
Development of a urea-bond cleavage reaction induced by nitric oxide for fluorescence imaging†
Yuqing
Zhang‡
,
Shushu
Wang‡
,
Lina
Zhang
and
Tao
Peng
 *
*
State Key Laboratory of Chemical Oncogenomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China. E-mail: tpeng@pku.edu.cn
First published on 10th September 2024
Abstract
Nitric oxide (NO) is a multifunctional signalling molecule with indispensable roles in physiological processes, but its abnormal production is implicated in various disease conditions. Detecting NO is crucial for interrogating its biological roles. Although many o-phenylenediamine-based fluorescent probes have been developed, only a small fraction has been employed in vivo. Moreover, these probes largely require direct modifications of the fluorophore backbones to render NO responsiveness, which restricts the general applicability of o-phenylenediamine-based probe designs to other types of fluorophores. Here, we report the rational development, optimization, and application of a NO-induced urea-bond cleavage reaction for selective fluorescence detection and imaging of NO in living systems. Through rational design and extensive screening, we identified a 2-aminophenylurea-derived functionality that can react with NO through N-nitrosation, acyltriazole formation, and hydrolysis to induce the cleavage of the urea bond and release of the amino-containing coumarin fluorophore. By caging different amino-containing fluorophore scaffolds with the 2-aminophenylurea-derived functionality, we modularly developed a series of NO fluorescent probes with different excitation and emission profiles for the detection of NO in aqueous solutions and live cells. Among these probes, the near-infrared probe has been demonstrated to enable in vivo fluorescence visualization of elevated endogenous levels of NO in a murine inflammation model. Overall, this study provides a NO-induced urea-bond cleavage reaction and establishes the utility of this reaction for the general and modular development of NO fluorescent probes, thus opening new opportunities for studying and manipulating NO in biological systems.
Introduction
Nitric oxide (NO) serves as a multifunctional signalling molecule and plays an essential role in various biological processes, such as vasodilation, cell proliferation, immune response, and neurotransmission.1–3 Endogenously synthesized from L-arginine (L-Arg) by NO synthases (NOSs),4 NO maintains a relatively steady concentration under normal physiological conditions.5 Increased NO production has been found during inflammation and immune stimulation conditions.6,7 The excessive production of NO in live organisms is known to induce the formation of highly reactive oxygen and nitrogen species, such as peroxynitrite,8 and thus has been implicated in various pathological conditions,3,9,10 including cardiovascular diseases,11 cancer,12 and neurodegeneration.13 The complex roles of NO in physiological and pathological processes have garnered intensive interest in developing tools for detecting and monitoring NO in living systems.14In recent decades, optical fluorescent and luminescent probes, featuring distinct advantages including high sensitivity, excellent selectivity, spatiotemporal resolution, and non-invasiveness, have emerged as powerful tools for the detection and imaging of NO in biological samples.15–17 By harnessing the chemical reactivity of NO, a series of chemical reactions has been exploited to develop NO-responsive fluorescent probes that exhibit fluorescence intensity and/or spectrum changes in the presence of NO.17 These reactions involve the use of distinct NO-reactive functionalities, such as o-phenylenediamine,18,19 aromatic primary and secondary amines,20–23 Hantzsch ester,24–26 thiosemicarbazide,27,28 and copper complexes (Fig. 1A).29,30 Among them, the o-phenylenediamine moiety has been the most commonly used, leading to numerous NO fluorescent probes.31–50 The underlying mechanism relies on the reaction of o-phenylenediamine with NO under aerobic conditions, forming triazole derivatives that effectively inhibit the photoinduced electron transfer (PeT) process and thus activate the fluorescence of appended fluorophores (Fig. 1A). Despite the significant progress in developing o-phenylenediamine-based NO fluorescent probes, these probes still have some limitations,51,52 especially suffering from potential interference from oxidants,53 methylglyoxal (MGO),54 dehydroascorbic acid (DHA),55,56 and ascorbic acid (AA).55,56 Recently, the reactions of aromatic primary and secondary amines with NO, generating diazo rings, deamination products, and N-nitrosation products, have been utilized to develop NO fluorescent probes (Fig. 1A), leading to improved selectivity for NO detection.21–23,57–65 However, these current probe design strategies are all based on NO-induced bond-formation reactions and heavily rely on direct modifications of fluorophore backbones to incorporate NO-reactive o-phenylenediamine or aromatic amine moieties. As such, these reactions and strategies cannot be readily generalized to existing fluorophore backbones of distinct photophysical properties through simple organic synthesis operations. Our ability to modularly develop NO fluorescent probes with varying excitation/emission profiles, cellular localization, and targeting properties has thus been seriously limited.
 | ||
| Fig. 1 The reactions and approaches used for developing NO-responsive fluorescent probes. (A) Previously reported chemical reactions of o-phenylenediamines, aromatic primary and secondary amines, Hantzsch ester, thiosemicarbazide, and copper complexes with NO for developing NO fluorescent probes. (B) The present work proposes and explores a NO-induced bond cleavage reaction for the general development of NO-responsive fluorescent probes. | ||
To address this issue, we herein report the development of a NO-induced bond cleavage reaction that can be generally incorporated into various amino-containing fluorophore scaffolds, enabling the easy construction of a range of fluorescent probes for NO detection. Through rational design and iterative optimization, we identify an optimal 2-aminophenylurea-derived functionality that can react with NO through N-nitrosation, followed by triazole formation and hydrolysis, to induce the urea-bond cleavage and release of the aminocoumarin model fluorophore. We then expand this NO-induced urea-bond cleavage reaction to commonly used amino-containing fluorophores, delivering a set of NO fluorescent probes with varying excitation and emission profiles for the highly selective detection of NO in aqueous solutions. We further demonstrate the utility of these probes for visualizing NO in live cells and showcase that the near-infrared (NIR) probe WNO28 allows in vivo fluorescence imaging of endogenous NO in living mice.
Results and discussion
Design and proof-of-concept of a NO-induced bond cleavage reaction
Building on our extensive experience in developing responsive sensing probes and therapeutic agents targeting reactive species and enzymes,66–76 we envisioned that separating the fluorophore scaffold from the NO-reactive moiety into independent modules should allow for a general and modular strategy for developing NO-responsive fluorescent probes. For this purpose, a NO-induced bond-cleavage reaction that can effectively disconnect the fluorophore scaffold from the NO-reactive moiety is imperative. To explore such a reaction, we sought to harness the N-nitrosation reactivity of NO toward aromatic amino groups under aerobic conditions and conceived the incorporation of the NO-reactive aniline functionality into phenol- and amino-containing fluorophores via carbamate and urea linkers, respectively (Fig. 1B). It was expected that the free aromatic amino group could undergo direct N-nitrosation by NO under aerobic conditions, followed by an intramolecular cyclization with the nearby N–H or N–Me group to form acylbenzotriazole and subsequent hydrolysis to break the urea bond and liberate the free fluorophore upon CO2 release (Fig. 1B). A NO-induced urea-bond cleavage reaction could thus be realized and in principle be applied to cage any phenol- or amine-based fluorophore to generally develop NO fluorescent probes.To examine this idea, we designed a series of coumarin-based probes, named WNO1–6 (Fig. 2A), which contain the 7-hydroxy-4-methylcoumarin or 7-amino-4-methylcoumarin fluorophore, an m-methoxyaniline-derived NO-reactive moiety, and unsubstituted or N-methyl substituted carbamate or urea linkers. These probes were generally synthesized through reactions of coumarin chloroformate or coumarin carbamoyl chloride with the m-methoxyaniline derivative followed by deprotection (Scheme S1, ESI†). The synthetic routes and procedures for these probes are detailed in the ESI.†
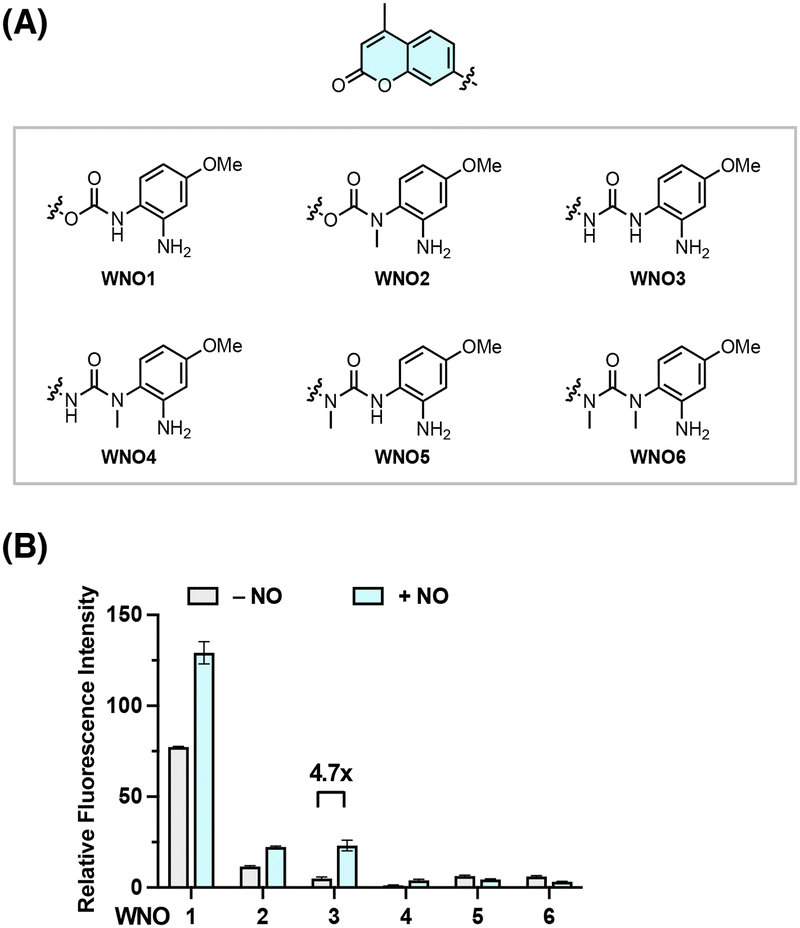 | ||
| Fig. 2 Screening of carbamate and urea linkers for the NO-induced bond cleavage reaction. (A) Chemical structures of coumarin-based probes bearing a NO-reactive moiety and carbamate or urea linkers. (B) Fluorescence responses of WNO1–6 toward NO. WNO1–6 (10 μM) were dissolved in PBS (20 mM, pH 7.4) and treated with NO (1 mM) at 37 °C for 1 h. The fluorescence intensity at 450 nm was recorded with excitation at 360 nm. Data are shown as mean relative fluorescence intensity before and after NO treatment ± standard deviation (n = 3). | ||
With WNO1–6 in hand, we evaluated their fluorescence properties in response to NO. In this regard, the probes (10 μM) were dissolved in aqueous phosphate-buffered saline (PBS) solutions at a physiological pH of 7.4 and treated with NO (delivered in the form of a NO stock solution prepared from NO donor DEA NONOate77) at 37 °C. The fluorescence intensities of WNO1–6 at 450 nm, corresponding to the maximal wavelength of coumarin fluorescence, before and after treatment with NO were measured and compared. The results showed varying fluorescence responses of WNO1–6 toward NO (Fig. 2B). Among them, WNO3 exhibited the most significant fluorescence increase with a ca. 4.7-fold enhancement. As a comparison, WNO1 and WNO2 displayed weaker fluorescence increases than WNO3, and WNO4, WNO5, and WNO6 did not exhibit any fluorescence increase upon treatment with NO. It is worth mentioning that WNO1 had a very strong background fluorescence in the absence of NO, due to carbamate hydrolysis that occurs by deprotonation of the N–H group in aqueous solutions and elimination of 7-hydroxy-4-methylcoumarin to give the isocyanate.78 In line with this, WNO2 containing N-methyl substituted carbamate is less prone to hydrolysis and thus has a lower background fluorescence. The weaker fluorescence response of WNO4 than that of WNO3 toward NO may be attributed to the N-methyl substituent in the NO-reactive moiety that could affect the formation of the acylbenzotriazole intermediate. In addition, the differential fluorescence responses of WNO3 and WNO5 to NO indicate that the N-methyl substituent in the fluorophore scaffold may hinder the hydrolysis of the acylbenzotriazole intermediate.
Together, these data demonstrate that WNO3 containing the urea linker shows good stability in aqueous solutions and exhibits an obvious fluorescence turn-on response to NO (Fig. 2). More importantly, the observed fluorescence increase of WNO3 upon treatment with NO is consistent with our proposed NO-induced bond cleavage reaction (Fig. 1B) to convert WNO3 into highly fluorescent 7-amino-4-methylcoumarin.
Structure–reactivity studies to optimize the NO-induced urea-bond cleavage reaction
The above studies suggest that the urea unit serves as a viable linker for the NO-induced bond cleavage reaction. We next sought to investigate the structure–reactivity relationship to optimize the reactivity of the aniline-derived reactive moiety toward NO. For this purpose, we designed and synthesized a series of coumarin-based probes, i.e., WNO7–16, by modifying the NO-reactive moiety structure with different substituents at varying positions (Fig. 3A and Scheme S2 in the ESI†). Their fluorescence responses to NO were then examined as described above in aqueous solutions (Fig. 3B). WNO9, WNO11, WNO13, and WNO15 exhibited moderate to strong background fluorescence before treatment with NO, indicating that substituents on the reactive moiety may affect the probe stability in aqueous solutions and/or the ability to quench coumarin fluorescence. WNO7, WNO10, and WNO14 showed strong fluorescence enhancement upon treatment with NO (Fig. 3B). Among them, WNO14 exhibited the most significant fluorescence response to NO with an approximately 10-fold fluorescence increase. Interestingly, WNO8, without the reactive aromatic amino group, showed no fluorescence response to NO, suggesting the essential role of the amino group in the responsiveness to NO. In addition, the introduction of a fluorine atom in the phenyl ring (i.e., WNO12) greatly attenuated the probe response to NO compared to the non-substituted and methyl-substituted analogous probes (i.e., WNO7 and WNO10, respectively), probably due to the lower reactivity of the aromatic amino group in the presence of the electron-withdrawing fluorine atom. Together, these results demonstrate that substituents of different properties in the NO-reactive moiety significantly affect the responses of probes toward NO and that WNO14 bearing a dimethylamino substituent at the para position of the amino group exhibits a relatively strong fluorescence response to NO (Fig. 3B).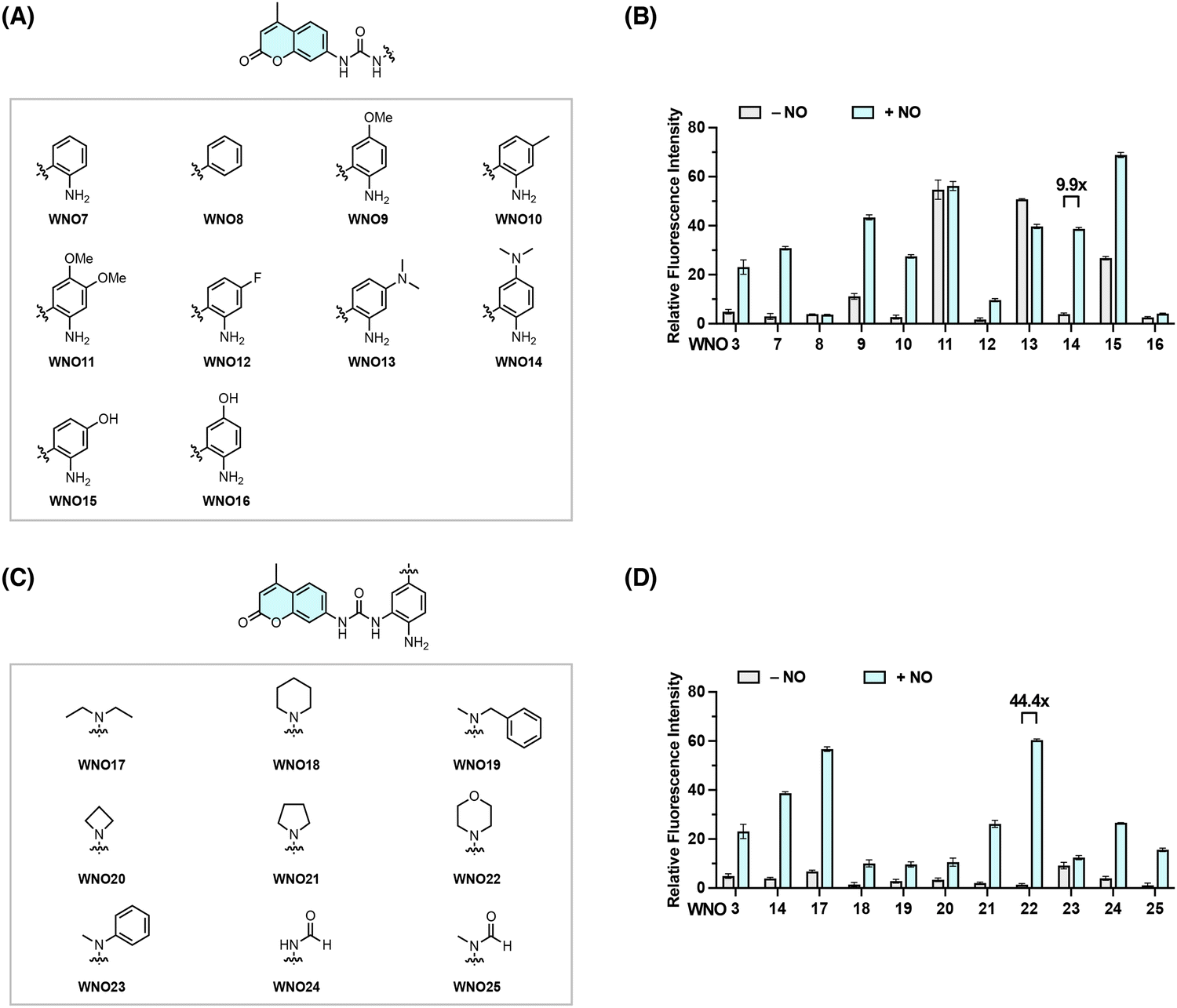 | ||
| Fig. 3 Screening of various coumarin-based probes for optimizing the NO-induced urea-bond cleavage reaction. (A) Chemical structures of coumarin-based probes bearing different substituents at varying positions. (B) Fluorescence responses of WNO7–16 toward NO. (C) Chemical structures of coumarin-based probes containing diverse N-substituents. (D) Fluorescence responses of WNO17–25 toward NO. The probes (10 μM) were dissolved in PBS (20 mM, pH 7.4) and treated with NO (1 mM) at 37 °C for 1 h. The fluorescence intensity at 450 nm was recorded with excitation at 360 nm. Data are shown as mean relative fluorescence intensity before and after NO treatment ± standard deviation (n = 3). | ||
Based on the structure of WNO14, we proceeded to explore various substituted amino groups at the para position of the amino group to further enhance the reactivity of the NO-reactive moiety and increase the fluorescence response to NO. Specifically, we designed and synthesized a second series of probes, i.e., WNO17–25, that contain diverse N-substituents ranging from acyclic and cyclic alkyl to phenyl and formyl groups with different steric and electronic effects (Fig. 3C and Scheme S3 in the ESI†). The fluorescence assays showed that these probes exhibited varying degrees of fluorescence enhancement after treatment with NO (Fig. 3D). Particularly, WNO22, containing a morpholinyl group at the para position of the amino group, displayed a minimal background fluorescence before treatment with NO and more importantly an approximately 44-fold fluorescence increase upon treatment with NO, substantially higher than those of WNO3 and WNO14. Therefore, incorporating different substituted amino groups into the NO-reactive moiety can improve the reactivity of the NO-reactive moiety toward NO.
Collectively, these structure–reactivity studies identify 5-morpholinyl-2-aminophenylurea as an optimal NO-reactive moiety to induce the cleavage of the urea bond and release of the free coumarin fluorophore, providing new opportunities for further developing new NO fluorescent probes.
Development of a series of NO fluorescent probes with the optimal NO-induced urea-bond cleavage reaction
Our prototype coumarin-derived probe WNO22 exhibited a maximal emission wavelength of 450 nm in an aqueous buffer upon NO treatment. We envisioned that the optimal NO-reactive moiety in WNO22 could be expanded to cage other amino-containing fluorophore scaffolds via the urea linker, generating new NO fluorescent probes with different fluorescence excitation and emission wavelengths. In this regard, we chose amino-containing 1,8-naphthalimide and hemicyanines as the core fluorophores. Specifically, 1,8-naphthalimide was derivatized with the 5-morpholinyl-2-aminophenylurea moiety to provide probe WNO26 (Fig. 4A). The hemicyanine fluorophore79 was derivatized similarly to prepare probe WNO27 (Fig. 4B). The optimized 5-morpholinyl-2-aminophenylurea moiety was also incorporated into the NIR hemicyanine fluorophore80 to generate probe WNO28 (Fig. 4C). Notably, these probes possess distinct structural scaffolds but can be synthesized using similar conjugation strategies (Scheme 1). Specifically, the amino group of the core fluorophore was activated to generate the corresponding 4-nitrophenyl carbamate, which was conjugated with the protected NO-reactive moiety to form the urea functionality. Subsequent deprotection yielded the final probe. The synthetic details for WNO26, WNO27, and WNO28 are described in Schemes S4–S6, respectively, in the ESI.† | ||
| Fig. 4 Photophysical characterization of NO fluorescent probes in chemical systems. (A)–(C) Chemical structures of NO fluorescent probes WNO26, WNO27, and WNO28. (D)–(F) Fluorescence emission spectra of (D) WNO26, (E) WNO27, and (F) WNO28 upon treatment with different amounts of NO at 37 °C for 1 h. (G)–(I) Fluorescence response kinetics of (G) WNO26, (H) WNO27, and (I) WNO28 toward NO. (J)–(L) Relative fluorescence intensities of (J) WNO26, (K) WNO27, and (L) WNO28 upon treatment with various potentially interfering biological species at 37 °C for 1 h. All analytes were present in 10-fold excess (100 μM) relative to the probe. Data were acquired in PBS (20 mM, pH 7.4) with 10 μM probe concentrations. WNO26, WNO27, and WNO28 were excited at 440 nm, 492 nm, and 674 nm, respectively, with fluorescence intensities recorded at 554 nm, 559 nm, and 700 nm. Data in (J)–(L) are shown as mean relative fluorescence intensity ± standard deviation (n = 3). | ||
 | ||
| Scheme 1 General synthesis of a series of NO-responsive fluorescent probes from amino-bearing fluorophores and their reaction with NO to produce the free fluorophores. | ||
We then assessed the fluorescence responses of these probes to NO in PBS buffer at physiological pH. WNO26, WNO27, and WNO28 were weakly fluorescent in the absence of NO, but exhibited dose-dependent fluorescence increases upon NO treatment with emission maxima at 554 nm, 559 nm, and 700 nm when excited at 440 nm, 492 nm, and 674 nm, respectively (Fig. 4D–F). The maximal fluorescence emission intensities of WNO26, WNO27, and WNO28 were linearly correlated with NO concentrations at low levels (Fig. S1–S3 in the ESI†). The detection limits of WNO26, WNO27, and WNO28 for NO were calculated to be 250, 160, and 320 nM, respectively. It should be mentioned that the detection limits of WNO26, WNO27, and WNO28 are higher than those of many previously reported NO fluorescent probes.17 For instance, the detection limits of 4,5-diaminofluorescein31 and fluoresceinamine22 for NO are 5 nM and 44 nM, respectively. The relatively high detection limits observed for WNO26, WNO27, and WNO28 may be attributed to the background fluorescence of the probes, as N-amido groups may not completely suppress the fluorescence of amino-containing fluorophores.81 Future efforts to improve the sensitivity of our probes should focus on incorporating self-immolative linkers and quenching moieties to enhance fluorescence quenching and reduce background signals. We also examined the time-dependent responses of WNO26, WNO27, and WNO28 toward NO. The results showed that their fluorescence signals almost reached the plateaus in approximately 5–15 min (Fig. 4G–I), suggesting that the probes reacted with NO at rates similar to those of most previously reported o-phenylenediamine-based NO probes.17 The response time of WNO26, WNO27, and WNO28 is generally longer than that of N-nitrosation-based NO probes,23,64,65 which react with NO in a single step to produce fluorescence increases. This difference is understandable due to the cascade reactions required by our probes (Fig. 1B). We envision that the response kinetics of our probes could be improved in future work by tuning the reactivity of the urea moiety on the fluorophore to enhance the reaction efficiency and reduce the response time.
To confirm the mechanisms of reactions between the probes and NO, we performed liquid chromatography–mass spectrometry (LC-MS) analyses. Indeed, we observed the generation of corresponding amino-containing core fluorophores in the reaction mixtures of WNO26, WNO27, and WNO28 with NO as the major products (Fig. S4–S6 in the ESI†). More importantly, we also detected the hydrolysed benzotriazole compound in these reaction mixtures as the byproduct. Therefore, these data corroborate the proposed reaction mechanism and the NO-induced urea-bond cleavage reaction (Scheme 1). Although WNO26, WNO27, and WNO28 may undergo amine loss via deamination in reaction with NO, a pathway previously utilized to develop NO fluorescent probes,22,57,63 our LC-MS analyses suggest that this reaction is at least not predominant, especially given the presence of the proximal N–H group that traps the N-nitrosation intermediate.
We proceeded to investigate the selectivity of these probes for detecting NO. As expected, NO led to significant fluorescence turn-on responses of WNO26, WNO27, and WNO28 (Fig. 4J–L). By contrast, other biological species, such as oxidants (e.g., hypochlorite, H2O2, hydroxyl radical, and peroxynitrite) and reductants (e.g., cysteine and glutathione), had minimal impact on the fluorescence signals of WNO26, WNO27, and WNO28. More importantly, potentially interfering bioanalytes, such as MGO, DHA, and AA, did not induce obvious fluorescence increases of these probes (Fig. 4J–L). Additionally, we evaluated the influence of pH on NO detection using these probes. WNO26 and WNO27 showed significant enhancements in fluorescence intensities upon exposure to NO across a wide pH range from 4 to 10 (Fig. S7 and S8 in the ESI†). WNO28 exhibited optimal fluorescence responses to NO at physiological pH levels close to 7.4, but was less sensitive to NO at low and high pH values (Fig. S9 in the ESI†), probably due to the pH-dependent fluorescence of the hemicyanine core fluorophore.72
Taken together, these results demonstrate that the NO-reactive moiety bearing the 4-morpholinyl substituent can be generally incorporated into diverse fluorophore scaffolds via the urea linker to enable fluorescence detection of NO, thus validating the NO-induced urea-bond cleavage reaction and a feasible approach to modularly develop NO-responsive fluorescent probes.
Fluorescence imaging of NO in living cells
After characterizing the probes for detecting NO in vitro, we further examined the capabilities of WNO27 and WNO28 for fluorescence imaging of NO in living cells. Specifically, HeLa cells were incubated with WNO27 or WNO28, treated with NO donor DEA NONOate, and monitored using confocal fluorescence microscopy. As shown in Fig. 5, HeLa cells loaded with WNO27 or WNO28 exhibited generally low fluorescence signals. In contrast, treatment of probe-loaded HeLa cells with DEA NONOate resulted in significant increases of fluorescence signals of both WNO27 and WNO28 in a dose-dependent manner (Fig. 5). Co-localization analyses using organelle markers indicated that WNO27 and WNO28 mainly localized in mitochondria (Fig. S10 in the ESI†). We also assessed the cytotoxicity of WNO27 and WNO28 using the MTT assay (Fig. S11 in the ESI†), which showed that they were largely non-toxic to cells at working concentrations. Therefore, these data demonstrate the suitability of WNO27 and WNO28 for imaging intracellular NO levels in living cells.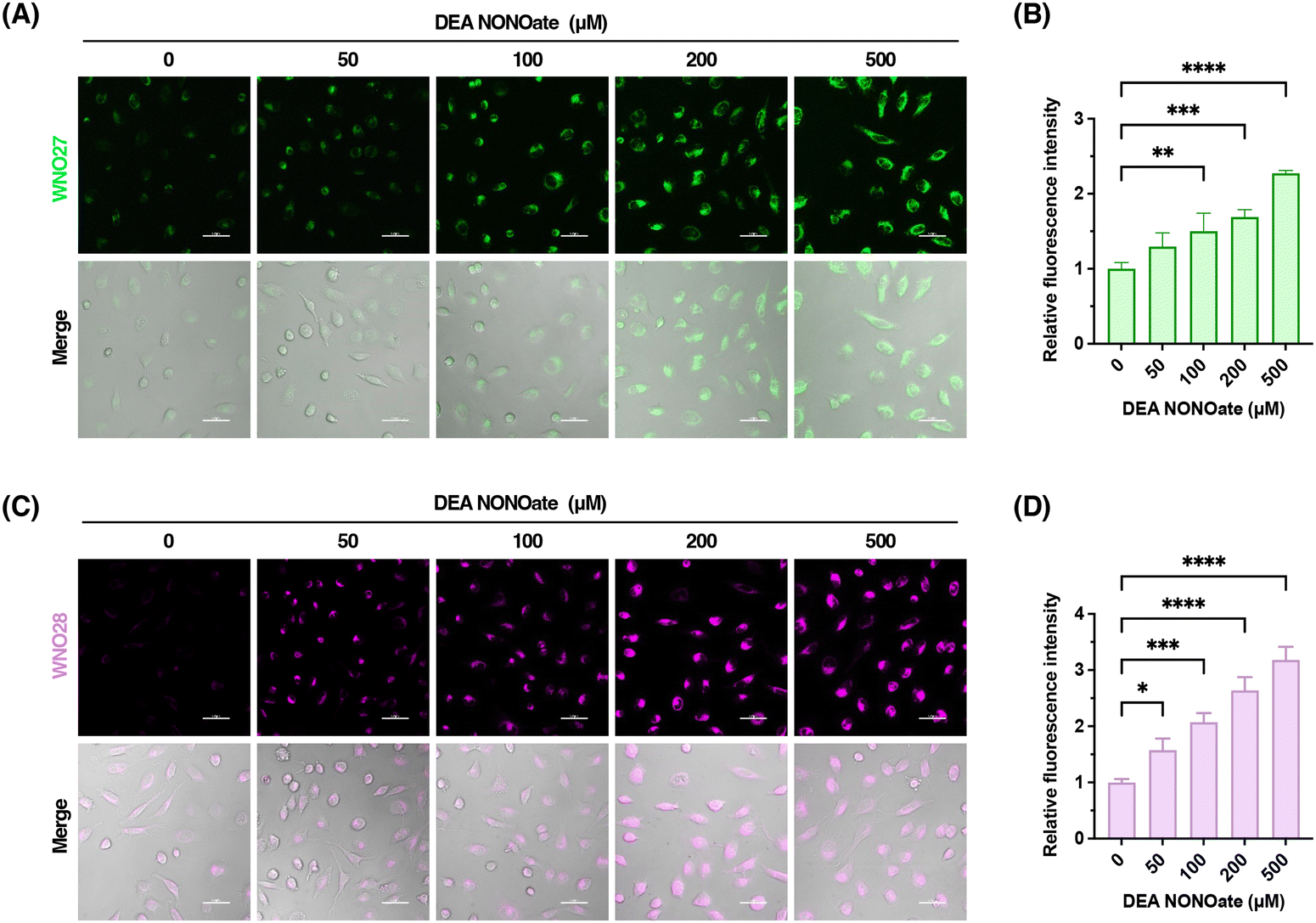 | ||
| Fig. 5 Confocal fluorescence imaging of exogenous NO with WNO27 and WNO28. (A) HeLa cells were loaded with WNO27 (5 μM) for 30 min and treated with DEA NONOate at different concentrations for 60 min. (B) Relative fluorescence intensities of fluorescence images shown in (A). (C) HeLa cells were loaded with WNO28 (2 μM) for 30 min and treated with DEA NONOate at different concentrations for 60 min. (D) Relative fluorescence intensities of fluorescence images shown in (C). Scale bars are 50 μm. Data in (B) and (D) are shown as mean ± standard deviation (n = 3). Statistical analyses for multiple comparisons were performed using one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
To further examine the utility of WNO27 and WNO28 in imaging endogenously produced NO, we focused on RAW264.7 macrophage cells that are known to produce high levels of NO upon immune stimulation.82,83 RAW264.7 cells loaded with WNO27 or WNO28 showed weak fluorescence signals, probably due to basal levels of intracellular NO (Fig. 6A and C). As a comparison, treatment of RAW264.7 cells with lipopolysaccharide (LPS) and L-Arg, which stimulate inducible NOS (iNOS) expression and subsequent NO production, respectively, substantially increased intracellular fluorescence intensities of WNO27 (Fig. 6A and B) and WNO28 (Fig. 6C and D). In addition, pretreatment of cells with iNOS inhibitors, such as aminoguanidine (AG)84 and N-(3-(aminomethyl)benzyl)acetamidine (1400 W),85 attenuated intracellular fluorescence signals of WNO27 and WNO28 (Fig. 6 and Fig. S12 in the ESI†), indicating that selective inhibition of iNOS led to reduced NO levels. These results thus demonstrate that WNO27 and WNO28 can be used to visualize endogenously produced NO in RAW264.7 cells under stimulation conditions.
 | ||
| Fig. 6 Confocal fluorescence imaging of endogenous NO with WNO27 and WNO28. (A) RAW264.7 cells were treated with LPS (20 μg mL−1) and L-Arg (5 mg mL−1) in the absence or presence of AG (0.5 mM) for 12 h and loaded with WNO 27 (2 μM) for 60 min. (B) Relative fluorescence intensities of fluorescence images shown in (A). (C) RAW264.7 cells were treated with LPS (20 μg mL−1) and L-Arg (5 mg mL−1) in the absence or presence of AG (0.5 mM) for 12 h and loaded with WNO 28 (1 μM) for 60 min. (D) Relative fluorescence intensities of fluorescence images shown in (C). Nuclei were stained with Hoechst 33342. Scale bars are 50 μm. Data in (B) and (D) are shown as mean ± standard deviation (n = 3). Statistical analyses were performed using one-way ANOVA. **p < 0.01, ***p < 0.001. | ||
In vivo fluorescence imaging of NO in an inflammation mouse model
Finally, we sought to apply the NIR probe WNO28 to image endogenous NO in vivo. We established an LPS-induced inflammatory mouse model by intravenously injecting LPS into mice followed by intraperitoneal injection of L-Arg for 24 h to induce inflammation and excessive production of NO, especially in the bowel.45 The mice were then intravenously injected with WNO28 in saline and imaged on an in vivo fluorescence imaging system with fluorescence signals collected at 720 ± 10 nm and excitation at 675 nm. As shown in Fig. 7A, no fluorescence was observed in control mice without injection of WNO28, and strong fluorescence was observed in the abdominal regions of LPS/L-Arg-stimulated mice following the administration of WNO28, with a 6-fold increase in fluorescence intensity compared to control mice without receiving the probe (Fig. 7B). In contrast, weak fluorescence was recorded in non-stimulated normal mice receiving an equal amount of WNO28 (Fig. 7A and B, and Fig. S13 in the ESI†), suggesting that LPS and L-Arg can stimulate the endogenous production of NO in living mice. We further confirmed the enhanced fluorescence signals and elevated NO levels in LPS/L-Arg-stimulated mice compared to non-stimulated mice using WNO28 at different concentrations (Fig. S14 in the ESI†). | ||
| Fig. 7 In vivo imaging of endogenous NO with WNO28 in an inflammatory mouse model. (A) Representative fluorescence images of normal and LPS/L-Arg-stimulated mice intravenously injected with WNO28 (250 μM, 200 μL) or saline. (B) Relative fluorescence intensities of mice shown in (A). Statistical analysis for multiple comparisons was performed using one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001. (C) Representative fluorescence images of major organs harvested from normal and LPS/L-Arg-stimulated mice receiving WNO28. (D) Fluorescence intensities of major organs shown in (C). (E) Time-dependent fluorescence imaging of normal and inflammatory mice after intravenous injection of WNO28 (250 μM, 200 μL). | ||
To investigate the distribution of fluorescence signals, major organs were dissected and imaged. The results demonstrated that LPS/L-Arg-stimulated mice exhibited the most intense fluorescence in the intestines (Fig. 7C and Fig. S15 in the ESI†), showing an 8-fold increase in fluorescence intensity compared to control mice that did not receive the probe, and a 2.8-fold increase compared to non-stimulated normal mice that received the probe (Fig. 7D and S16 in the ESI†). Additionally, moderate fluorescence signals were observed in the livers, while almost no fluorescence was detected in other organs (Fig. 7C and D, and Fig. S15 in the ESI†). These findings suggest that endogenous NO is predominantly produced in the intestines of mice in our murine inflammation model.
To examine the capability of WNO28 for time-dependent imaging of NO in vivo, we performed a series of imaging experiments on the mice at 0–24 h post-injection of WNO28. As shown in Fig. 7E, the LPS/L-Arg-stimulated mice exhibited detectable fluorescence at 45 min and maximal fluorescence at 1.5 h post-injection of WNO28. The fluorescence signals then decreased gradually with time. As a comparison, non-stimulated normal mice generally displayed weaker fluorescence, especially in the first several hours (Fig. 7E), suggesting that WNO28 enables the differentiation of inflammatory mice from normal mice. Interestingly, the normal mice receiving WNO28 showed detectable fluorescence, which could be attributed to the intrinsic NO in the gastrointestinal system with important roles in intestinal mucosal function.86
Together, these results suggest that NO is endogenously produced at high levels under LPS/L-Arg-stimulation conditions and demonstrate that the NIR probe WNO28 represents a promising imaging tool for direct visualization of endogenous NO in live animals.
Conclusions
To summarize, we have reported the development of a NO-induced urea-bond cleavage reaction and its application to develop fluorescent probes for the detection and imaging of NO in living cells and animals. Using coumarin model fluorophore scaffolds, we initially screened a series of coumarin-based probes (i.e., WNO1–6) that contain unsubstituted or N-methyl substituted carbamate or urea linkers and a NO-reactive aniline-derived moiety, revealing that the simple unsubstituted urea structure can be efficiently cleaved upon reaction of the aromatic amino group with NO through N-nitrosation, acyltriazole formation, and hydrolysis. We further examined a series of NO-reactive aniline derivatives bearing various substituents (i.e., WNO7–25) and found that the morpholinyl-containing aniline derivative afforded the most efficient reactivity in reaction with NO to induce the urea-bond cleavage. By installing this optimal NO-reactive moiety onto currently available amino-based fluorophore scaffolds, we constructed a set of NO fluorescent probes (i.e., WNO26–28) featuring different fluorescence excitation and emission wavelengths and selective fluorescence turn-on detection of NO in chemical systems. Among them, WNO27 and WNO28 were demonstrated for fluorescence imaging of endogenous NO in live cells. Moreover, we have successfully applied the NIR probe WNO28 to the in vivo imaging of endogenously produced NO in a murine LPS-induced inflammation model. We expect that the NO-induced urea-bond cleavage reaction developed herein is broadly applicable to any amino-based fluorescent and chemical scaffold, generating new chemical tools and strategies for NO imaging, manipulation, and therapeutic application. Overall, our work provides new opportunities for harnessing the chemical reactivity of NO for imaging and interrogating NO in various biological contexts.Author contributions
Y. Z.: investigation, writing – original draft, writing – review and editing; S.W.: investigation; L.Z.: investigation; T.P.: conceptualization, funding acquisition, project administration, supervision, writing – original draft, writing – review and editing.Data availability
The data supporting this article have been included as part of the Electronic ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was approved by the Institutional Animal Care and Use Committee of Peking University Shenzhen Graduate School. We thank the Laboratory Animal Centre at Peking University Shenzhen Graduate School for animal experiments. T. P. acknowledges grant support from the National Natural Science Foundation of China (22277008), the Shenzhen Science and Technology Innovation Committee (GXWD20201231165807007-20200814103057002), and the Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (2023SHIBS0004). Y. Z. acknowledges grant support from the China Postdoctoral Science Foundation (2023M730087).Notes and references
- A. R. Butler and D. L. H. Williams, Chem. Soc. Rev., 1993, 22, 233–241 RSC.
- D. S. Bredt and S. H. Snyder, Annu. Rev. Biochem., 1994, 63, 175–195 CrossRef CAS PubMed.
- J. O. Lundberg and E. Weitzberg, Cell, 2022, 185, 2853–2878 CrossRef CAS PubMed.
- U. Förstermann and W. C. Sessa, Eur. Heart J., 2011, 33, 829–837 CrossRef.
- C. N. Hall and J. Garthwaite, Nitric oxide, 2009, 21, 92–103 CrossRef CAS.
- C. Bogdan, Nat. Immunol., 2001, 2, 907–916 CrossRef CAS PubMed.
- S. Shreshtha, P. Sharma, P. Kumar, R. Sharma and S. P. Singh, J. Clin. Diagn. Res., 2018, 12, BE01–BE05 CAS.
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS PubMed.
- S. Moncada, R. M. J. Palmer and E. A. Higgs, Pharmacol. Rev., 1991, 43, 109–142 CAS.
- B. N. Gantner, K. M. LaFond and M. G. Bonini, Redox Biol., 2020, 34, 101550 CrossRef CAS PubMed.
- C. Farah, L. Y. M. Michel and J.-L. Balligand, Nat. Rev. Cardiol., 2018, 15, 292–316 CrossRef CAS PubMed.
- D. Fukumura, S. Kashiwagi and R. K. Jain, Nat. Rev. Cancer, 2006, 6, 521–534 CrossRef CAS.
- V. Calabrese, C. Mancuso, M. Calvani, E. Rizzarelli, D. A. Butterfield and A. M. Giuffrida Stella, Nat. Rev. Neurosci., 2007, 8, 766–775 CrossRef CAS PubMed.
- B. Almeida, K. E. Rogers, O. K. Nag and J. B. Delehanty, ACS Sens., 2021, 6, 1695–1703 CrossRef CAS PubMed.
- T. Nagano and T. Yoshimura, Chem. Rev., 2002, 102, 1235–1270 CrossRef CAS.
- L. E. McQuade and S. J. Lippard, Curr. Opin. Chem. Biol., 2010, 14, 43–49 CrossRef CAS PubMed.
- Y. Chen, Nitric oxide, 2020, 98, 1–19 CrossRef CAS PubMed.
- T. P. Misko, R. J. Schilling, D. Salvemini, W. M. Moore and M. G. Currie, Anal. Biochem., 1993, 214, 11–16 CrossRef CAS.
- H. Kojima, K. Sakurai, K. Kikuchi, S. Kawahara, Y. Kirino, H. Nagoshi, Y. Hirata, T. Akaike, H. Maeda and T. Nagano, Biol. Pharm. Bull., 1997, 20, 1229–1232 CrossRef CAS PubMed.
- T. Itoh, Y. Matsuya, K. Nagata and A. Ohsawa, Tetrahedron Lett., 1996, 37, 4165–4168 CrossRef CAS.
- Y. Yang, S. K. Seidlits, M. M. Adams, V. M. Lynch, C. E. Schmidt, E. V. Anslyn and J. B. Shear, J. Am. Chem. Soc., 2010, 132, 13114–13116 CrossRef CAS PubMed.
- T.-W. Shiue, Y.-H. Chen, C.-M. Wu, G. Singh, H.-Y. Chen, C.-H. Hung, W.-F. Liaw and Y.-M. Wang, Inorg. Chem., 2012, 51, 5400–5408 CrossRef CAS.
- J. Miao, Y. Huo, X. Lv, Z. Li, H. Cao, H. Shi, Y. Shi and W. Guo, Biomaterials, 2016, 78, 11–19 CrossRef CAS PubMed.
- T. Itoh, K. Nagata, M. Okada and A. Ohsawa, Tetrahedron Lett., 1995, 36, 2269–2272 CrossRef CAS.
- T. Itoh, K. Nagata, Y. Matsuya, M. Miyazaki and A. Ohsawa, J. Org. Chem., 1997, 62, 3582–3585 CrossRef CAS.
- S. Ma, D.-C. Fang, B. Ning, M. Li, L. He and B. Gong, Chem. Commun., 2014, 50, 6475–6478 RSC.
- A. S. M. Islam, R. Bhowmick, K. Pal, A. Katarkar, K. Chaudhuri and M. Ali, Inorg. Chem., 2017, 56, 4324–4331 CrossRef CAS PubMed.
- Q. Han, J. Liu, Q. Meng, Y.-L. Wang, H. Feng, Z. Zhang, Z. P. Xu and R. Zhang, ACS Sens., 2019, 4, 309–316 CrossRef CAS PubMed.
- M. H. Lim and S. J. Lippard, J. Am. Chem. Soc., 2005, 127, 12170–12171 CrossRef CAS.
- M. H. Lim, D. Xu and S. J. Lippard, Nat. Chem. Biol., 2006, 2, 375–380 CrossRef CAS PubMed.
- H. Kojima, N. Nakatsubo, K. Kikuchi, S. Kawahara, Y. Kirino, H. Nagoshi, Y. Hirata and T. Nagano, Anal. Chem., 1998, 70, 2446–2453 CrossRef CAS.
- H. Kojima, Y. Urano, K. Kikuchi, T. Higuchi, Y. Hirata and T. Nagano, Angew. Chem., Int. Ed., 1999, 38, 3209–3212 CrossRef CAS.
- H. Kojima, M. Hirotani, Y. Urano, K. Kikuchi, T. Higuchi and T. Nagano, Tetrahedron Lett., 2000, 41, 69–72 CrossRef CAS.
- H. Kojima, M. Hirotani, N. Nakatsubo, K. Kikuchi, Y. Urano, T. Higuchi, Y. Hirata and T. Nagano, Anal. Chem., 2001, 73, 1967–1973 CrossRef CAS PubMed.
- Y. Gabe, Y. Urano, K. Kikuchi, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2004, 126, 3357–3367 CrossRef CAS PubMed.
- E. Sasaki, H. Kojima, H. Nishimatsu, Y. Urano, K. Kikuchi, Y. Hirata and T. Nagano, J. Am. Chem. Soc., 2005, 127, 3684–3685 CrossRef CAS PubMed.
- H. Yu, Y. Xiao and L. Jin, J. Am. Chem. Soc., 2012, 134, 17486–17489 CrossRef CAS.
- G. K. Vegesna, S. R. Sripathi, J. Zhang, S. Zhu, W. He, F.-T. Luo, W. J. Jahng, M. Frost and H. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 4107–4112 CrossRef.
- H.-X. Zhang, J.-B. Chen, X.-F. Guo, H. Wang and H.-S. Zhang, Anal. Chem., 2014, 86, 3115–3123 CrossRef CAS PubMed.
- W. Liu, C. Fan, R. Sun, Y.-J. Xu and J.-F. Ge, Org. Biomol. Chem., 2015, 13, 4532–4538 RSC.
- Z. Mao, W. Feng, Z. Li, L. Zeng, W. Lv and Z. Liu, Chem. Sci., 2016, 7, 5230–5235 RSC.
- M. Weng, X. Yang, Y. Ni, C. Xu, H. Zhang, J. Shao, N. Shi, C. Zhang, Q. Wu, L. Li and W. Huang, Sens. Actuators, B, 2019, 283, 769–775 CrossRef CAS.
- N. Gupta, S. Imam Reja, V. Bhalla, M. Gupta, G. Kaur and M. Kumar, Chem. – Asian J., 2016, 11, 1020–1027 CrossRef CAS.
- Q. Wang, X. Jiao, C. Liu, S. He, L. Zhao and X. Zeng, J. Mater. Chem. B, 2018, 6, 4096–4103 RSC.
- S. Liu, Y. Zhu, P. Wu and H. Xiong, Anal. Chem., 2021, 93, 4975–4983 CrossRef CAS.
- S. J. Li, D. Y. Zhou, Y. Li, H. W. Liu, P. Wu, J. Ou-Yang, W. L. Jiang and C. Y. Li, ACS Sens., 2018, 3, 2311–2319 CrossRef CAS PubMed.
- Y.-Q. Sun, J. Liu, H. Zhang, Y. Huo, X. Lv, Y. Shi and W. Guo, J. Am. Chem. Soc., 2014, 136, 12520–12523 CrossRef CAS.
- H. Zheng, G.-Q. Shang, S.-Y. Yang, X. Gao and J.-G. Xu, Org. Lett., 2008, 10, 2357–2360 CrossRef CAS.
- Y. Huo, J. Miao, L. Han, Y. Li, Z. Li, Y. Shi and W. Guo, Chem. Sci., 2017, 8, 6857–6864 RSC.
- J. Tang, Z. Guo, Y. Zhang, B. Bai and W.-H. Zhu, Chem. Commun., 2017, 53, 10520–10523 RSC.
- P. Wardman, Free Radical Biol. Med., 2007, 43, 995–1022 CrossRef CAS PubMed.
- B. Kalyanaraman, V. Darley-Usmar, K. J. A. Davies, P. A. Dennery, H. J. Forman, M. B. Grisham, G. E. Mann, K. Moore, L. J. Roberts and H. Ischiropoulos, Free Radical Biol. Med., 2012, 52, 1–6 CrossRef CAS PubMed.
- D. Jourd’heuil, Free Radical Biol. Med., 2002, 33, 676–684 CrossRef PubMed.
- T. Wang, E. F. Douglass, Jr., K. J. Fitzgerald and D. A. Spiegel, J. Am. Chem. Soc., 2013, 135, 12429–12433 CrossRef CAS PubMed.
- X. Zhang, W. S. Kim, N. Hatcher, K. Potgieter, L. L. Moroz, R. Gillette and J. V. Sweedler, J. Biol. Chem., 2002, 277, 48472–48478 CrossRef CAS PubMed.
- X. Ye, S. S. Rubakhin and J. V. Sweedler, J. Neurosci. Methods, 2008, 168, 373–382 CrossRef CAS PubMed.
- A. Beltrán, M. Isabel Burguete, D. R. Abánades, D. Pérez-Sala, S. V. Luis and F. Galindo, Chem. Commun., 2014, 50, 3579–3581 RSC.
- C.-G. Dai, J.-L. Wang, Y.-L. Fu, H.-P. Zhou and Q.-H. Song, Anal. Chem., 2017, 89, 10511–10519 CrossRef CAS PubMed.
- Z. Mao, H. Jiang, Z. Li, C. Zhong, W. Zhang and Z. Liu, Chem. Sci., 2017, 8, 4533–4538 RSC.
- Z. Mao, H. Jiang, X. Song, W. Hu and Z. Liu, Anal. Chem., 2017, 89, 9620–9624 CrossRef CAS PubMed.
- H. Li, Y.-H. Hao, W. Feng and Q.-H. Song, J. Mater. Chem. B, 2020, 8, 9785–9793 RSC.
- M. Y. Lucero, A. K. East, C. J. Reinhardt, A. C. Sedgwick, S. Su, M. C. Lee and J. Chan, J. Am. Chem. Soc., 2021, 143, 7196–7202 CrossRef CAS PubMed.
- Y. Huo, J. Miao, Y. Li, Y. Shi, H. Shi and W. Guo, J. Mater. Chem. B, 2017, 5, 2483–2490 RSC.
- Y. Huo, J. Miao, J. Fang, H. Shi, J. Wang and W. Guo, Chem. Sci., 2019, 10, 145–152 RSC.
- C. Parisi, A. Pastore, M. Stornaiuolo and S. Sortino, J. Mater. Chem. B, 2024, 12, 5076–5084 RSC.
- T. Peng and D. Yang, Org. Lett., 2010, 12, 4932–4935 CrossRef CAS PubMed.
- T. Peng, N.-K. Wong, X. Chen, Y.-K. Chan, D. H.-H. Ho, Z. Sun, J. J. Hu, J. Shen, H. El-Nezami and D. Yang, J. Am. Chem. Soc., 2014, 136, 11728–11734 CrossRef CAS PubMed.
- T. Peng, X. Chen, L. Gao, T. Zhang, W. Wang, J. Shen and D. Yang, Chem. Sci., 2016, 7, 5407–5413 RSC.
- D. Yang, Z.-N. Sun, T. Peng, H.-L. Wang, J.-G. Shen, Y. Chen and P. K.-H. Tam, in Live Cell Imaging: Methods and Protocols, ed. D. B. Papkovsky, Humana Press, Totowa, NJ, 2010, pp. 93–103 DOI:10.1007/978-1-60761-404-3_5.
- Y. Zhang, Y. Du, M. Li, D. Zhang, Z. Xiang and T. Peng, Angew. Chem., Int. Ed., 2020, 59, 16352–16356 CrossRef CAS PubMed.
- S. Wang, X. Wu, Y. Zhang, D. Zhang, B. Xie, Z. Pan, K. Ouyang and T. Peng, Org. Biomol. Chem., 2021, 19, 3469–3478 RSC.
- Y. Du, Y. Zhang, M. Huang, S. Wang, J. Wang, K. Liao, X. Wu, Q. Zhou, X. Zhang, Y.-D. Wu and T. Peng, Chem. Sci., 2021, 12, 13857–13869 RSC.
- Y. Sun, Y. Chen, Y. Xu, Y. Zhang, M. Lu, M. Li, L. Zhou and T. Peng, Chem. Commun., 2022, 58, 8544–8547 RSC.
- M. Li, F. Wang, L. Yan, M. Lu, Y. Zhang and T. Peng, Chem. Commun., 2022, 58, 10186–10189 RSC.
- Y. Chen, L. Zhang, L. Fang, C. Chen, D. Zhang and T. Peng, JACS Au, 2024, 4, 2564–2577 CrossRef CAS.
- Y. Zhang, Y. Du, K. Liao and T. Peng, Anal. Methods, 2024, 16, 3646–3653 RSC.
- C. M. Maragos, D. Morley, D. A. Wink, T. M. Dunams, J. E. Saavedra, A. Hoffman, A. A. Bove, L. Isaac, J. A. Hrabie and L. K. Keefer, J. Med. Chem., 1991, 34, 3242–3247 CrossRef CAS PubMed.
- F. Xue and C. T. Seto, Org. Lett., 2010, 12, 1936–1939 CrossRef CAS PubMed.
- D. Lee, M.-K. Jung, J. Yin, G. Kim, C.-G. Pack and J. Yoon, Dyes Pigm., 2017, 136, 467–472 CrossRef CAS.
- L. Yuan, W. Lin, S. Zhao, W. Gao, B. Chen, L. He and S. Zhu, J. Am. Chem. Soc., 2012, 134, 13510–13523 CrossRef CAS PubMed.
- T. Peng and D. Yang, Org. Lett., 2010, 12, 496–499 CrossRef CAS PubMed.
- Y. Xia and J. L. Zweier, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6954–6958 CrossRef CAS PubMed.
- R. B. Lorsbach, W. J. Murphy, C. J. Lowenstein, S. H. Snyder and S. W. Russell, J. Biol. Chem., 1993, 268, 1908–1913 CrossRef CAS PubMed.
- M. J. D. Griffiths, M. Messent, R. J. MacAllister and T. W. Evans, Br. J. Pharmacol., 1993, 110, 963–968 CrossRef CAS PubMed.
- E. P. Garvey, J. A. Oplinger, E. S. Furfine, R. J. Kiff, F. Laszlo, B. J. R. Whittle and R. G. Knowles, J. Biol. Chem., 1997, 272, 4959–4963 CrossRef CAS PubMed.
- M. Guslandi, Dig. Dis., 2008, 12, 28–36 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S16, experimental procedures for chemical synthesis and biological assays, and characterization of new compounds. See DOI: https://doi.org/10.1039/d4tb01462f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |