
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUpper rim-bridged calixarenes
Pavel Lhoták *
*
Department of Organic Chemistry, University of Chemistry and Technology, Prague (UCTP), Technická 5, 166 28 Prague 6, Czech Republic. E-mail: lhotakp@vscht.cz
First published on 24th July 2024
Abstract
Calix[n]arenes represent a very attractive family of macrocyclic compounds with many potential supramolecular applications. Due to their well-established chemistry and many different synthetic strategies, enabling practically any derivatization of the basic skeleton, calixarenes are among the very popular building blocks used for the design and construction of various receptors, sensors and other sophisticated supramolecular systems. Regio- and/or stereo-selective derivatization of calixarenes currently represents a very extensive set of reactions, the overview of which would fill many thick books. Therefore, this review deals with only a small part of the above-mentioned reactions, specifically describes possible ways of bridging the upper rim of calixarenes, often leading to interesting rigidified structures, and also briefly mentions the potential use of these compounds.
Preface
Although modern supramolecular chemistry uses a wide range of macrocyclic compounds due to their increased preorganization compared to noncyclic analogues, calixarenes occupy an absolutely essential position in this context.1 Above all, they enable a simple multigram preparation from cheap starting materials (Fig. 1), while providing the option of choosing the size of the cavity depending on the reaction conditions. The resulting macrocycles (calix[4]arene to calix[8]arenes) then show considerable variability in terms of their subsequent derivatization, enabling the introduction of suitable functional groups into selected positions on the basic skeleton.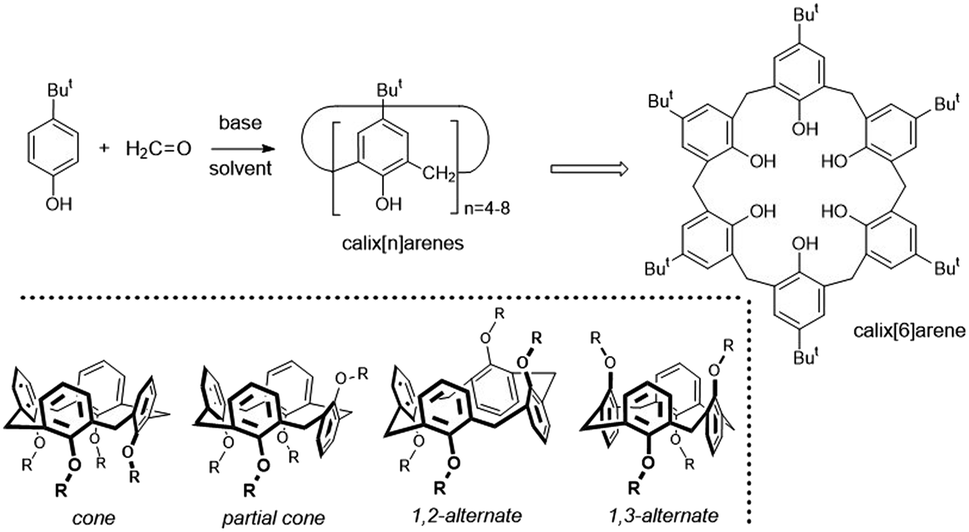 | ||
| Fig. 1 Preparation of calix[n]arenes; calix[8]arene as an example of the structure; four basic conformations (atropisomers) of calix[4]arene. | ||
Calixarenes are therefore almost ideal candidates for the design and synthesis of various receptors intended for catching cations, anions or neutral molecules (depending on the substitution of the macrocycle).2 If we focus on the smallest of them, calix[4]arene, we have a unique possibility to “tune” the three-dimensional structure of the molecule by simple alkylation/acylation of phenolic hydroxyls. It is the possibility to fix the basic skeleton in one of the four basic conformations (atropisomers) that makes these molecules indispensable as molecular scaffolds in the design of new receptors and building blocks. It is precisely the cone conformation of calix[4]arene, with a shape reminiscent of the ancient calix crater vase (Fig. 2), to which we owe the general name of this family of macrocycles.1b
 | ||
| Fig. 2 Lower rim and upper rim of calix[n]arenes and the upper rim-bridged derivatives (schematically). | ||
Calix[4]arenes in particular have become very popular building blocks for the design and synthesis of new receptors, with an extensive literature on “shaping” their cavity through conformational immobilization. In this context, by far the most common methods are simple alkylation/acylation (including bridging) of the phenolic functions of calixarenes (the so-called lower rim). As the title suggests, this review is only focused on derivatives with a bridged upper rim, a topic that, to the best of my knowledge, has not been covered before.
Although the introduction of substituents into the lower rim of the macrocycle leads to immobilization of the system (in the case of calix[4]arenes derivatives), at the same time it excludes the possibility of targeted use of free phenolic groups, either for subsequent derivatization or for relevant supramolecular applications. In addition, alkylation of the lower rim only works for calix[4]arene, whereas higher macrocycles cannot be fixed in this simple way. Therefore, it may be interesting to turn attention to the opposite side of calixarenes – the aromatic part of the molecule, usually called the upper rim (Fig. 2).
This part is primarily amenable to various types of aromatic electrophilic substitution, such as halogenation, acylation, nitration, sulfonation, etc. The chemistry of the upper rim of calixarenes therefore offers a much more diverse range of downstream derivatizations than that of the lower rim. A suitable bridging of the upper rim therefore leads to a rigidification of the molecule without the need for the substitution of phenolic groups, which are thus still available.
Furthermore, dimerization of calixarenes by linking their upper rims leads to larger cavities that are preorganized for various predominantly supramolecular applications. The combination of suitable functional groups together with aromatic cavities of macrocycles gives rise to interesting receptors capable of complexation using a whole range of non-covalent interactions. Simple bridging of calixarene leads to confined spaces, enabling complexation of the guest species within the rigidified cavity. Last but not least, upper-rim meta-bridging of calixarenes gives rise to a new class of (often inherently chiral) calixarene derivatives with an extremely rigid and distorted cavities, with possible applications in chiral recognition of suitable guests.
Because the number of reports dealing with calixarenes is huge, only selected examples of upper rim-bridged systems are described in this review. As shown schematically in Fig. 2, these are systems with different aromatic subunits arbitrarily connected by a spacer(s) or by a direct bond. In addition, selected systems based on bis-calixarenes, which are connected to each other by upper rims, are also included (see e.g. Fig. 4). However, it should be noted that this overview is not an exhaustive summary of all existing systems. Instead, it is intended to serve more as a guide, showing possible approaches to the synthesis of this interesting subgroup of calix[n]arene derivatives. The review is focused on demonstrating numerous synthetic approaches leading to bridging of the upper rim, so emphasis is placed on synthetic procedures and the selection of suitable starting systems, rather than on the actual use of compounds prepared in this way. Potential applications of bridged systems are usually only briefly indicated, if interested, the reader is referred to the original primary literature for a full description.
Overview of possible strategies
Condensation of suitable fragments
The oldest approach to the upper rim-bridged calix[4]arenes is based on the condensation of suitably substituted starting phenols as indicated in Fig. 3.3 The condensation of bisphenol 1 with 2,6-bis(bromomethy)-4-methylphenol 2 under Lewis acid (TiCl4) catalysis led directly to macrocycle 3. Interestingly, the authors found that it is not necessary to use high dilution conditions for the macrocyclization reaction as the desired calixarene 3 (R = Me, n = 8, Y = H) was isolated in 20% yield when the reaction was carried out at normal concentrations (30 h at 100 °C). Alkali metal ions transport experiments (from an alkaline source – phase through a liquid membrane to a neutral receiving phase) with Cs+ ions showed a sharp maximum of the transport ability for this compound (n = 8), suggesting complexation of the caesium inside the cavity. | ||
| Fig. 3 The first example of upper rim-bridged calix[4]arene. | ||
The conformational behaviour of similar systems 3 possessing different bridges (Fig. 3) was studied by CIDNP NMR technique4 and by single crystal X-ray analysis.5 Both techniques confirmed that the macrocycles adopt a cone conformation both in the solid state and in solution. A wide range of derivatives bearing various substituents on the unconnected phenolic units (3, R = methyl, octyl, dodecyl, octadecyl, tert-butyl, cyclohexyl, phenyl and chloro groups) was prepared6 by the aforementioned synthetic approach to show its general applicability. The yields of products were usually in the range of 5–25%, occasionally reaching 35–40%.7 The lower limit for the length of the bridge seems to be n = 5.
On the other hand, a unique cage molecule 4 was isolated in very low yield8 by the condensation of bisphenol 1 possessing a shorter spacer (n = 4) with bis-bromomethyl derivative 2 (Fig. 4).
 | ||
| Fig. 4 Calixarene cages by fragment condensation. | ||
A similar approach based on the tetrakis-bromomethylated compound 2a gave a fully bridged cage system 5, but again only in very low yield. In this case, a longer spacer was used for both reaction components 1 and 2a (n = 10) – see Fig. 4.8 Using the aforementioned strategy, even calix[4]arenes with bridged neighbouring aromatic groups have been reported, yet without any experimental details.7
Intramolecular linking via ether/thioether or ester functions
While bridging of lower rim with polyether chain is very common procedure in calixarene chemistry (giving rise to so called calixcrowns), analogous derivatives bridged on the upper rim are very rare. One of the examples is the alkylation of oligoethylene glycols 7 with the bis-chloromethyl compound 6 in refluxing THF in the presence of NaI and NaH which gave the corresponding bridged products 8a–e in 11–95% yields (Fig. 5), strictly depending on the length of the glycol.9 As the lower rim was alkylated with methyl groups, the resulting macrocycles are not conformationally rigidified and the upper rim crowns 8a–e were used for study of conformational interconversions between the individual conformers (cone – partial cone – 1,3-alternate) formed by the rotation of unbridged aromatic subunits.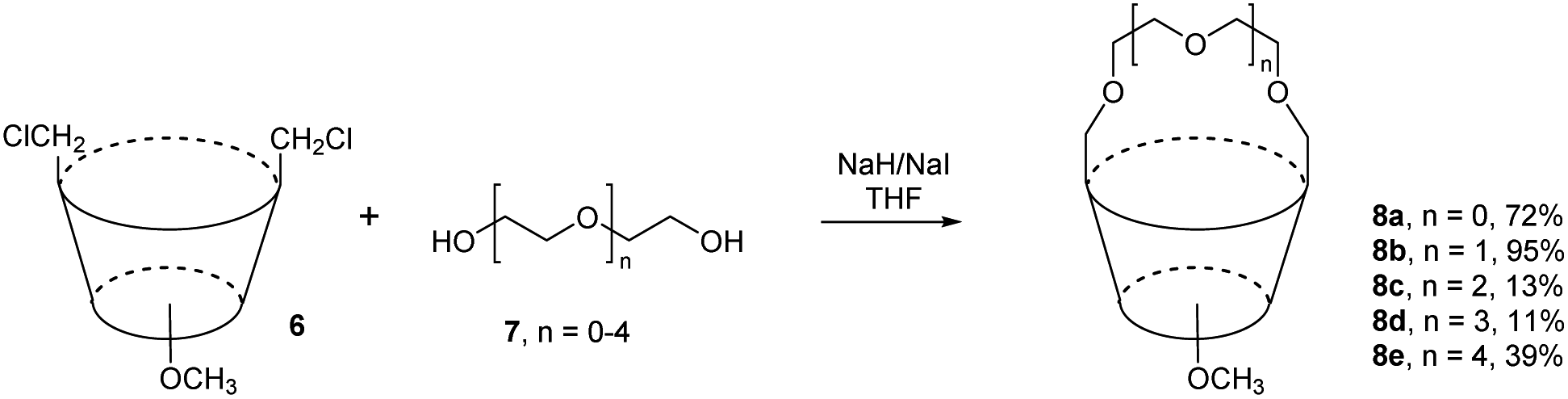 | ||
| Fig. 5 Upper rim calix[4]arene crowns. | ||
Tetrachloromethylated calix[4]arene 9b was transformed into a double calix-crown 13 in 50% yield by reaction with ethylene glycol in DMF/NaH (Fig. 6).10 The reverse approach relied on the bis-hydroxymethyl derivative 9a, which was allowed to react with α,α′-dibromo-p-xylene 10 or its anthracene analogue. The corresponding bridged products 11 and 12 were obtained in 30 and 35% yields, respectively.10 The absolute and relative ability of these compounds to undergo host–guest complexation with neutral molecules (AcO-iPr, AcOEt, AcO-tBu, AcO-nPr etc) was investigated in the gas-phase using a mass spectrometric technique.
 | ||
| Fig. 6 Upper rim calix[4]arene ethers. | ||
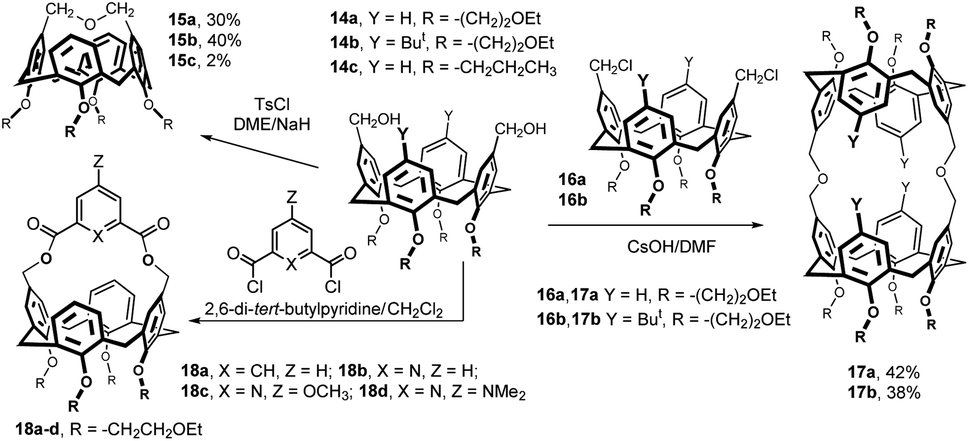
Bis(hydroxymethylated) calixarenes 14a and 14b (obtained by reduction of the corresponding dialdehydes) were reacted with tosyl chloride under basic conditions (Fig. 7). The main products 15a (30%) and 15b (40%), representing the results of an intramolecular cyclization, possess very short bridge chain (only three atoms). The 1H NMR spectra of these compounds are in an agreement with a highly distorted flattened cone conformation of these products.11 Interestingly, the coupling between the calixarene 14c and thioethyl tetrabenzoyl-β-D-galactopyranoside in the presence of Cu(OTf) in MeCN provided compound 15c (2%) as a byproduct to the expected bis-β-galactosyl calixarene (not shown).12 The 1H NMR properties of compound 15c were studied both experimentally and computationally.13
 | ||
| Fig. 7 Upper rim calix[4]arene ethers. | ||
Bis(hydroxymethylated) calixarenes 14a and 14b were also reacted with bis(chloromethyl) derivatives 16a and 16b in different conditions (Fig. 7). The best results were achieved using CsOH in DMF leading to bis-calix[4]arene product 17a and 17b in 42 and 38% yield, respectively.11
The new upper rim bridged hosts 18a–d, bearing pyridine or benzene rings within the cavity, were synthesized from diol 14a and the corresponding diacid chlorides under high dilution conditions (Fig. 7). The shape, rigidity, and chemical structure of the bridge control the host–guest complexation properties of these cavitands. The 1H NMR titrations suggested the formation of endo-complexes with selected neutral guest molecules having acidic C–H bonds (CH3CN, CH3NO2, CH2(CN)2).14
An interesting approach to bridging the upper rim using an ether bond is shown in Fig. 8. The initial bis(hydroxymethyl) derivative 14a was first alkylated using propargyl bromide to form the corresponding diether 19 in 70% yield (Fig. 8).15 The triple bonds were then oxidatively coupled using Cu(OAc)2 to provide a butadiyne-bridged compound 20 in 60% yield. The ability of this cavitand to undergo host–guest complexation with neutral molecules was investigated in the gas-phase using a MS technique.
 | ||
| Fig. 8 Upper rim calix[4]arene ethers via coupling of terminal alkynes. | ||
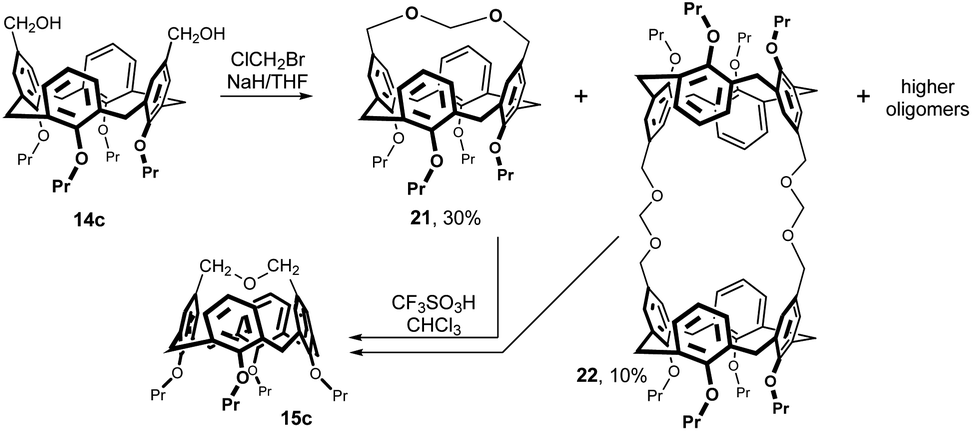
The reaction of 14c with bromochloromethane in the presence of NaH in THF (Fig. 9) provided intramolecularly bridged compound 21 (30%), 2 + 2 reaction product 22 and some higher cyclic oligomers.16 The chloroform solutions of pure monomer 21 or pure dimer 22 were treated with a catalytic amount of triflic acid (TfOH) at 25 °C. Under these conditions, both cyclic products were cleaved to form the short bridged calix[4]arene 15c as the main product (up to 60%).
 | ||
| Fig. 9 Upper rim calix[4]arene ethers via coupling of terminal alkynes. | ||
Chloromethyl derivative of calix[6]arene 24 (available by direct chloromethylation of starting 23) was reacted with thiourea at room temperature in DMSO followed by the treatment with aqueous NaOH solution (Fig. 10). This reaction afforded the mercaptomethyl calix[6]arene 25 in 52% yield. Equimolar amounts of 24 and 25 were then mixed in N-methylformanilide-THF mixture containing Cs2CO3 and NaBH4 under high dilution conditions to give “carcerand” 26.17 The MS and elemental analysis showed that 26 possesses a molecular cavity in which N-methylformanilide is trapped noncovalently as a guest.
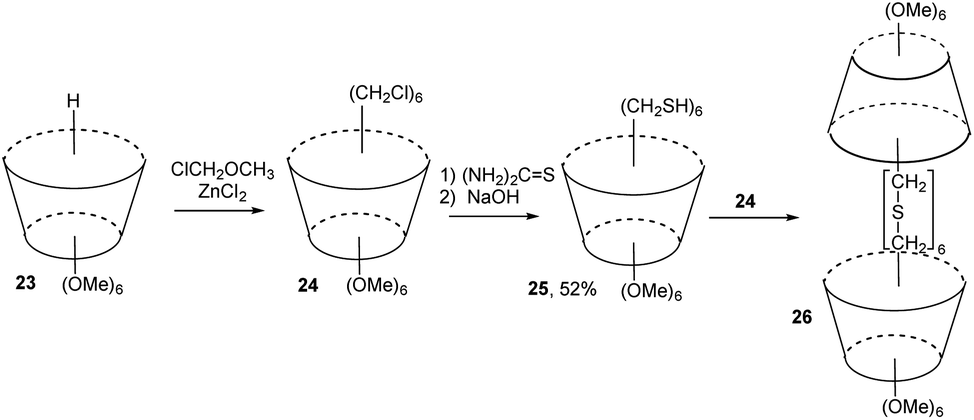 | ||
| Fig. 10 Molecular capsule based on calix[6]arene. | ||
Calix[4]arene-tetrathiafulvalene (TTF) conjugates were prepared by reaction of TTF dithiolates generated in situ from 2-cyanoethylthio TTF derivatives 27a–d (Fig. 11).18 The corresponding dithiolates were then alkylated by tetrakis(chloromethyl) calixarene 9c. The upper rim TTF-calixarene conjugates 28a–d were obtained in good yields (59–64%) as bright orange-yellow solids showing moderate binding with pyridinium cations in solution. Interestingly, the same reaction with calix[4]arene in the 1,3-alternate conformation (analogous to 9c), completely failed and no trace of the expected product 28e was observed in the reaction mixture.19 A binding study (1H NMR) in CDCl3 using compound 28b as a host and N-methylpyridinium derivatives as guests revealed poor binding efficiency under fast-exchange conditions due to the cation–π interactions.
 | ||
| Fig. 11 Calix[4]arene-TTF conjugates. | ||
Calix[4]arene-TTF conjugates (Z)-30 and (E)-30 were prepared using a similar strategy (Fig. 11) – the cyclisation of 2,6 (2,7)-TTF dithiolates (generated from (Z)-29 and (E)-29) with bis-bromomethylated calix[4]arene 16c.19 The (Z)-isomer (Z)-30 was found to be a stable compound. On the other hand, the (E)-isomer (E)-30 isomerised rather fast in solution, affording predominantly the thermodynamically more stable (Z)-30, as well as minor amounts of decomposition products. The 2 + 2 cyclisation product (Z/E)-31 was isolated as a mixture of four possible (Z/E)-stereoisomers. Redox properties of new calix[4]arene-TTF conjugates were characterised using cyclic voltammetry.
The octathiabis(calixarene) cage 31a was isolated in 30% yield from the reaction of tetrathiol 9d with methylene diiodide in DMA under high-dilution conditions using Cs2CO3 as a base (Fig. 12).20 An analogous bis(calixarene) cage 31b was prepared from the tetrakis(chloromethyl) derivative 9b and 1,2-ethanedithiol using K2CO3/DMF in 20% yield. Moreover, when 9d was reacted with α,α-dibromo-o-xylene, only a basket-shaped molecule 32 was formed.
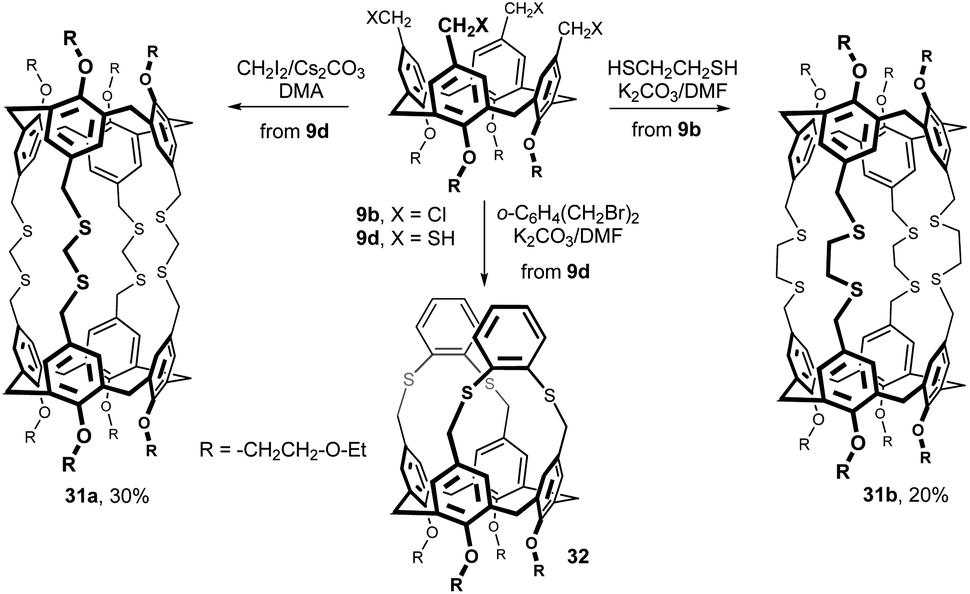 | ||
| Fig. 12 Molecular capsules based on thioether functions. | ||
Bridging via a hydrocarbon spacer
The reaction of dialdehydes 33a and 33b in THF with low-valent titanium species generated from Mg/Hg amalgam and TiCl4 (McMurry reaction) was studied21 (Fig. 13). In both cases, the intramolecularly bridged alkenes 34a and 34b were obtained as the main products in relatively low yields (25–30%).22 To verify the general applicability of intramolecular reductive coupling, the reactivity of the tri- and tetra-aldehydes 33c and 33d was also studied. As expected, only the aldehyde groups on the opposite aromatic subunits were coupled to yield the ethylene bridged products 34c (27%) and 34d (15%). The remaining aldehydes were at the same time reduced to methyl groups. Interestingly, in the case of starting calixarenes with four substituents on the upper rim (33b and 33d) the alkene bridged products 34b and 34d were accompanied by small amount of alkane analogues 35b (10%) and 35d (15%). A short reaction time also led to the isolation of the corresponding diol 36 as a mixture of two diastereomers. | ||
| Fig. 13 McMurry reaction of calix[4]arenes. | ||
The McMurry coupling of tetrapropoxydialdehyde 33e was carried out under different reaction conditions (TiCl3/Zn in refluxing THF).23 The resulting ethylene bridged product 34e was obtained in 30% yield (Fig. 12). The same reaction carried out at room temperature yielded two diastereomeric structures 36e in 43% yield. Since the aforementioned reaction always connects aldehydes on the opposite aromatic subunits, the use of tetraaldehyde in the 1,3-alternate conformation could lead to bridging on both sides of the molecule. Indeed, the reaction of starting compound 33f provided double bridged cage product 34f in 30% yield.23
Cyclization of suitable building blocks afforded calix[5]arene derivative which was transformed in multiple step procedure into diethynyl calix[5]arene 35 (Fig. 14). Eglinton coupling with copper acetate in pyridine at 60 °C, and the following deprotection of the acetyl groups provided the desired double calix[5]arene 36 in excellent yield (75%).24 Two sets of signals in 1H NMR spectra revealed a mixture of two conformational isomers syn 36A and anti 36B in a ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. At 130 °C, the relative population of the two isomers became almost equal and remained unchanged after cooling to room temp., indicating high energy barrier of interconversion between the isomers. Both atropisomers were finally isolated and the complexation ability of isomer 36A toward commercially available fullerenes (C60, C70, C76, C78 and C84) was studied. The syn isomer 36A showed very strong binding abilities for higher fullerenes and even precipitated as host–guest complexes. The captured fullerenes were released by heating the complex to afford the anti isomer 36B.24
:3. At 130 °C, the relative population of the two isomers became almost equal and remained unchanged after cooling to room temp., indicating high energy barrier of interconversion between the isomers. Both atropisomers were finally isolated and the complexation ability of isomer 36A toward commercially available fullerenes (C60, C70, C76, C78 and C84) was studied. The syn isomer 36A showed very strong binding abilities for higher fullerenes and even precipitated as host–guest complexes. The captured fullerenes were released by heating the complex to afford the anti isomer 36B.24
 | ||
| Fig. 14 Upper rim bridged calix[5]arenes. | ||
Although the Claisen rearrangement is quite often used in the chemistry of calixarenes for upper rim derivatization, its application for bridging is really rare. Thus, when tetrabutylammonium iodide (1 equiv.) was added to the reaction mixtures containing 1 equiv. of calix[4]arene and 0.5 equiv. of the alkylating agent (1,4-dichloro-2-butene or 3-chloro-2-chloromethyl-1-propene) in CH2Cl2 solution with NaH as the base, the bis-ethers 37a and 37b were obtained in yields of 84% and 51%, respectively (Fig. 15).25 A subsequent study of the tandem Claisen rearrangement revealed that the addition of bis-(trimethylsilyl)urea (BTSMU) (2 equiv. per phenolic unit) is a key prerequisite for a successful reaction. By heating the starting compounds in N,N-diethylaniline (DEA), the doubly bridged derivatives 38a and 38b were isolated in 15 and 22% yield, respectively. The same strategy was applied to analogous calix[6]arene derivatives (not shown here), where the Claisen rearrangement was performed in even higher yields (33% and 70%) than those reported for calix[4]arene derivatives.25 The complexation behaviour of these bis-calixarenes toward fullerenes C60 and C70 has been studied.
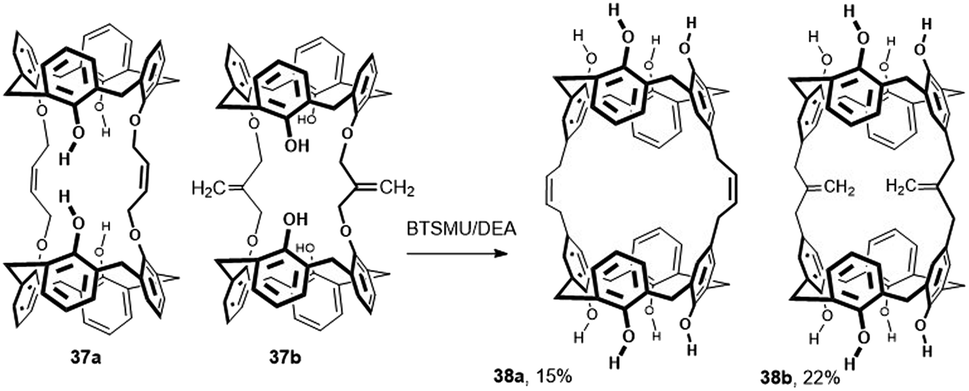 | ||
| Fig. 15 Tandem Claisen rearrangement of calix[4]arenes, BSTMU = 1,3-bis(trimethylsilyl)urea, DEA = N,N-diethylaniline. | ||
As shown in Fig. 16, ring-closing metathesis study within the calixarene series was carried out with Grubbs catalyst (Cl2Ru(PCy2)2 = CHPh). Diallyl calix[4]arene 39a, obtained by Claisen rearrangement of its lower rim counterpart, provided small amount of a cyclic dimer 38 (5%) accompanied by linear dimer 40a and trimer 40b in 25% and 20% yields in benzene. Surprisingly, the same reaction performed in DCM gave the cyclic trimer 42 as the only product that was obtained in virtually quantitative yield.26 While the ring-closing metathesis of the lower rim-unsubstituted compound 39a gave no intramolecularly bridged product, the ester group-bearing analogue 39b (–CH2COOMe) led smoothly to the cage structure 41 (84%).
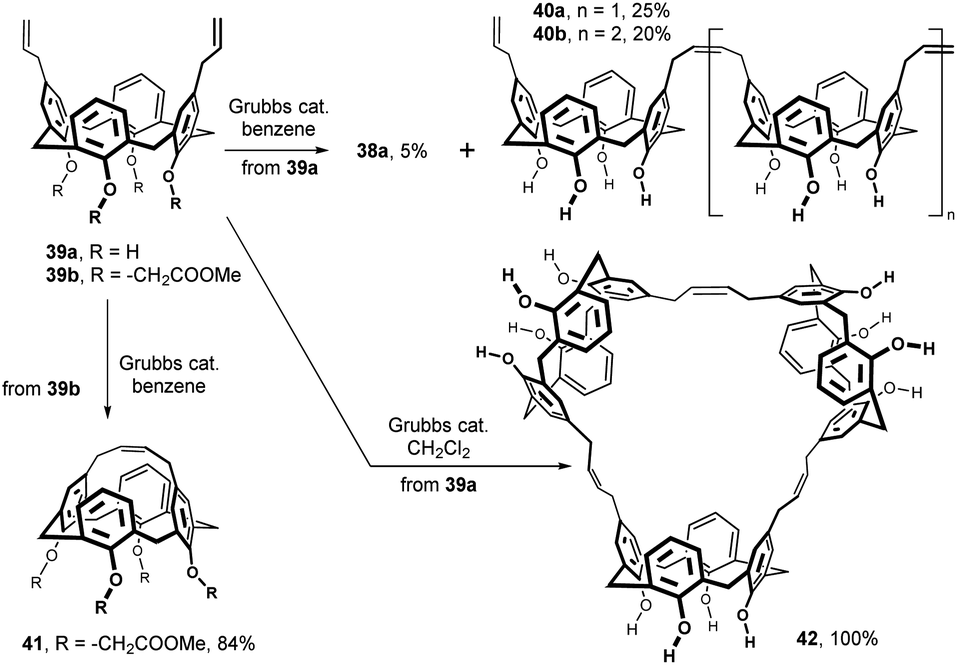 | ||
| Fig. 16 Upper rim-bridged calix[4]arenes by ring closing metathesis. | ||
Bridging via imine or hydrazone moiety
Because the introduction of the amine function (reduction of nitro group) or the direct formylation of the upper rim are well-established synthetic procedures in the chemistry of calixarenes, the application of the imine bonds in the construction of bridged systems is offered.The condensation of diamines 43a and 43b with dialdehydes 44a–c (reflux in DCM in the presence of molecular sieves) allowed for the isolation of the corresponding bis-calix[4]arenes 45a–c in good yields (74–92%) (Fig. 17). The two imine bridges were then easily reduced with an excess of NaBH4 at rt (EtOH-THF mixture) to the more flexible amines 46a and 46b, in good yields (87% and 95%, respectively).27 The presence of short bridges (2 atoms) results in a relatively rigid head-to-head arrangement of the two calix[4]arene groups and compounds 45a and 45b showed a very high affinity for Ag+ ions, with the complexation constants up to 9.5 × 105 M−1 (1H NMR titration) in CDCl3. Membrane transport experiments and CHEMFET studies revealed the selective transport and complexation of silver(I) ions.
 | ||
| Fig. 17 Bridging of calix[4]arenes via Schiff base formation. | ||
A very similar synthetic strategy was used for the preparation of molecular containers 47 and 48. The Schiff base derivative 47 was obtained in 69% yield by condensation of diamine 43b with the corresponding dialdehyde (Fig. 17). Subsequent reduction with NaBH3(CN) afforded amine 48 in 89% yield.28 A series of thermodynamic and structural studies on the host–guest complexation between molecular containers 48 and 49 (amide analogue) and the N-methylpicolinium salts were carried out. It has been shown that in these systems the organic cation is encapsulated within the molecular containers and therefore is separated from its counter anion.
The condensation reactions of diaminocalix[4]arene 43b with various aromatic dialdehydes (24 h reflux in DCM:MeOH) in the presence of MgSO4 provided the corresponding bis-calix[4]arenes 50a–d in excellent yields (>95%; Fig. 17). Interestingly, in the case of terephthalaldehyde, the yield of bis-calixarene 50e was only 19%. This indicates that the macrocyclization via imine bonds strongly depends on the geometry of the starting aldehyde (compare the meta-vs. para-substitution, 50d vs. 50e).29 The binding studies between the bis-calix[4]arenes and viologen-type guest molecules revealed that thiophene-based derivative 50a exhibited good binding affinities toward biologically interesting viologen guests even in relatively polar media (CDCl3:CD3OD = 2:1 v/v).
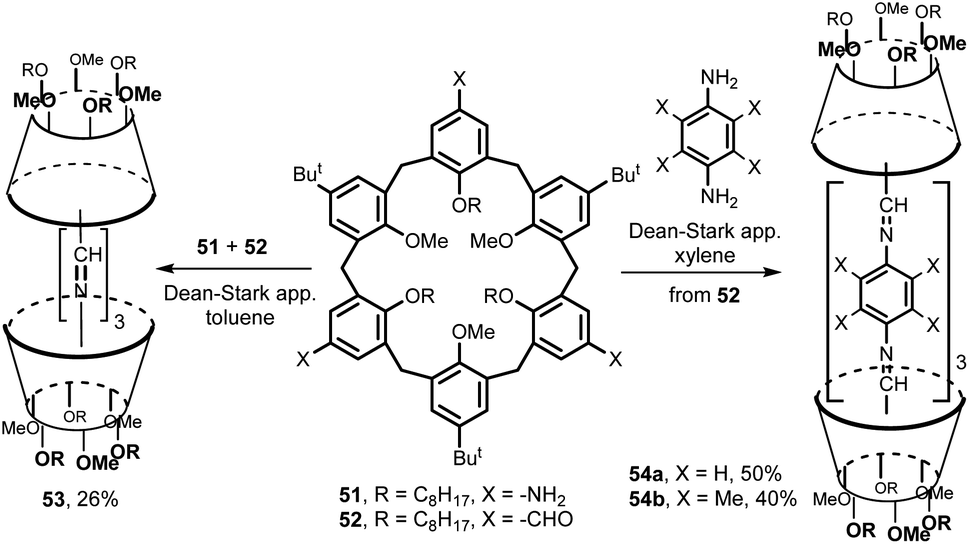
An extension of this strategy to the chemistry of higher calixarenes is shown in Fig. 18. A 1,3,5-trisubstituted triamine 51 and the corresponding trialdehyde 52 were prepared by a multi-step synthesis from the starting tert-butylcalix[6]arene. Their mutual condensation in boiling toluene (Dean–Stark trap) led to the cage-like structure 53 in 26% yield. Similarly, the aldehyde 52 was reacted with para-phenylenediamines to give double calix[6]arenes 54a and 54b with extended cavity in 50% and 40% yield, respectivelly.30 The binding studies towards N-methylpyridinium, N-methyl-2-picolinium and N-methyl-4-picolinium iodides as guests were carried out in CDCl3/CD3CN = 9:1 v/v. While cages 53 and 54b did not show any complexation ability, compound 54a was found to complex pyridinium salts with association constants up to 890 M−1.
 | ||
| Fig. 18 Bridging of calix[6]arenes via Schiff base formation. | ||
Calix[4]arene with intramolecularly bridged upper rim was prepared by reaction of dialdehyde 55a with 1,8-diamino-3,6-dioxaoctane (Fig. 19). The resulting macrocycle was isolated in 59% yield. Subsequent alkylation with ethyl bromoacetate afforded ionophore 57 (68% yield), which was studied using extraction experiments for its ability to bind/extract the dichromate anion.31
 | ||
| Fig. 19 Imine- or hydrazone-bridged calix[4]arenes. | ||
Dialdehyde 55b was bridged by reaction with carbohydrazide (reflux in EtOH) to give the corresponding carbohydrazone-based bis-calixarene 58 (72% yield) (Fig. 19).32 According to the authors, this compound shows a selective enhancement of fluorescence intensity in response to Fe3+ ions suggesting its application as an iron selective chemosensor. The hydrazone structural motif can also be found in compounds 60a–c, formed by the condensation of dialdehyde 33e with the corresponding bis-acyl hydrazides 59a–c (Fig. 19).33 The biological activity screening showed that these compounds exhibit high antimicrobial activity against both Gram-positive and Gram-negative bacteria.
Intramolecular cyclization via amidic function (from aminocalix[n]arenes)
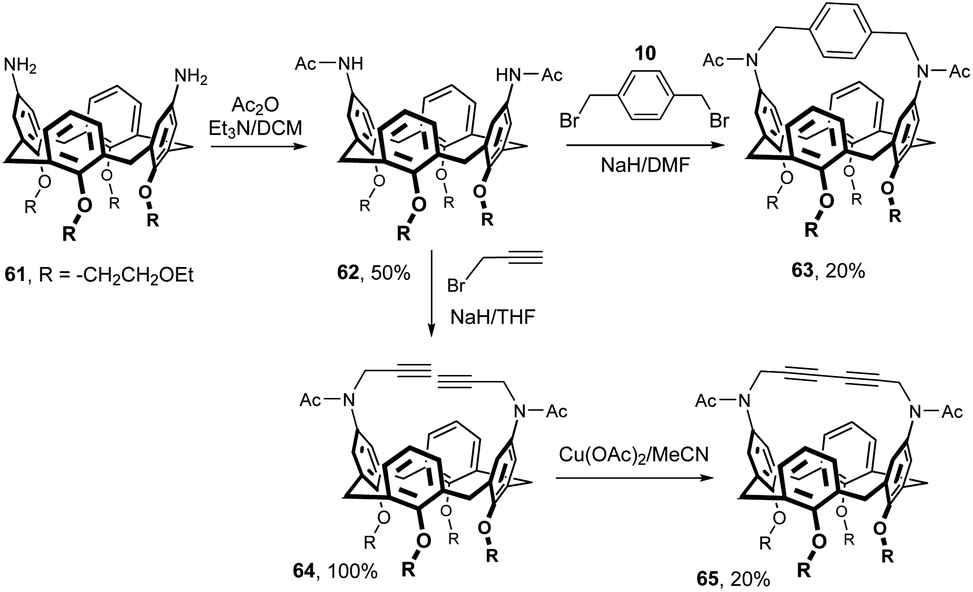
As already mentioned, introduction of an amino group into the upper rim of calixarenes is a well-established synthetic procedure in calixarene chemistry. It is therefore not surprising that a substantial part of the derivatives with a bridged upper rim is based on the use of the amide bond.In this context, the first example shown in Fig. 20 is somewhat unusual, since the authors used an amide functional group for alkylation reactions at the nitrogen atom. The key intermediate bis(acetamino) derivative 62 was obtained in 50% yield by the acylation of starting diamine 61. Di-sodium salt of 62 formed by deprotonation with NaH was reacted with α,α′-dibromo-p-xylene 10 to provide an intramolecularly bridged cavitand 63 (20%).15 A similar alkylation with propargyl bromide led to dialkynyl derivative 64 in essentially quantitative yield. Oxidative coupling of acetylene functions with Cu(OAc)2 provided cavitand 65 in 20% yield. All these cavitands, together with the structurally related compounds from Fig. 8, were studied for the complexation of neutral substances in the gas phase.
 | ||
| Fig. 20 Upper rim-bridged calix[4]arenes via alkylation of amidic functions. | ||
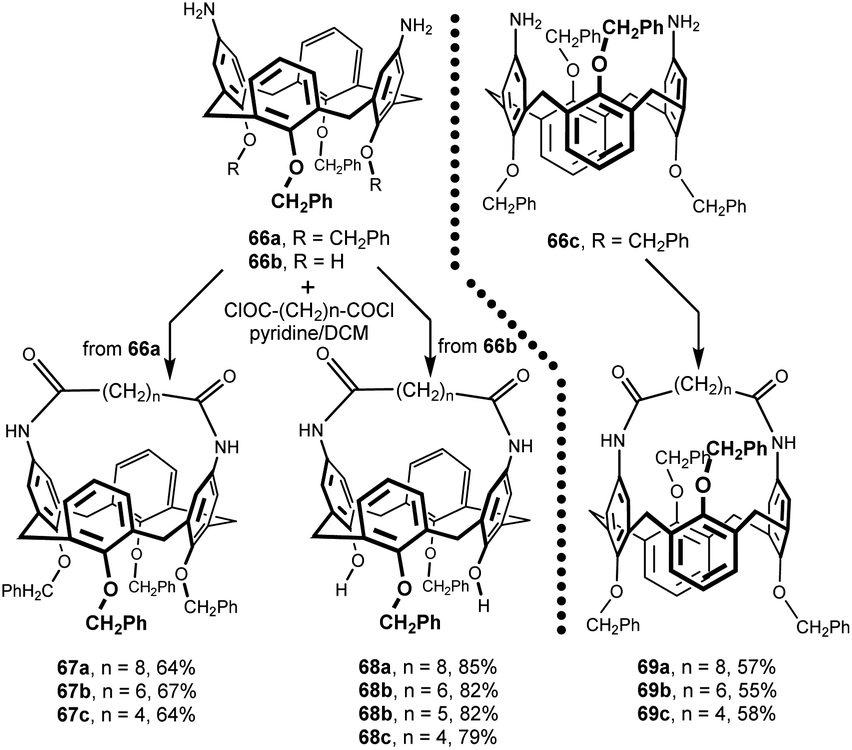
Treatment of 66a and 66b with aliphatic diacid dichlorides containing four to eight methylene groups between the carboxyl moieties resulted in intramolecular bridging of calix[4]arene diamines. Reaction carried out under high dilution conditions yielded the corresponding cone conformers 67a–c and 68a–d in very good yield (Fig. 21). The 1,3-alternate congeners 69a–c were obtained analogously from 66c.34
 | ||
| Fig. 21 Upper rim calix[4]arene via alkylation of amidic functions. | ||
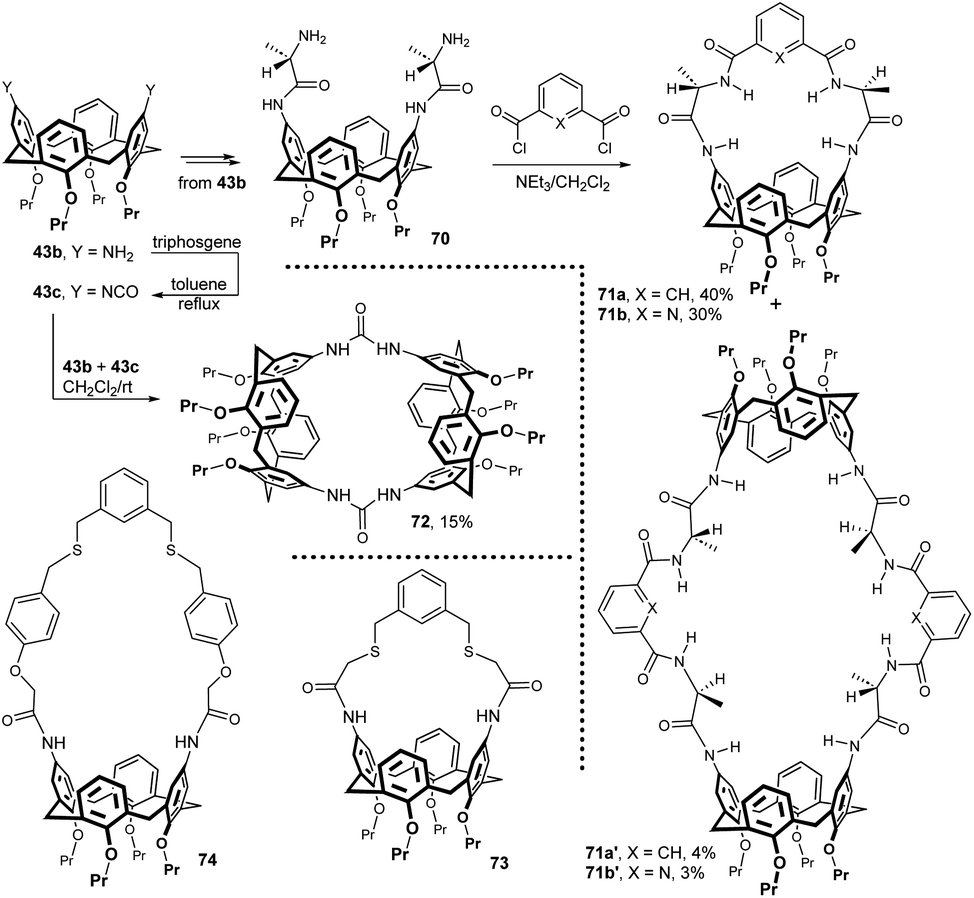
Starting from diamino derivative 43b, the L-alanine moiety was introduced into the upper rim to form compound 70 in high yield. The following reaction with aromatic diacyl dichlorides then gave compounds 71a and 71b in 40 and 30% yields.35 The formation of expected products was accompanied by [2 + 2] condensation leading to giant byproducts 71a′ and 71b′ in small amounts (Fig. 22). ESI-MS and 1H NMR experiments revealed that 71a and 71b behaved as macrocyclic receptors for carboxylate anion recognition in acetone solution. The combination of HB interactions and π–π stacking resulted in high complexation constants towards benzoate anion (Kc = 4 × 104 mol−1).
 | ||
| Fig. 22 Upper rim-bridged calix[4]arenes via amidic functions. | ||
Calixarene 43b was also transformed into the corresponding isocyanate 43c by reaction with triphosgene (CCl3OC(O)OCCl3) in toluene under reflux. The reaction between difunctional compounds 43b and 43c, carried out under high dilution conditions, afforded the bridged bis-calixarene 72 in 15% yield.36 Again, titration experiments showed the ability to complex selected anions (benzoate) in solution. The preparation of bis-amide analogue of 72 (not shown, 10% yield) by reacting the corresponding diacid dichloride with diamine 43b was also reported.
A similar general approach (bridging of amidic intermediates) was used in the synthesis of ligands 73 and 74 (Fig. 22). However, in this case, the last step was the dialkylation of the corresponding chloromethyl derivatives using α,α′-meta-xylene dithiol.37
The synthesis of macrocyclic peptido-calixarene derivatives was carried out by the application of the azide or mixed anhydride coupling methods (Fig. 23). Thus, the reaction of bis-aminomethyl calix[4]arene 75 with free diacid derivative in the presence of ethyl chloroformate (mixed anhydride method) afforded the corresponding chiral calix[4]arene 77 bearing L-tyrosine moieties in 65% yield.38 The same product was also obtained with diacyl diazide in 45% yield. Analogous procedures were used for the synthesis of the structurally related compound 78 starting from diaminocalixarene 61. A series of similar compounds containing valine or leucine residue was also reported.33 While the aminomethyl derivative 75 was easily bridged with 2,6-pyridinedicarboxylic acid to give compound 76, calixarene 61 bearing amino groups directly on the upper rim does not react in this way because the distance and geometry do not match the amide bonds in the intended product 79.38
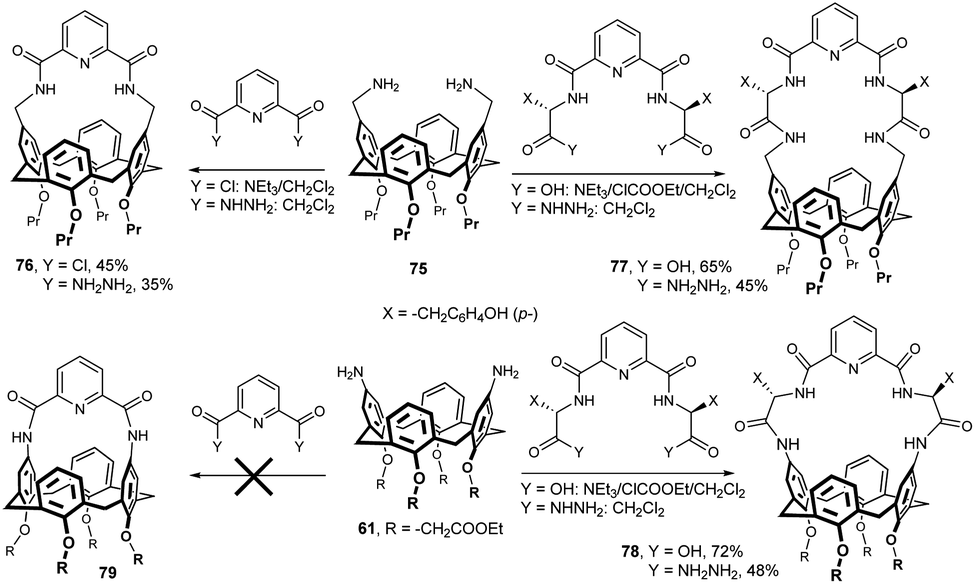 | ||
| Fig. 23 Synthesis of peptidocalix[4]arenes. | ||
Carcerand 85 based on a combination of calix[4]arene and resorcin[4]arene structural motifs was prepared by a very complex multi-step approach (Fig. 24). The key step was the reaction of the two building blocks 80 and 81 leading to diamide 82 in 40% yield.39 Subsequent deprotection of the phthalimide groups (Phth) with methylhydrazine provided the diamino derivative 83, which was acylated with chloroacetyl chloride to give intermediate 84 in essentially quantitative yield. The final carcerand 85 was formed quantitatively by the intramolecular alkylation of 84 induced by Cs2CO3/KI in DMF at 80 °C.
 | ||
| Fig. 24 Synthesis of carcerands and hemicarcerands, the example of carceroisomerism. | ||
The 1H NMR spectrum confirmed that molecule of DMF is incarcerated within the inner space of the carcerand (dramatic upfield shifts of DMF signals by up to 3.5 ppm), thus forming so called carceplex 85 DMF. The following study of formation of similar carceplexes40 with different guest molecules41 led eventually to the discovery that, due to the non-symmetrical cavity, two different orientations of the guest molecule with respect to the host are possible (Fig. 24). For this new type of isomerism the name carceroisomerism was proposed.42 A slightly modified synthetic strategy also led to the synthesis of hemicarcerands represented by formula 86.43 Using a synthetic strategy similar to that employed for the preparation of compound 85, a 2 + 1 system 87 (ref. 44) or even a huge 2 + 2 macrocycle45 (not shown) were obtained.
Calix[4]arenes containing ferrocene moiety bonded via amidic function across the upper rim have been synthesized. As shown in Fig. 25, coupling of diamino derivatives 88a, 88b and 61 with freshly synthesized 1,1-bis(chlorocarbonyl)ferrocene in the presence of NEt3 gave the corresponding products 89a–c in good yields (42–50%).46 1H NMR anion-binding studies in CDCl3 showed some selectivity of 89b for the chloride anion (over Br− and I−) and the ability to bind carboxylates for all three mentioned receptors. Moreover, the presence of ferrocene unit enabled the application of cyclic voltammetry and square wave voltammetry for anion recognition.47
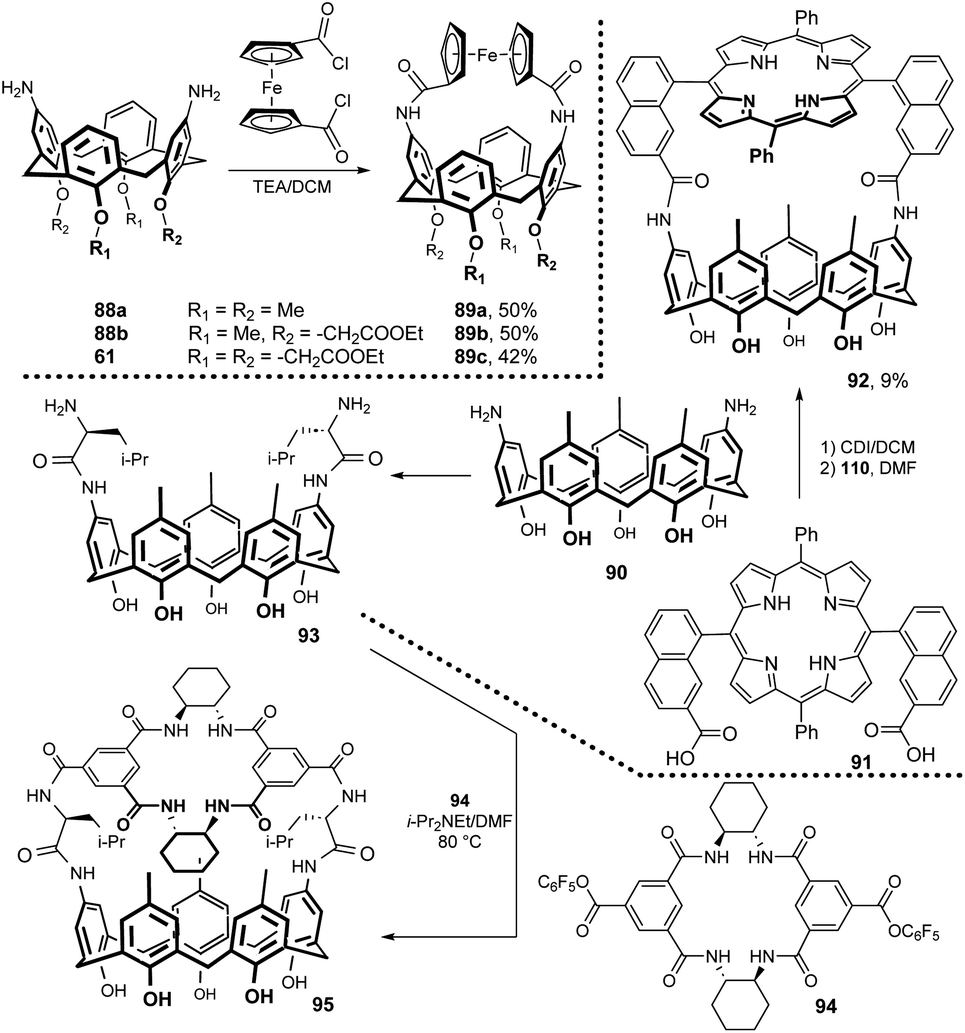 | ||
| Fig. 25 Intramolecularly bridged calix[4]- and calix[5]arenes. | ||
5,15-Bis(7-carboxyl-1-naphthyl)-10,20-diphenylporphyrin 91 possessing the syn arrangement of two naphthalene moieties was used for bridging diaminocalix[5]arene 90 (Fig. 25). The resulting rigidified receptor 92 was transformed into Zn-porphyrin derivative and studied for binding ability towards various pyridine derivatives.48 A similar motif, i.e. porphyrin bridged with diaminocalix[4]arene 43b, was studied as a receptor for various nitrogenous ligands. The confined rigid cavity showed high selectivity towards imidazole (versus pyridine).49
The coupling reaction of 94 with calix[5]arene derivative 93 furnished a chiral receptor 95 in 32% yield.50 The 1H NMR titration study confirmed some enantioselective recognition of chiral ammonium guest molecules. Regarding chiral bridged calixarene systems, the reaction of permethylated 6A,6D-diamino-β-cyclodextrin with calix[4]arene derivatives immobilised in the 1,3-alternate conformation was reported (not shown).51 The application of succinylamido spacer enabled the preparation of 1:1 or 1:2 (calixarene:cyclodextrin) upper rim-bridged conjugates.
An interesting synthetic route to bis-calixarene is shown in Fig. 26 and relies on multiple cycloadditions between bifunctional dipoles and bifunctional dipolarophiles. The starting diamine 43b was acylated with acryloyl chloride or methyl propiolate to give the corresponding amides 96 and 97, both in 79% yield. The quadruple cycloaddition with bifunctional hydroxamic acid chloride then provided bis-calix[4]arenes 98 (27%) or 99 (26%).52 The conformational studies of these compounds were carried out by solution NMR experiments and X-ray crystallography.53
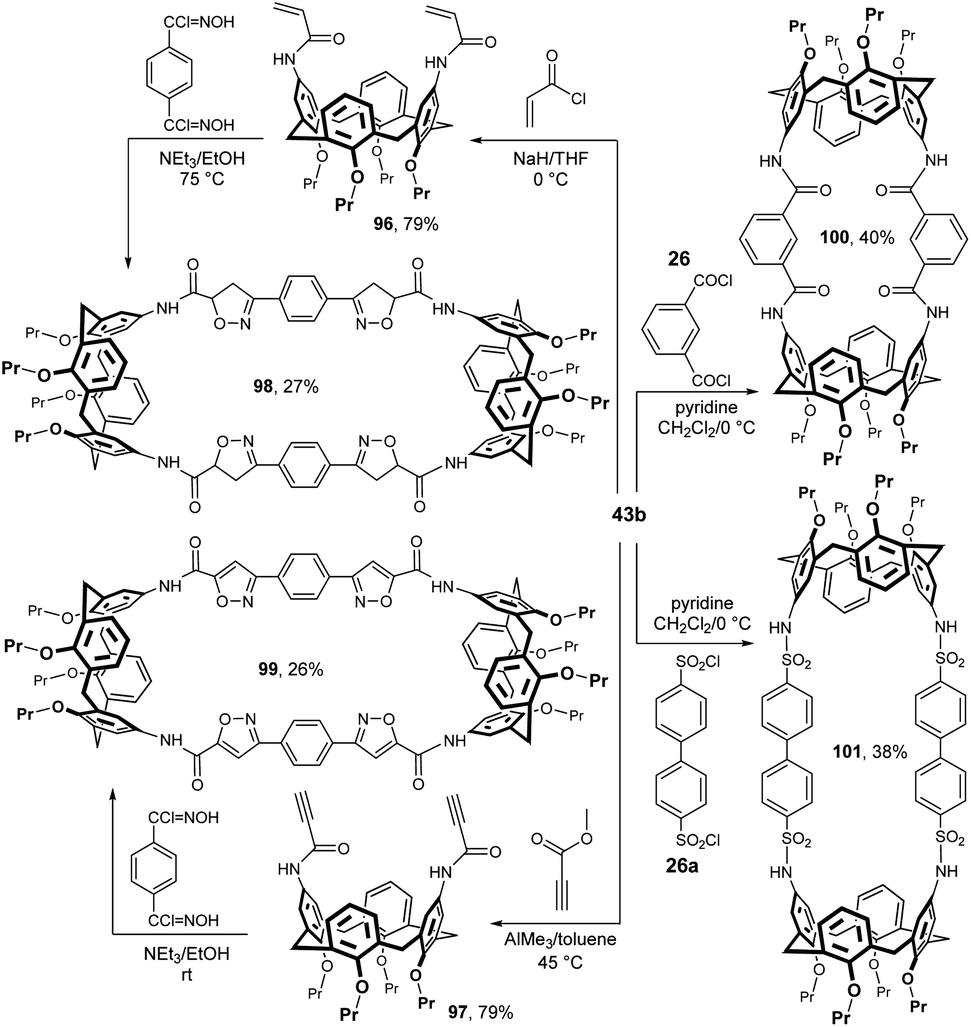 | ||
| Fig. 26 Synthesis of peptidocalix[4]arenes. | ||
When isophthaloyl dichloride 26 was treated with diaminocalix[4]arene 43b in DCM in the presence of pyridine, bis-calix[4]arene 100 was obtained in 40% yield.29b A similar reaction of biphenyl-4,4′-disulfonyl chloride 26a provided bis-calixarene 101 (38%), which also demonstrates the overall utility of the starting diamine 43b in the synthesis of this type of compounds.
A calix[6]arene-based ligand possessing a tren unit capping the upper rim was prepared from triamino derivative 102 (Fig. 27). The macrocyclization with nitrilotriacetic acid in the presence of PyBOP and NEt3 led to tris-amide 103 in 45% yield. The reduction of amide bonds with BH3·THF finally gave the ligand 104 in good yield (75%).54 The corresponding Zn2+ complex 105 was studied both in solution and in the solid state. A similar synthetic strategy also led to the corresponding Mo analogues of 105 or to Mo complex 106, where the three MeO moieties are replaced with hydrogens. These complexes possessing trigonally symmetric, sterically protected, and rigid substrate-binding pocket were studied for their ability to reduce the molecular nitrogen N2.55
 | ||
| Fig. 27 Calix[6]arene derivatives bridged via tren moiety. | ||
Cyclization via amidic function (from calixarene carboxylic acids)
While aminocalixarenes are readily available via nitration and subsequent reduction of intermediate nitro compounds, the corresponding calixarene-carboxylic acids are much less frequent. Several examples of amide bond formation are given in Fig. 28. The starting dicarboxylic acid 107a was activated via pentafluorophenol (PFP) ester (85% yield) and treated with the corresponding amine moiety (2-(4-(2-azidoethoxy)phenoxy)-ethanamine) in the presence of TEA. The intermediate bis-azide (isolated in 50% yield) was then reduced to diamine 108 in overall 42% yield. Reaction with pyridine-3,5-dicarbonyl chloride under high dilution conditions gave the macrocycle 109 in 50% yield. The N methylation of pyridine moiety provided macrocycle which was able to form pseudorotaxane with isophthalimide-derived thread and finally led to rotaxane (not shown).56 | ||
| Fig. 28 Amidic bridges starting from calix[4]arene carboxylic acids. | ||
Diacid 107a was converted to chloride using SOCl2 and then coupled with anthracene based diamine to give the fluorescent anion receptor 110 in 30% yield (Fig. 28). The fluorescence spectroscopy showed that this compound can selectively recognise AcO− anion over other anions examined (F−, Cl−, Br−, H2PO4− and HSO4−).57
Diacid 107b (available by oxidation of the corresponding aldehyde) was transformed into diacyl dichloride (SOCl2) and reacted with pseudopeptides possessing two terminal NH2 functions (Fig. 28). The cyclization reaction carried out under high dilution conditions provided the L-Phe and L-Thr derivatives 111a and 111b in 50 and 42% yield. Selective hydrolysis of the phosphoric methyl ester gave macrocycles 112a and 112b. These compounds were studied as carbohydrate receptors with good selectivity for β-glucosides.58
A rare example of a bridged calix[6]arene derivative is shown in Fig. 29. The starting tricarboxylic acid 113, prepared by a multistep procedure, was treated with SOCl2 under reflux and reacted with 2-aminopyridine in the presence of DMAP to afford 114 in 50% yield.59 Highly selective recognition of this receptor towards fluoride ion was demonstrated by fluorescent and 1H NMR titration experiments. A calix[6]crown-derived hydroxamic acid 115 (no synthetic details reported) was studied for the liquid–liquid extraction and sequential separation of some selected metal cations Cr(III), Mo(VI) and W(VI).60
 | ||
| Fig. 29 Calix[6]arene derivatives bridged via amidic bonds. | ||
Template synthesis of bridged calixarenes using tetraurea dimers
Tetraurea derivatives of calix[4]arenes in the cone conformation are known to spontaneously form dimeric capsules in nonpolar solvents. These supramolecular self-assemblies are held together by a cyclic array of hydrogen bonds (Fig. 30). It was shown that the tetratosylurea 116 cannot form homodimers, but instead exclusively forms heterodimers61 with the corresponding tetraarylurea calix[4]arene 117. This heterodimer, where 116 plays the role of template, can be used for preorganisation of alkenyl moieties of 117 in such a specific way, that the metathesis reaction between its double bonds proceeds in very high yields.62 Application of this procedure to a mixture of 116 + 117a and 117b (benzene, Grubbs catalyst) followed by the hydrogenation of alkene intermediate (H2/PtO2) gave macrocyclic compounds 118a and 118b in high yield (>90% according to the 1H NMR). The analogous reaction without the presence of template 116 was unsuccessful which indicates the essential role of the template for the correct preorganization of the respective functional groups. | ||
| Fig. 30 Macrocyclization via metathesis of tetraurea dimers. | ||
This approach proved so effective that it was used to synthesize a number of calixarene derivatives with unusual topologies. Thus, the heterodimerisation of tetraarylurea calix[4]arenes 119 with tetratosylurea 116 followed by metathesis and hydrogenation were used to provide products 120 bearing huge macrocycles on the upper rim (Fig. 31). Incredibly, the respective multimacrocyclic calix[4]arene derivatives 120 were the only identifiable products (yields of 60–95% after purification).63
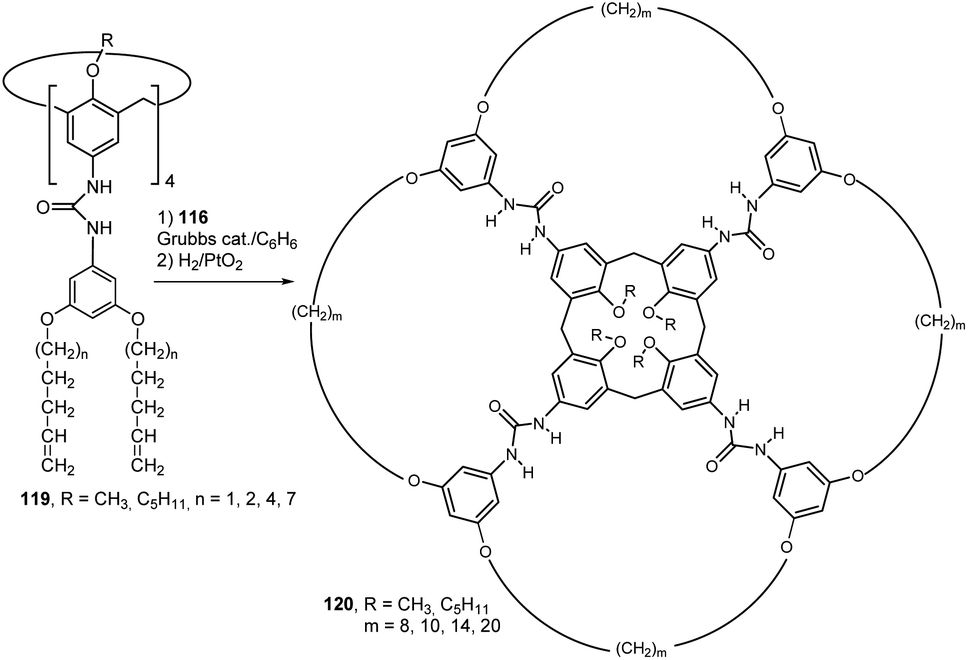 | ||
| Fig. 31 Synthesis of calixarene-based huge macrocycles. | ||
This behaviour of tetratosylurea 116 has been shown to be general and it always forms only heterodimeric structures with other ureas. Based on this general strategy, a number of previously unknown topologies were prepared, such as [8]catenanes,64 multicyclic bis[2]catenanes,65 fourfold [2]rotaxanes,66 combination of structural elements of rotaxanes and catenanes,67 bis- and trisloop tetraurea calix[4]arenes,68 self-assembled dendrimers,69 multiple catenanes based on tetraloop calix[4]arenes,70 or molecular capsules consisting of calix[4]arene and calix[4]pyrrole moieties.71
Direct bridging of the upper rim
As a part of ongoing effort to develop novel derivatisation procedures of basic calixarene skeleton, we reported the first example calix[4]arenes with intramolecularly bridged meta positions at the upper rim.72The starting iodocompound 121 was transformed into sulfide 122 (55%) employing the reaction with 2-pyridine disulfide and a CuI/Mg/2,2-bipyridine/DMF system (Fig. 32). Subsequent oxidation to sulfoxide 123 was smoothly accomplished by meta-chloroperoxybenzoic acid in chloroform (87% yield). The presence of the 2-pyridyl moiety then enabled Pd-catalyzed double C–H activation offering an access to unprecedented derivatives with intramolecular single bond bridge between the meta positions of the two neighbouring aromatic subunits. Thus, the reaction of 123 in the presence of Pd(OAc)2 (10 mol%), Ag2CO3 + benzoquinone (oxidants) in a DCE–benzene mixture provided a mixture of two products. As the introduction of a single bond leads to inherently chiral system, the combination with a stereogenic centre on sulfur atom results in the mixture of diastereomers 124a and 124b which were isolated in 43 and 29% yield, respectively.72 Interestingly, the replacement of the pyridine group with phenyl or pyrimidin-2-yl moieties did not give any bridged product. Only in the case of the ethyl sulfoxide derivative the bridged compound was obtained in 13% yield (not shown).73
 | ||
| Fig. 32 Direct bridging of calix[4]arenes via a single bond bridge, X-ray structure of 124a. | ||
As shown in Fig. 32, the cavity of 124a is substantially distorted from the symmetrical square arrangement typical for calix[4]arene derivatives. While the distance between two opposing CH2 bridges is typically around 7.15 Å, the lengths of the corresponding diagonals in 124a are dramatically different, 5.6 Å and 8.1 Å.72
Very recently, a synthesis of inherently chiral calixarenes by a catalytic enantioselective cross dehydrogenative coupling was reported.74 The Friedel–Crafts reaction of alkylated calix[4]arenes with (substituted) 2-bromobenzoyl chlorides afforded starting compounds 125 (Fig. 33). The application of Pd chemistry did not lead to the expected product 126, but the upper rim-bridged calix[4]arene was obtained instead. The enantioselective desymmetrization reaction of 125 was initially investigated by varying the Pd-sources, the ligands, the solvents, and the temperature to find the optimised reaction conditions. As shown in Fig. 32, under optimized conditions [PdBr2 (10 mol%), ligand L (20 mol%), Rb2CO3 (3.0 equiv.), THF/110 °C in a sealed tube] compounds 125 were successfully transformed into series of compounds 127 bearing different substituents on benzoyl moiety with ee values of up to 93%. The exclusive formation of 127 instead of 126 indicates that the 1,5-palladium migration (from intermediate A to intermediate B) takes place during the reaction (Fig. 33), eventually leading to the transannular arylation of B.74 Compounds 127 represent new family of inherently chiral calixarenes possessing unique chiroptical properties due to their highly rigid structure, induced by the 9H-fluorene segment, and they were found to be useful scaffolds for the fabrication of CPL materials.
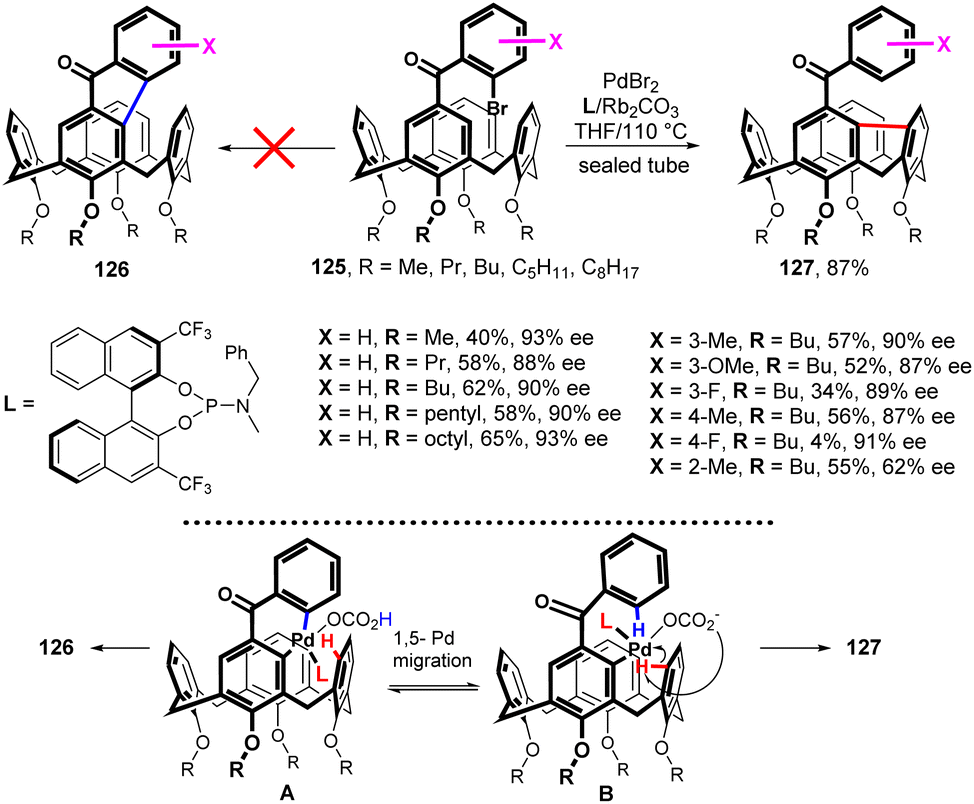 | ||
| Fig. 33 Direct bridging of calixarenes, 1,5-palladium migration in Pd complex. | ||
Bridging via organomercurial derivatives
Due to their structure, calixarenes are subject to a number of electrophilic substitutions on the upper rim. The electronic effects of the lower rim alkoxy groups (or free OH groups) result in regioselective substitution into the para position of aromatic subunits, including the ipso-substitution of tert-butyl groups.1 However, about a decade ago, a reaction leading to selective substitution of the meta position (relative to the OR group) of the aromatic skeleton of calix[4]arene was discovered – the mercuration of calix[4]arenes. In this way, the meta position of calixarenes, which was practically unavailable at that time, was made available and a number of previously unfeasible derivatizations and substitution patterns were made possible.As shown in Fig. 34, reaction of the starting di-tert-butyl derivative 128a with 1 equiv. of Hg(II) trifluoroacetate (Hg(TFA)2) afforded the unexpected meta-substituted isomer 129a, which was isolated in 44% yields (after conversion to chloromercurio derivative) as the only substitution product.75 The same reaction with two equiv. of Hg(TFA)2 provided meta,meta-disubstituted product 130a in 45% yield. Analogously, the reaction of a simple tetrapropoxy derivative 128b gave under similar condition even higher yield of monochloromercurio derivative 129b (70%) without any para-substituted isomer. The disubstitution of compound 128b resulted in the isolation of two main products, meta,meta-isomer 130b and meta,para-isomer 131b, both in 20% yields.76
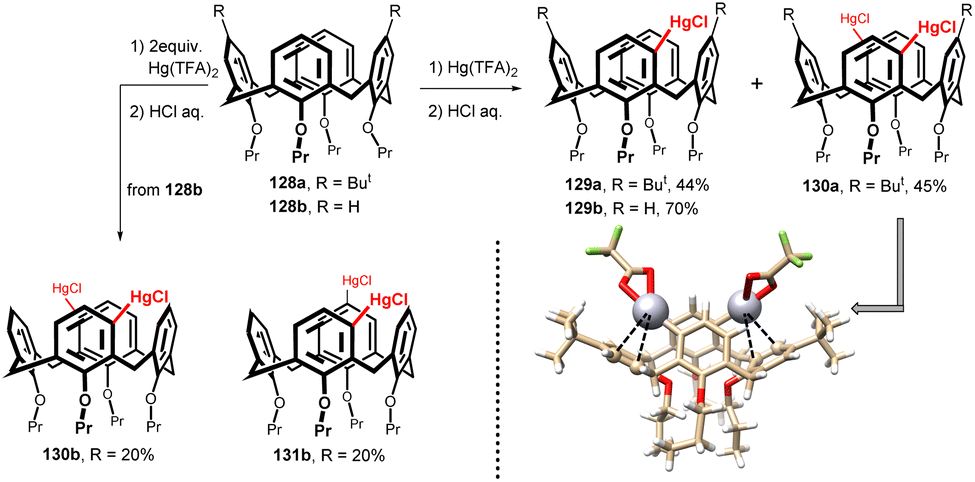 | ||
| Fig. 34 Mercuration of tetrapropoxycalix[4]arenes (cone), X-ray structure of compound 150a (HgTFA form). | ||
The corresponding quantum-chemical calculations revealed that the meta-substitution product 129b is favored over the para analogue by more than 3.5 kcal mol−1. This indicates that the regioselectivity is thermodynamically driven. Moreover, the X-ray analysis confirmed75,77 the stabilization of the meta product by neighbour aromatic subunit via intramolecular Hg⋯π interactions – see Fig. 34 for the X-ray structure of 130a (TFA form). It is clear that the corresponding para-analogue could not provide similar stabilization effect.
The unexpected regioselectivity of mercuration laid the foundation for an entirely new type of substitution in calixarene chemistry. Thus, the meta-chloromercurio derivative 129b was subjected to an intramolecular cross-coupling under Pd catalysis. The best reaction conditions [Pd(OAc)2/AsPh3/Cs2CO3 in DCM) provided a bridged product 132b in 64% yield (Fig. 35). In order to ascertain the general applicability of this synthetic approach, the conformationally mobile derivatives 128c and 128d were also reacted in the same way. Both compounds were successfully converted to bridged systems 132c and 132d via the HgCl intermediates 129c and 129d.78 The VT 1H NMR study revealed that methoxy derivative 132d exists as a dynamic mixture of two conformations at rt (cone – 1,2-alternate). A similar equilibrium was observed for Et derivative 132c at 130 °C. On the other hand, the increased rigidity of the system enabled the separation of the two conformers at room temperature.
 | ||
| Fig. 35 Synthesis of single bond-bridged calix[4]arenes. | ||
The presence of fluorene moiety within the bridged molecule 132b enables the regioselective alkylation of calix[4]arenes furnishing compounds 133 in good yield (Fig. 35). As the CH2 group of fluorene moiety is known for its enhanced acidity, deprotonation of 132b occurs exclusively at this position. Moreover, based on the X-ray analysis, the alkylation takes place stereoselectively at the equatorial position of the CH2 bridging unit. The same reaction with 128b as a model compound does not proceed at all.79
These single bond-bridged compounds possess rigidified and highly distorted cavities. Due to the high internal strain, these systems are susceptible to reactions otherwise unknown in calixarene chemistry. Recently, the cleavage of such systems with various electrofiles leading to linear oligophenols was described.80
To determine the extent of possible application of meta-substitution in calix[4]arenes, the mercuration of all remaining conformers was performed. Thus, the reaction of the partial cone conformer 134 using one equivalent of Hg(TFA)2 in CHCl3 at room temperature afforded regioisomer 135 (of seven theoretically possible) as the main product (Fig. 36). Heating the toluene solution overnight at 110 °C in the presence of Pd(OAc)2/AsPh3 as the catalyst and Cs2CO3 as the base provided the meta-bridged compound 136 in 88% yield.81 This compound was also regio- and stereo-selectively alkylated as evidenced by the formation of compound 137 (70%).
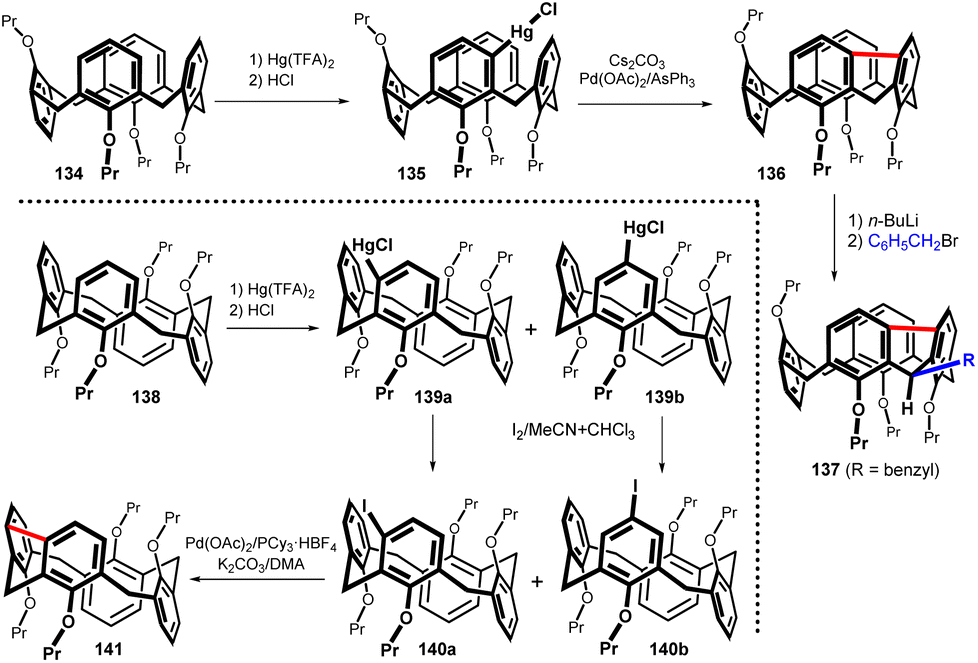 | ||
| Fig. 36 Regioselective mercuration of the partial cone 134 and 1,2-alternate 138 including the subsequent bridging reactions. | ||
The mercuration of the 1,2-alternate conformer 138 using a standard procedure provided an inseparable mixture of regioisomers 139a and 139b (40:60) together with 26% of unreacted starting compound (Fig. 36). Conversion of this mixture to iodo derivatives by reaction with I2 unfortunately led to another inseparable mixture of products 140a and 140b. Despite the isolation problems, the final Pd-catalysed cyclization followed by preparative TLC on silica gel afforded the bridged product 141 in 13% overall yield (over three steps from 138).82
The bis-chloromercurio derivative 130b (meta,meta) or the corresponding diiodo compound 143, easily available by the reaction with I2 (Fig. 37), represent a perfect starting point for the synthesis of double bridged system. Unfortunately, despite our great efforts, compound 142 was never prepared, probably due to the extreme internal strain introduced into the cavity by two short bridges.
 | ||
| Fig. 37 Synthesis of upper rim single atom-bridged calix[4]arenes. | ||
On the other hand, diiodo intermediate 143 was smoothly transformed into the corresponding carboxylic acid 144 via the lithiation with BuLi and the treatment with CO2 (Fig. 37). The doubly carbonyl-bridged derivative 145 was obtained in 32% yield (from 143) by the intramolecular Friedel–Crafts acylation, demonstrating the possibility of double bridging of the cavity using single-atom bridges. The ketone groups react by nucleophilic addition reaction with PhLi or with LiAlH4 to give the corresponding alcohols 146a and 146b in good yields. It is interesting that in both cases the reaction proceeds stereospecifically with access of the nucleophile from the outside of the cavity.83
Diketone derivative 145 with two monoatomic bridges possessing sp2 hybridization represents an extremely distorted calixarene cavity, as can be demonstrated by the exceptionally short/long main diagonals (distance between the opposite CH2 bridges) which are 5.43 and 8.27 Å (Fig. 38a). A highly preorganised cavity of 145 was found to form a complex with CH2Cl2 molecule (Fig. 38b) held by hydrogen bonding interactions between the phenolic oxygens and the C–H bonds of DCM (C–H⋯O distance of 2.66 Å). At the same time, the Cl atoms (of DCM) interact with carbonyl oxygens from the neighbouring 145 via halogen bonding interactions (Cl⋯O = 3.183 Å).83
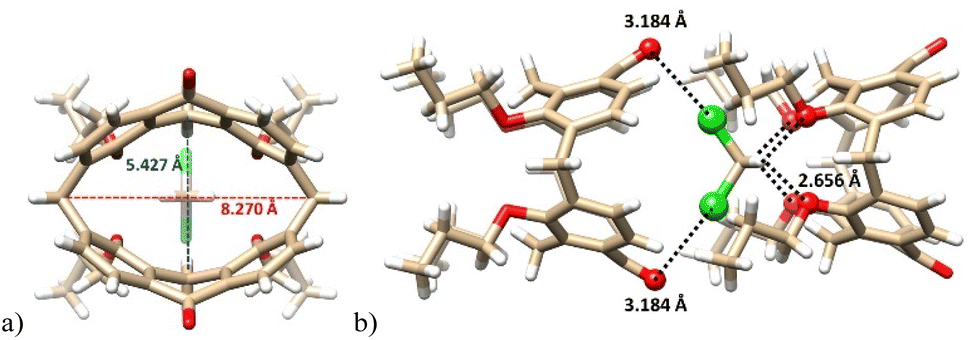 | ||
| Fig. 38 The X-ray structure of 145 DCM complex (a): highly distorted cavity of 145, (b) supramolecular interactions of DCM within the complex. | ||
The same type of chemistry was carried out starting with monochloromercurio derivative 129b which was reacted with I2 to provide the iodo calix[4]arene 147 (86%) and transformed into the corresponding carboxylic acid 148 (96%) via the lithiation with BuLi and the reaction with CO2 (Fig. 37). The final ketone-bridged macrocycle 149 was obtained in high yield (95%) by intramolecular Friedel–Crafts acylation, and subjected to nucleophilic addition (RLi or LiAlH4) to provide the corresponding alcohols 150a–e in good yields.84 Rigidified skeleton together with a hydroxyl group pointing into the cavity make these compounds suitable for the interaction with neutral guest molecules (MeOH, MeCN, MeNO2). An example of such a complex is shown in Fig. 37, where MeCN is captured by the CH–π interactions of the methyl group with the aromatic cavity and simultaneously by the HB interaction of the OH group with the nitrile nitrogen (O–H⋯N distance 2.257 Å).
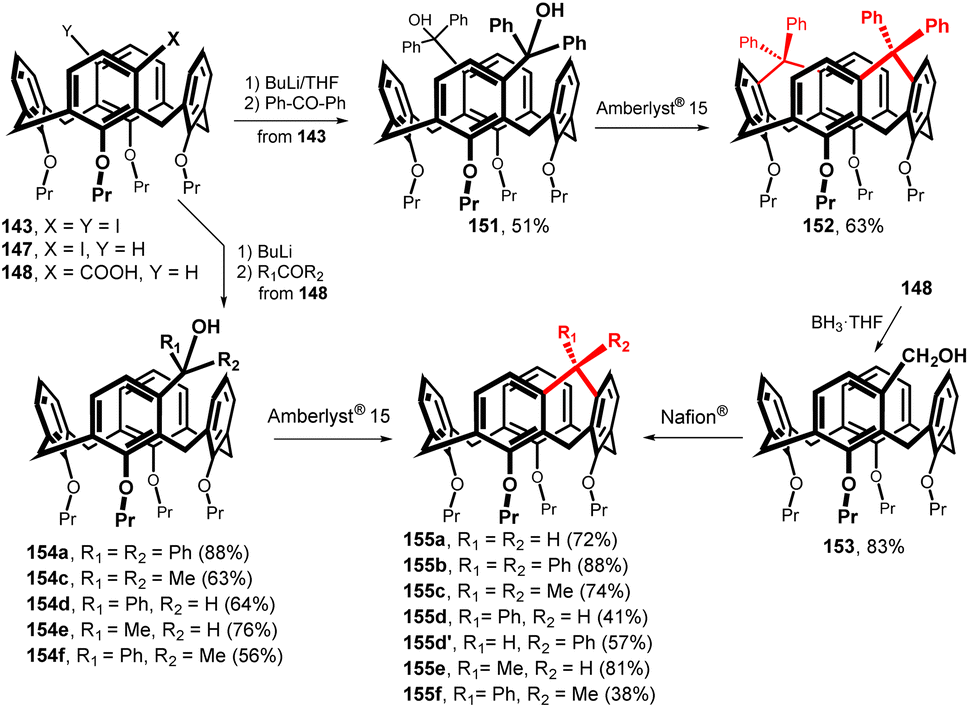
A similar strategy for the synthesis of single-atom bridges is based on the use of Friedel–Crafts intramolecular alkylation (Fig. 39). The initial iodo derivatives 143 and 147 were lithiated (BuLi/THF, −78 °C) and allowed to react with ketones or aldehydes to give alcohols 151 and 154a–f in good yields.85 Final cyclization using Amberlyst® 15 then provided the respective bridged macrocycles 155b–f.86 The utility of this approach for double bridged calix[4]arenes was demonstrated by the preparation of derivative 152, which was smoothly obtained in 63% yield. The methylene-bridged derivative 155a was prepared from the corresponding carboxylic acid 148 by reduction with borane (to furnish 153 in 83% yield) and subsequent cyclization with Nafion® which, in this particular case, gave higher yield (72%). All the bridged calixarenes 152 and 155a–f described above represent highly rigid cavities capable of complexing quaternary ammonium cations or neutral substances by cation–π or CH–π interactions.
 | ||
| Fig. 39 Upper rim-bridged calix[4]arenes by intramolecular Friedel–Crafts alkylation. | ||
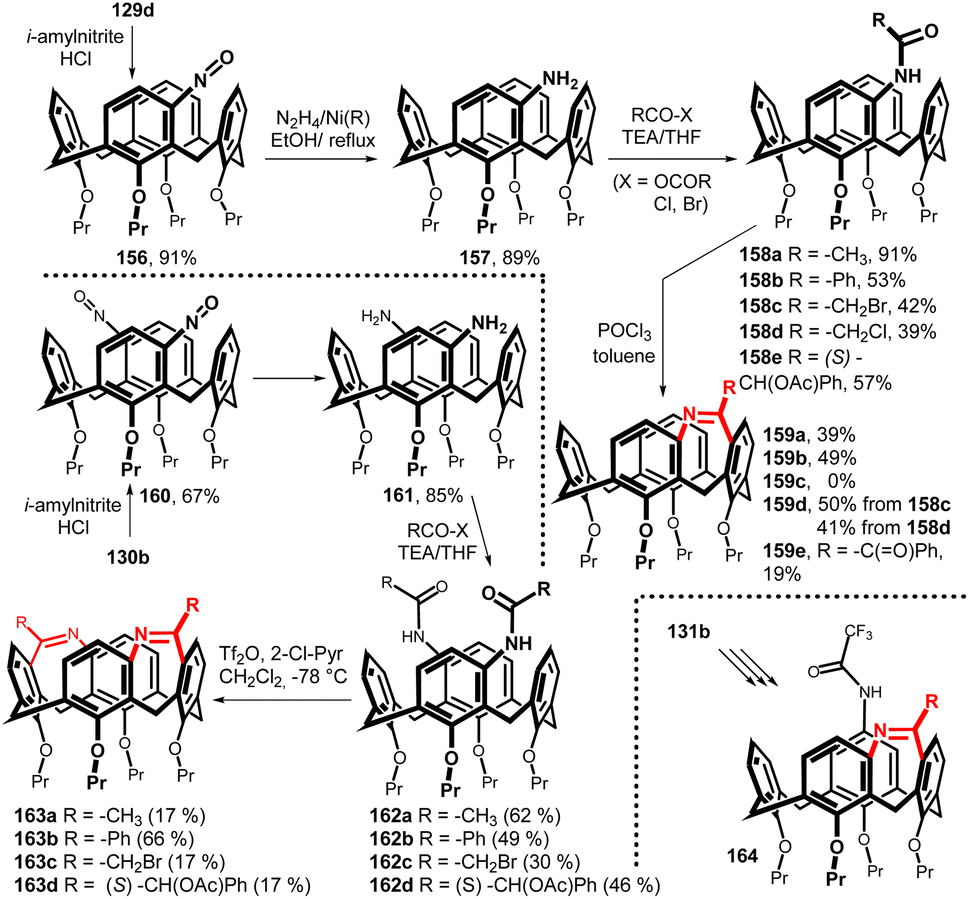
When the chloromercurio derivatives 129d and 130b were reacted with nitrosyl chloride, generated in situ from isopentyl nitrite and HCl at low temperature (0 °C), the corresponding nitroso calixarenes 156 and 160 were obtained in good yields.87 These key intermediates were then reduced to meta-amino derivatives 157 (89%) and 161 (85%) using RANEY® nickel and hydrazine hydrate (Fig. 40). The acylation with various carboxylic acid derivatives (chloride, anhydride, bromide) provided the corresponding monoamides 158a–e and diamides 162a–d. The intramolecular Bischler–Napieralski cyclization of monoamides 158 (reflux with POCl3 in toluene) led to a novel type of bridged calixarene containing a seven membered ring (diatomic bridge). The racemic product 159a was then successfully separated into pure enantiomers on a chiral column (Chiral Art Amylose-SA). The 1H NMR titration experiments indicated that the cavity of 159a was preorganised for the complexation of selected guest molecules (N-methylpyridinium salts) including some enantioselectivity towards N-methylnicotinium iodide.88 The use of mild reaction conditions for Bischler–Napieralski cyclization of diamides 162a–d (Tf2O/2-chloropyridine in CH2Cl2 at −78 °C) afforded the double-bridged product 163a–d. However, it turned out that this type of double bridging does not lead to very stable compounds and their decomposition occurs relatively quickly. A similar strategy starting from the meta, para isomer 131b led to the bridged systems 164 (Fig. 40).89
 | ||
| Fig. 40 The intramolecular Bischler–Napieralski cyclization of calix[4]arenes. | ||
A new strategy to bridge the upper rim of calix[4]arene was recently published.90 The starting ketone 149 (one-atom bridge) was subjected to Baeyer–Villiger reaction (Fig. 41) with meta-chloroperoxybenzoic acid in toluene to form a cyclic lactone 165 (58%) (two-atom bridge). Thus, expansion of the bridge led to an inherently chiral product (in the form of a racemate). The attempts to carry out the reaction enantioselectively using enzymes have failed, unfortunately. Conversion of the ketone 149 to the corresponding oxime 166 (74%) and subsequent Beckmann rearrangement led analogously to lactam 167 (59%). 1H NMR titration experiments with quaternary ammonium salts revealed some unusual complexation stoichiometries depending on the chirality of 167 (racemic mixture versus pure enantiomers).
 | ||
| Fig. 41 Transformation of ketone-bridged calix[4]arenes. | ||
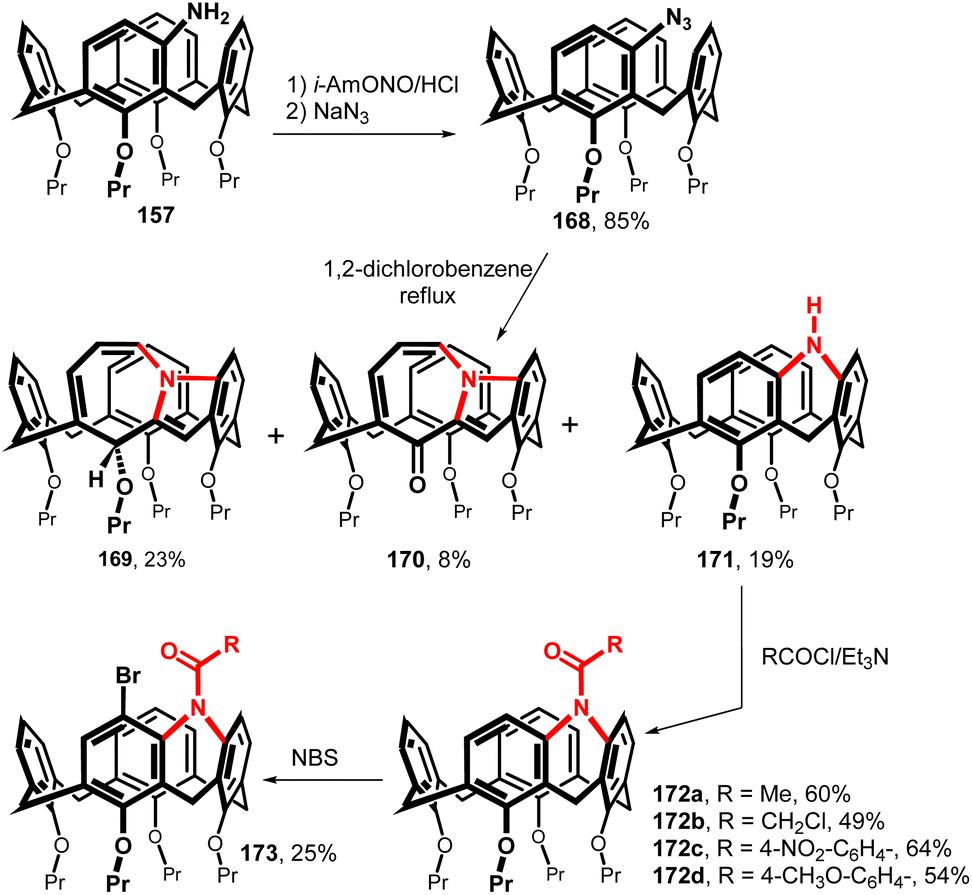
The amine bridged calix[4]arene was synthesised starting from meta-amino derivative 157 (Fig. 42). The reaction with isoamyl nitrite in HCl/THF at 0 °C generated a diazonium salt which was immediately reacted with NaN3 to provide an azide 168 in 85% yield. The thermal decomposition (1,2-dichlorobenzene/reflux) furnished a rather complex reaction mixture from which the three main products 169–171 were isolated. In addition to the expected amine 171 (19%), structures 169 and 170 with a seven-membered ring resulting from the rearrangement of the primary forming nitrene were also obtained.91 The amine-bridged calix[4]arene 171 was acylated under standard conditions (RCOCl/TEA) to afford the corresponding amides 172a–d. As shown by 1H NMR spectroscopy, the restricted rotation around the amidic C–N bond led finally to the desymmetrization of the whole molecule (atropisomerism) at low temperatures (slow exchange conditions). The introduction of bromine atom into the para-position of the neighbour ring (compound 173) led to inherently chiral system with hindered rotation at rt.92
 | ||
| Fig. 42 Synthesis of amine-bridged calix[4]arene derivatives. | ||
Conclusions and outlook
In this review, I have attempted to show the basic strategies leading to the bridging of the upper rim of calix[n]arenes. Although a substantial part of the structures comes from the chemistry of calix[4]arenes, where the exceptionally well-developed chemistry of these compounds can be applied, there are also examples showing the successful bridging of larger calixarenes. Using relatively diverse synthetic approaches, many structurally and functionally interesting systems with a rigidified cavity capable of various supramolecular applications (depending on the specific chemistry used) can be prepared.Much attention has been paid to the synthesis of systems with bridged meta positions of adjacent aromatic subunits. Although organomercury compounds are not among the most popular chemicals, their use in calixarene chemistry is proving to have a completely irreplaceable role, as it allows direct access to derivatives that are otherwise essentially unattainable. The bridged derivatives formed in this way show extremely rigid and often inherently chiral structures with many possible applications, e.g. in the field of chiral recognition. In addition, the presence of the fluorene moiety within the calixarene skeleton gives us the possibility to apply some procedures known from the chemistry of this compound (e.g. regioselective deprotonation of the methylene bridge followed by stereoselective alkylation).
As shown by some recent examples, a similar way of bridging calixarene systems (meta bridging) can be achieved by direct C–H activation (intramolecular coupling) using transition metal chemistry. It can be assumed that the further development of this strategy may lead to new macrocyclic systems with a hitherto unknown topology. Especially in the case of larger calixarenes, where system rigidification cannot be achieved by simple alkylation of the lower rim, new methods of bridging the upper rim could provide conformationally immobilized systems, thereby enabling greater use of these macrocycles in supramolecular chemistry.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Czech Science Foundation (23-07154S).Notes and references
- (a) C. D. Gutsche, Calixarenes Revisited, Royal Society of Chemistry, Cambridge, U. K., 1998 RSC; (b) C. D. Gutsche, Calixarenes: an Introduction, RSC Publishing, Cambridge, U. K., 2nd edn, 2008 Search PubMed.
- (a) L. Mandolini and R. Ungaro, Calixarenes in Action, Imperial College Press, London, 2000 CrossRef; (b) V. M. Mirsky and A. Yatsimirsky, Artificial Receptors for Chemical Sensors, Wiley, Weinheim, 2010 CrossRef; (c) P. Neri, J. L. Sessler and M. X. Wang, Calixarenes and beyond, Springer, Cham, Switzerland, 2016 CrossRef.
- V. Böhmer, H. Goldmann and W. Vogt, The first example of a new class of bridged calixarene, J. Chem. Soc., Chem. Commun., 1985, 667–668 RSC.
- V. Böhmer, H. Goldmann, R. Kaptein and L. Zetta, Photo-CIDNP studies on calixarenes and bridged calixarenes, J. Chem. Soc., Chem. Commun., 1987, 1358–1360 RSC.
- (a) V. Böhmer, H. Goldmann, W. Vogt, E. F. Paulus, F. L. Tobiason and M. J. Thielman, Bridged calix[4]arenes; x-ray crystal and molecular structures and spectroscopic studies, J. Chem. Soc., Perkin Trans. 2, 1990, 1769–1775 RSC; (b) E. Paulus, V. Böhmer, H. Goldmann and W. Vogt, The crystal and molecular structure of two calix[4]arenes bridged at opposite para positions, J. Chem. Soc., Perkin Trans. 2, 1987, 1609–1615 RSC.
- H. Goldmann, W. Vogt, E. Paulus and V. Böhmer, A series of calix[4]arenes, having two opposite para positions connected by an aliphatic chain, J. Am. Chem. Soc., 1988, 110, 6811–6817 CrossRef CAS.
- V. Böhmer and W. Vogt, Calix[4]arenes, bridged at the upper rim, Pure Appl. Chem., 1993, 65, 403–408 CrossRef.
- V. Böhmer, H. Goldmann, W. Vogt, J. Vicens and Z. Asfari, The synthesis of double-calixarenes, Tetrahedron Lett., 1989, 30, 1391–1394 CrossRef.
- J. D. Van Loon, L. C. Groenen, S. S. Wijmenga, W. Verboom and D. N. Reinhoudt, Upper rim calixcrowns: elucidation of the mechanism of conformational interconversion of calix[4]arenes by quantitative 2-D EXSY NMR spectroscopy, J. Am. Chem. Soc., 1991, 113, 2378 CrossRef CAS.
- A. Arduini, A. Casnati, M. Fabbi, P. Minari, A. Pochini, A. R. Sicuri and R. Ungaro, New artificial receptors from selectively functionalized calix[4]arenes, Supramol. Chem., 1993, 1, 235–246 CrossRef CAS.
- A. Arduini, S. Fanni, G. Manfredi, A. Pochini, R. Ungaro, A. R. Sicuri and F. Ugozzoli, Direct regioselective formylation of tetraalkoxycalix [4] arenes fixed in the cone conformation and synthesis of new cavitands, J. Org. Chem., 1995, 60, 1448–1453 CrossRef CAS.
- A. Dondoni, A. Marra, M.-C. Scherrmann, A. Casnati, F. Sansone and R. Ungaro, Synthesis and properties of O-glycosyl calix[4]arenes (calixsugars), Chem.–Eur. J., 1997, 3, 1774–1782 CrossRef CAS.
- H. D. Banks, A. Dondoni, M. Kleban and A. Marra, Computational and experimental studies of Di- and tetrasubstituted calix[4]arenes, Chirality, 2002, 14, 173–179 CrossRef CAS PubMed.
- A. Arduini, W. M. McGregor, A. Pochini, A. Secchi, F. Ugozzoli and R. Ungaro, New Upper Rim Pyridine-Bridged Calix[4]arenes: Synthesis and Complexation Properties toward Neutral Molecules and Ammonium Ions in Organic Media, J. Org. Chem., 1996, 61, 6881–6887 CrossRef CAS PubMed.
- A. Arduini, M. Cantoni, E. Graviani, A. Pochini, A. Secchi, A. R. Sicuri, R. Ungaro and M. Vincenti, Gas-phase complexation of neutral molecules by upper rim bridged calix[4]arenes, Tetrahedron, 1995, 51, 599–606 CrossRef CAS.
- R. Cacciapaglia, S. Di Stefano and L. Mandolini, On the 'livingness' of a dynamic library of cyclophane formaldehyde acetals incorporating calix[4]arene subunits, J. Phys. Org. Chem., 2008, 21, 688–693 CrossRef CAS.
- T. Arimura, S. Matsumoto, O. Teshima, T. Nagasaki and S. Shinkai, Calixarene-based molecular capsule, Tetrahedron Lett., 1991, 32, 5111–5114 CrossRef CAS.
- M. H. Düker, R. Gómez, C. M. L. Vande Velde and V. A. Azov, Upper rim tetrathiafulvalene-bridged calix[4]arenes, Tetrahedron Lett., 2011, 52, 2881–2884 CrossRef.
- M. H. Düker, F. Kutter, T. Dülcks and V. A. Azov, Calix[4]arenes with 1,2- and 1,3-upper rim tetrathiafulvalene bridges, Supramol. Chem., 2014, 26, 552–560 CrossRef.
- M. T. Blanda and K. E. Griswold, Synthesis of a Symmetric Octathio Bis(calix[4]arene) Cage Molecule, J. Org. Chem., 1994, 59, 4313–4315 CrossRef CAS.
- T. Takeda and A. Tsubouchi, Synthesis of alkenes by McMurry coupling and related reductive dimerization reactions, Sci. Synth., 2010, 47a, 247–325 Search PubMed.
- A. Arduini, S. Fanni, A. Pochini, A. R. Sicuri and R. Ungaro, Highly distorted Cone calix[4]arenes through intramolecular mc murry coupling reaction, Tetrahedron, 1995, 51, 7951–7958 CrossRef CAS.
- P. Lhotak and S. Shinkai, Structurally unusual calix[4]arene derivatives generated by intra- and intermolecular McMurry reactions, Tetrahedron Lett., 1996, 37, 645 CrossRef CAS.
- T. Haino, C. Fukunaga and Y. Fukazawa, A New Calix[5]arene-Based Container: Selective Extraction of Higher Fullerenes, Org. Lett., 2006, 8, 3545–3548 CrossRef CAS PubMed.
- J. Wang and C. D. Gutsche, Complexation of Fullerenes with Bis-calix[n]arenes Synthesized by Tandem Claisen Rearrangement1, J. Am. Chem. Soc., 1998, 120, 12226–12231 CrossRef CAS.
- M. Pitarch, V. McKee, M. Nieuwenhuyzen and M. A. McKervey, Synthesis of bridged, multifunctional calixarenes via ring closing metathesis, J. Org. Chem., 1998, 63, 946–951 CrossRef CAS.
- O. Struck, L. A. J. Chrisstoffels, R. J. W. Lugtenberg, W. Verboom, G. J. van Hummel, S. Harkema and D. N. Reinhoudt, Head-to-Head Linked Double Calix[4]arenes: Convenient Synthesis and Complexation Properties, J. Org. Chem., 1997, 62, 2487–2493 CrossRef CAS PubMed.
- H. Iwamoto, K. Niimi, T. Haino and Y. Fukazawa, Energetics of guest binding to calix[4]arene molecular containers, Tetrahedron, 2009, 65, 7259–7267 CrossRef CAS.
- (a) G. T. Hwang and B. H. Kim, Bis-calix[4]arenes with imine linkages: synthesis and binding study of thiopheno bis-calix[4]arene with viologens, Tetrahedron Lett., 2000, 41, 5917–5921 CrossRef CAS; (b) G. T. Hwang and B. H. Kim, Synthesis and binding studies of multiple calix[4]arenes, Tetrahedron, 2002, 58, 9019–9028 CrossRef CAS.
- A. Arduini, R. Ferdani, A. Pochini and A. Secchi, Synthesis of Upper Rim Covalently Linked Double Calix[6]arenes, Tetrahedron, 2000, 56, 8573–8577 CrossRef CAS.
- A. Yilmaz, S. Memon and M. Yilmaz, Synthesis and study of allosteric effects on extraction behavior of novel calixarene-based dichromate anion receptors, Tetrahedron, 2002, 58, 7735–7740 CrossRef CAS.
- H. M. Chawla and T. Gupta, Novel bis-calix [4] arene based molecular probe for ferric iron through colorimetric, ratiometric, and fluorescence enhancement response, Tetrahedron Lett., 2015, 56, 793–796 CrossRef CAS.
- A. E.-G. E. Amr, A. M. Mohamed and A. A. Ibrahim, Synthesis of Some New Chiral Tricyclic and Macrocyclic Pyridine Derivatives as Antimicrobial Agents, Z. Naturforsch., B, 2003, 58, 861–868 CrossRef CAS.
- S. K. Sharma and C. D. Gutsche, Upper Rim Substitution of Calix[4]arenes via Their Upper Rim A,C Dinitro Compounds1, J. Org. Chem., 1999, 64, 998–1003 CrossRef CAS PubMed.
- F. Sansone, L. Baldini, A. Casnati, M. Lazzarotto, F. Ugozzoli and R. Ungaro, Biomimetic macrocyclic receptors for carboxylate anion recognition based on C-linked peptidocalix[4]arenes, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4842–4847 CrossRef CAS PubMed.
- V. Šťastný, P. Lhoták, V. Michlová, I. Stibor and J. Sýkora, Novel biscalix[4]arene-based anion receptors, Tetrahedron, 2002, 58, 7207–7211 CrossRef.
- S. J. Loeb and B. R. Cameron, Calixarenes for Separations, Am. Chem. Soc., 2000, 757(ch. 21), 283–295 CAS.
- A. E.-G. E.-S. Amr, M. Abo-Ghalia and M. M. Abdalah, Synthesis of Novel Macrocyclic Peptido-calix[4]arenes and Peptidopyridines as Precursors for Potential Molecular Metallacages, Chemosensors and Biologically Active Candidates, Z. Naturforsch., B, 2006, 61, 1335–1345 CrossRef CAS.
- B.-H. Huisman, D. M. Rudkevich, A. Farrán, W. Verboom, F. C. J. M. van Veggel and D. N. Reinhoudt, Synthesis of (Hemi)Carceplex Adsorbates for Self-Assembly on Gold, Eur. J. Org Chem., 2000, 2000, 269–274 CrossRef.
- A. M. van Wageningen, W. Verboom, X. Zhu, J. A. Ripmeester and D. N. Reinhoudt, Solid state NMR spectroscopy of calix [4] arene-based carceplexes, Supramol. Chem., 1998, 9, 31–36 CrossRef CAS.
- A. M. A. van Wageningen, J. P. M. van Duynhoven, W. Verboom and D. N. Reinhoudt, Synthesis and functionalization of calix[4]arene-based carceplexes, J. Chem. Soc., Chem. Commun., 1995, 1941–1942 RSC.
- P. Timmerman, W. Verboom, F. C. J. M. van Veggel, J. P. M. van Duynhoven and D. N. Reinhoudt, A Novel Type of Stereoisomerism in Calix[4]arene-Based Carceplexes, Angew. Chem., Int. Ed., 1994, 33, 2345–2348 CrossRef.
- A. M. van Wageningen, P. Timmerman, J. P. van Duynhoven, W. Verboom, F. C. van Veggel and D. N. Reinhoudt, Calix [4] arene-Based (Hemi) carcerands and Carceplexes: Synthesis, Functionalization, and Molecular Modeling Study, Chem.–Eur. J., 1997, 3, 639–654 CrossRef CAS.
- P. Timmerman, E. A. Brinks, W. Verboom and D. N. Reinhoudt, Synthetic receptors with preorganized cavities that complex prednisolone-21-acetate, J. Chem. Soc., Chem. Commun., 1995, 417–418 RSC.
- P. Timmerman, W. Verboom, F. C. van Veggel, W. P. van Hoorn and D. N. Reinhoudt, An organic molecule with a rigid cavity of nanosize dimensions, Angew. Chem., Int. Ed., 1994, 33, 1292–1295 CrossRef.
- B. Tomapatanaget and T. Tuntulani, Lower rim tetra-substituted and upper rim ferrocene amide calix[4]arenes: synthesis, conformation and anion-binding properties, Tetrahedron Lett., 2001, 42, 8105–8109 CrossRef CAS.
- B. Tomapatanaget, T. Tuntulani and O. Chailapakul, Calix[4]arenes Containing Ferrocene Amide as Carboxylate Anion Receptors and Sensors, Org. Lett., 2003, 5, 1539–1542 CrossRef CAS PubMed.
- H. Iwamoto, Y. Yukimasa and Y. Fukazawa, Synthesis and binding behavior of a Zn(II)-porphyrin having calix[5]arene cap, Tetrahedron Lett., 2002, 43, 8191–8194 CrossRef CAS.
- H. Iwamoto, S. Nishi and T. Haino, Highly shape-selective guest encapsulation in the precisely defined cavity of a calix[4]arene-capped metalloporphyrin, Chem. Commun., 2011, 47, 12670–12672 RSC.
- (a) T. Haino, H. Fukuoka, H. Iwamoto and Y. Fukazawa, Synthesis and Enantioselective Recognition of a Calix[5]arene-based Chiral Receptor, Supramol. Chem., 2008, 20, 51–57 CrossRef CAS; (b) T. Haino, H. Fukuoka, M. Katayama and Y. Fukazawa, Enantioselective Recognition of Calix[5]arene-based Artificial Receptor Bearing Chiral Macrocycle for Chiral Ethyltrimethylammonium Salts, Chem. Lett., 2007, 36, 1054–1055 CrossRef CAS.
- C. Hocquelet, C. K. Jankowski and L. Mauclaire, New tubular products from calixarene–cyclodextrin coupling, Tetrahedron, 2007, 63, 10834–10839 CrossRef CAS.
- B. H. Kim, E. J. Jeong, G. T. Hwang and N. Venkatesan, Multiple Cycloadditive Macrocyclization: An Efficient Method for Crown Ether-Type Cyclophanes, Bis-Calix[4]arenes and Silamacrocycles, Synthesis, 2001, 2191–2202 CrossRef CAS.
- G. T. Hwang and B. H. Kim, Cyclophane-type bis-calix[4]arenes: efficient synthesis via quadruple cycloadditive macrocyclization and conformational study, Tetrahedron Lett., 2000, 41, 10055–10060 CrossRef CAS.
- S. Zahim, L. A. Wickramasinghe, G. Evano, I. Jabin, R. R. Schrock and P. Müller, Calix[6]azacryptand Ligand with a Sterically Protected Tren-Based Coordination Site for Metal Ions, Org. Lett., 2016, 18, 1570–1573 CrossRef CAS PubMed.
- L. A. Wickramasinghe, R. R. Schrock, C. Tsay and P. Müller, Molybdenum Complexes that Contain a Calix[6]azacryptand Ligand as Catalysts for Reduction of N2 to Ammonia, Inorg. Chem., 2018, 57, 15566–15574 CrossRef CAS PubMed.
- D. E. Phipps and P. D. Beer, A [2]catenane containing an upper-rim functionalized calix[4]arene for anion recognition, Tetrahedron Lett., 2009, 50, 3454–3457 CrossRef CAS.
- R. Miao, Q.-Y. Zheng, C.-F. Chen and Z.-T. Huang, A novel calix[4]arene fluorescent receptor for selective recognition of acetate anion, Tetrahedron Lett., 2005, 46, 2155–2158 CrossRef CAS.
- M. Segura, B. Bricoli, A. Casnati, E. M. Muñoz, F. Sansone, R. Ungaro and C. Vicent, A Prototype Calix[4]arene-Based Receptor for Carbohydrate Recognition Containing Peptide and Phosphate Binding Groups, J. Org. Chem., 2003, 68, 6296–6303 CrossRef CAS PubMed.
- J. H. Mai, J. M. Liu, S. Y. Li and H. F. Jiang, A fluorescent probe for fluoride ion based on 2-aminopyridyl-bridged calix[6]arene, Chin. Chem. Lett., 2009, 20, 1191–1194 CrossRef CAS.
- Y. K. Agrawal and K. R. Sharma, Speciation, liquid–liquid extraction, sequential separation, preconcentration, transport and ICP-AES determination of Cr(III), Mo(VI) and W(VI) with calix-crown hydroxamic acid in high purity grade materials and environmental samples, Talanta, 2005, 67, 112–120 CrossRef CAS PubMed.
- M. O. Vysotsky, A. Bogdan, L. Wang and V. Böhmer, Template synthesis of multi-macrocycles by metathesis reaction, Chem. Commun., 2004, 1268–1269 RSC.
- O. Molokanova, A. Bogdan, M. O. Vysotsky, M. Bolte, T. Ikai, Y. Okamoto and V. Böhmer, Calix[4]arene-Based Bis[2]catenanes: Synthesis and Chiral Resolution, Chem.–Eur. J., 2007, 13, 6157–6170 CrossRef CAS PubMed.
- Y. Cao, L. Wang, M. Bolte, M. O. Vysotsky and V. Böhmer, Synthesis of huge macrocycles using two calix[4]arenes as templates, Chem. Commun., 2005, 3132–3134 RSC.
- L. Wang, M. O. Vysotsky, A. Bogdan, M. Bolte and V. Böhmer, Multiple Catenanes Derived from Calix[4]arenes, Science, 2004, 304, 1312–1314 CrossRef CAS PubMed.
- (a) A. Bogdan, M. O. Vysotsky, T. Ikai, Y. Okamoto and V. Böhmer, Rational Synthesis of Multicyclic Bis[2]catenanes, Chem.–Eur. J., 2004, 10, 3324–3330 CrossRef CAS PubMed; (b) M. O. Vysotsky, M. Bolte, I. Thondorf and V. Böhmer, New Molecular Topologies by Fourfold Metathesis Reactions within a Hydrogen-Bonded Calix[4]arene Dimer, Chem.–Eur. J., 2003, 9, 3375–3382 CrossRef CAS PubMed.
- (a) C. Gaeta, M. O. Vysotsky, A. Bogdan and V. Böhmer, Fourfold [2]Rotaxanes Based on Calix[4]arenes, J. Am. Chem. Soc., 2005, 127, 13136–13137 CrossRef CAS PubMed; (b) O. Molokanova, M. O. Vysotsky, Y. Cao, I. Thondorf and V. Böhmer, Fourfold [2]Rotaxanes of Calix[4]arenes by Ring Closure, Angew. Chem., Int. Ed., 2006, 45, 8051–8055 CrossRef CAS PubMed.
- A. Bogdan, M. Bolte and V. Böhmer, Molecules with New Topologies Derived from Hydrogen-Bonded Dimers of Tetraurea Calix[4]arenes, Chem.–Eur. J., 2008, 14, 8514–8520 CrossRef CAS PubMed.
- Y. Rudzevich, Y. Cao, V. Rudzevich and V. Böhmer, Stepwise Synthesis and Selective Dimerisation of Bis- and Trisloop Tetra-urea Calix[4]arenes, Chem.–Eur. J., 2008, 14, 3346–3354 CrossRef CAS PubMed.
- Y. Rudzevich, V. Rudzevich, C. Moon, G. Brunklaus and V. Böhmer, Self-assembled dendrimers with uniform structure, Org. Biomol. Chem., 2008, 6, 2270–2275 RSC.
- O. Molokanova, G. Podoprygorina, M. Bolte and V. Böhmer, Multiple catenanes based on tetraloop derivatives of calix[4]arenes, Tetrahedron, 2009, 65, 7220–7233 CrossRef CAS.
- M. Chas and P. Ballester, A dissymmetric molecular capsule with polar interior and two mechanically locked hemispheres, Chem. Sci., 2012, 3, 186–191 RSC.
- J. Holub, V. Eigner, L. Vrzal, H. Dvořáková and P. Lhoták, Calix [4] arenes with intramolecularly bridged meta positions prepared via Pd-catalysed double C–H activation, Chem. Commun., 2013, 49, 2798–2800 RSC.
- F. Stejskal, P. Cuřínová and P. Lhoták, Unexpected formation of disulfide-based biscalix[4]arenes, Tetrahedron, 2016, 72, 760–766 CrossRef CAS.
- X. Zhang, S. Tong, J. Zhu and M.-X. Wang, Inherently chiral calixarenes by a catalytic enantioselective desymmetrizing cross-dehydrogenative coupling, Chem. Sci., 2023, 14, 827–832 RSC.
- P. Slavík, M. Dudič, K. Flídrová, J. Sýkora, I. Císařová, S. Böhm and P. Lhoták, Unprecedented Meta-Substitution of Calixarenes: Direct Way to Inherently Chiral Derivatives, Org. Lett., 2012, 14, 3628–3631 CrossRef PubMed.
- K. Flídrová, S. Böhm, H. Dvořáková, V. Eigner and P. Lhoták, Dimercuration of Calix[4]arenes: Novel Substitution Pattern in Calixarene Chemistry, Org. Lett., 2014, 16, 138–141 CrossRef PubMed.
- P. Slavík, K. Flídrová, H. Dvořáková, V. Eigner and P. Lhoták, Meta-arylation of calixarenes using organomercurial chemistry, Org. Biomol. Chem., 2013, 11, 5528–5534 RSC.
- K. Flídrová, P. Slavik, V. Eigner, H. Dvořáková and P. Lhoták, meta-Bridged calix[4]arenes: a straightforward synthesis via organomercurial chemistry, Chem. Commun., 2013, 49, 6749–6751 RSC.
- P. Slavík, H. Dvořáková, V. Eigner and P. Lhoták, Regioselective alkylation of a methylene group via meta-bridging of calix[4]arenes, Chem. Commun., 2014, 50, 10112–10114 RSC.
- (a) P. Slavík, H. Dvořáková, M. Krupička and P. Lhoták, Unexpected cleavage of upper rim-bridged calix 4 arenes leading to linear oligophenolic derivatives, Org. Biomol. Chem., 2018, 16, 838–843 RSC; (b) P. Slavík and P. Lhoták, Unusual reactivity of upper-rim bridged calix[4]arenes - Friedel-Crafts alkylation via cleavage of the macrocyclic skeleton, Tetrahedron Lett., 2018, 59, 1757–1759 CrossRef; (c) P. Slavík, M. Krupička, V. Eigner, L. Vrzal, H. Dvořáková and P. Lhoták, Rearrangement of meta-Bridged Calix 4 arenes Promoted by Internal Strain, J. Org. Chem., 2019, 84, 4229–4235 CrossRef PubMed.
- P. Slavík, M. Kohout, S. Böhm, V. Eigner and P. Lhoták, Synthesis of inherently chiral calixarenes via direct mercuration of the partial cone conformation, Chem. Commun., 2016, 52, 2366–2369 RSC.
- P. Slavík, V. Eigner, S. Böhm and P. Lhoták, Mercuration of calix[4]arenes immobilized in the 1,2- and 1,3-alternate conformations, Tetrahedron Lett., 2017, 58, 1846–1850 CrossRef.
- P. Slavík, V. Eigner and P. Lhoták, Shaping of calix[4]arenes via double bridging of the upper rim, CrystEngComm, 2016, 18, 4964–4970 RSC.
- P. Slavík, V. Eigner and P. Lhoták, Intramolecularly Bridged Calix[4]arenes with Pronounced Complexation Ability toward Neutral Compounds, Org. Lett., 2015, 17, 2788–2791 CrossRef PubMed.
- P. Slavík, H. Dvořáková, V. Eigner and P. Lhoták, Meta-Bridged calix[4]arenes prepared by Friedel-Crafts alkylation, New J. Chem., 2017, 41, 14738–14745 RSC.
- P. Slavík, V. Eigner and P. Lhoták, meta-Bridged calix[4]arenes with the methylene moiety possessing in/out stereochemistry of substituents, New J. Chem., 2018, 42, 16646–16652 RSC.
- L. Vrzal, K. Flídrová, T. Tobrman, H. Dvořáková and P. Lhoták, Use of residual dipolar couplings in conformational analysis of meta-disubstituted calix[4]arenes, Chem. Commun., 2014, 50, 7590–7592 RSC.
- M. Tlustý, P. Slavík, M. Kohout, V. Eigner and P. Lhoták, Inherently Chiral Upper-Rim-Bridged Calix[4]arenes Possessing a Seven Membered Ring, Org. Lett., 2017, 19, 2933–2936 CrossRef PubMed.
- M. Tlustý, V. Eigner, M. Babor, M. Kohout and P. Lhoták, Synthesis of upper rim-double-bridged calix[4]arenes bearing seven membered rings and related compounds, RSC Adv., 2019, 9, 22017–22030 RSC.
- M. Tlustý, D. Spálovská, M. Kohout, V. Eigner and P. Lhoták, Ketone transformation as a pathway to inherently chiral rigidified calix[4]arenes, Chem. Commun., 2020, 56, 12773–12776 RSC.
- M. Tlustý, V. Eigner and P. Lhoták, Azide Decomposition as a Pathway to Intramolecularly Upper-Rim-Bridged Calix[4]arenes, Synthesis, 2022, 54, 1301–1308 CrossRef.
- M. Tlustý, V. Eigner, H. Dvořáková and P. Lhoták, The atropisomeric dynamic behaviour of symmetrical N-acylated amine-bridged calix[4]arenes, New J. Chem., 2022, 46, 11774–11781 RSC.
| This journal is © The Royal Society of Chemistry 2024 |