
Interaction mechanism between water molecules and perovskites
Yujia
Gao†
a,
Dongxu
Lin†
ab,
Pengyi
Liu
a,
Tingting
Shi
 *a and
Weiguang
Xie
*a
*a and
Weiguang
Xie
*a
aSiyuan Laboratory, Guangzhou Key Laboratory of Vacuum Coating Technologies and New Energy Materials, Guangdong Provincial Engineering Technology Research Center of Vacuum Coating Technologies and New Energy Materials, Department of Physics, Jinan University, Guangzhou 510632, China. E-mail: wgxie@email.jnu.edu.cn
bSUSTech Energy Institute for Carbon Neutrality, Department of Mechanical and Energy Engineering, Southern University of Science and Technology, Shenzhen 518055, China
First published on 9th November 2023
Abstract
In recent years, significant progress has been made in the application of perovskite materials in the field of photovoltaics. However, their stability is still affected by the presence of water molecules. Investigating the interaction mechanism between water and perovskites is of great importance for enhancing their stability and advancing commercialization. This review, based on theoretical calculations, summarizes the mechanisms underlying the interaction between water and perovskites. These mechanisms encompass water molecule diffusion, perovskite decomposition induced by a high concentration of water, phase transitions, as well as the dual impact of water on defects. Furthermore, it discusses the influence of water on the quality and performance of perovskite devices in experimental settings. By comprehensively elucidating the theoretical mechanisms governing the interaction between water molecules and perovskites, this review offers both a theoretical foundation and practical guidance for enhancing the stability and optoelectronic performance of perovskite photovoltaic devices, while also facilitating further exploration of the underlying mechanisms.
1. Introduction
In recent years, research on perovskite materials has been thriving. The generic molecular formula of perovskites is AMX3. In this formula, the A-site cation is typically represented by organic cation groups such as CN3NH3+ (MA+), CH(NH2)+ (FA+), or larger inorganic cations like Cs+. The A-site cation can also be substituted by long-chain organic cations, such as butylamine (BA+) and phenethylamine (PEA+), resulting in a dimensional reduction from 3D to 2D perovskite structures.1 The M-site is typically occupied by metal cations such as Pb2+ and Sn2+, while the X-site is filled with halide anions such as Cl−, Br−, and I−. Perovskite materials exhibit numerous advantages, including high charge carrier mobility and lifetime, high conversion efficiency, tunable optical and electronic properties, cost-effectiveness, and straightforward fabrication processes.2,3 Consequently, they have found extensive applications in the field of optoelectronic devices. However, the stability of perovskite materials under humid conditions remains a challenge, limiting their practical applications. Numerous studies have shown that perovskite solar cells are prone to degradation in ambient environments, with water being one of the main factors triggering material and device degradation.4–7 In earlier research, water was widely considered harmful to MAPbI3 perovskites as it irreversibly degrades the material into PbI2 and MAI, leading to loss of light absorption and severe electron–hole recombination.8,9 However, some reports suggest that annealing under humid conditions can actually significantly improve film quality and electronic properties, demonstrating the positive role of water.10,11 Subsequent studies have indicated both beneficial and detrimental effects of H2O on perovskites.12–17 Therefore, understanding the fundamental interaction mechanisms between water and perovskites is of great importance for their commercialization.Currently, most of the existing reviews on the influence of water on perovskite materials predominantly approach the subject from an experimental perspective. These reviews encompass the effects of water on perovskites, including degradation mechanisms,18–20 impacts on thin film quality and device performance,19,21,22 as well as strategies to enhance humidity stability.23–26 However, comprehensive summaries regarding theoretical computational investigations on the subject are scare. With this goal in mind, we primarily summarize the theoretical and computational studies on the physical and chemical processes between perovskites and water. Scheme 1 summarizes the main interactions between water and perovskites, along with the resulting outcomes of these interactions. Generally, the dominant interaction between perovskites and water is the hydrogen bonding between water molecules and organic cations as well as the inorganic framework, and the consequences of this interaction are closely related to the amount of water or the molar ratio of water to perovskites. In the case of trace amounts of water, water molecules tend to adsorb on the perovskite surface and undergo short-range diffusion, while in the presence of a large amount of water, excess water molecules diffuse to deeper positions and cause structural distortion of perovskites. The presence of surface defects promotes water adsorption and diffusion, but during the fabrication process, water can passivate defects and enhance film quality. Furthermore, a certain amount of water affects the energy barriers for the phase transition of the FA-system and Cs-system, thus influencing the material's phase transition behavior. In this review, we discuss the interaction mechanisms between water and perovskite ions as well as defects, and explore the effects of these mechanisms on perovskite materials.
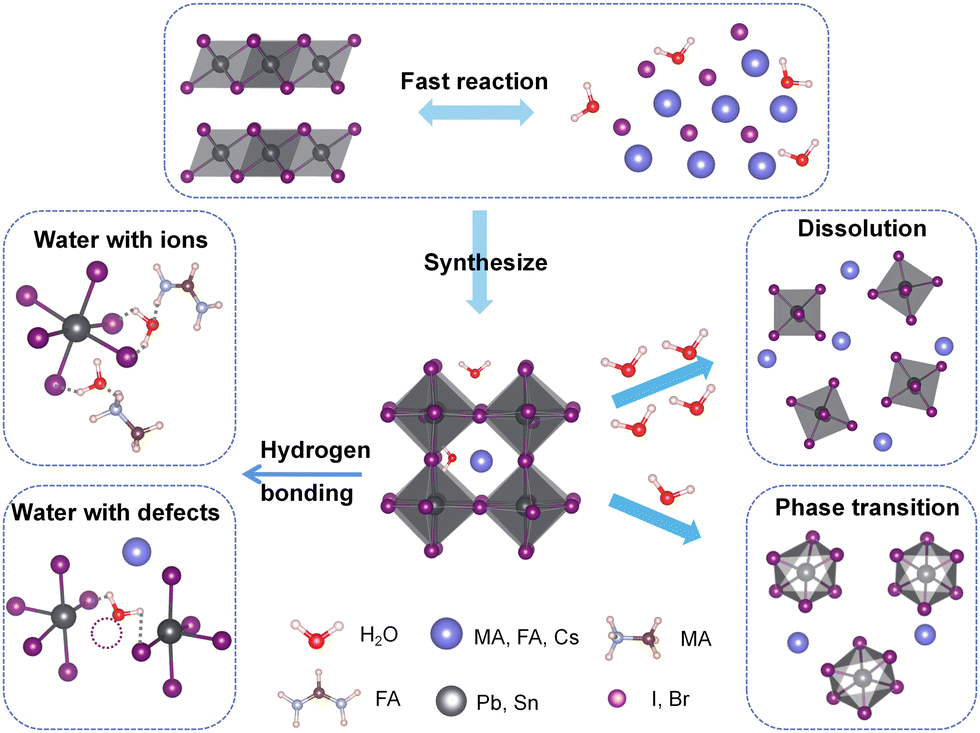 | ||
| Scheme 1 Schematic illustration of the interaction between water molecules and perovskites. | ||
2. Interaction between water molecules and perovskites
A water molecule consists of one oxygen atom and two hydrogen atoms. Oxygen has two pairs of lone electrons, giving it a bent molecular geometry. The electronegativity difference between oxygen and hydrogen results in oxygen having a partial negative charge and hydrogen having a partial positive charge. This charge difference leads to polarity. The polarity of water allows it to form hydrogen bonds. Additionally, as a polar solvent, water readily dissolves polar solutes. Therefore, theoretically, for organic–inorganic hybrid perovskites, water molecules can form hydrogen bonds with electronegative atoms, such as nitrogen (N) and halogen atoms that possess lone pairs of electrons. In this section, we verify the formation of hydrogen bonds between water and perovskite ions through a theoretical study that summarizes the interactions between water and perovskite ions, primarily including O–HN bonds with organic cations and X–HW bonds with halide ions. Moreover, when there are a sufficient number of water molecules, hydrogen bonds can also form between water molecules. These interactions have different effects on perovskite, including structural distortion, decomposition, phase transition, and electronic properties.2.1 Model description
In theoretical calculations, the construction of the model plays a crucial role in the accuracy of the computations. When modeling the interaction between water and perovskite, the following factors are generally considered: the quantity and proportion of water molecules relative to the perovskite, the adsorption sites of water molecules on different surfaces of the perovskite, and the relative positions and distances between water molecules and different atoms within the perovskite.In terms of the model for water on the perovskite surface, it is necessary to consider different surfaces, primarily encompassing two types as illustrated in Fig. 1. One is the AX-terminated surface represented by the blue and orange regions, and the other is the MX2-terminated surface represented by the green region. When studying the interaction between a single water molecule and the perovskite surface, the specific adsorption site needs to be taken into account. For AX-terminated surfaces, the potential adsorption sites for H2O can be chosen as near A+ or X−. For MX2-terminated surfaces, the potential adsorption sites for H2O can be chosen as near M2+, the M–X bond, or above A+. Previous studies have employed the calculation of adsorption energy, Eads = Esurface − H2O − (Ebare-surface + EH2O), to determine the most favorable adsorption configurations. For both AX-terminated and MX2-terminated surfaces, the specific Eads with different atomic species at each site are shown in Table 1. For AX-terminated surfaces, when considering the polarity of the MA+ species in the MA+-system, the lowest adsorption energy is obtained when a single H2O molecule is positioned near X− above the surface with the –NH3 moiety of MA+ pointing towards the surface.27,28 For MX2-terminated surfaces, the lowest adsorption energy is obtained when a single water molecule is positioned above M2+ with the –CH3 moiety of the surface MA+ pointing towards the surface. In both cases, when A, M, and X are different ions, the adsorption energies are negative, indicating that water readily adsorbs onto the perovskite surface.29
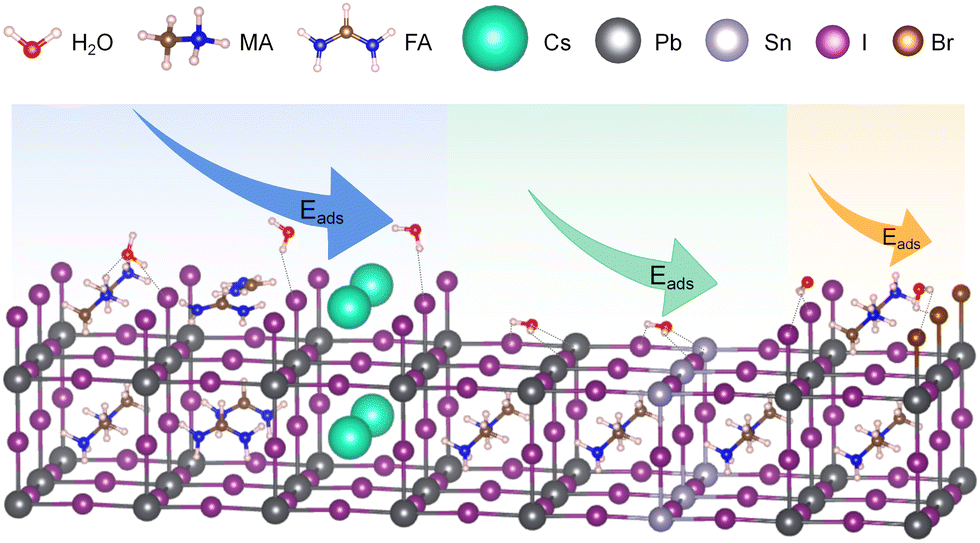 | ||
| Fig. 1 Model of water adsorption on perovskite surfaces. The blue region represents water adsorption on AX-terminated surfaces, from left to right, with MA+, FA+, and Cs+ at the A-site, respectively. The green region indicates water adsorption on MX2-terminated surfaces, from left to right, with Pb2+ and Sn2+ at the M-site. The orange region represents water adsorption on MAX-terminated surfaces, from left to right, with I− and Br− at the X-site. | ||
| The lowest Eads (eV) | ||
|---|---|---|
| AX-terminated | MAI | −0.48 |
| FAI | −0.32 | |
| CsI | −0.19 | |
| MABr | −0.48 | |
| MX2-terminated | PbI2 | −0.51 |
| SnI2 | −0.44 | |
| PbBr2 | −0.50 |
2.2 Impact of hydrogen bonding on water adsorption on perovskites
Firstly, concerning the A-site cation, the most common one is MA+. Density functional theory (DFT) calculations have revealed that on MAX-terminated surfaces, regardless of the adsorption site, the O of H2O tends to interact more with one of the H in –NH3 than with the –CH3. In other words, in MA+, the –NH3 moiety is hydrophilic, while the –CH3 moiety is hydrophobic.27,30,31 The overlap between the Opy orbital of H2O and the s orbital of HN, as indicated by the calculated projected density of states (PDOS), suggests the presence of hydrogen bonding interactions between them.32 Further investigations have found that this is due to the weaker N–H bond and the higher polarity in MA+ compared to the C–H bond. For FA+ and Cs+, their AX surfaces exhibit some hydrophobicity, manifested by higher surface adsorption energies compared to MA+. This is because in FA+, the strength of the N–H bond is similar to that of the C–H bond and both are stronger than in MA+, while the polarity of FA+ and Cs+ is weaker than that of MA+.27 Therefore, experimentally, the stability of perovskite can be enhanced by doping with A-site cations. By partially replacing MA+ with FA+ or Cs+, devices with improved humidity stability can be achieved.33,34Furthermore, for the M-site cations, common examples include Pb2+, Sn2+, and other metal cations. Calculations have revealed that the interaction between H2O and M-site cations differs from the hydrogen bonding interaction with the A-site. H2O interacts with the M-site ions through a combination of covalent and ionic interactions. Due to the closer electronegativity between Pb and I, the ionic interaction between H2O–MAPbI3 is stronger than H2O–MASnI3.27 Guo Xie et al. conducted a comparative analysis of the impact of H2O on MASnI3 and MAPbI3, and found that H2O has a significant influence on the Sn–I bonds on the surface of MASnI3, suggesting that, in a H2O atmosphere, MAPbI3 is more stable than MASnI3.29 This conclusion contradicts the stability analysis based solely on adsorption energy, emphasizing the necessity in practical research to consider multiple factors rather than relying on a single energy-based indicator to assess structural stability. On MI2-terminated surfaces, the O of H2O forms an O–M bond with the M2+ acceptor due to its lone pair of electrons. Other factors such as light illumination also influence the interaction between H2O and M2+. Lu Ying-Bo et al. found that above-band-gap light illumination of MAPbI3 induces a significant photo-induced lattice expansion effect, increasing the electron density around Pb2+ and enhancing the bonding interaction between H2O and Pb2+.35
Lastly, regarding the X-site anions, on AX-terminated surfaces, the H of H2O forms an H–X hydrogen bond with the adjacent X−.31 On MX2-terminated surfaces, Shuxia Tao suggests a covalent interaction between H2O and X−,27 while Bin Hu suggests that the interaction between I− on the (011) surface and the H of H2O is close to an ionic bond.29 The electronic density of the H–I bond is 0.05, indicating a relatively weak covalent interaction, and the H and I− form an antibonding state. However, regardless of the specific bonding analysis, the conclusions regarding Br− and I− are consistent, indicating that the interaction with Br− is weaker than with I−.27,36 Due to the weaker adsorption characteristics of H2O on Br−, experimental studies have improved the humidity stability of solar cells by partially replacing I− with Br−.37 Weiguang Zhu et al. investigated the dissolution and degradation mechanisms of isomorphous Cs2SnCl6 and Cs2SnI6 in direct contact with water. Cs2SnI6 undergoes direct dissolution in water, decomposing into CsI and SnI4, leading to the formation of an intermediate hydrolysis product, Sn(OH)4. In comparison to Cs2SnI6, Cs2SnCl6 exhibited greater stability and formed a smaller quantity of amorphous phase due to their different interaction with water molecules. The authors attribute this behavior to the smaller ionic radius of chloride, with chloride having a higher ionic potential than iodide, thereby forming stronger atomic bonds.38
2.3 Water molecule abundance and increased water diffusion depth on perovskites
How do water molecules diffuse into the interior of the perovskite? To elucidate this question, the perovskite region has been divided as depicted in Fig. 2(a) for illustrative purposes. The upper area of the (001) surface is denoted as O, F, and S regions. Region O signifies water adsorption on the perovskite surface, region F represents water molecules residing on the upper side of the first layer, and region S indicates water molecules staying on the lower side of the first layer. Additionally, the number of water molecules is represented by n, and On, Fn, and Sn denote the presence of n water molecules in these respective regions.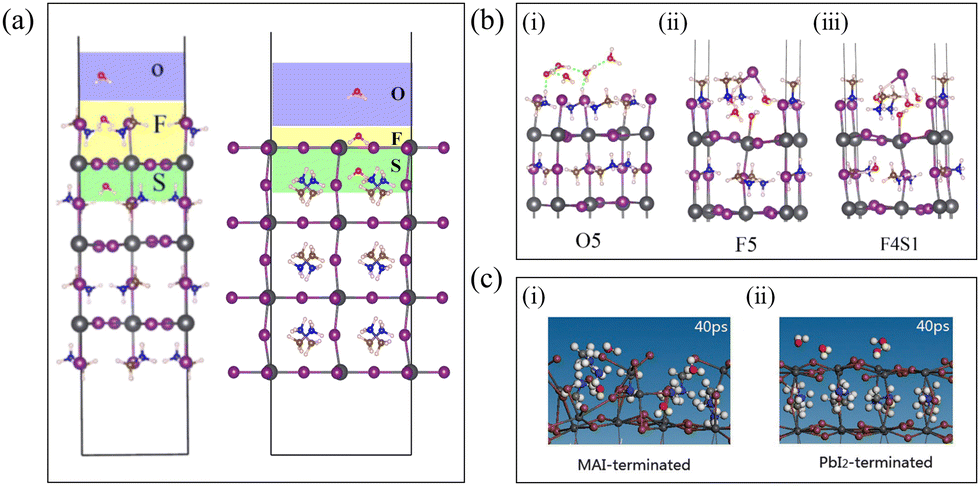 | ||
| Fig. 2 (a) Imaginary typical structure of three-layer (001) surface containing water named O1F1S1. The colored regions in the surface divisions of MAI-terminated (left) and PbI2-terminated (right) represent the possible locations of water molecules and are named O, F, and S, respectively. Reproduced with permission (left figure).39 Copyright 2015, American Chemical Society. (b) An illustration showing water adsorbed in the (i) O (O5) region, with water residing in the inner regions ((ii) F5 and (iii) F4S1). Reproduced with permission.39 Copyright 2015, American Chemical Society. (c) Snapshots of the key steps for a few water molecules absorbed on MAI-terminated (i) and PbI2-terminated surfaces (ii). Reproduced with permission.40 Copyright 2016, Royal Society of Chemistry. | ||
Firstly, regarding MAPbI3, whether a single water molecule can diffuse from its surface to the interior was investigated through computational simulations, and the answer to this question is affirmative. Specifically, it was observed that the energy of the system when H2O is in region F is lower than when it is in region O, indicating that H2O tends to stay energetically favorable on MAPbI3. Simultaneously, the diffusion barrier from region O to F is merely 0.04 eV, suggesting a highly facile process with minimal resistance. This behavior can be attributed to the unique structural characteristics of MAPbI3, featuring a large framework composed of Pb and I atoms with relatively spacious interstices, despite the presence of organic MA+ molecules. Furthermore, H2O diffuses from region F to region S, with a diffusion barrier of 0.31 eV, slightly higher than the barrier from O1 to F1, yet still relatively small, thereby providing the opportunity for water molecules to diffuse from region F to region S. Additionally, compared to water molecules in region F, those in region S exhibit a lower system energy. During this process, H2O needs to overcome the [PbI6]4− framework. Throughout this entire process, due to the interaction between O in H2O and HN in MA+, the orientation of MA+ in regions F and S changes as water molecules diffuse.38 Koocher Nathan et al. discovered through simulations that on the PbI2-terminated surface, as the –NH3 groups in MA+ in region S are directed towards the surface, water molecules begin to stay within the plane. O interacts with the HN in MA+ in region S through hydrogen bonding, while the two HW atoms interact with the surface I−. The penetration of H2O increases the cavity size, weakening the octahedral rotation due to the interaction between O and HN, thereby opening adjacent hollow spaces.28 This indicates that water adsorption beneath the surface has a collective influence on neighboring structures. Once H2O fully enters region S, strong hydrogen bonds form between O and HN, and the barrier for this process is smaller than that for the reverse process, indicating that once H2O molecules penetrate the hollow sites, they cannot easily leave.
As the number of water molecules increases, how does the diffusion model differ from that of a single water molecule? For MAPbI3, simulation studies have found that under high humidity conditions, whether on the MAI-terminated or PbI2-terminated surfaces, water molecules exhibit strong adsorption at all sites, with hydrogen bonding interactions forming between water molecules.
Firstly, for the MAI-terminated surface, similar to the case of a single water molecule, when water molecules are located in region O, as depicted in Fig. 2(b)(i), the hydrogen bonding interaction between water and MAPbI3 is weak, and there is no significant structural change after water adsorption. Subsequently, water molecules diffuse to region F, as shown in Fig. 2(b)(ii), where the interaction between water and MAPbI3 strengthens, leading to disruption of the perovskite structure in region F. Occasionally, certain water molecules can further diffuse to region S, disrupting the structure of region S, as shown in Fig. 2(b)(iii). From an energy perspective, compared to region O, H2O tends to stay in region F. The changes in MA+ positions and the distortion of Pb–I bonds in region F increase with increasing n. The oxygen atom (O) of H2O forms strong bonding with the remaining exposed Pb, breaking the Pb–I bonds until the structure of region F is completely disrupted. Driven by the high coverage of water molecules, they can further diffuse from region F to S, causing greater distortion of Pb–I bonds.39 Similarly, Lei Zhang et al. using ab initio molecular dynamics (AIMD) simulations, found that the presence of H2O on the MAI surface within MAPbI3 not only altered the orientation of MA+ but also led to larger fluctuations in Pb–Pb distances and dihedral angles, as illustrated in Fig. 2(c)(i). This indicates that the MAI surface lacks chemical stability due to interactions with water molecules. Furthermore, by extracting snapshots from the AIMD trajectories, it was observed that over time, the ordered structure of the outermost layer of the MAI-terminated surface was disrupted, resulting in a relatively wide diffusion range of water molecules on the surface.40
For the PbI2-terminated surface, some simulation results differ from the MAI-terminated case. Although H2O exhibits strong interactions with the surface's Pb2+, the PbI2-terminated surface does not undergo decomposition, as illustrated in Fig. 2(c)(ii). This might be attributed to the PbI2-terminated surface possessing the shortest and consequently strongest Pb–I bonds. A few individual water molecules may enter region S, forming hydrogen bonds with MA+ and becoming trapped within the cavity for the remaining simulation time. However, during this process, the inorganic framework of the perovskite undergoes only slight deformation while maintaining its integrity. Yet, when a certain quantity of water molecules accumulates in region S, the perovskite lattice elongates perpendicular to the surface. This suggests that higher water concentrations could weaken the interlayer binding strength in halide perovskites, thereby promoting the decomposition of MAPbI3.40 As the quantity of water molecules further increases, decomposition also occurs on the PbI2-terminated surface. Claudia Caddeo et al.'s simulation results of water and MAPbI3 interactions reveal that at a 75% water coverage rate on the MAI surface, an I atom is attracted and lifted from the surface by surrounding H2O molecules.41 This forms a hydrogen bond network between water molecules and surface I atoms, causing each molecule's hydrogen bond donor count to approach its ideal value.42 Consequently, Pb–I bond rupture occurs, enabling two water molecules to infiltrate the crystal interior, marking the first step of water degradation.
Lu Ying-Bo et al. discovered that under light illumination, the lattice of MAPbI3 undergoes expansion due to the photo-induced lattice expansion effect, resulting in an increase in the Pb–I–Pb angles. This expansion reduces the hydrogen bonding interactions between the organic and inorganic components. It is well-known that the hydrogen bonding interactions between the [PbI6]4− framework and the MA molecules maintain the unique structure of MAPbI3.35,43 Therefore, when water molecules penetrate into the interior of MAPbI3 under light illumination, they encounter weaker resistance compared to non-illuminated conditions. In experimental studies, Nurul Ain Manshor et al. compared the air stability of MAPbI3 stored at >70% humidity under dark and light conditions.44 It was found that exposure to humidity in the dark also led to slight decomposition of the perovskite into its initial precursors. When exposed to light, the thin film exhibits a degradation rate over 10 times higher in the presence of humidity. This further underscores that the larger voids within the perovskite structure are also one of the contributing factors enabling water molecules to penetrate the interior of the perovskite.
2.4 Disruption of hydrogen bonds on perovskites by water molecules
In the preceding section, we discussed the process of water molecule penetration into the perovskite lattice, which involves lattice distortions and the breaking and reforming of hydrogen bonds. These events lead to a certain level of structural disruption within the perovskite. When water enters the interior of the perovskite, how exactly do hydrogen bond interactions between different atoms contribute to the structural deterioration of the perovskite? Furthermore, it has been observed in the study that during the interaction between H2O and the perovskite, a hydrated intermediate phase MAPbI3·H2O emerges prior to structural disruption. The total energy of this system is lower by 0.526 eV compared to the total energy of MAPbI3 and H2O molecules, implying that water can be readily adsorbed onto MAPbI3.45Dong Xu et al. investigated the neighbor interactions in MAPbI3 using natural bonding orbital (NBO) analysis and identified two main types of neighboring interactions: (1) 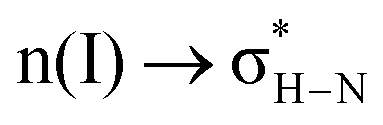 (interaction between the lone pair of I atom and the antibonding orbital of H–N bond) and (2)
(interaction between the lone pair of I atom and the antibonding orbital of H–N bond) and (2)  (interaction between the lone pair of I atom and the antibonding orbital of H–C bond).45 The stability energy of stereoelectronic interaction (1) is approximately 19 kcal mol−1, which is significantly higher than the stability energy of interaction (2). Upon encountering a water molecule, the weaker donor–acceptor interaction (2) within the MAPbI3 molecule is disrupted, and the two stereoelectronic interactions (1) are weakened, with the stability energy decreasing from ∼19 kcal mol−1 to ∼13 kcal mol−1. Furthermore, two additional interactions are established between MAPbI3 and water molecules: (3)
(interaction between the lone pair of I atom and the antibonding orbital of H–C bond).45 The stability energy of stereoelectronic interaction (1) is approximately 19 kcal mol−1, which is significantly higher than the stability energy of interaction (2). Upon encountering a water molecule, the weaker donor–acceptor interaction (2) within the MAPbI3 molecule is disrupted, and the two stereoelectronic interactions (1) are weakened, with the stability energy decreasing from ∼19 kcal mol−1 to ∼13 kcal mol−1. Furthermore, two additional interactions are established between MAPbI3 and water molecules: (3) 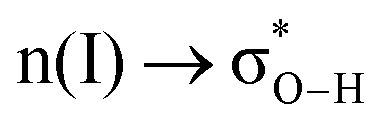 and (4)
and (4)  . The stability energy of the donor–acceptor interaction (4) between the lone pair of O atom in water and the antibonding orbital of N–H bond in the organic MA unit is 21.8 kcal mol−1, which is significantly higher than the stability energy between PbI3 and MA units. As a result, the presence of a water molecule decreases the stability of MAPbI3. Although one water molecule weakens the interactions between [PbI6]4− and MA units in MAPbI3, the structure of MAPbI3 is not completely destroyed. However, when two water molecules encounter a MAPbI3 molecule, the hydrogen bonds between [PbI6]4− and MA units are almost broken. The interaction (4) between the MA unit and H2O is much stronger than the interaction between [PbI6]4− and MA units, resulting in the complete decomposition of MAPbI3. Kakekhani Arvin et al. suggested that the polarization of water's lone pair induces more electron charge density closer to the proton-like H*N, leading to its interaction with the HN group on MA+.32 This interaction, in addition to hydrogen bonding, has a small covalent component manifested as a slight charge transfer from water to MA+. For a relatively low concentration of H2O
. The stability energy of the donor–acceptor interaction (4) between the lone pair of O atom in water and the antibonding orbital of N–H bond in the organic MA unit is 21.8 kcal mol−1, which is significantly higher than the stability energy between PbI3 and MA units. As a result, the presence of a water molecule decreases the stability of MAPbI3. Although one water molecule weakens the interactions between [PbI6]4− and MA units in MAPbI3, the structure of MAPbI3 is not completely destroyed. However, when two water molecules encounter a MAPbI3 molecule, the hydrogen bonds between [PbI6]4− and MA units are almost broken. The interaction (4) between the MA unit and H2O is much stronger than the interaction between [PbI6]4− and MA units, resulting in the complete decomposition of MAPbI3. Kakekhani Arvin et al. suggested that the polarization of water's lone pair induces more electron charge density closer to the proton-like H*N, leading to its interaction with the HN group on MA+.32 This interaction, in addition to hydrogen bonding, has a small covalent component manifested as a slight charge transfer from water to MA+. For a relatively low concentration of H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MA = 0.25, the water is oriented such that most of the highest occupied molecular orbital (HOMO) lone pairs interact with the H*N, lowering the system's energy, while hydrogen bonding exists between H2O and nearby I− anions. At this point, water causes some distortions in the lattice structure. When the ratio of H2O:MA ranges from 0 to 1, each H2O molecule binds to the –HN on MA+. Initially, each H2O perturbs (distorts) the inorganic framework to maximize hydrogen bonding attraction with I− and minimize electrostatic repulsion between O-2p lone pairs and I−. At H2O:MA = 1, as shown in Fig. 3(a), two water molecules form hydrogen bonds with each other, creating a connecting chain of (H2O–MA–H2O–H2O–MA), while the fourth water molecule interacts with two adjacent MA+ ions. At high water concentrations (H2O:MA > 1), due to continuous distortion of water molecules in the presence of MAPbI3, the system reaches an energetically favorable point for Pb bonding upon embedding. This results in the structure with broken octahedra becoming the actual ground state when sufficient water is present. Adequate water molecules form strong bonds with the ionic components of MAPbI3 and stabilize this distorted structure through a network of hydrogen bonds with other H2O molecules, as shown in Fig. 3(b). Experimental observations also indicate that MAPbI3 films are stable in dry air with humidity at or below 15%. However, in air with humidity as high as 60%, degradation can occur. During the degradation process, water initially decomposes MAPbI3 into crystalline MAI and PbI2, followed by the transformation of crystalline MAI into its amorphous form.
:MA = 0.25, the water is oriented such that most of the highest occupied molecular orbital (HOMO) lone pairs interact with the H*N, lowering the system's energy, while hydrogen bonding exists between H2O and nearby I− anions. At this point, water causes some distortions in the lattice structure. When the ratio of H2O:MA ranges from 0 to 1, each H2O molecule binds to the –HN on MA+. Initially, each H2O perturbs (distorts) the inorganic framework to maximize hydrogen bonding attraction with I− and minimize electrostatic repulsion between O-2p lone pairs and I−. At H2O:MA = 1, as shown in Fig. 3(a), two water molecules form hydrogen bonds with each other, creating a connecting chain of (H2O–MA–H2O–H2O–MA), while the fourth water molecule interacts with two adjacent MA+ ions. At high water concentrations (H2O:MA > 1), due to continuous distortion of water molecules in the presence of MAPbI3, the system reaches an energetically favorable point for Pb bonding upon embedding. This results in the structure with broken octahedra becoming the actual ground state when sufficient water is present. Adequate water molecules form strong bonds with the ionic components of MAPbI3 and stabilize this distorted structure through a network of hydrogen bonds with other H2O molecules, as shown in Fig. 3(b). Experimental observations also indicate that MAPbI3 films are stable in dry air with humidity at or below 15%. However, in air with humidity as high as 60%, degradation can occur. During the degradation process, water initially decomposes MAPbI3 into crystalline MAI and PbI2, followed by the transformation of crystalline MAI into its amorphous form.
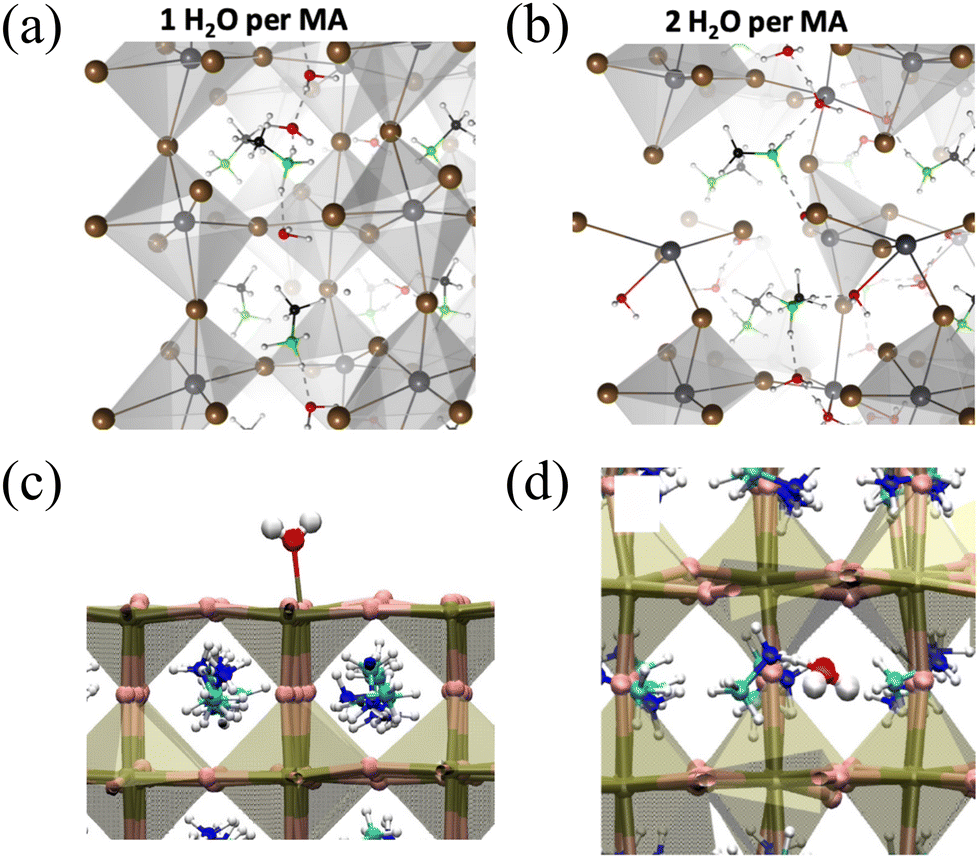 | ||
| Fig. 3 (a) and (b) Ground state water structure for different H2O:MA concentrations. Color code is O (red), H (white), C (black), N (green), I (brown), and Pb (dark gray). Reproduced with permission.32 Copyright 2019, American Institute of Physics. (c) Water molecule bound to the PbI2-terminated perovskite slab showing the formation of the Pb–O bond. (d) Water molecule infiltrated within the MAPI crystal showing the formation of the H–O link between the methylammonium and water. Red is O, white is H, pink is I, dark gold is Pb, cyan is C, and blue is N. Reproduced with permission.41 Copyright 2017, American Chemical Society. | ||
In contrast to liquid water, the case of water vapor is different. In the gas phase, individual water molecules placed in front of the surface are attracted to the crystal, with their oxygen eventually binding to the surface Pb atoms, forming polar Pb–O connections. Once the molecule is anchored in this manner, it does not undergo extensive diffusion on the nanosecond timescale at room temperature, thus water cannot disrupt the [PbI6]4− framework, as depicted in Fig. 3(c). Therefore, it can be concluded that water, as isolated molecules, gradually infiltrates and diffuses without disrupting the crystal until hydrated phases are formed. Conversely, as illustrated in Fig. 3(d), when water accumulates on the surface, irreversible lattice degradation may occur.41 These simulation results are consistent with the experimental observations by Aurélien M. A. Leguy et al. In their experiments, it was observed that when MAPbI3 single crystals and films were exposed to water vapor at room temperature, hydrated crystalline phases were formed.46 Importantly, this transition can be reversed under dry conditions. In contrast, when exposed to liquid water, MAPbI3 degrades and forms PbI2, which is an irreversible process. Under such conditions, degradation is highly efficient, and only minimal amounts of liquid, such as condensed droplets on the film surface or rapid immersion in water, have been observed.46,47 Similarly, Jong Un-Gi et al., through calculations, found that water interacts with PbX6 and MA through hydrogen bonding, with the interaction of water intercalation gradually increasing as X changes from I to Br and then Cl, while the hydration interaction remains nearly unchanged. Water-induced hydrogen bonding interactions significantly affect vacancy-mediated ion migration, as water intercalation in halide perovskites reduces the activation barrier. These results indicate that the degradation of perovskite solar cells (PSCs) following exposure to humidity arises from a multi-step process: the formation of water intercalation and hydrated compounds, followed by the decomposition of these compounds.36
3. Interaction of water molecules with defects in perovskite
In general, defect states are commonly present within perovskite materials. Defects in perovskite materials influence non-radiative carrier recombination and ion migration, thereby significantly constraining the energy conversion efficiency and stability of devices. The ionic nature of perovskites renders defect states charged, with deep-level defects arising from these charged defects being principal non-radiative recombination centers.48,49 Shallow-level defects, on the other hand, may not impact non-radiative recombination but can induce band bending, interface reactions, and phase separation, affecting carrier mobility.50–52 What then is the interaction between defects in perovskites and water molecules, and what are the resultant outcomes? To address this question, we provide the following summary and analysis.3.1 Defects in perovskites accelerate their degradation in water
In comparison to the PbI2-terminated surface, the PbI2-defective surface structure exhibits a distribution of H–O pairs within the range of 1.5–4 Å, indicating a stronger mutual interaction between H2O and the PbI2-defective surface. Consequently, under this interaction, the surface becomes disordered and tumultuous, with the absorbed H2O on the PbI2-defective surface able to traverse the PbI2 layers and reach the cavities formed between the outermost and the second PbI2 layers, forming hydrated compounds.40 Filippo De Angelis et al. also examined the potential role of surface defects in determining the stability of the water/perovskite interface.30 Computational analysis revealed that the PbI2-defective surface undergoes a rapid degradation process. During this process, two under-coordinated Pb atoms rapidly desorb from the perovskite surface, leading to the formation of solvated species. One of the Pb atoms is initially anchored to the perovskite surface through coordination with four I atoms, while a water molecule is added to the coordination sphere, as shown in Fig. 4(a). The other surface Pb atom is initially bound to three I atoms on the perovskite surface and two interface water molecules, as depicted in Fig. 4(a). Throughout the Car–Parrinello Molecular Dynamics (CPMD) trajectory, both surface Pb atoms depart from the perovskite surface, forming octahedral [PbI2(H2O)4] and [PbI(H2O)5]+ complexes in solution. In conclusion, defects on the perovskite surface may potentially trigger further degradation of MAPbI3. Hence, the growth of defect-free crystals and films seems crucial for stabilizing perovskite materials and maintaining their integrity in the presence of humidity.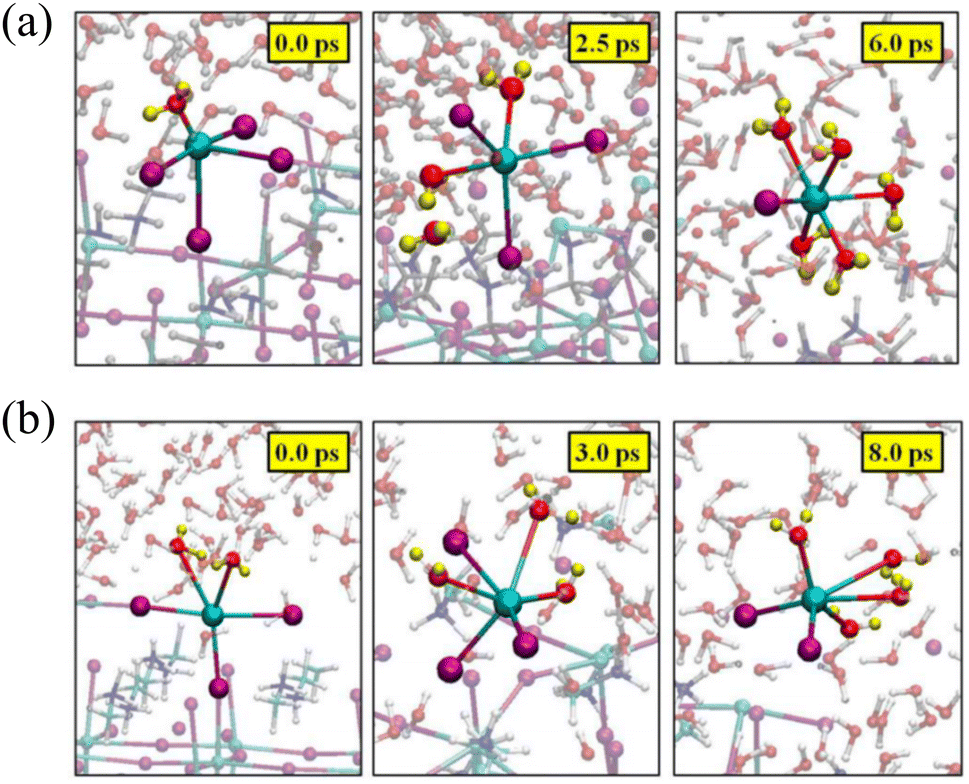 | ||
| Fig. 4 Representative geometrical structures of the formation of (a) [PbI2(H2O)4] and (b) [PbI(H2O)5]+ complexes. Reproduced with permission.30 Copyright 2015, American Chemical Society. | ||
Accelerated degradation of the perovskite structure invariably leads to deterioration in device performance. Pezhman Darvishzadeh et al. modeled the variation of open circuit voltage (VOC) over time, demonstrating that ion migration and PbI2 defects are the primary causes for the degradation of VOC exposed to humidity.53 Sohrabpoor Hamed et al. also concluded that the defect profile plays a pivotal role in the instability of the device.54
3.2 Influence of water molecules on defect formation
In their study, Run Long et al. investigated how the interaction between lead vacancies and water influences non-radiative electron–hole recombination in MAPbI3.55 Computational results indicate that the presence of water reduces the formation energy of both neutral and charged lead vacancies to values below −2 eV, facilitating their spontaneous creation. Notably, MAPbI3 with lead vacancies introduces localized shallow energy levels for holes, which merge with the valence band edge, attenuating the electron–phonon coupling between ground and excited states. Consequently, this leads to a reduction in non-radiative electron–hole recombination rates. When water molecules coexist with vacancies, unsaturated iodine atoms near lead vacancies form iodine dimers, introducing deep energy levels for electron capture. This accelerates electron–hole recombination, shortening the recombination time to approximately one-fourth of the original MAPbI3 system. Furthermore, they found through calculations of electron–hole recombination times that relative humidity plays a significant role in determining the excited state lifetime.54 Simulations suggest that moderate humidity prolongs the excited state lifetime, while high humidity reduces it. A small amount of water localizes electrons on the surface, simultaneously avoiding deep trap states and decoupling electrons from holes. Additionally, the quantum coherence time decreases. Both effects reduce the nonradiative charge recombination. Under high humidity conditions, water aggregates on the surface of perovskite, forming a continuous hydrogen bonding network. The interaction between perovskite and water is weaker than water–water interactions, thus the disturbance caused by continuous water layers on the perovskite surface is less pronounced compared to isolated water molecules. With increasing water content, the delocalization of electrons into the bulk and the overlap of electron–hole pairs recover, resulting in a nonadiabatic coupling that exceeds the value of the bare surface, accelerating recombination. These theoretical studies provide valuable insights into seemingly contradictory experimental phenomena, with some measurements indicating that increased humidity enhances the excited state lifetime of MAPbI3, thereby improving the performance of solar cells.12Yun-Hyok Kye et al. investigated the defect processes in MAPbI3, water-intercalated MAPbI3_H2O, and monohydrated phase MAPbI3·H2O.56 They considered the formation energies of isolated and clustered vacancy defects with different charge states under iodine-rich and iodine-poor conditions. The study found that the formation of VPbI2 from its isolated vacancy defect is spontaneous. However, due to the significantly lowered kinetic barrier for I− migration upon hydration, the concentration of VI should be reduced to prevent this formation, which can be achieved by imposing iodine-rich conditions. In hydrated compounds, the formation of individual point defects VI and VMA is easier than that of VMAI. Therefore, during decomposition, I2 or CH3NH2 or HI can be formed instead of MAI. Unlike in pristine MAPbI3, all vacancy defects lead to the creation of deep transition energy levels in hydrated compounds. This is attributed to electrostatic interactions with water molecules, suggesting the degradation of halide lead perovskites when exposed to humidity.
Yun-Hyok Kye et al. also calculated the thermodynamic transition levels ε(q1/q2) between defect states of different charge states q1 and q2. Fig. 5(a) depicts the potential transition levels along with the relative positions of MAPbI3_H2O and MAPbI3·H2O with respect to MAPbI3. Water molecules inserted through the membrane surface extract electrons, causing the valence band maximum (VBM) to shift to lower energies and the conduction band maximum (CBM) to higher energies, consequently altering the bandgap from the original phase to the water-intercalated and monohydrated phases. Evidently, in the case of MAPbI3, all vacancy defects possess shallow transition levels, while in the context of hydrated compounds, vacancies exhibit deep trap behavior. This can promote charge carrier recombination, leading to the degradation of solar cell performance. Specifically, in MAPbI3, VI and VMA act as shallow donors and acceptors, as evidenced by their respective transition levels ε(1+/0) and ε(0/1−) near the CBM and VBM. In the case of hydrated compounds, transition levels are situated deep within the bandgap, with the transition level for VMA not far from the valence band. Notably, VPb in MAPbI3 lacks a transition level within the band (ε(0/2−)), but in hydrated compounds, it could possess two deep transition levels, ε(0/1−) and ε(1−/2−). Both VMAI (with two transition levels) and VPbI2 (with two/four transition levels in pristine/hydrated MAPbI3) exhibit such characteristics.
 | ||
| Fig. 5 (a) Band alignment and thermodynamic transition levels in MAPbI3, water-intercalated MAPbI3_H2O, and monohydrate MAPbI3·H2O, where deep-lying Pb 5d levels are used as a reference for the VBM and CBM of each phase. Reproduced with permission.56 Copyright 2018, American Chemical Society. (b) Minimum energy level of π*2p of O2 absorbed at the interstitial space on a PbI2-terminated surface as a function of water content. Reproduced with permission.59 Copyright 2019, John Wiley and Sons. | ||
Water not only influences defect formation but also passivates defects. Water molecules passivate deep-level defects by forming hydrogen bonds with I atoms in the perovskite structure.57 Wenke Zhou et al. verified the defect passivation effect of water molecules on MAPbI3 thin films using thermal admittance spectroscopy. Initially, the authors measured control MAPbI3 perovskite films without water treatment and identified deep-level defects at 0.295 eV, typically associated with Pb–I anti-site defects. After appropriate water treatment, the deep-level position shifted closer to the conduction band, indicating effective passivation of Pb–I anti-site defects by water molecules. Upon further drying the water, the defect level position deepened. This outcome suggests that an appropriate amount of water can effectively passivate deep-level defects, and the process is reversible.
Apart from 3D perovskites, H2O can also passivate defects in 2D perovskites. Xinlu Guo et al., through DFT calculations, discovered that a suitable number of water molecules can preferentially occupy VI defects in Dion–Jacobson (DJ) phase perovskites, reducing trap centers, forming hydrogen bonds with surrounding atoms, enhancing the structural stability, and making them more robust. Concurrently, experimental observations showed that devices prepared under humid conditions exhibit improved photovoltaic performance and stability.58 However, compared to 3D perovskites, there has been limited research on the interaction of water and defects in 2D perovskites, and theoretical investigations in this context are particularly lacking. Conducting research on issues related to 2D perovskites could significantly promote the advancement of this field.
Light- and oxygen-induced degradation of MAPbI3 is a primary factor limiting the lifespan of PSCs. Yixin Ouyang et al. revealed through calculations that due to the weak affinity between oxygen and MA, as well as the capture of iodine vacancies, O2 cannot spontaneously diffuse from the surface into the internal lattice of MAPbI3.59 The slow diffusion properties lead to photodegradation concentrating on the perovskite surface. Induced by the MAPbI3 surface, the degenerate π*2p energy level of O2 slightly splits into two bands. When H2O approaches O2, the π*2p energy level of O2 decreases, moving it closer to the valence band of MAPbI3. The pronounced polarization effect of water enhances oxygen's electron affinity, causing a reduction in the O2 energy level. Changes in H2O content also impact this process. With more H2O molecules progressively surrounding adsorbed O2, the oxygen state shifts further downward. For O2 adsorbed at hollow sites on the PbI2-terminated surface, the transition of oxygen states is significantly influenced by water content. As illustrated in Fig. 5(b), as water content increases, more charge is transferred from surface I atoms to O2 due to water molecules. This results in a deceleration of the reduction in the O2 energy level. H2O acts as a bridge on VMA points, facilitating the transmission of changes between I atoms and absorbed O2. In conclusion, stable perovskite thin films necessitate crystal surfaces terminated with defect-free MAI.
4. The influence of water molecules on the morphology and performance of perovskite devices
The inevitable interactions between water molecules and perovskites also inevitably influence their fabrication processes, the resulting film quality, and device performance. Currently, experimental evidence has indicated that specific humidity conditions favor the preparation of perovskite thin films.60,61 In light of these findings, it is imperative to elucidate the role of water in this context. We present the following summary and analysis.4.1 The effect of moderate water on phase transitions in FAPbI3 and CsPbX3
Unlike MAPbI3, for FAPbI3, it is prone to undergo a phase transition from the α phase to the δ phase under humid atmospheric conditions. How do water molecules impact this phase transition process? The hydrogen bond interactions between water molecules and the perovskite structure of FAPbI3 are considered the primary factors influencing its phase transition.62,63 Dongxu Lin et al. through DFT calculations, demonstrated that water molecules can form both O⋯H–N and I⋯H–O hydrogen bonds with FAPbI3. Within a certain concentration range, this leads to anomalous contraction of the α phase and lowers the energy barrier between the two phases' transition states. At an appropriate water concentration, when the energy barrier is minimized, the α phase can easily and completely transform into the δ phase, and vice versa. The entire phase transition process is depicted in Fig. 6(a). Concurrently, experimental findings also reveal that under high humidity conditions, pure δ-FAPbI3 can be obtained, and further annealing produces high-quality α-FAPbI3 films.64 Chunqing MA et al., based on XPS and EXAFS data, concluded that water molecules adsorb on the surface of FAPbI3 perovskite through the formation of –OH groups, resulting in structural distortions with weakened Pb–I bonds. These distortion and weakening processes promote the δ phase transition at room temperature. Further theoretical calculations of water molecule interaction energy with the (100) and (111) crystal facets were performed. The adhesion energies for the two surfaces with H2O were found to be 93.08 and 85.12 mN m−1, respectively. Thus, the (100) surface is more favorable for H2O adhesion in terms of energy, indicating its susceptibility to degradation caused by humidity. Therefore, when α-FAPbI3 is exposed to H2O, the (111) surface is thermodynamically more stable.65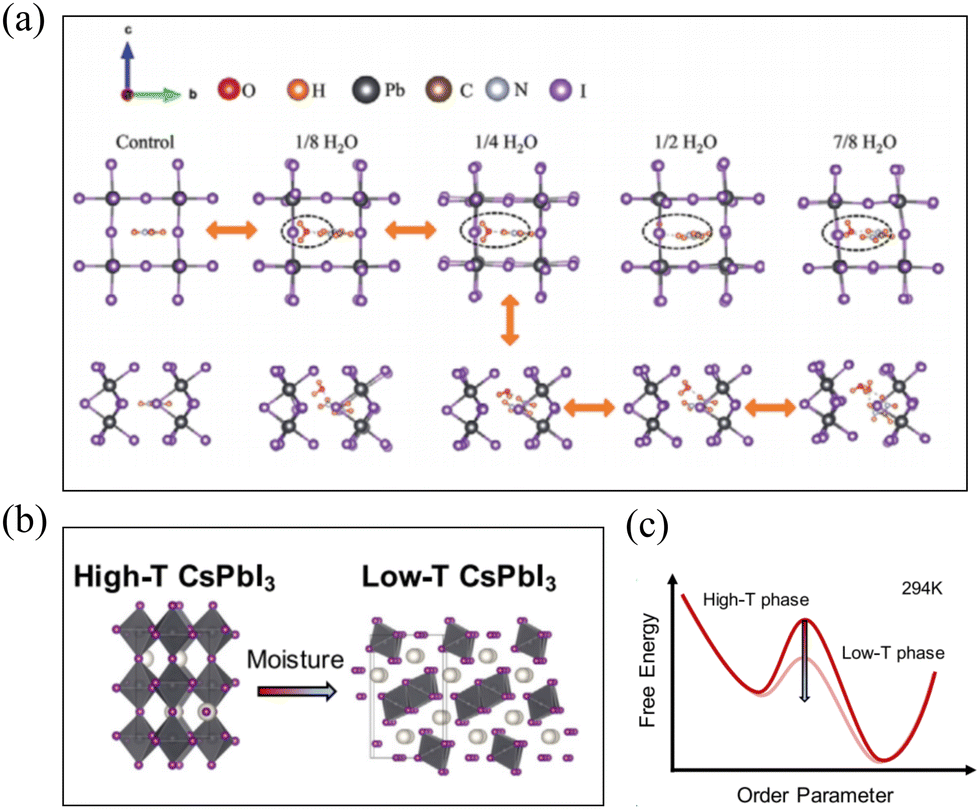 | ||
| Fig. 6 (a) Lattice evolution of α- and δ-FAPbI3 with increasing H2O concentration and an indication of phase transition path. Reproduced with permission.64 Copyright 2022, John Wiley and Sons. (b) Schematic diagrams of high-T CsPbI3 (left, orthorhombic structure with three-dimensionally connected corner-sharing octahedra) to low-T CsPbI3 (right, orthorhombic structure with one-dimensional chains of edge-sharing octahedra) phase transformation triggered by moisture. (c) Illustrative energy diagram of CsPbI3 (thick red line) comparison with common moisture induced modification (faint red line) of the energy scheme. Reproduced with permission.69 Copyright 2021, Elsevier. | ||
Similar to FAPbI3, CsPbI3 is also prone to undergo a phase transition from the high-temperature (high-T) γ phase to the low-temperature (low-T) δ phase under atmospheric conditions,66–68 as shown in Fig. 6(b). Zhenni Lin et al. employed molecular dynamics simulations and found that when a thin layer of water contacts the metastable perovskite surface, the large solvation enthalpy of I− and its associated low formation energy of vacancies enhance the halide vacancies at the water–perovskite interface. These halide vacancies can be considered as defect sites that lower the activation energy barrier (PT barrier) between the metastable γ- and stable δ- of CsPbI3, as illustrated in Fig. 6(c), effectively catalyzing the phase transition.
Further increasing the relative humidity (RH) results in an exponential increase in vacancy equilibrium concentration. Based on this, it can be predicted that elevated RH levels lead to an exponential increase in the nucleation rate of δ phase CsPbI3.69 Moreover, Kyoung E. Kweon et al. utilized DFT calculations to study the water-induced effects of H2O molecules and their dissociation species (H+/OH−) during the γ- to δ-transition of CsPbI3. They discovered that the reaction energy barrier is significantly lowered when H2O or OH- is present within CsPbI3, while the influence of H+ on kinetics can be neglected.70 Corresponding phenomena have also been experimentally observed and quantitatively validated the computational conclusions. Daniel B. Straus et al. experimentally observed the vapor-induced transformation of γ-CsPbI3 to δ-CsPbI3 through synthetic variations and in situ PXRD measurements.71 Similarly, for CsPbBr3, Jiayu Di et al. found through calculations that the presence of water during crystal growth reduces the formation energy of γ-CsPbBr3 and promotes the reaction in that direction.72 This can explain recent observations that atmospheric humidity accelerates the phase transition kinetics without significantly altering the transition enthalpy.
4.2 Optimization of crystallization
Beyond its impact on phase transitions, water molecules can influence the crystallization process of perovskites through specific interactions. Weili Fan et al., through DFT simulations, proposed that in the synthesis of FAPbI3, the introduction of a certain amount of H2O facilitates the ionization of FAI due to the high solubility of the organic salt FAI in water.73 This leads to enhanced mobility of FA+ and I− ions. The activation energy for FAPbI3 synthesis, both with and without water, was computed. The presence of water resulted in a negative activation energy, indicating a spontaneous reaction. Water also affects PbI2, as revealed by calculations of water molecule adsorption on the PbI2(001) and (102) surfaces. Water molecules tend to adsorb on the (102) surface, retarding its growth and thereby promoting oriented growth of the PbI2(001) plane. Similarly, Kaikai Liu et al. proposed that in hybrid organic–inorganic perovskites, water molecules enhance the mass mobility of hydrated salts through hydrogen bonding with organic salts.74 This facilitates greater penetration of organic salts into the PbI2 thin film, generating more crystallization sites for perovskite formation and ultimately favoring the development of high-quality perovskite films.75 A schematic representation of its crystal structure is shown in Fig. 7(a). Experimental observations also corroborate that under controlled humidity conditions, modulation of the crystallization process through water manipulation leads to the fabrication of higher quality perovskite films with reduced defects.12,76–78 Furthermore, Yangyang Du et al. found that after participating in the completion of crystallization regulation, water molecules migrate to the grain boundaries of perovskite thin films. This facilitates effective passivation of crystallization defects, ultimately leading to the formation of high-quality perovskite thin films.79 | ||
| Fig. 7 (a) Schematic illustration of H2O in regulating the crystallization process. Reproduced with permission.75 Copyright 2022, Springer Nature. (b) The PDOSs for three surfaces. Reproduced with permission.40 Copyright 2016, Royal Society of Chemistry. (c) Illustration of the perovskite–moisture interaction, leading to electron transfer, hydration, and degradation. Reproduced with permission.81 Copyright 2019, American Chemical Society. | ||
4.3 Impact of water on perovskite band structure and photoelectric performance
The interaction between H2O and perovskite materials generally has an impact on the electronic structure of perovskites and the optoelectronic performance of devices. Lei Zhang et al. simulated the effects of H2O on the band structure of MAPbI3.40 When water is adsorbed on the surface of the perovskite, for both the water-absorbing MAI-terminated and PbI2-terminated surfaces, the calculated band gaps fluctuate around 1.879 eV and 1.578 eV, corresponding to standard deviations of 0.210 eV and 0.094 eV, respectively. Relative to the bare surface, induced by water molecules, the strongly surface-reconstructed water-absorbed MAI-terminated surface exhibits a broader distribution of energy gaps, while its average gap remains nearly identical to that of the dry surface. Furthermore, as shown in Fig. 7(b), the composition of the VBM and CBM is not predominantly derived from the outermost layers’ states. For the PbI2-terminated surface, the main contribution to the VBM comes from the outermost layer of the surface. Charge transfer induced by water molecules adsorbed on the surface causes the surface's VBM to shift downward, resulting in a wider band gap. The water-absorbing PbI2-terminated surface's PDOS remains similar to that of the bare surface, due to the preservation of surface structure. On the water-absorbing MAI-terminated surface, surface reconstruction generates shallow surface states, which might capture photo-generated holes. These surface states are confined to the vicinity of the VBM without intruding into the bandgap of MAPbI3, implying intrinsic defect tolerance in MAPbI3.Andrew M. Rappe et al. discovered that when the H2O:MA ratio typically ranges from 0 to 1, and the addition of water generally leads to an increase in the bandgap.32,80 This gap variation is caused by structural deformation and lattice expansion. Generally, the introduction of water reduces the connectivity of the primary structure of MAPbI3, weakening bonds, narrowing bands, and increasing the bandgap. When the ratio of H2O:MA falls within the range of 1 to 2, water molecules exhibit a “chemical” effect in determining the bandgap. At higher water concentrations such as H2O:MA = 2, the orbitals of water more significantly contribute to the VBM and CBM due to increased covalency with the inorganic framework's I− and Pb2+. This is a consequence of the fractured octahedra, leading to direct binding of water with the inorganic framework (i.e., Pb2+).
The interaction of water molecules alters the geometry and electronic structure of MAPbI3, and undoubtedly influences the material's optical properties. Lei Zhang et al. computed snapshots of the absorption spectra for molecular dynamics (MD) trajectories, and at the beginning of the simulation, the initial structure exhibited optical responses similar to the bulk phase.40 As the simulation progressed and water molecules penetrated the interior layers of the MAI-terminated surface, significant changes in the perovskite structure occurred, resulting in a pronounced decrease in absorption in the visible range, particularly around 550 nm. However, the situation is slightly different on the water-adsorbed PbI2-terminated surface. Initially, the absorption decreases significantly and then remains stable over time, indicating the relative stability of the PbI2-terminated surface. The evolution of the absorption with time suggests that interacting water molecules gradually disrupt the perovskite structure, subsequently diminishing the performance of PSC devices. Ali Kachmar et al. calculated the absorption spectra of MASnI3 before and after water infiltration into the crystal structure, showing that water molecules enhance the absorption of photon energies within the range of approximately 1–2 eV. However, as the photon energy further increases, the absorption coefficient of the monohydrated perovskite system decreases. Moreover, the introduction of water molecules into the system eliminates two distinct peaks in the absorption curve at 2.5 and 3.5 eV. Only at photon energies starting from 4.5 eV (i.e., after the maximum absorption peak) do the absorption coefficients become nearly equal.31
Md Azimul Haque et al. fabricated MAPbI3 microstrips on a flexible substrate and observed a four-order-of-magnitude decrease in the microstrip resistance as the relative humidity levels varied from 10% to 95%. Additionally, the photocurrent response decreased with increasing humidity, and at a relative humidity level of 85%, the perovskite device no longer exhibited a photoresponse. The authors demonstrated a potential scenario for the interaction between perovskite and humidity. As shown on the left side in Fig. 7(c), moderate humidity or short-term exposure to high humidity conditions led to electron transfer from water molecules to the perovskite lattice. However, as depicted on the right side in Fig. 7(c), prolonged exposure to high humidity conditions resulted in degradation through different pathways in the perovskite.81
Thermalization of hot carriers is one of the main sources of efficiency loss in solar cells. Ali Kachmar et al. investigated the thermal electron cooling in both MASnI3 and MASnI3·H2O within the tetragonal phase. In the case of the monohydrated perovskite, the cooling time is less than 1 ps. The overall trend is that the cooling process of hot electrons is faster in the MASnI3·H2O system compared to the MASnI3 system. For energies higher than 2 eV, the cooling time of hot electrons appears to be less influenced by water molecules. This is because, in the case of the monohydrated system, the electron–phonon coupling (coupling between states in the conduction band) is stronger compared to the pristine system.31
5. Conclusions and outlook
In conclusion, the primary interaction between water molecules and perovskites is through hydrogen bonding between H2O and the perovskite. This interaction has been summarized as various effects on the perovskite lattices and defects, which ultimately affect the crystal quality and the optoelectronic performance of devices. With an understanding of the diverse and significant effect of water molecules, component doping,82,83 surface modification,84,85 optimizing photovoltaic device encapsulation,86 and process optimization87,88 have been employed to enhance the stability of perovskite materials and devices. For example, Hongjun Chen et al. achieved robust and durable monolithic perovskite-based devices with the aid of rare-earth co-catalysts and effective charge transfer engineering at the co-catalyst interface. This resulted in a single-piece photoelectrode displaying a remarkable total solar-to-hydrogen efficiency of 8.54% without external assistance.89We observe that water's influence on perovskites presents both beneficial and detrimental aspects. The advantageous effects predominantly manifest at lower water concentrations, while escalated water concentrations or typical environmental conditions tend to elicit adverse repercussions on perovskite stability. This culminates in material degradation, significantly compromising device performance and ultimately impeding the commercialization trajectory of perovskite devices. Consequently, a more profound exploration of the interactions and mechanisms between water and perovskites is imperative. This endeavor aims to guide experimental endeavors in effectively enhancing perovskite humidity stability, thereby expediting their commercialization endeavors.
By employing these methods, the humidity stability of perovskite optoelectronic devices has been improved to a certain extent. However, further optimization of these approaches is still required to enhance stability under various environmental conditions.
From the perspective of theoretical calculations, there are several challenges in studying the influence of water molecules on perovskites:
(1) Complex interactions: the interactions between perovskites and water involve numerous chemical bonds and forces, making theoretical calculations highly complex. Consideration of factors such as charges, ion radii, bond lengths, and bond angles is necessary, posing significant challenges for computer simulations and theoretical analyses.
(2) Dynamics: the interaction between perovskites and water molecules exhibits high dynamics. Water molecules can form various complexes on the surface and within the perovskite, leading to continuous structural changes. This implies that theoretical calculations need to account for a large number of structural configurations and interactions, increasing computational difficulties.
(3) Time scale: the influence of water on perovskites is a long-time scale process under realistic conditions. Theoretical calculations require simulation over sufficiently long time-scales to capture various possible interactions and structural changes. This may result in computationally intensive tasks that require substantial computing resources.
(4) Temperature: in practical applications, perovskites may be exposed to different temperature conditions. These conditions can affect the interactions between water molecules and perovskites, thereby influencing their stability. When performing theoretical calculations, these factors need to be considered to accurately simulate real-world scenarios.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grants No. 62174072, 61674070, 12104345 and 11804117), the Natural Science Foundation of Guangdong Province (Grant No. 2022A1515010276) and the Uniqueness and Innovation Projects for the Universities in Guangdong Province (2022KTSCX010).References
- Z. Lu, X. Xu, Y. Gao, Z. Wu, A. Li, Z. Zhan, Y. Qu, Y. Cai, X. Huang, J. Huang, Z. Zhang, T. Luo, L. Peng, P. Liu, T. Shi and W. Xie, The effect of spacer cations on optoelectronic properties of two-dimensional perovskite based on first-principles calculations, Surf. Interfaces, 2022, 34, 102343 CrossRef CAS
.
- Best Research-Cell Efficiency Chart, https://www.nrel.gov/pv/assets/pdfs/best-research-cell-efficiencies.pdf.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS
- Y. Han, S. Meyer, Y. Dkhissi, K. Weber, J. M. Pringle, U. Bach, L. Spiccia and Y.-B. Cheng, Degradation observations of encapsulated planar CH3NH3PbI3 perovskite solar cells at high temperatures and humidity, J. Mater. Chem. A, 2015, 3, 8139–8147 RSC
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, Investigation of CH3NH3PbI3 Degradation Rates and Mechanisms in Controlled Humidity Environments Using in Situ Techniques, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed
- T. M. Koh, K. Thirumal, H. S. Soo and N. Mathews, Multidimensional Perovskites: A Mixed Cation Approach Towards Ambient Stable and Tunable Perovskite Photovoltaics, ChemSusChem, 2016, 9, 2541–2558 CrossRef CAS
- Q. Shan, J. Li, J. Song, Y. Zou, L. Xu, J. Xue, Y. Dong, C. Huo, J. Chen, B. Han and H. Zeng, All-inorganic quantum-dot light-emitting diodes based on perovskite emitters with low turn-on voltage and high humidity stability, J. Mater. Chem. C, 2017, 5, 4565–4570 RSC
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Thermal degradation of CH3NH3PbI3 perovskite into NH3 and CH3I gases observed by coupled thermogravimetry–mass spectrometry analysis, Energy Environ. Sci., 2016, 9, 3406–3410 RSC
- W.-C. Lin, H.-Y. Chang, K. Abbasi, J.-J. Shyue and C. Burda, 3D In Situ ToF-SIMS Imaging of Perovskite Films under Controlled Humidity Environmental Conditions, Adv. Mater. Interfaces, 2017, 4, 1600673 CrossRef
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-B. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Interface engineering of highly efficient perovskite solar cells, Science, 2014, 345, 542–546 CrossRef CAS PubMed
- S. R. Raga, L. K. Ono and Y. Qi, Rapid perovskite formation by CH3NH2 gas-induced intercalation and reaction of PbI2, J. Mater. Chem. A, 2016, 4, 2494–2500 RSC
- G. E. Eperon, S. N. Habisreutinger, T. Leijtens, B. J. Bruijnaers, J. J. van Franeker, D. W. deQuilettes, S. Pathak, R. J. Sutton, G. Grancini, D. S. Ginger, R. A. J. Janssen, A. Petrozza and H. J. Snaith, The Importance of Moisture in Hybrid Lead Halide Perovskite Thin Film Fabrication, ACS Nano, 2015, 9, 9380–9393 CrossRef CAS PubMed
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Formamidinium and Cesium Hybridization for Photo- and Moisture-Stable Perovskite Solar Cell, Adv. Energy Mater., 2015, 5, 1501310 CrossRef
- L. Ling, S. Yuan, P. Wang, H. Zhang, L. Tu, J. Wang, Y. Zhan and L. Zheng, Precisely Controlled Hydration Water for Performance Improvement of Organic–Inorganic Perovskite Solar Cells, Adv. Funct. Mater., 2016, 26, 5028–5034 CrossRef CAS
- X. Zhang, X. Bai, H. Wu, X. Zhang, C. Sun, Y. Zhang, W. Zhang, W. Zheng, W. W. Yu and A. L. Rogach, Water-Assisted Size and Shape Control of CsPbBr3 Perovskite Nanocrystals, Angew. Chem., Int. Ed., 2018, 57, 3337–3342 CrossRef CAS PubMed
- Z. Liu, L. Qiu, L. K. Ono, S. He, Z. Hu, M. Jiang, G. Tong, Z. Wu, Y. Jiang, D.-Y. Son, Y. Dang, S. Kazaoui and Y. Qi, A holistic approach to interface stabilization for efficient perovskite solar modules with over 2,000-hour operational stability, Nat. Energy, 2020, 5, 596–604 CrossRef CAS
- J. Peng, C. Q. Xia, Y. Xu, R. Li, L. Cui, J. K. Clegg, L. M. Herz, M. B. Johnston and Q. Lin, Crystallization of CsPbBr3 single crystals in water for X-ray detection, Nat. Commun., 2021, 12, 1531 CrossRef CAS PubMed
- B. Kim and S. I. Seok, Molecular aspects of organic cations affecting the humidity stability of perovskites, Energy Environ. Sci., 2020, 13, 805–820 RSC
- B. Chen, S. Wang, Y. Song, C. Li and F. Hao, A critical review on the moisture stability of halide perovskite films and solar cells, Chem. Eng. J., 2022, 430, 132701 CrossRef CAS
- C. Zhou, A. B. Tarasov, E. A. Goodilin, P. Chen, H. Wang and Q. Chen, Recent strategies to improve moisture stability in metal halide perovskites materials and devices, J. Energy Chem., 2022, 65, 219–235 CrossRef CAS
- J. Huang, S. Tan, P. D. Lund and H. Zhou, Impact of H2O on organic–inorganic hybrid perovskite solar cells, Energy Environ. Sci., 2017, 10, 2284–2311 RSC
- C.-X. Zhang, T. Shen, D. Guo, L.-M. Tang, K. Yang and H.-X. Deng, Reviewing and understanding the stability mechanism of halide perovskite solar cells, InfoMat, 2020, 2, 1034–1056 CrossRef CAS
- M. Li, H. Li, J. Fu, T. Liang and W. Ma, Recent Progress on the Stability of Perovskite Solar Cells in a Humid Environment, J. Phys. Chem. C, 2020, 124, 27251–27266 CrossRef CAS
- M. F. Mohamad Noh, N. A. Arzaee, I. N. Nawas Mumthas, N. A. Mohamed, S. N. F. Mohd Nasir, J. Safaei, A. R. B. M. Yusoff, M. K. Nazeeruddin and M. A. Mat Teridi, High-humidity processed perovskite solar cells, J. Mater. Chem. A, 2020, 8, 10481–10518 RSC
- Y. Zhang, A. Kirs, F. Ambroz, C.-T. Lin, A. S. R. Bati, I. P. Parkin, J. G. Shapter, M. Batmunkh and T. J. Macdonald, Ambient Fabrication of Organic–Inorganic Hybrid Perovskite Solar Cells, Small Methods, 2021, 5, 2000744 CrossRef CAS
- R. Azmi, S. Zhumagali, H. Bristow, S. Zhang, A. Yazmaciyan, A. R. Pininti, D. S. Utomo, A. S. Subbiah and S. De Wolf, Moisture-Resilient Perovskite Solar Cells for Enhanced Stability, Adv. Mater., 2023, 2211317 CrossRef
- Q. Li, Z. Chen, I. Tranca, S. Gaastra-Nedea, D. Smeulders and S. Tao, Compositional effect on water adsorption on metal halide perovskites, Appl. Surf. Sci., 2021, 538, 148058 CrossRef CAS
- N. Z. Koocher, D. Saldana-Greco, F. Wang, S. Liu and A. M. Rappe, Polarization Dependence of Water Adsorption to CH3NH3PbI3 (001) Surfaces, J. Phys. Chem. Lett., 2015, 6, 4371–4378 CrossRef CAS PubMed
- G. Xie, L. Xu, L. Sun, Y. Xiong, P. Wu and B. Hu, Insight into the reaction mechanism of water, oxygen and nitrogen molecules on a tin iodine perovskite surface, J. Mater. Chem. A, 2019, 7, 5779–5793 RSC
- E. Mosconi, J. M. Azpiroz and F. De Angelis, Ab Initio Molecular Dynamics Simulations of Methylammonium Lead Iodide Perovskite Degradation by Water, Chem. Mater., 2015, 27, 4885–4892 CrossRef CAS
- A. Kachmar, G. Berdiyorov and M. E.-A. Madjet, Effect of Water on the Structural, Optical, and Hot-Carrier Cooling Properties of the Perovskite Material MASnI3, J. Phys. Chem. C, 2019, 123, 4056–4063 CrossRef CAS
- A. Kakekhani, R. N. Katti and A. M. Rappe, Water in hybrid perovskites: Bulk MAPbI3 degradation via super-hydrous state, APL Mater., 2019, 7, 041112 CrossRef
- C. Yi, J. Luo, S. Meloni, A. Boziki, N. Ashari-Astani, C. Grätzel, S. M. Zakeeruddin, U. Röthlisberger and M. Grätzel, Entropic stabilization of mixed A-cation ABX3 metal halide perovskites for high performance perovskite solar cells, Energy Environ. Sci., 2016, 9, 656–662 RSC
- K. Poorkazem and T. L. Kelly, Compositional Engineering To Improve the Stability of Lead Halide Perovskites: A Comparative Study of Cationic and Anionic Dopants, ACS Appl. Energy Mater., 2018, 1, 181–190 CrossRef CAS
- Y.-B. Lu, W.-Y. Cong, C. Guan, H. Sun, Y. Xin, K. Wang and S. Song, Light enhanced moisture degradation of perovskite solar cell material CH3NH3PbI3, J. Mater. Chem. A, 2019, 7, 27469–27474 RSC
- U.-G. Jong, C.-J. Yu, G.-C. Ri, A. P. McMahon, N. M. Harrison, P. R. F. Barnes and A. Walsh, Influence of water intercalation and hydration on chemical decomposition and ion transport in methylammonium lead halide perovskites, J. Mater. Chem. A, 2018, 6, 1067–1074 RSC
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Chemical Management for Colorful, Efficient, and Stable Inorganic–Organic Hybrid Nanostructured Solar Cells, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed
- W. Zhu, T. Yao, J. Shen, W. Xu, B. Gong, Y. Wang and J. Lian, In situ Investigation of Water Interaction with Lead-Free All Inorganic Perovskite (Cs2SnIxCl6–x), J. Phys. Chem. C, 2019, 123, 9575–9581 CrossRef CAS
- C.-J. Tong, W. Geng, Z.-K. Tang, C.-Y. Yam, X.-L. Fan, J. Liu, W.-M. Lau and L.-M. Liu, Uncovering the Veil of the Degradation in Perovskite CH3NH3PbI3 upon Humidity Exposure: A First-Principles Study, J. Phys. Chem. Lett., 2015, 6, 3289–3295 CrossRef CAS
- L. Zhang, M. G. Ju and W. Liang, The effect of moisture on the structures and properties of lead halide perovskites: a first-principles theoretical investigation, Phys. Chem. Chem. Phys., 2016, 18, 23174–23183 RSC
- C. Caddeo, M. I. Saba, S. Meloni, A. Filippetti and A. Mattoni, Collective Molecular Mechanisms in the CH(3)NH(3)PbI(3) Dissolution by Liquid Water, ACS Nano, 2017, 11, 9183–9190 CrossRef CAS PubMed
- W. L. Jorgensen and J. D. Madura, Temperature and size dependence for Monte Carlo simulations of TIP4P water, Mol. Phys., 1985, 56, 1381–1392 CrossRef CAS
- B. Akbali, G. Topcu, T. Guner, M. Ozcan, M. M. Demir and H. Sahin, CsPbBr3 perovskites: Theoretical and experimental investigation on water-assisted transition from nanowire formation to degradation, Phys. Rev. Mater., 2018, 2, 034601 CrossRef CAS
- N. A. Manshor, Q. Wali, K. K. Wong, S. K. Muzakir, A. Fakharuddin, L. Schmidt-Mende and R. Jose, Humidity versus photo-stability of metal halide perovskite films in a polymer matrix, Phys. Chem. Chem. Phys., 2016, 18, 21629–21639 RSC
- X. Dong, X. Fang, M. Lv, B. Lin, S. Zhang, J. Ding and N. Yuan, Improvement of the humidity stability of organic–inorganic perovskite solar cells using ultrathin Al2O3 layers prepared by atomic layer deposition, J. Mater. Chem. A, 2015, 3, 5360–5367 RSC
- A. M. A. Leguy, Y. Hu, M. Campoy-Quiles, M. I. Alonso, O. J. Weber, P. Azarhoosh, M. van Schilfgaarde, M. T. Weller, T. Bein, J. Nelson, P. Docampo and P. R. F. Barnes, Reversible Hydration of CH3NH3PbI3 in Films, Single Crystals, and Solar Cells, Chem. Mater., 2015, 27, 3397–3407 CrossRef CAS
- B. Hailegnaw, S. Kirmayer, E. Edri, G. Hodes and D. Cahen, Rain on Methylammonium Lead Iodide Based Perovskites: Possible Environmental Effects of Perovskite Solar Cells, J. Phys. Chem. Lett., 2015, 6, 1543–1547 CrossRef CAS
- R. Li, B. Li, X. Fang, D. Wang, Y. Shi, X. Liu, R. Chen and Z. Wei, Self-Structural Healing of Encapsulated Perovskite Microcrystals for Improved Optical and Thermal Stability, Adv. Mater., 2021, 33, 2100466 CrossRef CAS PubMed
- W. Liu, H. Yu, Y. Li, A. Hu, J. Wang, G. Lu, X. Li, H. Yang, L. Dai, S. Wang and Q. Gong, Mapping Trap Dynamics in a CsPbBr3 Single-Crystal Microplate by Ultrafast Photoemission Electron Microscopy, Nano Lett., 2021, 21, 2932–2938 CrossRef CAS PubMed
- W.-J. Yin, T. Shi and Y. Yan, Unique Properties of Halide Perovskites as Possible Origins of the Superior Solar Cell Performance, Adv. Mater., 2014, 26, 4653–4658 CrossRef CAS
- E. Aydin, M. De Bastiani and S. De Wolf, Defect and Contact Passivation for Perovskite Solar Cells, Adv. Mater., 2019, 31, 1900428 CrossRef PubMed
- T. A. S. Doherty, A. J. Winchester, S. Macpherson, D. N. Johnstone, V. Pareek, E. M. Tennyson, S. Kosar, F. U. Kosasih, M. Anaya, M. Abdi-Jalebi, Z. Andaji-Garmaroudi, E. L. Wong, J. Madéo, Y.-H. Chiang, J.-S. Park, Y.-K. Jung, C. E. Petoukhoff, G. Divitini, M. K. L. Man, C. Ducati, A. Walsh, P. A. Midgley, K. M. Dani and S. D. Stranks, Performance-limiting nanoscale trap clusters at grain junctions in halide perovskites, Nature, 2020, 580, 360–366 CrossRef CAS PubMed
- P. Darvishzadeh, M. Babanezhad, R. Ahmadi and N. E. Gorji, Modeling the degradation/recovery of open-circuit voltage in perovskite and thin film solar cells, Mater. Des., 2017, 114, 339–344 CrossRef CAS
- H. Sohrabpoor, G. Puccetti and N. E. Gorji, Modeling the degradation and recovery of perovskite solar cells, RSC Adv., 2016, 6, 49328–49334 RSC
- L. Qiao, W. H. Fang and R. Long, The Interplay Between Lead Vacancy and Water Rationalizes the Puzzle of Charge Carrier Lifetimes in CH3NH3PbI3: Time-Domain Ab Initio Analysis, Angew. Chem., Int. Ed., 2020, 59, 13347–13353 CrossRef CAS PubMed
- Y. H. Kye, C. J. Yu, U. G. Jong, Y. Chen and A. Walsh, Critical Role of Water in Defect Aggregation and Chemical Degradation of Perovskite Solar Cells, J. Phys. Chem. Lett., 2018, 9, 2196–2201 CrossRef CAS PubMed
- W. Zhou, Y. Zhao, C. Shi, H. Huang, J. Wei, R. Fu, K. Liu, D. Yu and Q. Zhao, Reversible Healing Effect of Water Molecules on Fully Crystallized Metal–Halide Perovskite Film, J. Phys. Chem. C, 2016, 120, 4759–4765 CrossRef CAS
- X. Guo, Y. Gao, Q. Wei, K. Ho Ngai, M. Qin, X. Lu, G. Xing, T. Shi, W. Xie, J. Xu and M. Long, Suppressed Phase Segregation in High-Humidity-Processed Dion–Jacobson Perovskite Solar Cells Toward High Efficiency and Stability, Sol. RRL, 2021, 5, 2100555 CrossRef CAS
- Y. Ouyang, L. Shi, Q. Li and J. Wang, Role of Water and Defects in Photo-Oxidative Degradation of Methylammonium Lead Iodide Perovskite, Small Methods, 2019, 3, 1900154 CrossRef
- M. L. Petrus, Y. Hu, D. Moia, P. Calado, A. M. A. Leguy, P. R. F. Barnes and P. Docampo, The Influence of Water Vapor on the Stability and Processing of Hybrid Perovskite Solar Cells Made from Non-Stoichiometric Precursor Mixtures, ChemSusChem, 2016, 9, 2699–2707 CrossRef CAS
- B. Wang, Z.-G. Zhang, S. Ye, H. Rao, Z. Bian, C. Huang and Y. Li, Room-temperature water-vapor annealing for high-performance planar perovskite solar cells, J. Mater. Chem. A, 2016, 4, 17267–17273 RSC
- F. Cordero, F. Craciun, F. Trequattrini, A. Generosi, B. Paci, A. M. Paoletti and G. Zanotti, Influence of Temperature, Pressure, and Humidity on the Stabilities and Transition Kinetics of the Various Polymorphs of FAPbI3, J. Phys. Chem. C, 2020, 124, 22972–22980 CrossRef CAS
- B. A. Rosales, L. E. Mundt, T. G. Allen, D. T. Moore, K. J. Prince, C. A. Wolden, G. Rumbles, L. T. Schelhas and L. M. Wheeler, Reversible multicolor chromism in layered formamidinium metal halide perovskites, Nat. Commun., 2020, 11, 5234 CrossRef CAS
- D. Lin, Y. Gao, T. Zhang, Z. Zhan, N. Pang, Z. Wu, K. Chen, T. Shi, Z. Pan, P. Liu and W. Xie, Vapor Deposited Pure α-FAPbI3 Perovskite Solar Cell via Moisture-Induced Phase Transition Strategy, Adv. Funct. Mater., 2022, 32, 2208392 CrossRef CAS
- C. Ma, F. T. Eickemeyer, S.-H. Lee, D.-H. Kang, S. J. Kwon, M. Grätzel and N.-G. Park, Unveiling facet-dependent degradation and facet engineering for stable perovskite solar cells, Science, 2023, 379, 173–178 CrossRef CAS PubMed
- R. J. Sutton, M. R. Filip, A. A. Haghighirad, N. Sakai, B. Wenger, F. Giustino and H. J. Snaith, Cubic or Orthorhombic? Revealing the Crystal Structure of Metastable Black-Phase CsPbI3 by Theory and Experiment, ACS Energy Lett., 2018, 3, 1787–1794 CrossRef CAS
- H. Yao, J. Zhao, Z. Li, Z. Ci and Z. Jin, Research and progress of black metastable phase CsPbI3solar cells, Mater. Chem. Front., 2021, 5, 1221–1235 RSC
- Z. Yao, W. Zhao and S. Liu, Stability of the CsPbI3 perovskite: from fundamentals to improvements, J. Mater. Chem. A, 2021, 9, 11124–11144 RSC
- Z. Lin, Y. Zhang, M. Gao, J. A. Steele, S. Louisia, S. Yu, L. N. Quan, C.-K. Lin, D. T. Limmer and P. Yang, Kinetics of moisture-induced phase transformation in inorganic halide perovskite, Matter, 2021, 4, 2392–2402 CrossRef CAS
- K. E. Kweon, J. Varley, T. Ogitsu, L. F. Wan, M. Shelby, Z. Song, R. J. Ellingson, Y. Yan and J. R. I. Lee, Influence of External Conditions on the Black-to-Yellow Phase Transition of CsPbI3 Based on First-Principles Calculations: Pressure and Moisture, Chem. Mater., 2023, 35, 2321–2329 CrossRef CAS
- D. B. Straus, S. Guo and R. J. Cava, Kinetically Stable Single Crystals of Perovskite-Phase CsPbI3, J. Am. Chem. Soc., 2019, 141, 11435–11439 CrossRef CAS
- J. Di, H. Li, J. Su, H. Yuan, Z. Lin, K. Zhao, J. Chang and Y. Hao, Reveal the Humidity Effect on the Phase Pure CsPbBr3 Single Crystals Formation at Room Temperature and Its Application for Ultrahigh Sensitive X-Ray Detector, Adv. Sci., 2022, 9, 2103482 CrossRef CAS PubMed
- W. Fan, K. Deng, Y. Shen, Y. Bai and L. Li, Moisture-Accelerated Precursor Crystallisation in Ambient Air for High-Performance Perovskite Solar Cells toward Mass Production, Angew. Chem., Int. Ed., 2022, 61, e202211259 CrossRef CAS
- M. K. Gangishetty, R. W. J. Scott and T. L. Kelly, Effect of relative humidity on crystal growth, device performance and hysteresis in planar heterojunction perovskite solar cells, Nanoscale, 2016, 8, 6300–6307 RSC
- K. Liu, Y. Luo, Y. Jin, T. Liu, Y. Liang, L. Yang, P. Song, Z. Liu, C. Tian, L. Xie and Z. Wei, Moisture-triggered fast crystallization enables efficient and stable perovskite solar cells, Nat. Commun., 2022, 13, 4891 CrossRef CAS
- K. Zhang, Z. Wang, G. Wang, J. Wang, Y. Li, W. Qian, S. Zheng, S. Xiao and S. Yang, A prenucleation strategy for ambient fabrication of perovskite solar cells with high device performance uniformity, Nat. Commun., 2020, 11, 1006 CrossRef CAS PubMed
- L. Li, X. Zhang, H. Zeng, X. Zheng, Y. Zhao, L. Luo, F. Liu and X. Li, Thermally-stable and highly-efficient bi-layered NiOx-based inverted planar perovskite solar cells by employing a p-type organic semiconductor, Chem. Eng. J., 2022, 443, 136405 CrossRef CAS
- C. Zhang, A. Zhang, G. Zhang, Y. Fang, J. Cheng, L. Liang, J. Shi, Z. Li, T. Meng and D. Wang, Moisture-dependent room-temperature perovskite crystallization in vacuum flash-assisted solution processed intermediate phase films, Org. Electron., 2022, 111, 106652 CrossRef CAS
- Y. Du, H. Cai, X. Bao, Z. Xing, Y. Wu, J. Xu, L. Huang, J. Ni, J. Li and J. Zhang, Flexible Perovskite Solar Cells onto Plastic Substrate Exceeding 13% Efficiency Owing to the Optimization of CH3NH3PbI3–xClx Film via H2O Additive, ACS Sustainable Chem. Eng., 2017, 6, 1083–1090 CrossRef
- Z.-K. Tang, Y.-N. Zhu, Z.-F. Xu and L.-M. Liu, Effect of water on the effective Goldschmidt tolerance factor and photoelectric conversion efficiency of organic–inorganic perovskite: insights from first-principles calculations, Phys. Chem. Chem. Phys., 2017, 19, 14955–14960 RSC
- M. A. Haque, A. Syed, F. H. Akhtar, R. Shevate, S. Singh, K.-V. Peinemann, D. Baran and T. Wu, Giant Humidity Effect on Hybrid Halide Perovskite Microstripes: Reversibility and Sensing Mechanism, ACS Appl. Mater. Interfaces, 2019, 11, 29821–29829 CrossRef CAS
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Surface passivation of perovskite film for efficient solar cells, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS
- J. Yuan and Q. Wang, Nontoxic bismuth-based hybrid inorganic-organic double perovskites with high air stability, Surf. Interfaces, 2021, 22, 100821 CrossRef CAS
- X. Kong, Z. Li, Y. Jiang, Z. Xu, S.-P. Feng, G. Zhou, J.-M. Liu and J. Gao, Improving stability and efficiency of perovskite solar cells via a cerotic acid interfacial layer, Surf. Interfaces, 2021, 25, 101163 CrossRef CAS
- P. Su, Y. Huang, Y. Li, C. Hu and W. Shi, Improving photoluminescent water-stability of CsPbBr3 perovskite nanocrystals via constructing nanocrystals/polymer composites with hydrophobic surfaces for LED applications, Surf. Interfaces, 2023, 37, 102719 CrossRef CAS
- T. Wang, J. Yang, Q. Cao, X. Pu, Y. Li, H. Chen, J. Zhao, Y. Zhang, X. Chen and X. Li, Room temperature nondestructive encapsulation via self-crosslinked fluorosilicone polymer enables damp heat-stable sustainable perovskite solar cells, Nat. Commun., 2023, 14, 1342 CrossRef CAS PubMed
- W. Kong, G. Wang, J. Zheng, H. Hu, H. Chen, Y. Li, M. Hu, X. Zhou, C. Liu, B. N. Chandrashekar, A. Amini, J. Wang, B. Xu and C. Cheng, Fabricating High-Efficient Blade-Coated Perovskite Solar Cells under Ambient Condition Using Lead Acetate Trihydrate, Sol. RRL, 2018, 2, 1700214 CrossRef
- Y. Gao, W. Xu, F. He, T. Fan, W. Cai, X. Zhang and G. Wei, Boosting Performance of CsPbI3 Perovskite Solar Cells via the Synergy of Hydroiodic Acid and Deionized Water, Adv. Energy Sustainability Res., 2021, 3, 2100149 CrossRef
- H. Chen, M. Zhang, T. Tran-Phu, R. Bo, L. Shi, I. Di Bernardo, J. Bing, J. Pan, S. Singh, J. Lipton-Duffin, T. Wu, R. Amal, S. Huang, A. W. Y. Ho-Baillie and A. Tricoli, Integrating Low-Cost Earth-Abundant Co-Catalysts with Encapsulated Perovskite Solar Cells for Efficient and Stable Overall Solar Water Splitting, Adv. Funct. Mater., 2021, 31, 2008245 CrossRef CAS
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |