
A metal-coordination stabilized small-molecule nanomedicine with high drug-loading capacity and synergistic photochemotherapy for cancer treatment†
Han
Chen
ab,
Haijing
Qu
ab,
Beike
Lu
a,
Yuqing
Pan
ab,
Jiaojiao
Yang
a,
Zhiran
Duan
ab,
Jie
Wu
ab,
Yanjun
Wang
ac,
Chao
Wang
a,
Rong
Hu
*c and
Xiangdong
Xue
 *ab
*ab
aShanghai Frontiers Science Center of Drug Target Identification and Delivery, School of Pharmaceutical Sciences, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: xuexd@sjtu.edu.cn
bNational Key Laboratory of Innovative Immunotherapy, Shanghai Jiao Tong University, Shanghai 200240, China
cShanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Centre for Specialty Strategy Research of Shanghai Jiao Tong University China Hospital Development Institute, Shanghai 200011, China. E-mail: hur1152@sh9hospital.org.cn
First published on 8th July 2024
Abstract
Conventional nanomedicines typically employ a significant amount of excipients as carriers for therapeutic delivery, resulting in generally low drug-loading and compromised anti-cancer efficacy. Here, we propose a small-molecule nanomedicine (CMC NP) directly assembled using a chemotherapeutic drug (chlorambucil, CBL) and a phototherapeutic agent (chlorin e6, Ce6), and stabilized by metal coordination. The CMC NP exhibits exceptionally high drug loading (89.21%), robust stability, and smart disassembly in response to glutathione (GSH). Such a straightforward yet multifunctional delivery strategy could be a better alternative to overcome the above shortcomings of conventional nanomedicines while achieving enhanced efficacy. The CMC NP not only directly induces CBL-induced chemotherapy but also elicits synergistic antitumor responses through Ce6-mediated photodynamic and photothermal therapies. Owing to the multifaceted efforts from photodynamic, photothermal and chemo-therapies, the CMC NP exhibits excellent antitumor efficacy with negligible systemic toxicity which is untenable in traditional CBL-induced chemotherapy. Therefore, this study provides a feasible strategy for overcoming existing challenges and presents a potential opportunity to augment the clinical therapeutic effectiveness associated with conventional nanomedicine.
 Xiangdong Xue | Dr Xiangdong Xue serves as an Associate Professor and Principal Investigator at the School of Pharmaceutical Sciences and National Key Laboratory of Innovative Immunotherapy, Shanghai Jiao Tong University. He obtained his Ph.D. degree from the National Center for Nanoscience and Technology, Chinese Academy of Sciences, followed by a 5-year postdoctoral fellowship at the University of California, Davis. As of July 2024, Dr Xue has authored over 60 high-impact papers with the total citation count exceeding 4100 and an H-index of 31. Dr Xue's research interests encompass (1) self-adaptive nanomedicine for cancer treatment and (2) immunoregulation and immunotherapy. |
1. Introduction
Nanomedicines have shown immense promise in enhancing the in vivo efficacy of therapeutic and diagnostic agents, owing to their exceptional structural controllability, multifunctionality, and disease-targeting capability.1,2 Conventional nanomedicines based on polymeric nanoparticles, micelles and liposomes have been successfully developed for cancer treatment; however, they suffer from complicated synthesis of building blocks and limited drug loading, typically less than 20 wt%.3,4 Consequently, up to 80 wt% of carrier materials are introduced into our body and may lead to undesirable toxicity upon metabolism. To cover these shortcomings, Cui and co-workers introduced the concept of “one-component nanomedicine” by directly conjugating therapeutics to peptides that self-assemble into nanoparticles.5–7Yan and coworkers have advanced this concept by developing drug–drug amphiphiles that conjugate two drugs with opposite polarities, facilitating their self-assembly into nanoparticles that markedly augment antitumor efficacy.8–10 Our group also developed a series of photosensitizer–drug conjugates designed to achieve high drug loading while enabling imaging-guided and synergistic therapy.11–13 Although these drug-conjugate-assembled nanomedicines can dramatically increase drug loading, their synthesis involves specialized chemical processes, complicating the production process. Furthermore, the cost of nanomedicines can escalate especially when the production yield is low and the parent drugs are expensive.
To minimize the production complexity, nanomedicines composed of pure drugs that self-assemble into nanostructures through supramolecular assembly have been developed. Luo and co-workers developed a series of small-molecule nanomedicines by physically mixing different drugs and assembling them into nanostructures.14,15 We also developed a series of such nanomedicines by directly mixing photosensitizers and chemotherapeutics, which then self-assemble into nanostructures driven by non-covalent interactions.16,17 These small-molecule nanomedicines feature a straightforward preparation process, obviating the need for chemical synthesis, and effectively improve tumor accumulation and anticancer efficacy while mitigating the adverse effects of chemotherapy. However, the stability of these non-covalently self-assembled nanostructures may not be sufficient for maintenance during blood circulation, potentially leading to premature drug release and non-specific accumulation. We have previously stabilized pure drug-assembled nanomedicines using an “on-surface” crosslinking strategy18 with PEG polymers, enhancing circulation stability and optimizing biodistribution.19–21 Nonetheless, this approach still requires additional chemical synthesis to add surface cross-linkages, and the inclusion of PEG reduces drug loading. Hence, our goal is to develop small-molecule nanomedicines with high drug loading, a simple production process, and stable nanostructures.
Given the aggressive and malignant characteristics of many cancers, nanomedicines consisting solely of chemotherapeutics may be insufficient for effectively inhibiting tumor growth. Clinical treatment typically combines chemotherapy with other modalities such as surgery and radiotherapy to strengthen effectiveness. Phototherapy, encompassing photodynamic therapy (PDT) and photothermal therapy (PTT), offers a minimally invasive therapeutic approach that utilizes reactive oxygen species (ROS) and local hyperthermia to ablate tumor lesions, respectively.22,23 By co-assembling chemotherapeutics with phototherapeutic agents, nanomedicines can be readily engineered to exhibit high drug loading capacity, with a simplified preparation process, as well as synergistic multi-modal therapeutic effects for effective cancer treatment. Metal ions have been reported to form stable yet stimuli-sensitive metal–organic bonds through coordination24,25 with different chemical groups, making them an optimal choice for stabilizing the aforementioned small-molecule nanomedicines. Therefore, combining chemo- and photo-therapeutic agents with metal-coordination would yield small-molecule nanomedicines demonstrating high drug loading capacity, robust stability, multifunctionality, and responsive drug release potential, thus exhibiting promise in improving conventional nanomedicines characterized by low drug loading and complex preparation procedures.
Herein, we propose a straightforward yet multifunctional small-molecule nanomedicine (CMC NP), which involves the coordination of a chemotherapeutic drug (chlorambucil, CBL) and a phototherapeutic agent (chlorin e6, Ce6) with manganese II (Mn2+), in an attempt to realize synergistic effects from PDT, PTT and chemotherapy (Scheme 1). The CMC NP features metal-coordination mediated robust stability, high loading capacity (up to 89.21%) and desirable tumor accumulation. Furthermore, the metal-coordination makes the CMC NP labile to high concentration GSH in tumor cells, unleashing the loaded CBL specifically in tumor cells to alleviate off-target toxicity. The CMC NP is designed to act against multi-individual mechanisms to induce apoptosis or necrosis of tumor cells, involving CBL-induced DNA alkylation and improved Ce6-mediated phototherapy performance. Collectively, amalgamating the features of superb drug loading capacity, GSH-responsive drug release and DNA alkylation characteristics, the new self-assembling strategy driven by Mn2+ overcomes existing challenges in nitrogen mustard-related delivery, further providing a potential opportunity to enhance the clinical therapeutic efficacy of these lipophilic antitumor agents.
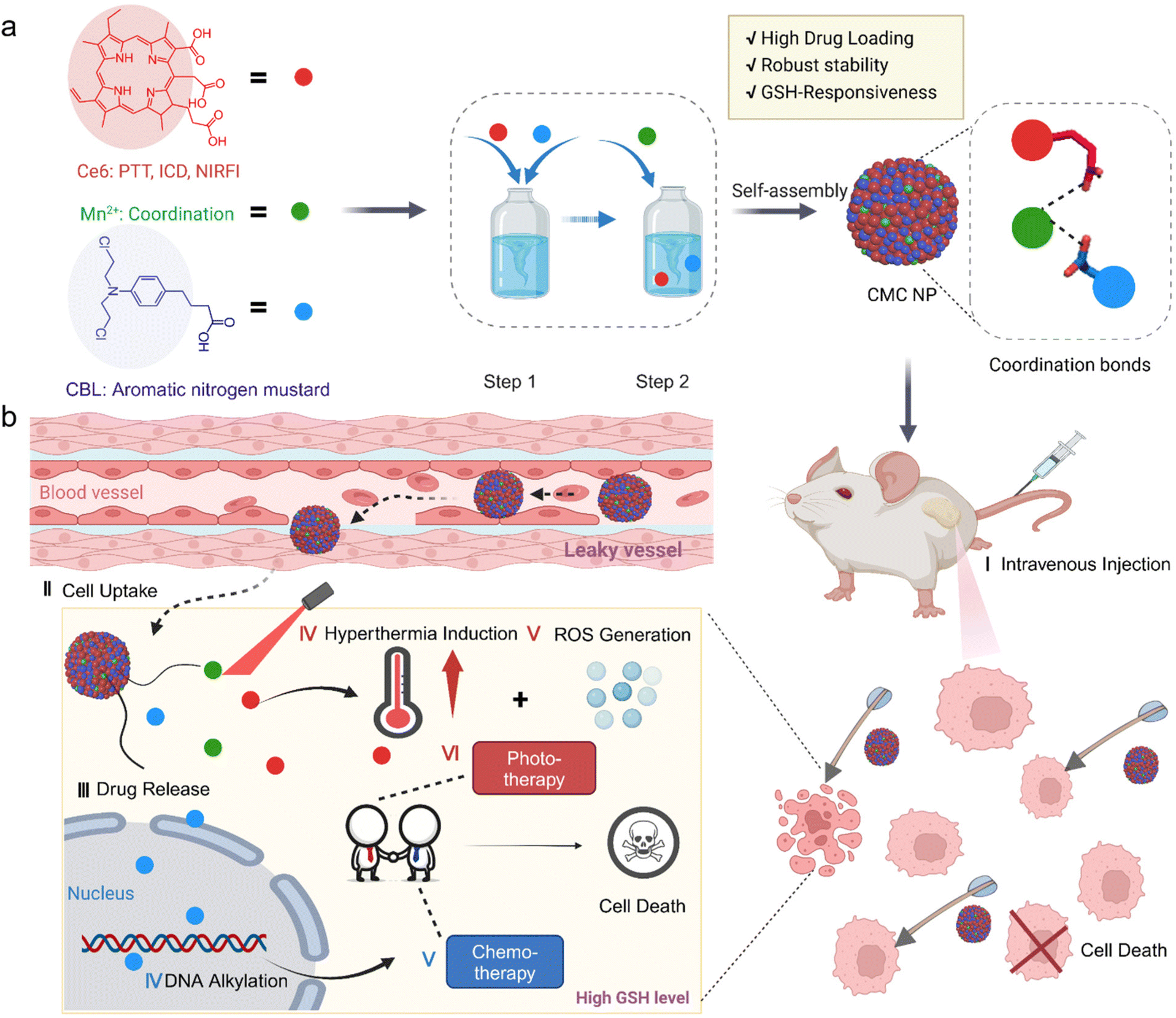 | ||
| Scheme 1 Schematic illustration of CMC NPs and their sequential release process for synergistic photo-/chemo-therapy. (a) The self-assembly routes of CMC NPs. (b) Proposed synergistic mechanism of the CMC NPs in mice. Image created with Biorender. | ||
2. Results and discussion
2.1. Self-assembly and characterization of CMC NPs
CMC NPs were prepared through a two-step process in aqueous solution, leveraging the formation of amorphous coordination-bonding networks between Mn2+ and –COOH groups of CBL and Ce6 (Fig. 1a). In order to obtain an optimal particle size and performance of CMC NPs, we screened the reactant proportions by synthesizing nanoparticles at different molar ratios of Ce6/CBL, specifically 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:3, 1:5, and 1:7. As shown in Fig. 1b and Fig. S1,† self-assembled nanoparticles could be readily formed at different ratios, and the particle size increased when more CBL was added, in which the ratio of 1:5 yielded uniformly dispersed nanoparticles as evidenced by the lowest PDI. Upon the addition of an increasing ratio of CBL, the surface negative charge of the resulting CMC NPs exhibited a declining trend (Fig. 1c). Moreover, the fluorescence quenching effect enhanced with higher levels of CBL (Fig. 1d and Fig. S1†), implying that more Ce6 molecules formed aggregation. The drug loading potential of the nano-systems constructed with different Ce6/CBL ratios was further assessed, in which the ratio of 1:5 achieved the highest encapsulation efficiency (EE%) for CBL at approximately 64.56%, coupled with almost 100% encapsulation of Ce6 (Fig. S2†). Considering the promising performance mediated by the EE% of CBL and the PDI, the final molar ratio of Ce6 to CBL was set at 1:5 in CMC NPs.
:1, 1:3, 1:5, and 1:7. As shown in Fig. 1b and Fig. S1,† self-assembled nanoparticles could be readily formed at different ratios, and the particle size increased when more CBL was added, in which the ratio of 1:5 yielded uniformly dispersed nanoparticles as evidenced by the lowest PDI. Upon the addition of an increasing ratio of CBL, the surface negative charge of the resulting CMC NPs exhibited a declining trend (Fig. 1c). Moreover, the fluorescence quenching effect enhanced with higher levels of CBL (Fig. 1d and Fig. S1†), implying that more Ce6 molecules formed aggregation. The drug loading potential of the nano-systems constructed with different Ce6/CBL ratios was further assessed, in which the ratio of 1:5 achieved the highest encapsulation efficiency (EE%) for CBL at approximately 64.56%, coupled with almost 100% encapsulation of Ce6 (Fig. S2†). Considering the promising performance mediated by the EE% of CBL and the PDI, the final molar ratio of Ce6 to CBL was set at 1:5 in CMC NPs.
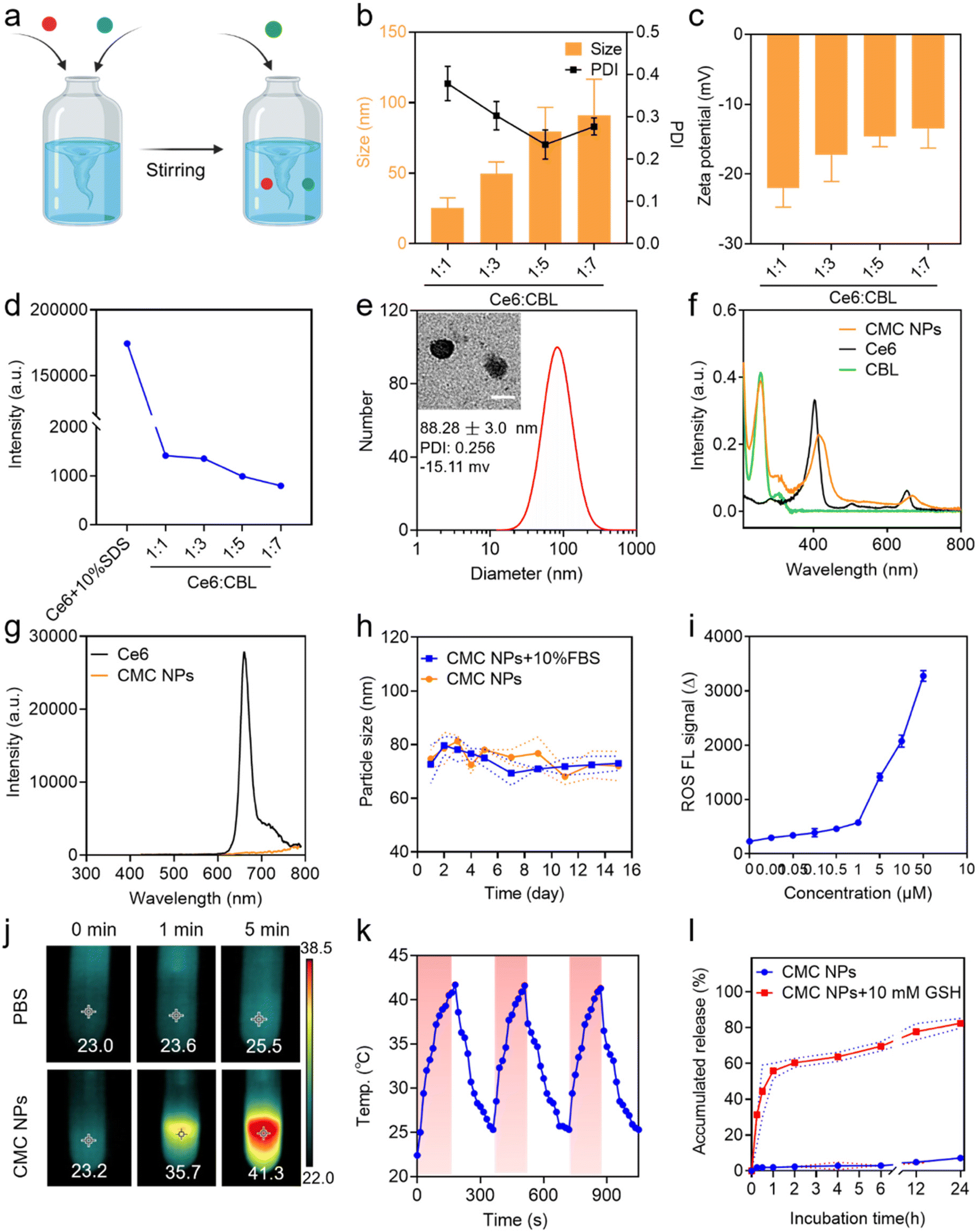 | ||
| Fig. 1 Preparation and characterization of CMC NPs. (a) Scheme of the preparation process of CMC NPs. (b) Size distribution, PDI and (c) zeta potential of CMC NPs assembled with Ce6 and CBL at various molar ratios. (d) The quantitative analysis of fluorescence intensity of CMC NPs assembled with Ce6 and CBL at various molar ratios. (e) The size distributions and PDI of CMC NPs; the inset shows a TEM micrograph of CMC NPs. Scale bar: 100 nm. (f) UV-vis spectra of free Ce6, free CBL and CMC NPs. (g) Fluorescence spectra of free Ce6 and CMC NPs. Excitation was at 425 nm. (h) Stability of CMC NPs in the physiological environment (PBS or 10% FBS). (i) Measurement of ROS generation via SOSG probe indication. (j) In vitro thermal images of PBS containing CMC NPs and PBS alone exposed to 680 nm laser irradiation (0.4 W cm−2) for 1 or 5 min. The maximum temperature was automatically recorded via an infrared thermal camera. (k) Photothermal curves of CMC NPs for three heating/cooling cycles. (l) The evaluation of the GSH-responsive drug release profile of CMC NPs in vitro (n = 3). | ||
After successful preparation and optimization, we proceeded to perform systematic characterization of the CMC NPs. Initially, the morphology and particle size of the self-assembled nanoparticles were determined by TEM and DLS studies, respectively, which revealed that all CMC NPs have a homogeneously dispersed spherical morphology with a hydrodynamic size of ∼80 nm (Fig. 1e). Moreover, the optical properties were investigated to confirm successful fabrication of CMC NPs. The self-assembled CMC NPs exhibited an additional absorption peak at 257 nm assigned to CBL and an ∼10 nm red shift in the Ce6 Q-band (670 nm) compared to that of free Ce6 (Fig. 1f). The fluorescence intensity of Ce6 in CMC NPs was in a quenched state, further supporting the self-assembly of nanostructures (Fig. 1g). To assess their suitability for biomedical applications, the stability of CMC NPs was monitored in a physiological environment (PBS and 10% FBS). As depicted, the CMC NPs were able to retain a stable size distribution (Fig. 1h) and PDI (Fig. S3†) over a period of 14 days. Along with this stable nanostructure, the CMC NPs also had an excellent drug loading capacity (approximately 89.21%, specifically 54.49% CBL and 34.72% Ce6), which markedly exceeded that of conventional drug delivery systems that generally only load 20% drugs. In addition, quantification of drug loading revealed that the actual ratio of CBL, Ce6, and Mn2+ in the resulting CMC NPs was 5.5:3.5:1. Collectively, our coordination-stabilized self-assembly strategy would be a highly potent way to achieve nitrogen mustard-related delivery.
2.2. Phototherapeutic effects of CMC NPs
By virtue of the high loading of Ce6 (a phototherapeutic agent), the CMC NPs were anticipated to exhibit potent photodynamic and photothermal effects. Hence, the ROS-generating and hyperthermia-inducing properties of CMC NPs were evaluated. As expected, laser-irradiated CMC NPs induced abundant ROS generation in a concentration-dependent manner (Fig. 1i). In addition, due to the laser-induced hyperthermia of the CMC NP solution the temperature immediately increased from 23.2 to 35.7 °C during the 1 min of laser irradiation, even up to 41.3 °C through a 5 min exposure. In comparison, the temperature of Milli-Q water alone showed an imperceptible change (from 23.0 to 23.6 °C) under the same conditions (Fig. 1j). Simultaneously, Fig. S4† shows the photothermal performance of CMC NPs in a concentration-dependent manner under 680 nm laser exposure for 3 min. To further check the photostability of CMC NPs, the temperature of CMC NP solution was recorded during three cycles of laser on/off, whose results suggested their excellent photothermal stability (Fig. 1k). The remarkable phototherapeutic effects and photothermal stability endowed the CMC NPs with possibilities to be a powerful weapon to directly ablate tumors.2.3. GSH-responsive drug release
The competitive coordination of glutathione (GSH) with Mn2+ made the Mn2+-coordinated nanostructure labile to high concentration GSH in the TME, leading to responsive drug release.26 To validate the property of GSH-responsive release of the CMC NPs, we fostered a gradient GSH environment (5, 10, and 15 mM GSH PBS solutions) to break the nanostructures. A gradually enlarged particle size of the self-assembled nanoparticles was observed in a 10 mM GSH PBS solution, along with a blue shift in the Ce6 Q-band (670 nm), both indicating the GSH responsiveness of the CMC NPs (Fig. S5†). Additionally, visual evidence from Fig. S6† strongly reaffirmed that the CMC NPs effectively achieved drug release in high-GSH environments. Consequently, we investigated the cumulative GSH-responsive drug release of CMC NPs. This experiment was intentionally designed to mimic the two-stage in vivo drug release, mirroring conditions in the bloodstream (PBS) and in the TME (PBS containing 10 mM GSH). Consistent with the above excellent stability of nanostructures, only 7% of Ce6 was released within 24 h in PBS solution. Upon the addition of 10 mM GSH to PBS solution, the CMC NPs exhibited an irruptive drug release profile with almost 60% DOX released within the initial 1 h (Fig. 1l). The release profile supported the stability of CMC NPs during blood circulation and their responsive therapeutic release in high-GSH intracellular environments within tumor cells, indicating that the CMC NPs enabled tumor-specific killing while minimizing off-target toxicity.2.4. Cellular uptake and subcellular distribution
Due to the exceptional phototherapeutic properties, we further assessed their propensity for cellular internalization and subcellular distribution. To achieve visual interrogation of the processes of subcellular distribution in 4T1 cells, the nuclei and lysosomes were stained with Hoechst 33342 and Lysotracker, respectively, followed by observation using confocal laser scanning microscopy (CLSM). It was observed that the intracellular fluorescence intensity of Ce6 increased in CMC NP-treated cells compared to that in those treated with free Ce6, indicating improved cellular uptake facilitated by the nano-formulation (Fig. 2a and c). When the incubation time was extended to 4 h, intracellular Ce6 fluorescence in 4T1 cells was distinctly stronger up to 2 h of treatment, suggesting that the cellular uptake occurred in a time-dependent manner within the initial 4 h (Fig. 2d and Fig. S7†). In addition, the time-dependent dislocations of Ce6 fluorescence proved that the CMC NPs would escape from the lysosomes into the cytoplasm (Fig. S7†).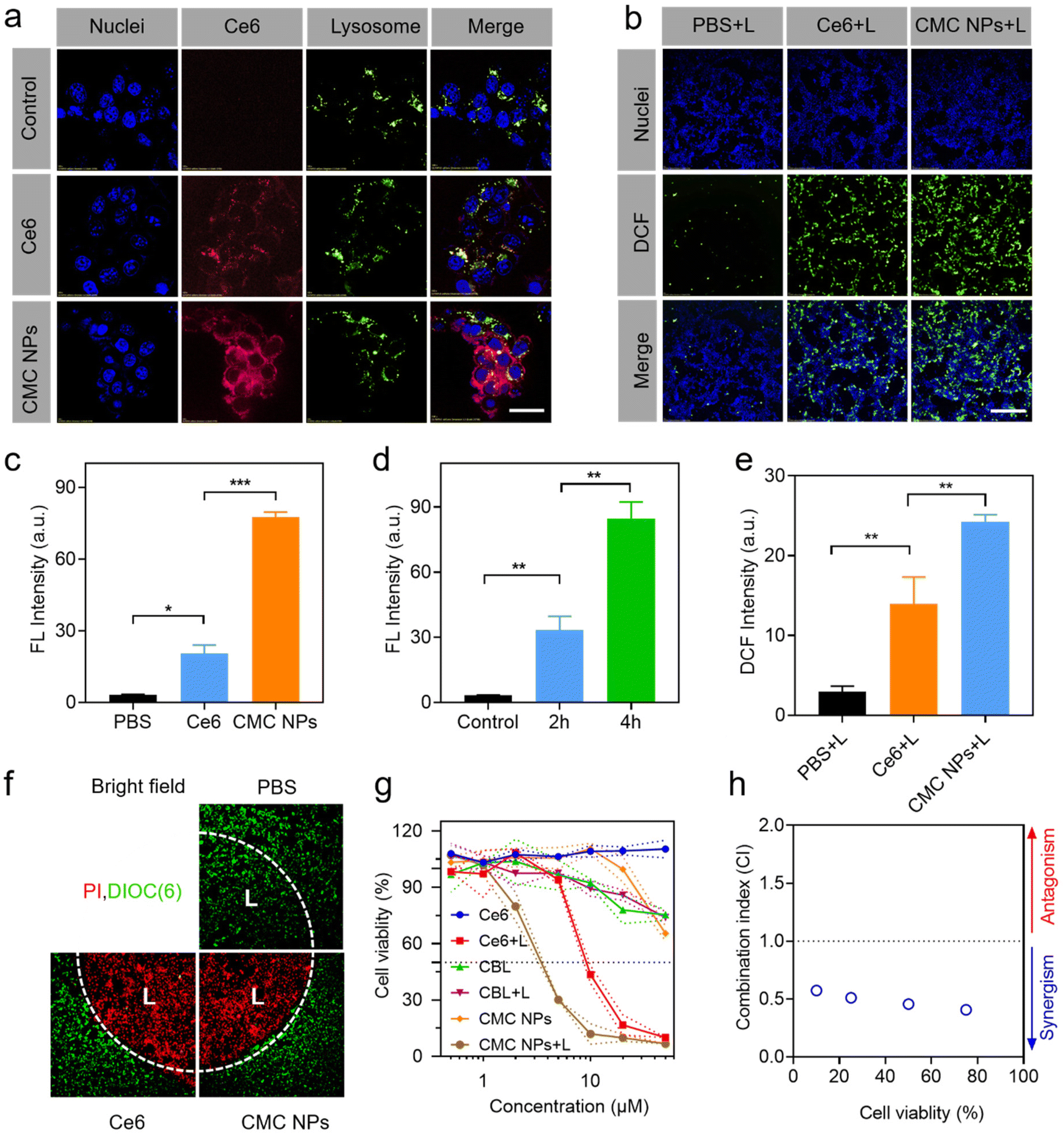 | ||
| Fig. 2 In vitro evaluation of the antitumor efficacy of CMC NPs in 4T1 cells. (a) Cellular uptake and subcellular distribution of free Ce6 and CMC NPs after 2 h incubation. Scale bar: 20 μm. (b) Intracellular ROS production when treated with CMC NPs and the control materials under laser irradiation. Scale bar: 200 μm. (c) The quantification of cellular uptake based on (a). (d) The quantification of CMC cellular uptake for different incubation times. (e) The quantification of intracellular ROS production based on (b). (f) Laser-directed phototherapeutic effect on cells, and the laser-treated areas are marked with ‘L’. (g) Cell viability of 4T1 cells treated with CMC NPs and the control materials. (h) The CI of the CBL-induced DNA alkylation and Ce6-mediated cell apoptosis. CI < 1 was considered a synergistic effect. **P ≤ 0.01; ***P ≤ 0.001. | ||
2.5. Intracellular ROS generation
The ROS generation of CMC NPs at the cellular level was evaluated via the DCFH-DA probe, which emitted green fluorescence under CLSM observation (Fig. 2b, e and Fig. S8†). Control PBS-treated cells produced little amount of ROS under laser exposure, and the cells treated with Ce6 and CMC NPs exhibited comparable ROS levels in the absence of laser irradiation. In contrast, considerable green fluorescence was detected in the Ce6-containing treated cells after 1 min laser irradiation. Quantitative analysis further demonstrated the superiority of CMC NPs + L in ROS generation, which was well in accordance with the enhanced intracellular uptake.2.6. In vitro controllable phototherapy with CMC NPs
Controllability is one of the prerequisites for the clinical application of phototherapeutic agents. Therefore, the live–dead cell staining assay was performed to verify the highly controllable and accurate therapeutic effect of CMC NPs. We irradiated a discrete area of 4T1 cells pre-incubated with CMC NPs or free Ce6 and intuitively evaluated cell death between the laser-treated and non-treated cells. Live–dead imaging results demonstrated that the dead cells (PI, red) were predominant in the irradiated area (“L”) and live cells (DiOC6(3), green) prevailed over dead cells in the non-irradiated area after 3 min laser irradiation (Fig. 2f). This well-defined cell death supported that the CMC NPs possessed excellent controllability for further phototherapy-induced tumor elimination.2.7. Cytotoxicity studies of CMC NPs
The enhanced PDT effect and the excellent photothermal performance of CMC NPs prompted us to further assess the cytotoxicity to cancer cells in vitro (Fig. 2g). Inconspicuous cytotoxicity was observed after incubation with free Ce6 even at high concentrations, demonstrating the excellent biocompatibility of free Ce6. After co-incubation with CBL for 24 h, the cell viability decreased to nearly 70% with or without laser irradiation, which could be attributed to the fact that CBL as a small lipophilic molecule entered the cells through passive diffusion. Notably, the CMC NPs + L treatment showed lower cell viability than the Ce6 + L treatment due to the synergistic effects from PDT, PTT, and chemotherapy. The combination index (CI) in Fig. 2h further supports that the CMC NPs could have a noticeable synergistic effect through CBL-induced DNA alkylation and Ce6-mediated phototherapy, hence holding great potential for cancer treatment.2.8. Tumor accumulation and biodistribution of CMC NPs
After demonstrating the in vitro antitumor efficacy of CMC NPs, we proceeded to assess their in vivo performance. Initially, we employed an IVIS imaging system to track the dynamic fluorescence variations (Ce6 signal) and examine the biodistribution of CMC NPs in 4T1 tumor-bearing mice. As shown in Fig. 3a, free Ce6 quickly migrated into tumors at 2 h post-injection but dissipated quickly thereafter. In contrast, mice treated with CMC NPs showed higher Ce6 signals within tumors than those treated with free Ce6, and they were sustained at a high level for up to 8 h (Fig. 3b). These findings suggested that our nanoformulation could improve tumor accumulation and retention compared to free drugs. Ex vivo fluorescence images (Fig. 3c and d) revealed that the CMC NPs exhibited stronger tumor accumulation but weaker organ distribution than free Ce6, supporting that our nano-formulation would potentially alleviate concerns regarding off-target toxicity associated with toxic therapeutics. In addition, the exceptional accumulation of CMC NPs was confirmed by fluorescence images of ex vivo tumor cryo-sections. As shown in Fig. 3e, the intratumor signal of CMC NPs was much higher than that of free Ce6, further supporting the superior tumor accumulation of our nano-formulation. Collectively, the CMC nano-formulation could efficiently achieve substantial drug accumulation at tumor sites, laying a solid foundation for the subsequent synergistic anti-tumor efficacy.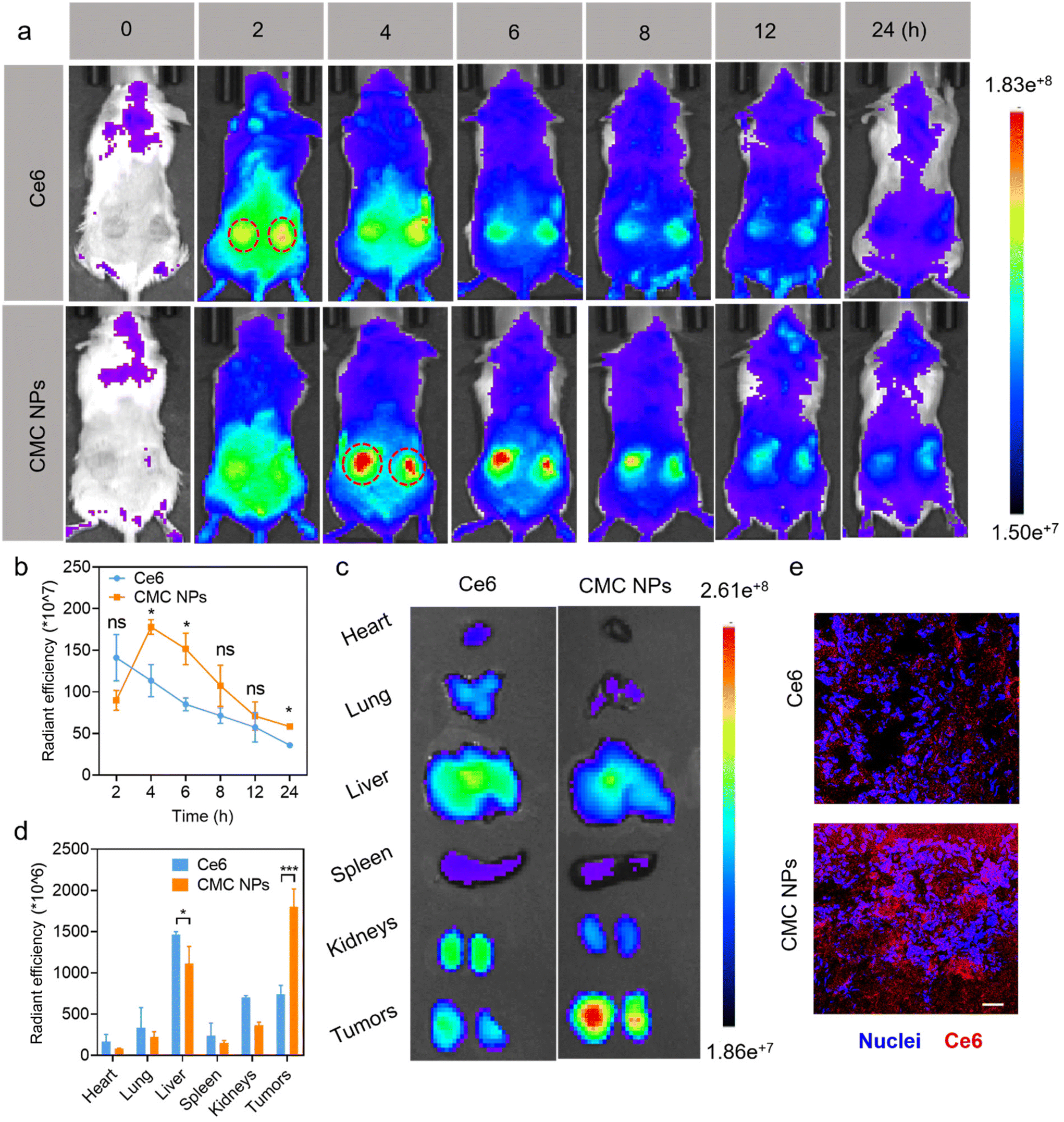 | ||
| Fig. 3 In vivo CMC NP-mediated improved drug delivery efficiency. (a) In vivo fluorescence images and (b) quantitative analysis (n = 3) of the 4T1 tumor-bearing mice after being injected with CMC NPs (10 mg kg−1). (c) Ex vivo IVIS fluorescence images and (d) quantitative analysis (n = 3) of the tumors and major organs excised from 4T1 tumor-bearing mice after being injected with CMC NPs (10 mg kg−1). (e) CLSM images of tumor slices stained with Hoechst 33342 showing the tumor infiltration and accumulation of CMC NPs. Scale bar: 100 μm. *P ≤ 0.05; ***P ≤ 0.001; ns, not significant. | ||
2.9. In vivo synergistic chemo-/photo-therapy
To further evaluate the anti-tumor effect of our nano-formulation in vivo, we administered various materials into 4T1 tumor-bearing mice via i.v. injection on days 1, 4, 7, 10, and 13 after tumor implantation. The mice were divided into 5 cohorts (n = 6), including PBS-, Mix-, CMC NP-, Mix + L-, and CMC NPs + L-treated groups (Fig. 4a). The PBS treatment group served as the negative control that did not receive any treatment. The Mix treatment represented a physical mixture of Ce6 and CBL for free drug treatment while the CMC NP treatment aimed to highlight the potential of drug delivery. Laser irradiation (0.4 W cm−2, 5 min) was introduced at 6 h post-injection (determined from the in vivo imaging results in Fig. 3a) in the Mix + L and CMC NP + L treatments. Upon laser irradiation, the intratumoral temperature increase in the CMC NP-treated group was much larger than that in the PBS group with laser treatment (Fig. 4b), and the CMC NP group displayed the highest temperature (even up to 57.2 °C) at the tumor site after 5 min laser irradiation (Fig. 4c and S9†). Such an excellent phototherapeutic performance could be ascribed to the enhanced delivery efficiency of Ce6 and effective accumulation within tumors.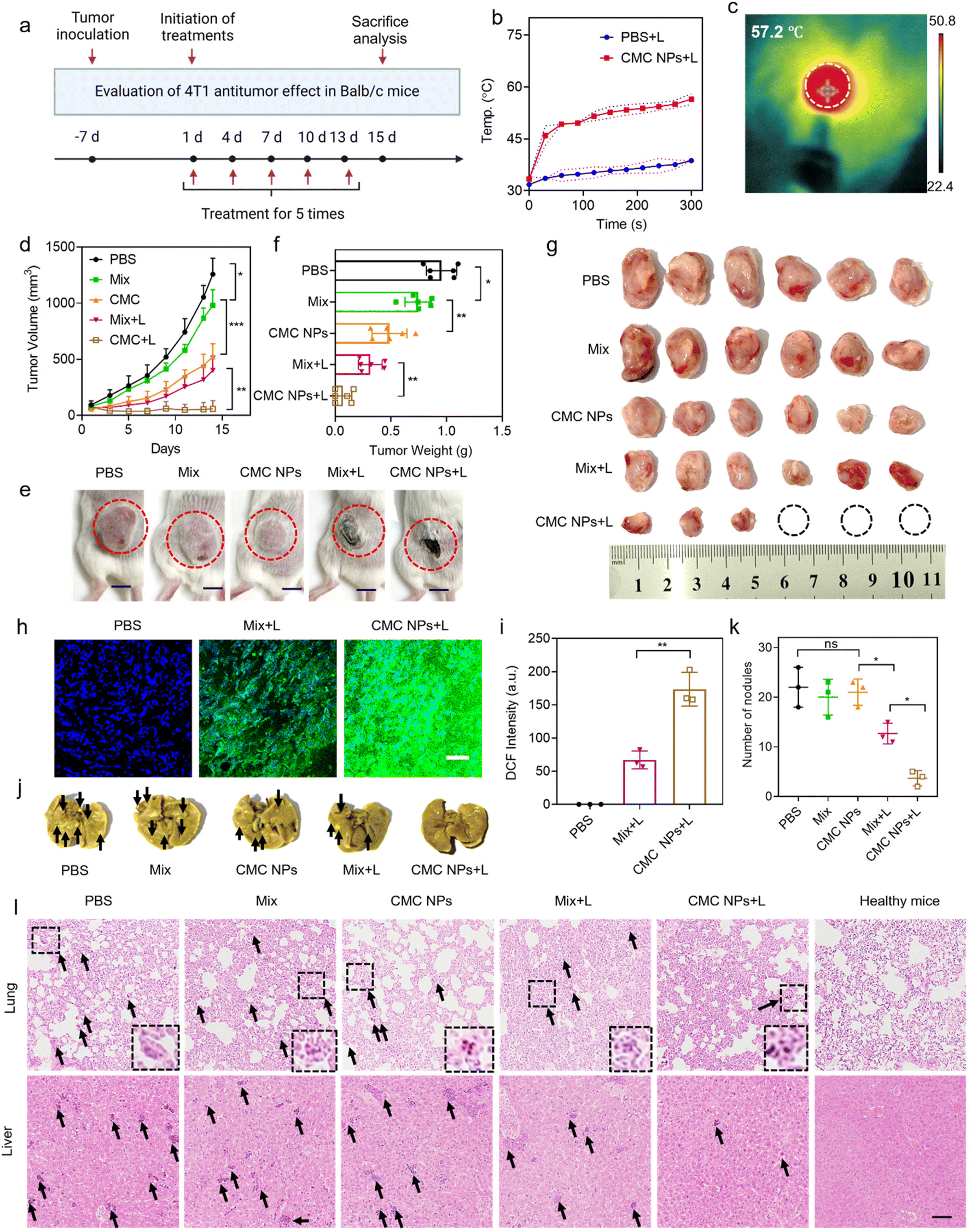 | ||
| Fig. 4 In vivo synergistic chemo-/photo-antitumor effect of CMC NPs. (a) Schematic illustration of the timeline of antitumor studies in 4T1 tumor-bearing mice. (n = 6). (b) Tumor temperature changes in the CMC NP group were monitored during a 5-minute laser irradiation period. (c) Thermal images of the 4T1 tumor-bearing mice after 6 h post-injection of CMC NPs upon exposure to 680 nm laser irradiation (0.4 W cm−2) for 5 min. (d) Tumor growth curves of the 4T1 tumor-bearing mice were monitored over a 14-day treatment period for the various treatment groups. (e) Representative photographs of the treated mice at predetermined time points. (f) The weight and (g) photographs of the tumors on day 14 after the different treatments. (h) CLSM images of tumor slices stained with DCF and (i) quantitative analysis of DCF intensity for evaluating intratumoral ROS generation. The higher intensity corresponded to greater ROS generation. Scale bar: 200 μm. (j) Representative photographs of harvested lung tissues with tumor metastasis at the endpoint of experiments. The black arrows indicate micrometastasis. (k) Quantitative analysis of tumor metastatic nodules in lung tissues. (l) H&E staining of ex vivo lungs and livers at the end of experiments. The black arrows indicate micrometastasis. The insets show enlarged details of tumor metastatic nodules in lung tissues. Scale bar: 100 μm. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ns, not significant. | ||
The therapeutic efficacy of CMC NPs was primarily evaluated based on tumor growth inhibition and anti-metastatic properties. Specifically, the tumor volumes were monitored over a 14-day treatment period for the various treatment groups. As shown in Fig. 4d, the tumor in the PBS group grew fast due to the high malignancy of 4T1 tumors. The Mix group showed limited inhibition of tumor growth as it solely relied on the chemotherapeutic effect of free CBL. In striking contrast, the CMC NPs demonstrated significantly greater effectiveness than the Mix treatment, indicating that this nanoformulation possessed potent in vivo drug delivery capabilities that substantially enhanced the chemotherapeutic effect of CBL. Furthermore, treatments involving phototherapy greatly improved anti-tumor effects. Notably, compared to its non-laser treated counterpart (Mix), the Mix + L treatment exhibited a pronounced tumor-suppressive effect. Impressively, the CMC NPs + L treatment exhibited the most powerful therapeutic effect, which is attributed to increased drug accumulation at the tumor sites and the synergism of chemotherapy, PTT, and PDT. The images of the treated mice (Fig. 4e) exhibited a consistent pattern of tumor volume change at the endpoint of experiments. In addition, all tumors were harvested and weighed to evaluate therapeutic efficacy. As shown in Fig. 4f and g, CMC + L effectively impeded tumor progression with complete eradication observed in approximately 50% of tumors. Further hematoxylin and eosin (H&E) staining was employed to assess the anti-tumor effects of various treatments, revealing substantial destruction of tumor cells in the CMC NP group (Fig. S10†). To further confirm Ce6-mediated ROS production, we cryo-sectioned tumor tissues to analyze the ROS levels in Ce6-containing groups under laser irradiation. As expected, the ROS generation in the CMC + L group surpassed that of the Mix + L group (Fig. 4h and i), providing an additional and more formidable arsenal for successful cancer treatment.
Given the propensity of breast cancer to metastasize to organs such as the lungs and the liver, we conducted further analysis of metastatic nodules in these organs. As illustrated in Fig. 4j and k, mice treated with CMC NPs + L exhibited the lowest level of lung metastasis among all treatment groups. The organ sections underwent histological analysis through H&E staining to further assess metastasis (Fig. 4l and Fig. S11†). Similarly, treatment with CMC NPs plus laser irradiation showed the most effective inhibition of tumor metastasis to the liver, as reflected by its lowest metastatic nodules. The considerable anti-metastatic potential further substantiated the anti-tumor efficacy of our nanoformulation (CMC NPs) and its synergistic photo-/chemo-therapies.
2.10. Biocompatibility of CMC NPs
Additionally, we assessed the biocompatibility of the nanoformulation through evaluation of body weight changes, blood cell counts, biochemical parameters, and histopathological analysis. Throughout the treatment, there were no notable changes in the body weights of tumor-bearing mice in all groups, confirming that these treatments could efficiently suppress 4T1 tumor growth without significant acute toxicity (Fig. 5a). At the endpoint of the experiment, further complete blood count (CBC) and biochemical examinations were conducted to assess the potential long-term biosafety of all treatments (Fig. 5b–l). As shown in Fig. 5m, all the treatments did not show a negative effect on normal organs at the tissue level, including the heart, the kidneys and the spleen. The white blood cells (WBCs) of the mice treated with the materials exhibited a significant increase compared to those of healthy mice, which is attributed to the presence of malignant tumors. The minimal impact of CMC NPs + L on white blood cells implied that the CMC NP-mediated synergistic strategies could mitigate systemic inflammation. Furthermore, no significant fluctuations were observed in the other hematological and biochemical parameters of tumor-bearing mice in all treatment groups compared with those of normal mice. Organ coefficients exhibited minimal variations after different treatments, except for the spleen (Fig. S12†). The spleens of tumor-bearing mice were noted to be swollen, and the spleen weight in the CMC NPs + L group confirmed its efficacy in alleviating systemic inflammation (Fig. S13†).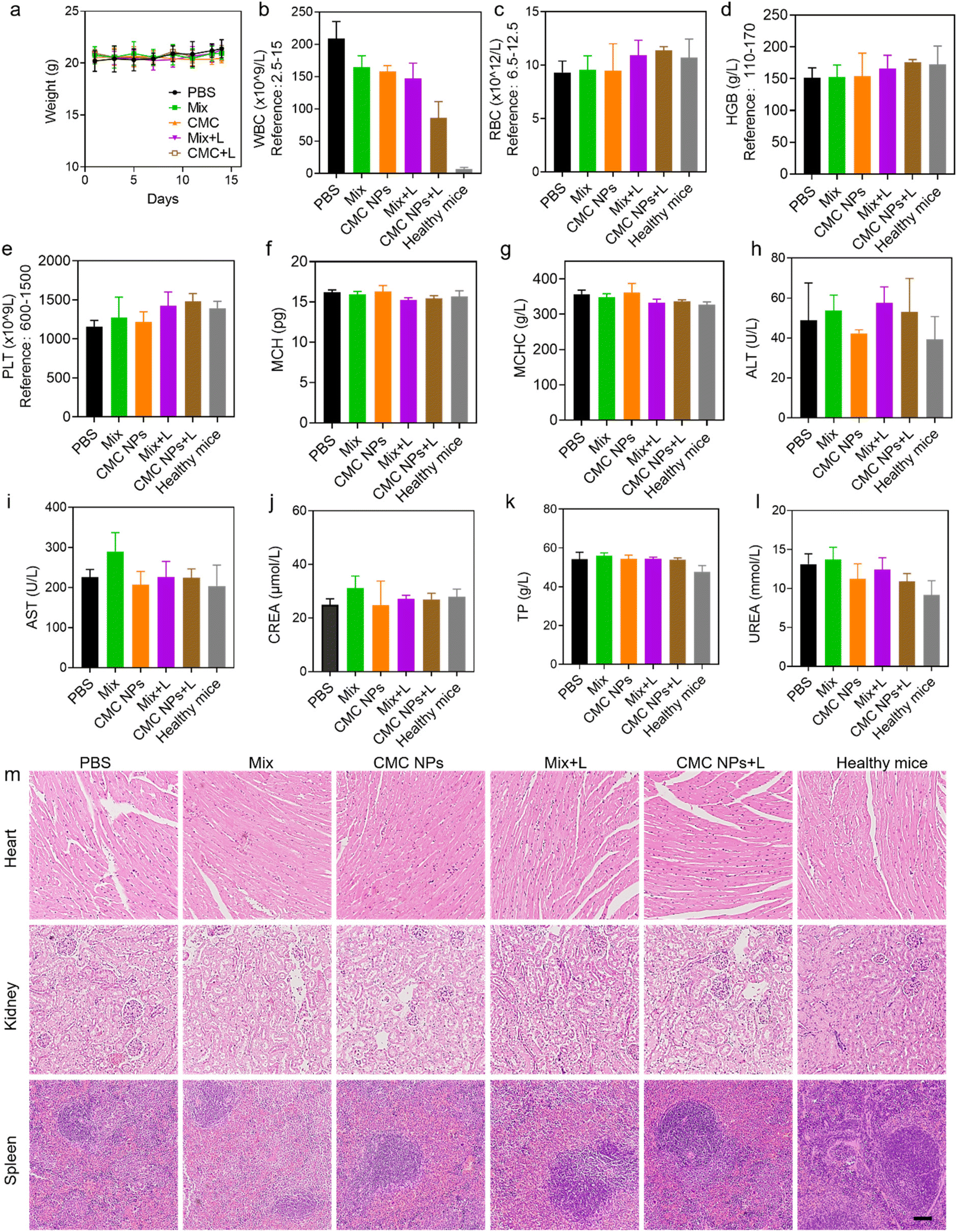 | ||
| Fig. 5 Biocompatibility evaluation of CMC NPs. (a) Body weight change of 4T1 tumor-bearing mice during treatment. (b–g) A comparison of CBC parameters of mice in different treatment groups at the endpoint of the experiment, including white blood cell (WBC) count, red blood cell (RBC) count, hemoglobin (HGB) level, hematocrit value (HCT), platelet (PLT) count, mean corpuscular hemoglobin (MCH) and mean corpuscular hemoglobin concentration (MCHC). (h–l) A comparison of biochemical parameters of mice in different treatment groups at the endpoint of the experiment, including alanine transaminase (ALT), aspartate aminotransferase (AST), creatinine (CREA), total protein (TP) and urea (UREA). (m) Histopathological results of mice in different treatment groups. Scale bar: 100 μm. | ||
To further evaluate the biosafety of CMC NPs, we conducted additional assessments in healthy mice. Red blood cells (RBCs) obtained from healthy mice were subjected to incubation with CMC NPs at various concentrations. Fig. S14† illustrates that CMC NPs induced imperceptible hemolysis even at high concentrations and microscopy images provided additional confirmation of the insignificant hemolytic effects of CMC NPs. Furthermore, the blood parameters of the mice exhibited negligible change during the two weeks after administration of CMC NPs (Fig. S15†). Overall, the aforementioned results thoroughly validated the favorable biosafety profile of the CMC nanoformulation for cancer therapy.
3. Conclusions
In summary, we successfully developed a metal-coordination nanoparticle (CMC NP) nanomedicine, which featured high drug loading capacity and robust stability. Leveraging the potent coordination between Mn2+ and COOH groups (provided by Ce6 and CBL molecules), the CMC NPs exhibited remarkable stability under physiological conditions and smart disassembly in response to glutathione (GSH) at tumor sites. Moreover, these nanoparticles manifested a multi-faceted antitumor effect, involving CBL-induced DNA alkylation and Ce6-mediated phototherapy. In vitro and in vivo results consistently validated that the CMC NPs exhibited potent therapeutic efficacy, which is attributed to the enhanced intracellular uptake and the synergistic chemo-/photo-treatment. Notably, CMC NPs exerted significant tumor-suppressing effects while maintaining minimal systemic toxicity that is unsustainable in traditional CBL-chemotherapy. Collectively, the new self-assembling delivery system driven by Mn2+ would provide a viable and promising avenue for addressing challenges associated with nitrogen mustards and other lipophilic antitumor agents, including poor pharmacokinetic characteristics and non-specific drug distribution.4. Materials and methods
4.1. Experimental materials and reagents
Chlorin e6 was acquired from Bide Pharmatech Ltd (Shanghai, China). Chlorambucil and the reactive oxygen species detection kit DCFH-DA (2′,7′-dichlorofluorescein diacetate) were procured from Sigma-Aldrich (St Louis, USA). Milli-Q water (18.2 MΩ cm) was obtained from a Milli-pore gradient system (Billerica, MA, USA) and employed for the preparation of all aqueous solutions. LysoTracker Green DND-26, Hoechst 33342, ROS assay kit-singlet oxygen sensor green (SOSG), and RPMI 1640 medium were from Thermo Fisher Scientific, Inc. (USA). All chemicals and experimental reagents were from commercial sources and were used as received.4.2 Fabrication and characterization of CMC NPs
Ce6 was dissolved in NaOH solution (1 M, pH = 14) at a concentration of 1 mM. CBL was dissolved in NaOH solution (0.1 M, pH = 13) and then diluted to a final concentration of 16.44 mM with Milli-Q water. Initially, Ce6 (100 mM, 10 μL) and CBL (16.44 mM) were premixed at various molar ratios: 1:1, 1:3, 1:5 and 1:7. Then, the drug mixture was added to Milli-Q water with a total reaction volume of 800 μL. Subsequently, a MnCl2 aqueous solution (10 mM, 200 μL) was added dropwise to the mixture under agitation, followed by the addition of an HCl aqueous stock solution (0.1 M, pH = 1.0) to achieve a final neutral pH. The mixture was further vortexed for 48 h to yield CMC NPs. The resultant CMC suspension was dialyzed (MWCO = 3500 Da) to remove non-coordinated molecules.
The size distribution, polydispersity index (PDI) and zeta potential of the CMC NPs suspended in 1× PBS were measured using a dynamic light scattering instrument (DLS, Brookhaven, USA). The morphology of the as-prepared CMC NPs was characterized using a transmission electron microscope (TEM, Talos-L120C, USA). The ultraviolet-visible (UV-vis) and fluorescence spectra of CMC NPs were recorded using a UV-vis spectrophotometer (UV-1800, Shimadzu, Japan) and a fluorescence spectrophotometer (RF-6000, Shimadzu, Japan), respectively. The amount of Mn2+ in CMC NPs was quantified using an inductively coupled plasma optical emission spectrometer (ICP-OES, Avio 500 series, Singapore).
4.3 The stability of CMC NPs
To simulate CMC NPs under different environments and evaluate their stability, the stock solution was diluted to 200 μM using either PBS or 10% fresh fetal bovine serum (FBS). Subsequently, the solution was incubated at 4 °C, and DLS measurements were conducted over a period of 14 days to monitor any changes in the size or PDI of the nanoparticles.4.4 Phototherapeutic effect of CMC NPs
For photothermal evaluation, CMC NPs at different concentrations (0.01, 0.05, 0.1, 0.5, 1, 5, 10 and 50 μM) were irradiated with a 680 nm laser (0.4 W cm−2, 3 min). The temperature changes were continuously recorded throughout the experiment using an FLIR thermal camera. To assess the photothermal stability of CMC NPs, the temperature was measured every 15 s during three cycles between “heat-up” and “cool-down” progress. Specifically, 50 μM CMC NPs were continuously exposed with the aforementioned laser parameters for 3 min to reach steady-state conditions before removing the laser source, subsequently allowing the natural cooling down of the solution containing CMC NPs to ambient temperature over several minutes. A method previously established by Roper et al.27 was introduced to calculate the photothermal conversion efficiency of CMC NPs. ROS generation in the system was detected by using SOSG as the probe according to the manufacturer's instructions.4.5 Release profile of CMC NPs
To evaluate the GSH-triggered release profile of CMC NPs in vitro, the stock solution was diluted to 200 μM using Milli-Q water. Subsequently, 2 mL of CMC NPs (n = 3) was added to a dialysis tube (MWCO 3500 Da) and immersed in 60 mL of buffer solution at 37 °C under agitation, with or without GSH supplementation. At specific time intervals, 1 mL of the released buffer was withdrawn for analysis using UV-vis spectroscopy, while an equal volume of fresh buffer solution was replenished to maintain continuous dialysis over time. Ce6 served as a surrogate to monitor the drug release, and its release profile was determined according to a standard curve. Prior to conducting the release experiments, dialysis was performed to remove any free drugs.4.6 Cell culture
The 4T1 murine breast cancer cell line was cultured in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin–streptomycin at 37 °C in a standard cell incubator with 5% CO2. The 4T1 cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA).4.7 Cellular uptake and intracellular ROS generation
4T1 cells (8 × 104 per well) were seeded in 4-well microscopy chambers for 12 h incubation, followed by culture in a fresh medium containing CMC NPs or free Ce6 for an additional 2 or 4 h, respectively. At predesignated time points, the supernatant was removed and the cells were then washed with PBS several times. The fluorescence intensities of Ce6 in cells represented the cellular uptake of CMC NPs. To further confirm the intracellular distribution, the cells were stained with Hoechst 33342 (2 μg mL−1) and Lysotracker (500 nM) for 10 min before being observed by CLSM.DCFH-DA was employed as a fluorescent probe to assay the intracellular ROS generation resulting from CMC NPs. After being seeded in 4-well microscopy chambers and precultured for 12 h, the cells were treated with different materials for another 4 h. Next, the material-containing medium was removed and subsequently replenished withfresh culture medium. The cells were irradiated with and without 680 nm laser exposure (0.4 W cm−2, 1 min) and immediately stained with DCFH-DA (10 μM) in a dark environment for 30 min. Then, the fluorescent probe was washed off and fresh culture medium was promptly replaced in each chamber. Cellular ROS generation was assessed by observing the DCF fluorescence intensity with confocal laser scanning microscopy (CLSM). The aforementioned treating concentration of CMC NPs was 20 μM based on Ce6.
4.8 In vitro cytotoxicity assay
The in vitro cytotoxicity effect was examined using the MTT assay. 4T1 cells (1 × 104 cells per well) were plated in 96-well plates and cultured overnight. Thereafter, fresh medium containing various concentrations of Ce6, CBL and CMC NPs (0.5, 1, 2, 5, 10, 20, and 50 μM, based on Ce6) was added to the dish and cocultured with cells for a duration of 24 h. Next, the material-containing medium was washed off and replenished with fresh medium followed by exposure to 680 nm laser irradiation (0.4 W cm−2, 90 s) and incubation for 2 h. Afterward, 30 μL of MTT solution (3 mg mL−1 in fresh medium) was added to each well and co-incubated with cells for another 4 h in the dark. At the end of incubation, the medium was substituted with dimethyl sulfoxide (DMSO, 150 μL) before the absorbance of solution was measured at 490 nm using a microplate reader. The combination index (CI) between CBL-induced chemotherapy and Ce6-mediated phototherapy (Ce6 + L) was assessed via CompuSyn software.4.9 Live–dead cell staining assay
The live–dead cell staining assays were further conducted to confirm the cytotoxicity of CMC NPs. 4T1 cells (2 × 105 cells per well) were seeded in 4-well microscopy chambers and precultured for 12 h. The culture medium was replaced with a medium containing 20 μM CMC NPs or free Ce6. After incubation for 4 h, the cells were washed with PBS several times and then irradiated with or without 680 nm laser irradiation (0.4 W cm−2, 3 min). To identify and visualize the viability of cells, the material-treated cells were co-stained with DiOC6(3) (40 nM) and PI (4 μM) in fresh medium for 15 min, followed by CLSM observation.4.10 The establishment of a 4T1 mouse model
Healthy female Balb/c mice were procured from Vital River Laboratory Animal Technology Co., Ltd (Shanghai, China), and were all 6 weeks old. These mice were then acclimatized under specific pathogen-free (SPF) conditions for 7 days prior to the commencement of the experiments. All animal procedures were handled under the protocol approved by the Institutional Animal Use and Care Administrative Advisory Committee of Shanghai Jiao Tong University (Approval number: A2024064). To establish the 4Tl tumor-bearing mouse model, murine 4T1 breast cancer cells (2 × 106) suspended in a 50 μL mixture containing 10% Matrigel were subcutaneously injected into the left flank of each mouse. The animal studies were initiated on day 0 when the tumors reached a volume of approximately 50–60 mm3.4.11 Tumor accumulation and biodistribution
Free Ce6 and CMC NPs were intravenously administered to tumor-bearing mice (n = 3) separately for in vivo quantitative biodistribution analysis. Fluorescence images (Ex 675 nm, Em 720 nm) were acquired at predesignated time points (0, 2, 4, 6, 8, 12, and 24 h) using an IVIS imaging system (PerkinElmer, USA). To investigate organ distribution, a new set of tumor-bearing mice were intravenously injected with free Ce6 or CMC NPs. At 6 h post-injection (based on tumor accumulation results), the mice were sacrificed. Subsequently, the tumors and major organs such as the heart, liver, spleen, lung, and kidney were harvested and the following ex vivo imaging analysis was conducted. To assess the infiltration of free Ce6 or CMC NPs in tumor tissues, visual frozen sections that could be used for CLSM observation were prepared. Specifically, harvested tumors were embedded in OCT compound and frozen at −80 °C, followed by sectioning at ∼20 μm and staining with Hoechst 33342.4.12 Evaluation of the antitumor effect
The 4T1 tumor-bearing mice were randomly divided into five groups (n = 6), when the tumor reached an approximate size of 50 mm3. Subsequently, tail-vein injection was implemented separately every three days (five doses in total) with different materials (PBS, Mix and CMC NPs) at an equivalent dose of 10 mg kg−1 Ce6. To evaluate the curative effect of phototherapy in combinational therapy, 680 nm laser irradiation (0.4 W cm−2, 5 min) was applied at 6 h post-injection, guided by tumor accumulation results. Tumor volume and body weights were strictly monitored during the evaluation period. Then, tumor size was assessed using a caliper, and tumor volume was calculated using the following formula: (width2 × length)/2. After 14 days of treatment, all the mice were euthanized. The tumors and major organs (heart, liver, spleen, kidneys, and lung) were dissected and preserved in a 4% formaldehyde solution for histological analysis. Tumor nodules in the lungs were also observed and counted via H&E staining.4.13 Intratumoral ROS detection
Intratumoral ROS detection was performed by following a similar protocol to that described for intracellular ROS generation. Specifically, the tumors were irradiated with 680 nm laser exposure (0.4 W cm−2, 5 min) at 6 h post-injection, guided by tumor accumulation results. The mice were all intratumorally injected with DCFH-DA (200 μM) before various treatments. Subsequently, they were sacrificed, and their tumors were harvested for freezing microtome sectioning and CLSM observation.4.14 Systemic toxicity of CMC NPs
Prior to initiating the antitumor studies, biocompatibility evaluation was first conducted. The red blood cells (RBC) from healthy female Balb/c mice were collected and incubated with different concentrations (0.1, 0.5, 1, 5, 10 and 50 μM) of CMC NPs at 37 °C for 30 min. Subsequently, centrifugation was performed for direct hemolysis observation. The RBC in PBS solution (1×) and the RBC in Triton X-100 (1%) served as negative and positive controls, respectively. Meanwhile, the healthy female Balb/c mice were injected intravenously with CMC NPs at an equivalent dose of 10 mg kg−1 Ce6. The body weights of mice were monitored strictly every other day. Blood samples from these mice were collected in two weeks to carry out hematological analysis using an automatic blood analyzer (Sysmex Japan). Furthermore, the major organs (heart, liver, spleen, kidneys, and lung) in the healthy mice were harvested and fixed with paraformaldehyde for further H&E assays.Next, the biosafety was also further evaluated in tumor-bearing mice. The measurement of the body weights of mice was performed during the evaluation period. At the indicated time, blood sample (approximately 200 μL) was collected through the tail vein to perform hematology and biochemistry assays (n = 3). The biochemistry assays were performed using an automatic biochemical analyzer (Mindray, China). Then, the mice were euthanized and their major organs (heart, liver, spleen, kidneys, and lung) were fixed with paraformaldehyde and then stained with H&E. Mice that did not receive any material administration served as the control group.
4.15 Statistical analysis
All experimental data are presented in this article as mean ± standard deviation (SD). Statistical analysis was carried out with GraphPad Prism 8.0 software. Statistical evaluation between different groups was conducted using the Student's t-test or one-way ANOVA as appropriate. Asterisks indicate significant differences in comparison between different groups (*P < 0.01, **P < 0.005, and ***P < 0.001).Author contributions
Conceptualization, Xue, X.; methodology, Chen, H. and Qu, H.; validation, Xue, X. and Hu, R; formal analysis, Chen, H. and Lu, B.; investigation, Chen, H., Lu, B., Qu, H., Pan, Y., Yang, J., Wu, J., Wang, C. and Wang, Y; resources, Xue, X. and Hu, R; data curation, Chen, H and Xue, X.; writing—original draft preparation, Chen, H.; writing—review and editing, Xue, X.; visualization, Chen, H., Lu, B., Qu, H., and Duan, Z.; supervision, Xue, X. and Hu, R.; project administration, Xue, X. and Hu, R; funding acquisition, Xue, X. and Hu, R.Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the support from the National Natural Science Foundation of China (82172084, 81803002, and 32171246), STI2030-Major Projects (2022ZD0212500) and Shanghai Frontiers Science Center of Drug Target Identification and Delivery (ZXWH2170101). The Scheme was drawn using Biorender.com.References
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2016, 1, 16075 CrossRef.
- X. Sun, S. Zhang, S. Jiang, J. Shen, Y. Wu, H. Zhang, M.-R. Zhang, R. Wang and K. Hu, iRADIOLOGY, 2024, 2, 103–112 CrossRef.
- Y. Zhang, T. Ren, J. Gou, L. Zhang, X. Tao, B. Tian, P. Tian, D. Yu, J. Song, X. Liu, Y. Chao, W. Xiao and X. Tang, J. Controlled Release, 2017, 261, 352–366 CrossRef CAS PubMed.
- R. X. Zhang, T. Ahmed, L. Y. Li, J. Li, A. Z. Abbasi and X. Y. Wu, Nanoscale, 2017, 9, 1334–1355 RSC.
- H. Cui and X. Chen, Adv. Drug Delivery Rev., 2017, 110–111, 1–2 CrossRef CAS PubMed.
- H. Su, J. M. Koo and H. Cui, J. Controlled Release, 2015, 219, 383–395 CrossRef CAS PubMed.
- A. G. Cheetham, R. W. Chakroun, W. Ma and H. Cui, Chem. Soc. Rev., 2017, 46, 6638–6663 RSC.
- P. Huang, J. Ao, L. Zhou, Y. Su, W. Huang, X. Zhu and D. Yan, Bioconjugate Chem., 2016, 27, 1564–1568 CrossRef CAS PubMed.
- W. Qu, Q. Yang, G. Wang, Z. Wang, P. Huang, W. Huang, R. Zhang and D. Yan, RSC Adv., 2020, 10, 8958–8966 RSC.
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef CAS PubMed.
- Y. Yuan, R. Bo, D. Jing, Z. Ma, Z. Wang, T.-y. Lin, L. Dong, X. Xue and Y. Li, Nano Res., 2020, 13, 503–510 CrossRef CAS.
- X. Xue, Y. Huang, X. Wang, Z. Wang, R. P. Carney, X. Li, Y. Yuan, Y. He, T.-y. Lin and Y. Li, Biomaterials, 2018, 161, 203–215 CrossRef CAS PubMed.
- H. Qu, L. Li, H. Chen, M. Tang, W. Cheng, T.-y. Lin, L. Li, B. Li and X. Xue, J. Controlled Release, 2023, 363, 361–375 CrossRef CAS PubMed.
- Z. Wang, S. Zhang, Z. Kong, S. Li, J. Sun, Y. Zheng, Z. He, H. Ye and C. Luo, Cell Rep. Med., 2023, 4, 101014 CrossRef CAS PubMed.
- H. Zhang, Z. Zhao, S. Sun, S. Zhang, Y. Wang, X. Zhang, J. Sun, Z. He, S. Zhang and C. Luo, Nat. Commun., 2023, 14, 255 CAS.
- H. Qu, H. Chen, W. Cheng, Y. Wang, Y. Xia, L. Zhang, B. Ma, R. Hu and X. Xue, Acta Biomater., 2023, 164, 407–421 CrossRef CAS PubMed.
- X. Xue, A. Lindstrom, H. Qu and Y. Li, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 12, e1607 Search PubMed.
- X. Xue, H. Qu and Y. Li, Exploration, 2022, 20210134 CrossRef PubMed.
- X. Xue, H. Qu, R. Bo, D. Zhang, Z. Zhu, B. Xiang, L. Li, M. Ricci, C.-X. Pan, T.-Y. Lin and Y. Li, Biomaterials, 2023, 299, 122145 CAS.
- H. Qu, H. Chen, W. Cheng, Y. Pan, Z. Duan, Y. Wang, X.-J. Liang and X. Xue, Nano Res., 2023, 16, 13267–13282 CAS.
- X. Xue, Y. Huang, R. Bo, B. Jia, H. Wu, Y. Yuan, Z. Wang, Z. Ma, D. Jing, X. Xu, W. Yu, T.-y. Lin and Y. Li, Nat. Commun., 2018, 9, 3653 CrossRef PubMed.
- R. Zhang, Y. Duan and B. Liu, Nanoscale, 2019, 11, 19241–19250 RSC.
- H. Chen, P. Timashev, Y. Zhang, X. Xue and X.-J. Liang, RSC Adv., 2022, 12, 9725–9737 RSC.
- J. Yan, G. Wang, L. Xie, H. Tian, J. Li, B. Li, W. Sang, W. Li, Z. Zhang and Y. Dai, Adv. Mater., 2022, 34, 2105783 CrossRef CAS PubMed.
- H. Tian, G. Wang, W. Sang, L. Xie, Z. Zhang, W. Li, J. Yan, Y. Tian, J. Li, B. Li and Y. Dai, Nano Today, 2022, 43, 101405 CrossRef CAS.
- H. Zhang, K. Liu, S. K. Li, X. Xin, S. L. Yuan, G. H. Ma and X. H. Yan, ACS Nano, 2020, 14, 16157–16157 CrossRef CAS PubMed.
- D. K. Roper, W. Ahn and M. Hoepfner, J. Phys. Chem. C, 2007, 111, 3636–3641 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02101k |
| This journal is © The Royal Society of Chemistry 2024 |