
Recent progress in atomically precise metal nanoclusters for photocatalytic application
Yuanxin Du*,
Chengqi Li,
Yali Dai,
Haijiao Yin and
Manzhou Zhu *
*
Department of Materials Science and Engineering, Centre for Atomic Engineering of Advanced Materials, Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Key Laboratory of Functional Inorganic Material Chemistry of Anhui Province, Anhui University, Hefei 230601, China. E-mail: duyuanxin@ahu.edu.cn; zmz@ahu.edu.cn
First published on 24th June 2024
Abstract
Photocatalysis is a widely recognized green and sustainable technology that can harness inexhaustible solar energy to carry out chemical reactions, offering the opportunity to mitigate environmental issues and the energy crisis. Photocatalysts with wide spectral response and rapid charge transfer capability are crucial for highly efficient photocatalytic activity. Atomically precise metal nanoclusters (NCs), an emerging atomic-level material, have attracted great interests owing to their ultrasmall size, unique atomic stacking, abundant surface active sites, and quantum confinement effect. In particular, the molecule-like discrete electronic energy level endows them with small-band-gap semiconductor behavior, which allows for photoexcitation in order to generate electrons and holes to participate in the photoredox reaction. In addition, metal NCs exhibit strong light-harvesting ability in the wide spectral UV–near IR region, and the diversity of optical absorption properties can be precisely regulated by the composition and structure. These merits make metal NCs ideal candidates for photocatalysis. In this review, the recent advances in atomically-precise metal NCs for photocatalytic application are summarized, including photocatalytic water splitting, CO2 reduction, organic transformation, photoelectrocatalytic reactions, N2 fixation and H2O2 production. In addition, the strategy for promoting photostability, charge transfer and separation efficiency of metal NCs is highlighted. Finally, a perspective on the challenges and opportunities for NCs-based photocatalysts is provided.
1. Introduction
Nanocatalyst research has been flourishing over the past few decades. With the improvement of catalysts’ controllable synthesis methods and fine characterization analysis techniques,1 the current nanocatalyst family can be classified into relatively large sized nanoparticles (NPs), mid-sized nanoclusters (NCs) with a size less than 2 nm, and ultrasmall single-atom catalysts (SACs). The study of NPs, the starting point of nanocatalysis, reveals the contribution of the low coordination environment and quantum size effect on the metal surface to the ultra-high catalytic activity. However, just as nanochemists have been disturbed by the fact that no two NPs are identical, the polydispersity of NPs (i.e. the heterogeneity in size, morphology, and surface environment) makes it difficult to achieve tailored catalytic performance based on the structure–activity relationship. Metal NCs are composed of few to a few hundred metal atoms and bridge the gap between NPs and SACs. With the advantages of the ultrasmall size, abundant surface unsaturated coordinative sites, unique atomic stacking mode, and quantum confinement effect, metal NCs have received great attention in catalysis. Unlike the continuous energy band in NPs and single atomic orbital in SACs, metal NCs exhibit a discrete energy level and behave like molecules, and some larger ones even span nonmetallic-to-metallic regimes. Especially, in such a small size range, the quantum confinement effect becomes much more significant, and even one atom change will bring about a huge change in the catalytic activity. In addition, metal NCs with a precise composition, structure, and surface environment provide the opportunity to investigate in depth the catalytic mechanism and build the catalyst structure–property relationship.2–5 A clear structure and high atom utilization are also the advantages of SACs;6 however, there is no single atom in the true sense because a single atom cannot exist without a support. Besides, it is difficult to simultaneously activate multiple reactants by SACs to achieve high activity in a complex reaction. Recently, some studies have reported that few-atom catalysts show superior activity than SACs in certain reactions.In terms of photocatalysis, the ideal catalyst needs to have strong light-harvesting ability, can effectively generate photoinduced electrons and holes, and rapidly transfer and separate charge carriers.7–9 Due to the surface plasmon resonance (SPR) effect induced by the collective oscillations of electrons on the surface of the metal NPs, conventional Au and Ag NPs exhibit unique size or morphology-dependent optical absorbance property. However, the optical absorption range is usually limited to the visible light region, for example, the typical SPR absorption peak of Au and Ag NPs is in the range of 520–570 nm and 450–500 nm, respectively. Unlike the narrow visible light absorption of plasmonic metal NPs, metal NCs exhibits wide light absorption range from the UV to the near IR region. Moreover, the absorption property of metal NCs is various and diverse, which can be precisely regulated by the composition and structure.10,11 On the other hand, the ultrasmall size endows metal NCs with discrete electronic energy level-like molecular behavior different from the metal-like behavior of large metal NPs.12 Metal NPs themselves cannot be excited to generate electrons and holes and usually act as the co-catalyst to improve the photocatalytic activity. For example, the Schottky junction is formed at the interface when metal NPs contact with the semiconductor, and it can build an effective one-way electron transfer channel for facilitating the photogenerated charge transfer and separation. However, metal NCs with the highest occupied molecular orbital (HOMO) and the lowest occupied molecular orbital (LUMO) gap can serve as a small-band-gap semiconductor and exhibit the capability of generating electrons and holes under light excitation.13–15 Theoretically, metal NCs themselves can be used as the photocatalyst or it can be combined with another semiconductor to construct a heterojunction photocatalyst. Therefore, metal NCs with the merits of strong light response and semiconductor-like characteristic show great application potentials in photocatalysis (Scheme 1).
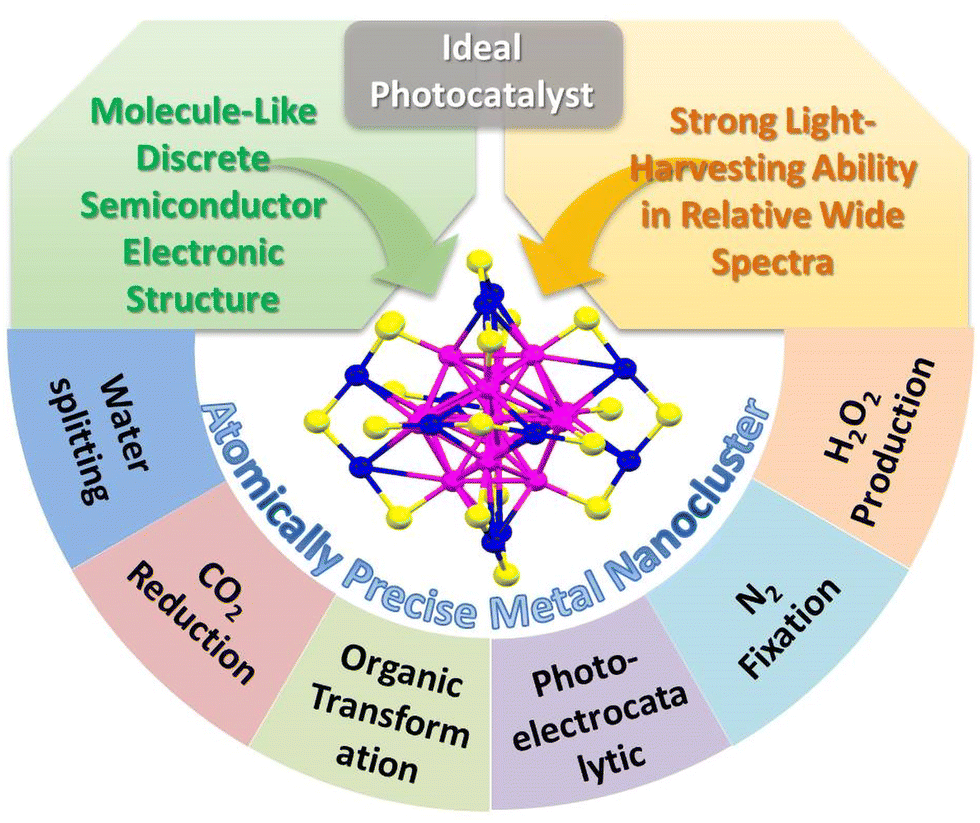 | ||
| Scheme 1 The merits and applications of atomically-precise metal NCs as ideal candidates for photocatalysis. | ||
The application of metal NCs in catalysis has developed rapidly and is in a state of explosion in recent years; however, the application in photocatalysis is still lagging behind the development of electrocatalysis, thermal catalysis, liquid phase catalysis, and so on.11,16–20 The ultrasmall size is a double-edged sword, although it contributes to the abundant catalytic active sites and unique electronic structure; it causes high surface energy, leading to the unfavorable agglomeration of metal NCs during the photocatalytic reaction. In addition to the poor photostability, the ultrasmall size makes charge carrier recombination easy, resulting in a short carrier lifetime. Recently, various synthesis strategies have been developed to construct NCs-porous support composites to avoid the aggregation of NCs and provide a synergic effect for enhanced catalysis; especially, NCs encapsulated in metal organic frameworks (MOFs) is a classical and effective method for improved catalytic stability and activity.21–23 Inspired by these NCs-based composites works, many researches propose a strategy of coupling with the other material to fabricate the NCs-based heterostructure to improve the photostability and prolong the carrier lifetime of NCs in order to overcome the inherent defects.
This review includes the recent progress of atomically-precise metal NCs in photocatalytic water splitting, CO2 reduction, organic transformation, photoelectrocatalytic reaction, and other reactions such as N2 fixation and H2O2 production over the last five years (Scheme 1). The strategy of constructing the NCs-based heterostructure with improved photostability and charge transfer and separation efficiency is summarized and highlighted. In addition, the current challenges and future opportunities faced by NCs-based photocatalysts are provided.
2. Photocatalytic applications of NCs
2.1 Photocatalytic water-splitting
With the proposal of the carbon emission reduction plan and the deepening of the green economy plan, hydrogen energy has become the focus of the world's attention. Due to the low density, high calorific value, and zero-pollution during combustion, hydrogen is called the “ideal energy source” in the 21st century. With the development of fuel cells and new energy vehicles, hydrogen energy is expected to replace fossil energy and officially enter the world energy stage. Photocatalytic water splitting technology can use ready-made solar energy to produce hydrogen from water and has been considered as a clean and environment-friendly hydrogen production method.24–29 Generally, photocatalytic water splitting includes three main steps: firstly, the formation of photogenerated electron–hole pairs, secondly, the separation and migration of the generated electron and hole, finally, the participation in water splitting to produce H2 and O2. From the aspect of thermodynamics, the energy position of the conduction band (CB) and valence band (VB) of the photocatalyst is required to be more negative and positive than that of H+/H2 (0 V vs. NHE) and O2/H2O (1.23 V vs. NHE) for photocatalytic H2 and O2 evolution, respectively.2,25 In 2013, Negishi et al. loaded glutathione-protected Au25(GSH)18(GSH = Glutathione) NCs on BaLa4Ti4O15 as co-catalysts, which increased the photocatalytic water splitting activity by 2.6 times.30 Since then, atomically precise metal NCs have been widely applied to photocatalytic water splitting. Here, only the related researches in the last 5 years are summarized.Pt NCs-based photocatalyst
Due to the excellent photogenerated carrier separation ability, Pt-based materials have been regarded as one of the best co-catalysts. However, the high cost limits the wide application. Pt NCs with small size and high mass activity attract great attention. Lu et al. synthesized Pt5(GSH)10 NCs and immobilized them on multi-armed CdS nanorods (NRs).31 The presence of Pt-S4 active sites were confirmed by X-ray absorption fine structure (XAFS) spectroscopy. Based on the results of ultrafast transient absorption (TA) spectroscopy, Pt NCs could extract the photoinduced electrons of CdS NRs, leading to effective charge separation. Therefore, the Pt5–CdS composite exhibited an improved photocatalytic H2 production activity of 13.0 mmol h−1 g−1 (under 300 W xenon lamp irradiation with λ > 420 nm), 5.9 times higher than that of CdS NRs.The ultrasmall size of NCs endows not only high activity but also poor stability. The porous material can act as a support to confine the NCs, preventing their growth or aggregation. Huang et al. utilized MIL-125-NH2 as a support to encapsulate Pt NCs based on the in situ auto-reduction method.32 Firstly, the amino groups of MIL-125-NH2 reacted with HCHO to form MIL-125-CH2OH with reduction ability. Secondly, MIL-125-CH2OH was immerged in Pt salt precursor solution. Finally, the Pt NCs were obtained by in situ auto-reduction without any additional reducing agent (Fig. 1(a)). The obtained Pt NCs were uniformly dispersed in the cavities of MIL-125-CH2OH with the size of 1–2 nm. With the optimal Pt NCs amount, the Pt(1.5)/MIL-125-NH-CH2OH showed the highest photocatalytic H2 production rate of 4496.4 μmol g−1 h−1 under λ > 420 nm visible light irradiation in the mixture of CH3CN and H2O system with triethanolamine (TEOA) as the sacrificial agent. Due to the high reactive property of the amino groups, it is speculated that other metal NCs-based composites can be synthesized through the in situ auto-reduction method for other applications.
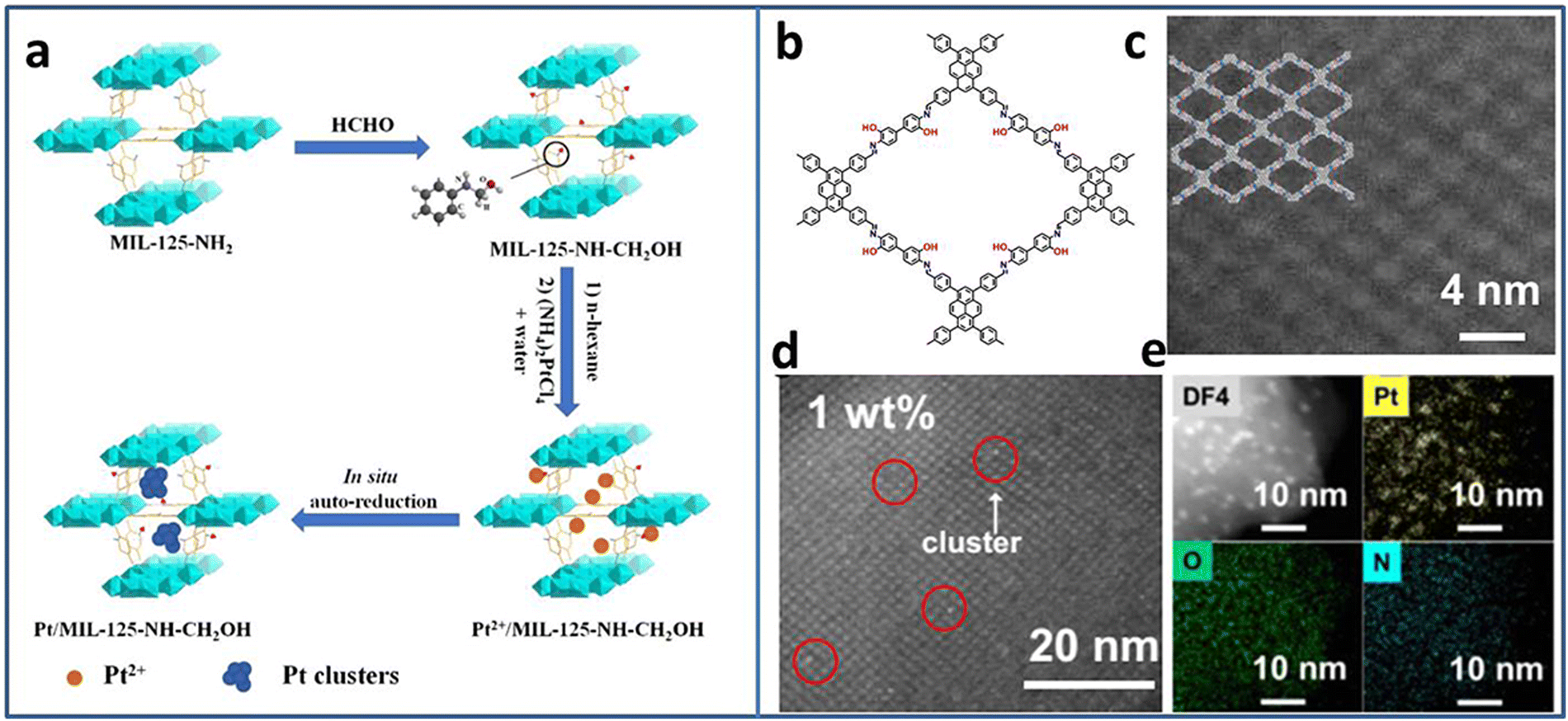 | ||
| Fig. 1 (a) Steps for the synthesis of Pt/MIL-125-NH-CH2OH. (b) The structure and (c) the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of PY-DHBD-COF. (d) HAADF-STEM image and (e) EDX mapping of 1 wt% Pt-loaded PY-DHBD-COF. Reproduced with permission from ref. 32,33, copyright Springer Nature. | ||
Similar to MOFs, covalent organic frameworks (COFs) are another kind of porous functional support consisting of various ligands connected by covalent bonds. Due to the adjustability of the structure, it provides the opportunity to rationally design and control the deposition of metal NCs. Li et al. synthesized a novel COF named PY-DHBD-COF with an adjacent hydroxyl group and imine bond in each constitutional unit (Fig. 1(b)–(e)).33 The periodically distributed –OH and imine-N sites created an adsorption environment for Pt species. Under light irradiation, the adsorbed Pt ions were in situ reduced to Pt NCs by photogenerated electrons. The optimized Pt NCs-COF with 1 wt% Pt NCs loading exhibited a high photocatalytic H2 generation activity of 42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 432 μmol g−1 h−1 (under λ > 420 nm visible light irradiation in H2O with ascorbic acid as the sacrificial agent) due to the exposed large active surface.
432 μmol g−1 h−1 (under λ > 420 nm visible light irradiation in H2O with ascorbic acid as the sacrificial agent) due to the exposed large active surface.
Metal NCs can serve as a photosensitizer to enhance the photocatalytic activity; however, sometimes the poor stability limits their application. Dai et al. reported that metal NCs could spontaneously transform to metal NPs under light irradiation, which may result in reduced photoredox activity.35 Fu et al. compensated the photosensitization loss of metal NCs by judiciously utilizing self-transformed metal NPs as interfacial Schottky-type charge mediators to build unidirectional charge flow for enhancing the charge transfer efficiency.36 The CdS@CdTe@PDDA@Aux (PDDA = poly(dialyldimethylammonium chloride)) multilayered core–shell heterostructure was fabricated in the following three steps: first, partial transformation of CdS nanowires (NWs) to ultrathin CdTe layer, then the modification of the PDDA interim layer to convert the surface charge state, and finally, the electrostatic self-assembly of Aux@GSH (Fig. 2(a)). The electron-trapping ability of the PDDA layer and Schottky-type unidirectional electron flow of self-transformed metal NPs form a long-range cascade charge transfer chain to synergistically accelerate the electron separation on CdS@CdTe; therefore, the photocatalytic H2 generation activity under the visible light irradiation of CdS@CdTe@PDDA@Aux was enhanced.
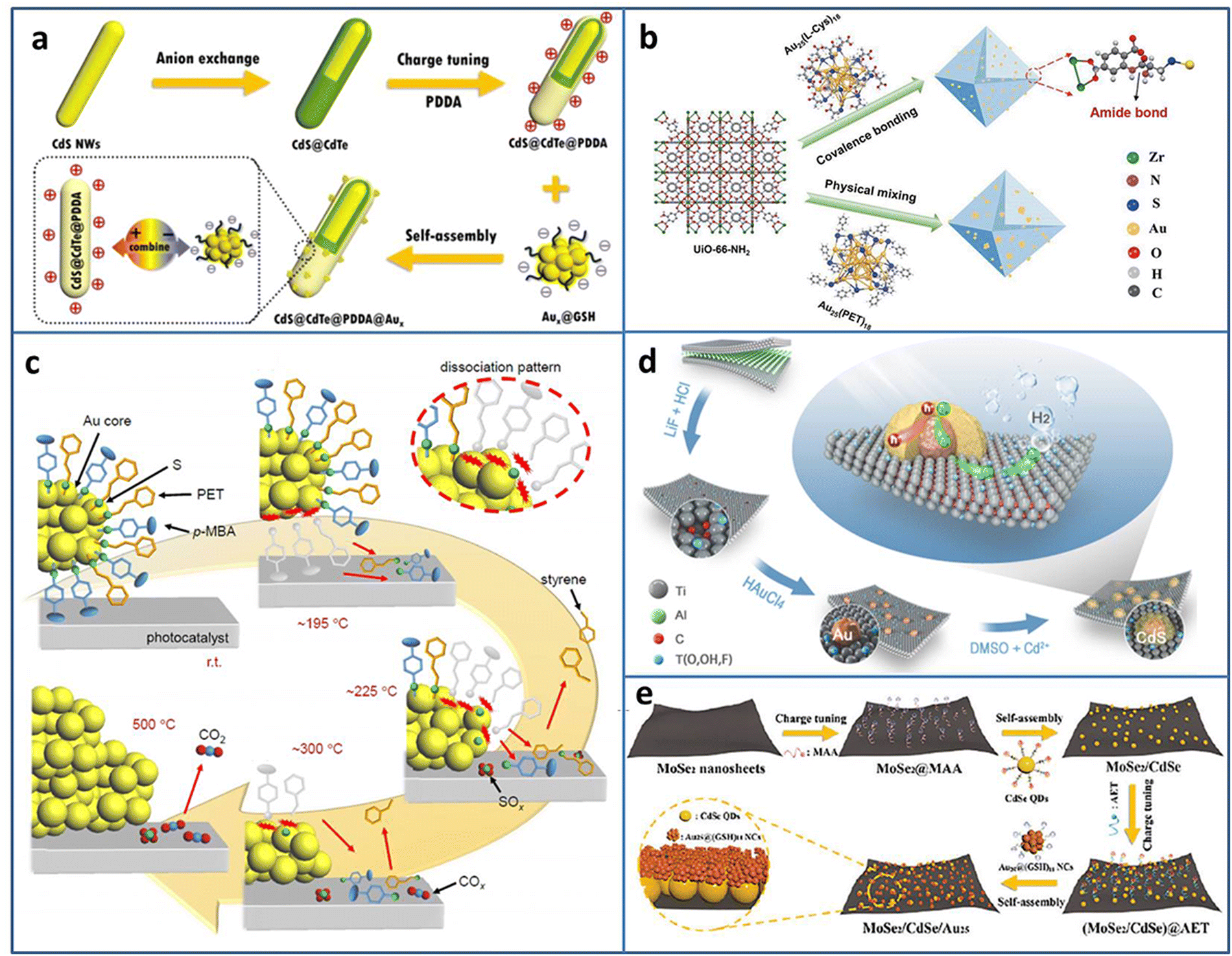 | ||
| Fig. 2 (a) Construction of CdS@CdTe@PDDA@Aux. (b) Covalent binding Au25(L-Cys)18 NCs and UiO-66-NH2. (c) Ligand desorption process during the calcination of Au25(PET, p-MBA)18/BaLa4Ti4O15. (d) Preparation of CdS@Au/MXene. (e) Fabrication of MoSe2/CdSe QDs/Au25 NCs. Reproduced with permission from ref. 36–40, copyright American Chemical Society, Springer Nature, and Wiley-VCH GmbH. | ||
Due to the high surface energy caused by the ultrasmall size of NCs, the easy aggregation induced by the light irradiation results in the catalytic activity loss. Coupling with a photoactive support with a porous structure can not only solve the problem of poor stability of NCs but also improves the catalytic activity by the synergy effect. Yao et al. stabilized Au25(L-Cys)18 NCs on photoactive porous UIO-66-NH2 by the formation of amide covalent bond (Fig. 2(b)).37 Due to the strong metal-support interaction, the obtained UiO-66-NH2-Au25(L-Cys)18 exhibited enhanced photostability and photocatalytic activity for H2 production, compared to the mixed UiO-66-NH2/Au25(PET)18 (PET = 2-phenylethanethiolate). A series of thermodynamic and kinetic experiments revealed that UiO-66-NH2-Au25(L-Cys)18 possessed lower reaction energy barrier, faster charge carrier generation and transfer efficiency, which was the underlying reason for the improved photocatalytic activity.
In the NCs-based heterostructure, the presence of ligands at the interface usually prevents the contact between the reactant and NCs, leading to a decrease in the catalytic activity. Calcination can remove some ligands. However, over calcination will lead to NCs aggregation and loss of the specific size-related activity of NCs. Kawawaki et al. explored the influence of calcination on the ligand removal process and the size change of NCs by taking the metal-oxide-supported 2-phenylethanethiolate protected Au25 NCs as the examples.38 Ligand desorption with the increase in temperature takes [lace in three steps: (1) the ligand dissociation on the NCs surface; (2) the generated compounds adsorption on the support; (3) the compounds desorption from the support (Fig. 2(c))]. By controlling the calcination process, a water-splitting photocatalyst based on NCs with high activity and stability was created.
Constructing metal NCs-based heterostructure is an effective way to promote carriers separation and improve the photocatalytic activity. In addition to the well-known wide-band-gap TiO2, some novel semiconductors with excellent photocatalytic activity have emerged in recent years. Li et al. utilized reductive Ti vacancy in Ti3−xC2Ty MXene to in situ construct core–shell CdS@Au (Fig. 2(d)).39 The ternary CdS@Au/MXene exhibited two Schottky barriers at the interface, and the charge transferred from CdS to Au NCs and Au NCs to MXene. With the optimal Au NCs and Mxene contents, the CdS@Au/MXene showed 5371 μmol g−1 h−1 H2 production rate, 26.6 times higher than that of CdS. Yan et al. fabricated MoSe2/CdSe quantum dots (QDs)/Au25(GSH)18 NCs Z-scheme heterostructure by cascade electrostatic self-assembly method (Fig. 2(e)).40 The possible photocatalytic H2 production pathway of MoSe2/CdSe QDs/Au25 NCs was followed was: CdSe QDs and Au25 NCs could generate photoinduced e− and h+, and an Z-scheme charge transfer was formed between CdSe QDs and Au25 NCs. Due to the metal-like property, MoSe2 served as a terminal electron acceptor to accelerate charge separation. The electron trapped by MoSe2 participated in the reduction of H+/H2O to H2.
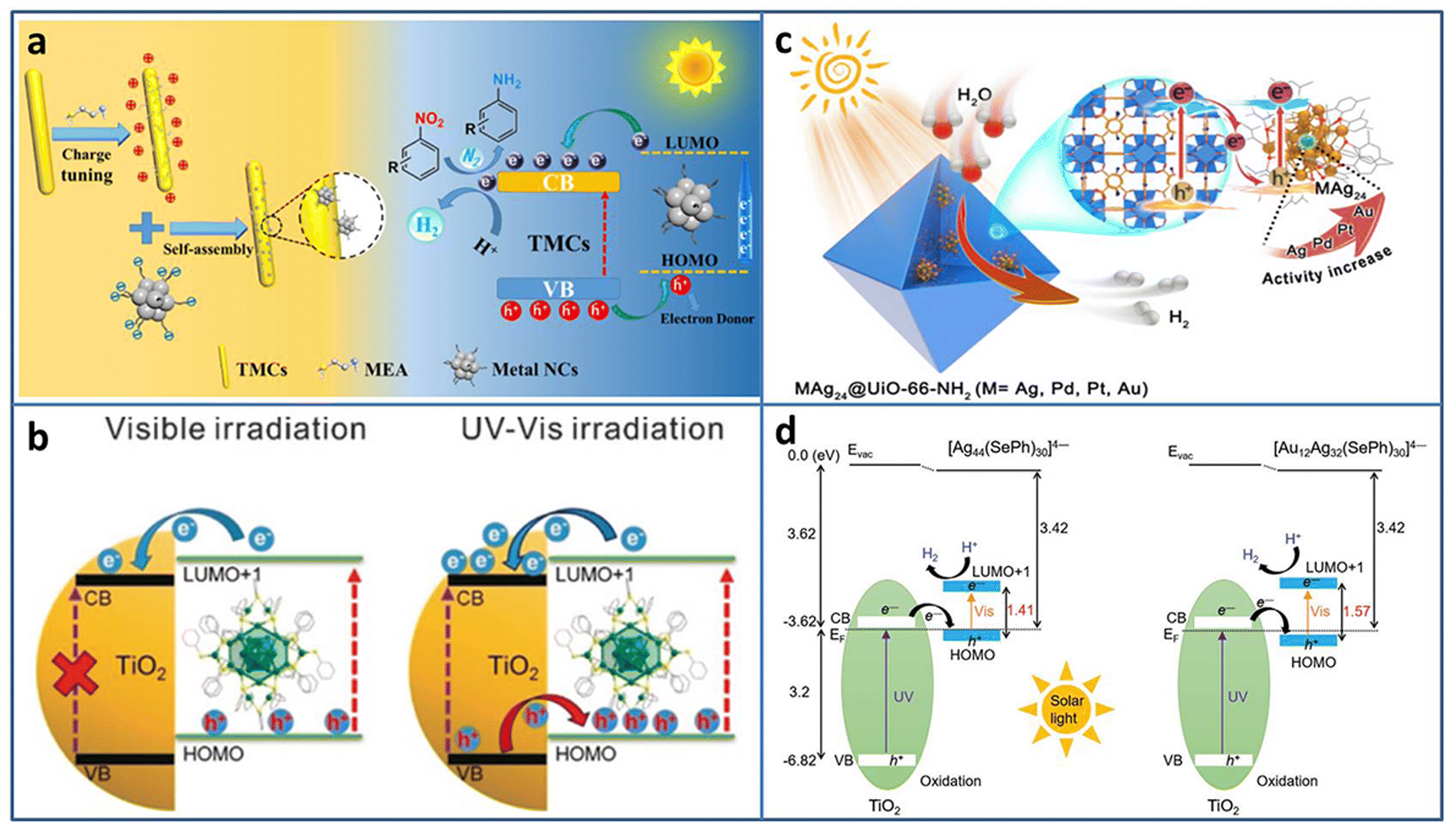 | ||
| Fig. 3 (a) Construction and photocatalytic mechanism of Agx NCs (Ag31, Ag16, Ag9)/TMC composites. (b) The excitation comparison of TiO2–Ag44 NCs under visible/UV-vis light irradiation. (c) Illustration of M1Ag24@UIO-66-NH2 (M = Ag, Pd, Pt, and Au) for photocatalytic H2 production. (d) Energy level diagram of Ag44/TiO2 and Au12Ag32/TiO2. Reproduced with permission from ref. 41–44; copyright American Chemical Society and Wiley-VCH GmbH. | ||
Besides acting as a photosensitizer to expand the light absorption region, metal NCs can be treated as a small-band-gap semiconductor to build a heterojunction. Wang et al. modified TiO2 with Ag44(SR)30 (SR = thiolate) NCs to study the charge transfer behavior at the interface.42 Changing the excitation light region from visible to full solar spectrum, the type-II charge transport pathway was constructed between TiO2 and Ag44 NCs, and thus 3 orders of magnitude increase in the photocatalytic H2 production rate (∼7.4 mmol h−1 gcatalyst−1) was obtained (Fig. 3(b)). By the analysis of ultrafast TA spectroscopy, the role of Ag44 NCs was assigned to the light-absorber and small-band-gap semiconductor co-catalyst under UV/Vis light irradiation. The enhanced light response and charge transfer both contributed to the enhanced photocatalytic activity of the Ag44–TiO2 composite.
Constructing an effective charge transfer channel and driving force is helpful to enhance the photocatalytic activity. Zhu et al. fabricated Pt1Ag28-BTT/CoP (BTT = 1,3,5-benzenetrithiol) NCs-based heterostructure through the Co–S interface.45 Due to the strong build-in electric field and Co–S interfacial charge transfer channel, Pt1Ag28-BTT/CoP shows an obvious improvement in photocatalytic H2 production with 24.89 mmol·h−1·g−1 H2 production rate, which is 1.49, 2.29, and 5.21 times higher than that of Pt1Ag28-BDT/CoP (BDT = 1,3-benzenedithiol), Ag29-BDT/CoP, and CoP. In Pt1Ag28-BTT, central Pt atom doping makes the band alignment between NC and CoP, and the surface S modification provides the opportunity to construct an effective interface charge transport channel, both contributing to the enhanced photocatalytic performance. In addition, the stable and intimate interface connection benefits in the long-term photostability of the NCs-based heterostructure; Pt1Ag28-BTT/CoP shows almost unchanged catalytic activity during five consecutive recycling tests.
To avoid metal NCs from aggregation, Jiang et al. encapsulated M1Ag24 (M = Ag, Pd, Pt, Au) NCs with 2,4-dimethylbenzenethiolate as the ligand into UIO-66-NH2 based on the electrostatic interaction.43 Compared to Ag NPs@UIO-66-NH2, Ag25 NCs@UIO-66-NH2 displayed 5.6 times enhancement in the photocatalytic H2 production rate (under λ > 380 nm light irradiation in the mixture of CH3CN and H2O system with TEOA as the sacrificial agent). The influence of the central heteroatom type on the photocatalytic activity was also investigated; the activity order was Au1Ag24 NCs@UIO-66-NH2 > Pt1Ag24 NCs@UIO-66-NH2 > Pd1Ag24 NCs@UIO-66-NH2 > Ag25 NCs@UIO-66-NH2 (Fig. 3(c)). Femtosecond time-resolved TA spectroscopy demonstrated that the order of charge carrier lifetime of the composite was consistent with that of the photocatalytic activity. X-ray photoelectron spectroscopy (XPS) under the conditions of dark and light irradiation revealed the formation of Z-scheme charge transfer between M1Ag24 NCs and UiO-66-NH2.
Thanks to the development of the synthesis with atomic-level control, the opportunity of investigation of the core–shell nanomaterial structure–property relationship is given. Bootharaju et al. synthesized selenolated Au12Ag32 NCs consisting of an Au icosahedron core and Ag dodecahedron shell by the galvanic exchange method.44 Compared to the homometal analog Ag44 NCs, Au12Ag32 NCs exhibited near-infrared-II photoluminescence (PL) and enhanced stability. Based on the interaction with the oxygen vacancy of TiO2, Au12Ag32 NCs were immobilized on TiO2. Owing to the favorable energy structure of the Au12Ag32 NCs that aligns well with TiO2 for enhanced photogenerated carrier separation, Au12Ag32/TiO2 showed significant enhancement in photocatalytic H2 evolution (6810 μmol g−1 h−1), about 6.2 times higher than that of Ag44/TiO2 (Fig. 3(d)).
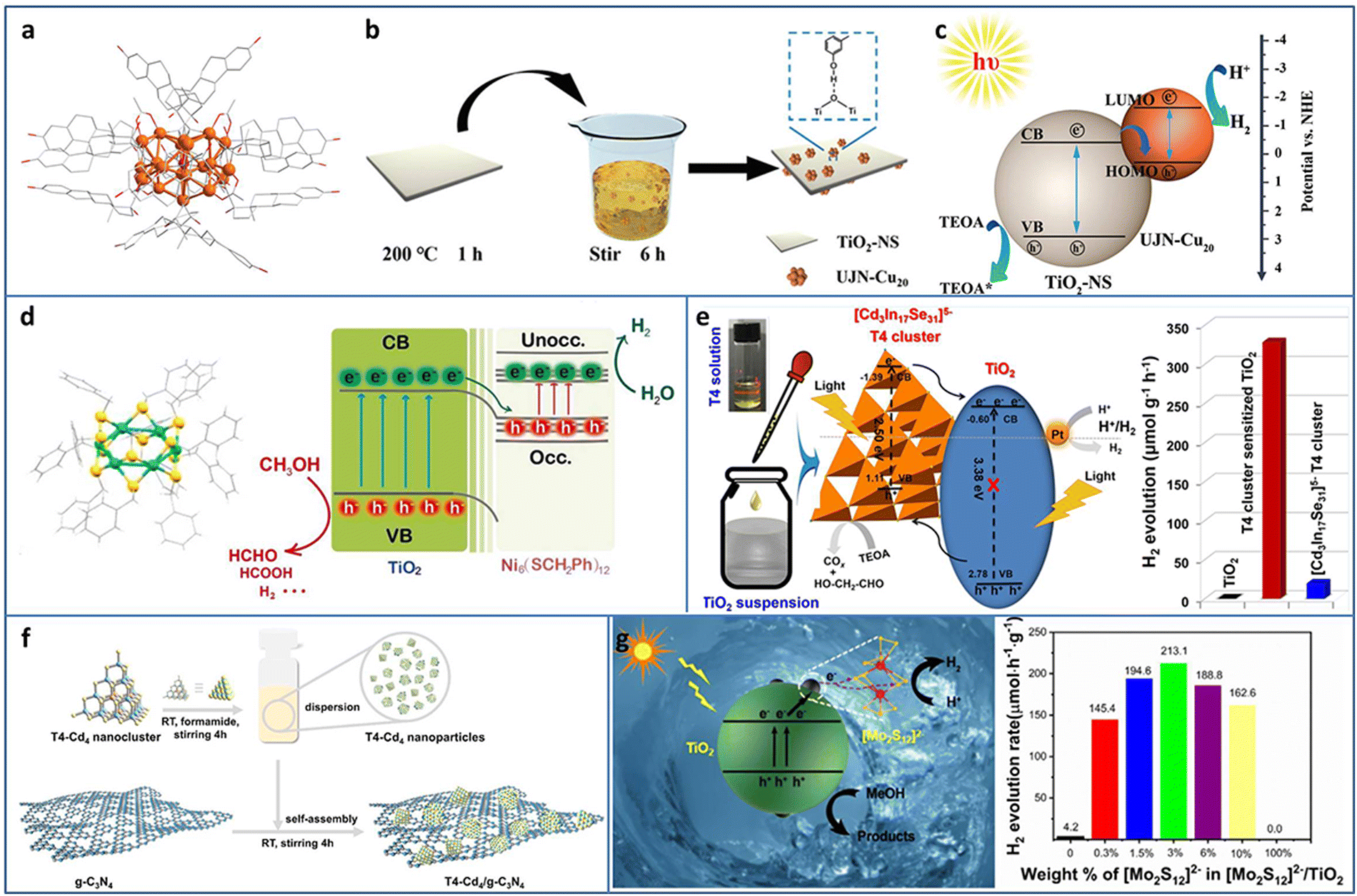 | ||
| Fig. 4 (a) The crystal structure of UJN-Cu20. (b) and (c) Synthesis and photocatalytic H2 evolution mechanism of UJN-Cu20@TiO2. (d) The crystal structure of Ni6 and the photocatalytic charge transfer of Ni6 and TiO2. (e) [Cd3In17Se31]5− T4 NCs/TiO2 for photocatalytic H2 production. (f) Synthesis of T4-Cd4/g-C3N4. (g) [Mo2S12]2− NCs and TiO2 composites for photocatalytic H2 production. Reproduced with permission from ref. 46–50; copyright the Royal Society of Chemistry, Elsevier B.V., and American Chemical Society. | ||
Tian et al. utilized double-crown Ni6(SCH2Ph)12 NCs protected by benzyl thiol ligands as the co-catalyst and deposited it on TiO2 by the solvent evaporation method for enhanced photocatalytic H2 evolution (Fig. 4(d)).47 In the mixture of methanol and water system, Ni6/TiO2 showed an H2 production rate of 5600 μmol gcat−1 h−1, turnover frequency (TOF) of 2800 s−1, and apparent quantum efficiency (AQE) of 7.8% at 365 nm excitation. TA spectroscopy was conducted to investigate the electron migration between Ni6 NCs and TiO2, and the results demonstrated that the photogenerated electron injection was from TiO2 to Ni6 NCs, which was consistent with the XPS and superoxide and hydroxyl radicals trapping results. In addition, the benzyl group may be the photogenerated electron–hole recombination center, which was important for the enhancement in the photocatalytic activity, confirmed by calcination removing ligands and phenethyl or butyl group replacement experiments.
Metal chalcogenide NCs, such as supertetrahedral Tn, Pn, and Cn NCs, can be regarded as the smallest QDs and considered as promising photosensitizers. Wang et al. synthesized supertetrahedral T4 NCs of [Cd3In17Se31]5− with excellent solubility (≥100 mg mL−1 in dimethyl sulfoxide).48 T4 NCs not only played the role of a visible light absorber but also served as a charge separator. The T4 NCs/TiO2 showed a remarkable improvement in the photocatalytic H2 production of 328.2 μmol g−1 h−1 in aqueous solution with TEOA as the sacrificial agent, compared to TiO2 (∼0) and T4 NCs (19.5 μmol g−1 h−1) (Fig. 4(e)). In addition, T4 NCs/TiO2 exhibited high stability with almost no change in the 50 h photocatalytic H2 production test. Wu et al. fabricated a series of T4-Cd4−xZnx/g-C3N4 (x = 0, 1, 2, 3, 4) heterostructures (Fig. 4(f)).49 By tuning Zn doping content, the optimized T4-Cd1Zn3/g-C3N4 with a suitable band structure showed the highest photocatalytic H2 production rate of 288 μmol g−1 h−1 in aqueous solution with TEOA as the sacrificial agent due to the enhanced interfacial charge transfer. Zhang et al. decorated [Mo2S12]2− NCs on TiO2 through a facile impregnation method.50 The intimate contact between [Mo2S12]2− NCs and TiO2 effectively promoted the charge transfer and the abundant bridging S in the [Mo2S12]2− NCs served as active sites; thereby, the [Mo2S12]2− NCs/TiO2 displayed ∼51 times enhancement in the photocatalytic H2 production rate compared with TiO2 (Fig. 4(g)).
At present, metal NCs have made encouraging progress in photocatalytic hydrogen evolution reaction. However, in most cases, hole sacrificial agents are added to consume oxidizing holes produced at the valence bands, which reduces the recombination of electron–hole pairs and effectively utilizes photogenerated electrons to obtain high hydrogen-producing activity. This operation is actually only a half reaction of photocatalytic water splitting. Although the hydrogen production activity can be improved, it will cause an increase in the actual reaction cost and additional environmental pollution. The highest pursuit of photocatalytic reaction is to achieve photocatalytic overall water splitting without sacrificial agents, and photogenerated electrons and holes can be used to produce hydrogen and oxygen with stoichiometric ratios, which is the future trend of photocatalytic development.
2.2 Photocatalytic CO2 reduction
The excessive emission of CO2 has caused serious environmental problems. The capture of CO2 has been researched for decades and led to some inspiring achievements. However, the effective utilization of CO2 is the ultimate goal of solving CO2 emission. Several works have focused on CO2 hydrogenation at high temperature and pressure without the consideration of hydrogen cost. Electrocatalytic CO2 reduction is a potential alternative strategy, but the required additional electricity is essentially another form of energy. As a green and facile method, photocatalytic CO2 reduction to valuable C-containing products has attracted great interests benefiting from the inexhaustible solar energy.51–54 The first two steps of photocatalytic CO2 reduction are the same as that of photocatalytic water splitting, except for the final step, due to the complex proton-coupled electron transfer processes and various C-containing products (i.e., 2 e− for CO and HCOOH, 6 e− for CH3OH, 8 e− for CH4 and CH3COOH, 12 e− for C2H5OH and C2H4, 14 e− for C2H6) during CO2 reduction. In addition, the energy position of CB of the photocatalyst required in theory is changed according to the targeted reduction product, as summarized by previous reported reviews.55,56Due to the accurate controllable composition and structure, atomically precise Au NCs exhibit size or structure-related light response, which makes them light harvesters in photocatalysis. However, the lack of catalytically active sites for CO2 reduction inhibits Au NCs applications in photocatalytic CO2 conversion. Cui et al. modified Au-GSH NCs with L-cys ligands to graft transition metal cations (Fe2+, Co2+, Ni2+, and Cu2+) (Fig. 5(a)).57 The Au NCs and connected metal ions acted as light-harvesting centers and catalytic centers for photocatalytic CO2 reduction, respectively. The photogenerated electrons transferred from the Au NCs to the metal ions through the metal–S bridge to participate in CO2 reduction. The metal ions type greatly influenced the photoinduced electron transfer efficiency, the donation ability of the electrons to the reactants, and the adsorption ability of the reactants, and thus led to different CO2 conversion activities and product selectivities. The Au NCs–Co2+ exhibited a production rate of 3.54 μmol gcat−1 h−1 and a selectivity of 65.2% in the photoreduction of CO2 to CO under λ > 420 nm light irradiation in water system with TEOA as the sacrificial agent.
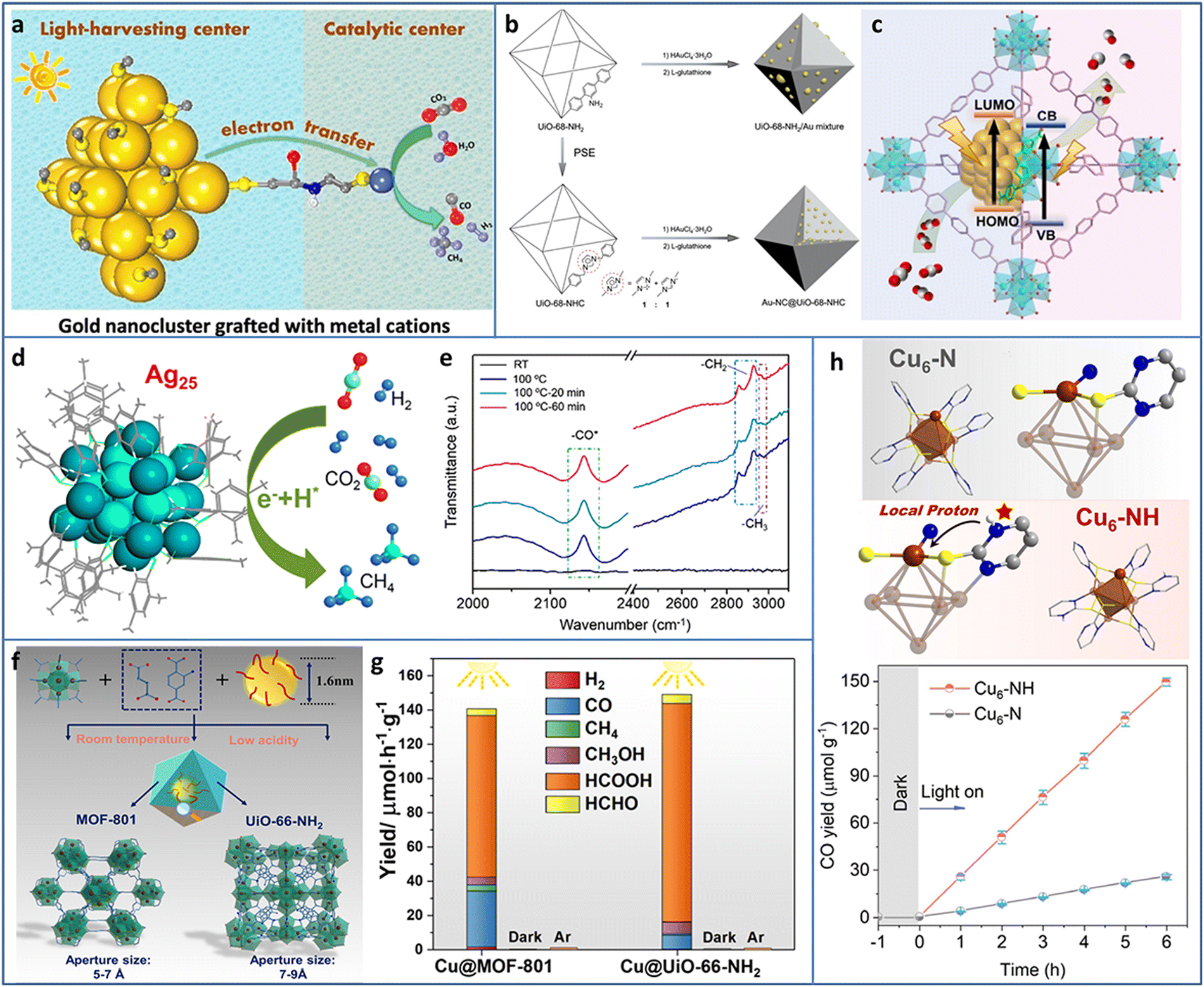 | ||
| Fig. 5 (a) Au NCs grafted with transition metal cations for photocatalytic CO2 reduction. (b) and (c) Synthesis and photocatalytic CO2 reduction mechanism of Au-NC@UiO-68-NHC. (d) Photoreduction of CO2 to CH4 and (e) time-resolved operando IR spectra of Ag25 NCs. (f) and (g) Synthesis and photocatalytic CO2 reduction of Cu NCs@MOFs. (h) The comparison of structure and photocatalytic CO2 reduction activity of Cu6–N and Cu6–NH. Reproduced with permission from ref. 57–61; copyright American Chemical Society and Wiley-VCH GmbH. | ||
Most of the previously synthesized Au NCs were protected by thiol ligands due to the stable Au–S bonds. Recently, N-heterocyclic carbenes (NHCs) were used in preparing Au NCs owing to the strong Au–carbene covalent bond. In addition, NHC ligands possessed the ability of CO2 activation, presenting reactivity in CO2 conversion. The wide diversity in the structure of NHC ligands offered the possibility to modify the Au NCs and expand their applications. Jiang et al. encapsulated Au NCs in UIO-68-NHC by the NHC-assisted nucleation method (Fig. 5(b)).58 The Au NCs were uniformly dispersed in UIO-68-NHC with a tunable size in the range of 1.3–1.8 nm. Due to the Au–carbene covalent bond at the interface of the heterostructure, Au-NC@UiO-68-NHC showed significantly enhanced photostability. In contrast, the Au species on the amine or imidazolium-functionalized MOF showed typical aggregation. In photocatalytic CO2 reduction, Au-NC@UiO-68-NHC showed a high CO production rate of 57.6 μmol g−1 h−1 under 300 W xenon lamp irradiation with 300 nm ≤ λ ≤ 800 nm in the mixture of CH3CN and CH3OH system (Fig. 5(c)), four times higher than that of the UiO-68-NH2/Au mixture.
Methane is a widely used fuel and chemical raw material with high value; however, photocatalytic CO2 reduction to CH4 is relatively difficult due to the involvement of the eight-electron transfer process. Xiong et al. utilized atomically precise Ag25 NCs, realizing nearly 100% selectivity in the photoreduction of CO2 to CH4 (Fig. 5(d)).59 Time-resolved operando IR spectroscopy was used to investigate the intermediates generated during CO2 reduction. The bands at 2142, 2854, and 2963 cm−1 were assigned to *CO, –CH2, and –CH3, respectively (Fig. 5(e)). CH4 was formed through an –H-assisted multielectron reaction pathway. In addition, the adsorption of CO2 on Ag25 NCs was energetically favorable, as confirmed by DFT calculation.
Cu NCs are considered as one of the most promising catalysts for CO2 conversion not only due to their relatively low cost but also because of the specific ability to generate multiple carbon products. Encapsulating metal NCs in the cavity of MOFs can avoid the aggregation of NCs but trigger some new problems, such as the hindered diffusion of the reactant and uncertain distribution of active sites. Dai et al. proposed a facile seed-mediated growth method to synthesize gram-scale core–shell Cu NCs@MOFs at room temperature in aqueous solution (Fig. 5(f)).60 Two benchmark Zr-based MOFs (MOF-801 and UIO-66-NH2) were selected as the templates. Compared to the Cu NCs/MOF (Cu NCs on the surface or in the pore of MOFs), the core–shell Cu NCs@MOFs showed three times higher catalytic activity. In addition, the polar functional group of MOF played an important role in tuning the catalytic activity and selectivity. Compared to the 94 μmol h−1 g−1 photocatalytic CO2 reduction rate of Cu NCs@MOF-801 (under 385 nm light irradiation with TEOA as the sacrificial agent), Cu NCs@UIO-66-NH2 exhibited a 36% higher catalytic rate (Fig. 5(g)). The HCOOH product selectivity of Cu NCs@UIO-66-NH2 was also higher than that of Cu NCs@MOF-801. X-ray absorption near edge structure/extended X-ray absorption fine structure (XANES/EXAFS) and in situ IR spectroscopy measurements were conducted to explore the catalytic mechanism and revealed that the Cu+ sites at the interface between Cu NCs and MOFs were the active sites for the photoreduction of CO2.
Constructing stable Cu NCs with suitable band structure and active catalytic sites for photocatalytic CO2 reduction remains a challenge. 2-Mercaptopyrimidine (PymSH) with thiol and thione tautomeric forms presents high affinity for multiple types of metal centers and is widely used as a protective ligand to stabilize NCs. In addition, the uncoordinated N atom can serve as a proton relay station, offering a local proton source for accelerating the reaction dynamics. Dong et al. synthesized a novel Cu6–NH NC containing local protonated N–H with high stability and semiconductor property.61 Under λ > 400 nm visible light irradiation, with H2O as an electron donor, Cu6–NH showed ∼100% selectivity for the photoreduction of CO2 to CO (Fig. 5(h)). The other isostructural Cu6–N NCs were further synthesized to systematically investigate the ligand effect on the catalytic activity. The underlying reason of the high activity of Cu6–NH was attributed to the protonated pyrimidine N, which provided a local proton to reduce the energy barrier for the formation of the key *COOH intermediate, as demonstrated by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements and DFT calculations.
In photocatalytic CO2 reduction reaction, noble metal NCs, such as Au and Ag NCs, have been studied more, mainly due to the relatively mature synthesis methods and the intrinsically better stability than that of non-noble metal NCs. However, the types of products for CO2 reduction using noble metal NCs as catalysts are relatively simple. For example, for Au NCs, the common CO2 reduction product is CO. In order to deeply reduce CO2 into more valuable chemicals, more types of metal NCs need to be explored. In addition, in the current studies, only a few works have reported using water as a proton source. To achieve cleaner and greener energy conversion in the future, it is necessary to explore CO2 reduction reaction in pure water systems. At the same time, how to efficiently transform the low concentration of CO2 in the atmospheric environment is also a meaningful topic worth exploring.
2.3 Photocatalytic organic transformation
Utilizing solar energy, catalytic pollutant degradation and organic transformations are ideal potential solutions for green chemistry.62–65 Metal NCs with molecular-like discrete energy band can serve as light-harvesting antenna. In addition, the unique atom packing mode, quantum size effect, and abundant surface active sites make them potential effective catalysts for multiple types of photoredox reactions.Atomically precise metal NCs can achieve the elaborate tuning of the catalytic performance due to the accurate and controllable size, composition, and structure. Among the various tunable strategies, metal doping is of significance for obtaining advanced catalytic activity. Cheng et al. synthesized serial Ag4M2(SPhMe2)8 (M = Ni, Pd, Pt) NCs (SPhMe2 = 2,4-dimethylbenzenethiol) with the same spatial geometric structure (Fig. 6(a)).66 The overall structure is similar to a distorted hexahedron, in which four Ag atoms were at the midpoints of the four side edges and two foreign metal atoms were at the center of the top and bottom planes. Ag and M atoms formed an electron donor–acceptor pair, which was important for photocatalysis. In the photocatalytic degradation of methyl orange, Ag4Pd2/TiO2 showed the best catalytic activity with ∼100% degradation in 18 minutes, followed by Ag4Pt2 (42 minutes) and Ag4Ni2 (54 minutes). However, in the photocatalytic degradation of rhodamine B, the order of catalytic activity was reversed, i.e., Ag4Ni2 > Ag4Pt2 > Ag4Pd2. The total difference in the catalytic performance of the three NCs in the degradation of methyl orange and rhodamine B was attributed to the alignment of the molecular orbitals near the HOMO–LUMO gap caused by different inter-molecular recombination mechanisms (complexation-induced recombination for methyl orange and collision-induced recombination for rhodamine B) (Fig. 6(b)).
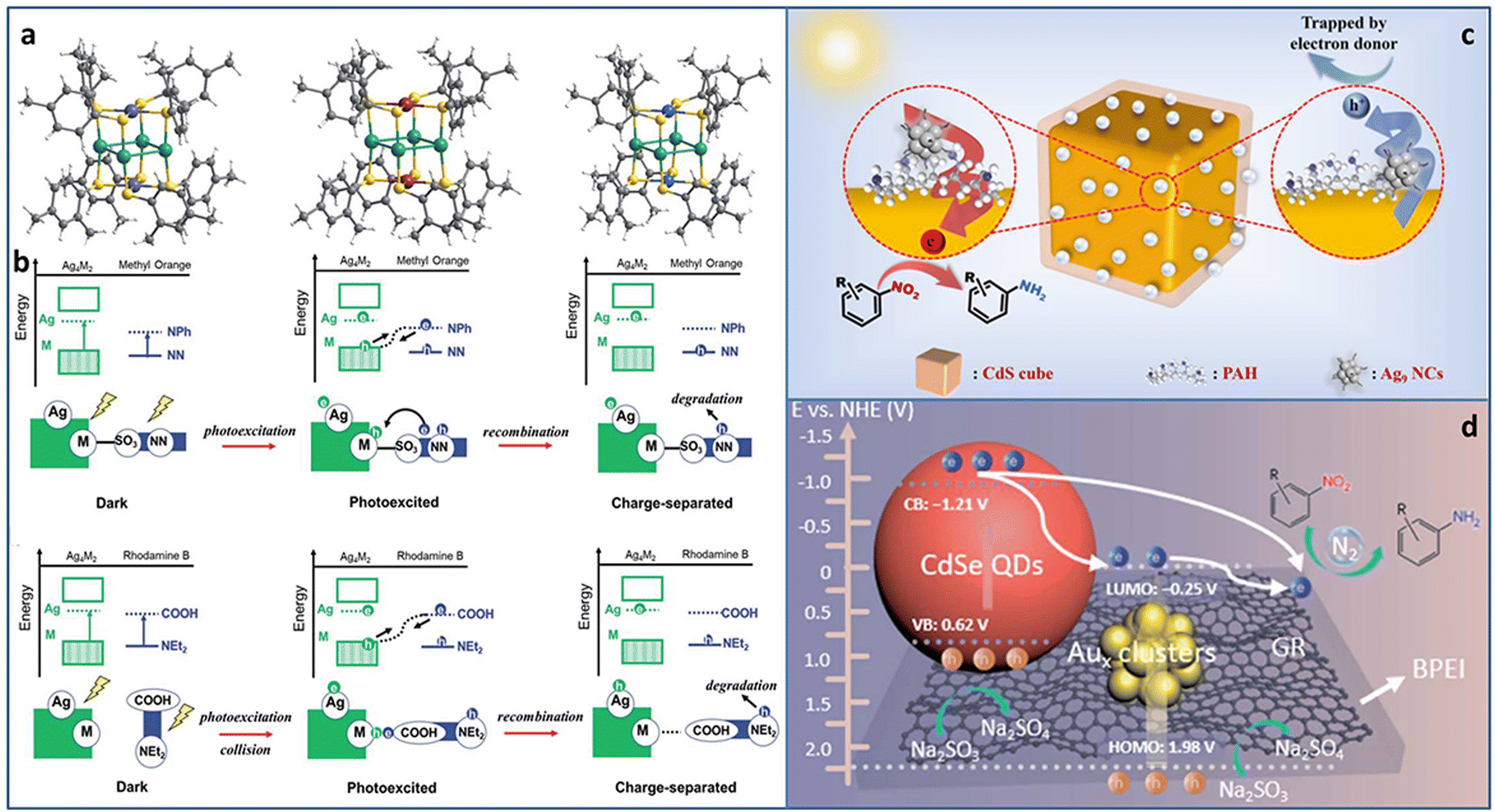 | ||
| Fig. 6 (a) The crystal structure of Ag4M2(SPhMe2)8 (M = Ni, Pd, and Pt) NCs. (b) The photocatalytic mechanisms of methyl orange and rhodamine B degradation over Ag4M2(SPhMe2)8 (M = Ni, Pd, and Pt) NCs. Photocatalytic reduction of nitroaromatics to amino derivatives over (c) CdS@PAH@Ag9 NCs and (d) CdSe–Aux NCs–graphene. Reproduced with permission from ref. 66–68 copyright the Royal Society of Chemistry anf the American Chemical Society. | ||
Rapid charge recombination and poor stability limit the photocatalytic application of metal NCs. Building a heterostructure with cascade charge transport pathway can effectively solve the dilemma. Xiao et al. designed two kinds of transition TMC–metal NC-based heterostructure (CdS@PAH@Ag9 NCs and CdSe–Aux NCs–graphene, PAH = poly(allylamine hydrochloride)) via step-by-step surface modification and electrostatic self-assembly. In the CdS@PAH@Ag9 NCs system, PAH played the dual roles of a surface charge modifier to connect Ag9(GSH)6 NCs and CdS and an interfacial charge transport mediator to accelerate charge separation. Under light irradiation, the photoinduced electrons generated on the Ag9 NCs flowed to CdS with the aid of PAH; thereby the charge lifetime was effectively prolonged, leading to an enhancement in the catalytic activity for the selective anaerobic photocatalytic reduction of nitroaromatics to amino derivatives (Fig. 6(c)).67 In the CdSe–Aux NCs–graphene system, Aux NCs and CdSe both acted as the photosensitizer. More importantly, Au NCs were used as an intermediate charge relay mediator, and graphene provided the electron-withdrawing capability, synergistically promoting interfacial charge transfer and separation, leading to enhanced photoactivity towards the selective reduction of aromatic nitro compounds to amino derivatives (Fig. 6(d)).68
COF is one of the most attractive porous supports due to its highly ordered structure and ultrasmall pores, offering an opportunity to uniformly load metal NCs. Deng et al. synthesized a two-dimensional (2D) COF with thiol chains, in which the –SH groups acted as the nucleation sites for the in situ growth of Au NCs in COF (Fig. 7(a)).69 Due to the strong Au–S binding force and ultrasmall pores of COF, the obtained Au NCs with uniform size were highly dispersed in the pores of the COF. Based on the formation of Au–S-COF bonding, a Z-scheme photocatalytic heterostructure was constructed for improved charge separation and photocatalytic activity. In a continuous flowing pollution system, the filter paper made by Au NCs@COF catalysts was applied in the degradation of rhodamine B and bisphenol A and showed satisfactory activity in five consecutive cycles (Fig. 7(b)).
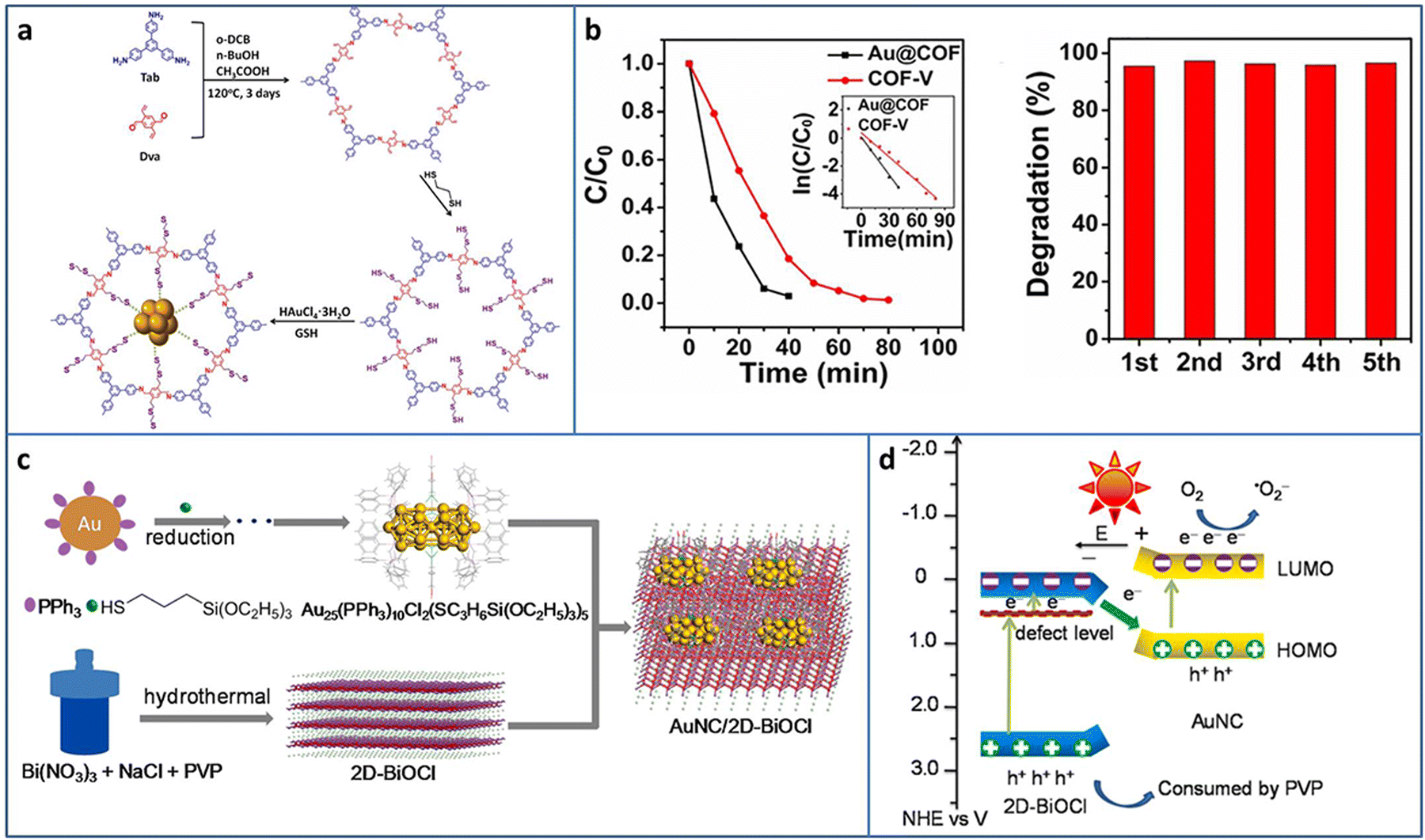 | ||
| Fig. 7 (a) Synthesis of Au@COF. (b) Activity and recycling ability of the degradation of RhB Au@COF. (c) and (d) Synthesis and photocatalytic charge transfer pathway of Au25 NCs/BiOCl. Reproduced with permission from ref. 69,70, copyright Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim and Elsevier B.V. | ||
Under light irradiation, the organic protective ligands on the surface of metal NCs may deplete by photogenerated holes, causing the coalescence or growth of NCs. Bismuth oxyhalides BiOX (X = Cl, Br, I), as new layer materials, are widely applied in the photocatalytic field due to their semiconductor features. As a typical example, BiOCl consists of [Bi2O2]2+ layers interleaved with two layers of Cl ions and exhibits a self-built internal electric field for improving the carrier separation. In addition, the layer structure can shorten the distance of carrier transportation from inside to the surface. Chen et al. fabricated Au25(PPh3)10(SC3H6Si(OC2H5)3)5Cl2 (PPh3 = triphenylphosphine, SC3H6Si(OC2H5)3 = mercaptopropyltriethoxysilane) NCs/BiOCl heterostructure via the wet-impregnation method (Fig. 7(c)).70 The 0D/2D heterostructure possessed a large amount of junctions, benefiting the effective charge transfer and separation. Besides this, the heterostructure was conducive to the stabilization of the Au NCs, preventing them from self-oxidation. Based on the analysis of the energy band structure and a series of photoelectrochemical measurements, a Z-scheme charge transfer pathway was confirmed in the Au25 NCs/BiOCl heterostructure (Fig. 7(d)). The Au25 NCs/BiOCl realized the photocatalytic oxidative coupling of various amine substrates and showed a high TOF of 1916 h−1 for benzylamine (under 100 W LED lamp irradiation in a mixture of ethanol and water system), which were 4.7 and 12773.3 times higher than that of Au25 NCs and 2D-BiOCl, respectively.
Currently, most photocatalytic reactions proceed under the irradiation of UV/visible light. However, nearly half of the solar spectrum is made up of the much lower energy infrared light. From the energy-saving perspective, the less energy to excite the electron transition is more attractive to researchers.71 Therefore, developing photocatalyst with near-infrared (NIR) light response is highly desired. Due to the precisely adjustable number, the type and arrangement of metal atoms, metal NCs exhibit tunable HOMO–LUMO gap, realizing the wide light spectrum utilization from UV to NIR. Wang et al. synthesized Au25(F-Ph)18− (F-Ph = 3,4-difluorothiophenol) NCs via surface modification on the Au25(PET)18− template (Fig. 8(a)).72 Due to the π–π stacking between the F-Ph ligands, the modified Au25(F-Ph)18− shows improved stability compared to Au25(PET)18−, reflecting the almost unchanged UV-vis characteristic peaks of Au25(F-Ph)18− in the presence of H2O2 and the remaining negatively charged state after irradiation. In addition, Au25(F-Ph)18− displays higher 1O2 production ability, indicating the enhanced catalytic activity than that of Au25(PET)18−. On the basis of these findings, Au25(F-Ph)18− NCs are used to realize the C–H functionalization of 2-aryl-1,2,3,4-tetrahydroisoquinolines (Fig. 8(b)). Under NIR light irradiation, [Au25]− is photoexcited to [Au25]−*, and then [Au25]−* transfers the energy for the formation of 1O2 from 3O2 for the subsequent C–H functionalization process. From the time-dependent synthesis yield curve, it can be seen that when using 850 nm NIR LED instead the 460 nm blue LED light source, the yield of the product is significantly improved from 69% in 48 h to 99% in 8 h (Fig. 8(c)). Due to the high activity of Au25(F-Ph)18− NCs for NIR photocatalytic oxidation, it is further extended to other useful oxidation reactions, i.e. the selective oxidation of sulfides to sulfoxides and aerobic oxidation of β-ketoesters (Fig. 8(d) and (e)), achieving excellent catalytic performance in oxidative functionalization.
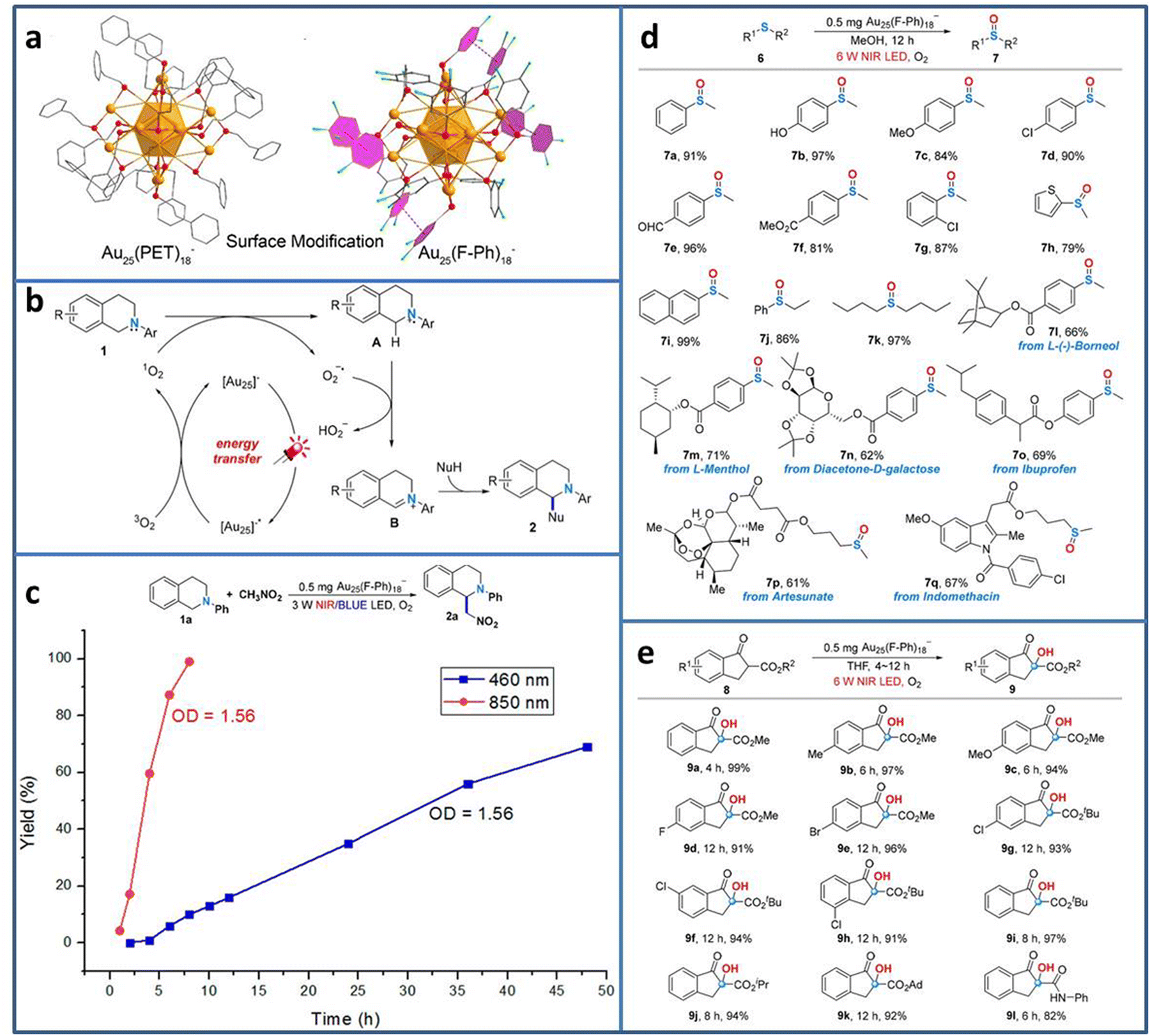 | ||
| Fig. 8 (a) Structure of Au25(F-Ph)18− and Au25(PET)18−. (b) C–H functionalization of 2-aryl-1,2,3,4-tetrahydroisoquinolines catalyzed by Au25(F-Ph)18−. (c) Time-dependent synthesis yield using Au25(F-Ph)18− under NIR and blue LED light irradiation. (d) Oxidation of sulfides to sulfoxides and (e) aerobic oxidation of β-ketoesters using Au25(F-Ph)18− under NIR light irradiation. Reproduced with permission from ref. 72, copyright American Chemical Society. | ||
In photocatalytic organic transformation, since most of the cases are for dye degradation, the transformation of nitroaromatics to amino derivatives model reactions, it is necessary to apply NCs to more practical reactions. In the case of organic reactants, whether NC ligands will participate in the reaction and what effect it has on the reaction activity is an attractive orientation. In addition, NCs themselves have the property of generating singlet oxygen by light excitation; thus, how to apply this property to the photocatalytic reaction is also a problem worth thinking about.
2.4 Photoelectrocatalytic reaction
Photoelectrocatalysis is a combination of photocatalysis and electrocatalysis, which can maximize the advantages of both the technologies. Compared with photocatalysis, photoelectrode can generate photogenerated electrons by sunlight irradiation, and the photogenerated charge can be separated applying a certain bias voltage to the photoelectrode, which improves the reaction activity and catalytic efficiency. Compared with electrocatalysis, the photoelectrocatalytic reaction greatly reduces the injection of external energy and can effectively reduce the energy consumption and environmental pollution. In addition, the catalyst that is not suitable for photocatalysis due to the mismatch of the energy band structure can be applied to photoelectrocatalysis under appropriate applied voltage conditions.73–75Atomically precise metal NCs can be used as photosensitizers in photoelectrochemical (PEC) cells. Wang et al. fabricated phenylacetylene (PA)-modified TiO2 nanotube array (TNA) and deposited Au24Ag20(PhCC)20(SPy)4Cl2 NCs (Py = pyridine) (PhCC = Phenylacetylene) based on the π–π interactions between the phenyl rings of NCs and that of the organic coating of TiO2 (Fig. 9(a)).76 The introduction of Au24Ag20 NCs enhanced the light harvesting due to its high molar extinction coefficient. The photocurrent of Au24Ag20-TNA is higher than that of Au25-TNA because of the higher light absorption ability and charge-separation efficiency of Au24Ag20 NCs. In addition, Au24Ag20-TNA shows much better photostability in both neutral and basic conditions compared to Au25-TNA.
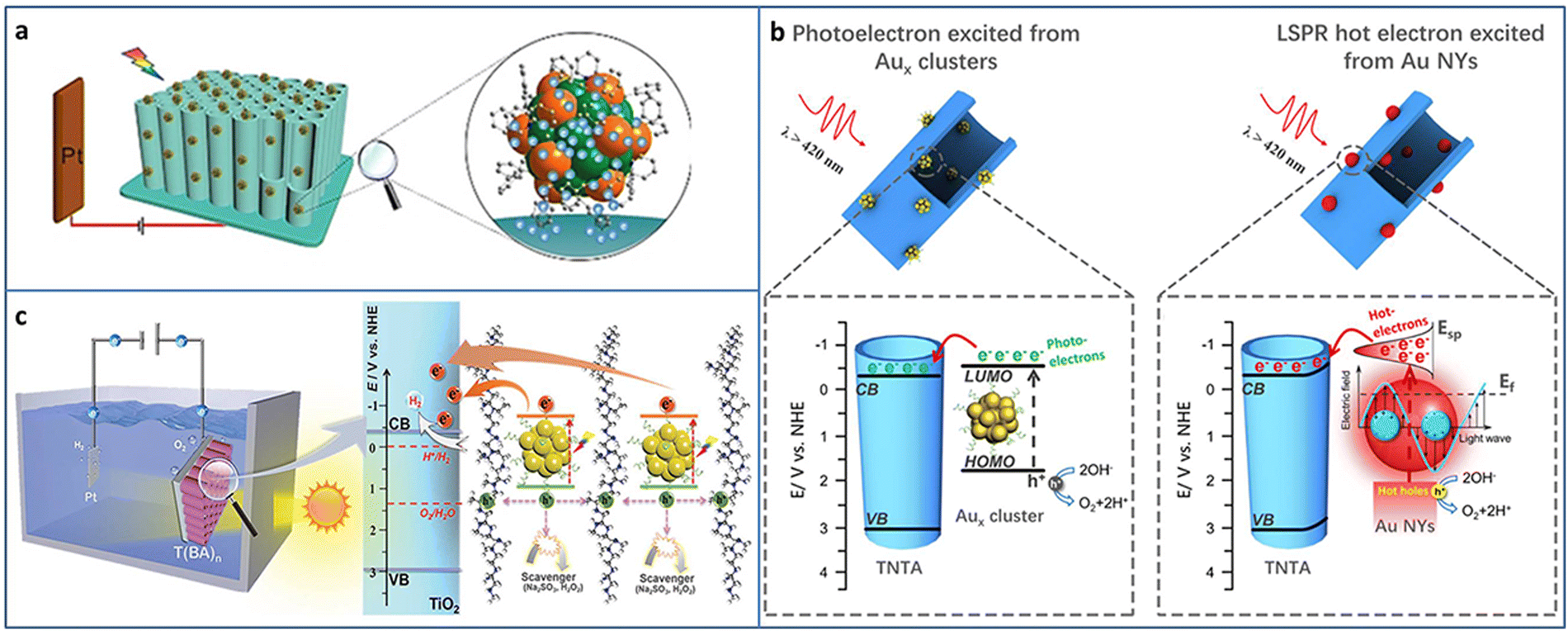 | ||
| Fig. 9 (a) Synthesis of Au24Ag20-TNA for PEC application. (b) The comparison of PEC water splitting by metal NCs and NPs. (c) PEC water splitting of the Aux NCs-BPEI-metal oxide system. Reproduced with permission from ref. 35, 76, 77; copyright Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, and American Chemical Society. | ||
It is well-known that there are great differences between atomically precise metal NCs and conventional metal NPs, such as physicochemical properties, quantum confinement effect, and photosensitization effect, especially, the electrons photoexcited from the metal NCs and the hot electrons from the surface plasmonic resonance (SPR) effect of metal NPs. Xiao et al. compared the performance of photoelectrochemical water splitting between metal NCs and NPs by strict control experiments (Fig. 9(b)).35 TNA-Aux NCs were fabricated by assembling Aux@GSH NCs on TNA based on electrostatic interaction. The corresponding TNA-Au NPs were obtained by the calcination of TNA-Aux NCs. A series of experiments such as LSV curves, applied bias photon-to-current efficiency, photocurrent response, electron impedance spectroscopy, Mott–Schottky plots and the charge carrier density calculation, open circuit voltage and the electron lifetime calculation, and incident-photon-to-current conversion efficiency was conducted to investigate the difference between the metal NCs and metal NPs-triggered charge transfer characteristics. In terms of interfacial charge separation and transfer, the photoelectrons from Aux@GSH NCs are superior to the hot electrons from plasmonic Au NPs. The PEC water splitting mechanisms of TNA-Aux NCs and TNA-Au NPs were further revealed. Aux@GSH NCs with HOMO–LUMO gap are able to serve as a small-band-gap semiconductor and can be photoexcited to generate electrons and holes. Due to the band alignment, the electrons in the LUMO of Aux NCs can transfer to the CB of TiO2 and then flow to the photocathode to produce hydrogen and generate photocurrent. Au NPs are able to be photoexcited due to the SPR effect and generate hot electrons. The hot electrons are injected into the CB of TiO2 by surpassing the Schottky barrier. The holes oxidize water to oxygen at the photoanode.
To improve the stability and carrier utilization efficiency of NCs-based photosystems, Xiao et al. constructed an NCs-based multilayer heterostructure with spatially separated charge transport pathways via the layer-by-layer assembly strategy. GSH-protected Aux NCs and branched poly-ethylenimine (BPEI) have opposite surface charge state, and they are alternately deposited on the metal oxide (i.e., TiO2, WO3, and Fe2O3). Aux NCs serve as lightharvesting antennas and generate photocarriers. BPEI interim layer contributes to the tandem hole transport. As a result, the charge transfer cascade is motivated, leading to enhanced PEC water splitting activity (Fig. 9(c)).77
When the particle size becomes extremely small, the space charge layer cannot be formed, which affects the carrier separation ability. However, in the PEC system, even if the very small particles do not form a space charge layer, the photogenerated charge carriers can also be driven by the applied potential from the photoelectrode material to the opposite electrode so as to achieve effective separation. There is no doubt that the photoelectrode catalyst is the most important component, which determines the reaction process and the catalytic efficiency of the PEC system. Metal NCs can act as catalysts, photosensitizers, charge transport media and other roles in the PEC process, but the NCs are small in size and easy to aggregate, requiring a suitable matrix as the support and stabilizer. In addition, the PEC process is also affected by various other factors, such as solution conductivity and pH, applied bias voltage, light intensity, and temperature. The PEC process is more complex than the photocatalytic process; thus, it is still very challenging to clearly identify the role of NCs in it and figure out the factors affecting its efficiency.
2.5 Other photocatalytic reaction
Metal NCs with excitonic behavior are capable of generating electrons induced by solar energy and donating electrons to N2 for its reduction. Sun et al. utilized TiO2 with oxygen vacancy (TiO2-Vo) to support Au4Ru2(PPh3)2(SC2H4Ph)8 NCs.82 Under light irradiation, the excited Au4Ru2 NCs generated photoinduced electron–hole pairs. The photogenerated carriers effectively separated between Au4Ru2 NCs and TiO2-Vo, in which the electrons transferred to the Ru atoms and then injected to N2 for its activation. TiO2-Vo was responsible for the water splitting to provide the proton for N2 hydrogenation (Fig. 10(a)). In comparison with a series of homometal Aun NCs (n = 8, 9, 11, 18, 23, 24, 25, 28, 36, 44), the alloyed Au4Ru2 NCs can effectively activate N2. The distance between N2 and NCs in the Aun NCs was larger than 3.2 Å, indicating the physical adsorption of N2 on these Aun NCs (Fig. 10(b)). On the contrary, N2 was closer to Au4Ru2 NCs with a distance of 2.14 Å and 1.84 Å for the side-on and end-on configuration, respectively, suggesting the chemical adsorption of N2 on Au4Ru2 NCs. Au4Ru2 NCs/TiO2-Vo showed a NH3 production rate of 44.5 μmol g−1 h−1, 3 times higher than that of Aun NCs/TiO2-Vo.
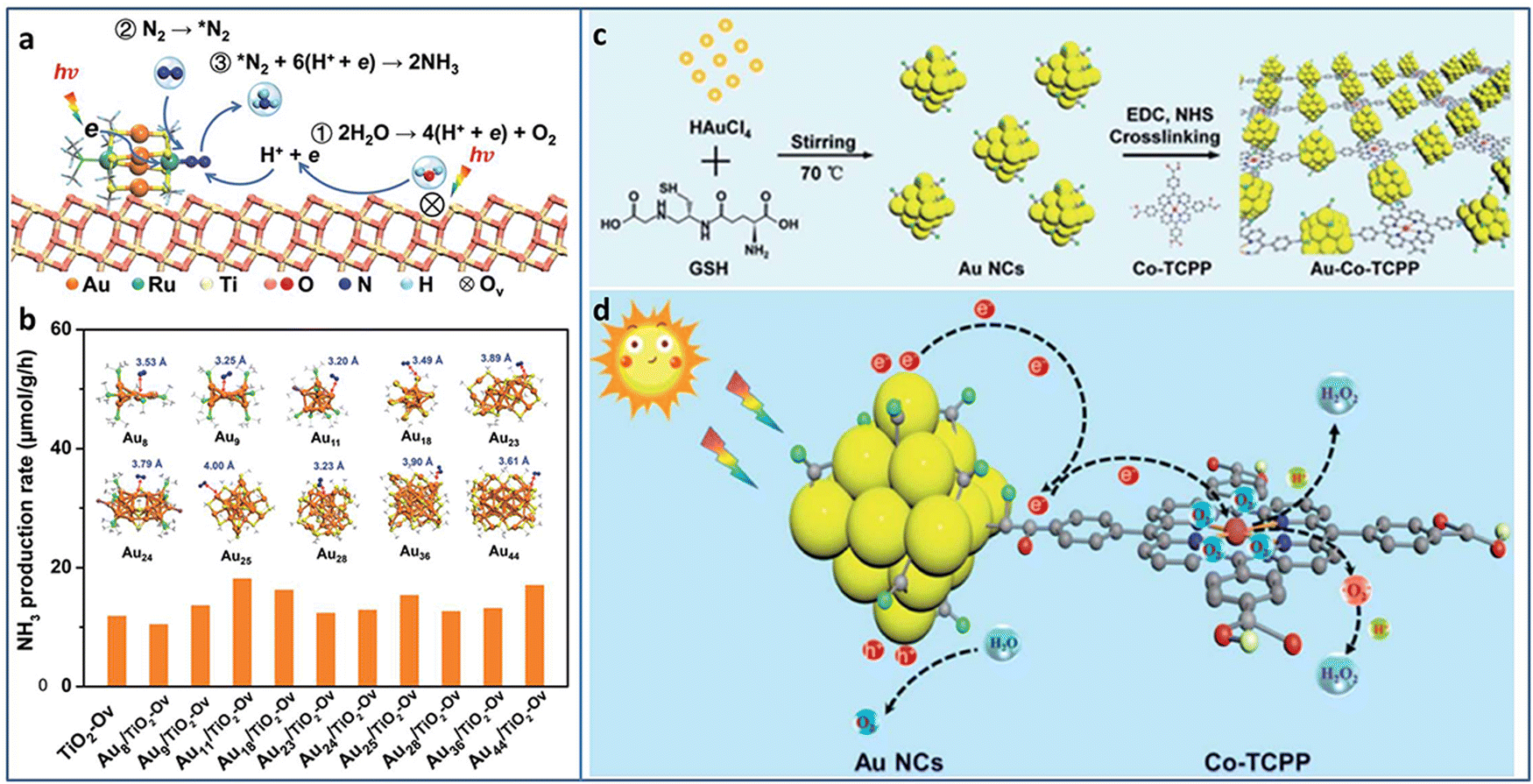 | ||
| Fig. 10 (a) Photocatalytic N2 reduction mechanism on Au4Ru2/TiO2–Ov. (b) The photocatalytic N2 reduction activity of homometal Aun NCs (n = 8, 9, 11, 18, 23, 24, 25, 28, 36, and 44) on TiO2-Ov. Inset: The structures of the Aun NCs, and the distance between N2 and these Aun NCs. (c) and (d) Preparation and photocatalytic H2O2 production of the Au–Co–TCPP. Reproduced with permission from ref. 82 and 83, copyright the Royal Society of Chemistry. | ||
Xue et al. designed and grafted multifunctional Co porphyrin molecule on the surface of Au NCs by ligand engineering strategy to obtain three-in-one functionalized Au–Co–TCPP (Fig. 10(c)).83 The Au–Co–TCPP exhibited enhanced visible light absorption, promoted charge separation efficiency, and more active sites for photocatalysis (Fig. 10(d)). Au–Co–TCPP showed an outstanding performance of 235.93 μmol in photocatalytic H2O2 production within 60 minutes, 2.2 times higher than that of Au NCs. By introducing the scavenger of O2·, the H2O2 production mechanism showed that the two-electron O2 reduction was the main pathway over Au–Co–TCPP. In addition, the high-spin state of Co2+ in Au–Co–TCPP was also beneficial to the two-electron O2 reduction, as confirmed by the control experiment with Au-2H-TCPP.
3. Conclusions and perspectives
Due to the strong wide spectra light response and molecule-like discrete energy levels, the atomically precise metal NCs as promising photocatalysts have been widely applied in photocatalytic water-splitting, CO2 conversion, organic transformation, N2 fixation, H2O2 production, and PEC application. For catalytic reactions stimulated by light as an energy source, the primary consideration is the effective use of light. Therefore, in order to construct an effective NCs-based photocatalyst, the premise is to expand the light absorption range, which can be achieved from the aspects of adjusting the size of the NCs, heteroatom doping, ligand engineering, etc. At the same time, coupling a semiconductor with a wide spectral response is also one of the feasible strategies. Secondly, photogenerated electrons and holes can effectively migrate to the surface of the photocatalyst and participate in the subsequent redox reaction rather than recombination inside the catalyst, which requires the photocatalyst to have high carrier separation efficiency. For the NCs-based photocatalytic system, the electrons and holes generated by isolated NCs are prone to recombination due to their small size, resulting in fewer carriers to take part in the reaction. Therefore, as summarized above, choosing semiconductors with appropriate energy level structures to form heterojunctions is a strategy that can effectively suppress the recombination of electrons and holes pairs, thereby improving the photocatalytic activity. In addition, this way of loading NCs on the support also reduces the possibility of NCs aggregation to a certain extent and improves the stability under light irradiation, which is very important for practical applications. Despite the above progress, there are still numerous fundamental mechanisms that need to be investigated in depth and application obstacles that need to be overcome with regard to the photocatalysis of metal NCs. From our perspective, the current challenges and future opportunities are provided below.1. Considering that most of the sunlight is infrared light, it is necessary to develop metal NCs that are responsive to infrared light to make full use of sunlight and achieve high photocatalytic conversion efficiency. In addition, Au, Ag, Pt and other noble metal NCs have been mainly applied in photocatalysis to date. In the long run, it is urgent to develop non-noble metal NCs with high reserves, low prices and photoactivity.
2. The property of metal NCs is dependent on various factors including size, core structure, surface motifs, and ligands. At present, several studies on the structure–activity relationships are mixed with changes in multiple parameters. Therefore, it is necessary to develop more series of atomically precise metal NCs with only a single parameter variable change so as to provide an ideal platform for finely exploring the relationship between the catalyst structure and activity.
3. Photocatalysis technology is a highly promising way of energy conversion involving all aspects. For now, the application of metal NCs in the field of photocatalysis is still limited. The feasibility of the photocatalytic reaction can be preliminarily speculated from the position of the electronic energy level of the catalyst and the redox potential of the reactant/product. Therefore, for specific target reactions, metal NCs with specific electronic structures can be synthesized by precisely regulating the size, doping, ligand and charge state, which is also of great significance for expanding the application range of metal NCs photocatalysis.
4. The poor photostability of metal NCs is a major issue in practical application. Especially, under strong light irradiation, will the ligands will fall off? How many ligands will fall off? If a ligand falls off, what is the fragment that falls off, is it the whole ligand, or some part of the ligand? What sites on the NCs are the true centers with catalytic activity? What kinds of ligands are favorable for photoinduced charge transfer? These fundamental problems require sophisticated characterization techniques for further analysis. In addition, the ligand is composed of organic molecules, which may be converted into a carbon layer outside the metal NCs, causing activity loss, and how to avoid this situation needs serious consideration.
5. Coupling with the other semiconductor to construct NCs-based heterostructures has been recently proposed as an effective strategy to improve the stability of NCs. At present, the synthesis methods of NCs-based composites mainly focus on wet impregnation, electrostatic attraction, coordination assembly, etc. It is necessary to develop simple, facile and general synthesis methods of NCs-based composites with intimate interface contact. In addition, the energy band alignment between the NCs and the other semiconductor needs to be paid attention to form directional charge flow. The determination of the reaction sites and the charge transfer pathway in NCs-based composite photocatalysts and the influence of the interface of the NCs-based composite on the charge transport efficiency need to be investigated in-depth.
Overall, atomically precise metal NCs have some incomparable advantages that make them potential photocatalysts. Although there are still some difficulties and challenges, a large number of studies have tried to solve these problems. It is believed that great contribution will be made in NCs photocatalysis in the near future.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
We acknowledge the financial support of the Natural Science Research Project of Universities in Anhui Province (KJ2021ZD0001), Natural Science Foundation of Anhui Province (2208085MB20).References
- C. Zhuang, H. Qi, X. Cheng, G. Chen, C. Gao, L. Wang, S. Sun, J. Zou and X. Han, Angew. Chem., Int. Ed., 2019, 58, 18627–18633 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2023, 123, 4855–4933 CrossRef CAS PubMed.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- X. Yuan and M. Zhu, Inorg. Chem. Front., 2023, 10, 3995–4007 RSC.
- C. Zhuang, W. Li, Y. Chang, S. Li, Y. Zhang, Y. Li, J. Gao, G. Chen and Z. Kang, J. Mater. Chem. A, 2024, 12, 5711–5718 RSC.
- M. Sayed, J. Yu, G. Liu and M. Jaroniec, Chem. Rev., 2022, 122, 10484–10537 CrossRef CAS PubMed.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- L. Zhang, J. Ran, S.-Z. Qiao and M. Jaroniec, Chem. Soc. Rev., 2019, 48, 5184–5206 RSC.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- X. Du and R. Jin, ACS Nano, 2019, 13, 7383–7387 CrossRef CAS PubMed.
- H. Liang, Q. Chen, Q.-L. Mo, Y. Wu and F.-X. Xiao, J. Mater. Chem. A, 2023, 11, 9401–9426 RSC.
- T. Kawawaki, Y. Kataoka, S. Ozaki, M. Kawachi, M. Hirata and Y. Negishi, Chem. Commun., 2021, 57, 417–440 RSC.
- Q.-L. Mo, B.-J. Liu and F.-X. Xiao, J. Phys. Chem. C, 2021, 125, 22421–22428 CrossRef CAS.
- R. Qin, K. Liu, Q. Wu and N. Zheng, Chem. Rev., 2020, 120, 11810–11899 CrossRef CAS PubMed.
- X. Cai, G. Li, W. Hu and Y. Zhu, ACS Catal., 2022, 12, 10638–10653 CrossRef CAS.
- D. Yang, J. Wang, Q. Wang, Z. Yuan, Y. Dai, C. Zhou, X. Wan, Q. Zhang and Y. Yang, ACS Nano, 2022, 16, 15681–15704 CrossRef CAS PubMed.
- H. Yan, H. Xiang, J. Liu, R. Cheng, Y. Ye, Y. Han and C. Yao, Small, 2022, 18, 2200812 CrossRef CAS PubMed.
- D. Yang, Y. Sun, X. Cai, W. Hu, Y. Dai, Y. Zhu and Y. Yang, CCS Chem., 2021, 4, 66–94 CrossRef.
- Q. You, H. Wang, Y. Zhao, W. Fan, W. Gu, H.-L. Jiang and Z. Wu, J. Am. Chem. Soc., 2024, 146, 9026–9035 CrossRef CAS PubMed.
- H. Wang, X. Liu, W. Yang, G. Mao, Z. Meng, Z. Wu and H.-L. Jiang, J. Am. Chem. Soc., 2022, 144, 22008–22017 CrossRef CAS PubMed.
- Y.-M. Li, J. Hu and M. Zhu, Coord. Chem. Rev., 2023, 495, 215364 CrossRef CAS.
- A. Dhakshinamoorthy, Z. Li, S. Yang and H. Garcia, Chem. Soc. Rev., 2024, 53, 3002–3035 RSC.
- C. Bie, L. Wang and J. Yu, Chem, 2022, 8, 1567–1574 CAS.
- S. Nishioka, F. E. Osterloh, X. Wang, T. E. Mallouk and K. Maeda, Nat. Rev. Methods Primers, 2023, 3, 42 CrossRef CAS.
- B.-J. Ng, L. K. Putri, X. Y. Kong, Y. W. Teh, P. Pasbakhsh and S.-P. Chai, Adv. Sci., 2020, 7, 1903171 CrossRef CAS PubMed.
- Y. Li, C. Zhuang, S. Qiu, J. Gao, Q. Zhou, Z. Sun, Z. Kang and X. Han, Appl. Catal., B, 2023, 337, 122881 CrossRef CAS.
- K. Qi, C. Zhuang, M. Zhang, P. Gholami and A. Khataee, J. Mater. Sci. Technol., 2022, 123, 243–256 CrossRef CAS.
- Y. Negishi, M. Mizuno, M. Hirayama, M. Omatoi, T. Takayama, A. Iwase and A. Kudo, Nanoscale, 2013, 5, 7188–7192 RSC.
- X. Lu, A. Tong, D. Luo, F. Jiang, J. Wei, Y. Huang, Z. Jiang, Z. Lu and Y. Ni, J. Mater. Chem. A, 2022, 10, 4594–4600 RSC.
- X. Huang, X. Li, Q. Luan, K. Zhang, Z. Wu, B. Li, Z. Xi, W. Dong and G. Wang, Nano Res., 2021, 14, 4250–4257 CrossRef CAS.
- Y. Li, L. Yang, H. He, L. Sun, H. Wang, X. Fang, Y. Zhao, D. Zheng, Y. Qi, Z. Li and W. Deng, Nat. Commun., 2022, 13, 1355 CrossRef CAS PubMed.
- C. Wang, P. Lv, D. Xue, Y. Cai, X. Yan, L. Xu, J. Fang and Y. Yang, ACS Sustainable Chem. Eng., 2018, 6, 8447–8457 CrossRef CAS.
- X.-C. Dai, M.-H. Huang, Y.-B. Li, T. Li, S. Hou, Z.-Q. Wei and F.-X. Xiao, J. Phys. Chem. C, 2020, 124, 4989–4998 CrossRef CAS.
- X.-Y. Fu, Z.-Q. Wei, S. Xu, X. Lin, S. Hou and F.-X. Xiao, J. Phys. Chem. Lett., 2020, 11, 9138–9143 CrossRef CAS PubMed.
- A. Yao, Y. Du, M. Han, Y. Wang, J. Hu, Q. Zhu, H. Sheng and M. Zhu, Nano Res., 2023, 16, 1527–1532 CrossRef CAS.
- T. Kawawaki, Y. Kataoka, M. Hirata, Y. Akinaga, R. Takahata, K. Wakamatsu, Y. Fujiki, M. Kataoka, S. Kikkawa, A. S. Alotabi, S. Hossain, D. J. Osborn, T. Teranishi, G. G. Andersson, G. F. Metha, S. Yamazoe and Y. Negishi, Angew. Chem., Int. Ed., 2021, 60, 21340–21350 CrossRef CAS PubMed.
- Z. Li, W. Huang, J. Liu, K. Lv and Q. Li, ACS Catal., 2021, 11, 8510–8520 CrossRef CAS.
- X. Yan, X.-Y. Fu and F.-X. Xiao, Adv. Funct. Mater., 2023, 33, 2303737 CrossRef CAS.
- H. Liang, B.-J. Liu, B. Tang, S.-C. Zhu, S. Li, X.-Z. Ge, J.-L. Li, J.-R. Zhu and F.-X. Xiao, ACS Catal., 2022, 12, 4216–4226 CrossRef CAS.
- Y. Wang, X.-H. Liu, Q. Wang, M. Quick, S. A. Kovalenko, Q.-Y. Chen, N. Koch and N. Pinna, Angew. Chem., Int. Ed., 2020, 59, 7748–7754 CrossRef CAS PubMed.
- H. Wang, X. Zhang, W. Zhang, M. Zhou and H.-L. Jiang, Angew. Chem., Int. Ed., 2024, e202401443 CAS.
- M. S. Bootharaju, C. W. Lee, G. Deng, H. Kim, K. Lee, S. Lee, H. Chang, S. Lee, Y.-E. Sung, J. S. Yoo, N. Zheng and T. Hyeon, Adv. Mater., 2023, 35, 2207765 CrossRef CAS PubMed.
- Q. Zhu, H. Shen, C. Han, L. Huang, Y. Zhou, Y. Du, X. Kang and M. Zhu, Nano Res., 2024, 17, 5002–5010 CrossRef CAS.
- Y.-D. Cao, H.-P. Hao, H.-S. Liu, D. Yin, M.-L. Wang, G.-G. Gao, L.-L. Fan and H. Liu, Nanoscale, 2021, 13, 16182–16188 RSC.
- F. Tian, J. Chen, F. Chen, Y. Liu, Y. Xu and R. Chen, Appl. Catal., B, 2021, 292, 120158 CrossRef CAS.
- Y. Wang, J. Li, Q. Hu, M. Hao, Y. Liu, L. Gong, R. Li and X. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 40562–40570 CrossRef CAS PubMed.
- Z. Wu, X.-L. Wang, X. Wang, X. Xu, D.-S. Li and T. Wu, Chem. Eng. J., 2021, 426, 131216 CrossRef CAS.
- R. Zhang, K. Gong, F. Du and S. Cao, Int. J. Hydrogen Energy, 2022, 47, 19570–19579 CrossRef CAS.
- S. Fang, M. Rahaman, J. Bharti, E. Reisner, M. Robert, G. A. Ozin and Y. H. Hu, Nat. Rev. Methods Primers, 2023, 3, 62 CrossRef.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CrossRef CAS.
- Z. Jiang, H. Sun, T. Wang, B. Wang, W. Wei, H. Li, S. Yuan, T. An, H. Zhao, J. Yu and P. K. Wong, Energy Environ. Sci., 2018, 11, 2382–2389 RSC.
- D. Li, M. Kassymova, X. Cai, S.-Q. Zang and H.-L. Jiang, Coord. Chem. Rev., 2020, 412, 213262 CrossRef CAS.
- K. Sun, Y. Qian and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202217565 CrossRef CAS PubMed.
- E. Nikoloudakis, I. López-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC.
- X. Cui, J. Wang, B. Liu, S. Ling, R. Long and Y. Xiong, J. Am. Chem. Soc., 2018, 140, 16514–16520 Search PubMed.
- Y. Jiang, Y. Yu, X. Zhang, M. Weinert, X. Song, J. Ai, L. Han and H. Fei, Angew. Chem., Int. Ed., 2021, 60, 17388–17393 Search PubMed.
- Y. Xiong, H. Chen, Y. Hu, S. Yang, X. Xue, L. He, X. Liu, J. Ma and Z. Jin, Nano Lett., 2021, 21, 8693–8700 Search PubMed.
- S. Dai, T. Kajiwara, M. Ikeda, I. Romero-Muñiz, G. Patriarche, A. E. Platero-Prats, A. Vimont, M. Daturi, A. Tissot, Q. Xu and C. Serre, Angew. Chem., Int. Ed., 2022, 61, e202211848 CrossRef CAS PubMed.
- J.-P. Dong, Y. Xu, X.-G. Zhang, H. Zhang, L. Yao, R. Wang and S.-Q. Zang, Angew. Chem., Int. Ed., 2023, 62, e202313648 CrossRef CAS PubMed.
- Y.-Z. Cheng, W. Ji, X. Wu, X. Ding, X.-F. Liu and B.-H. Han, Appl. Catal., B, 2022, 306, 121110 CrossRef CAS.
- S. Zhang, W. Huang, X. Fu, X. Zheng, S. Meng, X. Ye and S. Chen, Appl. Catal., B, 2018, 233, 1–10 CrossRef CAS.
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6198–6223 RSC.
- N.-Y. Huang, Y.-T. Zheng, D. Chen, Z.-Y. Chen, C.-Z. Huang and Q. Xu, Chem. Soc. Rev., 2023, 52, 7949–8004 RSC.
- X. Cheng, X. Sui, J. Xu, X. Liu, M. Chen and Y. Zhu, RSC Adv., 2021, 11, 32526–32532 RSC.
- Q. Chen, X.-Z. Ge, L. Yu and F.-X. Xiao, Inorg. Chem., 2023, 62, 19358–19365 CrossRef CAS PubMed.
- S. Hou, M.-H. Huang and F.-X. Xiao, J. Mater. Chem. A, 2022, 10, 7006–7012 RSC.
- Y. Deng, Z. Zhang, P. Du, X. Ning, Y. Wang, D. Zhang, J. Liu, S. Zhang and X. Lu, Angew. Chem., Int. Ed., 2020, 59, 6082–6089 CrossRef CAS PubMed.
- H. Chen, L. Peng, Y. Bian, X. Shen, J. Li, H.-C. Yao, S.-Q. Zang and Z. Li, Appl. Catal., B, 2021, 284, 119704 CrossRef CAS.
- C. Han, B. K. Kundu, Y. Liang and Y. Sun, Adv. Mater., 2024, 36, 2307759 CrossRef CAS PubMed.
- S. Wang, L. Tang, B. Cai, Z. Yin, Y. Li, L. Xiong, X. Kang, J. Xuan, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2022, 144, 3787–3792 CrossRef CAS PubMed.
- T. Yao, X. An, H. Han, J. Q. Chen and C. Li, Adv. Energy Mater., 2018, 8, 1800210 CrossRef.
- Y. Song, Y. Wu, S. Cao, Y. Zhang, D. Luo, J. Gao, Z. Li, L. Sun and J. Hou, Adv. Energy Mater., 2022, 12, 2201782 CrossRef CAS.
- M. Faraji, M. Yousefi, S. Yousefzadeh, M. Zirak, N. Naseri, T. H. Jeon, W. Choi and A. Z. Moshfegh, Energy Environ. Sci., 2019, 12, 59–95 RSC.
- Y. Wang, X.-H. Liu, S. A. Kovalenko, Q.-Y. Chen and N. Pinna, Chem. – Eur. J., 2019, 25, 4814–4820 CrossRef CAS PubMed.
- Q.-L. Mo, X.-C. Dai and F.-X. Xiao, Small, 2023, 19, 2302372 CrossRef CAS PubMed.
- Y. Zhao, L. Zheng, R. Shi, S. Zhang, X. Bian, F. Wu, X. Cao, G. I. N. Waterhouse and T. Zhang, Adv. Energy Mater., 2020, 10, 2002199 CrossRef CAS.
- S. Zhang, Y. Zhao, R. Shi, G. I. N. Waterhouse and T. Zhang, EnergyChem, 2019, 1, 100013 CrossRef.
- B. Sun, S. Lu, Y. Qian, X. Zhang and J. Tian, Carbon Energy, 2023, 5, e305 CrossRef CAS.
- S. Lin, X. Zhang, L. Chen, Q. Zhang, L. Ma and J. Liu, Green Chem., 2022, 24, 9003–9026 RSC.
- Y. Sun, W. Pei, M. Xie, S. Xu, S. Zhou, J. Zhao, K. Xiao and Y. Zhu, Chem. Sci., 2020, 11, 2440–2447 RSC.
- Q. Xue, Z. Wang, S. Han, Y. Liu, X. Dou, Y. Li, H. Zhu and X. Yuan, J. Mater. Chem. A, 2022, 10, 8371–8377 RSC.
- J. Qiu, D. Dai and J. Yao, Coord. Chem. Rev., 2024, 501, 215597 CrossRef CAS.
- W. Fang and L. Wang, Catalysts, 2023, 13, 1325 CrossRef CAS.
- X. Xu, Y. Sui, W. Chen, W. Huang, X. Li, Y. Li, D. Liu, S. Gao, W. Wu, C. Pan, H. Zhong, H.-R. Wen and M. Wen, Appl. Catal., B, 2024, 341, 123271 CrossRef CAS.
- C. Zhuang, Y. Chang, W. Li, S. Li, P. Xu, H. Zhang, Y. Zhang, C. Zhang, J. Gao, G. Chen, T. Zhang, Z. Kang and X. Han, ACS Nano, 2024, 18, 5206–5217 CrossRef CAS PubMed.
- C. Zhuang, W. Li, T. Zhang, J. Li, Y. Zhang, G. Chen, H. Li, Z. Kang, J. Zou and X. Han, Nano Energy, 2023, 108, 108225 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |