
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformal encapsulation of mammalian stem cells using modified hyaluronic acid†
Jack
Whitewolf
a and
Christopher B.
Highley
 *ab
*ab
aDepartment of Biomedical Engineering, University of Virginia, Charlottesville, VA 22903, USA. E-mail: highley@virginia.edu
bDepartment of Chemical Engineering, University of Virginia, Charlottesville, VA 22903, USA
First published on 1st July 2024
Abstract
Micro- and nanoencapsulation of cells has been studied as a strategy to protect cells from environmental stress and promote survival during delivery. Hydrogels used in encapsulation can be modified to influence cell behaviors and direct assembly in their surroundings. Here, we report a system that conformally encapsulated stem cells using hyaluronic acid (HA). We successfully modified HA with lipid, thiol, and maleimide pendant groups to facilitate a hydrogel system in which HA was deposited onto cell plasma membranes and subsequently crosslinked through thiol-maleimide click chemistry. We demonstrated conformal encapsulation of both neural stem cells (NSCs) and mesenchymal stromal cells (MSCs), with viability of both cell types greater than 90% after encapsulation. Additional material could be added to the conformal hydrogel through alternating addition of thiol-modified and maleimide-modified HA in a layering process. After encapsulation, we tracked egress and viability of the cells over days and observed differential responses of cell types to conformal hydrogels both according to cell type and the amount of material deposited on the cell surfaces. Through the design of the conformal hydrogels, we showed that multicellular assembly could be created in suspension and that encapsulated cells could be immobilized on surfaces. In conjunction with photolithography, conformal hydrogels enabled rapid assembly of encapsulated cells on hydrogel substrates with resolution at the scale of 100 μm.
 Christopher B. Highley | Christopher B. Highley is an Assistant Professor in the Departments of Biomedical Engineering and Chemical Engineering at the University of Virginia. His laboratory develops technology for fabricating material and cellular systems to address biomedical challenges. The lab aims to advance capabilities for building biological constructs and maintains a focus on the development and application of hydrogel biomaterials in this work and in addressing medical needs. He received his BSE in Biomedical Engineering at Duke University and his PhD in Biomedical Engineering at Carnegie Mellon University. He was a postdoctoral researcher at the University of Pennsylvania before beginning his independent career. |
1. Introduction
Encapsulation of cells in biomaterials has become an increasingly sophisticated method to isolate cells from environmental stresses that could alter cell behavior; to protect them from harsh processing conditions; and to influence cellular phenotypes.1–8 While cell encapsulation often occurs in bulk materials, encapsulation methods using reduced volumes of hydrogels presents opportunities to increase the exchange of nutrients and cell-secreted products across the hydrogel, as well as to improve cell delivery to confined anatomical locations in cell therapy. Technologies to reduce material volumes in cell encapsulation include microencapsulation and approaches that reduce material volumes even further such as nanoencapsulation and conformal coatings. Microencapsulation have been shown to have immunoisolating as well as mechanically protective effects, rendering them a promising tool in stem cell transplantations or deliveries.9–17 Compared to microencapsulation, conformal encapsulation takes a further step in minimizing the material used for encapsulation. In conformal encapsulation, hydrogel layers are generated on or adjacent to the plasma membrane that range from 100s nm–1000s nm thick.Many conformal encapsulation processes do not require microfluidic systems that are often used in cell microencapsulations. The reduction of material volumes also further improves mass transport to and from encapsulated cells.18,19 While immunoisolation can still be achieved with conformal encapsulations, the thin layers of encapsulation can be designed to allow cell surface ligands to interact with their environment.18,20–24
Conformal encapsulations can be achieved by sequentially depositing multiple layers of polymer through electrostatic interactions;24–30 using cell surface-initiated chemistry to build hydrogels from the surfaces of cells;13,31,32 utilizing non-specific adsorption of materials to cell surfaces;33–36 and through various means of localizing hydrogel crosslinking to space adjacent cell surfaces.18,27,37,38 These encapsulations have been shown to help mechanically isolate, reinforce, and protect the encapsulated cells, as well as to be modified to include ligands that provide instructive cues to the encapsulated cell.27,28,37,39,40 Degradation of the encapsulating materials can also be strategically designed to allow cells egress at desired time points or, if long-term encapsulation is desired, to more stably encapsulate cells.41,42
Additionally, spatial relationships between encapsulated cells or between cells and external environments might be guided by material design in conformal encapsulations. Encapsulations that present to their surroundings reactive moieties such as chemical reactive groups, peptides, and oligonucleotides, might be directed to react with complimentary reactive groups. In a prime example, microtissues have been precisely assembled in suspension and on patterned substrates through modifications of the cell surfaces with complementary oligonucleotides.43,44 Conformal encapsulation offers the potential to combine directed assembly via surface chemistry and control of cell fates in designing multicellular constructs and interactions.
Up to this point, materials used for conformal encapsulation include a range of both synthetic and natural polymers. Synthetic polyelectrolytes,20,24,45 PEG-based polymers,27,37,46 and naturally derived polymers including alginate,47,48 gelatin,39 chondroitin sulfate49,50 and hyaluronic acid (HA)25,26,28,45,51 have been used, often in conjunction, to encapsulate mammalian cells. Here, we report a material system for encapsulation based entirely on HA for the first time. HA, a major extracellular matrix (ECM) glycosaminoglycan, plays important roles in development and wound healing.52 HA is an attractive platform for engineering cellular microenvironments as it can be readily modified with diverse functionalities; actively promotes cell survival; and is biodegradable through the enzyme hyaluronidase.52,53 In this paper, we modified HA with lipid groups and click chemistry groups to enable HA deposition onto the cell membrane and to allow for subsequent click reactions. Using these modified HAs, we conformally encapsulated neural stem cells (NSCs) as well as mesenchymal stromal cells (MSCs) to demonstrate the encapsulation capability of modified HAs. By utilizing the chemical groups present on the encapsulation material, we were also able to demonstrate the potential for conformal encapsulations to direct assembly between encapsulated cells in suspension, as well as assembly of cells onto patterned surfaces using photochemistry.
2. Materials and methods
2.1. Lipid-HA-thiol synthesis
Lipid- and thiol-modified hyaluronic acid (lipHASH) was synthesized in three steps. First, lipid-modified HA (lipHA) was synthesized. Second, 3,3′-dithiopropionic acid (DTA) was conjugated to lipHA. Third, the disulfide in the DTA moiety was cleaved to yield lipHASH. In the first step, hyaluronic acid (HA) sodium salt (Lifecore, 82 kDa) was dissolved at 20 mg mL−1 in deionized (DI) water to achieve a concentration of 2% w/v. Exchange of Na+ for H+ was achieved by adding 3 g of Dowex 50 W × 8 ion exchange resin (Sigma Aldrich) to the solution for each 1 g of HA and stirring for 3 h. Resin was removed by filtration, and the remaining solution titrated to pH 7 with tert-butylammonium (TBA) hydroxide (Fisher Scientific, 0.4 M in water) to produce HA-TBA.54 HA-TBA solution frozen and lyophilized to yield dry polymer. Next, to synthesize lipHA, HA-TBA was dissolved in anhydrous dimethylsulfoxide (DMSO, Sigma Aldrich, anhydrous, >99.9%) at 20 mg mL−1 under nitrogen. For each molar of disaccharide unit of HA, 0.01 mol of 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE, Sigma Aldrich, >97%) was dissolved in reagent grade ethanol (Sigma Aldrich, <0.003% water) at 75 °C at 2 mg mL−1, then added to HA-TBA solution. Finally, a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP, Sigma Aldrich, 97%) to DPPE was dissolved in DMSO and added to the reaction mixture. The mixture was stirred at 40 °C overnight.
:1 molar ratio of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP, Sigma Aldrich, 97%) to DPPE was dissolved in DMSO and added to the reaction mixture. The mixture was stirred at 40 °C overnight.
After the reaction, the mixture was placed in a 5000 MWCO membrane (Cole Palmer) and dialyzed against deionized (DI) water for 1 day, then filtered to remove precipitated excess reagent. The solution was purified by ultrafiltration (Millipore Sigma, Amicon Stirred Cells). The resulting lipHA solution was converted to lipHA-TBA using the same ion exchange method described above and lyophilized. To synthesize the lipHA-dithiopropionic acid intermediate product, lipHA-TBA, 3,3′-dithiopropionic acid (Sigma Aldrich, 99%), and 4-(dimethylamino)pyridine (DMAP, Sigma Aldrich, >99%) were dissolved at 20 mg mL−1 in anhydrous DMSO under nitrogen at 45 °C. Di-tert-butyl decarbonate (Boc2O, Sigma Aldrich, >99%) was then added to the dissolved reaction mixture. The reaction proceeded for 20 h at 45 °C. The molar ratios of lipHA-TBA:DTA:DMAP:Boc2O were 1:3.75:1.875:0.75 for synthesizing lipHASH used in conformal encapsulation and cell assembly and 1:5:2.5:1 for synthesizing material used in photo-patterning. The reaction solution was then dialyzed and ultrafiltered as described above to yield DTA-modified lipHA. To produce lipHASH, 1,4-dithiothreitol (DTT, Sigma Aldrich, 97%) was added to the DTA-modified lipHA solution at 1:1 w/w DTT to HA. After the DTT was dissolved, the pH was adjusted to pH 8.5 using 1 M NaOH and maintained for 2 h. The pH was then adjusted to pH 3.5 to quench the reaction and prevent further disulfides from forming. The final mixture was ultrafiltered with starting pH 3.5 and finally flash frozen for later use.
2.2. MaHA, HASH, norHA syntheses
Maleimide-modified HA (MaHA) was made as previously described.55 Briefly, HA-TBA was synthesized as described above. HA-TBA and N-(2-Aminoethyl)maleimide trifluoroacetate salt (Sigma Aldrich, >95%) were dissolved in DMSO. HA-TBA was dissolved at 20 mg mL−1 of DMSO, and the molar ratio of HA disaccharide units to MA was 1:0.3. BOP was dissolved in DMSO and added dropwise into the reaction mixture. The reaction was allowed to proceed for 3 h at room temperature, then dialyzed and ultrafiltered as described above. Thiol-modified HA (HASH) was made as previously described.56 HA-TBA, 3,3′-dithiopropionic acid, and DMAP were dissolved in DMSO at 45 °C. Then, Boc2O was added and the reaction was allowed to proceed for 20 h at 45 °C. The molar ratio of HA-TBA:DTA:DMAP:Boc2O was 1:5:2.5:1. The conjugated DTA was subsequently reduced using DTT as described above. Norbornene-modified HA (norHA) was made as previously described.57 Briefly, 5-norbornene-2-carboxylic acid (Sigma Aldrich, mixture of endo and exo, predominantly endo, 98%) was coupled anhydrously to HA-TBA in DMSO via Boc2O conjugation to yield norHA, purification was done through dialysis in DI water.
2.3. Peptide synthesis
All peptides used in this study were synthesized using a Liberty Blue (CEM) automated, microwave-assisted solid phase peptide synthesizer using Fmoc chemistry. Briefly, Rink amide resin (Advanced Chemtech, 100–200 mesh, 1% DVB) was swollen with dimethylformamide (DMF, Aldrich, ACS reagent grade), and the immobilized Fmoc group removed with 20% (v/v) piperidine in dimethylformamide. Fmoc-protected amino acids (Advanced ChemTech, 0.2 M in DMF, 5 equivalents relative to theoretical available sites on the resin) and the coupling agents diisopropylcarbodiimide (DIC, Aldrich, 99%, 1 M in DMF) and Oxyma Pure (Advanced ChemTech, 1 M in DMF) were added to the reaction vessel and heated to 90 °C for 4 min. The Fmoc deprotection and coupling steps were repeated to build the peptide from the C-terminus to the N-terminus. For fluorescent peptides, 5(6)carboxyfluorescein (Sigma Aldrich, >95%) was added last onto the N-terminus. The resultant peptides were cleaved from the resin with a cocktail of 92.5% trifluoroacetic acid (TFA, Aldrich, 99%), 2.5% triisopropylsilane (TIPS, Aldrich, 99%), 2.5% 2,2(ethylenedioxy)diethanethiol (DODT, Aldrich, 95%), and 2.5% DI water, and then isolated by precipitation into cold diethyl ether (Aldrich, ACS reagent, contains butylated hydroxytoluene as inhibitor) and centrifugation. After removal of ether under vacuum, the peptides were resuspended in DI water, frozen in liquid nitrogen, lyophilized, and stored at 20 °C as powders until ready for use. High performance liquid chromatography (HPLC) was used to determine peptide purity.2.4. lipHA-fluor synthesis
lipHA, synthesized as described above, was functionalized with hydrazides to which fluorescein isothiocyanate (FITC) was directly conjugated. First, lipHA-hydrazide (lipHA-ADH) was synthesized by combining lipHA, N-hydroxysuccinimide (NHS, Thermo Fisher, 99%), and adipic acid dihydrazide (ADH, Sigma Aldrich, >98%) in DI water at a molar ratio of lipHA (disaccharide units):NHS:ADH 1:2:5. LipHA was dissolved at 10 mg mL−1. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, Sigma, Aldrich, >98%) was then dissolved in DI water and added to the solution mixture at equimolar ratio to NHS. The pH of the reaction mixture was then titrated to 6 using 1 M HCl and 1 M NaOH. The reaction was allowed to proceed overnight. To purify lipHA-ADH, the resulting solution was dialyzed for 3 days in DI water and lyophilized. To convert hydrazide groups to fluorescent moieties, lipHA-ADH was dissolved at 0.5 wt% in 0.1 M dibasic disodium phosphate buffer (Sigma Aldrich, ACS reagent grade) and an equimolar quantity of fluorescein isothiocyanate (FITC, Sigma Aldrich, isomer I, >90%) to single disaccharide units of HA was dissolved at 0.5 wt% in DMSO and added to lipHA-ADH solution. The reaction was allowed to proceed overnight. The mixture was then dialyzed for 3 days and lyophilized.
2.5. NSC and MSC culture
Rat hippocampal NSCs (Millipore Sigma) were cultured on tissue culture flasks coated with poly-L-ornithine (Sigma Aldrich, poly-L-ornithine hydrobromide, MW 30000–70000), then laminin (Sigma Aldrich, 1–2 mg mL−1 in Tris-buffered saline) to support NSC attachment to generate single-cell (non-spheroid) cultures. To establish NSC spheroids, we cultured NSCs on non-adherent plates, which results in NSC expansion in suspension and the formation of NSC clusters. The NSC culture medium was DMEM with reduced glutamine (Thermo Fisher), B27 supplement (Thermo Fisher, B27 50X), and 1× antibiotic–antimycotic (anti-anti, Thermo Fisher, Antibiotic–Antimycotic (100X)). Medium was changed every two days and bFGF (PeproTech, Recombinant Human FGF-basic) was maintained at a concentration of 20 ng mL−1 for each media change. MSCs (Lonza) were cultured in MEM alpha (Sigma Aldrich) medium supplemented with 10% fetal bovine serum (Thermo Fisher) and 1× anti–anti. Culture medium was changed every 2 days during the course of culture. All cell lines were cultured in a humidified incubator with 5% CO2 at 37 °C. To harvest NSCs, flasks were treated with 3 mL of Accutase™ (Biolegend) after media aspiration and were incubated for 3 min to allow enzymatic detachment. For MSCs, 4 mL of 0.05% Trypsin (Fisher Scientific) was added to each flask after media aspiration and washing with PBS, and a 5 min incubation was used. The harvested cells were subsequently centrifuged to form pellets and resuspended in medium for continued passaging or in desired solutions for experimental use.
2.6. Conformal encapsulation of NSCs, MSCs, and NSC spheroids
NSCs and MSCs were encapsulated by first incubating the cells in 0.5 mg mL−1 lipHASH solution for 30 min at 4 °C. 1 mL of lipHASH solution was used for each 1–2 million NSCs or MSCs. The cells were then centrifuged at 300 g for 5 min and washed with 10 mL of Dulbecco's phosphate buffered saline (DPBS, Fisher Scientific, no calcium, no magnesium) once. For the second layer of encapsulation, the washed cells were resuspended in a 5 mg mL−1 MaHA solution and incubated for 15 min at 4 °C. After incubation, the cells were centrifuged at 450 g for 3 min, then washed once with 10 mL of DPBS. For a third layer of encapsulating material, cells were incubated in 5 mg mL−1 HASH solution for 15 min at 4 °C, then centrifuged at 450 g for 3 min and washed with 10 mL DPBS once. For encapsulations of four layers or more, the steps described above were repeated with alternating incubations in MaHA or HASH solutions. All solutions were made by dissolving modified HA in DPBS, then filtering the solution through a 0.45 μm syringe filter followed by a 0.22 μm sterile filter for sterilization. To enable fluorescent imaging of multilayered coatings, MaHA was covalently modified by a cysteine-containing peptide that was end-conjugated with rhodamine B (GCGKKK-RhoB). The peptide was dissolved in DPBS at 10 mg mL−1 then added to a 5 mg mL−1 MaHA solution prior to sterilization. Peptide was added to give a 3% degree of substitution relative to total HA disaccharide units.For encapsulation of NSC spheroids, the spheroids were collected by centrifuging at 200 g for 5 minutes. All centrifuge spins after washes were adjusted to 200 g and 5 minutes. Spheroids were resuspended by gentle trituration via pipetting of the spheroid pellet. The rest of the encapsulation procedures were the same as in single NSC encapsulations.
2.7. Live/dead assay and encapsulation fraction analysis
After encapsulation, NSCs or MSCs were cultured in 24 well plates for 3 days. Fluorescence around cells – resulting from labeling MaHA as described above – as well as cell shape were used as metrics to assess if the cells stayed in encapsulation. Microscopy images were taken and encapsulated cells, as well as cells that appeared unencapsulated in culture, were counted over 3 days to track the fraction of cells that stayed in encapsulation. For live/dead staining, calcein AM/ethidium homodimer (LIVE/DEAD™ Viability/Cytotoxicity Kit, for mammalian cells) were used. Each day, multiple wide field microscopy (Leica DMi8 Widefield) images were taken for each well after live/dead staining, live and dead cells were counted for all the images, and the cells from that day were disposed.2.8. Assembly of NSCs via complementary chemistries
NSCs were split into two groups and encapsulated in either lipHASH (single-layer encapsulation) or lipHASH and MaHA (2-layer encapsulation). After washing with DPBS, the cells were allowed to assemble through centrifugation. The cells were mixed at a ratio of 1:5 MaHA to lipHASH. Cell suspensions were centrifuged at 300 g for 5 min, and the pellet was resuspended by gentle pipetting without replacing the medium in which they were suspended.
2.9. Photopatterning of NSCs onto a norHA hydrogel surface
NSCs were encapsulated in a layer of lipHASH as described above. After washing with DPBS, the encapsulated cells were suspended at a density of 5 million cells per mL in a solution consisting of a photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, Sigma Aldrich, >95%) at 1 mM in DPBS. 100 μL of the cell solution was then pipetted onto the norHA gel and a photomask was positioned on top of the solution. Fabrication of norHA hydrogel was conducted as previously described.58 Briefly, glass coverslips were functionalized with 3-(mercaptopropyl)trimethoxysilane (MTS, Sigma Aldrich, 95%) The coverslips were then washed sequentially in dichloromethane (DCM, Sigma Aldrich, >99.8%), 70% ethanol in water, and DI water. norHA hydrogels were created on coverslips from a solution consisting of 5% (w/v) norHA, 1 mM LAP and DTT. The solution was crosslinked by irradiation for 2 min at 365 nm (10 mW cm−2, Omnicure). The NSCs were allowed to settle to the hydrogel's surface over 10 min, then the NSCs on the gels were exposed under 20 mW cm−2 UV light through the photomask for 1 min. This induced crosslinking between pendant norbornene moieties on the hydrogel surface and thiols on the lipHASH coating. The photomask was then removed and the hydrogel washed with DPBS 3 times to remove non-attached cells.2.10. Macroscale degradation of MaHA – HASH gel
Macroscale gels were made by mixing HASH and MaHA solutions together to achieve a final concentration of 5 mg mL−1 for both materials. HASH and MaHA solutions were made separately at 10 mg mL−1. 100 μL of MaHA solution was added to the bottom of 96 well plates. Then, 100 μL of HASH solution was added to the MaHA solution and mixed with a pipette. Gelation occurred within 10 seconds. The gels were then transferred to transwell inserts and cultured for 7 days in a 24-well plate with enough PBS to cover the gel. The mass of gels was measured every day and compared to their original mass.3. Results
3.1. Material synthesis and conformal encapsulation of neural stem cells
LipHASH and MaHA were synthesized as illustrated in Scheme 1. HASH was synthesized from HA via the same Boc2O reaction used to synthesize lipHASH from lipHA. All HA derivatives were characterized using 1H NMR (Fig. S1–S3, ESI†). The lipid chosen for our modification has previously been shown to be able to insert into cell membranes;43 we modified HA with an average of 1 lipid per HA chain in aiming to maximize the conformational freedom of lipHA attached to the cell membrane. Our syntheses yielded a ∼0.3 degree of substitution for both HASH and MaHA. Our approach to creating a multilayered conformal encapsulation is illustrated in Scheme 2, where the pendant lipids that were conjugated to the HA backbone in lipHASH inserted into the plasma membrane to create a lipHASH first layer with free thiols available for reaction. Then, via maleimide-thiol click chemistry, MaHA could be conjugated to the first layer to create a two-layer encapsulation formed by the crosslinking of a thin HA-based hydrogel around the cell. Unreacted maleimide moieties in the second layer then served to crosslink to a subsequent layer of HASH, with continuing build-up of the hydrogel encapsulation possible through further conjugation of alternating layers of MaHA and HASH.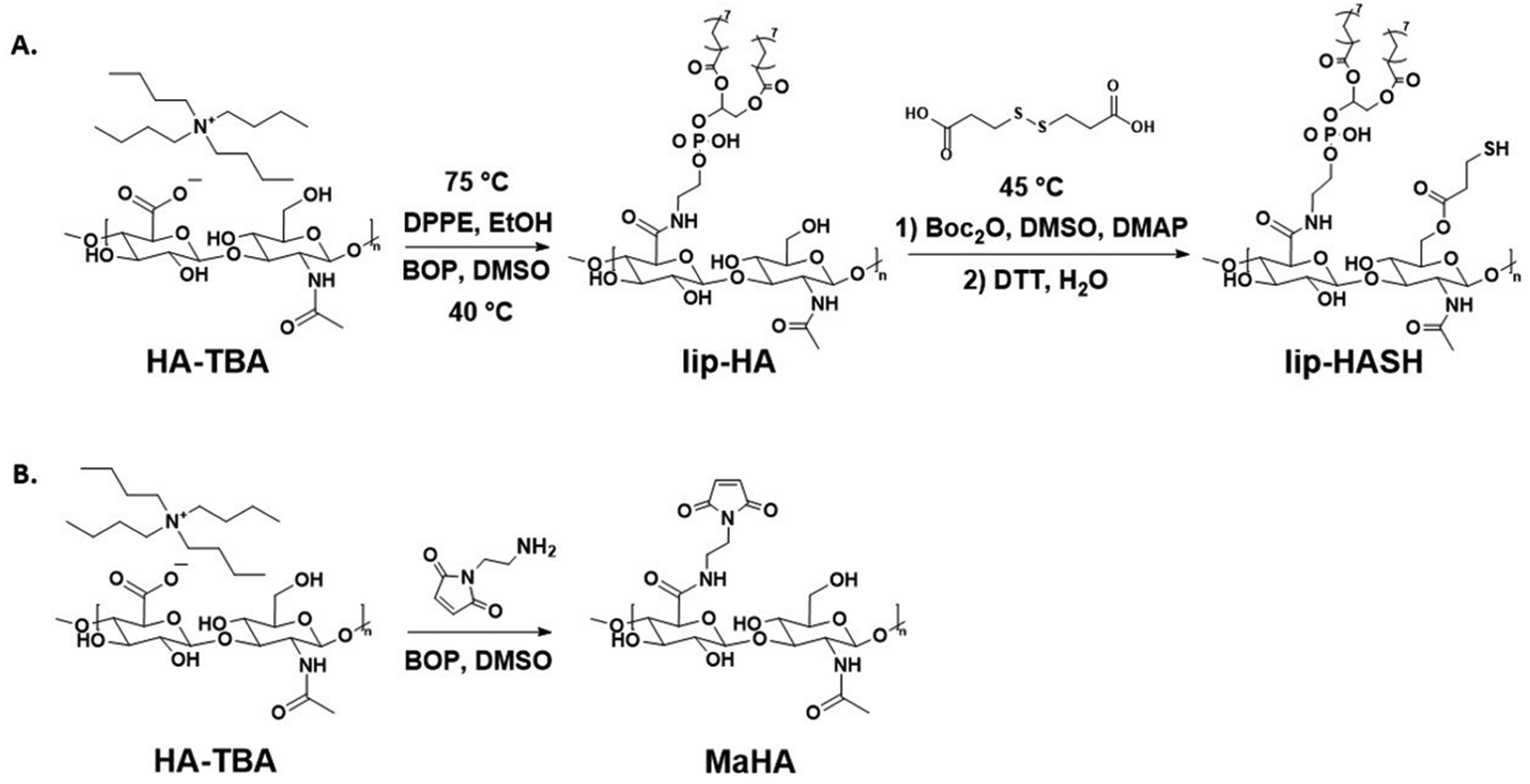 | ||
| Scheme 1 (A) Synthesis of lipHASH. HA-TBA is the tert-butyl ammonium salt of HA, generated through ion exchange as described above. lipHA is synthesized first, then lipHA-dithiol is made in the same way as HA-dithiol; and the dithiol (shown above the second step arrow) is subsequently cleaved using DTT to generate pendant thiol group. (B) Synthesis of MaHA. | ||
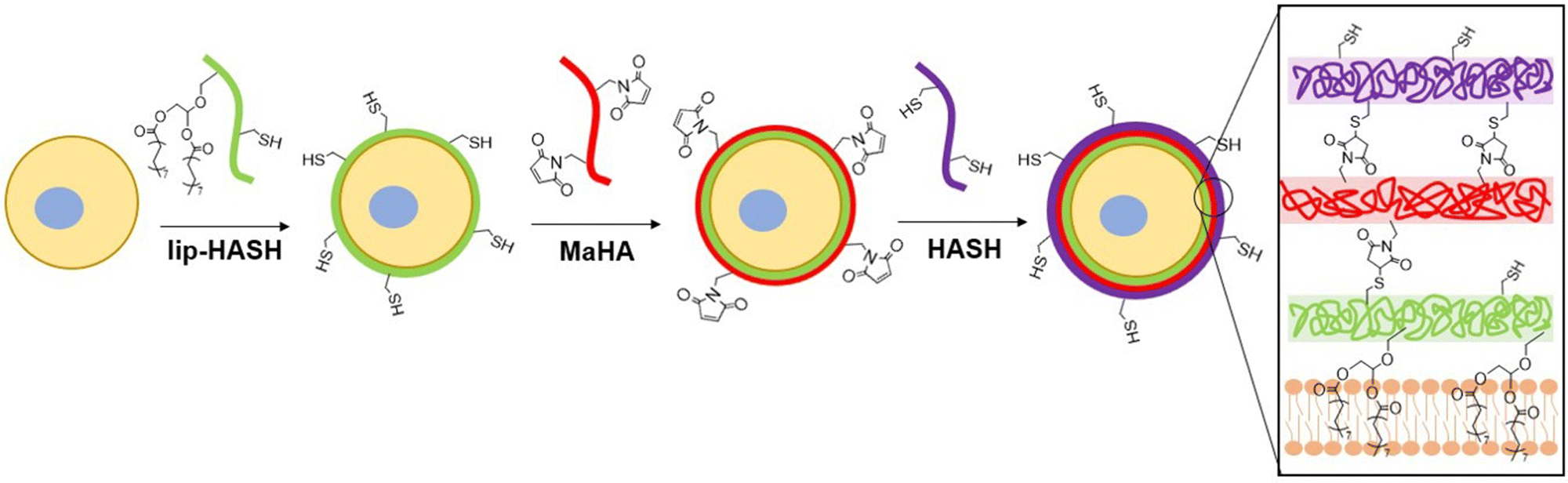 | ||
| Scheme 2 The process of conformal cell encapsulation. lipHASH is deposited onto the cell membrane by hydrophobic interaction, then maleimide-thiol click chemsitry is utilized for subsequent conjugations to deposit more layers of HA (final covalent bonds formed and the interaction of the conjugated lipids with the cell membrane are shown on the right). | ||
To confirm the association of lipHA with the cell membrane, we synthesized FITC-conjugated lipHA (lipHA-FITC) to enable visualization of lipHA by fluorescent microscopy. NSCs incubated with lipHA-FITC exhibited a clear fluorescent ring around cells indicative of lipHA-FITC localization at the cell surfaces (Fig. 1A). To confirm the addition of the second encapsulation layer using MaHA, we tagged MaHA with rhodamine B (RhoB-MaHA). Encapsulation of NSCs using non-fluorescent lipHASH and then RhoB-MaHA also showed clear fluorescence on NSCs (Fig. 1B). We next conducted experiments to rule out potential contributions of auto-fluorescence and non-specific interactions (Fig. 1C), such as adsorption of HA to the cell surface or the reaction of maleimides (in MaHA) to matters present on the cell surface. NSCs were first incubated in PBS (control), lipHA-FITC (non-thiol containing, fluorescent group), or an 80:20 mixture of lipHASH and lipHA-FITC (lipHASH for reactivity and lipHA-FITC for fluorescence). We expected the control group to show little or no fluorescence; and both lipHA-FITC containing groups to show strong fluorescence in the FITC channel due to lipid insertion of modified HAs into the plasma membrane. We also expected that NSCs incubated with only lipHA-FITC would show a stronger fluorescence due to the higher number of FITC-containing molecules. After the first incubation, NSCs were washed and incubated again in PBS (control) or RhoB-MaHA (all other groups). We expected that NSCs incubated in PBS to show little or no RhoB signal. For NSCs incubated in lipHA-FITC, if non-specific interactions were few, little or no RhoB signal should likewise be expected. However, for NSCs incubated in lipHASH and lipHA-FITC, stronger RhoB signal should be expected due to reactivity between RhoB-MaHA and lipHASH. We quantified fluorescence intensity of FITC and RhoB using ImageJ and saw that both lipHA-FITC-containing groups showed significant increases in fluorescence intensity compared to the PBS control. We also saw no significant increase in RhoB signal when RhoB-MaHA incubation followed lipHA-FITC incubation, suggesting minimal non-specific interactions. On the other hand, significantly higher RhoB signal was observed in NSCs incubated with lipHASH before RhoB-MaHA incubation.
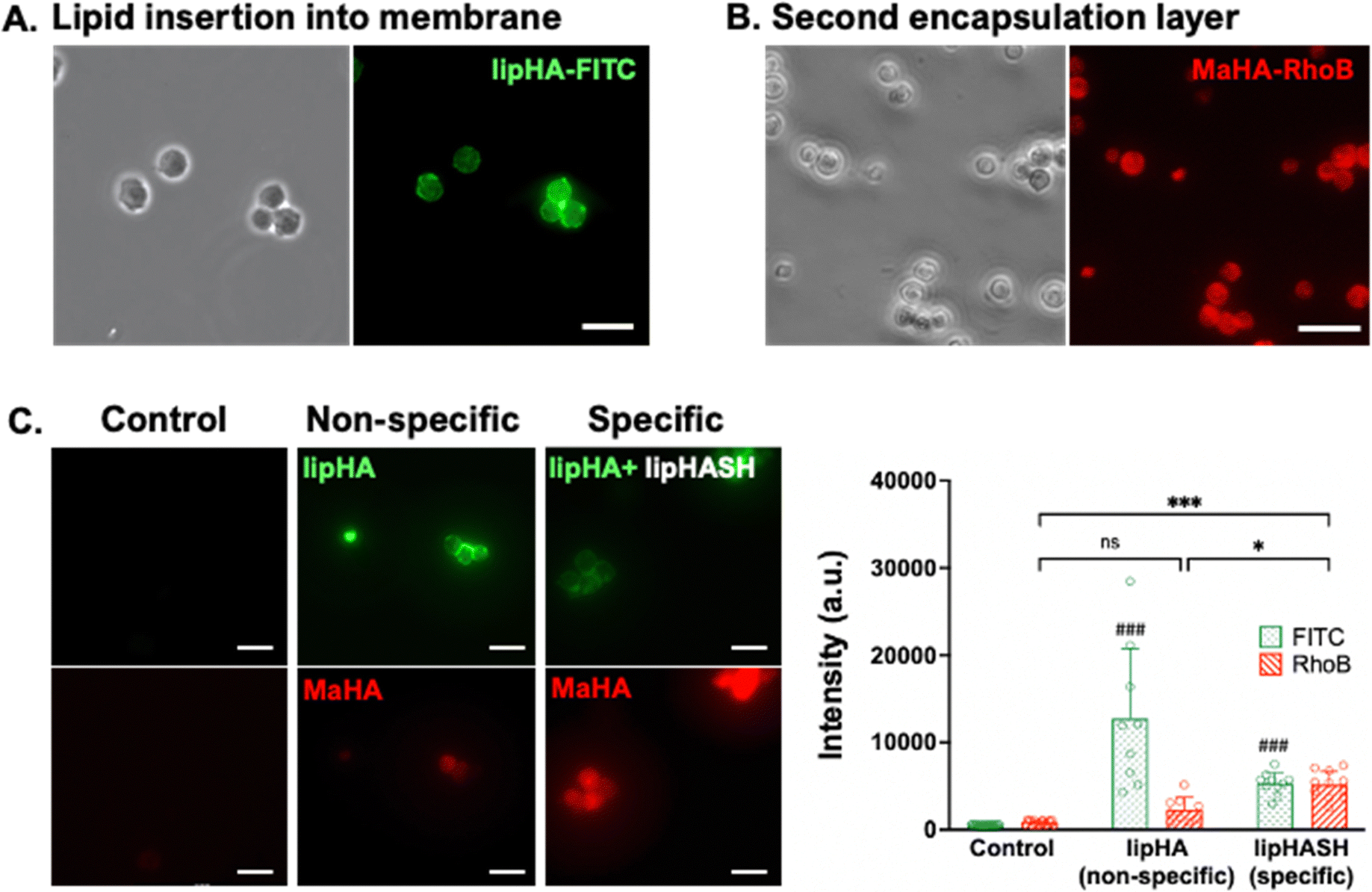 | ||
| Fig. 1 (A) NSCs encapsulated in a layer of FITC-tagged lipHA. (B) Fluorescence from MaHA around NSCs after 1 day of culture post encapsulation. (C) Left panels: Representative images of FITC (top row) and RhoB (bottom rjow) channels for NSCs incubated in PBS (left), lipHA-FITC then RhoB-MaHA (middle), and mixture of lipHA-FITC + lipHASH then RhoB-MaHA (right). Right: Quantification of standardized fluorescence among three groups. # Denotes significant differences of FITC when compared to control group. n = 88 cells were used for statistical testing using Welch's ANOVA and Dunnett's T3 test. Scale bars = 50 μm. For all figures: *p < 0.05, **p < 0.01, ***p < 0.001. | ||
3.2. Varying conformal encapsulation layers using NSCs and NSC spheroids
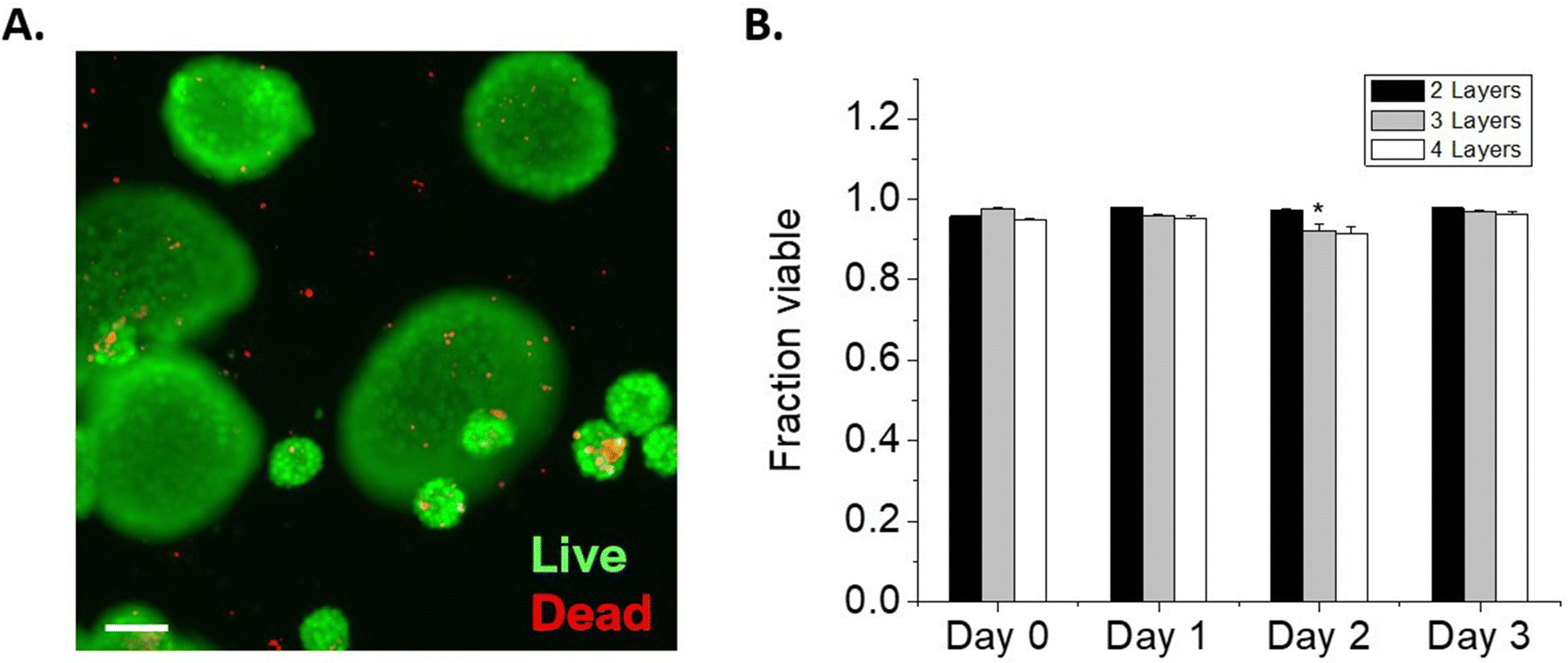
We next considered the conformal hydrogel's stability over time and effects on viability in NSC encapsulations. Since a minimum of 2 layers of material are needed to form a crosslinked network, NSCs were encapsulated with 2, 3, or 4 layers of modified HA and cultured over multiple days. To visualize the encapsulating hydrogel, we used RhoB-labeled MaHA and HASH tagged with Atto 488-maleimide. As expected, fluorescence in both channels were detected around the encapsulated NSCs after 2 layers of encapsulation (Fig. 2A). Next, NSCs were encapsulated with 3 layers and 4 layers of alternating MaHA and HASH. Live/dead stain using calcein AM and ethidium homodimer showed that over 94% cells remained viable immediately after encapsulation (Fig. 2B).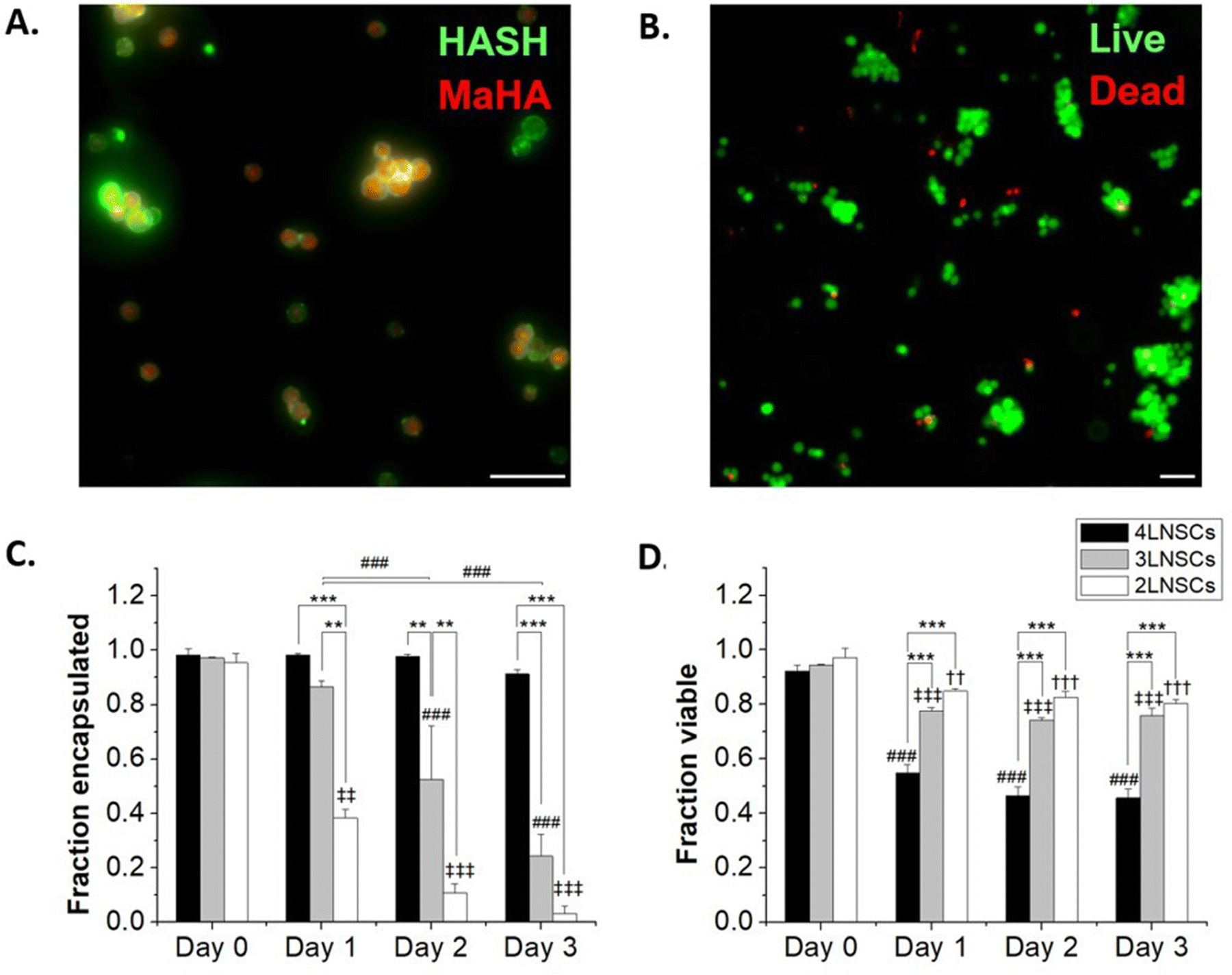 | ||
| Fig. 2 (A) NSCs encapsulated in RhoB-tagged MaHA and then Atto 488-tagged HASH. (B) Representative image of live/dead staining immediately after encapsulation. (C) The fraction of NSCs that remained encapsulated over 3-day culture periods. (D) The fraction of NSCs that remained viable over 3-day encapsulation periods. For both C and D, * denotes significant differences within groups. For C, and ‡ denotes significant differences of 3-layer and 4 layer-encapsulated NSCs compared to day 0, respectively. | ||
After encapsulations, NSCs were cultured in flasks that were surface-treated to support adherent NSC culture. We quantified cell egress from encapsulation based on the fraction of NSCs that lost the fluorescent HA at their plasma membranes, and we visually confirmed adherence to the planar surface by the extension of small processes and loss of spherical morphology seen in suspension culture while encapsulated (Fig S4, ESI†). We saw that for 2 layers of encapsulation, egress occurred within 24 hours. The time needed for egress increased for 3 layers of encapsulation, with the fraction of cells encapsulated remaining higher compared to 2 layers over subsequent days in culture (Fig. 2C). No significant cell egress occurred during multi-day culture when cells were surrounded by a 4-layer conformal hydrogel.
To further assess the stability of the encapsulation, macroscale hydrogels of MaHA and HASH were made at the same concentration used for cell encapsulation, and the degradation of the macroscale gel was tracked over a period of 7 days (Fig S5, ESI†). The degradation of the macroscale gel occurred slowly, with 80% of the gel remaining at day 3. Given the high surface area to volume ratio of the coatings, degradation occurring within them may lead to defects or disruptions that result in the coatings becoming unstable for encapsulation before the gels fully degrade. Because much of the gel may remain stable over days, as seen in the macroscale HA hydrogels, cell egress is likely also facilitated by other active mechanisms such as cytoskeletal movement and enzymatic degradation of HA.
We tracked NSC viability over 3 days in culture: in the group with a 2 layered HA hydrogel, NSCs retained >80% viability over 3 days as they were able to exit the encapsulation and proliferate. In groups with 3 layers and 4 layers of encapsulations, we observed viability decreases concomitant with reduced egress. NSCs exhibited 75.8 ± 2.8% and 45.7 ± 3.4% viability after 3 days in culture when encapsulated in 3 layers and 4 layers of HA, respectively.
One potential factor for the loss of NSC viability could be the reduced cell-to-cell contacts mediated by increasing layers of encapsulations. To elucidate the impact of cell–cell interactions on cell survival, we cultured NSCs on non-adherent plates to form NSC spheroids. These spheroids were then encapsulated similarly to NSCs, by incubation sequentially in solutions of lipHASH, MaHA, and HASH. NSC spheroid groups with 2 layers, 3 layers, and 4 layers of encapsulation material were established. Similar to individually encapsulated NSCs, the NSC spheroids showed strong fluorescence on Day 0 covering all spheroids (Fig. S6, ESI†). However, for all encapsulation groups, viability of the spheroids over the 3-day culture period was above 90% (Fig. 3), assessed using a live/dead kit. In comparison to the single cell encapsulation of NSCs (Fig. 2), these data (Fig. 3) suggest that cellular contacts established prior to encapsulation could enhance the survivability of certain encapsulated cells, such as NSCs. Interestingly, we observed that after 2 days, in 2-layer encapsulation groups, cells on the perimeter of the spheroids appeared to have no fluorescent signals, suggesting growth of NSC spheroids beyond the limits of the encapsulating hydrogel (Fig. S6 and S7, ESI†).
 | ||
| Fig. 3 (A) Representative image of a live/dead stain for D0 NSC spheroids with 2 layers of encapsulation. (B) Within encapsulated spheroids, the fraction of NSCs that remained viable over 3 days was >90% in all groups. * Denotes significant differences within the same group when compared to D0. There are at least 3 replicates for each group, and for each replicate, n > 3000 cells were counted. A total of 228605 cells were used to assess viability. Scale bar = 100 μm. | ||
3.3. Conformal encapsulation of mesenchymal stromal cells
NSCs are not contractile and have a reduced cytoskeletal structure compared to contractile cells, and thus are susceptible to damage from processing steps, particularly in single-cell suspension where they lack cell–cell contact that occurs in robust neurosphere cultures or in vivo.59 MSCs, on the other hand, readily spread on tissue culture plates and do not require significant cell–cell contact to survive.60 Furthermore, while both NSCs and MSCs have potential in therapeutic transplantation, MSCs are more widely studied as a candidate for cell therapy, due to their immunosuppressant abilities and trophic factor secretion61,62 and are employed in >1000 clinical trials.63 Therefore, towards assessing the generality of our encapsulation approach, we encapsulated MSCs in up to 4 layers of HA and assessed the effect of the conformal hydrogel on MSCs, as above for NSCs.MSCs were encapsulated using the same protocol as for NSCs. Up to 4 layers of encapsulations were established, where lipHASH was used as the first layer, RhoB-tagged MaHA as the second, HASH as the third, and MaHA as the fourth layer. Again, we observed fluorescence around MSCs after forming a 2 layered conformal hydrogel on their surfaces (Fig. 4A). When we looked at the fraction of cells encapsulated over time (Fig. 4B), we observed a faster rate of MSC egress from 3 layers and 4 layers of conformal encapsulations compared to NSCs. Interestingly, for 2-layer encapsulations, though MSCs started spreading as early as day 1, fluorescent coating remained on the surfaces of adherent cells. This indicates that MSCs were able to spread in the presence of the 2-layer conformal coating, perhaps spreading within the coating, through the coating, or in the presence of a coating which is partial disrupted (Fig. 4C).
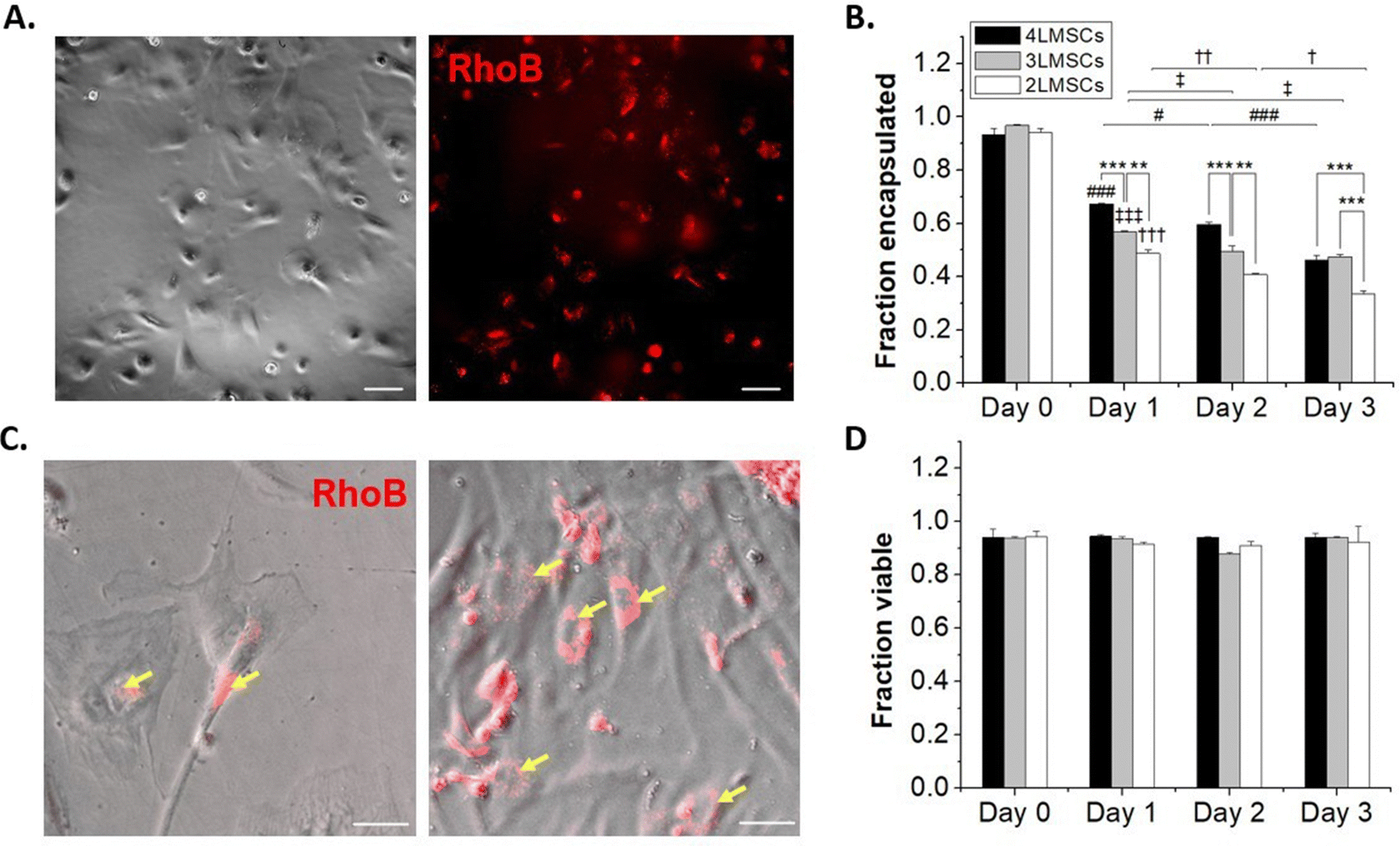 | ||
| Fig. 4 (A) MSCs encapsulated in lipHASH and then RhoB-tagged MaHA. (B) The fraction of MSCs that remained encapsulated over 3-day culture periods. (C) MSCs spread with MaHA still coating portions of the cell membrane. Yellow arrow denotes where MaHA localizes on the MSCs. (D) The fraction of MSCs that remained viable over 3-day encapsulation periods, no statistical significance were detected. * Denotes significant differences within groups, #, ‡, and † denotes significant differences of 2-layer, 3-layer, and 4-layer-encapsulated MSCs compared to Day 0, respectively. There are at least 3 replicates for each group, and for each replicate, at least 472 cells were counted. A total of 14103 cells and 12198 cells were used to assess encapsulation fraction and viability fraction, respectively. Two-way ANOVA and Tukey's Post hoc tests were run for statistical testing. All scale bars = 50 μm. | ||
To further investigate the dynamic of cell egress, we tracked the movement of MSCs encapsulated with 2 layers of material (Movie S1, ESI†). We saw that encapsulated MSCs (visualized by rhoB-conjugated MaHA) were able to spread as soon as 30 minutes after plating. However, we also saw that for some cells, the encapsulation prevented immediate adhesion to the plate. These cells underwent increased blebbing before adhering to the plate (indicated by yellow arrows in the movie). The encapsulating material, however, did not detach from the cell surface. Rather, the materials were rearranged and conformed to MSC movements. This observation is in line with previous work on MSC encapsulations.28 We also observed that while 4 layers of encapsulation prevented NSCs from egressing, it did not have the same effect on MSCs. The MSCs were able to spread and egress even from a 4-layer conformal encapsulation. Notably, we saw that the MSCs maintained high viability across groups and that viability did not significantly drop over multiple days in culture (Fig. 4D).
3.4. Assembly and photopatterning of encapsulated NSCs
To demonstrate the potential to leverage conformal encapsulation for multicellular assemblies, we utilized the same hydrogel materials used in the conformal encapsulation to generate multicellular clusters and to pattern cells onto a substrate with a resolution of 100 μm using photolithograpy. To direct the assembly of multicellular clusters, we used the complementary thiol-maleimide chemistry to organize multiple cells around a single cell. In one population of NSCs, we created a conformal coating in which the outer layer was lipHASH (thiol-NSCs), containing unreacted thiols. In a second population of NSCs, we assembled a coating whose outer layer was MaHA (mal-NSCs) that contained free maleimides. By mixing these populations in suspension with thiol-NSCs in excess, then centrifuging to bring them together, we were able to create assemblies of thiol-NSCs surrounding mal-NSCs in which the cells were in close proximity and held together by covalent bonds between conformal coatings in directed assembly (Fig. 5A). To assess the specificity of the interaction, we quantified the fraction of mal-NSCs and thiol-NSCs that formed clusters >2 cells. In the mixture of mal-NSCs with excess thiol-NSCs, almost all mal-NSCs formed clusters with thiol-NSCs. Thiol-NSCs, on the other hand, rarely aggregated to each other (Fig. 5B), confirming that maleimide-thiol reaction directed the assembly of NSCs. The assembled construct was stable and withstood vigorous mixing using a handheld pipette.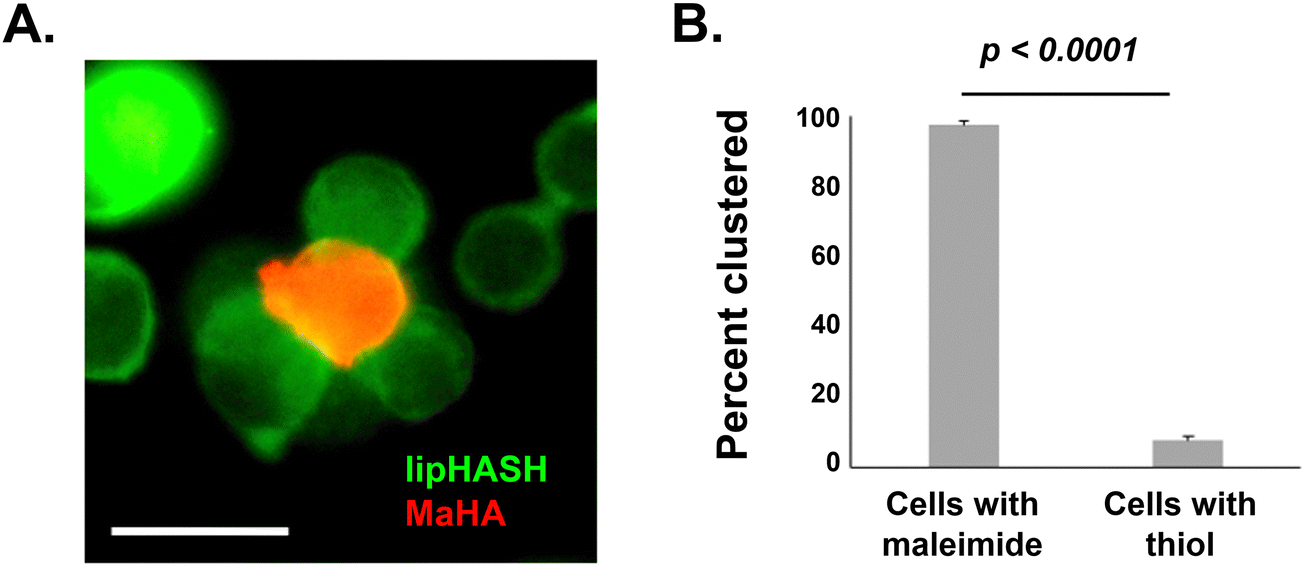 | ||
| Fig. 5 (A) NSCs encapsulated with MaHA (red cells in the center) were assembled together with excess lipHASH-encapsulated NSCs (green cells on the outside). Scale bar = 20 μm. (B) Significantly more maleimide-presenting cells clustered with thiol-presenting cells (97.5 ± 3.5%) than thiol-presenting cells with other thiol-presenting cells (7.67 ± 2.6%). A total of n = 1520 cells were used to perform statistical testing with student's t-test. | ||
Photolithography is often used to exert spatiotemporal control over properties of hydrogels that contain photoactive chemical functionalities. Photolithographic modification of a hydrogel offers high-resolution spatial control over crosslinking (e.g. photo-triggered crosslinking), degradation (e.g. using photolabile bonds), and presentation of bioactive molecules.64 Capabilities that enable cells to be assembled at resolutions that can be attained in light-based photopatterning, which include the length scales of single cells, may be useful in studying basic biology and in fabricating tissue structures.65,66 Because we could prepare conformal encapsulations that present reactive thiols to the surrounding environment, we investigated the photopatterning of thiol-presenting cells onto norHA hydrogels fabricated with remaining unreacted norbornene pendant groups.67 To demonstrate the ability to control the positions of conformally encapsulated cells, we encapsulated NSCs in a conformal hydrogel with an outermost layer of HASH or lipHASH. The process for photopatterning encapsulated NSCs is schematically illustrated in Fig. 6A. Briefly, we prepared the norHA substrate as a thin gel conjugated to a thiolated glass slide, with the upper surface of the hydrogel presenting unreacted norbornene groups.58 We incubated the cells in a dense suspension containing photoinitiator on top of the gel, then used a photomask placed on top of the cells to spatially control light exposure in 100 μm wide stripes. The photoinitiated thiol–ene reaction thus crosslinked the conformally encapsulated cells to the norHA substrate in 100 μm wide stripes with equal spacing, demonstrating the specificity and resolution of the interaction (Fig. 6B), the intensity profile across the striped pattern showed that the distance between patterns is ∼100 μm in size.
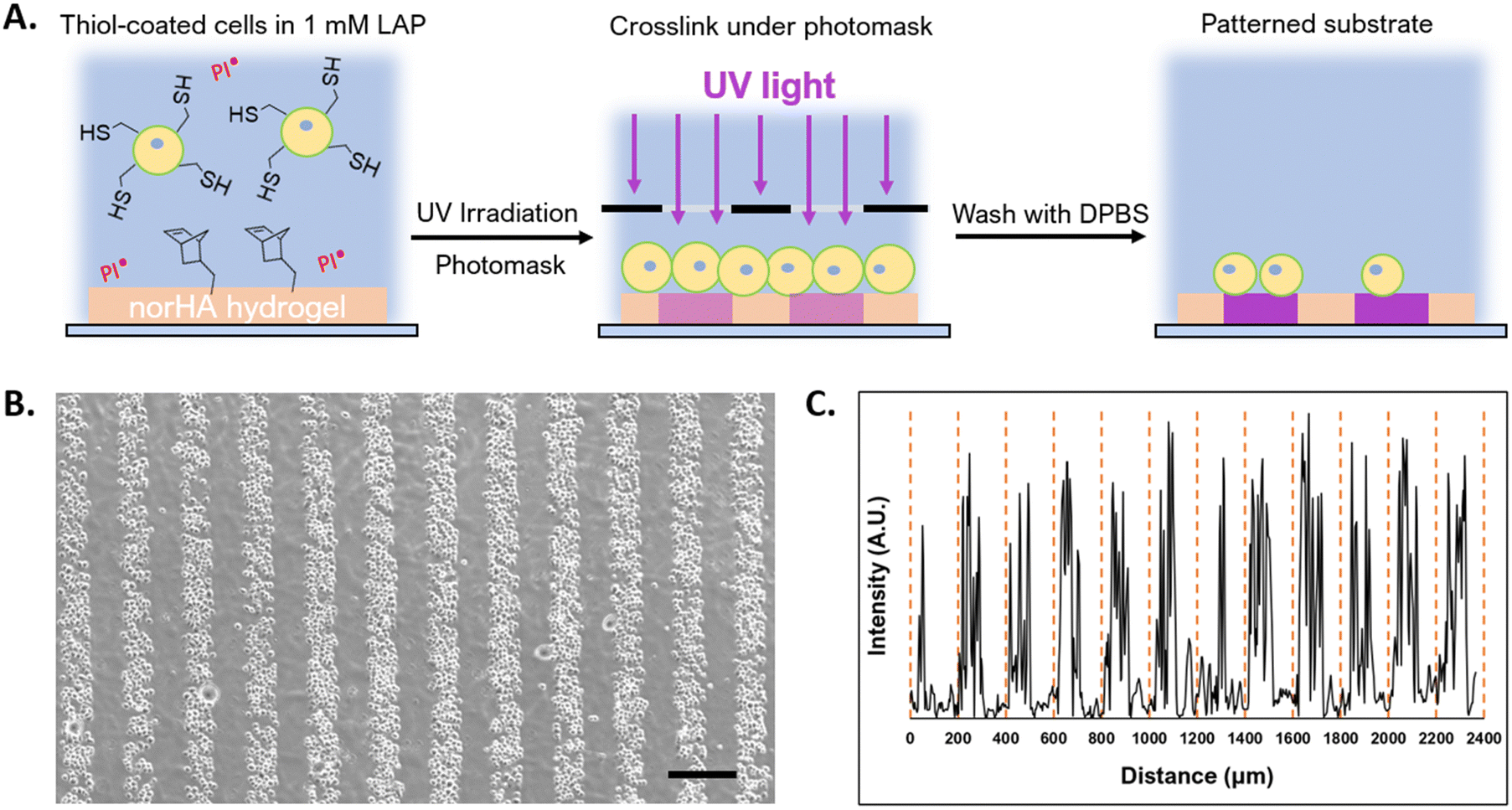 | ||
| Fig. 6 (A) Schematic of the photopatterning procedure, thiolated cells are allowed to settle on the norHA gel (orange in schematic) with a photoinitiator (LAP) present, a photomask (black slit in schematic) was then fitted to the surface of the solution. NSCs are then UV crosslinked to the substrate for 1 min, the striped pattern (purple in schematic) remained after washing with PBS. (B) NSCs encapsulated in lipHASH then photopatterned onto norHA substrate. Scale bar = 200 μm. (C) The intensity profile of the photopatterned substrate was quantified using imageJ, the distance between the yellow dashes is 200 μm. | ||
4. Discussion
The conformal encapsulation system we studied here was based on HA, which can be used to design hydrogels for many applications.53 We used a lipid modification to localize an initial layer of the HA material to the cell surface. This approach, based on the interaction between the pendant phospholipid and the cell membrane, has been used with synthetic glycopolymers,68 DNA,43 and PEG46,69 and appears generalizable for localizing material onto cell membranes. For our lipid material, our synthesis method allows us to control the degree of substitution for both lipid and thiol pendant groups, making it tunable: we observed that the degree of substitution (DoS) for lipid could be increased up to 5% before lipid-HA became almost insoluble, and we were able to achieve a DoS for thiol functionality of up to 40%. Concurrently, the use of a glycosaminoglycan (HA) reduced concerns about cytotoxicity present in some conformal encapsulation systems, as it is sometimes seen in synthetic polyelectrolyte-based encapsulations that are formed layering processes similar to the one used here.Using this new material system, we chose to encapsulate progenitor cells – NSCs and MSCs – towards the development of materials platforms that might ultimately enhance cell delivery or support cell survival and phenotype in therapies, as delivery of naïve cells through a needle often associates with low viability.5 With respect to NSCs, interactions with HA through the CD44 receptor play important roles in maintaining NSC stemness and differentiation.70,71 HA has also been shown to influence MSC phenotype in cell therapy for cartilage regeneration.72,73 Further material design to include instructive cues is possible using the same conjugation methods as those used to introduce fluorophores. We showed NSCs could be encapsulated in minimal coatings with high viability, although we observed significant decreases of viability in 4-layer conformal encapsulations in later time points: at 1 day and beyond. However, encapsulating NSCs as spheroids negated decreases in viability, suggesting that lack of cell–cell interaction when NSCs are encapsulated as single cells contributed to the loss of viability.
MSCs exhibited higher viability and tendency to escape encapsulation with time (Movie S1, ESI†) compared to NSCs, with NSCs are observed to grow beyond the limits of the encapsulating material only in the 2 layer coated spheroid group. The increased ability of MSCs to egress is likely due to differences in phenotype related to motility, where MSCs are more likely than NSCs to escape from defects in the nanoencapsulating hydrogel that appear as the result of hydrogel network degradation either by hydrolysis (Fig. S5, ESI†) or enzymatic activity. We did not optimize our system for long-term encapsulation. Cytoprotective and short-term surface modification may be exploited in certain applications, such as cell printing, where cells must be shielded from acute stress imposed by the extrusion process; and cellular assembly or patterning, where surface modification is needed for the initial positioning of cells. Additionally, expedient cell egress from encapsulation, as well as the ability for encapsulated cells to interact with their microenvironment and other cells, may prove beneficial in cell delivery applications, where therapeutic benefits rely on the activity of the delivered cells.
Studies of conformal encapsulations have demonstrated they convey cytoprotection from mechanical stresses, including shear during delivery by injection or extrusion in bioprinting applications.27,47,74 Towards biofabrication, while the work presented here did not directly evaluate mechanical protection, we showed the reactive moieties on the outer layer of encapsulation can be utilized to spatially assemble cells. Microtissues assembled through complementary oligonucleotides coatings have been demonstrated;43,75 here, our system offers similar capabilities, albeit limited to one complementary interaction: thiol-maleimide. The conformal hydrogel offers the potential for further cues to be engineered into the extracellular environments of assembled cells through HA modification and hydrogel design. Rapid elimination of engineered material may be desirable as cells begin to deposit their own ECM67 and as cells or microtissues establish interactions with their surroundings. Additionally, external cues engineered into coatings might be used to direct delivery of cells to, or influence responses by, the surrounding tissue into which cells and microtissues are implanted.
The reactive moieties, such as the thiol functionality used in this study, also allow encapsulated cells to be patterned onto substrates with complementary chemistry. In comparison to other approaches, such as cell adherence to patterned RGD islands, in this paper we utilized photochemistry to achieve cell attachment over a short period of time: 10 minutes for cells to settle to the substrate surface then 1 minute of crosslinking. With the use of a photomask, this method potentially allows for the patterning of multiple cell types onto overlapping shapes. This method can be carried out in culture medium at 37 °C, so that the cells remain in controlled and supportive culture conditions at all times. Going forward, the patterned cells might serve as another substrate for subsequent reactions; for example, thiol-coated cells patterned onto the norHA substrate can serve as reaction sites for the addition of maleimide-coated cells, allowing complex, cell-dense structures to be assembled.
5. Conclusions
Conformal hydrogels enable minimal volumes of engineered material to be used to protect encapsulated cells and influence phenotype and fate. These systems have potential applications in cell delivery, biofabrication, and systems designed to control interactions of the cells with their immediate surroundings or influence the interactions of the surroundings with the cell via the encapsulation material. The encapsulation system presented here featured an HA-based system that utilized a dual-modified HA, lipHASH, to encapsulate NSCs, for which few encapsulation systems have been demonstrated, and MSCs. We showed that the interactions of cells with the conformal hydrogel encapsulation was dependent both on cell type and material design, with the duration of cell encapsulation and cell viability being modulated by hydrogel properties. We also showed that conformal encapsulations could be used to direct cellular aggregation into multicellular assemblies and organization into designed, high-resolution patterns using photolithography. Given the extent of functionality which can be engineered into soft biomaterials, such as HA-based hydrogels, these systems offer opportunities for using biomaterials approaches to influence cell fate within minimal material environments, at the resolution of individual cells. This hydrogel represents a new, versatile material system that might be used in conformal encapsulations in the delivery of cells in cell-based therapies, in the assembly of cells 3D systems for expansion or culture in vitro, in directing interactions with tissue environments in vivo, and in assembling complex, cell-dense tissue constructs.Conflicts of interest
This author declares no conflicts of interest.Acknowledgements
This work was funded by National Institute of General Medical Sciences at the National Institutes of Health through grant R35GM147410 and the National Science Foundation through Future Manufacturing Seed grant 2036968.References
- G. D. Nicodemus and S. J. Bryant, https://home.liebertpub.com/teb, 2008, 14, 149–165.
- J. L. Wilson and T. C. Mcdevitt, Biotechnol. Bioeng., 2013, 110, 667–682 CrossRef CAS PubMed.
- O. Hasturk and D. L. Kaplan, Acta Biomater., 2019, 95, 3–31 CrossRef CAS.
- H. Lee, N. Kim, H. B. Rheem, B. J. Kim, J. H. Park and I. S. Choi, Adv. Healthcare Mater., 2021, 10, 2100347 CrossRef CAS.
- B. A. Aguado, W. Mulyasasmita, J. Su, K. J. Lampe and S. C. Heilshorn, Tissue Eng., Part A, 2012, 18, 806–815 CrossRef CAS PubMed.
- J. Lam, W. E. Lowry, S. T. Carmichael and T. Segura, Adv. Funct. Mater., 2014, 24, 7053–7062 CrossRef CAS.
- C. M. Madl, B. L. Lesavage, R. E. Dewi, C. B. Dinh, R. S. Stowers, M. Khariton, K. J. Lampe, D. Nguyen, O. Chaudhuri, A. Enejder and S. C. Heilshorn, Nat. Mater., 2017, 16, 1233–1242 CrossRef CAS PubMed.
- C. M. Madl, B. L. LeSavage, R. E. Dewi, K. J. Lampe and S. C. Heilshorn, Adv. Sci., 2019, 6, 1801716 CrossRef.
- R. G. Duff, Microencapsulation technology: A novel method for monoclonal antibody production, 1985.
- G. M. O’Shea and A. M. Sun, Diabetes, 1986, 35, 943–946 CrossRef.
- M. Y. Fan, Z. P. Lum, X. W. Fu, L. Levesque, I. T. Tai and A. M. Sun, Diabetes, 1990, 39, 519–522 CrossRef CAS.
- A. J. Hwa and G. C. Weir, Transplantation of Macroencapsulated Insulin-Producing Cells, Curr. Diab. Rep., 2018, 18, 50 CrossRef PubMed.
- G. Romero, J. J. Lilly, N. S. Abraham, H. Y. Shin, V. Balasubramaniam, T. Izumi and B. J. Berron, ACS Appl. Mater. Interfaces, 2015, 7, 17598–17602 CrossRef CAS.
- P. J. Wu, H. Peng, C. Li, A. Abdel-Latif and B. J. Berron, ACS Appl. Bio Mater., 2020, 3, 2930–2939 CrossRef CAS.
- H. F. Ding, R. Liu, B. G. Li, J. R. Lou, K. R. Dai and T. T. Tang, Biochem. Biophys. Res. Commun., 2007, 362, 923–927 CrossRef CAS.
- A. J. Ryan, H. S. O’Neill, G. P. Duffy and F. J. O’Brien, Curr. Opin. Pharmacol., 2017, 36, 66–71 CrossRef CAS.
- G. Basta and R. Calafiore, Curr. Diabetes Rep., 2011, 11, 384–391 CrossRef CAS.
- A. A. Tomei, V. Manzoli, C. A. Fraker, J. Giraldo, D. Velluto, M. Najjar, A. Pileggi, R. D. Molano, C. Ricordi, C. L. Stabler and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10514–10519 CrossRef CAS.
- J. L. Lilly, G. Romero, W. Xu, H. Y. Shin and B. J. Berron, Biomacromolecules, 2015, 16, 541–549 CrossRef CAS PubMed.
- M. Germain, P. Balaguer, J. C. Nicolas, F. Lopez, J. P. Esteve, G. B. Sukhorukov, M. Winterhalter, H. Richard-Foy and D. Fournier, Biosens. Bioelectron., 2006, 21, 1566–1573 CrossRef CAS PubMed.
- S. Zhao, L. Zhang, J. Han, J. Chu, H. Wang, X. Chen, Y. Wang, N. Tun, L. Lu, X. F. Bai, M. Yearsley, S. Devine, X. He and J. Yu, ACS Nano, 2016, 10, 6189–6200 CrossRef CAS.
- S. Krol, S. Del Guerra, M. Grupillo, A. Diaspro, A. Gliozzi and P. Marchetti, Nano Lett., 2006, 6, 1933–1939 CrossRef CAS PubMed.
- M. Borkowska, E. Godlewska, M. Antosiak-Iwanska, J. Kinasiewicz, M. Borkowska, E. Godlewska, M. AntosiakIwanskaiwanska, J. Kinasiewicz, M. Strawski, M. Szklarczyk and L. H. Granicka, J. Biomed. Nanotechnol., 2012, 8, 1–6 CrossRef.
- S. Mansouri, Y. Merhi, F. M. Winnik and M. Tabrizian, Biomacromolecules, 2011, 12, 585–592 CrossRef CAS PubMed.
- W. Li, T. Guan, X. Zhang, Z. Wang, M. Wang, W. Zhong, H. Feng, M. Xing and J. Kong, ACS Appl. Mater. Interfaces, 2015, 7, 3018–3029 CrossRef CAS.
- J. Hwang, D. Choi, M. Choi, Y. Seo, J. Son, J. Hong and J. Choi, ACS Appl. Mater. Interfaces, 2018, 10, 17685–17692 CrossRef CAS PubMed.
- J. Yang, J. Li, X. Li, X. Wang, Y. Yang, N. Kawazoe and G. Chen, Biomaterials, 2017, 133, 253–262 CrossRef CAS PubMed.
- D. Choi, H. Lee, H.-B. Kim, M. Yang, J. Heo, Y. Won, S. Soon Jang, J. Kuk Park, Y. Son, T. In Oh, E. Lee and J. Hong, Chem. Mater., 2017, 29, 2055–2065 CrossRef CAS.
- Y. Fukuda, T. Akagi, T. Asaoka, H. Eguchi, K. Sasaki, Y. Iwagami, D. Yamada, T. Noda, K. Kawamoto, K. Gotoh, S. Kobayashi, M. Mori, Y. Doki and M. Akashi, Biomaterials, 2018, 160, 82–91 CrossRef CAS.
- Y. Sasano, K. Fukumoto, Y. Tsukamoto, T. Akagi and M. Akashi, J. Biosci. Bioeng., 2020, 129, 749–755 CrossRef CAS.
- I. Koh, I. Yong, B. Kim, D. Choi, J. Hong, Y. M. Han and P. Kim, ACS Biomater. Sci. Eng., 2020, 6, 813–821 CrossRef CAS PubMed.
- S. Sakai and M. Taya, ACS Macro Lett., 2014, 3, 972–975 CrossRef CAS PubMed.
- M. R. Dzamukova, E. A. Naumenko, E. V. Rozhina, A. A. Trifonov and R. F. Fakhrullin, Nano Res., 2015, 8, 2515–2532 CrossRef CAS.
- J. H. Park, K. Kim, J. Lee, J. Y. Choi, D. Hong, S. H. Yang, F. Caruso, Y. Lee and I. S. Choi, Angew. Chem., Int. Ed., 2014, 53, 12420–12425 CrossRef CAS PubMed.
- J. Lee, J. Choi, J. H. Park, M.-H. Kim, D. Hong, H. Cho, S. H. Yang and I. S. Choi, Angew. Chem., Int. Ed., 2014, 53, 8056–8059 CrossRef CAS PubMed.
- B. J. Kim, T. Park, H. C. Moon, S. Y. Park, D. Hong, E. H. Ko, J. Y. Kim, J. W. Hong, S. W. Han, Y. G. Kim and I. S. Choi, Angew. Chem., Int. Ed., 2014, 53, 14443–14446 CrossRef CAS.
- J. Yang, Y. Yang, N. Kawazoe and G. Chen, Biomaterials, 2019, 197, 317–326 CrossRef CAS.
- Y. Teramura, O. P. Oommen, J. Olerud, J. Hilborn and B. Nilsson, Biomaterials, 2013, 34, 2683–2693 CrossRef CAS.
- M. Matsusaki, K. Kadowaki, Y. Nakahara and M. Akashi, Angew. Chem., Int. Ed., 2007, 46, 4689–4692 CrossRef CAS PubMed.
- K. Kadowaki, M. Matsusaki and M. Akashi, Langmuir, 2010, 26, 5670–5678 CrossRef CAS.
- S. Toda, A. Fattah, K. Asawa, N. Nakamura, K. N. Ekdahl, B. Nilsson and Y. Teramura, Micromachines, 2019, 10, 755 CrossRef.
- A. A. Stock, V. Manzoli, T. De Toni, M. M. Abreu, Y. C. Poh, L. Ye, A. Roose, F. W. Pagliuca, C. Thanos, C. Ricordi and A. A. Tomei, Stem Cell Rep., 2020, 14, 91–104 CrossRef CAS PubMed.
- Z. J. Gartner and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4606–4610 CrossRef CAS PubMed.
- J. S. Liu and Z. J. Gartner, Trends Cell Biol., 2012, 22, 683–691 CrossRef CAS.
- N. G. Veerabadran, P. L. Goli, S. S. Stewart-Clark, Y. M. Lvov and D. K. Mills, Macromol. Biosci., 2007, 7, 877–882 CrossRef CAS PubMed.
- S. Miura, Y. Teramura and H. Iwata, Biomaterials, 2006, 27, 5828–5835 CrossRef CAS PubMed.
- A. S. Mao, J. W. Shin, S. Utech, H. Wang, O. Uzun, W. Li, M. Cooper, Y. Hu, L. Zhang, D. A. Weitz and D. J. Mooney, Nat. Mater., 2017, 16, 236–243 CrossRef CAS PubMed.
- A. S. Mao, B. Özkale, N. J. Shah, K. H. Vining, T. Descombes, L. Zhang, C. M. Tringides, S. W. Wong, J. W. Shin, D. T. Scadden, D. A. Weitz and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15392–15397 CrossRef CAS.
- Z. L. Zhi, B. Liu, P. M. Jones and J. C. Pickup, Biomacromolecules, 2010, 11, 610–616 CrossRef CAS PubMed.
- D. Hachim, J. Melendez and R. Ebensperger, J. Encapsulation Adsorpt. Sci., 2013, 03, 1–12 CrossRef.
- G. Marchioli, L. Zellner, C. Oliveira, M. Engelse, E. D. Koning, J. Mano Karperien, A. V. Apeldoorn and L. Moroni, J. Mater. Sci.: Mater. Med., 2017, 28, 195 CrossRef PubMed.
- C. B. Highley, G. D. Prestwich and J. A. Burdick, Curr. Opin. Biotechnol, 2016, 40, 35–40 CrossRef CAS.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41–H56 CrossRef CAS PubMed.
- S. Sahoo, C. Chung, S. Khetan and J. A. Burdick, Biomacromolecules, 2008, 9, 1088–1092 CrossRef CAS PubMed.
- J. L. Holloway, H. Ma, R. Rai and J. A. Burdick, J. Controlled Release, 2014, 191, 63–70 CrossRef CAS PubMed.
- C. B. Rodell, J. W. MacArthur, S. M. Dorsey, R. J. Wade, L. L. Wang, Y. J. Woo and J. A. Burdick, Adv. Funct. Mater., 2015, 25, 636–644 CrossRef CAS PubMed.
- W. M. Gramlich, I. L. Kim and J. A. Burdick, Biomaterials, 2013, 34, 9803–9811 CrossRef CAS PubMed.
- M. G. Grewal, V. P. Gray, R. A. Letteri and C. B. Highley, Biomater. Sci., 2021, 9, 4374–4387 RSC.
- L. S. Campos, J. Neurosci. Res., 2004, 78, 761–769 CrossRef CAS PubMed.
- D. Mushahary, A. Spittler, C. Kasper, V. Weber and V. Charwat, Cytometry, Part A, 2018, 93, 19–31 CrossRef CAS PubMed.
- X. Wei, X. Yang, Z. P. Han, F. F. Qu, L. Shao and Y. F. Shi, Acta Pharmacol. Sin., 2013, 34, 747–754 CrossRef CAS PubMed.
- B. J. Jones and S. J. McTaggart, Exp. Hematol., 2008, 36, 733–741 CrossRef CAS PubMed.
- J. Galipeau and L. Sensébé, Cell Stem Cell, 2018, 22, 824–833 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- M. E. Todhunter, N. Y. Jee, A. J. Hughes, M. C. Coyle, A. Cerchiari, J. Farlow, J. C. Garbe, M. A. LaBarge, T. A. Desai and Z. J. Gartner, Nat. Methods, 2015, 12, 975–981 CrossRef CAS PubMed.
- X. Zhou, H. Wu, H. Wen and B. Zheng, Micromachines, 2022, 13, 80 CrossRef PubMed.
- C. Loebel, R. L. Mauck and J. A. Burdick, Nat. Mater., 2019, 18, 883–891 CrossRef CAS PubMed.
- D. Rabuka, M. B. Forstner, J. T. Groves and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 5947–5953 CrossRef CAS PubMed.
- K. Tatsumi, K. Ohashi, Y. Teramura, R. Utoh, K. Kanegae, N. Watanabe, S. Mukobata, M. Nakayama, H. Iwata and T. Okano, Biomaterials, 2012, 33, 821–828 CrossRef CAS PubMed.
- W. Su, S. C. Foster, R. Xing, K. Feistel, R. H. Olsen, S. F. Acevedo, J. Raber and L. S. Sherman, J. Biol. Chem., 2017, 292, 4434–4445 CrossRef CAS PubMed.
- Z. Z. Khaing and S. K. Seidlits, J. Mater. Chem. B, 2015, 3, 7850–7866 RSC.
- L. Bian, M. Guvendiren, R. L. Mauck and J. A. Burdick, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10117–10122 CrossRef CAS PubMed.
- M. Y. Kwon, C. Wang, J. H. Galarraga, E. Puré, L. Han and J. A. Burdick, Biomaterials, 2019, 222, 119451 CrossRef CAS PubMed.
- A. Matsuzawa, M. Matsusaki and M. Akashi, Langmuir, 2013, 29, 7362–7368 CrossRef CAS PubMed.
- M. E. Todhunter, N. Y. Jee, A. J. Hughes, M. C. Coyle, A. Cerchiari, J. Farlow, J. C. Garbe, M. A. Labarge, T. A. Desai, Z. J. Gartner, M. E. T. Conceived and J. C. G. Performed, Nat. Methods, 2015, 12, 975–981 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00223g |
| This journal is © The Royal Society of Chemistry 2024 |