. Yousef, Y. Mohammed, S. Namjoshi, J. Grice, W. Sakran and M. Roberts, AAPS J., 2017, 19, 180–190 CrossRef.
- D. D. Zhu, Q. L. Wang, X. B. Liu and X. D. Guo, Acta Biomater., 2016, 41, 312–319 CrossRef PubMed.
- Z. Wang, Z. Yang, J. Jiang, Z. Shi, Y. Mao, N. Qin and T. H. Tao, Adv. Mater., 2022, 34, 2106606 CrossRef PubMed.
- M. Rabiei, S. Kashanian, G. Bahrami, H. Derakhshankhah, E. Barzegari, S. S. Samavati and S. J. P. McInnes, Eur. J. Pharm. Sci., 2021, 167, 106040 CrossRef.
- B. Z. Chen, M. C. He, X. P. Zhang, W. M. Fei, Y. Cui and X. D. Guo, Drug Delivery Transl. Res., 2022, 12, 2730–2739 CrossRef.
- H. T. T. Duong, Y. Yin, T. Thambi, T. L. Nguyen, V. H. Giang Phan, M. S. Lee, J. E. Lee, J. Kim, J. H. Jeong and D. S. Lee, Biomaterials, 2018, 185, 13–24 CrossRef PubMed.
- M. J. Kim, S. C. Park and S.-O. Choi, RSC Adv., 2017, 7, 55350–55359 RSC.
- Y. X. Ou, H. Q. Wang, X. Ouyang, Y. Y. Zhao, Q. Zhou, C. W. Luo, Q. S. Hua, X. P. Ouyang and S. Zhang, Prog. Mater. Sci., 2023, 136, 101125 CrossRef.
- M. S. Roberts, Y. Dancik, T. W. Prow, C. A. Thorling, L. L. Lin, J. E. Grice, T. A. Robertson, K. König and W. Becker, Eur. J. Pharm. Biopharm., 2011, 77, 469–488 CrossRef PubMed.
- R. B. Sekar and A. Periasamy, J. Cell Biol., 2003, 160, 629–633 CrossRef PubMed.
- S. Ranjit, L. Malacrida, M. Stakic and E. Gratton, J. Biophotonics, 2019, 12, e201900156 CrossRef.
- R. Datta, T. M. Heaster, J. T. Sharick, A. A. Gillette and M. C. Skala, J. Biomed. Opt., 2020, 25, 1–43 Search PubMed.
- M. van de Weert and L. Stella, J. Mol. Struct., 2011, 998, 144–150 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01856g |
| This journal is © The Royal Society of Chemistry 2025 |