
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMedicinal chemistry perspective on the structure–activity relationship of stilbene derivatives†
Saghi Sepehri *ab,
Mina Khedmatic,
Faeze Yousef-Nejadd and
Mohammad Mahdavi*d
*ab,
Mina Khedmatic,
Faeze Yousef-Nejadd and
Mohammad Mahdavi*d
aPharmaceutical Sciences Research Center, Ardabil University of Medical Sciences, Ardabil, Iran. E-mail: saghisepehridr@gmail.com; s.sepehri@arums.ac.ir; Fax: +98-45-33522197; Tel: +98-45-33522437-39, ext. 164
bDepartment of Medicinal Chemistry, School of Pharmacy, Ardabil University of Medical Sciences, Ardabil, Iran
cStudents Research Committee, School of Pharmacy, Ardabil University of Medical Sciences, Ardabil, Iran
dEndocrinology and Metabolism Research Center, Endocrinology and Metabolism Research Institute, Tehran University of Medical Sciences, Tehran, Iran. E-mail: momahdavi@sina.tums.ac.ir
First published on 20th June 2024
Abstract
Stilbenes are a small family of polyphenolic secondary metabolites produced in a variety of closely related plant species. These compounds function as phytoalexins, aiding plant defense against phytopathogens and plants' adaptation to abiotic environmental factors. Structurally, some important phenolic compounds have a 14-carbon skeleton and usually have two isomeric forms, Z and E. Stilbenes contain two benzene rings linked by a molecule of ethanol or ethylene. Some derivatives of natural (poly)phenolic stilbenes such as resveratrol, pterostilbene, and combretastatin A-4 have shown various biological activities, such as anti-microbial, anti-cancer, and anti-inflammatory properties as well as protection against heart disease, Alzheimer's disease, and diabetes. Among stilbenes, resveratrol is certainly the most popular and extensively studied for its health properties. In recent years, an increasing number of stilbene compounds have been investigated for their bioactivity. This review focuses on the assessment of synthetic stilbene derivatives in terms of their biological activities and structure–activity relationship. The goal of this study is to consider the structural changes and different substitutions on phenyl rings that can improve the desired medicinal effects of stilbene-based compounds beyond the usual standards and subsequently discover biological activities by identifying effective alternatives of the evaluated compounds.
1. Introduction
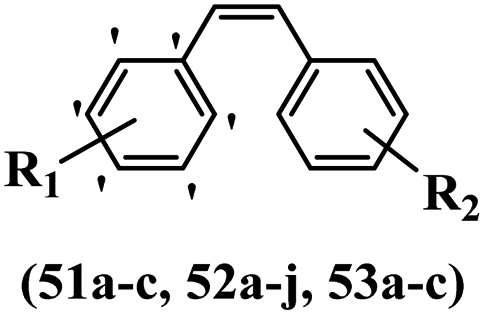
Stilbene, 1,2-diphenylethylene, gets its name from the Greek word “stilbos”, which means “shining”.1 It is a small molecule with a molecular weight of 180 g mol−1.2 The chemical structure of stilbene is composed of a 14-carbon skeleton. Stilbenes widely exist in nature and may act as phytoalexins, and some plants in response to pathogen attack and other stresses produce stilbenes as defense compounds.3,4 Stilbenes are abundant in plants with diverse vital biological activities.5 They are versatile structures composed of two aromatic rings linked by an ethylene moiety and exist in two diastereoisomeric forms, E-/Z-isomers; thus, they can undergo E/Z isomerization, altering their general configuration and decreasing their biological activity.6 Stilbenes show different biological activities such as anti-microbial, anti-oxidant, anti-leukemia, anti-platelet, protein tyrosine kinase inhibitor, anti-inflammatory, anti-cancer, and anti-HIV activities.7 Thus, stilbenes and their derivatives, which are vital groups of synthetic compounds and natural products, have attracted significant attention for their various pharmacological activities, complicated structures, and useful health properties.8 In addition to studying the various biological activities of stilbene analogues, the possible application of stilbene analogues as preservatives can be a new research direction.9 Moreover, these derivatives have attracted significant attention in diverse fields, including food biotechnology, drug discovery and development, and healthcare.10 The structure of stilbenes is not only limited to pharmaceutical and biological sciences but has also attracted attention from scientists because of their other vital properties, such as large geometrical alteration upon isomerization, high thermal stability of the Z isomer, high quantum yield for photochemical isomerization, and direct synthesis.11,12 Over the past years, increasing articles on stilbene have been found in the literature. Thus, it is a privileged structural scaffold belong to an enormous family of bioactive molecules, including synthetic molecules and natural products.11,13 More than 400 stilbene derivatives have been identified, which include different structures with various substituents at diverse positions.14 Hydroxylated stilbenes, such as resveratrol, pterostilbene, pinosylvin, and combretastatin A-4 (CA-4), are natural compounds that exhibit many biological activities (Fig. 1).12 Resveratrol and its analogues are well-known for their antioxidant properties against reactive oxygen species (ROS), which cause oxidative damage to biological substances and play a role in aging and inflammation.15 Resveratrol, a naturally occurring phytoalexin found in grapes and other plants, has been shown to inhibit carcinogenesis and tumor cell cycle progression, as well as interfere with intracellular signal transduction regulating cell survival and apoptosis (programmed cell death) in various human cancer cell lines.16,17 Piceatannol (PIC) (E-2,3′,4′,5-tetrahydroxystilbene) is a phenolic compound (stilbenoid) and a hydroxylated analogue of resveratrol (Fig. 1). Grapes, passion fruit, white tea, Japanese knotweed, Asian legume, and Korean rhubarb are some crucial sources of PIC. However, because the level of PIC in grapes is lower than that of resveratrol, it has received significantly less research attention compared to resveratrol. Scientists have reported that the seeds of passion fruit (Passiflora edulis) have a high content of PIC, which displays various biological activities such as protection of the skin from ultraviolet B irradiation, inhibition of melanogenesis, promotion of collagen synthesis, a vasorelaxant effect and Sirt1 induction activity. PIC possesses potent antioxidant activity and has chemopreventive and anti-cancer properties.18 Z-CA-4 is a polyphenol, which was first isolated from an African bush willow tree in 1982, Combretum caffrum. It is recognized for its potent anti-angiogenic and anti-tumor activities and potent depolymerizing agents as well as a strong tubulin polymerization inhibitor.13,19 Numerous derivatives have been developed to search for compounds with higher biological activity, such as ombrabulin, which shows higher activity than CA-4 (Fig. 1). Currently, it is being studied in phase III clinical trials for the treatment of advanced-stage soft-tissue carcinoma.20 In January 2013, Sanofi said it discontinued the development of ombrabulin after disappointing results from phase III clinical trials. Furthermore, phosphate derivatives have demonstrated good activity and have been used in clinical trials.21 Interestingly, in 2006, Li et al.3 synthesized stilbene derivatives with substituted hydroxyl groups and found that two of these compounds, E-2′,3,5′,5-tetrathydroxystilbene and E-3′,5,5′,6-tetrahydroxystilbene-2-nitrogen, inhibited SARS coronavirus replication using an in vitro model (Fig. 1). Thus, stilbene-based compounds can also be considered as promising anti-COVID-19 drug candidates due to their ability to disrupt the spike protein.6 Tamoxifen, a stilbene derivative, is currently used to treat several types of breast cancer in women, as well as a hormone treatment for male breast cancer (Fig. 1).20 Recently, many scientific institutions have been conducting research on stilbenes as alternative antibiotic growth stimulants. These compounds can be produced in plants by combining coumaric acid and cinnamic acid. Chalcones and flavanols can also be used for their synthesis. They have fungistatic properties (they inhibit fungi growth), toxic properties to fungi (they kill fungi), and estrogenic properties.22 Stilbenes may decrease obesity by regulating fat metabolism pathways such as adipogenesis, lipogenesis, lipolysis, and thermogenesis. Researchers are also investigating stilbene derivatives for cell proliferation and cytochrome P450 inhibitory activity.23,24 Natural product research, combined with the powerful possibilities provided by synthetic chemistry may provide an excellent method for discovering new structures and identifying therapeutic targets.13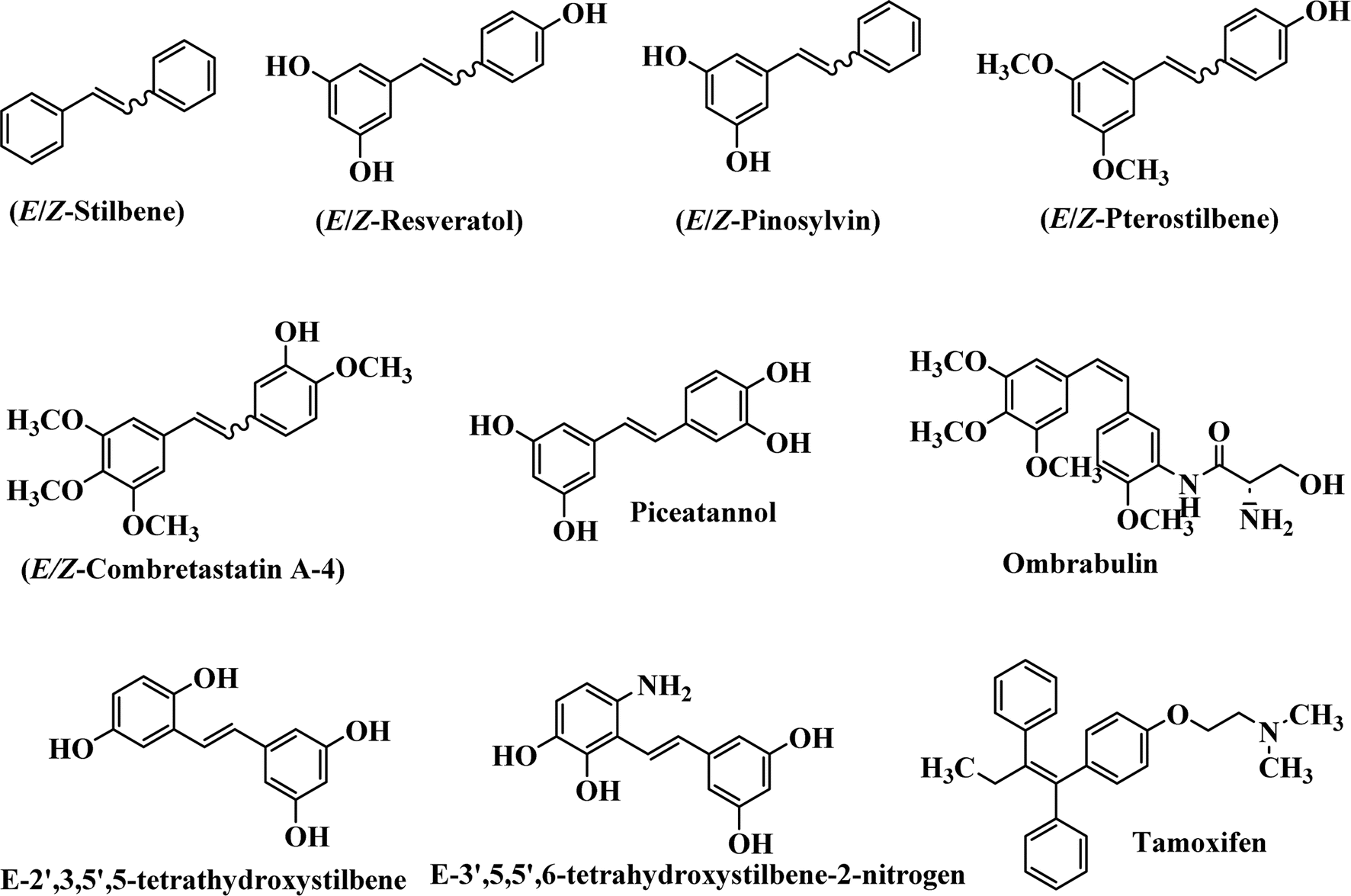 | ||
| Fig. 1 Some chemical structures of well-known stilbene-based derivatives and drugs. | ||
Other stilbene-based drugs that have been approved for use include dienestrol, toremifene, clomifene, tapinarof, raloxifene, ospemifene, and hydroxystilbamidine. Furthermore, some derivatives are under continuing clinical trials, such as cystic fibrosis-NCT04166396, chronic obstructive pulmonary disease-NCT03819517, chemoprevention-NCT04266353, combretastatin A1 di-phosphate/CA-1P, also known as OXI-4503 (acute myelogenous leukaemia and myelodysplastic syndromes-NCT02576301), pterostilbene or benvitimod, fispemifene, afimoxifene, droloxifene, and enclomiphene.11 Moreover, Ramizol, a first-generation stilbene-based antibiotic, is effective against 100 clinical isolates of C. difficile,25 and presently is under pre-clinical testing for the treatment of C. difficile-related diseases.26 Fosbretabulin (CA-4 phosphate) has been investigated in numerous clinical trials as monotherapy and combined therapy with other chemotherapeutic agents, such as carboplatin, paclitaxel, bevacizumab and pazopanib.27
However, despite all these recognized biological activities and advantages, the poor solubility of these analogues presents a critical problem in terms of their bioavailability, and thus prevents stilbene analogues from exhibiting the required activity. Some studies have reported that the complexation of polyphenols with cyclodextrins (CDs) and micellar systems results in a noticeable improvement in their aqueous solubility, and even increases their stability and bioactivity.10 CDs were shown to be more helpful for improving the solubility of stilbene with the benefit of being less toxic to humans.24 In addition, nanoformulation techniques have been newly applied to enhance the bioavailability and targeting ability of stilbene analogues.28 Some researchers conjugated stilbene analogues with mannose, glucose, and galactose to increase their solubility29 or yield phosphate and carbamate prodrug salts.30 Another strategy to increase their solubility is added or switching their groups with more polar groups based on the their SAR.31–34
This study provides an overview of the synthetic compounds derived from the stilbene scaffold as anti-microbial, anti-cancer, antioxidant, liver enzyme inhibitors, anti-Alzheimer's, anti-diabetes, and other agents. These compounds have undergone chemical modifications, such as the addition of substituents with varying electronic effects or the incorporation of heteroaromatic groups instead of phenyl rings. The biological activity of these compounds is mainly influenced by their chemical structure and the substituent groups attached to them.35 This comprehensive review mainly focuses on synthetic stilbene derivatives and the structure–activity relationship (SAR) studies on their various biological activities.
2. Anti-microbial activity
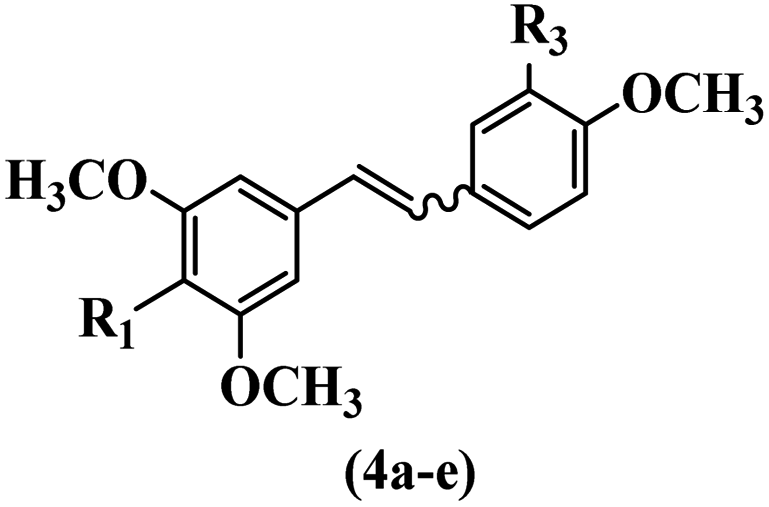
Pathogenic microorganisms have been a threat to humans throughout history, causing significant morbidity and mortality. Until the discovery of the first true antibiotic, penicillin, in 1928 and sulfa drugs in the 1930s, the only means of combating infectious diseases were various types of plant extracts, although their use yielded varying results.36 Stilbenes have long been recognized as potent anti-bacterial agents, and they continue to pique the interest of many research groups working with various bacteria. The discovery of antibiotic activity against bacteria and fungi sparked interest in stilbene derivative research.3,4 One example is resveratrol, which naturally exists in plants and has anti-microbial activity against both Gram-positive and Gram-negative bacteria.37 Anti-microbial activity of stilbene derivatives is dependent on the presence of a hydroxyl group in their primary phenyl ring (2-hydroxy, 3-hydroxy, and 4-hydroxy derivatives). If the primary phenyl ring lacks a hydroxyl substituent, 2,5-dihydroxy substituents in the secondary phenyl ring are required for anti-microbial activity. The effect of hydroxyl groups on anti-microbial activity is not surprising, given that phenol is one of the most important anti-microbial agents.38 The anti-microbial activity increases in derivatives with substituents (F, I, and Br). This can be explained by the change in the partition coefficient and increased permeability of cell membranes to fluoride derivatives, rather than the presence of the substituents themselves.39,40Pettit et al.41 described the synthesis and assessment of the anti-microbial activity of E/Z-CA-4 analogues. Most analogues were inactive against all strains. Among the synthesized compounds, Z-4b exhibited the highest anti-microbial activity against M. luteus. Also, E-4a having OH groups in the 4- and 3′-positions on its phenyl ring showed activity against C. neoformans; however, none of the compounds showed activity against this strain. Also, the N,N-diethylamino moiety in compound 4b in the 3′-position on the phenyl ring was replaced with N-pyrrolidinyl in 4c, which showed less activity against S. aureus, E. faecalis, and M. luteus. In addition, the presence of hydrophilic groups improved the anti-microbial activity compared to lipophilic groups (4d and 4e) against N. gonorrhoeae. The results showed that the anti-bacterial activity was greater than anti-fungal activity. According to the obtained results, the Z-isomers showed stronger anti-microbial activity than the E-isomers (Table 1).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Z/E | R1 | R3 | MIC (μg/disk) | |||||
| C. albicans | N. gonorrhoeae | M. luteus | S. aureus | E. faecalis | C. neoformans | ||||
| 4a | E | OH | OH | 50–100 | 12.5–25 | — | — | — | 6.25–12.5 |
| 4b | Z | OCH3 | 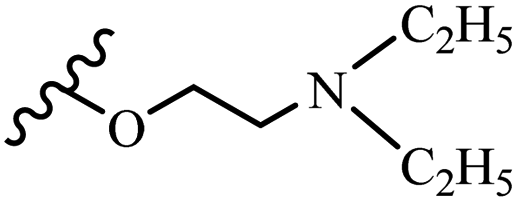 |
— | 50–100 | <6.25 | 25–50 | 50–100 | — |
| 4c | Z | OCH3 | 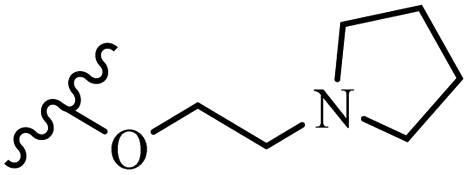 |
— | — | 12.5–25 | 50–100 | — | — |
| 4d | Z | OCH3 | 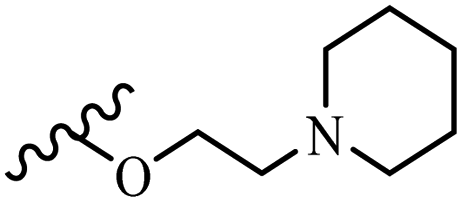 |
— | — | — | — | — | — |
| 4e | Z | OCH3 | 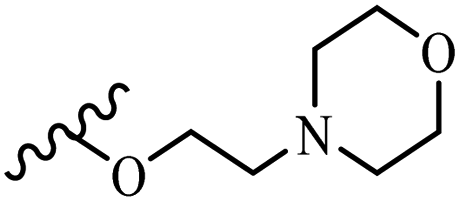 |
— | 50–100 | — | — | — | — |
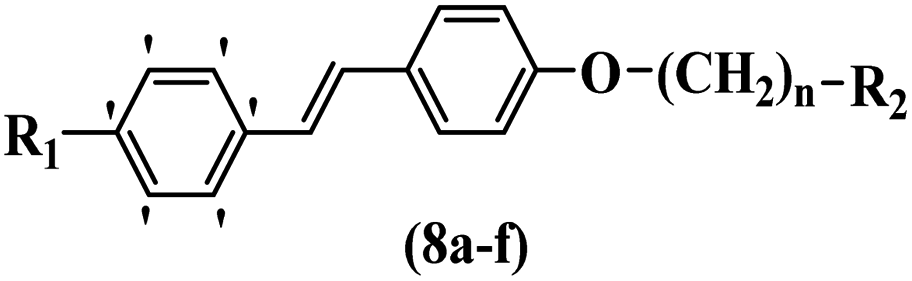
Wyrzykiewicz et al.42 synthesized E-piperidino and morpholino stilbenes and assessed their anti-microbial activity. Among the analogues, 8a showed an anti-microbial effect against all the tested strains (S. aureus, S. faecalis, B. subtilis, E. coli, C. albicans, and A. fumigatus). Among the compounds, 8b and 8c exhibited the highest activity against S. faecalis and B. subtilis (aerobic). These compounds showed almost similar activity as chloramphenicol against these two strains. Also, the presence of an NO2 group in the 4′-position on the phenyl ring in 8d compared to 8e (unsubstituted) displayed higher activity against S. aureus, S. faecalis, B. subtilis, and A. fumigatus. It seems that the presence of an electron-withdrawing group (EWG) such as NO2 in the 4′-position on the phenyl ring plays a vital role in anti-microbial activity. In addition, increasing or decreasing the linker length had no significant effect (e.g., 8a and 8f) against S. faecalis and B. subtilis. Compounds 8a–f were only endowed with weak anti-microbial activity against S. aureus and A. fumigatus compared to chloramphenicol and amphotericin B. Most of the screened compounds showed moderate to weak anti-fungal activity (C. albicans and A. fumigatus). All the tested compounds displayed an insignificant effect against K. pneumoniae and P. aeruginosa. According to the results, Gram-positive bacteria are more susceptible to the target compounds than Gram-negative bacteria due to the absence of an outer membrane (Table 2).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | n | R1 | R | MIC (μg mL−1) | |||||
| S. aureus | S. faecalis | B. subtilis | E. coli | C. albicans | A. fumigatus | ||||
| 8a | 2 | H | 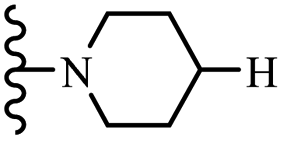 |
10 | 100 | 100 | 100 | 50 | 50 |
| 8b | 2 | H | 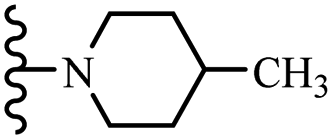 |
7.5 | 7.5 | 5 | — | 50 | 50 |
| 8c | 5 | NO2 | 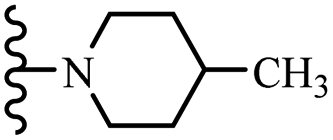 |
10 | 7.5 | 10 | — | — | 50 |
| 8d | 4 | NO2 | 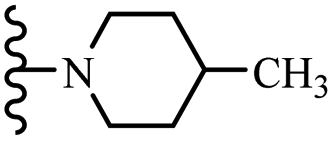 |
10 | 100 | 10 | — | 100 | 10 |
| 8e | 4 | H | 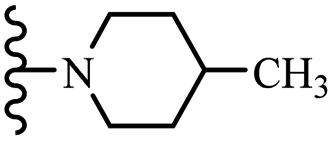 |
— | — | — | — | 50 | 50 |
| 8f | 4 | H | 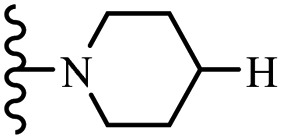 |
5 | 100 | 100 | — | — | 100 |
| Chloramphenicol | — | — | — | 5 | 5 | 5 | 5 | — | — |
| Amphotericin B | — | — | — | — | — | — | — | 10 | 1 |
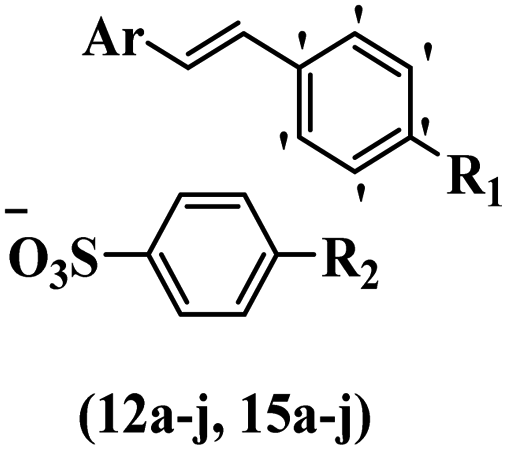
Chanawanno et al.43 synthesized pyridinium and quinolinium stilbene benzenesulfonate hybrids and evaluated their anti-bacterial activity. The quinolinium derivatives showed better activity compared to pyridinium derivatives. In the first series, quinolinium derivatives 12a–j exhibited higher activity against Gram-positive than Gram-negative bacteria. Derivatives 12a–c were the most potent compounds against all the tested Gram-positive bacteria. These compounds showed higher activity than that of the benzalkonium chloride drug and vancomycin. All quinolinium analogues were ineffective against P. aeruginosa and S. typhi. Also, the presence of EWG in the 4-position of the benzenesulfonate moiety resulted in higher anti-bacterial activity toward an electron-donating group (EDG) (e.g., 12a vs. 12d) against S. aureus and B. subtilis. Likewise, replacing OC2H5 in 12e with a (CH3)2NH group in 12c in the 4-position on the benzenesulfonate moiety increased the activity against all the strains. The presence of a substitution in the 4-position on the benzenesulfonate moiety increased the activity of the quinolinium derivatives by 2–4 times (12a–j). However, substitution in the 4-position of the benzenesulfonate moiety did not significantly improve the anti-bacterial activity of quinolinium derivatives (e.g., 12f) against S. aureus and B. subtilis. Compounds 12a–j were ineffective against all the tested Gram-negative bacteria. In the second series, the compounds containing a (CH3)2NH group in the 4′-position on the phenyl ring exhibited better activity than that containing an OC2H5 group in the same position (15a vs. 15b). All the pyridinium derivatives (15a–j) showed less potency than benzalkonium chloride and vancomycin. According to the results, both hydrophilic and lipophilic groups had a similar effect on anti-bacterial activity (Table 3).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Ar | R1 | R2 | MIC (μg mL−1) | |||||
| S. aureus | B. subtilis | E. faecalis | P. aeruginosa | S. typhi | S. sonnei | ||||
| 12a | 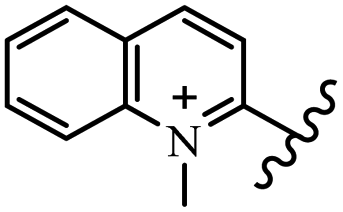 |
N(CH3)2 | Br | 2.34 | 2.34 | 2.34 | 300 | 300 | 2.34 |
| 12b | 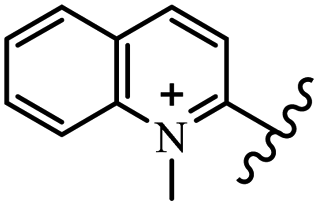 |
N(CH3)2 | Cl | 2.34 | 2.34 | 2.34 | 300 | 300 | 2.34 |
| 12c | 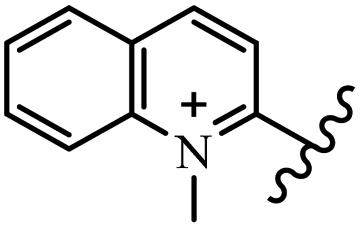 |
N(CH3)2 | CH3 | 2.34 | 2.34 | 2.34 | 300 | 300 | 2.34 |
| 12d | 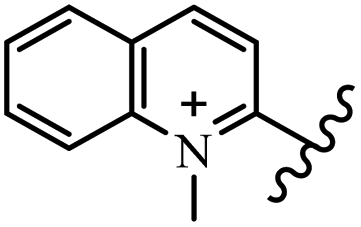 |
N(CH3)2 | OCH3 | — | 75 | 2.34 | 300 | 300 | 2.34 |
| 12e | 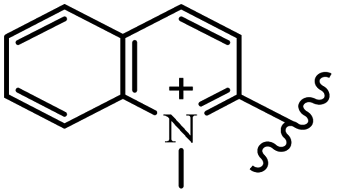 |
OC2H5 | CH3 | — | 9.37 | 37.5 | — | — | 150 |
| 12f | 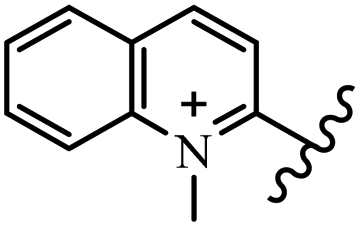 |
OC2H5 | Br | 300 | 18.75 | 37.5 | — | — | 75 |
| 12g | 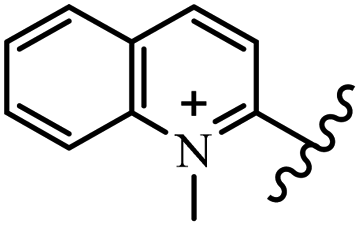 |
OC2H5 | OCH3 | — | 18.75 | 75 | — | — | 150 |
| 12h |  |
OC2H5 | Cl | — | 75 | 75 | — | — | 300 |
| 12i |  |
OC2H5 | NH2 | 37.5 | 37.5 | 37.5 | — | — | 75 |
| 12j |  |
N(CH3)2 | NH2 | 75 | 18.75 | 75 | 150 | 75 | 75 |
| 15a |  |
N(CH3)2 | OCH3 | 150 | 150 | 150 | 150 | 150 | 150 |
| 15b | 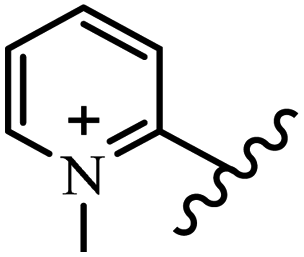 |
OC2H5 | OCH3 | 150 | — | — | — | — | — |
| 15c | 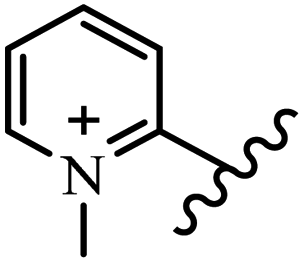 |
OC2H5 | NH2 | — | — | — | — | — | — |
| 15d | 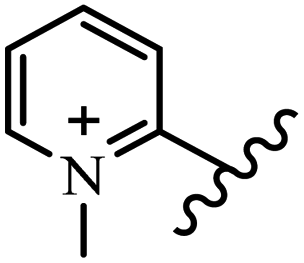 |
N(CH3)2 | Cl | — | 300 | — | 300 | 300 | — |
| 15e | 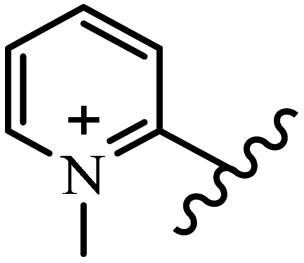 |
N(CH3)2 | NH2 | — | — | — | — | — | — |
| 15f | 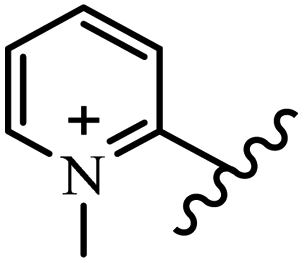 |
N(CH3)2 | CH3 | 300 | 150 | 300 | 150 | 300 | 300 |
| 15g | 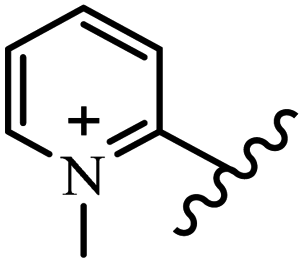 |
OC2H5 | CH3 | — | 300 | 150 | 37.5 | 150 | 150 |
| 15h | 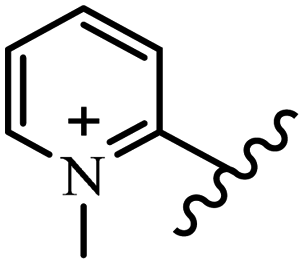 |
OC2H5 | Br | — | — | — | — | 300 | 300 |
| 15i | 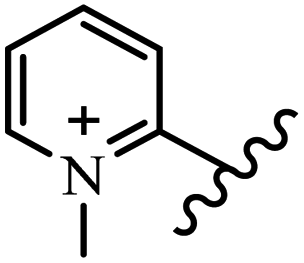 |
OC2H5 | Cl | — | — | — | — | — | — |
| 15j | 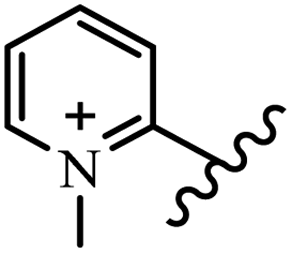 |
N(CH3)2 | Br | — | — | — | — | — | — |
| Benzalkonium chloride | — | — | — | <2.34 | 150 | 9.37 | 300 | 9.37 | — |
| Vancomycin | — | — | — | 9.37 | 2.34 | 9.375 | 2.34 | 2.34 | 2.34 |
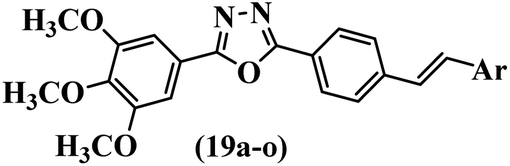
He et al.44 synthesized stilbene derivatives containing a 1,3,4-oxadiazole moiety and evaluated their fungicidal activity. Compound 19a inhibited cucumber P. cubensis with significant inhibitory activity comparable to fungicides. Most of the compounds exhibited moderate to weak control efficacy against S. cucurbitacearum (e.g., 19b). Also, changing the position of the NO2 group on the phenyl ring in 2′-position in 19c to the 3′- and 4′-positions in 19d and 19e enhanced the activity against P. cubensis and C. lagenarium, respectively. Moreover, among the halogenated compounds, the EWG and small size of F showed the highest activity (19f). The shifting of the nitrogen atom from the 2′- to 3′-position of the pyridine ring in compounds 19g and 19h increased the activity against P. cubensis and decreased the activity against C. lagenarium and S. cucurbitacearum, respectively. In addition, the presence of an anthracene ring in 19i showed lower activity than 19j with a naphthalene ring against P. cubensis and C. lagenarium. Also, the compounds containing EWG demonstrated better anti-fungal activity compared to that containing EDG (19k vs. 19l) against P. cubensis and C. lagenarium. The results showed that hydrophilic groups were more potent than lipophilic groups (19c vs. 19m). Also, the presence of substitution on the phenyl ring reduced the activity (e.g., 19n and 19b). In addition, the presence of an extra substitution on the phenyl ring decreased the activity (e.g., 19k and 19o) (Table 4).
|
||||
|---|---|---|---|---|
| Compound | Ar | Control efficacy (%) | ||
| P. cubensis | C. lagenarium | S. cucurbitacearum | ||
| 19a |  |
71.38 | 30.26 | 44.84 |
| 19b |  |
8.37 | 26.38 | 69.34 |
| 19c | 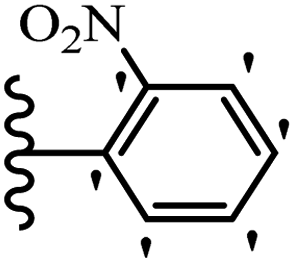 |
48.34 | 56.07 | 35.36 |
| 19d | 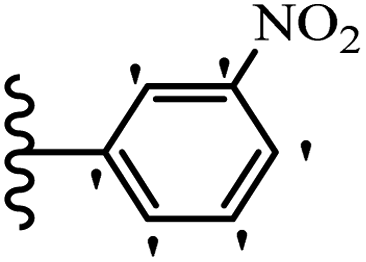 |
50.13 | 83.82 | 46.83 |
| 19e | 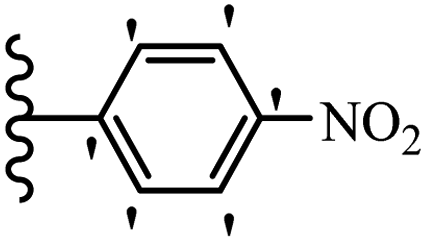 |
63.42 | 66.72 | 32.23 |
| 19f | 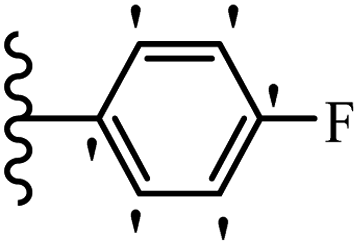 |
52.20 | 65.14 | 72.05 |
| 19g |  |
18.38 | 52.88 | 45.14 |
| 19h |  |
42.67 | 37.58 | 40.27 |
| 19i |  |
15.3 | 14.29 | 53.09 |
| 19j |  |
36.01 | 37.98 | 51.18 |
| 19k |  |
65.01 | 82.43 | 32.03 |
| 19l |  |
44.13 | 37.90 | 47.62 |
| 19m |  |
32.66 | 31.48 | −9.348 |
| 19n |  |
58.77 | 50.03 | 55.178 |
| 19o | 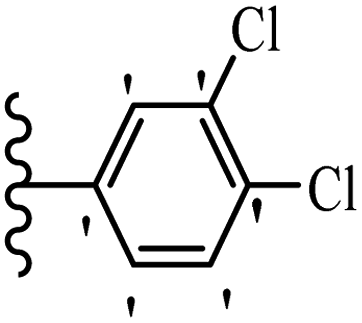 |
−4.08 | 44.81 | 8.03 |
| Fungicides | — | 70.13 | 81.57 | 69.02 |
Jian et al.45 synthesized fluorine-containing stilbene derivatives and assessed their anti-fungal activity. The synthesis method was the same as that in a previous study.44 Compounds 20a and 20b exhibited relatively high fungicidal potency against C. lagenarium and P. cubensis and were comparable to fungicides against both strains. Moreover, EDG showed higher activity than EWG (20c vs. 20d) against C. lagenarium and P. cubensis. In addition, compound 20e with two Cl atoms in the 3′- and 4′-positions on the phenyl ring showed lower activity compared to 20f having one Cl atom in the 3′-position against both strains. Also, the anti-fungal activity of the compounds containing hydrophilic groups was better compared to that bearing lipophilic groups (20b vs. 20f) (Table 5).
|
|||
|---|---|---|---|
| Compounds | Ar | Control efficacy (%) | |
| C. lagenarium | P. cubensis | ||
| 20a | 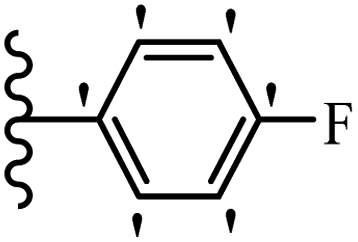 |
83.4 | 54.6 |
| 20b | 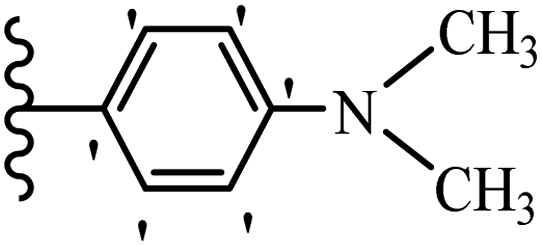 |
38.1 | 70.2 |
| 20c | 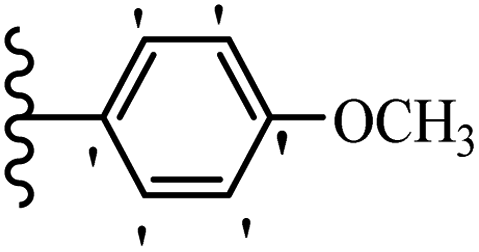 |
61.3 | 66.0 |
| 20d | 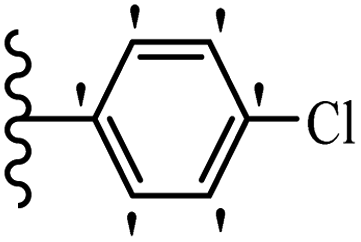 |
54.1 | 53.5 |
| 20e | 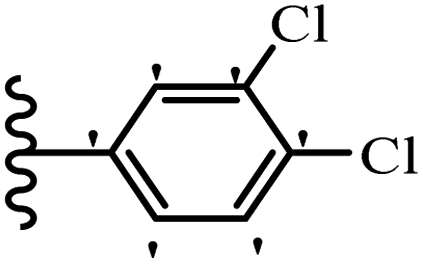 |
60.8 | 34.8 |
| 20f | 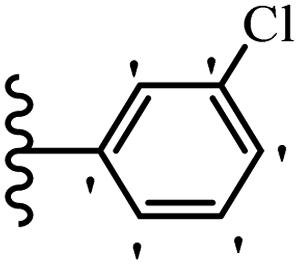 |
64.3 | 52.4 |
| Fungicides | — | 82.7 | 72.5 |
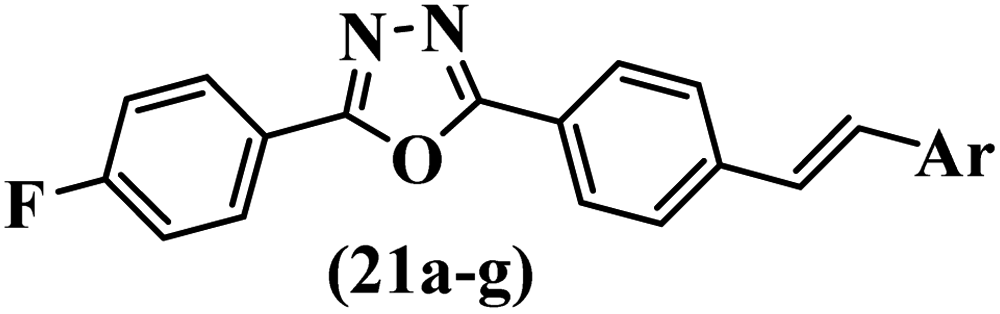
Similarly, a research group46 reported the synthesis of oxadiazole–stilbene hybrids and evaluated their efficacy against phytopathogenic fungi. The synthesis method was the same as in the previous study.44 Compounds 21a and 21b exhibited superior anti-fungal activity to that of resveratrol. Also, moving the OCH3 group from the 2′-position of 21c to the 3′-position on the phenyl ring in 21d increased the activity. Moreover, the presence of hydrophilic groups resulted in higher activity compared to the lipophilic groups (21e vs. 21f). Compound 21b with a pyridine ring in the 4′-position on the phenyl ring showed stronger activity than compound 21g with a pyridine ring in the 3′-position. This finding showed that the position of the nitrogen atom in the pyridine ring may play an important role in activity. Also, the compounds containing EWG and EDG showed equal activity (e.g., 21c and 21f) (Table 6).
|
||
|---|---|---|
| Compounds | Ar | EC50 (μg mL−1) |
| 21a | 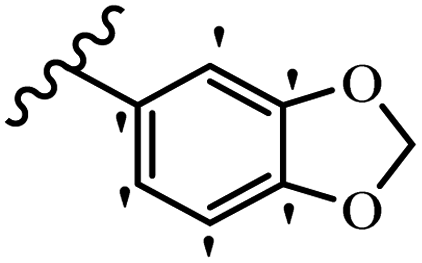 |
144.6 |
| 21b | 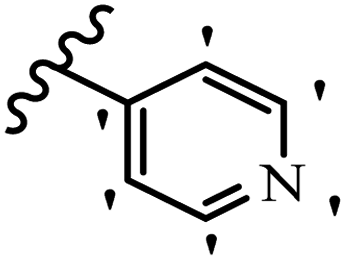 |
231.3 |
| 21c | 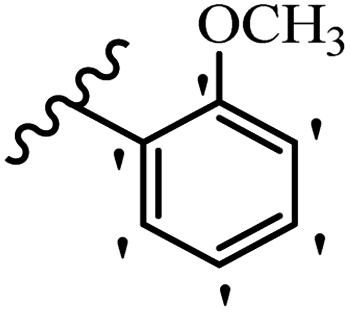 |
>400 |
| 21d | 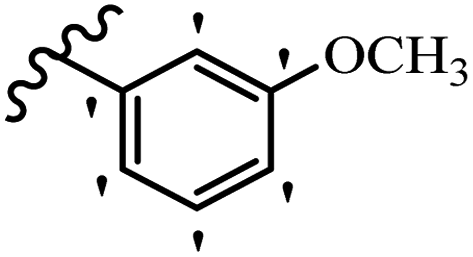 |
345.9 |
| 21e | 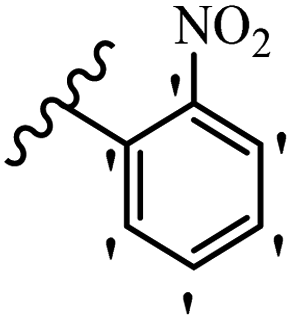 |
382.9 |
| 21f | 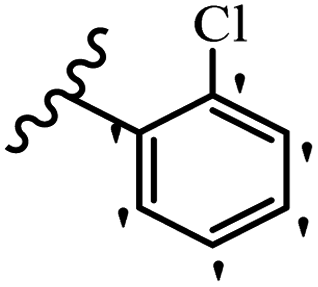 |
>400 |
| 21g | 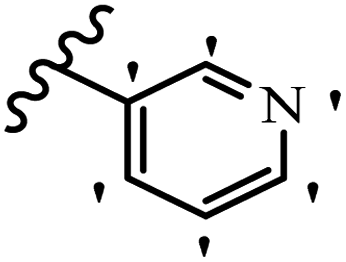 |
>400 |
| Resveratrol | — | 315.6 |
Wen et al.47 synthesized 1,3,4-oxadiazole–thiophene-based stilbene compounds and assessed their anti-fungal activity. In the previous study, the method of synthesized compounds was explained.44 Compound 22a exhibited higher activity compared to resveratrol but was weaker than carbendazim and fluopyram against B. cinerea and C. lagenarium. In addition, increasing the substitution on the phenyl ring displayed the maximum activity (e.g., 22b and 22c) against different strains. Furthermore, among the compounds with EWG in the 4′-position on the phenyl ring, compound 22d with a Br atom exhibited better activity compared to 22c having a Cl atom and 22e with an F atom in the same position against both strains. It seems that increasing the atom size improved the activity. Furthermore, the presence of the EDG in the 4′-position on the phenyl ring demonstrated greater activity than EWG in the same position (e.g., 22f and 22d) against P. cubensis. In addition, the anti-fungal activity of hydrophilic groups showed better activity than that of lipophilic groups (22f vs. 22c) (Table 7).
|
|||
|---|---|---|---|
| Compounds | Ar | EC50 (μg mL−1) | |
| B. cinerea | C. lagenarium | ||
| 22a | 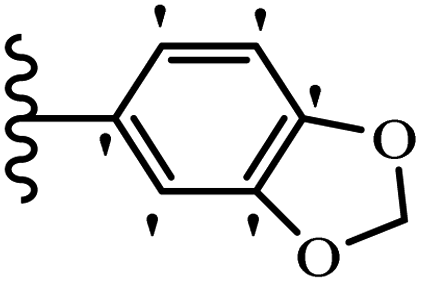 |
155.4 | 248.2 |
| 22b | 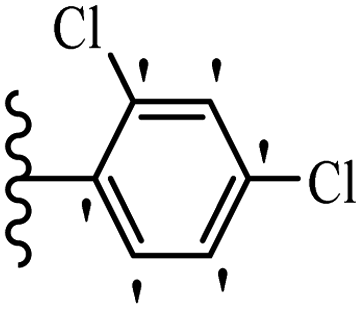 |
172.1 | 261.7 |
| 22c | 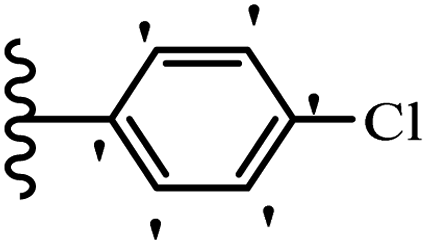 |
187.7 | 279.6 |
| 22d | 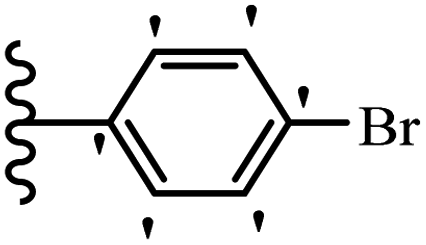 |
175.5 | 263.7 |
| 22e | 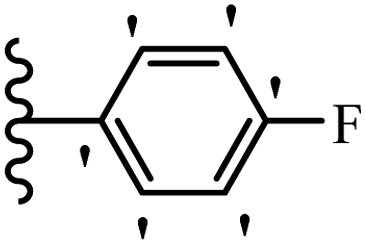 |
180.1 | 268.9 |
| 22f | 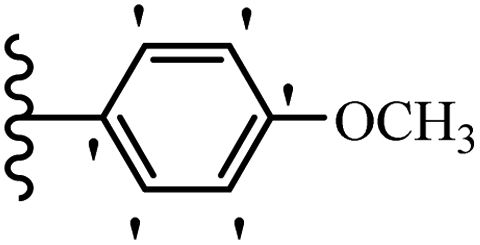 |
170.3 | 259.6 |
| Resveratrol | — | 263.1 | 342.6 |
| Carbendazim | — | 124.3 | 219.7 |
| Fluopyram | — | 117.9 | 223.5 |
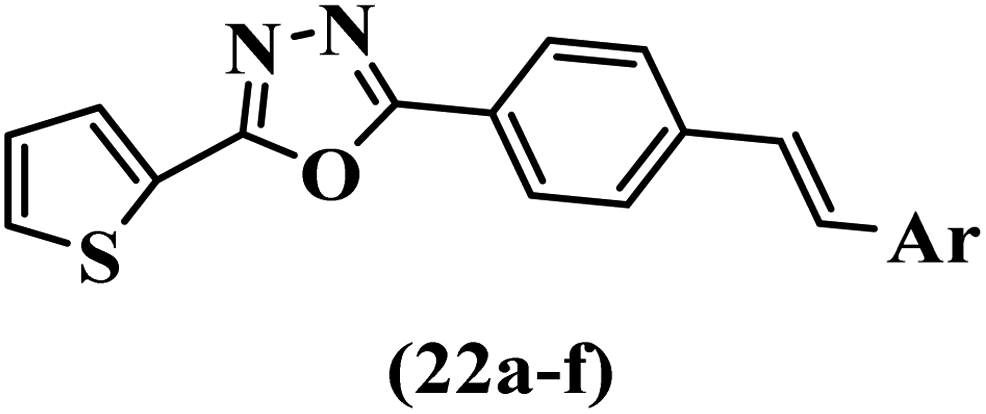
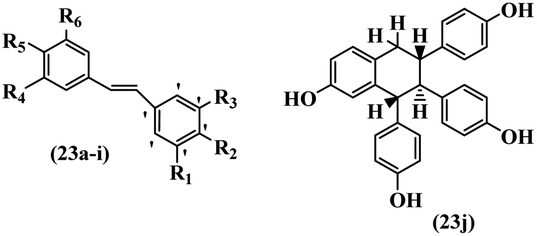
Singh et al.48 reported the anti-bacterial activity of resveratrol structural analogues. Compared to other bacteria tested, resveratrol was more effective against the enteric bacteria P. vulgaris and S. typhimurium. Compounds 23a and 23b exhibited higher inhibition than resveratrol, while compound 23c exhibited comparable activity against P. vulgaris, S. typhimurium, and E. coli. The other compounds showed lower activity than resveratrol against P. aeruginosa. The compounds that were impressive against Gram-negative bacteria were similarly impressive against Gram-positive bacteria (compounds 23a and 23b against S. aureus). In the case of 23d–f, no anti-bacterial activity was observed (wild-type or ΔtolC E. coli). Compounds 23g and 23j were also ineffective against wild type E. coli, while their activity was comparable to 23a and 23b against ΔtolC, the two analogues that were most active against wild-type E. coli. Also, the presence of a Br atom in the 4′-position on phenyl ring 23a showed excellent activity against S. aureus compared to 23b, which was unsubstituted. The antibacterial activity of compound 23c with an OH group in the 3-position on the phenyl ring was lower than that of 23g having an OCH3 group in the same position against S. aureus. The inhibitory activity of stilbenes against S. aureus reiterated the fact that analogues 23a, 23g, and 23h were more potent than resveratrol against S. aureus, while 23b and 23c showed comparable activity to resveratrol. When the OH groups in the molecule were replaced with acetoxy or methoxy groups, a significant reduction in anti-bacterial activity was observed (resveratrol vs. compounds 23e or 23f). This showed the importance of the OH group for the antibacterial activity. However, increasing the number of OH groups did not result in better antibacterial activity (resveratrol vs. 23h with OH groups in the 3′- and 4′-positions on the phenyl ring or 23i having OH groups in the 3′- and 5′-position on phenyl ring). The presence of an OH group in the 4′-position on the phenyl ring did not have a significant effect on the activity or inactivity of the molecule (23b, 23d, and 23h). However, it is worth noting that the best molecules in the series (23a–c, 23g, and 23j, as well as resveratrol) had an OH group in the 4′-position on the phenyl ring. Partially changing the OH group to other groups resulted in an improvement in antibacterial activity (resveratrol vs. 23a and 23c or 23g). The findings showed that the lipophilic groups had better activity than the hydrophilic groups (Table 8).
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | MIC (μg mL−1) | Inhibition percent (%) | ||||||||
| S. aureus | E. coli | ΔtolC | P. vulgaris | S. typhimurium | E. coli | ΔtolC | S. aureus | |||||||||
| 23a | H | Br | H | OH | H | OH | 25 | >100 | 25 | >80 | >80 | — | 90 | >80 | ||
| 23b | H | H | H | OH | H | OH | 100 | >100 | 100 | >80 | >80 | — | 90 | >80 | ||
| 23c | H | OH | H | OH | H | OCH3 | 100 | >100 | 100 | 40 | 40 | 40 | — | — | ||
| 23d | H | OH | H | H | OH | H | — | — | — | — | — | — | — | — | ||
| 23e | H | OCOCH3 | H | OCOCH3 | H | OCOCH3 | — | — | — | — | — | — | — | — | ||
| 23f | H | OCH3 | H | OCH3 | OH | OCH3 | — | — | — | — | — | — | — | — | ||
| 23g | H | OH | H | OCH3 | H | OCH3 | 25 | >100 | 25 | — | — | — | 90 | — | ||
| 23h | OH | OH | H | OH | OH | H | — | — | — | — | — | — | — | — | ||
| 23i | OH | OH | H | OH | H | OH | — | — | — | — | — | — | — | — | ||
| 23j | — | — | — | — | — | — | 10 | >100 | 10 | — | — | — | 80 | — | ||
| Resveratrol | — | — | — | — | — | — | 100 | >100 | 100 | — | — | — | — | — | ||
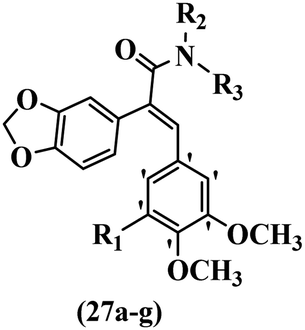
Song et al.21 synthesized and assessed the fungicidal activity of 1,3-benzodioxole-stilbene derivatives. Compound 27a showed the highest activity against G. theae-sinensis, A. tenuis Nees, F. graminearum, and R. solani. This analogue was stronger than piperine and azoxystrobine, but it showed lower activity compared to piperine against F. graminearum. Overall, the derivatives showed low inhibitory activity against all the tested strains, while the inhibition rates of the derivatives against most fungi of the tested fungi were not greater than 20%. Replacing propylsulfane linked to an amide moiety in 27a with methyl-4-(methylthio)butanoate in 27b showed lower activity against A. tenuis Nees. Also, the presence of an OCH3 group in the 3′-position on the phenyl ring compared to no substitution increased the fungicidal activity (e.g., 27c and 27d) against A. tenuis Nees, f. graminearum, and R. solani. Also, hydrophilic groups (e.g., 27e) and lipophilic groups (e.g., 27f) are suitable to improve the fungicidal activity against G. theae-sinensis and A. tenuis Nees. In addition, the presence of cycloalkane and aromatic rings linked to an amide moiety had the same effect on the activity (27g vs. 27d) against A. tenuis Nees and R. solani (Table 9).
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Fungicidal activity (%) | |||
| G. theae-sinensis | A. tenuis Nees | F. graminearum | R. solani | ||||
| 27a | H | H | 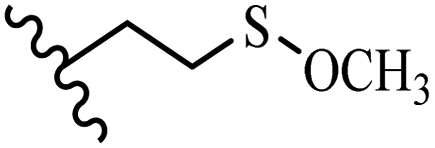 |
<10 | <40 | 0 | <10 |
| 27b | H | H | 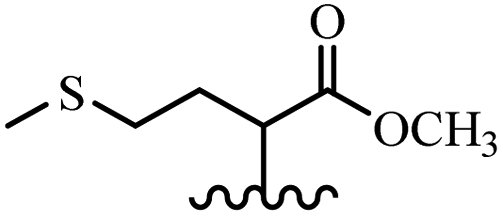 |
<10 | <20 | <20 | <20 |
| 27c | H | H | 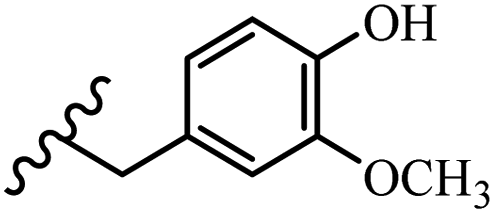 |
<20 | <10 | 0 | 0 |
| 27d | OCH3 | H | 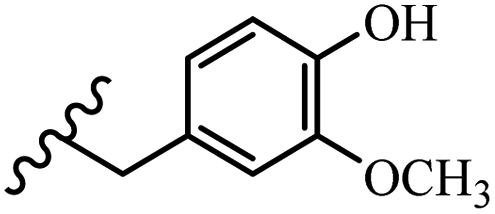 |
<20 | <20 | <20 | <20 |
| 27e | OCH3 | H | 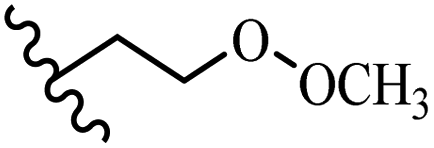 |
<30 | <30 | <20 | <20 |
| 27f | OCH3 | H | 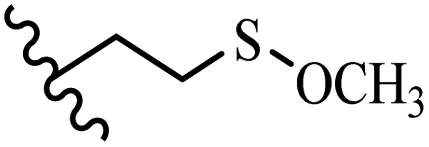 |
<30 | <30 | <10 | <10 |
| 27g | OCH3 | H | 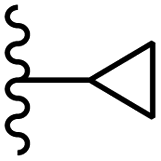 |
<10 | <20 | <30 | <20 |
| Piperine | — | — | — | 74 | 55 | <30 | 62 |
| Azoxystrobin | — | — | — | 69 | 55 | 64 | 55 |
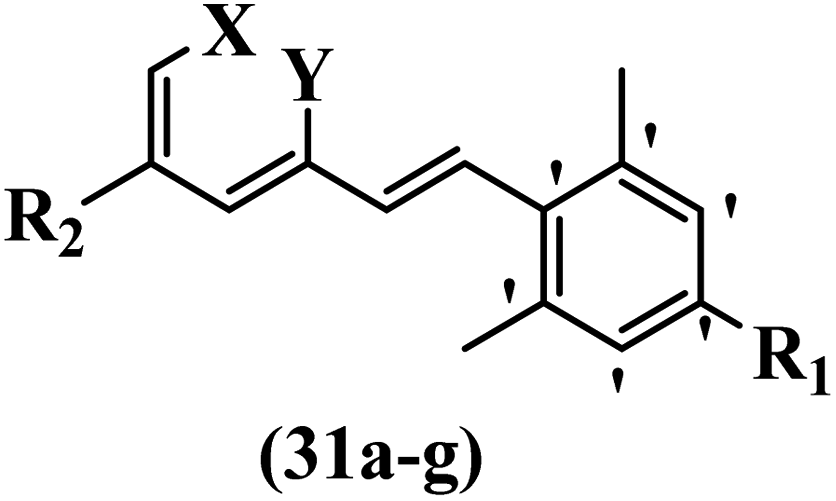
Hrast et al.49 described azastilbene derivatives as mur ligase inhibitors and anti-bacterial agents and evaluated the inhibition of four mur ligase subgroups (MurC, MurD, MurE, and MurF). Compound 31a was the most potent against subgroups MurD, MurE, and MurF. Compound 31b showed the highest activity against the MurC subgroup among the compounds. In addition, the lipophilic group in 31b displayed higher activity than the hydrophilic group in 31c against all the mur ligases. Most of the stilbene derivatives demonstrated poor anti-bacterial activity against both E. coli and S. aureus. This could be attributed to their low target activity or poor penetration into the bacterial cytoplasm. However, replacing oxazole linked to a pyridine ring on the 4-position phenyl ring in 31c with imidazole linked to a pyridine ring in same position of 31d resulted in the highest anti-bacterial activity against S. aureus and E. coli. Also, the presence of hydrophilic groups resulted in better activity than lipophilic groups against both strains (31d and 31b). Compound 31e with an OCH3 group in the 2′-position on the phenyl ring exhibited higher anti-bacterial activity compared to compound 31f having an OCH3 group in the 3′-position on the phenyl ring and 31g with an OCH3 group in the 4′-position on the phenyl ring against S. aureus and E. coli. It seems that changing the substitution on different positions of the phenyl ring had a positive effect on the anti-bacterial activity (the 2-position showed higher activity than the 4- and 3-positions on the phenyl ring) (Table 10).
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | X | Y | R1 | R2 | Inhibition percent (%) | MIC (mM) | ||||
| MurC | MurD | MurE | MurF | S. aureus | E. coli | |||||
| 31a | C | N |  |
 |
23 | 73 | 55 | 84 | >0.25 | >0.25 |
| 31b | N | C |  |
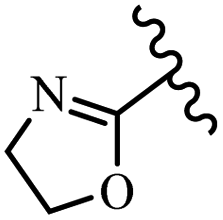
 |
56 | 70 | 46 | 58 | >0.25 | >0.25 |
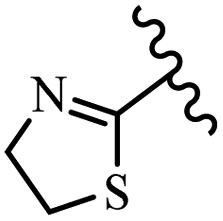
| 31c | N | C |  |
 |
44 | 45 | 32 | 44 | 0.125 | >0.25 |
| 31d | N | C |  |
 |
42 | 47 | 10 | 40 | 0.031 | 0.25 |
| 31e | N | C | 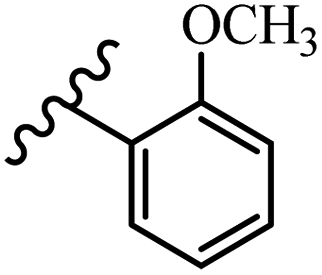 |
 |
35 | 42 | 33 | 0 | 0.125 | >0.25 |
| 31f | N | C | 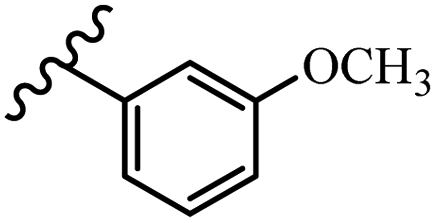 |
 |
15 | 23 | 21 | 18 | >0.25 | >0.25 |
| 31g | N | C | 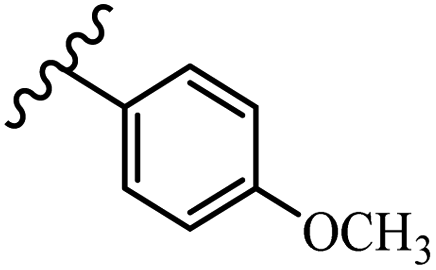 |
 |
30 | 30 | 37 | 25 | 0.25 | >0.25 |
3. Anti-cancer activity
Despite the huge effort to develop novel therapies, cancer remains the leading cause of death worldwide. One of the traditional and commonly used methods for cancer treatment is chemotherapy, which targets specific proteins and cellular structures or processes.50Resveratrol as an anti-cancer agent reduces angiogenesis and induces apoptosis via the suppression of VEGF and FGF-2.51 Piceatannol and pterostilbene, stilbene natural derivatives, are remarkably more potent than resveratrol against cancer cell lines.52 CA-4, a Z-stilbenoid analogue, is a strong inhibitor of tubulin polymerization, resulting in cancer cell death.53 In many experimental models in vitro and in vivo both Z- and E-stilbene analogues showed anti-cancer activity but with diverse mechanisms. Some Z-isomers of methoxylated stilbenes and their derivatives revealed higher antimetastatic or antiproliferative activity than their E-isomers; however, the E-isomer of resveratrol displayed higher antiproliferative activity. Moreover, they isomerize during storage, administration, and metabolism in liver microsomes.54,55 Z- and E-stilbene analogues have been studied for their cytotoxicity and anti-tubulin activities. The analysis of the results a wide range of stilbene analogues showed that the Z-isomer was useful for cytotoxicity and anti-mitotic activity. Tamoxifen, a stilbene analogue, is used for the treatment of some types of breast cancer.53
Lee et al.17 synthesized stilbene analogues and evaluated their cytotoxicity. Z-33a and b showed the highest cytotoxicity activity among the compounds. These compounds were stronger than resveratrol and oxyresveratrol against the A549 and Col2 cell lines. Especially, compound 33a having OCH3 and Br groups in the 4- and 4′-positions on the phenyl ring exhibited approximately 600-times and 1800-times, respectively, more potent cytotoxicity than resveratrol. In the E-isomers, the presence of an extra OCH3 group in 33c showed comparable activity to 33d. Also, replacing the aromatic ring of 33a with a heteroaromatic ring in 33b and 33e decreased the activity. In the Z/E-isomers, moving the OCH3 group from the 5′-position in 33f to the 6′-position on the phenyl ring in 33g increased the cytotoxicity. Based on the findings, the Z-isomers were more potent than their corresponding E-isomers (e.g., 33a and 33h). In addition, adding an OCH3 group to the phenyl ring (35a vs. 35b) reduced the cytotoxicity (Table 11).
|
|||||
|---|---|---|---|---|---|
| Compound | E/Z | R | R1 | IC50 (μg mL−1) | |
| A549 | Col2 | ||||
| 33a | Z | OCH3 |  |
0.01 | 0.01 |
| 33b | Z | OCH3 |  |
0.2 | 0.3 |
| 33c | E | H |  |
0.8 | 0.8 |
| 33d | E | H |  |
0.8 | 0.9 |
| 33e | Z | OCH3 | 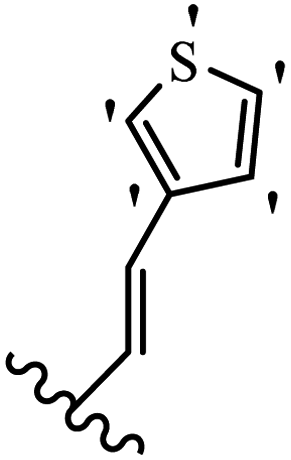 |
2.1 | 2.6 |
| 33f | Mix | H | 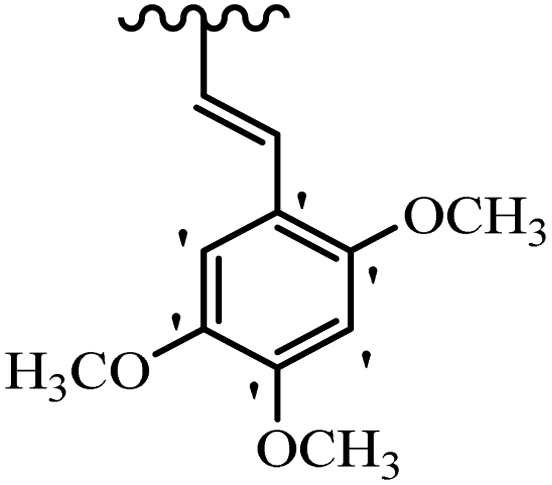 |
>20 | >20 |
| 33g | Mix | H | 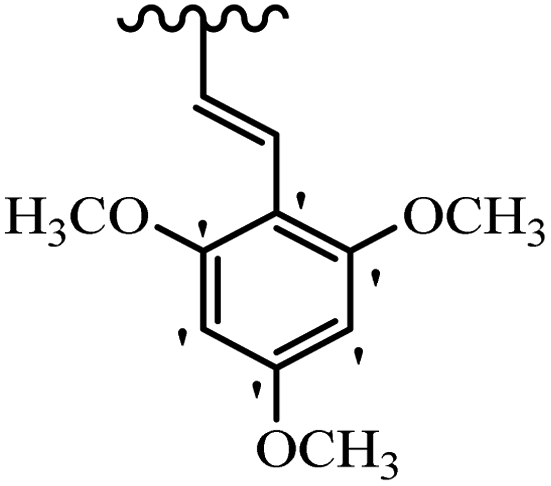 |
3.2 | 7.2 |
| 33h | E | OCH3 | 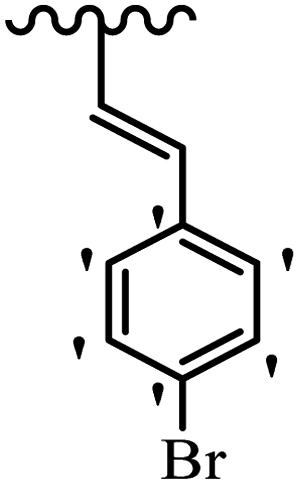 |
4.7 | 1.6 |
| 35a | — | H | 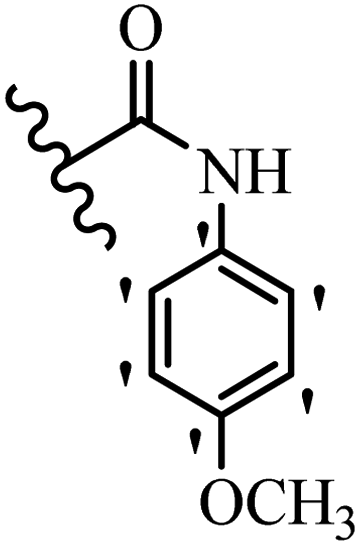 |
2.6 | 5.1 |
| 35b | — | H | 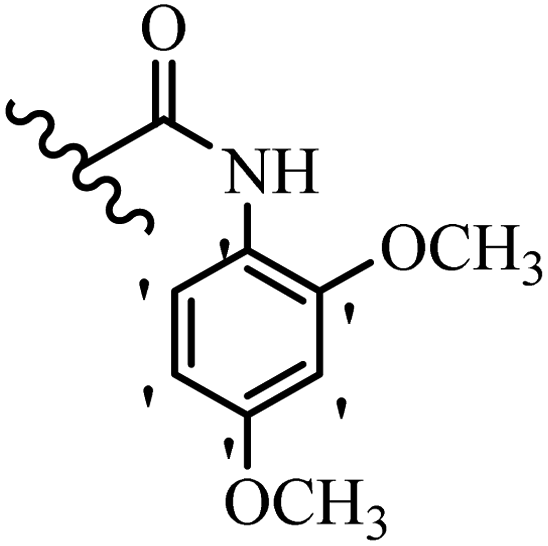 |
>20 | >20 |
| Resveratrol | E | — | — | 6.9 | 18.7 |
| Oxyresveratrol | E | — | — | >20 | >20 |
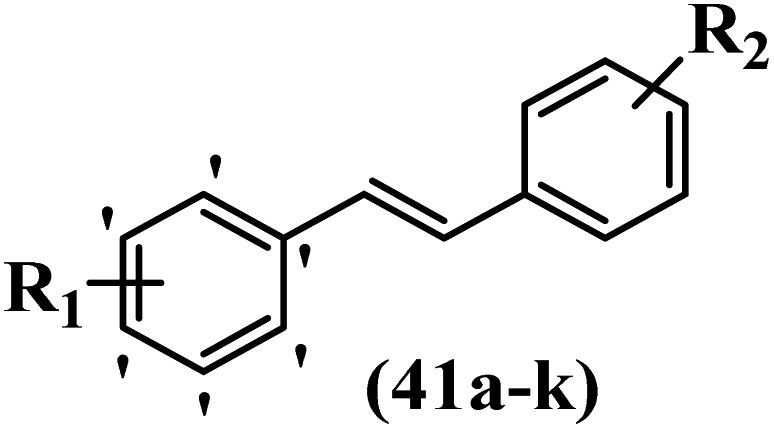
Lion et al.56 synthesized hydroxylated E-stilbenes and assessed their anti-tumor and apoptosis-inducing activity. Compounds 41a and 41b showed the highest activity among the compounds and resveratrol against MDA-MB-468 and HCT-116. Also, changing the position of the OH group in 41b from the 2- to 3- and 4-positions on the phenyl ring in 41c and 41d reduced the activity against both cell lines, respectively. Similarly, the compounds having EWG and lipophilic groups showed better activity than that having EDG and hydrophilic groups (e.g., 41e vs. 41f). Furthermore, the extra OCH3 group in the 5′-position on the phenyl ring of compound 41g caused a slight improvement in activity compared to 41h. This result showed that increasing the number of substitutes on the phenyl ring had a positive effect on activity. Substitution on the phenyl ring resulted in higher activity compared to compound with no substitution (e.g., 41i and 41j). With the exception of the relatively insensitive 41k, the percentage of apoptotic sub-G1/0 MDA-MB-468 cells following drug treatment was higher in all the stilbenes than in resveratrol. Following treatment with compounds 41a, 41e, and 41j, the highest percentage of cells was observed to be in early apoptosis. The induction of apoptosis was again associated with the anti-proliferative activity in MDA-MB-468 cells, particularly for the relatively potent compounds 41j and 41a. Particularly, compound 41j had a much higher percentage of cells in early apoptosis than resveratrol but a much lower percentage than camptothecin. Based on the findings, the presence of EDG and lipophilic groups resulted in the highest apoptosis activity (Table 12).
|
|||||
|---|---|---|---|---|---|
| Compounds | R1 | R2 | GI50 (μM) | Induced percent (%) | |
| HCT-116 | MDA-MB-468 | Apoptosis | |||
| 41a | 3,5-diOCH3 | 3-OH | 43.1 | 0.96 | 20 |
| 41b | 3,4-diF | 2-OH | 15.6 | 1.1 | — |
| 41c | 3,4-diF | 3-OH | 36.0 | 1.6 | — |
| 41d | 3,4-diF | 4-OH | 18.1 | 19.1 | — |
| 41e | 2-F | 4-OH | 38.5 | 2.8 | 10 |
| 41f | 2-OCH3 | 4-OH | 51.3 | 3.3 | — |
| 41g | 3,5-diOCH3 | 2-OH | 21.3 | 2.5 | — |
| 41h | 3-OCH3 | 2-OH | 58.0 | 3.1 | — |
| 41i | H | 3-OH | 57.4 | 7.8 | — |
| 41j | 2-OCH3 | 3-OH | 42.3 | 2.7 | 15 |
| 41k | H | 2-OH | 58.1 | 24.9 | 4 |
| Resveratrol | — | — | 49.6 | 41.1 | 4 |
| Camptothecin | — | — | — | — | 37 |
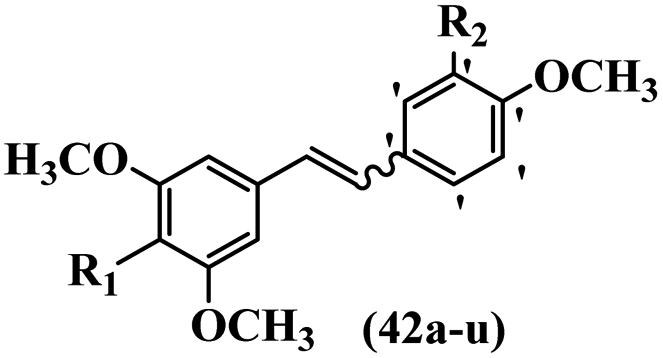
Pettit et al.41 evaluated Z/E-CA-4 derivatives for their neoplastic activity. Among the compounds, Z-42a showed the highest cytotoxicity activity against the BXPC-3, SK-N-SH, SW-1736, NCI-H460, DU-145, and FADU cell lines. In the Z-isomers, replacement of the OH group in the 3′-position on the phenyl ring of CA-4 with NO2, N(CH3)2, or Br groups in compounds 42b–d decreased their potency, respectively. In addition, the presence of hydrophilic groups improved the activity compared to lipophilic groups (42e and 42f). The replacement of the bulky group, such as OSO2CH3 in 42e, instead of OH in 42g reduced the activity. Likewise, the replacement of pyrrolidinyl in the 3′-position on the phenyl ring with piperidinyl in the same position resulted in better activity (42h vs. 42i) against the BXPC-3, SK-N-SH, SW-1736, NCI-H460, DU-145, and FADU cell lines. In the E-isomers, replacement of the NO2 group in the 3′-position on the phenyl ring in 42j with Br in the same position of 42k increased the activity against P-388, BXPC-3, SK-N-SH, DU-145, and FADU. Also, replacing the OCH3 group with an OH group in the 4′-position on the phenyl ring (Z/E-42l) resulted in a remarkable decrease (about 100-times) in activity. Subsequently, the compounds were tested for their ability to inhibit tubulin protein. Compound 42d was as active as CA-4, while 42b and 42e were about half as active. The Z-isomers with bulkier substituents at the 3′-position on the phenyl ring were much less active as inhibitors of assembly (e.g., 42f; Cl, 42g; SO3CH3 and 42m; CO2NC4H8Cl2 showed minor or no inhibitory activity). Nevertheless, Z-42n having an O(CH2)3OH group in the 3′-position on the phenyl ring showed better activity than 42o with 1H-imidazol-1-yl in the 3′-position on the phenyl ring. Alternatively, 42p having N-pyrrolyl in the 3′-position on the phenyl ring showed weaker inhibitory activity compared to 42o and 42n. This suggests that the aromatic substituent dominated the steric effect that restricts the stilbene–tubulin interaction indirectly based on the inactivity of 42f–i, 42q–t, and 42m. Surprisingly, the more effective tubulin inhibitor (42o) had little cytotoxicity, whereas the less effective 42p was highly cytotoxic in four of the seven cell lines tested. E-42k showed comparable activity to E-CA-4. E-42u with an OH group in the 3′- and 4-positions on the phenyl ring showed more than double activity. The results showed that the Z-isomers were stronger than the E-isomers (Table 13).
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | E/Z | R1 | R2 | GI50 (μg mL−1) | IC50 (μM) | ||||||
| P-388 | BXPC-3 | SK-N-SH | SW1736 | NCI-H460 | DU-145 | FADU | Tubulin | ||||
| 42a | Z | OCH3 | NH2 | >0.010 | 0.00043 | 0.00023 | 0.00080 | 0.00033 | 0.00033 | 0.00053 | — |
| 42b | Z | OCH3 | NO2 | 2.4 | 0.029 | 0.014 | 0.0067 | 0.0038 | 0.047 | 0.047 | 2.6 |
| 42c | Z | OCH3 | N(CH3)2 | 0.9 | 0.015 | 0.010 | 0.80 | 0.4 | 0.11 | 0.090 | 1.1 |
| 42d | Z | OCH3 | Br | 0.16 | 0.007 | 0.002 | 0.034 | 0.033 | 0.027 | 0.0058 | — |
| 42e | Z | OCH3 | 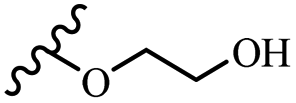 |
0.195 | 0.045 | 0.028 | 0.28 | 0.14 | 0.32 | 0.069 | 2.8 |
| 42f | Z | OCH3 | 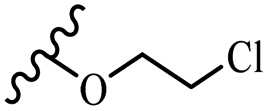 |
2.9 | 2.7 | 2.0 | 4.7 | 3.7 | 3.4 | 4.6 | >40 |
| 42g | Z | OCH3 | 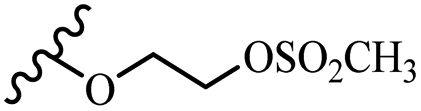 |
2.87 | 2.7 | 3.0 | 1.0 | 0.42 | 0.59 | 0.41 | >40 |
| 42h | Z | OCH3 | 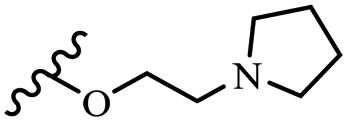 |
0.421 | 2.0 | 0.22 | 0.60 | 0.34 | 0.35 | 0.49 | >40 |
| 42i | Z | OCH3 | 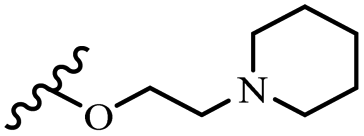 |
0.348 | >10.0 | 1.6 | 5.1 | 3.3 | 3.4 | 4.2 | >40 |
| 42j | E | OCH3 | NO2 | >10 | >10 | 7.3 | 2.8 | >0.0 | >10 | 3.8 | — |
| 42k | E | OCH3 | Br | 3.08 | 0.34 | 0.40 | 10.8 | 0.36 | 0.46 | 11.4 | 31 |
| 42l | E | OCH3 | O(CH2)3OH | 4.9 | >10.0 | 8.1 | 11.6 | >10.0 | 4.5 | 2.3 | — |
| 42m | Z | OCH3 |  |
0.074 | 2.8 | 0.14 | 0.72 | 0.37 | 0.31 | 0.15 | >40 |
| 42n | Z | OCH3 | O(CH2)3OH | 0.63 | 0.26 | 0.18 | 0.38 | 0.37 | 0.43 | 0.63 | 6.5 |
| 42o | Z | OCH3 |  |
19.0 | 0.82 | 0.31 | 1.5 | 0.56 | 1.5 | 0.67 | 7.6 |
| 42p | Z | OCH3 |  |
0.0232 | 2.3 | 0.0064 | 4.9 | 0.0033 | 0.56 | 0.0030 | 20 |
| 42q | Z | OCH3 | 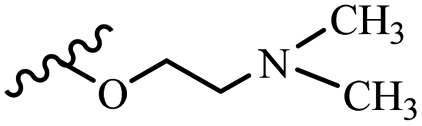 |
0.523 | >10.0 | 0.21 | 0.64 | 0.34 | 0.38 | 0.47 | >40 |
| 42r | Z | OCH3 | 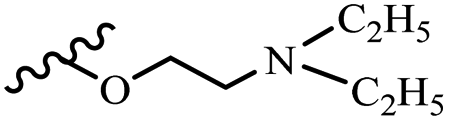 |
0.255 | >10.0 | 1.5 | 1.1 | 3.3 | 3.4 | 1.1 | >40 |
| 42s | Z | OCH3 | 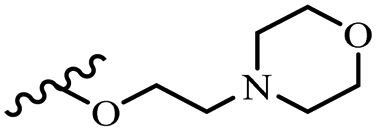 |
0.634 | 2.9 | 0.22 | 0.97 | 0.41 | 0.40 | 0.53 | >40 |
| 42t | Z | OCH3 | 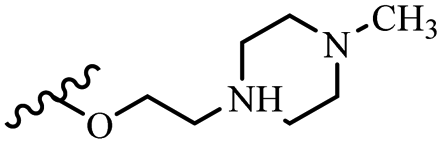 |
1.60 | >10.0 | 2.8 | >10.0 | 3.4 | 4.1 | 2.9 | >40 |
| 42u | E | OH | OH | 4.49 | 5.0 | 5.5 | >10.0 | 4.9 | 6.3 | 3.2 | 14 |
| Combretastatin A-4 | E | — | — | 0.0029 | 0.23 | 0.00025 | 0.00061 | 0.00035 | 0.00072 | 0.00045 | 33 |
| Combretastatin A-4 | Z | — | — | 0.0026 | >0.1 | 0.00026 | 0.00026 | 0.00056 | 0.00076 | 0.00065 | 1.2 |
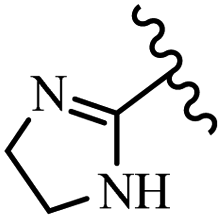
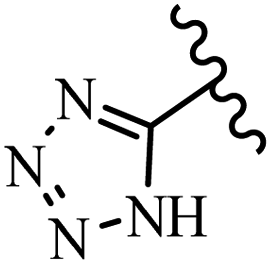
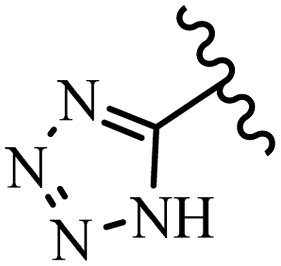
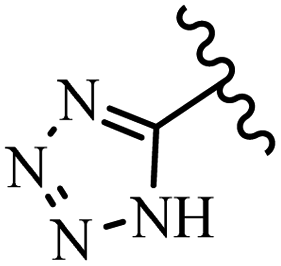
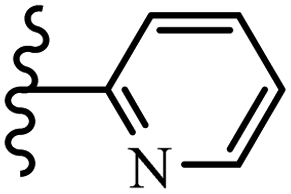
McDonald et al.57 synthesized aza-stilbenes and assessed their c-RAF inhibitor activity. Compound 45a showed the highest c-RAF inhibitory activity. In addition, removing the OH group in the 4′-poition on the phenyl ring of 45a produced 45b, which had a slight effect on the enzyme activity. Also, the compounds that had a hydrogen-bond acceptor or donor in the 3-position on the pyridine ring, such as compounds 45c with a COOH group, 45d having a CONH2 group, or 45b with tetrazole, showed higher activity against MEK/ERK. Moreover, replacing the OH group in the 4′-position on the phenyl ring with N-methyl carbamate caused a remarkable increase in c-RAF inhibitory activity (45d vs. 45e). Also, replacement of the compounds containing tetrazole (45a and 45b) with a 2-pyridine ring (45f) resulted in only a minor decrease in c-RAF activity. According to the primary findings, the aza-stilbene scaffold with the two CH3 groups in the 2′- and 6′-positions on the phenyl ring in compound 45g was kept to obtain potent c-RAF inhibitors. Also, compound 45h was 100-times less potent than 45g, demonstrating a size limitation. In addition, changing the CH3 group in 45i with a Cl atom in the 2-position on the phenyl ring of 45j resulted in a compound that was equal to 45g. Likewise, the presence of CH3 or Cl in the 2′-position on the phenyl ring, 45k and 45l, showed 10-times less activity than 45g. When only one of CH3 group was swapped with a Cl atom on the phenyl ring, as in 45h, the activity increased by about 3-times compared to 45g (Table 14).
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | n | R1 | R2 | R3 | R4 | pIC50 (μM) c-RAF/MEK/ERK | c-RAF |
| 45a | 10 | 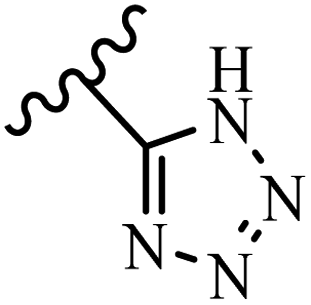 |
— | — | OH | 8.4 | 0.004 |
| 45b | 5 | 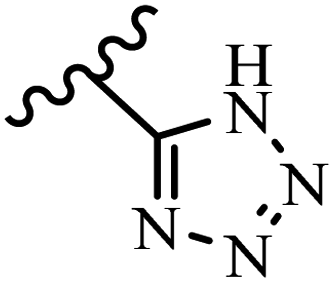 |
— | — | H | 7.9 | — |
| 45c | 3 | COOH | — | — | — | 8.2 | — |
| 45d | 4 | CONH2 | — | — | OH | 6.8 | — |
| 45e | 4 | CONH2 | — | — | — | 8.1 | — |
| 45f | 4 | 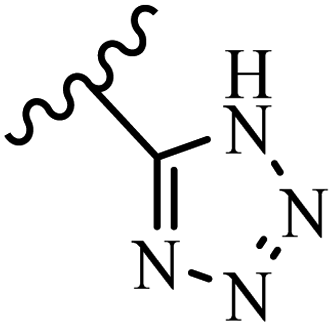 |
— | — |  |
7.0 | — |
| 45g | 4 | — | CH3 | CH3 | — | 6.6 | — |
| 45h | 1 | — | C2H5 | C2H5 | — | 4.6 | — |
| 45i | 4 | — | CH3 | Cl | — | 7.1 | — |
| 45j | 4 | — | Cl | Cl | — | 6.6 | — |
| 45k | 1 | — | CH3 | H | — | 5.5 | — |
| 45l | 1 | — | Cl | H | — | 5.7 | — |
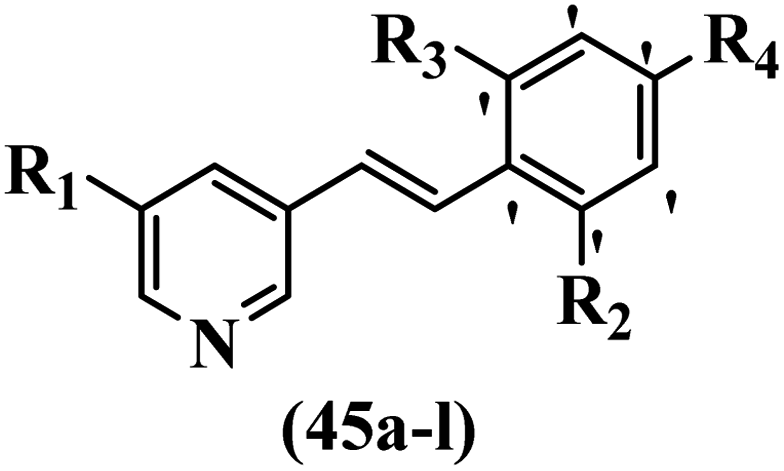
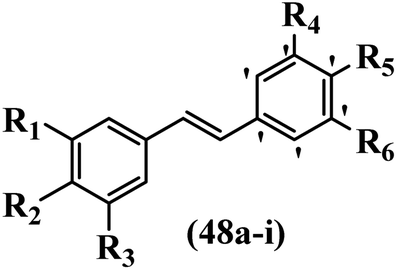
Gosslau et al.19 reported that E/Z-stilbene polyphenols induced p53-independent apoptosis and rapid perinuclear mitochondrial clustering. Among the Z-isomers, 48a and 48b were more than 1000-times effective compared to resveratrol against the WI38VA cell line. Furthermore, the addition of an NH2 group at the 3′-position on the phenyl ring in 48b resulted in lower activity compared to 48c. However, attaching an NO2 group in the 3′-position on the phenyl ring in 48d reduced the activity compared to 48c. In the E-isomers, the addition of an NO2 group in the 3′-position on phenyl ring decreased activity (48e vs. 48f). As well, appending OCH3 group in 3′-position on the phenyl ring in 48g drastically reduced the anti-proliferative activity compared to 48f. The addition of an OCH3 group in the 4′-position on the phenyl ring enhanced its anti-proliferative activity at least 10-times (48h vs. 48i). Attaching an OCH3 group in the 4′-position enhanced the activity for both the Z- and E-isomers by at least 4-times (48i vs. 48f, and 48c vs. 48a). Also, the hydrophilic group showed less activity compared to the lipophilic group (e.g., 48j and 48i). In addition, the presence of a Br atom in the 4′-position on the phenyl ring in 48k resulted in better activity than no substitution in 48l. E-48f, the most potent derivative in this series, was 200-times less potent than Z-48a. The Z-isomer analogues were the most potent E-isomer analogues, with IC50 values comparable to or better than resveratrol against WI38VA cells. Apoptosis revealed that 48a activated caspase 3/7 in the WI38VA cells. Similar to 48f, this compound activated apoptosis in the transformed cells. The findings recommend that mitochondrial clustering can happen in the absence of a main change in microtubule dynamics, implying that microtubule depolymerization is not directly responsible for perinuclear mitochondrial clustering. 48a and 48f both inhibited the growth of wild-type and p53-null cells. These findings imply that the proapoptotic activity of 48a and 48f against cancer cells was not dependent on p53 (Table 15).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | E/Z | R1 | R2 | R3 | R4 | R5 | R6 | IC50 (μM) WI38VA |
| 48a | Z | OCH3 | OCH3 | OCH3 | — | OCH3 | — | 0.02 |
| 48b | Z | OCH3 | — | OCH3 | NH2 | OCH3 | — | 0.05 |
| 48c | Z | OCH3 | — | OCH3 | — | OCH3 | — | 0.03 |
| 48d | Z | OCH3 | — | OCH3 | NO2 | OCH3 | — | 0.5 |
| 48e | E | OCH3 | OCH3 | OCH3 | NO2 | OCH3 | — | 10 |
| 48f | E | OCH3 | OCH3 | OCH3 | — | OCH3 | — | 2 |
| 48g | E | OCH3 | OCH3 | OCH3 | OCH3 | OCH3 | — | — |
| 48h | E | OCH3 | OCH3 | — | — | — | — | — |
| 48i | E | OCH3 | — | OCH3 | — | OCH3 | — | 25–50 |
| 48j | E | OH | — | OH | — | OH | — | 50 |
| 48k | E | OCH3 | OCH3 | OCH3 | — | Br | — | 25–50 |
| 48l | E | OCH3 | OCH3 | OCH3 | — | — | — | 80 |
| Resveratrol | E | — | — | — | — | — | — | 50 |
Simoni et al.58 designed stilbene-based derivatives and assessed their anti-tumor activity. Compound 52a was the most active compound among the derivatives, while it showed weaker or equal activity compared to vincristine, colchicine, and CA-4 against UCI-101, SNU-423, MDA-MB231, and MiaPaCa-2. Most of the compounds showed weak cytotoxicity against SNU-423, MDA-MB231, and MiaPaCa-2. In addition, compound 52b with N-methylpyrrole in the 3- and 4-positions on the phenyl ring exhibited weaker cytotoxicity activity compared with 52a having NH3+Cl− and OCH3 groups in the same position against MDA-MB231 and MiaPaCa-2. Also, replacement of the OCH3 in 52a with an OH group in the 3′-position on the phenyl ring in 52c decreased the activity by more than 10-times against the UCI-101 and MDA-MB23 cell lines. In contrast, when the OCH3 group in the 3-position on the phenyl ring was replaced by bulkier substituents, i.e., ethoxy or isopropoxy groups, as in compounds 52d and 52e compared to 52a, respectively, the activity decreased. Moreover, compound 52f, with a 2-hydroxyethyl group in the 3′-position on the phenyl ring, still retained its activity. However, the addition of a carboxyl group, as in compound 52g against UCI-101, completely eliminated its activity, representing unequivocally that the environment cannot tolerate excessive hydrophilicity. Also, the replacement of the OCH3 group with N-methylpyrrole in the 3′- and 4′-positions on the phenyl ring dramatically reduced the activity for 52h against UCI-101. This result showed that a hydrophobic environment is required for activity. The derivatives with an indole or imidazole ring in 51a, 51b, and 51c showed equal activity against this cell line. Shifting the nitrogen atom in the indole ring from the 1-position in compound 52b to the 3-position in compound 52i resulted in weaker activity against UCI-101. It is worth noting that 52b and 52i have diverse biological effects, therefore demonstrating a favorable interaction for the CH3 group in compound 52b. Among compounds 53a–c, only 53a with carbamic acid morpholin-4-yl-ethyl ester in the 3-position on the phenyl ring was active in suppressing the growth of tumor cells, which was 170-times less active than stilbene 52a. Both 53b with morpholin-4-yl-ethylureahydrochloride salt in the 3-position on the phenyl ring and 53c with carbamic acid (2-hydroxyethoxy)ethoxyethyl ester in the same position were not capable of causing remarkable inhibition of cell growth, demonstrating that their activity is lower than 53a. This could be because compounds 53a–c were not changed into the active form, stilbene 53a, under the tissue culture conditions. The tubulin inhibitory activity of the four derivatives 52 that changed substitution in the 3-position on the phenyl ring showed potency following the order of 52f > 52c > 52d > 52j. Compound 52c, in particular, has the second-best tubulin inhibitory activity but significantly weaker cytotoxic activity compared to derivatives 52f, 52d, and 52j. When selective second-ring derivatives were tested for tubulin inhibitory activity (52b > 52i > 52a), their cytotoxicity followed the order of 52b > 52i > 52a. As a result, in addition to tubulin depolymerization, stilbene derivatives may have other mechanisms for inducing cell death (Table 16).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R1 | R2 | R2 | IC50 (nM) | |||
| UCI-101 | SNU-423 | MDA-MB231 | MiaPaCa2 | |||||
| 51a | 3-OCH3 | 3-OCH3 |  |
— | 250 | 700 | 250 | 200 |
| 51b | 3-OCH3 | 3-OCH3 |  |
— | 250 | 700 | 250 | 200 |
| 51c | 3-OCH3 | 3-OCH3 |  |
— | 260 | 1000 | 800 | 800 |
| 52a | 3-OCH3 | 3-OCH3 | 3-OCH3 | 4-NH3+Cl− | 30 | 30 | 30 | 40 |
| 52b | 3-OCH3 | 3-OCH3 |  |
— | 40 | 30 | 80 | 80 |
| 52c | 3-OH | 3-OCH3 | 3-OCH3 | 4-NH3+Cl− | 800 | 2000 | 800 | 700 |
| 52d | 3-OCH2CH3 | 3-OCH3 | 3-OCH3 | 4-NH3+Cl− | 80 | — | — | — |
| 52e | 3-OCH(H3C)2 | 3-OCH3 | 3-OCH3 | 4-NH3+Cl− | 100 | — | — | — |
| 52f | 3-O(H2C)2OH | 3-OCH3 | 3-OCH3 | 4-NH3+Cl− | 150 | 200 | 80 | 180 |
| 52g | 3-OCH2COOH | 3-OCH3 | 3-OCH3 | 4-NH2 | >10.000 | — | — | — |
| 52h |  |
— | 3-OCH3 | NH2x(COOH)2 | 5000 | — | — | — |
| 52i | 3-OCH3 | 3-OCH3 |  |
— | 220 | — | — | — |
| 52j |  |
— | 3-OCH3 | NH2 | 200 | — | — | — |
| 53a | 3-OCH3 | 3-OCH3 | OCH3 | 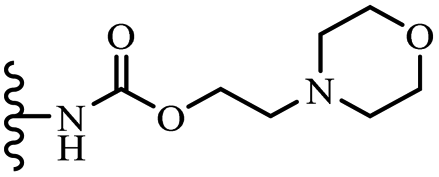 |
5000 | 5000 | — | — |
| 53b | 3-OCH3 | 3-OCH3 | OCH3 | 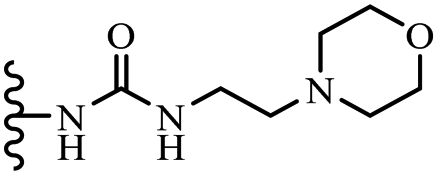 |
>10.000 | — | — | — |
| 53c | 3-OCH3 | 3-OCH3 | OCH3 |  |
>10.000 | — | — | — |
| Colchicine | — | — | — | — | 30 | 25 | 20 | 25 |
| Vincristine | — | — | — | — | 20 | 10 | 15 | 15 |
| Combretastatin A | — | — | — | — | 2 | 6 | 4 | 8 |
Moon et al.59 synthesized resveratrol derivatives and evaluated their cytotoxicity activity. Compounds 59a and 59b were the most potent compounds among them against the XF-498, SK-OV-3, HCT-15, A-549, and SK-MEL-2 cell lines. Furthermore, compound 59a showed the highest activity in all the cell lines in terms of cytotoxicity. However, none of the compounds exhibited stronger activity than adriamycin against all the cell lines. Besides, compound 59c with N-(4-benzylpiperidine)carbonyl in the 4′-position on the phenyl ring exhibited higher activity than 59d having N-(4-methylpiperidine)carbonyl in the same position against SK-MEL-2, A-549, and HCT-15. However, compound 59d was inactive against these cell lines. Also, replacement of the aromatic ring in 59e with a heteroaromatic ring in 59f decreased the activity against A549, SK-OV-3, SK-MEL-2, and XF-498. In addition, replacing N-decylaminocarbonyl in 59g in the 4′-position with N-cyclohexylaminocarbonyl in 59b on the same position enhanced the activity against all the cell lines (Table 17).
|
||||||
|---|---|---|---|---|---|---|
| Compound | R1 | IC50 (μM) | ||||
| A-549 | SK-OV-3 | SK-MEL-2 | XF-498 | HCT-15 | ||
| 59a |  |
8.7 | 5.7 | 10.4 | 11.4 | 14.4 |
| 59b |  |
10.4 | 6.8 | 12.4 | 13.6 | 17.2 |
| 59c | 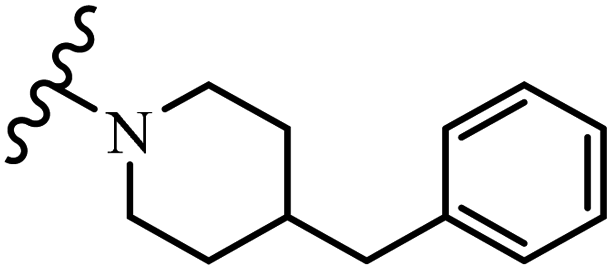 |
45.7 | 26.8 | 43.0 | 25.4 | 47.4 |
| 59d | 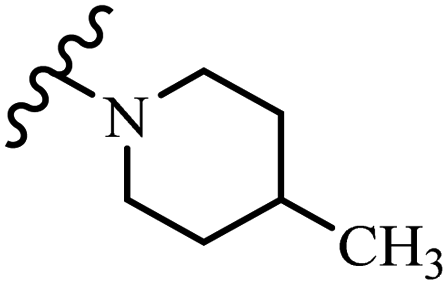 |
— | 21.6 | — | 21.6 | — |
| 59e | 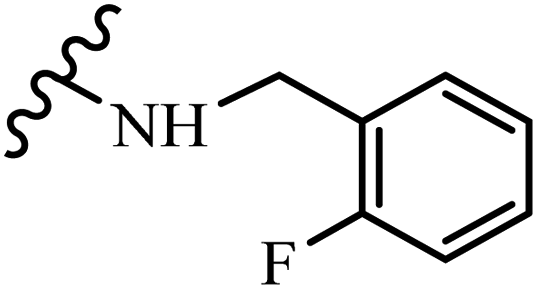 |
12.7 | 9.1 | 16.2 | 15.7 | 21.5 |
| 59f | 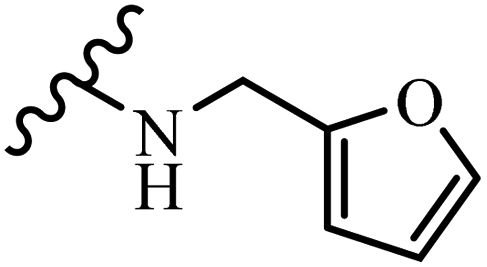 |
25.6 | 26.2 | 22.4 | 28.0 | 16.4 |
| 59g |  |
16.4 | 21.7 | 14.2 | 19.7 | 21.2 |
| Resveratrol | — | 36.9 | 42.6 | 41.4 | 32.1 | 35.6 |
| Adriamycin | — | 2.8 | 4.3 | 2.6 | 2.1 | 7.6 |
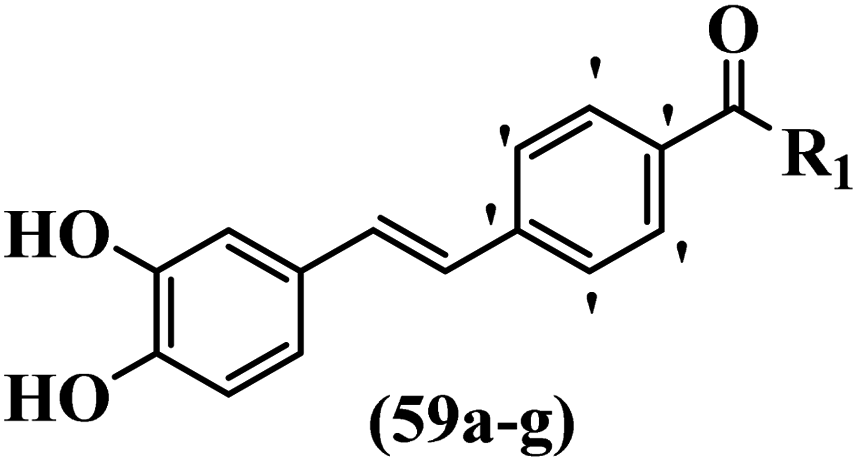
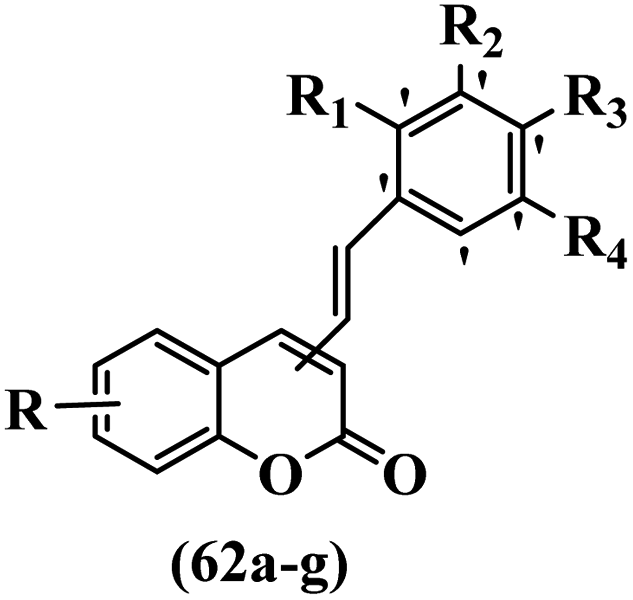
Belluti et al.60 synthesized stilbene–coumarin hybrids and evaluated their cytotoxicity. Compounds 62a and 62b displayed higher activity compared to resveratrol against the H460, A431, and JR8 cell lines and were the most potent compounds among the studied compounds. All the compounds were inactive against the A431 and JR8 cell lines except 62a and 62b. In this series, compound 62b appeared to be one of the most active compounds, thus presenting proof for the significance of the OCH3 group in the 4-position on the coumarin ring as a scaffold. On moving the OCH3 group from the 4- to 3-position on the coumarin ring in derivative 62b, resulting in compound 62c, a dramatic drop in potency was observed, suggesting that the 4-position insertion is also crucial. Furthermore, when the OCH3 groups of 62b were replaced with OH, derivative 62g was 60-times less active than 62b and 2-times less active than resveratrol against H460. Considering these results, the extra structural changes were made in 62b focusing on the phenyl ring, a significant decrease in anti-proliferative activity was detected for compound 62d with an OCH3 group in the 5′-position and 62e bearing an OCH3 group in the 2′-position on the phenyl ring. Besides, the removal of one OCH3 group in 62b resulted in a decrease in activity. Also, 62f showed comparable activity to resveratrol against H460. These findings revealed that the OCH3 groups in the 3′- and 5′-positions on the phenyl ring had an important role, and their exchange with CH3, 62a, showed somewhat greater potency than the corresponding molecule 62b. Subsequently, 62b and 62a was investigated for inducing apoptosis. In particular, the proapoptotic effect of 62b showed a level of apoptosis similar to that of cisplatin and substantially higher than that of resveratrol. Caspase 3 and PARP cleavage were investigated. This effect was linked to a partial cell tumor during the G2/M phase of the cell cycle. In summary, the existence of specific substituents in different positions of both moieties of the hydride compound was confirmed to be serious in discussing the anti-proliferative activity; substitutions shaped the 4-position on the coumarin ring and the 3′- and 5′-positions on the phenyl ring were revealed to be a satisfactory feature for both the anti-tumor and proapoptotic activities (Table 18).
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Insertion position | IC50 (μM) | Induced apoptosis (%) | |||||||
| R | R1 | R2 | R3 | R4 | H460 | A431 | JR8 | |||
| 62a | 4 | 7-OCH3 | H | CH3 | H | CH3 | 0.29 | 3.5 | 3.5 | 32 |
| 62b | 4 | 7-OCH3 | H | OCH3 | H | OCH3 | 0.45 | 3.44 | 3.2 | 27 |
| 62c | 3 | 7-OCH3 | H | OCH3 | H | OCH3 | >10 | — | — | — |
| 62d | 4 | 7-OCH3 | H | OCH3 | OCH3 | OCH3 | >100 | — | — | — |
| 62e | 4 | 7-OCH3 | OCH3 | OCH3 | OCH3 | H | >50 | — | — | — |
| 62f | 4 | 7-OCH3 | H | OCH3 | H | H | 12.9 | — | — | — |
| 62g | 4 | 7-OH | H | OH | H | OH | 27 | — | — | — |
| Resveratrol | — | — | — | — | — | — | 12.9 | — | — | — |
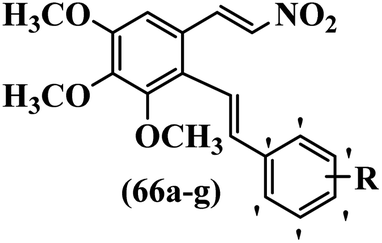
Reddy et al.61 designed resveratrol-based nitrovinylstilbenes and tested their anti-mitotic and anti-tubulin activities. Compound 66a was the most potent compound among them and showed higher activity than resveratrol against the MCF-7 cell line. However, 66a showed weaker or equal anti-proliferative activity compared to resveratrol against the SK-N-SH, A-549, and HeLa cell lines. Compound 66b showed higher anti-proliferative activity compared to resveratrol against MCF-7, SK-N-SH, A549, and HeLa. Also, EDG showed better activity compared to EWG (e.g., 66b vs. 66c) against SK-N-SH, A-549, and HeLa. Likewise, changing the dioxane ring in the 3′- and 4′-positions (66d) with a phenyl ring in the 4′-position on the phenyl ring (66a) reduced the activity against all the tested cell lines. In contrast, 66a showed stronger activity than 66d against the MCF-7 cell line. Furthermore, increasing the number of OCH3 groups in different positions on the phenyl ring of 66e resulted in lower anti-proliferative activity compared to 66f against the tested cell lines. Also, the presence of a lipophilic group had more favorable anti-proliferative activity than a hydrophilic group on the phenyl ring (66c vs. 66g) against the MCF-7, SK-N-SH, and A-549 cell lines. Compounds 66b, 66d, and 66g had higher cytotoxicity activity, which correlated well with their ability to effectively inhibit tubulin. Compound 66b, as expected, demonstrated the greatest inhibition of tubulin assembly. All the compounds were less potent than colchicine. The findings showed that the compounds inhibited tubulin assembly in the order of 66b > 66g > 66d. The anti-mitotic effects of active compounds 66b, 66g, and 66d were tested. These compounds showed higher activity in the G2/M phase of HeLa cells compared to resveratrol. Most of the cells were arrested at the G2/M phase by compounds 66b, 66d, and 66g. These findings support the inhibitory activity of nitrovinylstilbene derivatives (66b and 66g) against the HeLa cell line. The activation of caspase-3 by compounds 66b and 66g in HeLa cells was examined. Compared to resveratrol, treatment with compound 66b resulted in a 12-times increase in caspase-3 activity. In contrast, resveratrol did not cause a significant increase in caspase-3 activation (Table 19).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | R | IC50 (pM) | IC50 (μM) | Inhibition percent (%) | Fold-increase in | |||
| MCF-7 | SK-N-SH | A549 | HeLa | Tubulin | G2/M | Caspase-3 | ||
| 66a |  |
7.2 | 35.8 | 44.4 | — | — | — | — |
| 66b | 4-CH3 | 42.5 | 12.5 | 19.0 | 4.4 | 4.27 | 66.43 | 10 |
| 66c | 4-F | 19.0 | 18.8 | 35.9 | 12.4 | — | — | — |
| 66d | 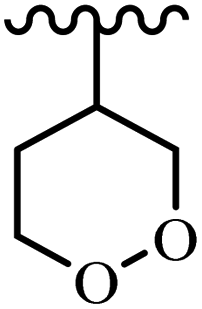 |
19.2 | 20.9 | 16.3 | 5.4 | 8.02 | 50.66 | — |
| 66e | 3,4,5-triOCH3 | 13.2 | 60.9 | 27.1 | — | — | — | — |
| 66f | 3,5-diOCH3 | 19.2 | 36.2 | 15.0 | 10 | — | — | — |
| 66g | 4-OCH3 | 21.4 | 20.7 | 40.4 | 7.8 | 4.90 | 60.40 | 11.5 |
| Resveratrol | — | 79.1 | 40.3 | 44.7 | 22.5 | — | 21.29 | — |
| Colchicine | — | — | — | — | — | 1.96 | — | — |
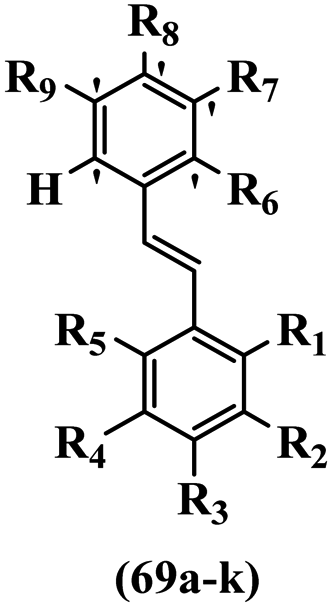
Csuk et al.20 synthesized E-stilbene-based derivatives and evaluated their anti-tumor activity. Compounds 69a and 69b showed the highest activity against 518A2, 850C, A253, A549, A2780, DLD1, Lipo, MCF-7, and NiH3T3 cell lines. These compounds were also more potent than tamoxifen. Among the compounds, 69c exhibited the lowest activity. Switching the OCH3 group from the 5′- to 4′-position on the phenyl ring (69d vs. 69e) decreased the activity against all the tested cell lines. In addition, adding an F atom in the 4′-position on the phenyl ring compared to no substitution resulted in lower activity (69f vs. 69g) against the tested cell lines. Also, changing OCH3 with an OH group in the 2′- and 5′-positions on the phenyl ring reduced the cytotoxicity activity (69h vs. 69i). It seems that lipophilic groups show higher activity than hydrophilic groups. Moreover, moving the OH group in the 4- to the 2-position on the phenyl ring showed almost equal cytotoxicity activity (e.g., 69j and 69k) against all the cell lines. In addition, the apoptotic activity of the compounds was almost similar to the cytotoxic activity against the A549 cell line (Table 20).
|
|||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | R9 | IC50 (μM) | |||||||||
| 518A2 | 850C | A253 | A549 | A2780 | DLD1 | Lipo | MCF7 | NiH3T3 | Induced apoptosis (%) | ||||||||||
| 69a | — | OH | OCH3 | — | F | OCH3 | — | OCH3 | — | 0.03 | 0.03 | 0.03 | 0.03 | 0.01 | 0.04 | 0.03 | 0.06 | 0.05 | 77.3 |
| 69b | — | OH | OCH3 | — | F | — | OCH3 | F | OCH3 | 0.20 | 0.13 | 0.48 | 0.18 | 0.11 | 0.20 | 0.21 | 0.22 | 0.19 | 64.1 |
| 69c | — | — | OH | — | — | — | OH | — | OH | >30 | >30 | >30 | >30 | >30 | >30 | >30 | >30 | 24.22 | — |
| 69d | — | OH | OCH3 | — | — | OCH3 | — | — | OCH3 | 0.72 | 0.86 | 0.80 | 0.96 | 0.87 | 0.91 | 0.65 | 0.54 | 1.54 | — |
| 69e | — | OH | OCH3 | — | — | OCH3 | — | OCH3 | — | 1.33 | 1.33 | 1.53 | 1.80 | 1.27 | 2.00 | 1.41 | 1.64 | 2.08 | — |
| 69f | — | OCH3 | OCH3 | — | — | — | OCH3 | F | OCH3 | 18.04 | 17.72 | 15.83 | 22.16 | 23.92 | 14.95 | 16.51 | 11.20 | 6.76 | — |
| 69g | — | OCH3 | OCH3 | — | — | — | OCH3 | — | OCH3 | 2.81 | 2.46 | 2.35 | 3.04 | 2.06 | 2.86 | 2.86 | 1.89 | 3.68 | 77.1 |
| 69h | — | OH | OCH3 | — | F | OCH3 | — | — | OCH3 | 1.74 | 2.26 | 1.34 | 2.21 | 2.01 | 2.26 | 1.98 | 2.09 | 2.66 | 72 |
| 69i | — | OH | OCH3 | — | F | OH | — | — | OH | 19.43 | 16.77 | 9.88 | 15.77 | 10.47 | 18.00 | >30 | 10.63 | 21.06 | — |
| 69j | — | — | OH | — | — | OH | — | — | OH | 28.36 | 18.70 | 15.16 | 12.66 | 16.62 | 22.91 | 17.37 | 14.17 | 9.59 | — |
| 69k | OH | — | — | — | — | OH | — | — | OH | 22.00 | 24.45 | 14.53 | 19.01 | 12.42 | 18.11 | 24.07 | 13.18 | 12.38 | — |
| Tamoxifen | — | — | — | — | — | — | — | — | — | 7.62 | 11.09 | 8.92 | 9.66 | 7.77 | 4.78 | 8.64 | 7.10 | 7.26 | — |
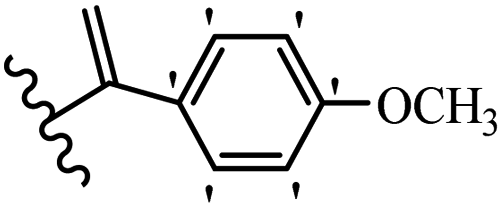
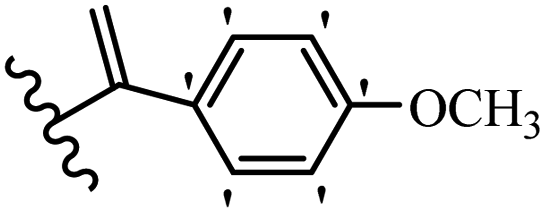
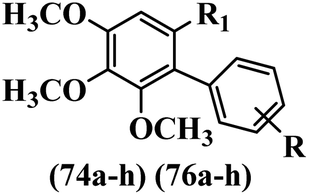
Kumar et al.62 synthesized biaryl stilbenes/ethylenes and assessed their anti-microtubule activity. Among the synthesized compounds in the first series, 74a and 74b exhibited the highest activity against the A549, HeLa, SK-N-SH, and DU-145 cell lines. Attaching OCH3 (74c) and Cl groups (74d) to the 4-position on the phenyl ring compared to 74e, which was unsubstituted resulted in lower anti-proliferative activity against the SK-N-SH cell line. In addition, an extra OCH3 group in the 3-position on phenyl ring decreased the activity (74c vs. 74f) against the HeLa and SK-N-SH cell lines. Also, the addition of a CH3 group in the 4′-position on the phenyl ring of 74h compared to no substitution (74g) enhanced the activity against the A549, HeLa, and SK-N-SH cell lines. Similarly, compounds 76a and 76b demonstrated the highest activity in the second series against the A549, HeLa, SK-N-SH, and DU145 cell lines. The compound with no substitution on the phenyl ring (76c) displayed higher activity than 76d having a phenyl on the phenyl ring against HeLa and DU145. However, 76c showed weaker activity than 76d against A549 and SK-N-SH. Also, attaching a Cl group in the 4′-position on the phenyl ring compared to no substitution resulted in better activity (76e and 76f). Moreover, according to the SAR studies of both series, it was concluded that the substitutions with a small size and strong electronegativity in the 3- and 4-positions on the phenyl ring may be important for effective anti-proliferative activity (e.g., 74b and 76b). Also, the presence of EDG in the 4-position on phenyl showed better activity compared to EWG against A549 (76h vs. 76g). The results showed that the second series exhibited stronger activity compared to the first series. Also, 74a demonstrated the maximum inhibition of tubulin. None of the compounds showed stronger activity than colchicine. 74a, 74b, 76a, and 76b arrested the majority of the population of cells at the G2/M phase. Overall, these findings indicated that compounds 74a, 74b, 76a, and 76b inhibited tubulin polymerization more effectively than the other compounds in both series (Table 21).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R | R1 | IC50 (μM) | Inhibition mitosis percent (%) | ||||
| A549 | Hela | SK-N-SH | DU145 | Tubulin | ||||
| 74a | 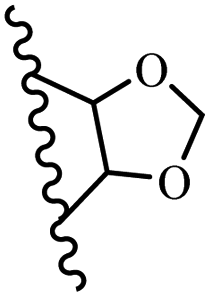 |
 |
6.3 | 8.7 | 8.0 | 7.3 | 6.8 | 73.02 |
| 74b | 3,4-diF |  |
7.5 | 9.2 | 6.9 | 7.5 | 9.2 | 71.16 |
| 74c | 4-OCH3 |  |
35 | 19 | 36 | 34 | — | — |
| 74d | 4-Cl |  |
30 | 49 | 15 | 35 | — | — |
| 74e | H |  |
36 | 26 | 11 | 51 | — | — |
| 74f | 3,4-diOCH3 |  |
28 | 45 | 42 | 34 | — | — |
| 74g | H |  |
79 | 89 | 63 | 82 | — | — |
| 74h | H |  |
54 | 45 | 31 | 101 | — | — |
| 76a | 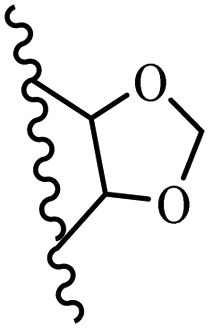 |
 |
5.0 | 5.4 | 6.1 | 6.0 | 8.6 | 73.00 |
| 76b | 3,4-diF | 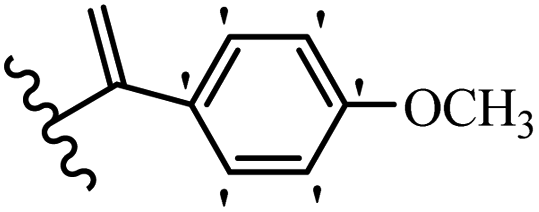 |
6.5 | 5.9 | 7.5 | 6.8 | 11.2 | 70.02 |
| 76c | H | 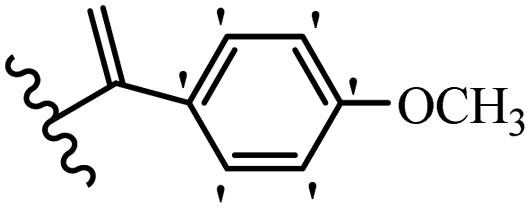 |
22 | 11 | 31 | 8 | — | — |
| 76d |  |
 |
15 | 35 | 6 | 46 | — | — |
| 76e | H | 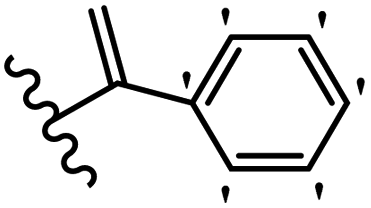 |
17 | 42 | 34 | 36 | — | — |
| 76f | H | 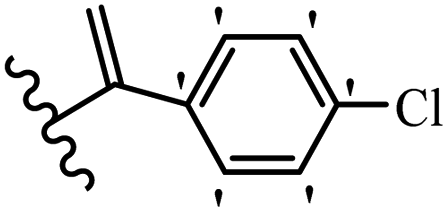 |
40 | 34 | 25 | 29 | — | — |
| 76g | 4-Cl |  |
29 | 30 | 15 | 33 | — | — |
| 76h | 4-OCH3 |  |
14 | 72 | 75 | 37 | — | — |
| Colchicine | — | — | — | — | — | — | 1.7 | 19.4 |
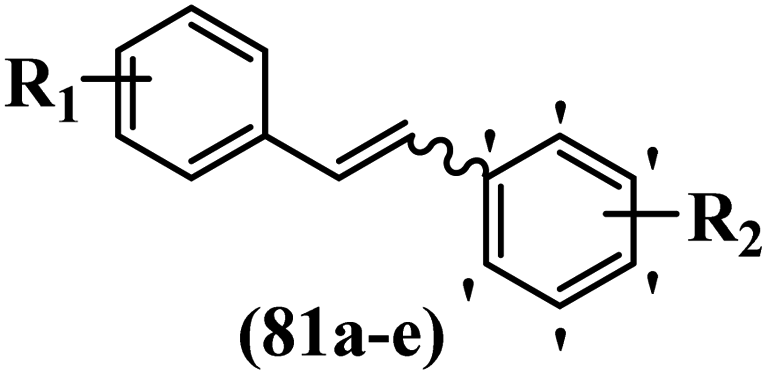
Roman et al.63 designed some E/Z-stilbenes and tested their anti-invasive activity. Among the synthesized compounds, E-81a and Z-81b exhibited the highest activity against MCF-7/6. In both isomers, attaching substitutions in the 4′-position phenyl ring enhanced the activity (e.g., 81a and 81c). The Z-isomers showed better activity than the E-isomers (e.g., Z-81d and E-81d). Analogues E-81e and Z-81e, combining both decoration patterns, displayed very weak potency. Overall, the results showed that two-atom spacers are well tolerated between the aromatic moieties (Table 22).
|
||||
|---|---|---|---|---|
| Compound | E/Z | R1 | R2 | IC50 (μM) MCF-7/6 |
| 81a | E | 4-OCH3 | 4-F | 0.01 |
| 81b | Z | 4-F | H | 0.01 |
| 81c | E | 4-OCH3 | H | 0.1 |
| 81d | E | 3,4,5-triOCH3 | H | 0.1 |
| 81d | Z | 3,4,5-triOCH3 | H | 0.01 |
| 81e | E | 3,4,5-triOCH3 | 4-F | 1 |
| 81e | Z | 3,4,5-triOCH3 | 4-F | 0 |
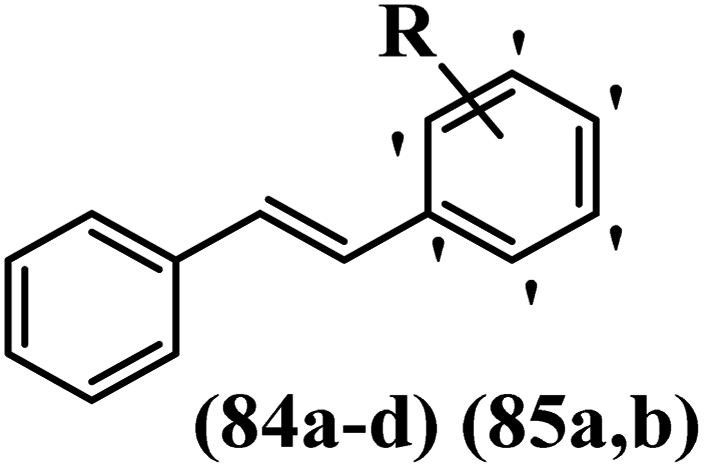
Centelles et al.64 synthesized stilbene derivatives and evaluated their cytotoxicity and inhibitory activity against VEGF. Compound 84a showed the highest activity against the BAE cell line among the synthesized compounds. Also, it was more potent than resveratrol. Likewise, moving the OH group in the 4′-position of compound 84a to the 2′- or 3′-position on the phenyl ring in 84b or 84c resulted in lower activity against BAE. Moreover, changing the OCH3 group in the 2′-position on the phenyl ring in 84d with an OH group in the same position in 84b resulted in an enhancement in activity against BAE. Further, moving the O-allyl moiety from 2′- to 4′-position on phenyl ring reduced activity against BAE (85a vs. 85b). In addition, the cytotoxicity of all the compounds was investigated against the HT-29 cell line. Among the synthesized compounds, 85b showed higher activity compared to resveratrol against HT-29. Moreover, compound 84a exhibited stronger activity than 84b and 84c. Compound 84d showed weaker cytotoxicity compared to 84b. Compound 85b had more potent activity than 85a. According to the results, changing the position of substitution on the phenyl ring improved the cytotoxicity activity against the HT-29 and BAE cell lines (4′ > 2′ > 3′). Resveratrol and some stilbene analogues reduced the VEGF expression in HT-29 cells. Compounds 84b and 85a decreased the VEGF expression to a higher extent than resveratrol and DMSO. Replacement of the OH in the 2′-position on the phenyl ring in 84b with an OCH3 group in the same position in 84d resulted in lower activity against VEGF. The results showed that hydrophilic groups displayed higher activity compared to lipophilic groups. Also, the presence of bulky groups decreased the activity against BAE and expression of VEGF. However, it increased the activity against the HT-29 cell line (Table 23).
|
||||
|---|---|---|---|---|
| Compound | R | IC50 (μM) | IC50 (ng mL−1) | |
| BAE | HT-29 | VEGF | ||
| 84a | 4-OH | 33.6 | 34.6 | — |
| 84b | 2-OH | 107 | 112 | 19 |
| 84c | 3-OH | 91.7 | 127 | — |
| 84d | 2-OCH3 | 152 | 42.8 | 39 |
| 85a | 2-Oallyl | 313 | 55 | 20 |
| 85b | 4-Oallyl | >400 | 23 | — |
| Resveratrol | — | 48 | 110 | 30 |
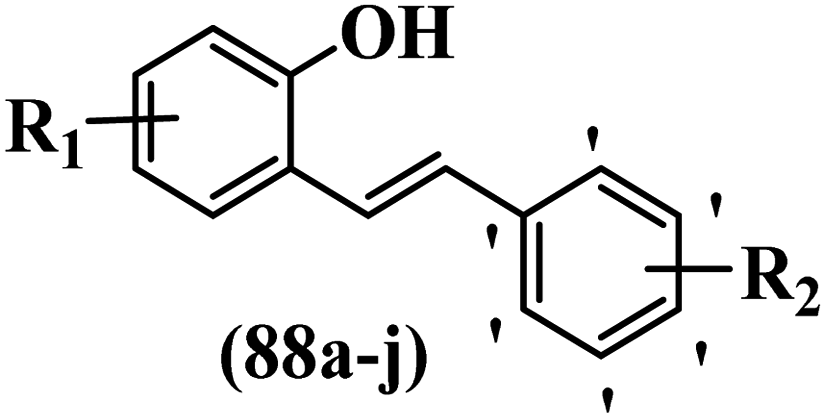
Zhang et al.65 synthesized 2-hydroxylated E-stilbenes and assessed their anti-proliferative activity. Among the synthesized compounds, 88a against Colo-205 and MGC80-3, 88b against HT-29, and 88c against MDA-468 showed the highest activity. Changing the Br atom in the 4′-position on the phenyl ring (88d) with CH3 (88e), OH (88f), and CN (88g) groups reduced cytotoxicity activity against all the cell lines. It seems that the compounds with a lipophilic group compared to a hydrophilic group showed stronger cytotoxicity activity. Also, the presence of an extra OCH3 group in the 4′-position on the phenyl ring in 88c enhanced the cytotoxicity activity compared to 88h against MDA-468 and MGC80-3. In addition, moving the OCH3 group from the 3- to 4-position on phenyl improved the cytotoxicity activity (88i vs. 88h) against MDA-468 and MGC80-3. Besides, the compounds with substitution on the phenyl ring showed better cytotoxicity activity than no substitution (88j vs. 88a) against all the cell lines. In addition, the compounds containing EWG exhibited the higher activity compared to EDG (e.g., 88e and 88d) (Table 24).
|
||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | IC50 (μM) | |||
| Colo-205 | MDA-468 | HT-29 | MGC80-3 | |||
| 88a | 4-OCH3 | 3,4,5-triOCH3 | 5.3 | 5.3 | 9.7 | 0.035 |
| 88b | 5-Br | 3,5-diOCH3 | 16.4 | 6.1 | 9.5 | 3.3 |
| 88c | 3-OCH3 | 3,4,5-triOCH3 | — | 2.6 | — | 1.1 |
| 88d | H | 4-Br | 21.1 | 7.4 | 14.4 | — |
| 88e | H | 4-CH3 | — | 21.9 | 22.9 | — |
| 88f | H | 4-OH | — | — | 33.5 | — |
| 88g | H | 4-CN | — | — | — | 3.1 |
| 88h | 3-OCH3 | 3,5-diOCH3 | — | — | — | — |
| 88i | 4-OCH3 | 3,5-diOCH3 | — | 8.4 | — | 0.8 |
| 88j | H | H | — | 19.3 | — | — |
| Resveratrol | — | — | 23.5 | 45.2 | 87.2 | 42 |
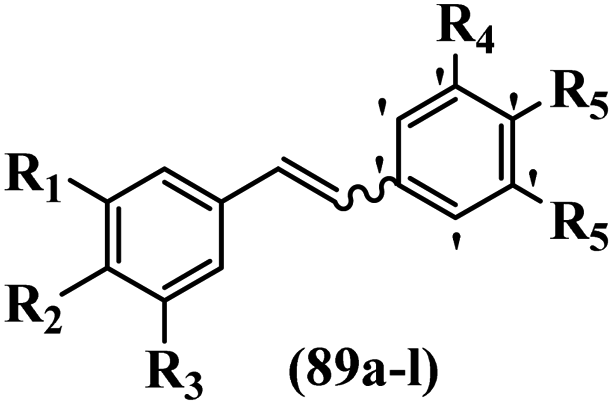
Morris et al.66 tested the anti-tumor properties of methylated Z-resveratrol. Compounds 89e and 89h showed lower activity than 89d against the proliferation of B16-F10. However, the inhibition potency of Z-89f and g was slightly higher than that of 89d. These results indicated that Z-tetra-methoxy inhibited motility more effectively than Z-tri- or penta-methoxy. However, isomers of 89a–c showed no visible activity on β-tubulin expression. It was discovered that 89d reduced the expression of tubulin in B16-F1 cells, while 89a–c exhibited no activity. Compared to DMSO, all the tested Z-isomers showed a dramatic decrease in intracellular tubulin protein, and in the case of 89f, the level of expression was below the detectable limit. When compounds 89a–d were compared, 89d was the most effective in inhibiting the proliferation of B16-F10 cells, B16-F1 cells, and melanocytes. These findings imply that these cells respond differently to the anti-proliferative effects of 89d. The Z-polymethoxy compounds were created to inhibit cell proliferation compared to DMSO-treated cells, with 89d being the most potent inhibitor, while analogues 89e, 89f, and 89h were significantly less potent. It is worth noting that neither the E-isomer (89a) nor any of the counterpart trans-polymethoxystilbenes (89c and 89i–l) had a significant effect on B16-F10 cell proliferation. In addition, the present lipophilic groups showed higher activity compared to hydrophilic groups (89d vs. 89b). Also, the Z-isomers exhibited superior activity in comparison to their corresponding E-isomers (Table 25).
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | E/Z | R1 | R2 | R3 | R4 | R5 | R6 | Inhibition percent (%) | ||
| B16-F10, B16-F1, melanocytes | Proliferation B16-F10 | Motility B16-F10 | ||||||||
| 89a | E | OH | H | OH | H | OH | H | — | — | — |
| 89b | Z | OH | H | OH | H | OH | H | — | — | — |
| 89c | E | OCH3 | H | OCH3 | H | OCH3 | H | — | — | — |
| 89d | Z | OCH3 | H | OCH3 | H | OCH3 | H | 75 | 71 | — |
| 89e | Z | OCH3 | OCH3 | H | H | OCH3 | H | — | 45 | 50–60 |
| 89f | Z | OCH3 | H | OCH3 | OCH3 | OCH3 | H | — | 45 | 80–90 |
| 89g | Z | OCH3 | OCH3 | OCH3 | H | OCH3 | H | — | 66 | 80–90 |
| 89h | Z | OCH3 | OCH3 | OCH3 | H | OCH3 | OCH3 | — | 45 | 50–60 |
| 89i | E | OCH3 | OCH3 | H | H | OCH3 | H | — | — | — |
| 89j | E | OCH3 | OCH3 | H | H | OCH3 | OCH3 | — | — | — |
| 89k | E | OCH3 | OCH3 | OCH3 | H | OCH3 | H | — | — | — |
| 89l | E | OCH3 | OCH3 | OCH3 | OCH3 | H | OCH3 | — | — | — |
| DMSO | — | — | — | — | — | — | — | 100 | 100 | — |
Scherzberg et al.67 evaluated resveratrol derivatives against tumor cells. The method for the synthesis of resveratrol derivatives 90a and 90b was the same as in the reported study.26 Compared to E-resveratrol, Z-90a showed 100-times higher anti-proliferative activity against HT-29. Compound Z-90a inhibited HepG2 cell growth. E-Resveratrol exhibited lower activity for HepG2 cells compared to Caco-2. In contrast to Caco-2, the IC50 value for Z-90a in HepG2 is the highest. However, in both proliferation assays, Z-90a had significantly lower IC50 values than E-resveratrol. These findings are comparable to that obtained in the Caco-2 cell line. In comparison to the results in tumor cell lines, Z-90a only had moderate effects on HUVEC cell growth. Compound Z-90a exhibited no cytotoxic effect on CaCo-2, HT-29, HepG2 cells, and HUVECs. Another distinction was revealed by the cell cycle analysis, where both E/Z-resveratrol arrested cells in the S-phase, whereas E/Z-90a arrested cells in the G2/M phase. According to the results, lipophilic groups displayed better activity than hydrophilic groups incorporation in BrdU (Table 26).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | E/Z | R1 | R2 | R3 | IC50 (μM) | |||
| HT-29 | CaCo-2 | HepG2 | BrdU incorporation | |||||
| Resveratrol | E | OH | OH | OH | 115.9 | 190.2 | 110.7 | 100 |
| Resveratrol | Z | OH | OH | OH | — | — | — | >200 |
| 90a | Z | OCH3 | OCH3 | OCH3 | 0.115 | 0.145 | 0.473 | 0.2–0.5 |
| 90a | E | OCH3 | OCH3 | OCH3 | — | — | — | 5–20 |
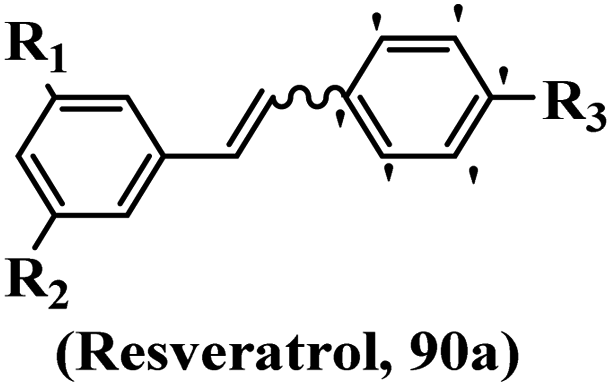
Yan et al.68 synthesized benzoselenazole-stilbene hybrids and assessed their cytotoxicity activity. Among the analogues, 95a showed the highest activity, and it was more active than resveratrol and ebselen against the Bel-7402, A549, HeLa, and MCF-7 cell lines. Also, moving the OCH3 group from the 4′-position in 95b to the 5′-position on the phenyl ring in 95c decreased the anti-proliferative activity against the tested cell lines. In addition, the extra OCH3 group in the 5′-position on the phenyl ring of 95b produced 95d, which showed reduced activity against the A549 and HeLa cell lines. However, 95b and 95d showed equal activity against the Bel-7402 and MCF-7 cell lines. When the number of OCH3 groups on phenyl ring decreased, the activity decreased markedly (95c vs. 95e). This exhibited that the OCH3 group is essential for an anti-cancer effect. Consequently, it can be concluded that the compound with two OCH3 groups, particularly at the 3′- and 4′-positions on the phenyl ring, was active. In contrast, the findings revealed that the OCH3 groups in the 4- and 5-positions on the phenyl ring had a negative effect on the activity of 95f against all the tested cell lines compared to the other compounds. When the group in the 4-position on the phenyl ring changed to F or Cl atoms, the anti-proliferative activity remarkably increased (95g and 95a). The findings exhibited that the E-isomer stilbene scaffold was favorable for cytotoxic activity compared to the corresponding Z-isomers (95b vs.98). Compared with 95b, the anti-proliferative activity of compound 102 was significantly lower (<10-times), which strongly demonstrated that the benzoselenazole-stilbene hybrids were beneficial for the activity. The in vitro inhibition of thioredoxin reductases (TrxR) was tested using the compounds. Compound 95b revealed the highest activity among the compounds, which was also better than that of ebselen. Compounds 95c and 95d exhibited better cell growth inhibition effects compared to 95e and 95h. Compounds 95g, 95a, 95i, and 95j, which have halogen atoms on the phenyl ring, exhibited the highest TrxR inhibitory activity. However, compound 95f, which demonstrated relatively good inhibitory activity against TrxR, showed less cell growth inhibition. Compound 102, which is characterized by the absence of a selenium atom, revealed no activity, further demonstrating the requirement of selenium in the derivatives. Furthermore, treatment with compound 95b resulted in significant cell cycle arrest at the G2/M phase. These findings showed that the compounds could halt cell cycle progression at the mitosis stage. The cell apoptosis assay results showed that resveratrol and compound 95b caused significant cell apoptosis. Compound 95b effectively induced cell apoptosis in Bel-7402 cells, eventually leading to cell death according to the data (Table 27).
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | E/Z | R1 | R2 | R3 | R | IC50 (μM) | Inhibition percent (%) | Induced apoptosis (%) | |||||
| Bel-7402 | A549 | HELA | MCF-7 | TrxR | TrxR | G2/M | |||||||
| 95a | E | OCH3 | H | OCH3 |  |
0.79 | 0.52 | 0.23 | 0.47 | — | 36.4 | — | — |
| 95b | E | OCH3 | OCH3 | H |  |
1.01 | 1.53 | 1.52 | 3.37 | 3.10 | — | 44.23 | 53.4 |
| 95c | E | OCH3 | H | OCH3 |  |
3.49 | 3.73 | 3.80 | 7.78 | 8.27 | — | — | — |
| 95d | E | OCH3 | OCH3 | OCH3 |  |
1.11 | 3.11 | 5.67 | 3.97 | 5.78 | — | — | — |
| 95e | E | OCH3 | H | H |  |
13.2 | 10.4 | 7.34 | 9.62 | 12.8 | — | — | — |
| 95f | E | OCH3 | H | OCH3 |  |
25.4 | 52.6 | 31.6 | 88.9 | 7.53 | — | — | — |
| 95g | E | OCH3 | H | OCH3 |  |
0.99 | 1.22 | 0.64 | 0.51 | — | 36.6 | — | — |
| 95h | E | H | H | H |  |
12.4 | 23.8 | 35.7 | 24.8 | — | 22.4 | — | — |
| 95i | E | OCH3 | H | OCH3 | 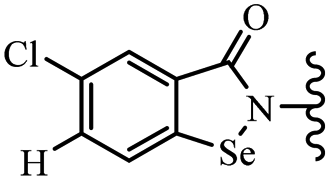 |
— | — | — | — | — | 31.2 | — | — |
| 95j | E | OCH3 | OCH3 | H | 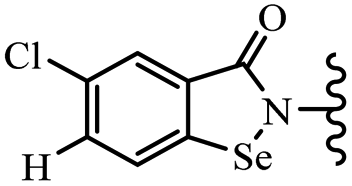 |
6.13 | 6.95 | 12.0 | 16.9 | — | 38.4 | — | — |
| 98 | Z | OCH3 | OCH3 | H | 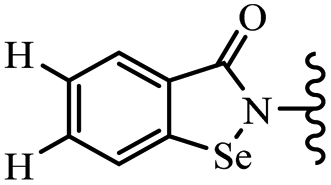 |
36.1 | 43.0 | 28.4 | 56.3 | 9.62 | — | — | — |
| 102 | E | OCH3 | OCH3 | H | 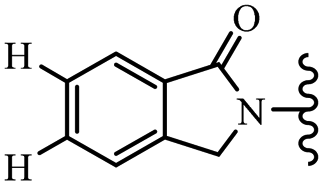 |
>10 | >10 | >10 | >10 | — | — | ||
| Resveratrol | — | — | — | — | 50.7 | >100 | 47.9 | >100 | — | 26.1 | — | — | |
| Ebselen | — | — | — | — | 68.2 | >100 | 78.5 | >100 | 8.54 | — | — | — | |
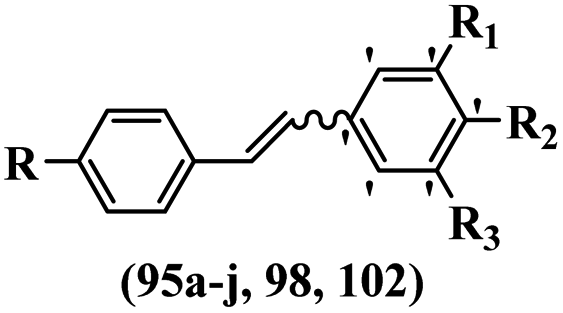
Mahdavi et al.69 designed N-substituted 2-arylquinazolinones and tested their cytotoxicity activity. Among the synthesized compounds, 109a against MCF-7 and T-47D and 109b against MDA-MB-231 showed the best cytotoxicity activity. They were also more potent than etoposide against MCF-7, MDA-MB-231, and T-47D. In addition, the compounds containing an aliphatic ring showed increased activity compared to that having an aromatic ring (e.g., 109c vs. 109d) against MCF-7, MDA-MB-231, and T-47D. Likewise, N-alkyl groups were more active than N-aryl and N-benzyl groups. Compound 109e with N-cyclopentyl was inactive against all the tested cell lines, while 109f with N-propyl showed activity against MCF-7. Most of the N-alkyl quinazolinones had similar sensitivity in all the tested cell lines. However, in the case of compound 109g, the sensitivity of MCF-7 was significantly lower than that of MDA-MB-231 and T-47D. The IC50 values of compounds 109d and 109h–l indicated that the aryl or benzyl substituents were not favorable for cytotoxicity; however, compound 109k with a 4-methylbenzyl moiety in the 3-position of quinazolinones showed mild activity against all the tested cell lines. Additionally, there is a similar difference against MCF-7 in 109g and 109a having isobutyl and sec-butyl groups, respectively. This could be because connecting diverse bonds of N-isobutyl and N-sec-butyl groups to the 3-position of quinazolinones caused diverse steric effects on the 2-aryl ring. Consequently, compounds 109a and 109c were tested compared to etoposide for the detection of apoptosis in the MDA-MB-231 and MCF-7 cell lines. Compounds 109c and 109a reduced the cell viability and induced apoptosis in MCF-7 and MDAMB-231 cells. Although the screened derivatives caused some necrosis in the treated cells, the results showed that 109a and 109c induced apoptosis in MDA-MB-231 cells. The ability of compound 109c to induce apoptosis in MDA-MB-231 cells was comparable to that of etoposide. The results also revealed that the percentage of MCF-7 cells undergoing apoptosis after exposure to compounds 109a and 109c was higher than MDA-MB-231. According to the findings, compounds 109c and 109a exhibited cytotoxic activity in the MDA-MB-231 and MCF-7 cell lines via apoptosis (Table 28).
|
||||||
|---|---|---|---|---|---|---|
| Compound | R | IC50 (μM) | Induce apoptosis | |||
| MCF-7 | MDA-MB-231 | T-47D | MCF-7 | MDA-MB-231 | ||
| 109a |  |
3.8 | 4.9 | 3.6 | 26.46 | 16.55 |
| 109b |  |
9.8 | 4.0 | 7.9 | — | — |
| 109c |  |
5.3 | 5.5 | 6.8 | 25.88 | 22.4 |
| 109d |  |
44.9 | >100 | >100 | — | — |
| 109e |  |
>100 | >100 | >100 | — | — |
| 109f |  |
29.4 | >100 | >100 | — | — |
| 109g |  |
23.3 | 6.9 | 9.3 | — | — |
| 109h | 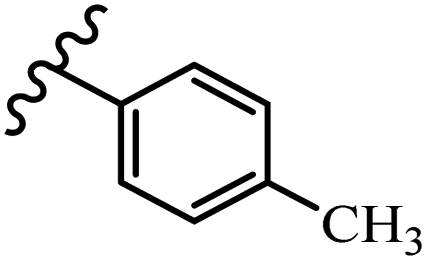 |
>100 | >100 | >100 | — | — |
| 109i | 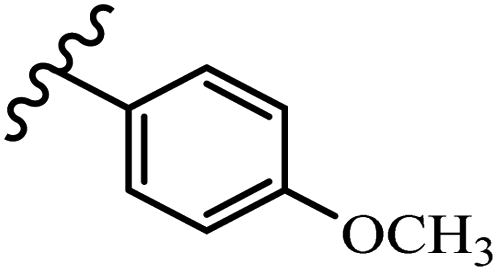 |
>100 | >100 | >100 | — | — |
| 109j | 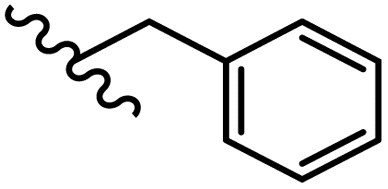 |
>100 | >100 | >100 | — | — |
| 109k | 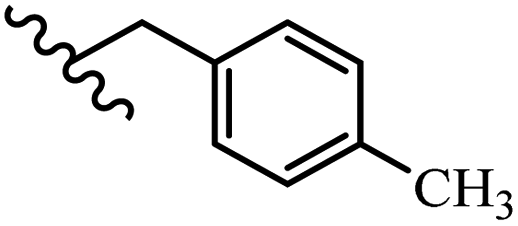 |
24.7 | 23.8 | 28.2 | — | — |
| 109l |  |
>100 | >100 | >100 | — | — |
| Etoposide | — | 7.6 | 10.3 | 8.9 | — | — |
Penthala et al.70 synthesized and evaluated the cytotoxicity of heteroaromatic analogues of resveratrol. Compound 112a was the most potent compound against most of the tested cell lines. Also, hydrophilic groups exhibited higher cytotoxicity activity than lipophilic groups against most of the tested cell lines (112a vs. 112b) (Table 29).
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | X | Y | R1 | R2 | GI50 (μM) | |||||||||||
| K562 | SR | HOP-92 | NCI-H226 | HT29 | KM12 | U251 | MDA-MB-435 | OVCAR-3 | NCI/ADR-RES | A498 | UO-31 | |||||
| 112a | N | S | OCH3 | OCH3 | 0.041 | 0.036 | 0.036 | 0.245 | 0.038 | 0.072 | 0.088 | 0.024 | 0.069 | 0.106 | 0.041 | 0.376 |
| 112b | CH | S | OCH3 | OCH3 | 0.088 | 0.120 | 0.322 | 0.442 | 0.234 | 0.213 | 0.338 | 0.036 | 0.224 | 0.070 | 0.213 | 0.513 |
Centelles et al.71 synthesized nitrogen-containing heterocyclic stilbene analogues and tested their inhibitory activity against hTERT, VEGF, and c-Myc. Among them, compounds 115a against HT-29, 115b against MCF-7, and 118a and 118b against HEK-293 showed the highest cytotoxicity activity. These compounds also exhibited stronger activity than resveratrol against the HEK-293, MCF-7, and HT-29 cell lines. Most of the compounds showed weaker cytotoxicity than resveratrol. Likewise, moving the OCH3 group from the 3′-position in 115a to the 4′-position on the phenyl ring in 115c reduced the cytotoxicity against all the tested cell lines. Also, moving the nitrogen atom from the 3-position to the 2-position in the pyridine ring mitigated the cytotoxicity activity (115d vs. 115e) against the HT-29 and MCF-7 cell lines. However, 115e had stronger activity than 115d against the HEK-293 cell line. Also, the conversion of pyridine ring 115f to pyrimidine ring 115g reduced the cytotoxicity against the three cell lines. In addition, the lipophilic groups showed greater cytotoxicity activity compared to hydrophilic groups (e.g., 115a and 118c) against all the tested cell lines. Also, the HT-29 tumoral cell line was used to study the effect of stilbene derivatives on VEGF secretion and VEGF gene inhibition. Compounds 115h and 115c demonstrated significant capability to inhibit VEGF expression. Surprisingly, they showed significantly higher activity than resveratrol. Compound 115a, the most potent compound, also revealed considerable inhibitory activity against VEGF expression. Compounds 115c and 115h, which had the strongest inhibitory activity against VEGF expression, showed significantly lower ability to inhibit VEGF gene expression. In contrast, compound 118c, which was previously only weakly active in inhibiting VEGF expression, had the strongest ability to inhibit VEGF gene expression. Compounds 115i, 115d and 115h all had the same capacity to inhibit hTERT gene expression. The remaining compounds, including 115a, had marginal activity that is slightly lower than the control. However, the trend of activities for inhibiting c-Myc gene expression was demonstrated to be noticeably diverse. Compounds 115i, 115d, and 115h showed significant inhibitory activity on hTERT gene expression, which were much less active in inhibiting c-Myc gene expression. Compounds 115a, 115c, 118c, and 118d, which previously had such poor activity against hTERT gene expression, were much more active against c-Myc gene expression, mainly 115a and 118d, which had the same activity with resveratrol (Table 30).
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R | A | B | C | D | E | IC50 (μM) | Inhibition percent (%) | |||||
| HT-29 | MCF-7 | HEK-293 | VEGF protein | VEGF gene | hTERT gene | c-Myc gene | |||||||
| 115a | 3-OCH3 | — | — | N | — | — | 44 | 88 | 56 | 52 | 95 | 95 | 50 |
| 115b | 2-OCH3 | N | — | — | — | — | 274 | 1.7 | 175 | — | — | — | — |
| 115c | 4-OCH3 | — | — | N | — | — | 61 | 142 | 114 | 42 | 91 | — | 63 |
| 115d | 3-OCH3 | — | N | — | — | — | 104 | 128 | 123 | — | — | 57 | 71 |
| 115e | 3-OCH3 | N | — | — | — | — | 223 | 204 | 68 | — | — | — | — |
| 115f | 4-OCH3 | — | N | — | — | — | 299 | 62 | 213 | — | — | — | — |
| 115g | 4-OCH3 | — | N | — | N | — | >500 | >500 | 259 | — | — | — | — |
| 115h | 2-OCH3 | — | — | N | — | — | 76 | 100 | 85 | 37 | 54 | 54 | 64 |
| 115i | 4-OCH3 | N | — | — | — | — | 114 | 147 | 199 | — | — | 52 | 87 |
| 118a | 4-OH | — | — | N | — | — | 168 | 16 | 13 | — | — | — | — |
| 118b | 2-NH2 | — | — | N | — | — | 255 | 383 | 13 | — | — | — | — |
| 118c | 3-NH2 | — | — | N | — | — | 168 | 235 | >500 | 93 | 48 | — | 58 |
| 118d | 4-NH2 | — | — | N | — | — | 100 | 20.6 | 41 | — | — | — | 49 |
| Resveratrol | — | — | — | — | — | — | 150 | 71 | 31 | 78 | 65 | 57 | 51 |
Centelles et al.72 synthesized resveratrol analogues and assessed their cytotoxicity. Analogue 121a showed the highest cytotoxicity and it was also better than resveratrol against the HEK-293, MCF-7, and HT-29 cell lines. Moreover, moving the OH group from the 2′- to 3′-position on the phenyl ring enhanced the cytotoxicity (121b vs. 121c) against HT-29 and HEK-293. Likewise, shifting the OCH3 group from the 4- in 121d to 2-position on the phenyl ring in 121e resulted in lower cytotoxicity against all the tested cell lines. Also, the comparison of compound 121f having an NH2 group in the 4-position, 121g bearing an NH2 group in the 3-position, and 121h having an NH2 group in the 2-position on the phenyl ring showed that 121f was more potent than 121g and 121h against HT-29. In addition, moving the NH2 group from the 2- to 3-position on the phenyl ring resulted in stronger activity (e.g., 121i and 121j) against the MCF-7 and HEK-293 cell lines. In addition, the presence of lipophilic groups compared to hydrophilic groups enhanced the cytotoxicity activity (121k vs. 121l) against all the tested cell lines. Also, EWG in 121m showed higher activity compared to EDG in 121f against MCF-7. According to the findings, different substitutions in the diverse positions of both phenyl rings had a positive effect on the cytotoxicity (4 > 2 > 3). In addition, compound 121a showed ability to suppress the expression of h-TERT and c-Myc genes. Compound 121o was the most active compound among the amide stilbenes. Among the amino stilbenes, those with a methoxy group (121i, 121j, 121n, and 121a) exhibited greater activity than that with a hydroxy group (Table 31).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | R1 | R2 | IC50 (μg mL−1) | % gene expression | ||||
| HT-29 | MCF-7 | HEK-293 | VEGF | hTERT | c-Myc | |||
| 121a | 4-NH2 | 4-OCH3 | 0.0036 | 0.0021 | 0.012 | 67 | 48 | 69 |
| 121b | 4-OH | 2-OH | 22 | 21 | 17.5 | — | — | — |
| 121c | 4-OH | 3-OH | 6.9 | 23 | 1.1 | — | — | — |
| 121d | 4-OCH3 | 4-OH | 17.4 | 11.3 | 3.9 | — | — | — |
| 121e | 2-OCH3 | 4-OH | 24 | 19 | 41 | — | — | — |
| 121f | 4-NH2 | H | 4.4 | 16 | 11.4 | 24 | 65 | 46 |
| 121g | 3-NH2 | H | 12.9 | 14 | 84 | — | — | — |
| 121h | 2-NH2 | H | 17 | 1.4 | 1.3 | — | — | — |
| 121i | 2-NH2 | 3-OCH3 | 21.2 | 6.1 | 0.5 | 100 | 35 | 79 |
| 121j | 3-NH2 | 3-OCH3 | 19.6 | 34 | 16 | 23 | 100 | 34 |
| 121k | 3-NH2 | 3-OCH3 | 16.3 | 23 | 11 | — | — | — |
| 121l | 3-NH2 | 2-OH | 30.4 | 24.7 | 96 | 38 | 52 | 37 |
| 121m | 3-NHCO(CH2)10CH3 | H | >100 | 3.5 | 11.6 | — | — | — |
| 121n | 3-NH2 | 2-OCH3 | 25.3 | 38.7 | 45 | 67 | 52 | 45 |
| 121o | 2-NHCO(CH2)10CH3 | H | 2.5 | 12 | 16 | 37 | 53 | 45 |
| Resveratrol | — | — | 34.1 | 16.1 | 7.1 | 65 | 57 | 51 |
Srivastava et al.73 synthesized quinolino-stilbene derivatives and evaluated their cytotoxicity activity. Among the synthesized E/Z-compounds, 127a showed the highest activity against the HeLa, MCF-7, MDA-MB231, MDA-MB468, and 184B5 cell lines. Most of the compounds exhibited better activity compared to paclitaxel, camptothecin, and chloroquine. In the Z-isomers, replacing the CF3 group with an F atom in the 4′-position on the phenyl ring reduced the activity by 2–5 times (127b vs. 127c) against five cell lines. In addition, moving the CF3 group from the 4′- in 127d to 3′-position on the phenyl ring in 127a improved the activity by 2-times. However, adding an OCH3 group in the 3′-position of the phenyl ring did not have any discernible effect on the anti-proliferative activity when combined with a CF3 group in the 3-position (127a vs. 127e). Moreover, an extra CF3 group on the phenyl ring had a negative effect on the activity (127d vs. 127f). Also, the presence of CF3 groups at the 3′- and 5′-positions resulted in 5–10 times better activity than the 2′- and 4′-positions (127f vs. 127g) and (127h vs. 127i) against the HeLa cell line. The addition of a Cl atom at the 4-position of the phenyl ring reduced the activity by 5-times (127f vs. 127j) against all the tested cell lines, whereas the addition of an OCH3 group in the 3-position on the phenyl ring improved the activity by 2-times (127f vs. 127h). A methylenedioxy group on the phenyl ring reduced the activity by 10-times against HeLa, MCF-7, MDA-MB231, MDA-MB468, and 184B5 (127a vs. 127k). The SAR studies showed that a CF3 group in the 3′- or 4′-position on the phenyl ring is the best position for anti-proliferative activity. Although an OCH3 group in the 4-position on the phenyl ring is tolerable, it did not contribute to an enhancement in activity. According to the findings, the stereochemistry and positions of various functional groups are likely to play vital roles in the cytotoxicity. E-127l with an F atom in the 4′-position on the phenyl ring was very active against the MDA-MB468 cell line and moderately active against the MCF7 cell line. However, it had a low level of activity against HeLa, MDA-MB231, and 184B5. Besides 127l, the Z-stilbene derivatives had higher activity and better anti-cancer effect than the E-stilbene derivatives. Compounds 127a and 127l inhibited cell cycle progression at the mitosis and S-phase, respectively, eventually leading to apoptosis. Compound 127a appeared to impede normal G2-M progression, eventually leading to cell death. In MDA-MB231 metastatic breast cancer cells, compound 127a caused a similar pattern of G2/M arrest and DNA fragmentation. Compound 127a did not exhibit significant G2/M arrest or DNA fragmentation against MCF10A. In comparison to DMSO, compound 127l significantly increased the S-phase population in MDA-MB-468 cells. According to the data, 127l causes DNA damage, which leads to S-phase arrest, and eventually cell death (Table 32).
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | E/Z | R1 | R2 | R3 | R4 | R5 | R6 | IC50 (μM) | Inhibition percent (%) | ||||
| HeLa | MCF-7 | MDA-MB231 | MDA-MB468 | 184B5 | S-phase | ||||||||
| 127a | Z | H | CF3 | H | H | H | H | 2.85 | 3.53 | 3.75 | 3.70 | 6.15 | 17 |
| 127b | Z | H | H | CF3 | H | H | Cl | 12.64 | 6.45 | 13.55 | 16.13 | 26.40 | — |
| 127c | Z | H | H | F | H | H | Cl | 38.62 | 16.89 | 42.56 | 10.91 | 26.89 | — |
| 127d | Z | H | H | CF3 | H | H | H | 4.22 | 4.26 | 5.36 | 7.38 | 7.94 | — |
| 127e | Z | H | CF3 | H | H | OCH3 | H | 3.03 | 2.78 | 3.28 | 3.91 | 5.58 | — |
| 127f | Z | H | CF3 | H | CF3 | H | H | 6.78 | 7.46 | 10.13 | 10.52 | 13.67 | — |
| 127g | Z | CF3 | H | CF3 | H | H | H | 35.89 | 10.45 | 41.76 | — | — | — |
| 127h | Z | H | CF3 | H | CF3 | OCH3 | H | 4.14 | 4.70 | 4.47 | 5.79 | 7.97 | — |
| 127i | Z | CF3 | H | CF3 | H | OCH3 | H | >50 | >50 | >50 | >50 | >50 | — |
| 127j | Z | H | CF3 | H | CF3 | H | Cl | 41.35 | 41.71 | >50 | >50 | 42.95 | — |
| 127k | Z | H | CF3 | H | H | OCH2O | H | 20.09 | 18.90 | 26.0 | >50 | 16.97 | — |
| 127l | E | H | H | F | H | H | H | >50 | 15.13 | >50 | 0.12 | 38.45 | — |
| Paclitaxel | — | — | — | — | — | — | — | 2.29 | 3.99 | 2.56 | 3.87 | 2.32 | 41.7 |
| Camptothecin | — | — | — | — | — | — | — | 6.13 | — | 5.77 | 14.76 | 4.96 | — |
| Chloroquine | — | — | — | — | — | — | — | 29.96 | — | 36.53 | 19.86 | 63.08 | — |
| DMSO | — | — | — | — | — | — | — | — | — | — | — | — | 16.9 |
Kachhadia et al.74 described the synthesis of stilbene derivatives and evaluated their anti-cancer activity. Compound 131a exhibited the highest inhibitory activity against histone deacetylase (HDAC). Moreover, the secondary amide in 131b slightly reduced the activity compared with the tertiary amide in 131c. In addition, the presence of cyclopropyl in compound 131d increased the activity by 5-times against HDAC compared to 131b. Also, changing the cyclopropyl ring in 131d to cyclooctyl in 134e significantly reduced the activity. Moreover, the HDAC activity decreased when the substitution changed from morpholine to pyrroline (e.g., 131f and 131g). Also, aromatic substitutions such as phenyl in 131h or benzyl in 131i decreased the activity. Other substitutions, such as benzyloxy 131j and cyclopentyloxy 131k, also showed excellent HDAC inhibitory activity. Likewise, replacing the OCH3 group in the 3-position on the phenyl ring of 131l with a Cl atom of 131m in the same position enhanced the activity. In addition, the F atom at the 3′-position of the phenyl ring had a positive effect on the activity when combined with an OCH3 group at the 4′-position on the phenyl ring (131n vs. 131o). In the compounds having a heteroaryl ring, the indol-3-yl in compound 138a had the highest activity, while the other rings significantly reduced the HDAC inhibitory activity. The effect of acrylo phenylacetic acids 139a–c on the position of the acid group was investigated. The HDAC inhibitory activity of positional isomers 139a–c was comparable to compounds 131d, 131p, 131h, and 131q. Encouragingly, compound 139a with an OCH3 group in the 4′-position on the phenyl ring was 62-times more potent than compound 131p. Alternatively, compound 139p was 38-times less potent than 131a. The carbonyl group of the amide bond in 131r reduced to the corresponding amine derivative 135 to produce a secondary amine that is more hydrophilic in nature. Compound 132a showed 8-times greater HDAC inhibitory activity compared to compound 131d, while compound 132b showed 10-times decrease in HDAC inhibitory activity compared to 131q. Also, the anti-proliferative activity of some compounds was investigated. Among the synthesized compounds, 132a showed the highest cytotoxicity activity against the NCI-H460, HCT-116, and U-251 cell lines. In addition, compound 131s with an isopropyl moiety exhibited lower activity than 131d having cyclopropyl moiety against the tested cell lines. In addition, moving the F atom from the 2′- to 3′-position on the phenyl ring reduced the cytotoxicity against all the tested cell lines (e.g., 131t and 131n). Furthermore, the aliphatic groups improved the cytotoxicity compared to aromatic groups (131u vs. 131k and 132c vs. 132b) against the tested cell lines. Also, increasing the number of F atoms in 4′-position of compound 131n produced 131v, which showed the highest cytotoxicity activity against all the tested cell lines. Furthermore, compound 131m with EWG on the phenyl ring showed higher cytotoxicity activity than 131l having EDG against the tested cancer cell lines (Table 33).
|
|||||
|---|---|---|---|---|---|
| Compounds | R | IC50 (nM) | GI50 (μM) | ||
| HDAC | NCI-H460 | HCT-116 | U251 | ||
| 131a |  |
3.7 | 0.65 | 0.22 | 0.05 |
| 131b |  |
350 | — | — | — |
| 131c |  |
390 | — | — | — |
| 131d |  |
48 | 1.8 | 1.4 | 0.02 |
| 131e |  |
990 | — | — | — |
| 131f |  |
430 | — | — | — |
| 131g | 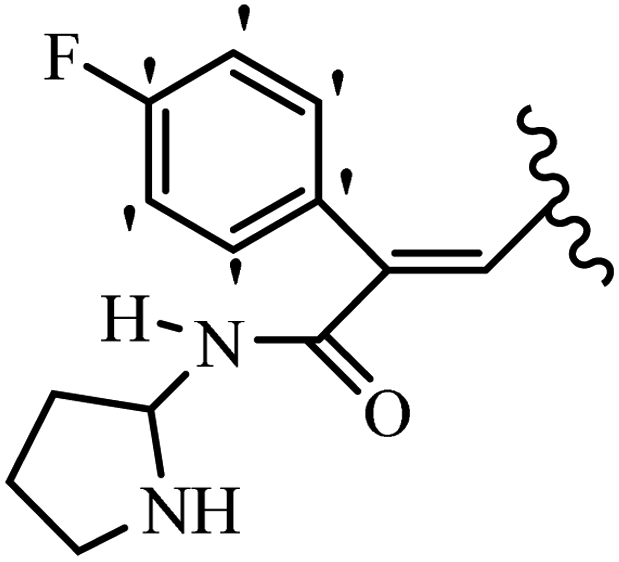 |
410 | — | — | — |
| 131h | 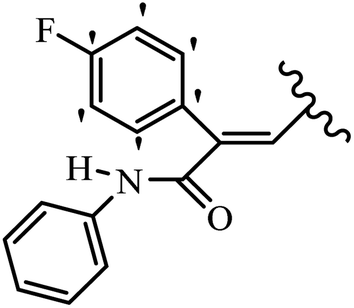 |
200 | — | — | — |
| 131i | 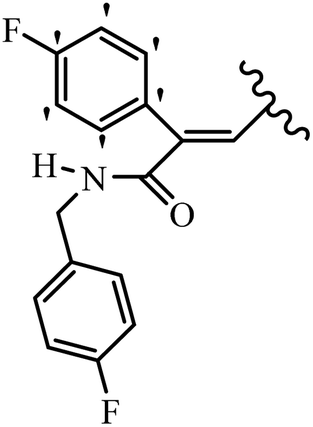 |
190 | — | — | — |
| 131j | 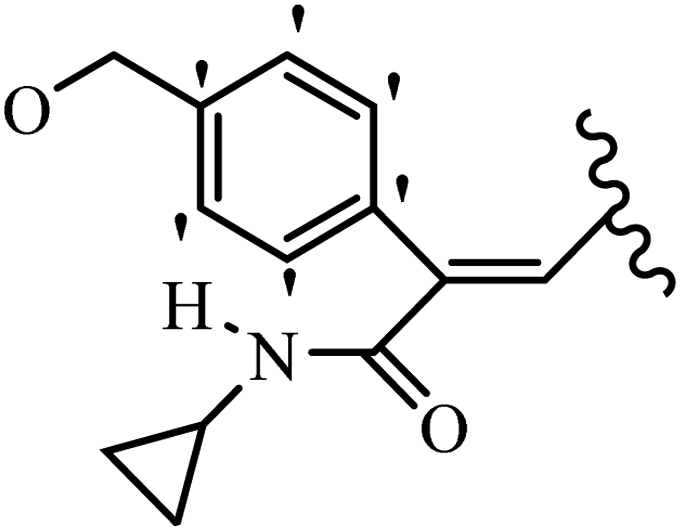 |
39 | 2.5 | 2 | 2 |
| 131k |  |
18 | 0.75 | 1 | 1 |
| 131l |  |
68 | 8 | 3 | 1.8 |
| 131m |  |
30 | 0.5 | 0.5 | 1 |
| 131n |  |
40 | 10 | 2 | 4.2 |
| 131o |  |
15 | 9.1 | 1.1 | 3 |
| 131p |  |
310 | — | — | — |
| 131q |  |
56 | 7 | 6 | 6 |
| 131r |  |
240 | — | — | — |
| 131s |  |
80 | 11 | 3 | 10.5 |
| 131t |  |
90 | 13 | 6 | 7 |
| 131u |  |
34 | 0.4 | 0.5 | 0.8 |
| 131v |  |
30 | 8 | 1.8 | 4 |
| 135a |  |
44 | 0.03 | 3.5 | 1.6 |
| 139a |  |
5 | 4 | 1 | 2.8 |
| 139b |  |
140 | — | — | — |
| 132a |  |
6 | 0.3 | 0.02 | 0.3 |
| 132b |  |
600 | >100 | >100 | >100 |
| 132c |  |
60 | 0.2 | 0.3 | 0.2 |
Duan et al.75 designed resveratrol derivatives and tested their activity against LSD1. Compounds 143c and d showed the highest activity against LSD1, and they were more potent than resveratrol and tranylcypromine. In addition, replacing the F atom with an OH group in the 3′-position on the phenyl ring decreased the inhibitory activity against LSD1 (e.g., 143e and 143f). Moreover, exchanging the phenyl ring with a pyridine ring (143g vs. 147) or indole ring (143h vs. 149) resulted in a significant decrease in activity, demonstrating the importance of the phenyl ring in retaining the activity. Also, increasing the atom size improved the activity (e.g., 143c and 143i). According to the comparison of 143h and 144, changing benzimidamide to benzimidohydrazide significantly reduced the inhibitory activity. Amidoxime in the 3-position on the phenyl ring was preferential; the amidoxime in the 3-position on the phenyl ring of derivatives such as 143g showed higher inhibitory activity than the corresponding amidoxime in the 4-position on the phenyl ring-substituted derivatives (143h) against LSD1. Treatment with 143c and 143d resulted in the remarkable buildup of H3K4me2, the substrate of LSD1, without influencing LSD1 expression (Table 34).
|
|||||
|---|---|---|---|---|---|
| Compound | Ar1 | Ar2 | IC50 (μM) | ||
| LSD1 | H3K4me2 | CD86 mRNA | |||
| 143a |  |
 |
— | — | — |
| 143b |  |
 |
— | — | — |
| 143c |  |
 |
0.121 | 6 | 7.70 |
| 143d |  |
 |
0.123 | 5.70 | 9 |
| 143e |  |
 |
0.492 | — | — |
| 143f |  |
 |
— | — | — |
| 143g |  |
 |
0.333 | — | — |
| 143h |  |
 |
0.739 | — | — |
| 143i |  |
 |
0.192 | — | — |
| 144 |  |
 |
— | — | — |
| 147 |  |
 |
3.61 | — | — |
| 149 |  |
 |
— | — | — |
| Resveratrol | — | — | 10.20 | — | — |
| Tranylcypromine | — | — | 26.31 | — | — |
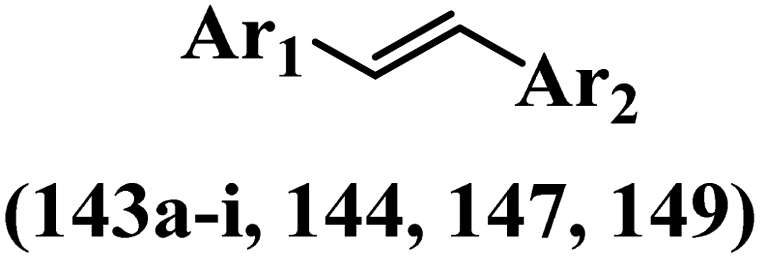
Ismail et al.76 synthesized stilbene derivatives and evaluated their tyrosinase inhibitory activity. Compound 152a showed the highest murine tyrosinase inhibitory activity. This compound exhibited 2-times stronger inhibitory activity than resveratrol against murine tyrosinase. Among the E-isomers, compounds 152b and 152c showed that fluorination of compound 152d increased its tyrosinase inhibitory activity. The F-substituted compounds compared to the other halo-substituted compounds in the 4′-position on the phenyl ring and unsubstituted compounds showed no improvement with an increase in molecular weight (F; 152b < Br; 152e < Cl; 152f < 152d, H). This implies that the effect of fluorination on the activity of stilbene derivatives cannot be attributed to its effect on lipophilicity but rather to the increase in favorable and specific dipolar interactions. The IC50 values of stilbenes substituted with NO2, CN, CO2CH3, OH, and OCH3 groups supported this hypothesis (152g, 152h, 152i, 152j, and 152k, respectively). Compounds 152k and 152l carrying one and four methoxy groups, respectively, reduced the inhibitory activity. Among the Z-isomers, compounds 152m, 152n, 152o, and 152p exhibited no activity compared to the inhibitory activity of stilbenes. It was observed that the introduction of methyl acetate on the linker carbon in 152p compared to 152o with a carboxylic acid group in the same position revealed no notable activity on their inhibition potential. The activity studies for compound 152q indicated that the pyridine ring decreased the inhibitory activity to an excessive range (Table 35).
|
||||||
|---|---|---|---|---|---|---|
| Compound | E/Z | X | R1 | R2 | R3 | IC50 (μM) |
| 152a | Z | CH | — | OH | — | 5.06 |
| 152b | E | — | 4-F | H | — | 79.44 |
| 152c | E | — | 3,5-diF | H | — | 56.16 |
| 152d | E | — | H | H | — | >500 |
| 152e | E | — | 4-Br | H | — | 276.56 |
| 152f | E | — | 4-Cl | H | — | 319.75 |
| 152g | E | — | 4-NO2 | H | — | 360.70 |
| 152h | E | — | 4-CN | H | — | 360.70 |
| 152i | E | — | 4-OCOCH3 | H | — | >500 |
| 152j | E | — | 4-OH | H | — | 17.42 |
| 152k | E | 4-OCH3 | H | — | 432.97 | |
| 152l | E | — | 4-OCH3 | 3,4,5-OCH3 | — | 393.99 |
| 152m | Z | — | 4-OCH3 | 3,4-OCH3 | COOH | — |
| 152n | Z | — | 4-OCH3 | 4-OCOCH3 | COOH | — |
| 152o | Z | — | 4-OCH3 | 3-OCH3, 4-OH | COOH | — |
| 152p | Z | — | 4-OCH3 | 3-OCH3, 4-OH | COOCH3 | >500 |
| 152q | Z | N | — | H | — | 270.22 |
| Resveratrol | — | — | — | — | — | 10.78 |
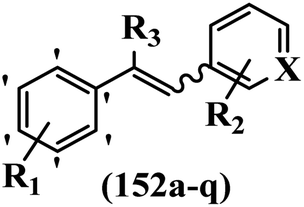
Katherine et al.77 synthesized stilbene analogues and evaluated their cytotoxicity activity. Compound 185a showed the highest activity among the synthesized compounds, which was stronger than that of novobiocin against the MCF-7, SK-Br-3, and HCT-116 cell lines. Also, replacing the cyclopropyl linker in compound 182 with a double bond in 181 decreased the activity. The triazole moiety was replaced by a biaryl amide, which resulted in a significant increase in anti-proliferative activity against all the tested cell lines (185b vs. 185c). When N-methyl-piperidine was replaced with 3-(dimethylamino)propane, the activity decreased as in 185d, while the activity of 185c was nearly identical to 185e against the MCF-7, SKBr3 and HCT-116 cell lines. Interestingly, when Z-2-(dimethylamino)ethane was added to the stilbene scaffold, both triazole-containing compounds (185g and 185f) showed improved anti-proliferative activity against all the cell lines. Because 185b is remarkably less active than the standard, it is clear that the improvement in anti-proliferative activity is due to the triazole moiety replacing the biaryl amide side chain (Table 36).
|
||||||
|---|---|---|---|---|---|---|
| Compound | R | R2 | R4 | IC50 (μM) | ||
| MCF-7 | SK-Br-3 | HCT-116 | ||||
| 181 |  |
— | — | 0.814 | 0.894 | 0.801 |
| 182 |  |
— | — | 2.64 | 1.31 | 2.90 |
| 185a | — |  |
 |
0.092 | 0.099 | 0.186 |
| 185b | — |  |
 |
5.86 | 9.68 | 5.51 |
| 185c | — |  |
 |
0.325 | 0.382 | 0.350 |
| 185d | — |  |
 |
0.494 | 0.184 | 0.740 |
| 185e | — |  |
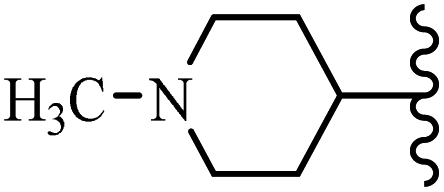 |
0.326 | 0.231 | 0.301 |
| 185f | — | 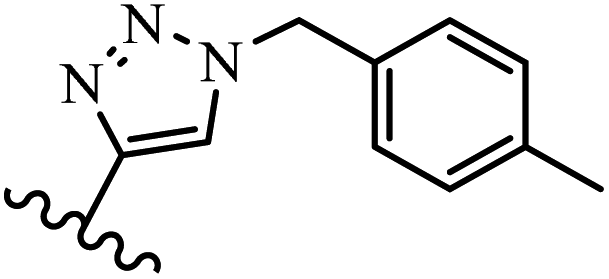 |
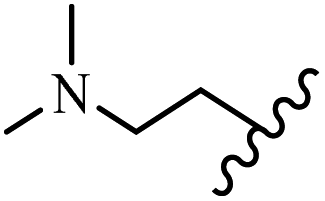 |
0.150 | 0.141 | 0.179 |
| 185g | — | 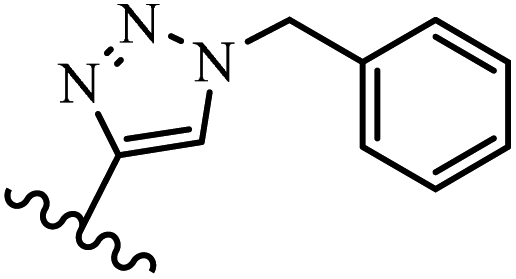 |
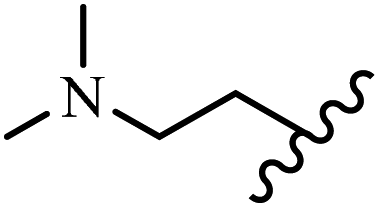 |
0.234 | 0.183 | 0.350 |
| Novobiocin | — | — | — | 1.3 | 0.68 | 3.68 |
Duan et al.78 designed stilbene derivatives as LSD1 inhibitors and evaluated their cytotoxicity. Among the synthesized compounds, 190a and 193a exhibited the highest activity, and they were more active than ORY-1001. Also, the replacement of the pyridine ring in 190b with a pyrimidine ring in 190c intensely reduced the anti-LSD1 activity. In addition, replacing the F in the 3′-position on the pyridine ring of compound 190d with OH in the same position of 190e enhanced the activity. Compounds 190f bearing amine in the 3-position on the phenyl ring showed reduced anti-LSD1 activity comparable to that of 190g having amidoxime in the same position. However, OH substitution (190h, 190i, and 193b) led to a dramatic reduction activity. Introducing diverse substituents on the pyridine ring (e.g., 190e) usually resulted in significantly lower activity compared to no substitution in 190a. This indicates that substituents at this position are unfavorable. The amidoxime or amino moiety placed in the 3-position on the phenyl ring displayed the highest activity. For example, compounds 190a, 190f and 193c were more potent against LSD1 than the corresponding 4-position-substituted derivatives (190b and 193d). The compounds bearing an OCH3 group in the 2′-position of the arylbenzene moiety (190j and 190k) showed no activity against LSD1 compared with compounds 190a and 190f. It seems that the presence of hydrophilic and small-size groups improved the activity compared to lipophilic and bulk groups, demonstrating the importance of the OH group in maintaining their activities. The synthesized compounds 190a, 190l, 193a, and 193c with the most potent LSD-inhibitory activity were assessed for their anti-proliferative activities against three MOLM-13, THP-1 and MV-4-11 cell lines. Compound 193a displayed the best anti-proliferative activity against MOLM-13 and THP-1. Compound 190l showed fairly higher anti-proliferative activity against MV-4-11, which was more potent than compound 193a (Table 37).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | X | R1 | R2 | R3 | R4 | IC50 (μM) | |||
| LSD1 | MOLM-13 | THP-1 | MV-4-11 | ||||||
| 190a | — | OH | H | H |  |
0.301 | 12.51 | 10.51 | 10.94 |
| 190b | — | OH | H | H |  |
3.57 | — | — | — |
| 190c | N | OH | H | H |  |
9.55 | — | — | — |
| 190d | — | OH | F | H |  |
— | — | — | — |
| 190e | — | OH | OH | H |  |
1.47 | — | — | — |
| 190f | N | OH | H | H |  |
0.859 | — | — | — |
| 190g | N | OH | H | H |  |
1.29 | — | — | — |
| 190h | — | OH | H | H |  |
4.24 | — | — | — |
| 190i | N | OH | H | H |  |
— | — | — | — |
| 190j | — | OCH3 | H | H |  |
— | — | — | — |
| 190k | N | OCH3 | H | H |  |
— | — | — | — |
| 190l | — | OH | H | H |  |
0.72 | 22.59 | 7.89 | 4.71 |
| 193a | — | — | H | F |  |
0.283 | 8.34 | 5.76 | 7.49 |
| 193b | — | — | H | H |  |
— | — | — | — |
| 193c | — | — | H | H |  |
0.364 | 9.05 | 13.72 | 15.85 |
| 193d | — | — | H | H |  |
0.764 | — | — | — |
| ORY-1001 | — | — | — | — | — | 11.26 | — | >20 | — |
Singh et al.48 reported the cytotoxicity of resveratrol structural analogues. The compounds showed no cytotoxicity activity against L929. All the compounds showed weaker activity than resveratrol. Compound 194a exhibited the highest activity against INT407. In addition, the presence of Br on the phenyl ring in 194a enhanced the activity compared to no substitution in 194b. According to the results, the compounds with hydrophilic groups on the phenyl rings had a positive effect on cytotoxicity activity (194c and 194d) (Table 38).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | IC50 (μg mL−1) | |
| INT 407 | L929 | |||||||
| 194a | H | Br | H | OH | H | OH | 28 | — |
| 194b | H | H | H | OH | H | OH | 38 | — |
| 194c | H | OH | H | OH | H | OCH3 | 52 | — |
| 194d | H | OH | H | OCH3 | H | OCH3 | 99 | — |
| Resveratrol | — | — | — | — | — | — | 21 | — |
Iqbal et al.79 synthesized E-stilbene hydrazides and tested their cytotoxicity activity. Analogue 199a showed the best cytotoxicity activity among the synthesized analogues. It was more potent than doxorubicin against MCF-7. Also, 199b having a Cl atom in the 4-position exhibited stronger cytotoxicity activity than 199c bearing an OH group in same position. Moreover, changing its position on the phenyl ring did not change the cytotoxicity activity (199d with OH group in 2-position vs. 199c). Likewise, the size of the atom on the phenyl ring did not change the activity (e.g., 199e and 199f having a Cl atom and I atom in the 2-position, respectively). All the compounds were more potent than the negative control. Some compounds were stronger than doxorubicin. Analogue 199c showed higher apoptosis activity than doxorubicin and other compounds. Substitution of the phenyl ring showed no effect on apoptosis activity (199g vs. 199e and 199c) (Table 39).
|
|||
|---|---|---|---|
| Compound | R | MCF-7 | Apoptosis (%) |
| 199a | 4-NO2 | 95 | 30.09 |
| 199b | 4-Cl | 78 | 55.98 |
| 199c | 4-OH | 56 | 80.09 |
| 199d | 2-OH | 60 | 75.09 |
| 199e | 2-Cl | 86 | 42.21 |
| 199f | 2-I | 84 | 48.31 |
| 199g | H | 73 | 60.98 |
| Control | — | 100 | 7.30 |
| Doxorubicin | — | 62 | 73.69 |
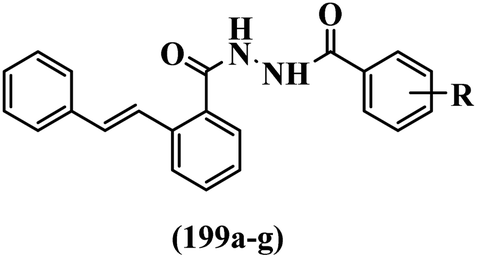
Song et al.21 assessed the cytotoxicity of stilbene derivatives containing a 1,3-benzodioxole moiety. Among the tested compounds, three compounds, 200a, 200b, and 200c, showed activity against HepG-2, but none of them showed activity against the A875 and MARC145 cell lines. These compounds revealed lower activity compared to 5-FU. Also, adding an OCH3 group in the 3′-position on the phenyl ring in 200c showed less activity compared to 200a with no substitution in the phenyl ring. In addition, large-size and lipophilic groups had a positive effect on activity (200a and 200b, respectively) (Table 40).
|
||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | IC50 (μg mL−1) | ||
| HepG-2 | A875 | MARC145 | ||||
| 200a | H | H |  |
16.53 | >40 | >40 |
| 200b | H | H |  |
22.57 | >40 | >40 |
| 200c | OCH3 | H |  |
25.65 | >40 | >40 |
| 5-FU | — | — | — | 9.41 | 10.66 | 10.09 |
Wong et al.80 synthesized stilbene long-chain fatty acid conjugates and assessed their cytotoxicity. Among the compounds, 203a and 206a exhibited the highest activity and they were stronger than colchicine against the KB-3-1b, NCI-H460c, and HEK-293 cell lines. All the compounds showed weaker cytotoxicity activity compared to colchicine against MCF-7. E-Stilbenes showed lower cytotoxicity than the Z-isomer (206b vs. 206c) against all four cell lines. Moreover, stilbene 206d showed considerably decreased cytotoxicity compared to compound 206e against the HEK-293 cell line. Compound 206f showed higher activity than 206g on all the tested cell lines. Compounds 206d and 206h exhibited weak activity against all the tested cell lines (Table 41).
|
||||||
|---|---|---|---|---|---|---|
| Compound | E/Z | Ar | IC50 (μM) | |||
| KB-3-1 | NCI-H460 | HEK-293 | MCF-7 | |||
| 203a | Z |  |
0.01 | 0.01 | 0.01 | 0.09 |
| 206a | Z |  |
0.03 | 0.01 | 0.023 | >10 |
| 206b | Z |  |
0.03 | 0.03 | 0.03 | 0.24 |
| 206c | E |  |
>10 | >10 | 2.90 | >10 |
| 206d | E |  |
>100 | >100 | >100 | >100 |
| 206e | E |  |
2.40 | >10 | 2.40 | >10 |
| 206f | Z |  |
3.10 | >10 | 3.90 | 7.70 |
| 206g | Z |  |
>100 | >100 | >100 | >100 |
| 206h | E |  |
>100 | >100 | >100 | >100 |
| Colchicine | — | — | 0.02 | 0.06 | 0.02 | 0.06 |
Das et al.81 synthesized and tested stilbene-linked 1,2,3-triazoles against six cell lines. All the synthesized compounds showed lower cytotoxicity activity than docetaxel and staurosporine against the HCT-116, Capan-1, K-562, DND-41, Z-138, and NCI-H460 cell lines. Compounds 213a and b exhibited the highest activity among them against the HCT-116, Capan-1, and NCI-H460 cell lines. Furthermore, the replacement of the OCH3 group in 213c with a Cl atom in 213d resulted in lower activity against NCI-H460, HCT-116, and Capan-1. It seems that hydrophilic groups displayed stronger activity than lipophilic groups. In addition, no substitution in 213e exhibited better activity compared to substitution on the phenyl ring in 213f against the Capan-1, NCI-H460, DND-41, and K-562 cell lines. Also, the presence of EDG compared to EWG on the phenyl or benzyl ring had a positive effect on cytotoxicity against Capan-1, HCT-116, and NCI-H460 (e.g., 213g vs. 213h) (Table 42).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | R1 | R | IC50 (μM) | |||||
| Capan-1 | HCT-116 | NCI-H460 | DND-41 | K-562 | Z-138 | |||
| 213a | F | NO2 | 40.2 | 12.2 | 11.6 | >100 | >100 | >100 |
| 213b | F | H | 30.7 | 46.7 | 31.7 | >100 | >100 | >100 |
| 213c | OCH3 | H | 55.3 | 13.5 | 35.1 | >100 | >100 | >100 |
| 213d | Cl | H | 96.4 | 36.1 | >100 | >100 | 19.3 | >100 |
| 213e | H | H | 46.7 | 29.1 | 34.3 | 61.4 | 39.9 | >100 |
| 213f | H | NO2 | >100 | 26.1 | 78.5 | 82.1 | >100 | >100 |
| 213g | CH3 | CH3 | 52.4 | 87.4 | 60.8 | >100 | >100 | >100 |
| 213h | F | CH3 | 73.4 | >100 | >100 | >100 | >100 | >100 |
| Docetaxel | — | — | 0.0063 | 0.0008 | 0.0001 | 0.0019 | 0.0034 | 0.0019 |
| Staurosporine | — | — | 0.0046 | 0.0003 | 0.0032 | 0.0064 | 0.0298 | 0.0003 |
4. Liver enzyme inhibitors
Kim et al.82 synthesized E-stilbene analogues and evaluated their inhibitory activity on human CYP1A1, CYP1A2, and CYP1B1. Compound 216a displayed the highest inhibitory activity on CYP1A1, CYP1A2, and CYP1B1. This compound was also more potent than oxyresveratrol. Replacing the OH groups of oxyresveratrol with OCH3 groups in 216b improved the inhibitory activity and produced deep variations in selectivity against CYP1A1, CYP1A2, and CYP1B1. A change in the position of the OCH3 groups on the phenyl ring of compounds 216c (in 3′-, 4′- and 5′-positions), 216d (in 3′- and 5′-positions), 216e (in 3′- and 4′-positions), and 216f (in 4′-position) resulted in a decrease in potency and selectivity. These findings suggested that the precise location of the OCH3 groups is a critical feature for selectivity. When the ethylene linker of compound 216b was replaced with an amide or an imine linker (220 and 218), there was no remarkable inhibitory activity on CYPs. In another modification, the phenyl ring was replaced with 4-pyridyl and 3-furanyl rings (216g and 216h), which reduced the activity compared to other compounds. However, 216a with 2-thiophenyl exhibited the highest inhibitory activity on CYPs, while none of them was as selective as 216b. Therefore, replacing the OCH3 group in the 2′-position on the phenyl ring of 216b with an OH or F in compounds 216i and 216j showed less inhibitory activity on CYP1A1, CYP1A2, and CYP1B1, respectively. Also, the presence of lipophilic groups and EWG in 216j showed higher inhibitory activity than hydrophilic groups and EDG in 216i (Table 43).
|
||||
|---|---|---|---|---|
| Compound | Ar | IC50 (nM) | ||
| 1A1 | 1A2 | 1B1 | ||
| 216a |  |
61 | 11 | 2 |
| 216b |  |
300 | 3100 | 6 |
| 216c |  |
140 | 930 | 3200 |
| 216d |  |
920 | 198![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
17600 |
| 216e |  |
750 | 570000 |
3000 |
| 216f |  |
830 | 6200 | 790 |
| 216g |  |
1100 | 290 | 460 |
| 216h |  |
6600 | 740 | 2100 |
| 216i | 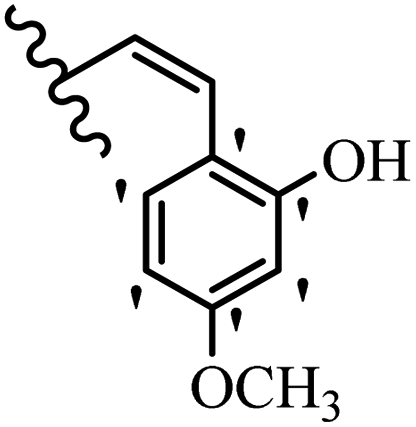 |
980 | 31100 |
390 |
| 216j | 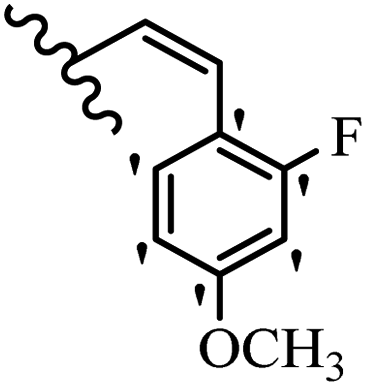 |
610 | 5800 | 97 |
| 218 | 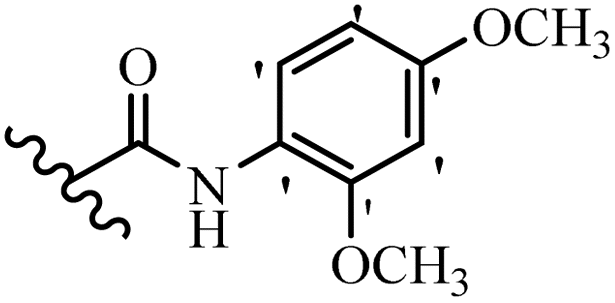 |
1500 | 64000 |
670 |
| 220 | 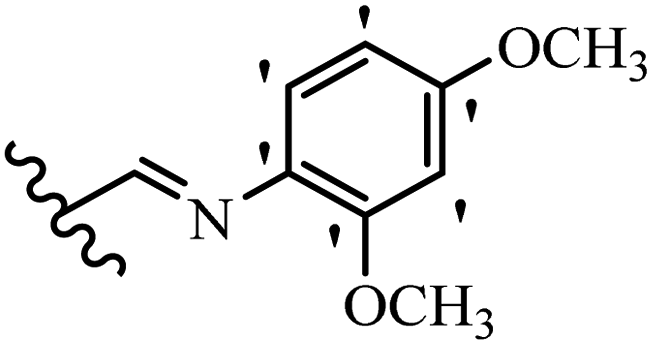 |
300 | >2 × 106 | 4900 |
| Oxyresveratrol | — | 15000 |
150000 |
34000 |
Das et al.83 synthesized pinacolyl boronate-substituted stilbenes and tested them as lipogenic inhibitors. Among the synthesized compounds, 227a showed the highest lipogenic inhibitory activity, which was greater than that of DMSO. Compound 227b exhibited the lowest activity among the compounds, which was equal to that of DMSO. Also, replacing the OCH3 group in the 4′-position on the phenyl ring with an OH group in the same position decreased the activity (227a vs. 227b). It seems that the hydrophilic group had a positive effect on activity. In addition, the presence of an OH group in the 6′-position on the phenyl ring in 227c displayed almost equal activity compared to 227d (Table 44).
|
|||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | FAS relative mRNA level |
| 223a | H | OH | H | H | 0.39 |
| 223b | H | OCH3 | H | H | 1.0 |
| 223c | Cl | H | Cl | OH | 0.57 |
| 223d | Cl | H | Cl | H | 0.58 |
| DMSO | — | — | — | — | 1.0 |
Mikstacka et al.23 synthesized E-resveratrol analogues and evaluated their inhibitory activity on some CYPs. Among the synthesized compounds, 226a and 226b showed the highest inhibitory activity against CYPs. Also, shifting the OCH3 group in the 4′- to 5′-position on the phenyl ring reduced the activity (226c vs. 226d) on CYP1A1 and CYP1A2. In contrast, 226d had better inhibitory activity than 226c on CYP1B1. Compounds having substituents in the 2′- and 4′-positions showed higher affinity to the CYP1A2 active site (226c). However, based on the results, CYP1A2 is the most sensitive to changes in the design of methoxy substituents. Compound 226e with OCH3 groups in the 3′-, 4′-, and 5′-positions on the phenyl ring did not reduce the affinity of these three OCH3 groups on the phenyl ring to CYP1A2, but it significantly increased the inhibitory potency against CYP1A1 and CYP1B1. In addition, increasing the number of OCH3 groups on the phenyl ring improved the inhibitory activity (226d vs. 226f). The results showed that the EWG and lipophilic groups had better activity compared to EDG and hydrophilic groups (Table 45).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | IC50 (μM) | ||
| CYP1A1 | CYP1A2 | CYP1B1 | ||||||
| 226a | OCH3 | H | OCH3 | H | OCH3 | 0.4 | 6.5 | 0.5 |
| 226b | Cl | H | H | H | H | 1.0 | 14.5 | 0.3 |
| 226c | OCH3 | H | OCH3 | H | H | 2.4 | 8.1 | 2.0 |
| 226d | OCH3 | H | H | OCH3 | H | 3.8 | 39.5 | 1.1 |
| 226e | H | OCH3 | OCH3 | OCH3 | H | 3.6 | 7.0 | 2.6 |
| 226f | OCH3 | H | OCH3 | OCH3 | H | 0.8 | 15.0 | 0.9 |
Mikstacka et al.24 designed E-stilbene derivatives and evaluated their inhibitory activity on CYP450. The synthesis method in this study was described in a previous report.18 Compounds 227a and b showed the highest activity on CYP1A1, CYP1A2, and CYP1B1 among the synthesized compounds. All the compounds strongly inhibited CYP1A1 activity. Compound 227b was a selective inhibitor of CYP1B1, showing a 90-times higher selectivity for CYP1B1 over CYP1A1 and 830-times higher selectivity for CYP1B1 over CYP1A2. In addition, moving the OCH3 group in the 3′- to the 5′-position on the phenyl ring enhanced the CYP1A1 inhibition activity (227c vs. 227d). Adding an OCH3 group in the 3′-position on the phenyl ring in 227b produced 227e, which exhibited lower activity against CYP1A1, CYP1A2, and CYP1B1. Moreover, attaching an OCH3 group in the 4′-position on the phenyl ring in 227f showed stronger activity than 227a against CYP1A2. In contrast, 227a and 227f showed almost equal activity for CYP1A1 and CYP1B1. Also, increasing the number OCH3 groups on the phenyl ring had a negative effect on activity (e.g., 227e and 227g). In addition, changing the position of the OCH3 group on the phenyl ring in 227f, 227d, and 227c improved the inhibitory activity against CYP1A1. Therefore, according to the results, the activity followed the order of 6′ > 5′ > 4′ (Table 46).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | IC50 (μM) | ||
| CYP1A1 | CYP1A2 | CYP1B1 | ||||||
| 227a | OCH3 | — | — | — | OCH3 | 0.23 | 2.31 | 0.31 |
| 227b | OCH3 | — | — | — | — | 0.36 | 3.32 | 0.0040 |
| 227c | OCH3 | OCH3 | OCH3 | — | — | 5.18 | >100 | 4.17 |
| 227d | OCH3 | — | OCH3 | OCH3 | — | 1.78 | >100 | 4.44 |
| 227e | OCH3 | OCH3 | — | — | — | 0.45 | 16.02 | 0.37 |
| 227f | OCH3 | — | OCH3 | — | OCH3 | 0.27 | >100 | 0.30 |
| 227g | OCH3 | — | — | OCH3 | — | 0.50 | >100 | 0.62 |
Wierzchowski et al.84 tested E-methylthio stilbene derivatives as CYP450 inhibitors. The synthesis method of this study is mentioned in the previous study.23 All the tested compounds inhibited the CYP1A1 and CYP1B1 activities, but only moderately inhibited CYP1A2. Compounds 228a–c showed the highest activity and they were stronger than the standard α-naphthoflavone on CYPs. Also, the addition of an OCH3 group in the 3′-position on the phenyl ring of 228c enhanced the inhibitory activity compared to no substitution in 228d. According to the results, both hydrophilic and lipophilic groups on both phenyl rings improved the inhibitory activity (Table 47).
|
||||||
|---|---|---|---|---|---|---|
| Compound | R | R1 | R2 | IC50 (μM) | ||
| CYP1A1 | CYP1A2 | CYP1B1 | ||||
| 228a | SCH3 | H | OCH3 | 0.076 | 8.38 | 0.034 |
| 228b | OCH3 | OCH3 | SCH3 | 0.76 | 3.54 | 0.029 |
| 228c | OCH3 | OCH3 | OCH3 | 0.36 | 4.93 | 0.0034 |
| 228d | OCH3 | H | OCH3 | 0.75 | 4.13 | 0.066 |
| ANF | — | — | — | 0.012 | 0.0038 | 0.0013 |
5. Anti-Alzheimer's activity
One of the most common neurodegenerative diseases is Alzheimer's disease (AD). It is assessed that there is one new case of dementia every 3 seconds around the world. Fifty million people worldwide were living with dementia in 2018, and this number is rapidly increasing in countries with an aging population.85 Although the biology of AD is very complex and not completely determined, some factors such as abnormal Aβ appearance and buildup, reduction of acetylcholine (ACh), tau hyperphosphorylation, dyshomeostasis of biometals, and oxidative stress have been determined to play significant roles in the pathophysiology of AD.86 In this case, resveratrol has health functional properties in neuronal degenerative pathologies such as AD.87 Trans-resveratrol and trans-piceatannol exhibited a good binding mode in silico against AChE. Scirpusin A and cassigarol E displayed inhibitory potential against butyrylcholinesterase (BChE) and AChE compared to tacrine and donepezil.88Lu et al.89 synthesized resveratrol derivatives and tested their Aβ-aggregation inhibitory activity. Compound 233a showed the highest inhibitory effect on Aβ fibrillization among the synthesized compounds. This compound exhibited stronger activity than curcumin and equal activity to resveratrol. Likewise, changing the position of the dimethylamino group in 233b from the 2′- to 4′-position on the phenyl ring in compound 233c resulted in increased activity. Among the four substituted groups (dimethylamino groups; 233a, pyridyl; 233d, Br; 233e and alkyl; 233f), the dimethylamino group showed the best result. In addition, the inhibitory activity of 237g was not significantly changed when the dimethylamino group was replaced with one- or two-substituted amino groups. Also, replacing the OCH3 groups in the 3- and 5-positions on the phenyl rings of 232a compared to 233c with an OH group in the same position reduced the inhibitory activity. These findings suggested that hydrogen bonds play an important role in the interaction of polyphenols and proteins. Compound 233h, whose carbon–carbon double bond is reduced compared to the structure of compound 233c, demonstrated lower activity (Table 48).
|
||||
|---|---|---|---|---|
| Compound | R2 | R2 | R3 | Inhibition percent (%) |
| Aβ-aggregation | ||||
| 232a | OCH3 | OCH3 |  |
7.02 |
| 233a | OH | OH |  |
71.65 |
| 233b | OH | OH |  |
59.40 |
| 233c | OH | OH |  |
65.20 |
| 233d | OH | OH |  |
13.56 |
| 233e | OH | OH |  |
25.23 |
| 233f | OH | OH |  |
43.56 |
| 233g | OH | OH |  |
65.50 |
| 233h | OH | OH |  |
48.54 |
| Resveratrol | — | — | — | 69.73 |
| Curcumin | — | — | — | 52.77 |
Andhare et al.90 designed E-distyrylbenzenes and tested their activity against Alzheimer's disease. Compound 236a showed the highest activity against Aβ-aggregation. Also, changing the position of the nitrostyrylbenzene moiety from the 3-position in 236b to the 4-position on the phenyl ring in 236c enhanced the activity. Likewise, the presence of substitution on the phenyl ring between two phenyl stilbene rings decreased the activity (239a and 239b). In addition, replacing the biphenyl ring in 239c with a phenyl ring in 239d reduced the activity. Moreover, compound 236d exhibited the highest inhibitory activity against AChE. Compound 236c showed better activity compared to 236b. According to the results, the presence of both lipophilic and hydrophilic groups enhanced the activity (236a, 236e, and 236f and 236d) against Aβ-aggregation and AChE (Table 49).
|
||||
|---|---|---|---|---|
| Compound | Ar1 | Ar2 | Aβ-aggregation (μM) | AChE (μM) |
| 236a |  |
 |
40 | 100 |
| 236b |  |
 |
75 | 180 |
| 236c |  |
 |
55 | 610 |
| 236d |  |
 |
45 | 70 |
| 236e |  |
 |
100 | 55 |
| 236f |  |
 |
65 | 80 |
| 239a |  |
 |
35 | 40 |
| 239b |  |
 |
70 | 40 |
| 239c |  |
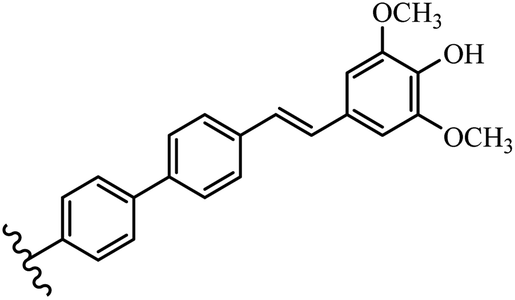 |
55 | 80 |
| 239d |  |
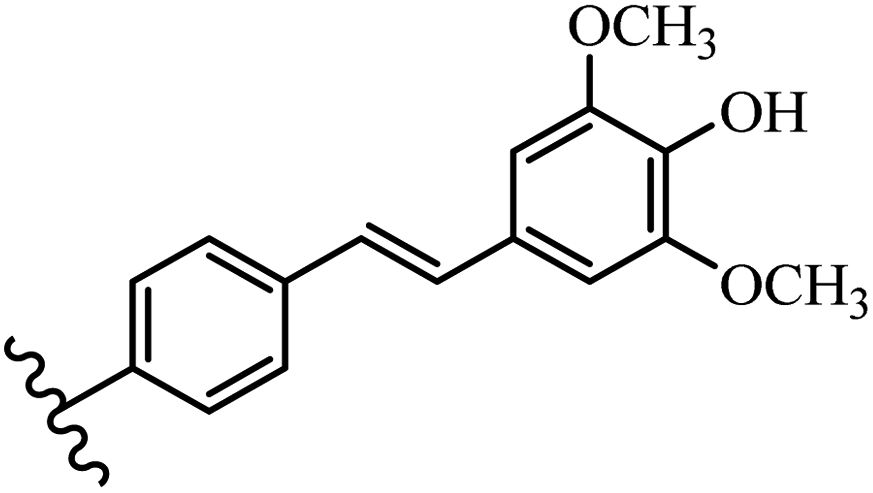 |
45 | 80 |
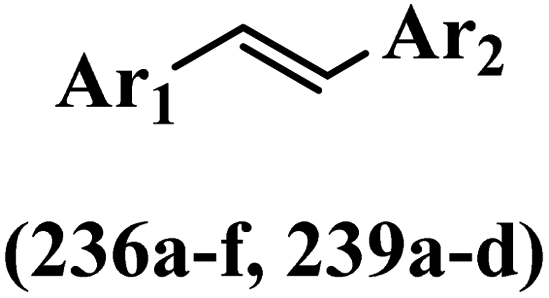
Patel et al.86 designed carbazole-stilbene hybrids and tested their anti-Alzheimer's activity. Among the compounds, 245a against hAChE and 246a against EqBuChE showed the highest activity. These compounds were weaker than tacrine and donepezil. They were also stronger than E-9-ethyl-3-styryl-9H-carbazole against hAChE and EqBuChE. In the first series, comparing the compounds with piperidine rings (245a, 245b, and 245c), compound 245a (n = 4) showed the best inhibitory activity against AChE, while compounds 245b (n = 3) and 245c (n = 2) exhibited the weakest inhibitory activity. A similar pattern was seen for the compounds with pyrrolidine rings (245d, 245e, and 245f). Compound 245b exhibited the strongest inhibitory activity against BuChE among them. When the pyrrolidinyl ring in 246b and piperidinyl ring in 246c were attached directly to provide urea derivatives, the inhibitory effect against both enzymes was reduced, particularly compared to the first series. When amide linkers (e.g., 245d) were replaced with urea linkers (e.g., 246d), there was no significant change in inhibitory activities. In the second series, a comparison of the inhibitory potential of compounds 251a, 251b, and 251c with a pyrrolidine ring revealed that compound 246b (n = 2) had the best profile of AChE and BuChE inhibitory activity, whereas compounds 251a (n = 1) and 251c (n = 3) showed slightly lower AChE and BuChE inhibitory activities. All the urea derivatives inhibited ChEs the most, while compounds 252a and 252b, in which the heterocyclic amine was directly attached to form urea, inhibited them the least. When the amide linkers in compound 251d were replaced with urea linkers in compound 252c, there was no significant change in AChE inhibitory activity, but the BuChE inhibitory activity increased by 2-times. All the thiourea derivatives demonstrated excellent inhibitory activity against ChEs. Among them, compound 252e (n = 2) was conferred with the highest inhibitory activity against AChE and BuChE. Then, the Aβ1–β2-aggregation inhibition activity was tested. Compound 252d showed the highest activity among the compounds, which was better than curcumin. Almost all the compounds showed relatively similar Aβ1–β2-aggregation inhibitory activity. According to the results, lipophilic groups improved the activity against AChE, BuChE, and Aβ1–β2-aggregation (Table 50).
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | n | X | A | NR1R2 | IC50 (μM) | Inhibition percent (%) | |
| hAChE | EqBuChE | Aβ1–β2-Aggregation | |||||
| 245a | 4 | — | — | 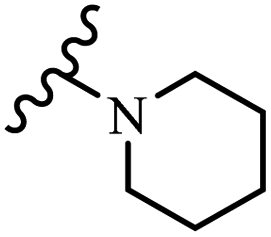 |
1.84 | 2.51 | 48.09 |
| 245b | 2 | — | — | 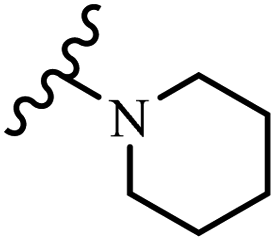 |
4.96 | 1.40 | 52.08 |
| 245c | 3 | — | — | 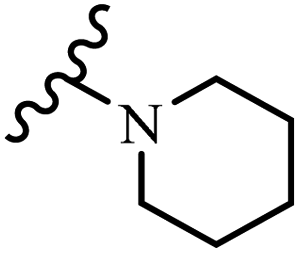 |
3.54 | 2.56 | 49.88 |
| 245d | 2 | — | — | 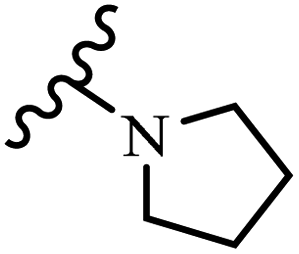 |
3.00 | 1.53 | 42.72 |
| 245e | 3 | — | — | 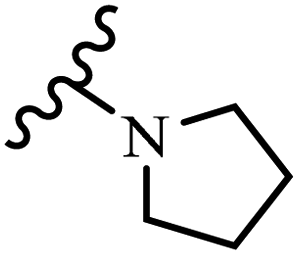 |
2.91 | 1.51 | 53.92 |
| 245f | 4 | — | — | 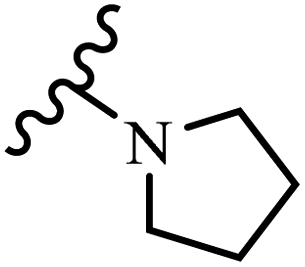 |
2.63 | 3.17 | 46.72 |
| 246a | 3 | — | NH | 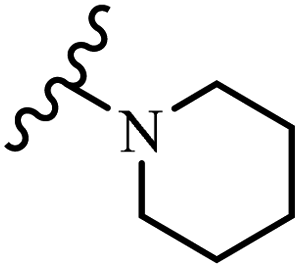 |
3.57 | 1.02 | 54.35 |
| 246b | — | — | — | 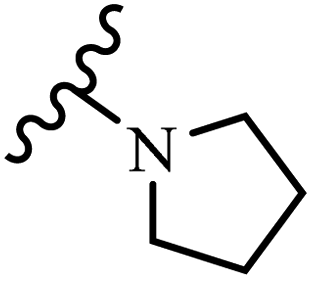 |
6.63 | 4.48 | 17.58 |
| 246c | — | — | — | 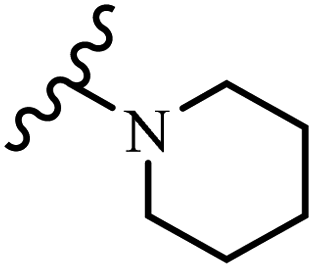 |
5.99 | 5.01 | 21.55 |
| 246d | 2 | — | NH | 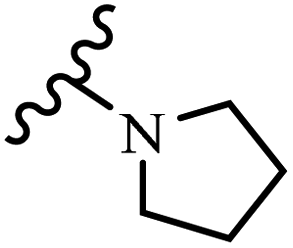 |
2.65 | 1.70 | 52.29 |
| 251a | 1 | O | — | 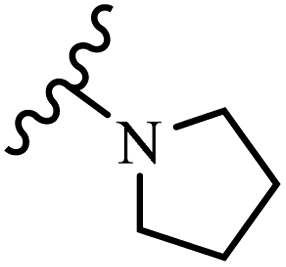 |
2.98 | 2.49 | 51.14 |
| 251b | 2 | O | — | 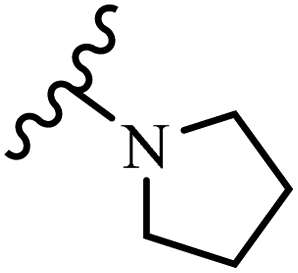 |
2.36 | 1.46 | 54.27 |
| 251c | 3 | O | — |  |
4.77 | 4.76 | 53.08 |
| 251d | 3 | O | — |  |
3.29 | 2.11 | 49.78 |
| 252a | — | O | — |  |
16.22 | 11.65 | 42.84 |
| 252b | — | O | — |  |
12.37 | 8.58 | 44.15 |
| 252c | 2 | O | NH |  |
4.71 | 2.32 | 38.90 |
| 252d | 2 | O | NH |  |
3.13 | 1.20 | 55.79 |
| 252e | 2 | S | NH |  |
2.64 | 1.29 | 51.29 |
| CS-1 | — | — | — | — | >100 | >100 | 27.52 |
| Tacrine | — | — | — | — | 0.056 | 0.008 | — |
| Donepezil | — | — | — | — | 0.023 | 1.87 | — |
| Curcumin | — | — | — | — | — | (IC50 = 20.43 μM) | |
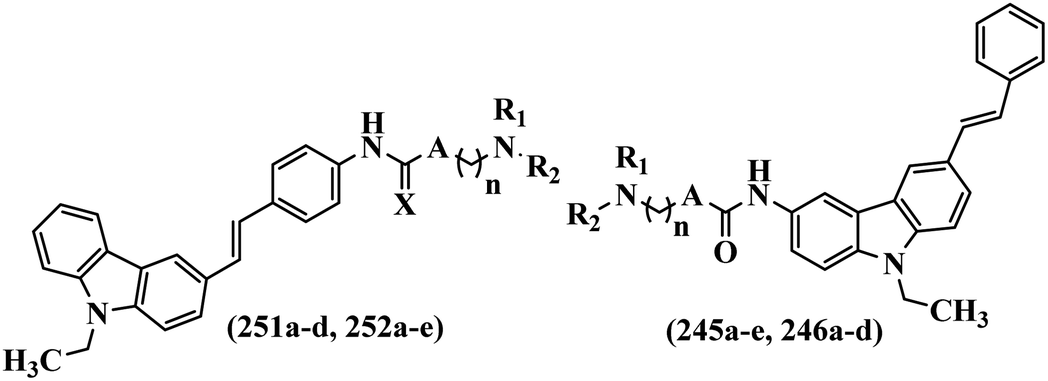
6. Antioxidant activity
The imbalance in antioxidant reactions caused by the buildup of free radicals in the body and oxidation results in oxidative stress.91 The highest free radicals in cells, mostly formed by mitochondria, are produced by reactive oxygen species (ROS).92 Excess ROS disturb metabolic function, break down cells and tissues, and lead to diverse health problems.93 Antioxidants are a group of compounds that aid in neutralizing and trapping free radicals, and thus they can reduce the harm to the body caused by free radicals.94,95 Natural phenolic derivatives are a vital group of antioxidants investigated and widely used in the nutritional and biopharmaceutical fields to inhibit oxidation processes.96 Stilbenes, as polyphenolic compounds,97 possess interesting antioxidant activity.96 The hydroxy group is important for antioxidant activity, which can be due to the stabilization via resonance in the double-bond linker.98 Resveratrol exhibited antioxidant activity in tumor initiation, promotion and progression, disturbing its progression by blocking the S and G2 phases of the cell cycle.99Jung et al.15 synthesized E-stilbene derivatives and evaluated their antioxidant activity. The synthesis method of this study was mentioned in a previous study.44 Most of the compounds showed equal antioxidant activity to resveratrol. Compounds 253a and 253b exhibited the highest antioxidant activity among the synthesized compounds. These compounds were also better than resveratrol. Compound 253c with an N-tetrahydrofuran-2-ylmethyaminocarbonyl moiety in the 4′-position on the phenyl ring showed lower activity compared to 253d having N-furan-2-ylmethylaminocarbonyl in the same position. In addition, the presence of N-(3-ethoxycarbonylphenyl)aminocarbonyl of 253e showed higher antioxidant activity than 253f with no substitution in the same position. The results showed that acyclic amine moieties displayed higher radical-scavenging activity than the cyclic amine moieties in the 4′-position on the phenyl ring (e.g., 253b, and 253g). Also, the presence of an aromatic ring compared to an aliphatic ring reduced the activity (253f and 253h) (Table 51).
|
|||
|---|---|---|---|
| Compound | R | R1 | IC50 (μM) |
| Antioxidant activity | |||
| 253a |  |
H | 47.87 |
| 253b |  |
H | 43.59 |
| 253c |  |
H | >200 |
| 253d |  |
H | 69.0 |
| 253e | 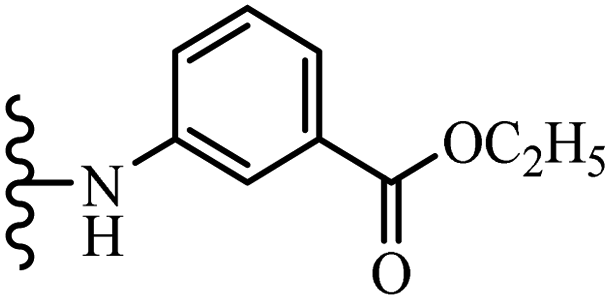 |
H | 126.63 |
| 253f |  |
H | >200 |
| 253g |  |
H | >200 |
| 253h |  |
H | 127.93 |
| Resveratrol | — | — | >200 |
Lu et al.89 investigated the antioxidant activity of resveratrol compounds. Among the compounds, 254a demonstrated the highest antioxidant activity, which was comparable to that of resveratrol. Furthermore, a secondary amine in the 4-position on the phenyl ring of 254b and 254c displayed higher antioxidant activity than a tertiary amine in 254d in the same position. The findings indicated that the amine groups in the 4-position on the phenyl ring play a significant role in the antioxidant effect. According to the results, the presence of two propyl chains on the tertiary amine and cyclohexyl ring on the secondary amine resulted in an increase in antioxidant activity (254e and 254f) (Table 52).
|
||
|---|---|---|
| Compound | Ar | ORAC |
| 254a |  |
5.54 |
| 254b |  |
5.19 |
| 254c |  |
4.33 |
| 254d |  |
3.31 |
| 254e |  |
2.23 |
| 254f |  |
2.25 |
| Resveratrol | — | 5.92 |
Patel et al.86 designed carbazole–stilbene hybrids and evaluated their antioxidant activity. Most of the compounds exhibited weak activity. Among them, 255a showed the highest activity, but weaker activity than ascorbic acid. In addition, replacing the sulfur atom with oxygen increased the antioxidant activity (255b vs. 255c). It seems that increasing the atom size and lipophilic groups improved the antioxidant activity. The other compounds showed almost equal activity (Table 53).
|
|||||
|---|---|---|---|---|---|
| Compound | n | X | A | NR1R2 | Inhibition percent (%) |
| DPPH | |||||
| 255a | 2 | S | NH |  |
72.36 |
| 255b | 3 | O | NH |  |
45.95 |
| 255c | 3 | S | NH |  |
70.36 |
| Ascorbic acid | — | — | — | — | 98.25 |
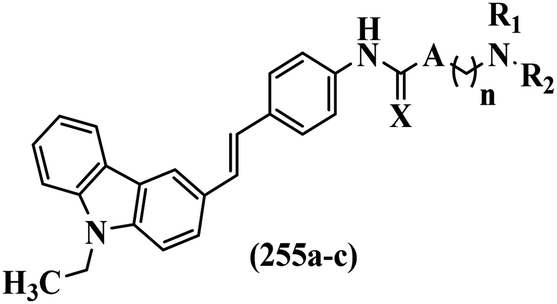
7. Antidiabetic activity
Diabetes mellitus is an endocrine metabolic disorder identified by abnormal levels of glucose in the blood stream. Chronic hyperglycemia can cause severe long-term problems including kidney failure, cardiovascular disease, and nerve damage.100 In this case, resveratrol and rosewood exhibited anti-diabetic activity.101 Also, stilbenes showed potent inhibitory activity against α-glucosidase.102,103 In mice nourished by a high-fat diet, they improved the insulin resistance. Resveratrol is also known to regularize hyperglycemia, and significantly improve hyperinsulinemia in diet-induced obese and diabetic mice.104Jung et al.105 synthesized stilbene derivatives and tested their inhibitory activity against protein tyrosine phosphatase 1B (PTP1B). Compound 270a showed the highest inhibitory activity against PTP1B. It was more potent than molybdate and RK-682. Also, replacing alcohol and aldehyde in 263a and 264a instead of methyl ester in 262 reduced the activity. Modifying the molecular structure of a compound by extending a conjugated system within it, from compound 262 to 270a, resulted in more potent inhibition of PTP1B. However, 3′,4′-dihydroxycinnamic acid 272 and its amide analogs, 274a and 274b, exhibited no activity. This indicated that the phenyl ring linked by the double bond is essential for PTP1B inhibition. According to the findings, it seemed that the OH groups in the 3′- and 4′-positions on the phenyl ring and EWGs in the 2- and 4-positions on the phenyl ring, such as ester (262 and 270a), aldehyde (264b), nitro (276a and 276b) and amides (264a) of E-stilbene analogues enhanced the inhibitory effect against PTP1B. It is important that E-isomers (270a) are relatively more potent than Z-isomers (271a), which may be due to their binding appropriateness to the PTP1B active site (Table 54).
|
||||||
|---|---|---|---|---|---|---|
| Compound | E/Z | R1 | R2 | R3 | R4 | IC50 (μM) |
| PTP1B | ||||||
| 270a | E | — | OH | OH |  |
14.9 |
| 263a | E | — | OH | OH |  |
260 |
| 264a | E | — | OH | OH |  |
27.2 |
| 264b | E | — | OH | OH |  |
101 |
| 272 | — | — | OH | OH | 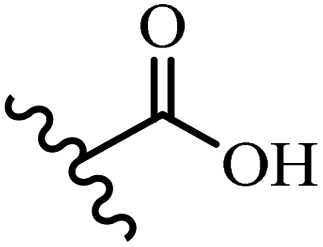 |
— |
| 274a | — | — | OH | OH | 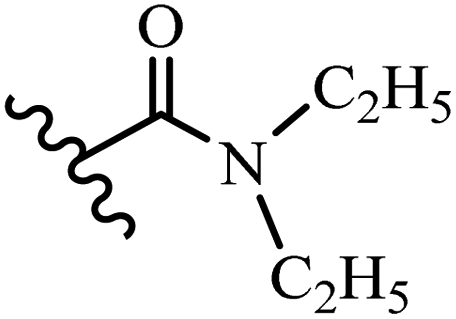 |
— |
| 274b | — | — | OH | OH | 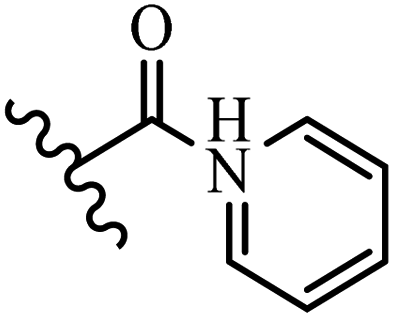 |
— |
| 262 | E | — | OH | OH | 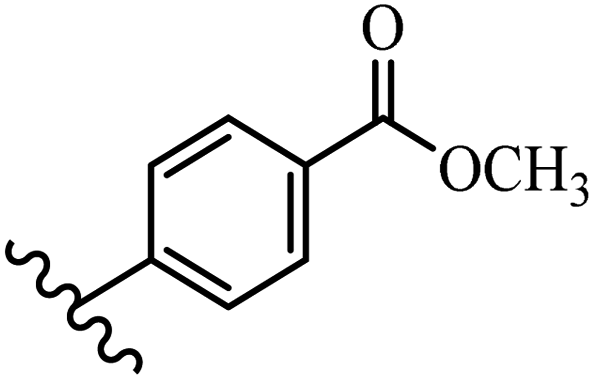 |
25.6 |
| 271a | Z | — | OH | OH |  |
— |
| 276a | E | — | OH | OH |  |
190 |
| 276b | E | OH | OH | — |  |
168 |
| Molybdate | — | — | — | — | — | 21 |
| RK-682 | — | — | — | — | — | 45 |
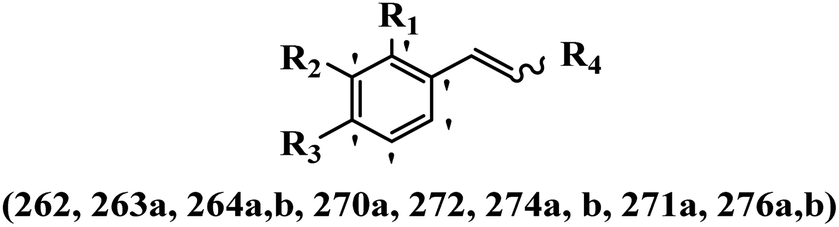
Mizuno et al.106 studied pterostilbene analogues for peroxisome proliferator-activated receptor α (PPARα) activation. The NO2 group in compounds E/Z-280a and NH2 group in E/Z-281a in the 4-position on the phenyl ring remarkably activated PPARα. Among the Z-isomers, compound 280c having an OCH3 group in the 4-position on the phenyl ring had significant activity as a PPARα agonist (>2-times). The introduction of an ester group (280b having COOCH3 in the 4-position on the phenyl ring) also resulted in an improvement in activity (>2-times). To study the role of the vinyl double bond of pterostilbene in the activation of PPARa, saturated compound 283a was evaluated. Saturation of the double bond changed the conformation of the molecule, resulting in the loss of activity. The most active compound in this series was phosphate derivative 283a. This displayed that the addition of an acidic group resulted in greater activity. It is possible that 283a with dihydrogen phosphate in the 4-position on the phenyl ring acts as a prodrug, resulting in the observed increase in activity. In general, compounds E-280b and E/Z-280c bearing an OCH3 group in the 4-position on the phenyl ring were also more effective at activating PPAR than ciprofibrate. Considering compounds 280a–c, 281a and b, and 283a, having OCH3 groups in the 3- and 5-positions on the phenyl ring, showed that presence of different groups at the 4′-position determined the activity; thus, methoxy, ester, and phosphate are preferable. Compound 289a with OH groups in the 3- and 5-positions on the phenyl ring did not show significant PPAR activation. Compound 289b with an OCH3 group in the 4′-position on the phenyl ring showed activation on PPARα (>2-times), indicating that the OH groups in the 3- and 5-positions on the phenyl ring are highly effective for activity. The data indicated that the E-isomer is more favorable than the Z-isomer for activating PPARα (Table 55).
|
||||
|---|---|---|---|---|
| Compound | E/Z | R | R1 | R2 |
| 280a | E | NO2 | OCH3 | OCH3 |
| 280a | Z | NO2 | OCH3 | OCH3 |
| 280b | E | COOCH3 | OCH3 | OCH3 |
| 280b | Z | COOCH3 | OCH3 | OCH3 |
| 280c | E | OCH3 | OCH3 | OCH3 |
| 280c | Z | OCH3 | OCH3 | OCH3 |
| 281a | E | NH2 | OCH3 | OCH3 |
| 281a | Z | NH2 | OCH3 | OCH3 |
| 281b | E | COOH | OCH3 | OCH3 |
| 281b | Z | COOH | OCH3 | OCH3 |
| 283a | E | OPO3H2 | OCH3 | OCH3 |
| 283b | E | OH | OCH3 | OCH3 |
| 289a | Z | OCH3 | OH | OH |
| 289b | E | OCH3 | OH | OH |
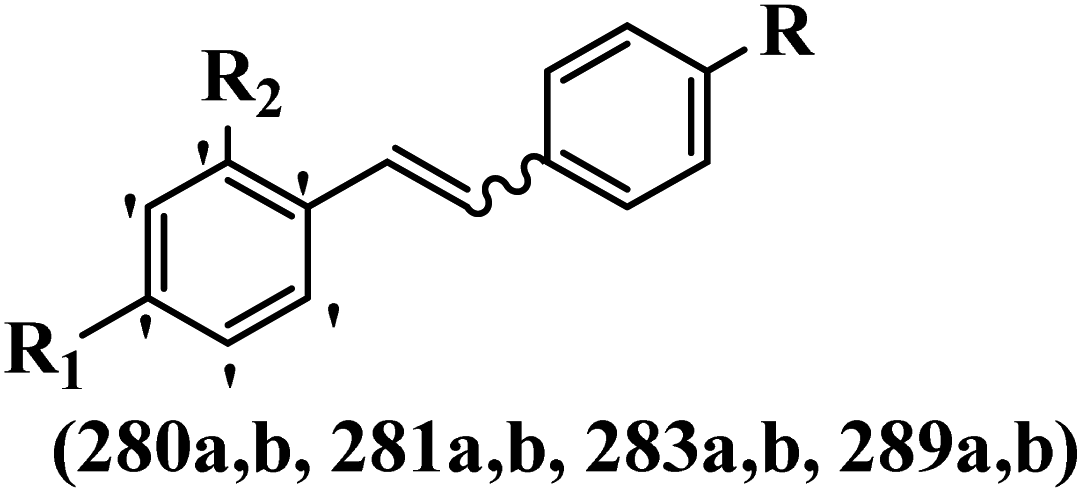
8. Miscellaneous studies
Jung et al.107 described the synthesis of stilbene compounds and assessed their antimalarial activity. The synthesis method of this research is similar to that in previous research.105 Among the synthesized compounds, 290a and b exhibited the highest activity, and they were stronger than resveratrol, while they had lower activity compared to chloroquine. Compounds 290a and b were significantly more active than 290c, which had N-(furan-2-ylmethyl)aminocarbonyl. When the carbon chain was replaced with cycloalkane or aromatic ring, the activity was enhanced (e.g., 290d and 290e). Also, the presence of N-(4-benzylpiperidine)carbonyl in 290f resulted in lower activity compared to 290g. In addition, 290c compared to 290h having N-(2-fluorobenzyl)aminocarbonyl showed a decrease in activity (Table 56).
|
||
|---|---|---|
| Compound | R | IC50 (μM) |
| Antiplasmodial | ||
| 290a | 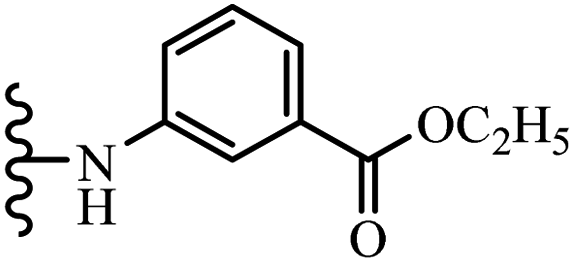 |
1.56 |
| 290b | 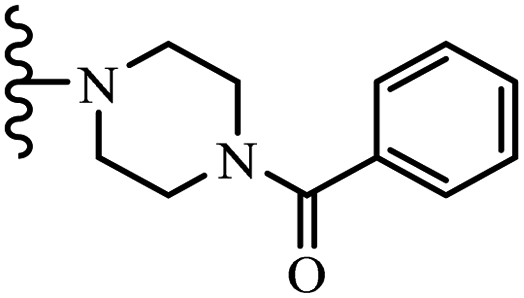 |
1.96 |
| 290c | 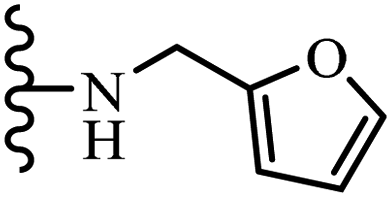 |
11.88 |
| 290d |  |
53.92 |
| 290e |  |
45.40 |
| 290f |  |
51.57 |
| 290g |  |
37.84 |
| 290h |  |
33.88 |
| Resveratrol | — | 0.02 |
| Chloroquine | — | 115.35 |
Kang et al.108 synthesized resveratrol analogues and reported their inhibitory activity against COX-1, COX-2, and NF-κB. Compounds 294a against COX-1, 294b against COX-2, and 294c against NF-κB showed the highest activity among the analogues and they were stronger than resveratrol. All the potent COX-1 inhibitors contained a resorcinol ring, although in 294d, the OH is replaced by OCH3 groups. This trend, combined with the observation that the two resveratrol derivatives 294e and 294f are equipotent, suggests that for resveratrol to bind COX-1, both rings must rotate out of the plane of the alkene. Analogues 294d, 294e, and 294f displayed potent inhibition against COX-1 and COX-2. This hypothesis is also supported by the SAR, where 294a with C2H5 on carbon number one linker on the alkene was the most potent COX-1 inhibitor, whereas 294g, which lacked a C2H5 substituent on the alkene, was 100-times less active. Although most of the active compounds had EDG in the R1 position (e.g., 294h and 294a), the COX-1 inhibitors showed that the phenol ring of resveratrol tolerates a wide variation of substitution. Thus, it seems reasonable that either the phenol of resveratrol occupies a hydrophobic and large cavity in the COX-1 active site or that area of resveratrol is solvent exposed in the complex, which improved the effect of 294h and 294a slightly to the hydrophobic interactions. However, COX-2, unlike COX-1, recognizes a wide range of substitution patterns on R3, all of which are EDG, indicating low COX-2 activity. R1 of the potent COX-2 inhibitors, similar COX-1, showed no particular electronic preference and can accommodate the steric bulk of a naphthalene ring. Also, it is worth noting that the COX-2 inhibitors discovered in the assay showed high (>15:1) selectivity for COX-2 over COX-1 (e.g., 294b and 294i). Based on the data, 294c was more potent than resveratrol against NF-κB. Although the pseudo-symmetry of the resveratrol analogues lacking an alkene substituent complicates drawing firm conclusions, it is fairly clear that the catechol occupies the R3 (resorcinol) site. In this case, it again appears that there is little selectivity for the R1 ring, beyond a general preference for large and electron-rich rings, again suggesting either a hydrophobic or a solvent-exposed binding site for R1 (Table 57).
|
||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | IC50 (μM) | ||
| COX-1 | COX-2 | NF-κB | ||||
| 294a |  |
C2H5 |  |
0.17 | 3.3 | — |
| 294b |  |
CH3 |  |
36.3 | 0.47 | — |
| 294c |  |
H |  |
— | — | 6.91 |
| 294d |  |
H |  |
0.7 | 0.82 | 19.5 |
| 294e |  |
CH3 |  |
1.9 | 1.57 | — |
| 294f |  |
CH3 |  |
1.9 | 1.78 | — |
| 294g |  |
H |  |
18.4 | — | — |
| 294h |  |
H |  |
0.29 | 21.3 | — |
| 294i |  |
CH3 |  |
— | 1.74 | — |
| Resveratrol | — | — | — | 0.83 | 0.99 | 16.1 |
9. Conclusion
Stilbene, a natural compound having anti-microbial, antioxidant, anti-cancer, and anti-Alzheimer properties, has some issues, such as poor water solubility, which reduce its clinical use. Thus, due to the biological importance of this compound, the structure–activity relationship of stilbene derivatives and their biological activities were investigated in this review. The introduction of different substituents on the stilbene scaffold, such as halogens and heterocycles, or the synthesis of hybrid molecules, affects the behavior of the compounds. The goal was to summarize the main structural changes and their associated activities that extend their activities compared to the standard compounds. The conclusions of the present investigations can be summarized as follows:1. According to the results, the presence of different substitutions on phenyl rings improved the TXRT, anti-microbial, anti-cancer, antioxidant, and CYP inhibitory activity compared to unsubstituted compounds. Alternatively, the presence of substitution on the carbon linker reduced the activity against cancer and COX.
2. The results indicated better antibacterial activity compared to antifungal activity. In addition, the findings showed that these compounds were more potent against Gram-positive bacteria than Gram-negative bacteria.
3. Z-Isomers displayed higher antibacterial, anti-cancer, Brdu incorporation and tubulin inhibitory activity compared to E-isomers. However, E-isomers showed better activity than Z-isomers on PPARα activation and PTB1B inhibition.
4. The presence of bulky groups on phenyl rings increased the antioxidant activity, while they decreased the anti-microbial and BAE, VEGF, AChE, and HADC inhibitory activities. However, bulky groups on the phenyl rings did not show any change in anti-cancer, and tubulin inhibitory activity.
5. Increasing the number of substitutions on the phenyl rings improved the inhibitory activity of CYPs, but it had a negative effect on the anti-cancer activity. This factor showed no change in anti-microbial activity.
6. The presence of EWG on phenyl rings exhibited the highest inhibitory activity against PTB1B and CYPs and PPARα activation. The presence of EDG on the phenyl rings showed the highest activity on induced apoptosis and against Aβ1–β2-aggregation. Moreover, both EDG and EWG improved the activity against HADC, LSD1, COX, and cancer, and microbial activity.
7. Results showed that lipophilic groups on the phenyl rings enhanced the activity against VEGF, h-TERT, AChE, BuChE, mur ligase, CYPs, and PTB1B, and on PPARα activation, antioxidant, and anti-cancer activity. Alternatively, hydrophilic groups increased the anti-microbe, induced apoptosis and BAE, LSD1, and COX inhibitory activity.
8. The aliphatic chain attached to the oxygen atom on the phenyl ring and the aliphatic chain on the substituted amide part of the phenyl ring showed better activity than the aliphatic ring on anti-microbial and antioxidant activity. Alternatively, the aliphatic ring on the substituted amide part of the phenyl ring exhibited higher activity than the aliphatic chain in terms of anti-malaria and anti-cancer activity.
9. Aromatic rings showed stronger anti-cancer activity and CYP inhibitory activity than heteroaromatic and aliphatic rings. However, heteroaromatic rings exhibited the highest antimalaria and antioxidant activity.
10. Aliphatic rings showed higher anti-microbial and antimalaria activity and inhibition of HADC than aromatic rings.
11. The presence of amide groups showed inhibitory activity higher than that of amine groups against CYPs.
12. The antioxidant activity and inhibition of HADC of secondary amides were better than that of tertiary amides, while both secondary and tertiary amides showed the highest anti-cancer activity.
13. Expanding a conjugated system increased the inhibitory activity on Aβ1–β2-aggregation; however, reduced PTB1B and PPARα activation, while this process had the opposite result in anti-cancer activity.
14. The nature and position of substituents on the phenyl ring were significant for Aβ1–β2-aggregation and AChE inhibitory activity. For example, substituents in the 4-position on the phenyl ring showed higher activity than in the 2- and 3-positions. Also, placing substituents on the phenyl or pyridine rings in all the positions resulted in the highest anti-microbial and anti-cancer activity and inhibition of CYPs and VEGF. The presence of groups in the 4-position was more potent than the 3-position on the coumarin ring anti-cancer activity. 3-Amidaxim on the phenyl ring displayed higher inhibitory activity than 4-amidaxim in the same position against LSD1. Changing the positions did not show any change in anti-microbial activity.
15. Increasing the chain length enhanced the AChE activity, but it had a negative effect on the BuChE inhibitory activity.
16. The presence of a bulky group on the phenyl ring showed strong antioxidant activity. However, the atom size had no effect on anti-microbial and anti-cancer activity.
Author contributions
Saghi Sepehri and Mohammad Mahdavi designed this review. Saghi Sepehri and Mina Khedmati drafted the manuscript. Faezeh Yousef-Nejad revised the manuscript. All authors contributed to the article and approved the submitted version.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- G. I. Likhtenshtein, Kirk-Othmer Encycl. Chem. Technol., 2000, 1–24 Search PubMed.
- K. A. Roupe, C. M. Remsberg, J. A. Yáñez and N. M. Davies, Curr. Clin. Pharmacol., 2006, 1, 81–101 CrossRef CAS PubMed.
- Y.-Q. Li, Z.-L. Li, W.-J. Zhao, R.-X. Wen, Q.-W. Meng and Y. Eur, J. Med. Chem., 2006, 41, 1084–1089 CAS.
- N. Rameau, B. Russo, S. Mangematin, C. Pinel and L. Djakovitch, Appl. Catal., A, 2018, 560, 132–143 CrossRef CAS.
- C. Rivière, A. D. Pawlus and J.-M. Mérillon, Nat. Prod. Rep., 2012, 29, 1317–1333 RSC.
- P. Pecyna, J. Wargula, M. Murias and M. J. B. Kucinska, Biomolecules, 2020, 10, 1111 CrossRef CAS PubMed.
- S. M. Nobre, M. N. Muniz, M. Seferin, W. M. da Silva and A. L. Monteiro, Appl. Organomet. Chem., 2011, 25, 289–293 CrossRef CAS.
- S. Henderson, Stilbene: Derivatives, Applications and Research (Chemistry Research and Applications), Nova Science Pub Inc., 2017, p. 88, ISBN-10: 1536109746, ISBN-13: 978-1536109740 Search PubMed.
- M. Kluska, J. Jablonska and W. Prukala, Molecules, 2023, 28, 4482 CrossRef CAS PubMed.
- F. Silva, E. Gallardo, C. Nerin and A. Figueiras, Food Chem., 2014, 145, 115–125 CrossRef CAS PubMed.
- P. Pecyna, J. Wargula, M. Murias and M. Kucinska, Biomolecules, 2020, 10, 1111 CrossRef CAS PubMed.
- D. Villaron and S. J. Wezenberg, Angew. Chem., Int. Ed., 2020, 59, 13192–13202 CrossRef CAS PubMed.
- B. De Filippis, A. Ammazzalorso, M. Fantacuzzi, L. Giampietro, C. Maccallini and R. Amoroso, ChemMedChem, 2017, 12, 558–570 CrossRef CAS PubMed.
- T. El Khawand, A. Courtois, J. Valls, T. Richard and S. J. P. R. Krisa, Phytochem. Rev., 2018, 17, 1007–1029 CrossRef CAS.
- J.-C. Jung, E. Lim, Y. Lee, J.-M. Kang, H. Kim, S. Jang, S. Oh and M. Jung, Eur. J. Med. Chem., 2009, 44, 3166–3174 CrossRef CAS PubMed.
- R. Chillemi, S. Sciuto, C. Spatafora and C. Tringali, Nat. Prod. Commun., 2007, 2, 499–513 CrossRef CAS.
- S. K. Lee, K. A. Nam, Y. H. Hoe, H.-Y. Min, E.-Y. Kim, H. Ko, S. Song, T. Lee and S. Kim, Arch. Pharmacal Res., 2003, 26, 253–257 CrossRef CAS PubMed.
- K. Banik, A. M. Ranaware, C. Harsha, T. Nitesh, S. Girisa, V. Deshpande, L. Fan, S. P. Nalawade, G. Sethi and A. B. Kunnumakkara, Pharmacol. Res., 2020, 153, 104635 CrossRef CAS PubMed.
- A. Gosslau, S. Pabbaraja, S. Knapp and K. Y. Chen, Eur. J. Pharmacol., 2008, 587, 25–34 CrossRef CAS PubMed.
- R. Csuk, S. Albert, B. Siewert and S. Schwarz, Eur. J. Med. Chem., 2012, 54, 669–678 CrossRef CAS PubMed.
- D. Song, X. Cao, W. Huang and S. Ke, ChemistrySelect, 2020, 5, 13563–13568 CrossRef.
- R. Chrząścik, Crit. Rev. Anal. Chem., 2009, 39, 70–80 CrossRef.
- R. Mikstacka, A. M. Rimando, Z. Dutkiewicz, T. Stefański and S. Sobiak, Bioorg. Med. Chem., 2012, 20, 5117–5126 CrossRef CAS PubMed.
- R. Mikstacka, M. Wierzchowski, Z. Dutkiewicz, A. Gielara-Korzańska, A. Korzański, A. Teubert, S. Sobiak and W. Baer-Dubowska, MedChemComm, 2014, 5, 496–501 RSC.
- C. Wolfe, P. Pagano, C. M. Pillar, D. L. Shinabarger and R. A. Boulos, Diagn. Microbiol. Infect. Dis., 2018, 92, 250–252 CrossRef CAS PubMed.
- K. Sibley, J. Chen, L. Koetzner, O. Mendes, A. Kimzey, J. Lansita and R. A. Boulos, Sci. Rep., 2019, 9, 158 CrossRef PubMed.
- A. Hamze, M. Alami and O. Provot, Eur. J. Med. Chem., 2020, 190, 112110 CrossRef CAS PubMed.
- Chapter 6 - Phytonutrients in the Management of Glucose Metabolism.
- Z. Huang, G. Li, X. Wang, H. Xu, Y. Zhang and Q. Gao, MedChemComm, 2017, 8, 1542–1552 RSC.
- C. J. Maguire, Z. Chen, V. P. Mocharla, M. Sriram, T. E. Strecker, E. Hamel, H. Zhou, R. Lopez, Y. Wang, R. P. Mason, D. J. Chaplin, M. L. Trawick and K. G. Pinney, MedChemComm, 2018, 9, 1649–1662 RSC.
- M. Gonzalez, Y. Ellahioui, R. Alvarez, L. Gallego-Yerga, E. Caballero, A. Vicente-Blazquez, L. Ramudo, M. Marin, C. Sanz, M. Medarde and R. Pelaez, Molecules, 2019, 24, 4319 CrossRef CAS PubMed.
- D. Simoni, F. P. Invidiata, M. Eleopra, P. Marchetti, R. Rondanin, R. Baruchello, G. Grisolia, A. Tripathi, G. E. Kellogg, D. Durrant and R. M. Lee, Bioorg. Med. Chem., 2009, 17, 512–522 CrossRef CAS PubMed.
- S. Yang, Z. Tang, C. Hu and Z. Dawei, Adv. Mater., 2019, 31, 1805955 CrossRef PubMed.
- N. Shen, J. Wu, C. Yang, H. Yu, S. Yang, T. Li, J. Chen, Z. Tang and X. Chen, Nano Lett., 2019, 19, 8021–8031 CrossRef CAS PubMed.
- B. De Filippis, A. Ammazzalorso, R. Amoroso and L. Giampietro, Drug Dev. Res., 2019, 80, 285–293 CrossRef CAS PubMed.
- I. Górniak, R. Bartoszewski and J. Króliczewski, Phytochem. Rev., 2019, 18, 241–272 CrossRef.
- M. Kluska, J. Jabłońska and W. Prukała, Molecules, 2023, 28, 4482 CrossRef CAS PubMed.
- S. Albert, R. Horbach, H. B. Deising, B. Siewert and R. Csuk, Bioorg. Med. Chem., 2011, 19, 5155–5166 CrossRef CAS PubMed.
- P. Jeandet, A.-C. Douillet-Breuil, R. Bessis, S. Debord, M. Sbaghi and M. Adrian, J. Agric. Food Chem., 2002, 50, 2731–2741 CrossRef CAS PubMed.
- S. N. Aslam, P. C. Stevenson, T. Kokubun and D. R. Hall, Microbiol. Res., 2009, 164, 191–195 CrossRef CAS PubMed.
- G. R. Pettit, M. R. Rhodes, D. L. Herald, E. Hamel, J. M. Schmidt and R. K. Pettit, J. Med. Chem., 2005, 48, 4087–4099 CrossRef CAS PubMed.
- E. Wyrzykiewicz, M. Wendzonka and B. Kędzia, Eur. J. Med. Chem., 2006, 41, 519–525 CrossRef CAS PubMed.
- K. Chanawanno, S. Chantrapromma, T. Anantapong, A. Kanjana-Opas and H.-K. Fun, Eur. J. Med. Chem., 2010, 45, 4199–4208 CrossRef CAS PubMed.
- D. He, W. Jian, X. Liu, H. Shen and S. Song, J. Agric. Food Chem., 2015, 63, 1370–1377 CrossRef CAS PubMed.
- W. Jian, D. He, P. Xi and X. Li, J. Agric. Food Chem., 2015, 63, 9963–9969 CrossRef CAS PubMed.
- W. Jian, D. He and S. Song, Sci. Rep., 2016, 6, 31045 CrossRef PubMed.
- L. Wen, W. Jian, J. Shang and D. He, Pest Manage. Sci., 2019, 75, 1123–1130 CrossRef CAS PubMed.
- D. Singh, R. Mendonsa, M. Koli, M. Subramanian and S. K. Nayak, Toxicol. Appl. Pharmacol., 2019, 367, 23–32 CrossRef CAS PubMed.
- M. Hrast, R. Frlan, D. Knez, I. Zdovc, H. Barreteau and S. Gobec, Bioorg. Med. Chem. Lett., 2021, 40, 127966 CrossRef CAS PubMed.
- F. Borys, P. Tobiasz, M. Poterała, H. Fabczak, H. Krawczyk and E. Joachimiak, Molecules, 2023, 28, 3558 CrossRef CAS PubMed.
- A. G. Linkous and E. M. Yazlovitskaya, Anticancer Res., 2012, 32, 1–12 CAS.
- J. Luo, C. Zhou, W. Zhang and L. Kong, Acta Pharm. Sin. B, 2013, 3, 174–179 CrossRef.
- B. De Filippis, A. Ammazzalorso, M. Fantacuzzi, L. Giampietro, C. Maccallini and R. Amoroso, ChemMedChem, 2017, 12, 558–570 CrossRef CAS PubMed.
- V. Cardile, R. Chillemi, L. Lombardo, S. Sciuto, C. Spatafora, C. Tringali and Z. Naturforsch C, J. Biosci., 2007, 62, 189–195 CAS.
- J. Lee, S. J. Kim, H. Choi, Y. H. Kim, I. T. Lim, H.-m. Yang, C. S. Lee, H. R. Kang, S. K. Ahn, S. K. Moon, D.-H. Kim, S. Lee, N. S. Choi and K. J. Lee, J. Med. Chem., 2010, 53, 6337–6354 CrossRef CAS PubMed.
- C. J. Lion, C. S. Matthews, M. F. Stevens and A. D. Westwell, J. Med. Chem., 2005, 48, 1292–1295 CrossRef CAS PubMed.
- O. McDonald, K. Lackey, R. Davis-Ward, E. Wood, V. Samano, P. Maloney, F. Deanda and R. Hunter, Bioorg. Med. Chem. Lett., 2006, 16, 5378–5383 CrossRef CAS PubMed.
- D. Simoni, F. P. Invidiata, M. Eleopra, P. Marchetti, R. Rondanin, R. Baruchello, G. Grisolia, A. Tripathi, G. E. Kellogg, D. Durrant and R. M. Lee, Bioorg. Med. Chem., 2009, 17, 512–522 CrossRef CAS PubMed.
- H.-I. Moon, I.-M. Chung, J.-C. Jung, E. Lim, Y. Lee, S. Oh and M. Jung, J. Enzyme Inhib. Med. Chem., 2009, 24, 328–336 CrossRef CAS PubMed.
- F. Belluti, G. Fontana, L. Dal Bo, N. Carenini, C. Giommarelli and F. Zunino, Bioorg. Med. Chem., 2010, 18, 3543–3550 CrossRef CAS PubMed.
- M. A. Reddy, N. Jain, D. Yada, C. Kishore, J. R. Vangala, R. P. Surendra, A. Addlagatta, S. V. Kalivendi and B. Sreedhar, J. Med. Chem., 2011, 54, 6751–6760 CrossRef CAS PubMed.
- A. S. Kumar, M. A. Reddy, N. Jain, C. Kishor, T. R. Murthy, D. Ramesh, B. Supriya, A. Addlagatta, S. V. Kalivendi and B. Sreedhar, Eur. J. Med. Chem., 2013, 60, 305–324 CrossRef CAS PubMed.
- B. I. Roman, L. M. De Coen, S. T. F. Mortier, T. De Ryck, B. W. Vanhoecke, A. R. Katritzky, M. E. Bracke and C. V. Stevens, Bioorg. Med. Chem., 2013, 21, 5054–5063 CrossRef CAS PubMed.
- R. Martí-Centelles, R. Cejudo-Marín, E. Falomir, J. Murga, M. Carda and J. A. Marco, Bioorg. Med. Chem., 2013, 21, 3010–3015 CrossRef PubMed.
- Y. Zhang, M. Shen, S. Cui and T. Hou, Bioorg. Med. Chem. Lett., 2014, 24, 5470–5472 CrossRef CAS PubMed.
- V. L. Morris, T. Toseef, F. B. Nazumudeen, C. Rivoira, C. Spatafora, C. Tringali and S. A. Rotenberg, Mol. Cell. Biochem., 2015, 402, 83–91 CrossRef CAS PubMed.
- M.-C. Scherzberg, A. Kiehl, A. Zivkovic, H. Stark, J. Stein, R. Fürst, D. Steinhilber and S. Ulrich-Rückert, Toxicol. Appl. Pharmacol., 2015, 287, 67–76 CrossRef CAS PubMed.
- J. Yan, Y. Guo, Y. Wang, F. Mao, L. Huang and X. J. Li, Eur. J. Med. Chem., 2015, 95, 220–229 CrossRef CAS PubMed.
- M. Mahdavi, K. Pedrood, M. Safavi, M. Saeedi, M. Pordeli, S. K. Ardestani, S. Emami, M. Adib, A. Foroumadi and A. Shafiee, Eur. J. Med. Chem., 2015, 95, 492–499 CrossRef CAS PubMed.
- N. R. Penthala, S. Thakkar and P. A. Crooks, Bioorg. Med. Chem. Lett., 2015, 25, 2763–2767 CrossRef CAS PubMed.
- R. Martí-Centelles, J. Murga, E. Falomir, M. Carda and J. A. Marco, MedChemComm, 2015, 6, 1809–1815 RSC.
- R. Martí-Centelles, E. Falomir, J. Murga, M. Carda and J. A. Marco, Eur. J. Med. Chem., 2015, 103, 488–496 CrossRef PubMed.
- V. Srivastava and H. Lee, Bioorg. Med. Chem., 2015, 23, 7629–7640 CrossRef CAS PubMed.
- V. Kachhadia, S. Rajagopal, T. Ponpandian, R. Vignesh, K. Anandhan, D. Prabhu, P. Rajendran, S. Nidhyanandan, A. M. Roy, F. A. Ahamed, N. Surendran, S. Rajagopal, S. Narayanan and B. Gopalan, Eur. J. Med. Chem., 2016, 108, 274–286 CrossRef CAS PubMed.
- Y.-C. Duan, Y.-Y. Guan, X.-Y. Zhai, L.-N. Ding, W.-P. Qin, D.-D. Shen, X.-Q. Liu, X.-D. Sun, Y.-C. Zheng and H.-M. Liu, Eur. J. Med. Chem., 2017, 126, 246–258 CrossRef CAS PubMed.
- T. Ismail, S. Shafi, J. Srinivas, D. Sarkar, Y. Qurishi, J. Khazir, M. S. Alam and H. M. S. Kumar, Bioorg. Chem., 2016, 64, 97–102 CrossRef CAS PubMed.
- K. M. Byrd, C. N. Kent and B. S. J. Blagg, ChemMedChem, 2017, 12, 2022–2029 CrossRef CAS PubMed.
- Y. Duan, W. Qin, F. Suo, X. Zhai, Y. Guan, X. Wang, Y. Zheng and H. Liu, Bioorg. Med. Chem., 2018, 26, 6000–6014 CrossRef CAS PubMed.
- A. Iqbal, Z. A. Khan, S. A. Shahzad, S. A. Khan, S. A. R. Naqvi, A. Bari, H. Amjad and M. I. Umar, J. Mol. Str., 2019, 1197, 271–281 CrossRef CAS.
- T. Wong, S. Narayanan, D. P. Brown and Z.-S. Chen, J. Nat. Prod., 2020, 83, 1563–1570 CrossRef CAS PubMed.
- A. Das, S. Kumar, L. Persoons, D. Daelemans, D. Schols, H. Alici, H. Tahtaci and S. S. Karki, Heliyon, 2021, 7, e05893 CrossRef CAS PubMed.
- S. Kim, H. Ko, J. E. Park, S. Jung, S. K. Lee and Y.-J. Chun, J. Med. Chem., 2002, 45, 160–164 CrossRef CAS PubMed.
- B. C. Das, X. Zhao, X.-Y. Tang and F. J. B. Yang, Bioorg. Med. Chem. Lett., 2011, 21, 5638–5641 CrossRef CAS PubMed.
- M. Wierzchowski, Z. Dutkiewicz, A. Gielara-Korzańska, A. Korzański, A. Teubert, A. Teżyk, T. Stefański, W. Baer-Dubowska and R. Mikstacka, Chem. Biol. Drug Des., 2017, 90, 1226–1236 CrossRef CAS PubMed.
- C. Patterson, World Alzheimer Report 2018, 2018 Search PubMed.
- D. V. Patel, N. R. Patel, A. M. Kanhed, D. M. Teli, K. B. Patel, P. D. Joshi, S. P. Patel, P. M. Gandhi, B. N. Chaudhary and N. K. Prajapati, Bioorg. Chem., 2020, 101, 103977 CrossRef CAS PubMed.
- A. Freyssin, G. Page, B. Fauconneau and A. R. Bilan, Neural Regener. Res., 2020, 15, 843–849 CrossRef PubMed.
- N. Mostefa, N. Djebli, P. N. Khanh, N. X. Ha, H. T. N. Anh, V. T. Ha, T. T. Huong, D. V. Anh and N. M. Cuong, Chem. Biodiversity, 2023, 20, e202201051 CrossRef CAS PubMed.
- C. Lu, Y. Guo, J. Li, M. Yao, Q. Liao, Z. Xie and X. Li, Bioorg. Med. Chem. Lett., 2012, 22, 7683 CrossRef CAS PubMed.
- N. H. Andhare, Y. Thopate, L. Kumar, T. Sharma, M. Siddiqi, A. K. Sinha and A. Nazir, Tetrahedron, 2018, 74, 1655–1667 CrossRef CAS.
- G. Pizzino, N. Irrera, M. Cucinotta, G. Pallio, F. Mannino, V. Arcoraci, F. Squadrito, D. Altavilla and A. Bitto, Oxid. Med. Cell. Longevity, 2017, 2017, 8416763 Search PubMed.
- K. H. Al-Gubory, C. Garrel, P. Faure and N. Sugino, Reprod. BioMed. Online, 2012, 25, 551–560 CrossRef CAS PubMed.
- H. Liu, Y. Liu, L. Hu, Y. Suo, L. Zhang, F. Jin, X. Feng, N. Teng and Y. Li, Poult. Sci., 2014, 93, 347–353 CrossRef CAS PubMed.
- A. Bouyahya, N. El Menyiy, L. Oumeslakht, A. El Allam, A. Balahbib, A. Rauf, N. Muhammad, E. Kuznetsova, M. Derkho and M. Thiruvengadam, Antioxidants, 2021, 10, 1553 CrossRef CAS PubMed.
- S. Mineo, N. Takahashi, M. Yamada-Hara, T. Tsuzuno, Y. Aoki-Nonaka and K. Tabeta, Arch. Oral Biol., 2021, 129, 105215 CrossRef CAS PubMed.
- S. Hamadouche, A. Ounissi, K. Baira, N. Ouddai, M. Balsamo, A. Erto and Y. Benguerba, J. Mol. Struct., 2021, 1229, 129496 CrossRef CAS.
- A. Benayahoum, H. Amira-Guebailia and O. Houache, Comput. Theor. Chem., 2014, 1037, 1–9 CrossRef CAS.
- S. Choiri, R. Fitriastuti, F. Z. Faradiva and W. V. Rahayu, Pharm. Res., 2021, 28, 365–375 Search PubMed.
- C. A. De La Lastra and I. Villegas, Biochem. Soc. Trans., 2007, 35, 1156–1160 CrossRef PubMed.
- A. M. Dirir, M. Daou, A. F. Yousef and L. F. Yousef, Phytochem. Rev., 2022, 21, 1049–1079 CrossRef CAS PubMed.
- A. Chakraborty, N. Gupta, K. Ghosh and P. Roy, In Vitro Toxicol., 2010, 24, 1215–1228 CrossRef CAS PubMed.
- A. C. Pereira, M. S. Arruda, E. A. da Silva, M. N. da Silva, V. S. Lemos and S. F. Cortes, Planta Med., 2012, 78, 36–38 CrossRef CAS PubMed.
- A. J. Zhang, A. M. Rimando, C. S. Mizuno and S. T. Mathews, J. Nutr. Biochem., 2017, 47, 86–93 CrossRef CAS PubMed.
- S. Sharma, C. S. Misra, S. Arumugam, S. Roy, V. Shah, J. A. Davis, R. K. Shirumalla and A. Ray, Phytother. Res., 2011, 25, 67–73 CrossRef CAS PubMed.
- M. Jung, Y. Lee, M. Park, H. Kim, H. Kim, E. Lim, J. Tak, M. Sim, D. Lee, N. Park, W. K. Oh, K. Y. Hur, E. S. Kang and H.-C. Lee, Bioorg. Med. Chem. Lett., 2007, 17, 4481–4486 CrossRef CAS PubMed.
- C. S. Mizuno, G. Ma, S. Khan, A. Patny, M. A. Avery and A. M. Rimando, Bioorg. Med. Chem., 2008, 16, 3800–3808 CrossRef CAS PubMed.
- M. Jung, W. H. Park, J. C. Jung, E. Lim, Y. Lee, S. Oh and H. I. Moon, Chem. Biol. Drug Des., 2009, 73, 346–354 CrossRef CAS PubMed.
- S. S. Kang, M. Cuendet, D. C. Endringer, V. L. Croy, J. M. Pezzuto and M. A. Lipton, Bioorg. Med. Chem., 2009, 17, 1044–1054 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02867h |
| This journal is © The Royal Society of Chemistry 2024 |