
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of the delocalization state on electrocatalytic CO2 reduction: a mini-review
Shuyi
Kong
 a,
Ximeng
Lv
*b and
Jiacheng
Wang
*cd
a,
Ximeng
Lv
*b and
Jiacheng
Wang
*cd
aState Key Lab of Metal Matrix Composites, School of Materials Science and Engineering, Shanghai Jiao Tong University, Dongchuan Road 800, 200240 Shanghai, China
bLaboratory of Advanced Materials, Department of Chemistry, Faculty of Chemistry and Materials Science, Fudan University, Shanghai 200438, China. E-mail: ximeng_lv@fudan.edu.cn
cState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China. E-mail: jiacheng.wang@mail.sic.ac.cn
dSchool of Materials Science and Engineering, Taizhou University, Zhejiang 318000, China
First published on 3rd January 2024
Abstract
The electrochemical carbon dioxide reduction reaction (CO2RR) is a way to alleviate environmental pollution and realize carbon recycling, which converts CO2 into value-added chemicals and fuels. Recently, the delocalization state regulation of catalysts has emerged as an effective method to evaluate the catalytic performance in CO2RR. The delocalization state of catalysts has been found to promote the stability and selectivity of CO2RR, which is essentially based on enhancing the electron conductivity of catalysts or regulating the adsorption of intermediates. In this mini-review, we first discuss how the delocalization state of catalysts affects their catalytic properties. Then, we summarize recent progress on the subject in two parts: stability and selectivity of CO2RR. In particular, we have emphasized the selectivity part, breaking it into two components: hydrogen adsorption and CO2RR intermediates. Finally, we conclude the review with some observations by outlining the challenges and presenting our viewpoints on the future research directions in this field.
Broader contextThe electrochemical carbon dioxide reduction reaction (CO2RR) is a way to alleviate environmental pollution and realize carbon recycling, which converts CO2 into value-added chemicals and fuels. Recently, the delocalization state regulation of catalysts has emerged as an effective method to evaluate the catalytic performance in CO2RR. The delocalization state of catalysts has been found to promote the stability and selectivity of CO2RR, which is essentially based on enhancing the electron conductivity of catalysts or regulating the adsorption of intermediates. In this mini-review, we first discuss how the delocalization state of catalysts affects the catalytic properties. Then, we summarize recent progress on the subject in two parts: stability and selectivity of CO2RR. In particular, we have emphasized the selectivity part, breaking it into two components: hydrogen adsorption and CO2RR intermediates. Finally, we conclude the review with some observations by outlining the challenges and presenting our viewpoints on the future research directions in this field. |
1. Introduction
In recent years, the electrochemical CO2 reduction reaction (CO2RR) has attracted much attention as a new energy technology due to the problem of energy shortage.1,2 By utilizing renewable electricity, this technology consumes CO2 captured from the atmosphere to accelerate the carbon cycle and reduce the amount of CO2 in the atmosphere (∼400 ppm), thus protecting the environment and producing valuable products.2–4 Most importantly, this catalytic reaction could change the way petroleum and coal are used in traditional organic synthesis industries.5 The electrochemical CO2 reduction technology could reduce the consumption of oil and coal if industries can produce organic matter using atmospheric CO2 and clean electricity.6,7The poor selectivity and low stability of CO2RR are the biggest issues faced during its industrial development.2,8 To solve these two issues, optimizing catalysts9,10 and reactors4,11 can be a cost-effective solution. Furthermore, theoretical research deepens the understanding of the reaction mechanism, which could guide the synthesis of efficient catalysts (e.g., volcano plots are a powerful tool for finding catalysts).2,12 To date, researchers have revealed that the intrinsic electron state (spatial configuration, bonding orbital type, etc.)13–15 and reaction microenvironment (surface species, CO2 pressure, hydrophobicity, etc.)3,16–21 are two major factors affecting the catalytic performance.
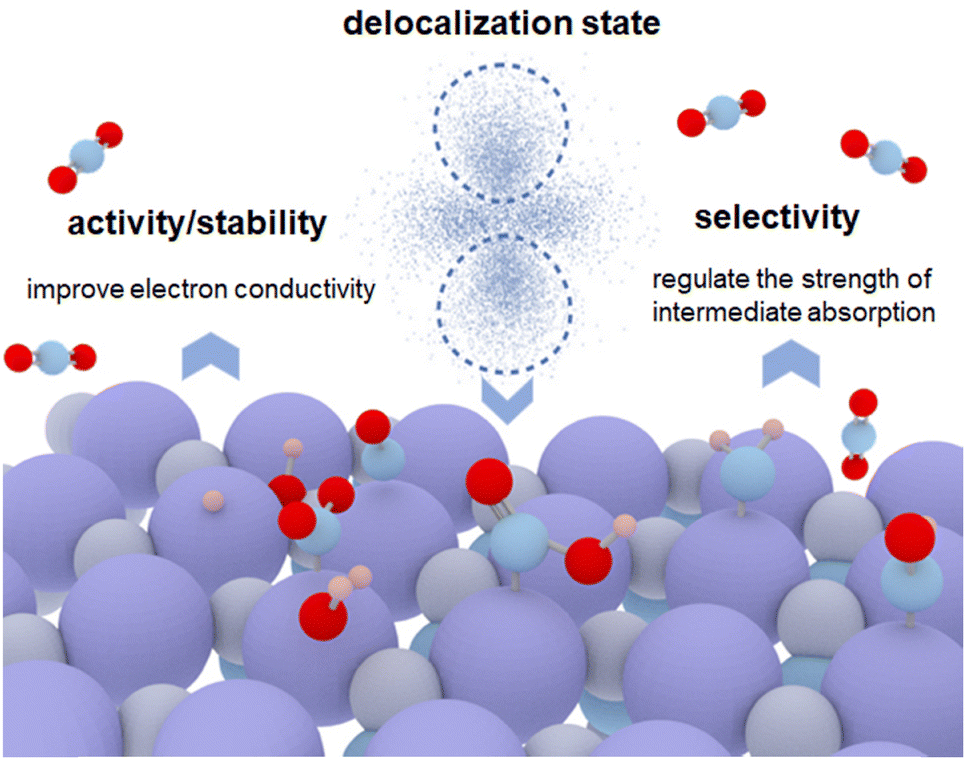
It is important to understand the mechanisms of the delocalization state affecting the performance of electrode materials.22 The delocalization state of catalysts affects the activity, stability, and selectivity, which is essentially based on improving the conductivity of the catalysts and regulating the adsorption of intermediates.2,3 This review discusses that CO2RR can be influenced by the delocalization states of the catalysts from the following aspects: (1) maintaining the stability of catalysts during CO2RR.23 It is well known that the CO2 reaction potential is a reduction potential. Under an overpotential, the catalyst is likely to be reduced if the reaction is prolonged. The electron conductivity of catalysts can be improved by delocalization, which inhibits the destruction of the material structure. (2) Regulating the adsorption of key intermediates.24 For example, the desorption of CO from the transition metal sites is inherently difficult. This is mainly because the localized 3d orbitals of transition metals tend to hybridize with the 5σ and 2π* orbitals of CO leading to a high energy barrier for CO desorption.25,26 The delocalization state of metal atoms decreases the energy barrier to improve the selectivity of the CO product. For instance, Xu et al. found that the partially delocalized MoSeS monolayers facilitate CO desorption due to the off-center charge around Mo atoms.27
In this mini-review, we first introduce the definition of a delocalization state. Then, we discuss the current research on how the delocalization state affects the catalytic performance of catalysts, such as electron conductivity and the energy barrier (Fig. 1). Moreover, the role of delocalization states of catalysts on the CO2RR stability is discussed. In addition, we focus on the selective adsorption/desorption of reaction intermediates during CO2RR caused by the delocalization states. In the end, we raise the unsolved challenges associated with the regulation of the delocalization state of catalysts and offer an outlook for future research in this exciting field.
 | ||
| Fig. 1 Schematic diagram of the influences of the delocalization state on the physical and chemical properties of catalysts toward CO2RR. | ||
2. The delocalization state of catalysts toward CO2RR
2.1 The effect of the delocalization state
Delocalization is a concept employed to describe the electron state.28,29 The appearance of delocalization of metal atoms in a compound can be described as the nephelauxetic effect, that is, the formation of the complex makes the electron cloud in a metal ion more diffuse than that of an uncoordinated metal ion. Electronic delocalization refers to the distribution of electrons over more than two atoms in a molecule or a solid material. The electrons, not localized around a specific atom, could spread out across multiple atoms or even throughout the entire molecule or material. This phenomenon is crucial in understanding the behavior and properties of various chemical and physical systems.In solid materials, the delocalized electron refers to the electron that is subjected to a weak periodic potential, and it can move from one potential minimum to another under the influence of a small electric field, thermal energy, or even the uncertainty principle.24,30 Generally, the electron density distribution can be employed to describe the delocalization effect. From the perspective of condensed matter physics, the delocalization state in crystal materials can be quantitatively described by the effective mass (m*) of the carriers at the conduction band minimum (eqn (1)).28 The detailed formula is as follows, where Ek is the energy of Bloch electrons, k is the wavevector, and ħ refers to the reduced Planck constant:
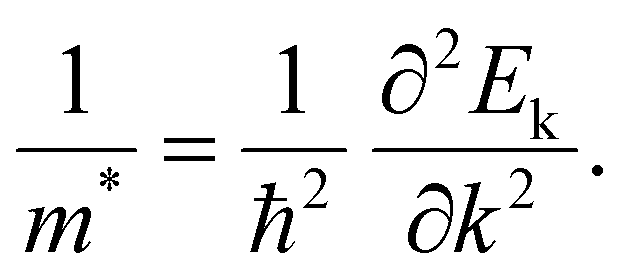 | (1) |
Electronic delocalization plays a significant role in determining the reactivity and efficiency of catalysts. Catalysts are substances that facilitate chemical reactions by lowering the activation energy required for the reaction to occur. The presence of delocalized electrons in catalysts allows them to participate actively in the reaction process. Another important aspect of electronic delocalization in catalysis is the formation of conjugated systems. Conjugation refers to the alternating pattern of single and multiple bonds between atoms, creating a system of overlapping p-orbitals. This results in electron delocalization along the entire conjugated structure. Conjugated systems are often found in organic molecules, such as aromatic compounds and polymers. They exhibit special electronic properties, including increased stability and enhanced π-electron interactions. These features make conjugated systems valuable in catalytic reactions.
From current research, we figured out some physical and chemical characteristics that were generated by the delocalization state and contributed to improving the CO2RR performance of catalysts. These studies regarded delocalization states as a useful tool for an in-depth understanding of the material properties. Herein, we discuss the influences of the delocalization state on the physical and chemical properties of catalysts from the following aspects (Fig. 1).
(1) High electron conductivity. The delocalization state leads to higher electron density at the metal atoms,31,32 which causes a high electron conductivity of catalysts. Recent researches33,34 have demonstrated that layered 2D conjugated MOFs with fully in-plane π-delocalization along 2D directions and weak out-plane π–π stacking exhibit higher density of exposed metal centers and improved electron conductivity (up to 2500 S cm−1) apart from the inherited features of traditional MOFs, suggesting a great potential in high-performance electrocatalysis.35
(2) Lower reaction energy barriers. The intensity of the adsorption/desorption of intermediates influences the selectivity of CO2RR products.6 For example, the electronic delocalization state of NiO clusters-decorated Ni-based single-atom catalysts reduces the free energy barriers for *COOH intermediates formation, leading to enhanced reaction kinetics for CO production.36
2.2 The regulation and characterization method of delocalization state
We first summarize how to regulate the delocalization state of materials. Some materials naturally possess the property of electron delocalization, such as compounds with special ligands.37,38 Introducing structural distortion to the atomic layers is also a way to regulate delocalized states.39 In addition, the method of constructing vacancy or the heterojunction or doping atom plays the role of electron rearrangement, which is likely to enhance electron delocalization.39–41 Besides, charge delocalization can be regulated by utilizing photo- or electro-driven switches.42In terms of the methods used to characterize the delocalization state, X-ray absorption near-edge structure (XANES) spectroscopy is one of the most effective techniques to investigate the spin configurations of transition metals.39 Therefore, XANES can identify the delocalization state by confirming the structural distortion. Density functional theory (DFT) is also a way to study the electronic structure by displaying the charge density of catalysts.
3. The effect of the delocalization state on CO2RR
3.1 The effect of the delocalization state on stability
Most catalysts face the degradation process during CO2RR, which rearranges structure, morphology, and electron distribution and then affects the activity and selectivity.43 Many researchers made efforts to improve the stability of the catalysts during CO2 reduction reactions, but using methods such as coating and doping.44–46 Delocalization is also a way to improve stability from the perspective of electron distribution.For instance, Liang et al. reported a nitride-based anti-perovskite CuNNi3 coated on a CuNi3 alloy core, which was first used in CO2RR.47 As shown in the stability test, the current density of the CuNNi3 catalyst decreases continuously within 10 hours while the CuNi3@CuNNi3 catalyst maintains steadily up to 10 hours (Fig. 2). XANES analysis confirmed the interfacial electron delocalization from the CuNi3 alloy core to the CuNNi3 nanoshell. DFT calculations revealed that interfacial delocalization enriches the electron density in the CuNNi3 nanoshell, increases the N escape energy in the face-centered cubic cell of CuNNi3, and regulates the catalytic behaviors of anti-perovskite CuNNi3 and enhances its electrochemical stability during CO2RR.
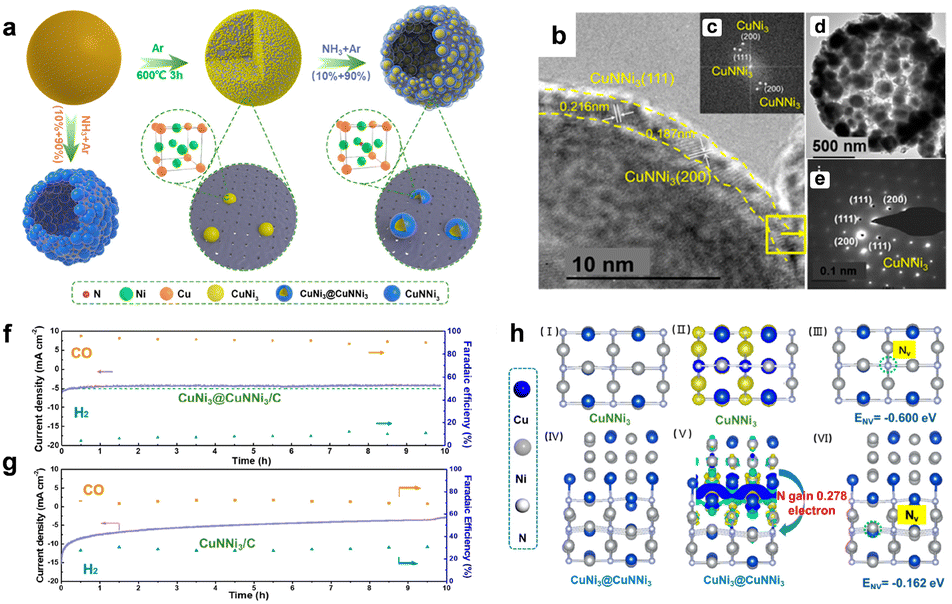 | ||
| Fig. 2 (a) Schematic diagram of the preparation process of core–shell-structured CuNi3@CuNNi3/C nanocomposites; (b) scanning electron microscopy (SEM) and a high angle annular dark field scanning transmission electron microscopy (HADDF-STEM) image of CuNi3@CuNNi3/C, (inset: (c) relevant FFT pattern). (d) TEM image and (e) SAED pattern of the surface area in (b); the comparison of stability obtained in KHCO3 solution in-saturated with CO2 by the potentiostatic stability analysis of (f) CuNi3@CuNNi3/C and (g) CuNNi3/C at −0.858 V (vs. RHE). (h) DFT calculation results of the N element escape energies in pure CuNNi3 crystals and CuNNi3 being interfaced with CuNi3: (I) top view of CuNNi3(110) and (IV) side view of CuNi3@CuNNi3. Bader charge difference calculation results of CuNNi3(110) and (V) CuNi3@CuNNi3 and N vacancy formation energy in (III) CuNNi3(110) and (VI) CuNi3@CuNNi3. Copyright ©2021 American Chemical Society. | ||
Moreover, Sun et al. investigated a semi-conductive metal–organic framework (MOF)–Cu3(HITP)2 adding Ketjen Black, which stabilized the faradaic efficiency of C2H4.48 Electrochemical reconstruction has always happened to MOFs as electrocatalysts, especially under high current conditions. This phenomenon is analogous to the process of dendrite formation observed on the anodes of metal ion batteries, which destroys the electrode and consequently decreases the cycle numbers of batteries. The way to improve charge delocalization on the less-conductive solid–electrolyte interface would suppress the dendrite growth and enhance chemical stability. The effect of Ketjen Black-promoted charge delocalization is one of the reasons that the Cu crystallites in Ketjen Black@Cu3(HITP)2 did not aggregate during the stability test.
Similar to the MOFs, the problems of cobalt phthalocyanine (CoPc) include instability and low conductivity during CO2RR due to its fast deactivation or structure changes at high reduction current density. Recently, Masana et al. reported a cobalt phthalocyanine (CoPc) with a nitrogen-rich carbon nitride electrocatalyst.49 The coordination of the nitrogen-rich carbon nitride can modulate the intrinsic electronic structure of CoPc and then induce electron delocalization of the Co–N4 site verified by XANES. After electrolyzing for 40 h, the current density of the CO product and the structure of CoPc remained stable. This is owing to electron delocalization, which prolonged carrier lifetime, increased electron density, and subsequently enabled rapid charge transfer.
As can be seen from the above examples, delocalization is mainly aimed at electron distribution and improving the conductivity of the catalysts to enhance the structure stability during CO2RR.
3.2 The effect of delocalization state on selectivity
In addition to the effect on chemical stability, the delocalization state also has an influence on product selectivity during CO2 reduction. The regulation of product selectivity by the delocalization state is mainly based on the different adsorption strengths of the intermediate on the catalyst surface. Therefore, the following part is divided into the regulation of hydrogen evolution reaction (HER) and the adsorption strength of CO2 reduction intermediates.For example, Liu et al. reported that S-doped Bi2O3 electrocatalysts coupled with carbon nanotubes (S-Bi2O3-CNTs) maintain formate production (FE > 90%) at high current density.53 The improved selectivity originated from the delocalization state of Bi atoms caused by the doping of S atoms. DFT calculations revealed that the electronic delocalization of Bi enhanced the binding of *CO2 and *HCOO and weakened the adsorption of *H, thus inhibiting the hydrogen evolution reaction and improving the activity of CO2 reduction.
Aside from doping elements, some defective materials also can cause the delocalization state of metal atoms. For example, Wang et al. designed the Se-defective CuInSe2 (V-CuInSe2), which has high faradaic efficiency (FE) of CO (>90%).54 The Se vacancies on the surface of CuInSe2 are found to redistribute the electrons around the Se atoms in pristine CuInSe2 and cause increased electron numbers around the surface of Cu and In atoms and thus promote the delocalization state of metal atoms. DFT calculations showed that the electron delocalization of V-CuInSe2 decreases the energy barrier of the RDS (CO2 → CO), indicating a more favorable CO2RR kinetics and inhibiting hydrogen adsorption.
The design of molecular catalysts makes it possible to increase the electronic delocalization to maximize selectivity for CO2 reduction. Derrick et al. recently reported an iron polypyridine complex [Fe(tpyPY2Me)(CH3CN)]2+([Fe1]2+), which has a high faradaic efficiency for CO production (>90%) at a low overpotential.55 Previous work from these laboratories has found that the PY5Me2 ligand and its molybdenum and cobalt derivatives have the catalytic activity for HER.56,57 In this work, it was possible to increase metal–ligand orbital mixing and delocalization of electron density away from the metal center to favor CO2 reduction.
For example, Kong et al. reported a new compound Cu2NCN,24 which has high methanol faradaic efficiency (>40%) and a current density of 92 mA cm−2. Theoretical calculations indicated that with the nephelauxetic effect and cation polarization by strong-coordinated NCN2− anions, the electron cloud dispersion around each Cu(I) was substantially enhanced (Fig. 3), thus changing the original electronic structure of Cu(I) into a more delocalized state (i.e., softer metal sites). The soft Cu(I) sites are favorable to breaking the Cu–O bonds and releasing *OCH3 intermediates, thus improving the selectivity of methanol.
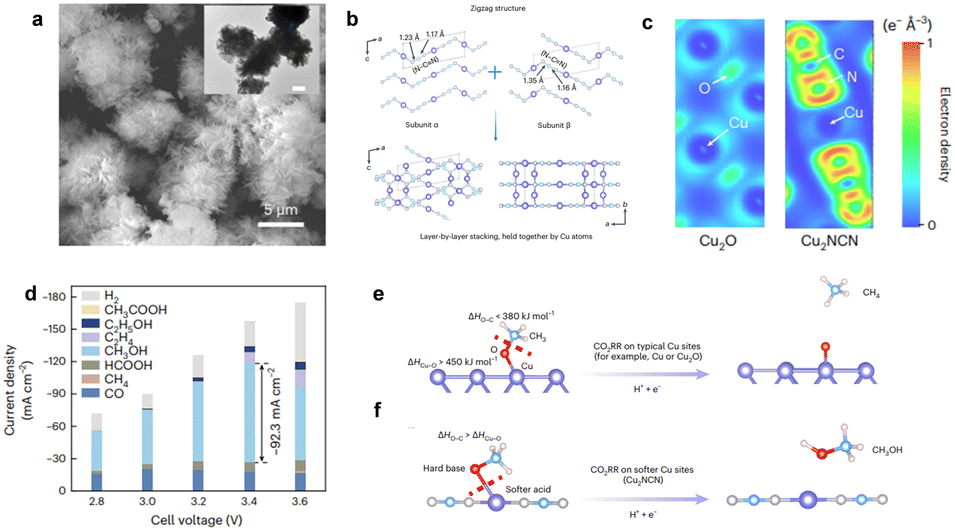 | ||
| Fig. 3 (a) SEM and TEM (inset, scale bar: 1 μm) images of Cu2NCN. (b) Schematic illustration of the Cu2NCN structure. (c) PDOS patterns of the Cu 3d orbitals in Cu2O and Cu2NCN. (d) CO2RR product distribution achieved on Cu2NCN in MEA-based electrolyzers. (e) and (f) Schematic illustration of the CO2RR mechanism. Typical Cu catalytic sites (for example, Cu or Cu2O) display a relatively strong Cu–O bond (that is, the bond dissociation enthalpy, ΔHCu–O is >450 kJ mol−1) compared with the O–C bond (ΔHO–C in OCH3 is <380 kJ mol−1), thus leading to the cleavage of the O–C bond to release *CH3 and then form CH4. Copyright © 2023 Springer Nature. | ||
In contrast to the easily tunable and delocalized electronic structure of d-orbitals in the transition metals, main-group metals exhibited localized p-orbital electron states.59 He et al. reported that the p-orbital delocalization of Bi atoms is conducive to the adsorption of intermediate *OCHO, which is the key intermediate in the formation of formate.60 DFT simulations revealed that the delocalization state of the Bi p-orbital can be adjusted by reducing the inter-layer Bi–Bi bond length. Therefore, they designed a bismuth catalyst with p-orbital delocalization by electrochemically reducing BiOCl nanosheets and verified the existence of a compressed inter-layer bond length by XAFS analysis. The Bi catalyst with p-orbital delocalization showed a high formate faradaic efficiency (>90%) and a current density of 500 mA cm−2.
Furthermore, Liu et al. reported a delocalized Bismuth catalyst co-deposited with Cu2+ ions, which showed the large part current density of formate (856 mA cm−2) and FEHCOOH (>85%).61 The calculated projected density of electronic states (DOS) proved the distinct delocalization of Bi p-orbitals. Moreover, they pointed out that the delocalization state of Bi electrons forms more anti-bonding orbitals via the orbital hybridization with *OCHO (or *COOH) intermediate. The orbital hybridization optimizes the interaction energies, lowering the thermodynamic barrier of the CO2-to-HCOOH reaction. On the other hand, the bond strength between *OCHO (or *COOH) intermediate and Bi atoms can be evidenced by the distance at the Fermi level. The total DOS center of BiCu-bimetal (delocalization state) is closer to the Fermi level while bulk Bi (more localization state) moves away from the Fermi energy level (Table 1).
| Catalysts | Electrolyte | Catalytic performancea |
|---|---|---|
| a FE: faradaic efficiency; J: current density; T: test time. | ||
| CuNi3@CuNNi3 nano-composite47 | 0.1 M KHCO3 | FE: 96% CO |
| J: −5 mA cm−2 | ||
| T: 10 h | ||
| Ketjen Black@Cu3(HITP)262 | KHCO3 | FE: 70% C2H4 |
| J: −26.3 mA cm−2 | ||
| T: 10 h | ||
| Co–N5/MPF49 | 0.5 M KHCO3 | FE: 99% CO |
| J: −5.3 mA cm−2 | ||
| T: 40 h | ||
| S2-Bi2O3-CNT53 | 0.5 M KHCO3 | FE: 97.06% formate |
| J: −48.6 mA cm−2 | ||
| T: 10 h | ||
[Fe(tpyPY2Me)]2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 55 55 |
Both organic and neutral aqueous electrolytes | FE: 94% CO |
| J: −3.6 mA cm−2 | ||
| T: 20 h | ||
| Se-defective CuInSe254 | 0.5 M KHCO3 | FE: 91.0% CO |
| J: −112 mA cm−2 | ||
| T: 40 h | ||
| Cu2NCN24 | 0.5 M KHCO3 | FE: 43% CH3OH |
| J: −93 mA cm−2 | ||
| T: 10 h | ||
| POD-Bi63 | 0.5 M KHCO3 | FE: 93% formate, |
| J: −100 mA cm−2 | ||
| T: 0.5 h | ||
| BiCu-bimetallic film on Cu foam61 | 1.0 M KOH | FE: 85% formate |
| J: −856 mA cm−2 | ||
| T: 10 h | ||
| Mg–C3N464 | KHCO3 | FE: 90% CO |
| J: −300 mA cm−2 | ||
| T: 12 h | ||
| NiO clusters-decorated Ni–N–C SACs65 | 0.1 M KHCO3 | FE: 96.5% CO |
| J: −18 mA cm−2 | ||
| T: 10 h | ||
| Zn–Ag–O@ultrahigh-surface-area carbon66 | 0.5 M KHCO3 | FE: 94% CO |
| J: −20 mA cm−2 | ||
| T: 150 h | ||
| Pyridine catalysts on Ag electrode67 | 0.1 M KHCO3 | FE: 74% CO |
| J: −150 mA cm−2 | ||
| T: 2 h | ||
| Porous N-doped carbon nanotubes stabilized Ni SACs68 | 0.5 M KHCO3 | FE: 100% CO |
| J: −34.3 mA cm−2 | ||
| T: 20 h | ||
Compared to the localized d-orbitals, delocalized s-orbitals can weaken CO adsorption ability, providing a workable deal to regulate the CO2RR pathway. For example, Liu et al. reported an atomically dispersed magnesium atom embedded in graphitic carbon nitride (Mg–C3N4), which has a high selectivity of CO (FECO ≥ 90%).64 The main group metal sites have a delocalized s/p band.69 Moreover, due to the strong hybridization between CO and 3d-orbitals, a delocalized 3s-orbital possesses a much weaker CO desorption energy barrier than that on 3d orbitals. Therefore, the nondirectional delocalization of Mg 3s-orbital results in weak interaction with CO.
In general, the effect of delocalization on selectivity is realized through the change of band structure, which can change the adsorption strength and induce the selective adsorption of reaction intermediates.
4. Perspective and conclusions
Electrochemical CO2 reduction provides an effective technological solution to both energy and environmental problems. In recent decades, the regulation method of delocalization state has gradually become an idea for understanding and analyzing the catalytic performance of the material towards CO2RR. At present, various types of CO2RR catalysts with delocalization states have been developed. Some CO2RR catalysts possibly show a delocalization state of metal atoms, especially in the field of molecular catalysts. With the in-depth exploration of the CO2RR mechanism, the delocalization state has become a useful tool for researching the role of atoms and electrons in chemical reactions from a microscopic perspective. Specifically, the delocalization state affects the catalytic reaction by regulating the conductivity of catalysts and the adsorption/desorption strength of reaction intermediates. Therefore, the catalysts with the delocalization state have a good performance on the product selectivity and reaction stability toward CO2RR.In this review, we have introduced the definition of delocalization state and discussed how it affects the CO2RR performances of catalysts, which is essentially based on the high electron conductivity of catalysts and the low energy barrier of intermediates. By summarizing the relevant studies, we divide them into two aspects (stability and selectivity) according to the different CO2RR performances, which are efficiently improved by delocalization state regulation. In addition to the aspects summarized in this review, enhancing reactant activation is one of the manifestations induced by the delocalization state.59
Though some notable outcomes have been achieved, it is still in the early stages of developing delocalization state regulation towards high-performance CO2RR catalysts. As we look ahead, the development of electronic delocalization in catalysis is expected to follow several key directions.
First, there is a continued focus on the design and synthesis of novel catalysts with enhanced electronic delocalization. For example, this may involve the incorporation of π-conjugated systems, especially used in organic frameworks with extended electron delocalization. These catalysts will not only facilitate electron transfer processes but also provide unique active sites for specific reactions.
Second, the exploration of new strategies to control and manipulate electronic delocalization in catalysts is crucial. For example, this could involve the use of light, heat, or electric fields to modulate the electronic structure of catalysts in real time as mentioned in Section 2.2. Researchers can fine-tune the catalysts, leading to improved catalytic performance.
Furthermore, the difficulty of characterizing the delocalization states makes it tough to study the catalyst design conveniently. In general, a reliable method to characterize the delocalization states is to use XANES. The cost of this test greatly limits the amount of material that can be studied. To solve this problem, theoretical calculation may be a good way. The use of theoretical calculations, such as density functional theory, could enable researchers to predict and understand the electronic properties of catalysts with great accuracy.70,71 Advancements in computational modelling and simulation techniques play a vital role in elucidating the intricate mechanisms underlying electronic delocalization in catalysis. The catalyst with the delocalization state could be selected by theoretical calculation, and then the experimental study could be carried out.
Overall, the future of electronic delocalization in catalysis holds great promise. By harnessing the power of electron delocalization, we can expect to see the development of highly selective and stable catalysts that drive important chemical transformations, leading to a more sustainable and greener future.
In the end, we believe that future research on the delocalization state is profound through the help of theoretical calculations and characteristic techniques. With these advances, delocalization state regulation will undoubtedly continue to spark more research excitement toward CO2RR as well as other reactions.
Author contributions
S. Kong and X. Lv designed and wrote this review, J. Wang supervised the project and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. W. thanks the National Natural Science Foundation of China (52072389, 92163117) and the Program of Shanghai Academic Research Leader (20XD1424300) for the financial support. X. L. thanks the National Natural Science Foundation of China (22379026) and the China Postdoctoral Science Foundation (2023M730637, 2023TQ0082) for the financial support.References
- I. E. L. Stephens, K. Chan, A. Bagger, S. W. Boettcher, J. Bonin, E. Boutin, A. K. Buckley, R. Buonsanti, E. R. Cave, X. Chang, S. W. Chee, A. H. M. da Silva, P. de Luna, O. Einsle, B. Endrődi, M. Escudero-Escribano, J. V. Ferreira de Araujo, M. C. Figueiredo, C. Hahn, K. U. Hansen, S. Haussener, S. Hunegnaw, Z. Huo, Y. J. Hwang, C. Janáky, B. S. Jayathilake, F. Jiao, Z. P. Jovanov, P. Karimi, M. T. M. Koper, K. P. Kuhl, W. H. Lee, Z. Liang, X. Liu, S. Ma, M. Ma, H.-S. Oh, M. Robert, B. R. Cuenya, J. Rossmeisl, C. Roy, M. P. Ryan, E. H. Sargent, P. Sebastián-Pascual, B. Seger, L. Steier, P. Strasser, A. S. Varela, R. E. Vos, X. Wang, B. Xu, H. Yadegari and Y. Zhou, JPhys: Energy, 2022, 4, 042003 Search PubMed.
- X. Lv, Z. Liu, C. Yang, Y. Ji and G. Zheng, Acc. Mater. Res., 2023, 4, 264–274 CrossRef CAS.
- P.-P. Yang and M.-R. Gao, Chem. Soc. Rev., 2023, 52, 4343–4380 RSC.
- X. Wang, X. Lv, G. Zheng and Y. Fu, Energy Chem., 2022, 4, 100086 CrossRef CAS.
- P. D. Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef PubMed.
- L. Shang, X. Lv, L. Zhong, S. Li and G. Zheng, Small Methods, 2022, 6, 2101334 CrossRef CAS.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustain., 2021, 4, 911–919 CrossRef.
- X. Lv, M. Chen, Z. Xie, L. Qian, L. Zhang and G. Zheng, Chin. J. Catal., 2022, 43, 92–103 CrossRef CAS.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- S. Overa, B. H. Ko, Y. Zhao and F. Jiao, Acc. Chem. Res., 2022, 55, 638–648 CrossRef CAS PubMed.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- Y. Y. Birdja, E. Perez-Gallent, M. C. Figueiredo, A. J. Gottle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- P. Yu, X. Lv, Q. Wang, H. Huang, W. Weng, C. Peng, L. Zhang and G. Zheng, Small, 2023, 19, 2205730 CrossRef CAS PubMed.
- X. Lv, L. Shang, S. Zhou, S. Li, Y. Wang, Z. Wang, T. K. Sham, C. Peng and G. Zheng, Adv. Energy Mater., 2020, 10, 2001987 CrossRef CAS.
- L. Qian, X. Lv, Y. Ren, H. Wang, G. Chen, Y. Wang and J. Shen, J. Chromatogr. A, 2013, 1322, 81–89 CrossRef CAS PubMed.
- Z. Liu, X. Lv, J. Zhang, A. Guan, C. Yang, S. Yan, Y. Chen, K. Liu and G. Zheng, Adv. Mater. Interfaces, 2022, 9, 2101956 CrossRef CAS.
- Y. Chen, M. Kan, S. Yan, J. Zhang, K. Liu, Y. Yan, A. Guan, X. Lv, L. Qian and G. Zheng, Chin. J. Catal., 2022, 43, 1703–1709 CrossRef CAS.
- Z. Liu, X. Lv, S. Kong, M. Liu, K. Liu, J. Zhang, B. Wu, Q. Zhang, Y. Tang, L. Qian, L. Zhang and G. Zheng, Angew. Chem., Int. Ed., 2023, 62, e202309319 CrossRef CAS.
- L. Yang, X. Lv, C. Peng, S. Kong, F. Huang, Y. Tang, L. Zhang and G. Zheng, ACS Cent. Sci., 2023, 9, 1905–1912 CrossRef CAS PubMed.
- P.-P. Yang, X.-L. Zhang, P. Liu, D. J. Kelly, Z.-Z. Niu, Y. Kong, L. Shi, Y.-R. Zheng, M.-H. Fan, H.-J. Wang and M.-R. Gao, J. Am. Chem. Soc., 2023, 145, 8714–8725 CAS.
- Z.-Z. Niu, L.-P. Chi, Z.-Z. Wu, P.-P. Yang, M.-H. Fan and M.-R. Gao, National Sci. Open, 2023, 2, 20220044 CrossRef.
- Y. Fang, X. Lv, Z. Lv, Y. Wang, G. Zheng and F. Huang, Adv. Sci., 2022, 9, 2205680 CrossRef CAS PubMed.
- B. Hou, H. Zheng, K. Zhang, Q. Wu, C. Qin, C. Sun, Q. Pan, Z. Kang, X. Wang and Z. Su, Chem. Sci., 2023, 14, 8962–8969 RSC.
- S. Y. Kong, X. M. Lv, X. Wang, Z. Z. Liu, Z. C. Li, B. Q. Jia, D. Sun, C. Yang, L. J. Liu, A. X. Guan, J. C. Wang, G. F. Zheng and F. Q. Huang, Nat. Catal., 2023, 6, 6–15 CrossRef CAS.
- S. S. Sung and R. Hoffmann, J. Am. Chem. Soc., 1985, 107, 578–584 CrossRef CAS.
- C. C. Leon, Q. Liu and S. T. Ceyer, J. Phys. Chem. C, 2019, 123, 9041–9058 CrossRef CAS.
- J. Q. Xu, X. D. Li, W. Liu, Y. F. Sun, Z. Y. Ju, T. Yao, C. M. Wang, H. X. Ju, J. F. Zhu, S. Q. Wei and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 9121–9125 CrossRef CAS PubMed.
- J. Bardeen and W. Shockley, Phys. Rev., 1950, 80, 72–80 CrossRef CAS.
- N. Sablon, F. De Proft and P. Geerlings, Chem. Phys. Lett., 2010, 498, 192–197 CrossRef CAS.
- A. D. McNaught and A. Wilkinson, Compendium of chemical terminology, Blackwell Science Oxford, 1997 Search PubMed.
- Z. Lv, B. Peng, X. Lv, Y. Gao, K. Hu, W. Dong, G. Zheng and F. Huang, Adv. Funct. Mater., 2023, 33, 2214370 CrossRef CAS.
- Z. Lv, C. Zhao, M. Xie, M. Cai, B. Peng, D. Ren, Y. Fang, W. Dong, W. Zhao, T. Lin, X. Lv, G. Zheng and F. Huang, Adv. Mater., 2023, n/a, 2309637 CrossRef PubMed.
- R. H. Dong, T. Zhang and X. L. Feng, Chem. Rev., 2018, 118, 6189–6235 CrossRef CAS PubMed.
- D. W. Feng, T. Lei, M. R. Lukatskaya, J. Park, Z. H. Huang, M. Lee, L. Shaw, S. C. Chen, A. A. Yakovenko, A. Kulkarni, J. P. Xiao, K. Fredrickson, J. B. Tok, X. D. Zou, Y. Cui and Z. A. Bao, Nat. Energy, 2018, 3, 30–36 CrossRef CAS.
- H. X. Zhong, M. Ghorbani-Asl, K. H. Ly, J. C. Zhang, J. Ge, M. C. Wang, Z. Q. Liao, D. Makarov, E. Zschech, E. Brunner, I. M. Weidinger, J. Zhang, A. V. Krasheninnikov, S. Kaskel, R. H. Dong and X. L. Feng, Nat. Commun., 2020, 11, 1409 CrossRef CAS PubMed.
- H. Q. Li, K. N. Gan, R. Li, H. W. Huang, J. B. Niu, Z. P. Chen, J. Zhou, Y. Yu, J. S. Qiu and X. J. He, Adv. Funct. Mater., 2023, 33, 2208622 CrossRef CAS.
- X. H. Liu, M. A. Wankeu, H. Lucken and R. Dronskowski, Z. Naturforsch. B, 2005, 60, 593–596 CrossRef CAS.
- H. P. Xiang, X. H. Liu and R. Dronskowski, J. Phys. Chem. C, 2009, 113, 18891–18896 CrossRef CAS.
- S. C. Chen, Z. X. Kang, X. Hu, X. D. Zhang, H. Wang, J. F. Xie, X. S. Zheng, W. S. Yan, B. C. Pan and Y. Xie, Adv. Mater., 2017, 29, 1701687 CrossRef.
- K. K. Ma, J. Wu, X. Wang, Y. Sun, Z. Z. Xiong, F. L. Dai, H. K. Bai, Y. Xie, Z. Kang and Y. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202211094 CrossRef CAS PubMed.
- C. X. Wang, M. Yang, S. Cao, X. X. Wang, H. Fu, Y. Bai, T. Lookman, P. Qian and Y. J. Su, Phys. Rev. Mater., 2023, 7, 085801 CrossRef CAS.
- G. T. Xu, B. Li, J. Y. Wang, D. B. Zhang and Z. N. Chen, Chem. – Eur. J., 2015, 21, 3318–3326 CrossRef CAS PubMed.
- S. Popović, M. Smiljanić, P. Jovanovič, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef PubMed.
- Y. P. Zang, T. F. Liu, P. F. Wei, H. F. Li, Q. Wang, G. X. Wang and X. H. Bao, Angew. Chem., Int. Ed., 2022, 61, e202209629 CrossRef CAS PubMed.
- D. H. Nam, P. De Luna, A. Rosas-Hernandez, A. Thevenon, F. W. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- T. Wang, J. D. Chen, X. Y. Ren, J. C. Zhang, J. Ding, Y. H. Liu, K. H. Lim, J. H. Wang, X. N. Li, H. B. Yang, Y. Q. Huang, S. Kawi and B. Liu, Angew. Chem., Int. Ed., 2023, 62, e202211174 CrossRef CAS PubMed.
- Q. Liang, Y. Zhao, J. D. Chen, J. J. Dai, X. Y. Ding, Z. Tong, S. J. Xie, J. Zhang, Z. H. Zhou, J. T. Li, J. F. Li and Y. Zhou, Chem. Mater., 2022, 34, 5607–5620 CrossRef CAS.
- H. Sun, L. Chen, L. K. Xiong, K. Feng, Y. F. Chen, X. Zhang, X. Z. Yuan, B. Y. Yang, Z. Deng, Y. Liu, M. H. Rummeli, J. Zhong, Y. Jiao and Y. Peng, Nat. Commun., 2021, 12, 6823 CrossRef CAS PubMed.
- J. J. Masana, J. Xiao, H. Zhang, X. Lu, M. Qiu and Y. Yu, Appl. Catal., B, 2023, 323, 122199 CrossRef CAS.
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed.
- M. C. O. Monteiro, F. Dattila, N. Lopez and M. T. M. Koper, J. Am. Chem. Soc., 2022, 144, 1589–1602 CrossRef CAS PubMed.
- A. Goyal, C. J. Bondue, M. Graf and M. T. M. Koper, Chem. Sci., 2022, 13, 3288–3298 RSC.
- S. Q. Liu, M. R. Gao, R. F. Feng, L. Gong, H. B. Zeng and J. L. Luo, ACS Catal., 2021, 11, 7604–7612 CrossRef CAS.
- J. J. Wang, X. R. Zheng, G. J. Wang, Y. H. Cao, W. L. Ding, J. F. Zhang, H. Wu, J. Ding, H. L. Hu, X. P. Han, T. Y. Ma, Y. D. Deng and W. N. Hu, Adv. Mater., 2022, 34, 2106354 CrossRef CAS.
- J. S. Derrick, M. Loipersberger, R. Chatterjee, D. A. Iovan, P. T. Smith, K. Chakarawet, J. Yano, J. R. Long, M. Head-Gordon and C. J. Chang, J. Am. Chem. Soc., 2020, 142, 20489–20501 CrossRef CAS PubMed.
- D. Z. Zee, T. Chantarojsiri, J. R. Long and C. J. Chang, Acc. Chem. Res., 2015, 48, 2027–2036 CrossRef CAS PubMed.
- Y. J. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212–9215 CrossRef CAS PubMed.
- Z. Liu, J. Cao, B. Wu, L. Qian, A. Guan, C. Yang, X. Lv, L. Zhang and G. Zheng, ACS Catal., 2022, 12, 12555–12562 CrossRef CAS.
- D. Mukherjee, B. D. Sahoo, K. D. Joshi and S. C. Gupta, J. Appl. Phys., 2014, 115, 053702 CrossRef.
- S. He, F. Ni, Y. Ji, L. Wang, Y. Wen, H. Bai, G. Liu, Y. Zhang, Y. Li, B. Zhang and H. Peng, Angew. Chem., Int. Ed., 2018, 57, 16114–16119 CrossRef CAS PubMed.
- B. Liu, Y. Xie, X. Wang, C. Gao, Z. Chen, J. Wu, H. Meng, Z. Song, S. Du and Z. Ren, Appl. Catal., B, 2022, 301, 120781 CrossRef CAS.
- H. Sun, L. Chen, L. Xiong, K. Feng, Y. Chen, X. Zhang, X. Yuan, B. Yang, Z. Deng, Y. Liu, M. H. Rümmeli, J. Zhong, Y. Jiao and Y. Peng, Nat. Commun., 2021, 12, 6823 CrossRef CAS PubMed.
- S. S. He, F. L. Ni, Y. J. Ji, L. E. Wang, Y. Z. Wen, H. P. Bai, G. J. Liu, Y. Zhang, Y. Y. Li, B. Zhang and H. S. Peng, Angew. Chem., Int. Ed., 2018, 57, 16114–16119 CrossRef CAS PubMed.
- Q. Y. Wang, K. Liu, J. W. Fu, C. Cai, H. J. W. Li, Y. Long, S. Y. Chen, B. Liu, H. M. Li, W. Z. Li, X. Q. Qiu, N. Zhang, J. H. Hu, H. Pan and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 25241–25245 CrossRef CAS PubMed.
- H. Li, K. Gan, R. Li, H. Huang, J. Niu, Z. Chen, J. Zhou, Y. Yu, J. Qiu and X. He, Adv. Funct. Mater., 2023, 33, 2208622 CrossRef CAS.
- Z. Zhang, G. B. Wen, D. Luo, B. H. Ren, Y. F. Zhu, R. Gao, H. Z. Dou, G. R. Sun, M. Feng, Z. Y. Bai, A. P. Yu and Z. W. Chen, J. Am. Chem. Soc., 2021, 143, 6855–6864 CrossRef CAS PubMed.
- M. Abdinejad, E. Irtem, A. Farzi, M. Sassenburg, S. Subramanian, H. P. I. Van Montfort, D. Ripepi, M. R. Li, J. Middelkoop, A. Seifitokaldani and T. Burdyny, ACS Catal., 2022, 12, 7862–7876 CrossRef CAS PubMed.
- Y. Hou, Y. L. Liang, P. C. Shi, Y. B. Huang and R. Cao, Appl. Catal., B, 2020, 271, 118929 CrossRef CAS.
- S. Liu, Z. D. Li, C. L. Wang, W. W. Tao, M. X. Huang, M. Zuo, Y. Yang, K. Yang, L. J. Zhang, S. Chen, P. P. Xu and Q. W. Chen, Nat. Commun., 2020, 11, 938 CrossRef CAS PubMed.
- Z. Sun, H. Yin, K. Liu, S. Cheng, G. K. Li, S. Kawi, H. Zhao, G. Jia and Z. Yin, SmartMat, 2022, 3, 68–83 CrossRef CAS.
- Y. Feng, N. Ran, X. Wang, Q. Liu, J. Wang, L. Liu, K. Suenaga, W. Zhong, R. Ma and J. Liu, Adv. Energy Mater., 2023, 13, 2302452 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |