
Investigating the properties of fatty acid-based ionic liquids: advancement in AMOEBA force field†
Sahar
Heidari
and
Hedieh
Torabifard
 *
*
Department of Chemistry and Biochemistry, The University of Texas at Dallas, 800 West Campbell Road, Richardson, Texas 75080, USA. E-mail: Hedieh.Torabifard@utdallas.edu; Tel: +1 972-883-4686
First published on 18th November 2024
Abstract
Developing the multipolar-polarizable AMOEBA force field for large molecules presents its own set of complexities. However, by segmenting the molecules into smaller fragments and ensuring that each fragment is transferable to other systems, the process of parameterizing large molecules such as fatty acids can be simplified without compromising accuracy. In this study, we present a fragment-based AMOEBA FF development for long-chain fatty acid ionic liquids (LCFA-ILs). AMOEBA enables us to incorporate polarization to measurably enhance the precision in modeling these large highly charged systems. This is of significant importance since the computational investigation of ILs needs accurate modeling. Additionally, to leverage the tunability of ILs, it is essential to test numerous anion and cation combinations to identify the most suitable formulation for each application. However, conducting such experiments can be resource-intensive and time-consuming, but accurate molecular modeling can expedite the exploration process. Here, the newly developed parameters were evaluated by comparing the decomposed intermolecular interaction energies for ion pairs with energies determined by quantum mechanics calculations as a reference. By employing this FF in molecular dynamics simulations, we predicted bulk and structural properties including density, enthalpy of vaporization, diffusion coefficient, structure factor and radial distribution function of diverse LCFA-ILs. Notably, the good agreement between the experimental data and those calculated using our parameters validates the accuracy of our methodology. Therefore, this new procedure provides an accurate approach to parameterizing large systems, paving the way for studying more complicated systems such as lipids, polymers, micelles and membrane proteins.
1 Introduction
Ionic liquids (ILs) are salts characterized by a melting point of less than 100 °C,1 generally formed of an organic cation and inorganic anion that remain liquid across a broad temperature range due to their low lattice energy.2,3 The first IL was introduced in 1914, with a surge of interest during the 1980s.4,5 ILs have unique physicochemical properties such as high thermal and electrochemical stability, good thermal and electrical conductivity, exceptional solubility across a wide range of substances, and low vapor pressure. These properties make them attractive for many applications in various fields.6–8 ILs are suitable alternatives as solvents, electrolytes, and catalysts in industry, which have made them an interesting topic for research in recent years.9–11 Despite the advantages of ILs, concerns about their usage have been raised in both industrial and health-related fields due to possible toxicity and low biodegradability.12 Since employing ILs as green solvents was one of the essential characteristics that scientists relied on in the initial stages of research, these concerns decreased the interest in their applications. However, the green character of ILs could be improved by making ions from bio-sources.13–16 A large number of task-specific ILs have been created from various combinations of environmentally friendly and low-cost bio-based ions, which has attracted researchers' attention.16,17 Bio-based ILs, derived from renewable and non-toxic bio-sources such as amino acids18 from proteins, sugar19 from cellulose and fatty acids20 from vegetables, offer promising solutions for addressing issues such as water pollution, oil depletion and medical applications.12,19,21Long-chain fatty acid ionic liquids (LCFA-ILs) are a particular class of bio-based ILs obtained from algae-derived oils or vegetables. LCFA-ILs are mostly comprised of ammonium, phosphonium, and imidazolium cations combined with different natural fatty acid carboxylates as anions.22–25 They are classified as hydrophobic ILs and are green alternatives to conventional ILs in applications such as liquid–liquid extraction, metal extraction,26,27 and lubricants.23 Due to their hydrophobicity, efficient and green manufacturing principles, and antimicrobial properties,22 LCFA-ILs can overcome the above concerns and deserve more investigations as bio-based ILs.
The synthesis of ILs can be carried out via different classes of cations combined with a wide range of anions. In other words, a single cation can be combined with one of the numerous available anions to form a new IL. Changes in the ion selection can dramatically affect the physical properties of an IL.28 Many combinations of anion–cation constituents with different characteristics should be tested to find a specific IL to address a particular issue. However, experimental investigations to find a suitable IL structure for a specific application among many anion–cation combinations can be a costly and time-consuming process. Instead, molecular dynamics (MD) simulation can be used as a powerful tool to design ILs, predict, and tune their properties before attempting experimental investigations.29–32 To perform a reliable MD simulation, we need accurate force fields (FFs). While some FFs utilize partial atomic charges to describe molecules' electrostatic interactions, they may fail to predict some properties correctly due to the lack of anisotropy, polarization, and the charge density penetration effect.33
To improve the representation of non-bonded interactions, multipolar-polarizable FFs such as Atomic Multipole Optimized Energetics for Biomolecular Applications (AMOEBA),34 have been introduced.35 AMOEBA utilizes explicit polarization through induced dipoles and a multipole expansion up to quadrupoles at the atomic level to provide a better description of charge density anisotropy and electrostatic interactions compared to conventional FFs that use point charges.36 Moreover, incorporating explicit polarization enhances the precision in representing non-bonded interactions. Due to the significant electron polarizability in IL systems,37 several studies showed that polarizable FFs like AMOEBA give an accurate description of the ILs systems.38–42
In this study, we use multipolar-polarizable AMOEBA FF to model various LCFA-ILs. We introduce a new approach based on AMOEBA FF development for amino acids and aliphatic compounds.43,44 We introduce a fragment-based multipolar polarizable AMOEBA FF specifically designed for n-butylammonium oleate ([C4NH3]+[OLE]−), but transferable to other FA-IL systems. Experimental liquid densities for this system at five different temperatures are reported in the literature,45 enabling the validation of the newly developed parameters. Through comprehensive analyses, we demonstrate the potential of our FFs in accurately predicting the properties of FA-ILs. We applied the parameters developed for [C4NH3]+[OLE]− to study two other FA-IL systems: ammonium palmitate ([NH4]+[PALM]−), with an existing experimental density at room temperature, was used to demonstrate the transferability of the newly developed FF to other FA-ILs. Additionally, 1,3-ethylmethylimidazolium palmitate ([EMIM]+[PALM]−) was modeled to predict its properties using the new FFs. This method facilitates accurate modeling of large molecules such as FAs, lipids, micelles and polymers by generating transferable fragment-based parameters, thereby enhancing the applicability of MD simulations. Section 2 outlines our parameterization approach and the validation process. We then describe the MD simulation methodology. In Section 3, we discuss the comparison of the calculated parameters with quantum mechanics (QM) and molecular mechanics (MM) data. We also discuss the findings derived from MD simulations of the three different FA-IL systems, which were created from the anions and cations represented in Fig. 1 at different temperatures.
 | ||
| Fig. 1 The structures of cations and anions studied in this work. Parameters developed for fragments shown in red in [C4NH3]+[OLE]− ionic pairs. | ||
2 Method
The parameterization details and a concise description of the parameter fitting methodology and MD simulation, followed by related analyses, are discussed in this section. Section 2.1 describes the parameterization of bonded and non-bonded interactions. Sections 2.2 and 2.3 describe the parameter evaluation and MD simulation method, respectively.2.1 Parameterization of the fatty acid-based ionic liquid
The parameterization procedure for the cation and anion follows the AMOEBA parameterization method.28 The AMOEBA potential contains both non-bonded and bonded terms to represent inter- and intra-molecular interactions (see eqn (1)).| U = Uband + Uangle + Utorsion + Uout + Ubθ + Ucoul + Upol + UvdW | (1) |
There are different functional forms to calculate the bond and angle parameters. In AMOEBA, these parameters are calculated using Hooke's law with higher-order terms34 to provide a better description of bond and angle energies. Since high energies are required to deform bonds and angles, most structural changes come from torsion and non-bonded terms. Due to the highly charged and polarized nature of ILs, our main focus is on non-bonded interactions i.e., electrostatic, polarization, and vdW.
Permanent atomic multipoles should be fitted for each atom from monopole up to quadrupole terms, oriented in the global XYZ coordinate frame. This can be generated via Stone's33 original DMA procedure. The methodology to get the distributed atomic multipoles for cations and anions from Gaussian distributed multipole analysis (GDMA) is well-established.33
Polarization is accounted by using the induced atomic dipoles approach,43 which is calculated by placing an inducible atomic polarizable point dipole moment, μi, on every atomic site using eqn (2).
| μi = αiEi | (2) |
The last term to be considered is vdW interactions. The vdW interactions are described by the buffered Halgren's46 potential shown in eqn (3).
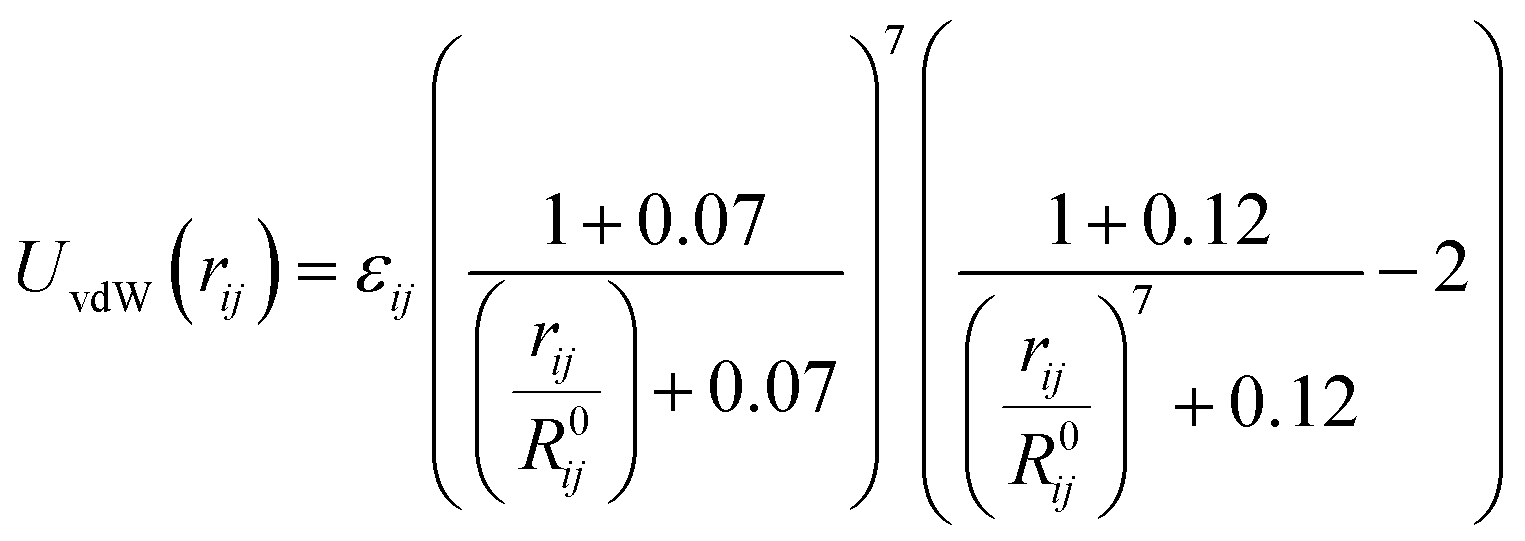 | (3) |
The parameterization process requires separate parameterization for cations and anions. However, this is impractical for large molecules such as fatty acids because QM calculations that are needed to get multipoles are computationally expensive. To address this challenge, we performed parametrization for molecule fragments instead of for the entire molecule. This method allows us to parameterize large molecules by breaking them down into smaller fragments. Additionally, it gives the advantage of transferring the parameters obtained from one molecule to other molecules containing similar fragments.
In this method, we aim to calculate parameters for the fragments of the molecule that do not exist in the AMOEBA FF or literature. In this study, –CH2–NH3+ from the n-butylammonium and –CH2–COO− and –CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2– from oleate were the fragments needed to be parameterized (Fig. 1). To parameterize these fragments, we capped them with an ethyl group as shown in Fig. S1 (ESI†). By incorporating the newly developed parameters for our new fragments into the original AMOEBA FF, we could effectively model various systems. N-butylammonium oleate was selected for parameterizing the required fragments due to the availability of the experimental densities at five different temperatures, which is crucial for the thorough validation of the obtained parameters.
CH–CH2– from oleate were the fragments needed to be parameterized (Fig. 1). To parameterize these fragments, we capped them with an ethyl group as shown in Fig. S1 (ESI†). By incorporating the newly developed parameters for our new fragments into the original AMOEBA FF, we could effectively model various systems. N-butylammonium oleate was selected for parameterizing the required fragments due to the availability of the experimental densities at five different temperatures, which is crucial for the thorough validation of the obtained parameters.
All intramolecular parameters and vdW values for both the anion and cation were initially taken from the original AMOEBA FF.34,47 However, missing bonded parameters, in the oleate, were determined using values calculated by Li and co-workers.48 For n-butylammonium, the missing bonded and vdW parameters were provided by Ponder et al.49
AMOEBA FF relies significantly on an enhanced electrostatic potential, which is built upon atomic multipoles and induced dipole moments. To establish permanent atomic multipoles and polarization for each molecule, we conducted initial QM calculations using the seven-step protocol outlined by Ren et al.44 on capped fragments (Fig. S1, ESI†). The ab initio geometry optimization followed by single-point energy calculation were carried out at the MP2 level of theory and 6-311G(1d,1p) and aug-cc-pVTZ basis sets, respectively, using Gaussian 16.50 GDMA computes the atomic multipole moments in the global frame. Following this, the POLEDIT program in TINKER51 was used to rotate the atomic multipoles into a local frame on each atomic site, enabling the generation of conformation-independent atomic multipole parameters for large molecules with various polarization groups.44 The dipole and quadrupole orientations were determined based on the local reference frames created by neighboring atoms.
Another crucial step in this process involves defining intramolecular direct polarization groups to separate the contribution of intramolecular polarization from the DMA multipole values.52 In the case of large molecules, polarization doesn't just come from the electric field of neighboring molecules but also from remote regions within the same molecule. Therefore, a group-based intramolecular polarization approach has been developed.47 These groups typically consist of functional groups, the ones that can rotate, and those with resonance. In our study, for example, –COO−, –CH2–, –CH3–, –HCCH– and –CH2–NH3+ were considered distinct polarization groups. Fig. S1 (ESI†) shows the polarization groups in each fragment. This approach ensures a more accurate representation of intramolecular polarization effects.
Ultimately, the TINKER51 package's potential program was employed to fine-tune the permanent atomic multipole parameters. The reference potential used in this fitting step was obtained through a single-point calculation at the MP2/aug-cc-pVTZ level. We selected five and three low-energy conformers for capped fragments of oleate and n-butylammonium, respectively, and used all conformers simultaneously in the fitting process. In this step, the partial charge values (monopoles) were kept intact, whereas the dipole and quadrupole moments were free to adjust or relax. Finally, the multipoles were fitted only to the fragments without the capping ethyl groups. The final multipole and polarization parameters for each fragment were transferred to the original AMOEBA parameter file where we added other bonded and non-bonded parameters. Once the fragments' parameters transferred, there were some non-integral charges on the molecule which were fixed manually.52
In AMOEBA, polarization effects are addressed using Thole's interaction induction model, incorporating distributed atomic polarizability.53,54 Under this induction model, induced dipoles originating from atomic centers reciprocally polarize all other atoms. To avoid the polarization catastrophe,33 a damping function with the damping factor of 0.39 is implemented at short range.44
Besides the parameters for [C4NH3]+[OLE]−, which were added to the AMOEBA FF, parameters for the other two systems were required for the purpose of evaluation and prediction. Parameters for [NH4]+ already exist in the AMOEBA FF. The parameters for [PALM]− are identical to those parameterized for [OLE]−, while the parameters for [EMIM]+ were previously calculated by our group. All the parameters are now available at Zenodo https://doi.org/10.5281/zenodo.13362550.
To validate the accuracy of our parameters, we compared the non-bonded energies obtained using our parameters with those from QM. Symmetry-adapted perturbation theory (SAPT)55 was used as the QM energy decomposition analysis method which directly computes intermolecular interactions through perturbation and separates the total interaction energy into components: Edisp (dispersion), Eexch (Pauli repulsion), Eelst (permanent electrostatics), and Eind (sum of polarization and charge transfer). While SAPT offers advantages by remaining unaffected by basis set superposition error, it lacks the capability to uniquely separate charge transfer energy due to uncertainties in decomposing higher-order contribution terms.56,57 A series of [C4NH3]+[OLE]− and [EMIM]+[PALM]− ion pairs were generated separately by varying the distance between the anion around the cation randomly using Avogadro58 to calculate non-bonded interaction energies for each ion pair. To account for varying distances between the ion pairs, we generated 82 pairs for [C4NH3]+[OLE]− and 135 pairs for [EMIM]+[PALM]−, ensuring comprehensive coverage of possible configurations. The energies calculated using SAPT0 with MP2 level of theory, employing the Psi4 package59 as a QM reference, were compared with the results obtained from the developed parameters using ANALYZE program in the TINKER package.51 Good agreement between the calculated energies for ion pairs from QM and MM validates the credibility of the generated parameters. Once the parameters were determined, we performed MD simulations and compared the calculated results with experimental data as a second step of parameter validation.
2.2 Molecular dynamics simulation methodology
The initial structures were created by Avogadro.58 Packmol60 was used to create a cubic simulation cell with 200 ion pairs containing optimized cations and anions. After preparing the initial structure of our system, MD simulations were conducted via Tinker-HP61 simulation package using our developed AMOEBA FF. The first step involved energy minimization using the conjugate gradient minimization algorithm to ensure the absence of any close contacts between the structures in the box. Since the minimization is computed at absolute zero, the temperature was gradually increased during the heating stage to the desired value. The system was heated up to 500 K and subsequently cooled down to the target temperature in the NVT ensemble. For each system, 3.5 ns of NVT simulations were conducted during the heating phase. To reach the equilibrium, we performed simulations on the NVT ensemble with an integration time step of 1.0 fs. The particle mesh Ewald method62,63 was employed by considering the vdW and Ewald cutoff radius of 9.0 Å to compute non-bonded and long-range electrostatic interactions. After equilibration, the production was carried out via the NPT ensemble at 1 atm with the same time step and cutoff. For temperature and pressure control, the Bussi thermostat64 and Berendsen barostat65 were employed. The same procedure was done for each IL combination at different temperatures. The total simulation time for all systems after equilibration is reported in Table S1 (ESI†). Using the simulation results, we calculated bulk and structural properties such as density (ρ), enthalpy of vaporization (ΔHvap), diffusion coefficient (D±), radial distribution function (RDF), and total structure factor S(q). Density was directly determined from the output of the NPT simulation, while other properties were calculated using obtained trajectories. More details on calculating ΔHvap, D±, RDF and S(q) are provided in Sections 3.2.2, 3.2.3 and 3.2.4, respectively.3 Results and discussion
3.1 Comparison of the decomposed energies calculated by molecular mechanics and quantum mechanics
We compared electrostatics, polarization, and vdW energies using our parameters (MM) with electrostatics, induction, and the combined dispersion and exchange-repulsion energies in SAPT (QM), respectively. Fig. 2 shows the comparison of decomposed energies calculated by QM and MM for [C4NH3]+[OLE]− IL system. The slope and square correlation coefficient of the total intermolecular interaction energy indicate strong agreement between QM and MM. The other decomposed energies exhibit good correlations, except for polarization. This discrepancy is attributed to induction energy in SAPT, which includes charge transfer along with polarization. The same comparison was reported in Fig. S2 (ESI†) for [EMIM]+[PALM]− IL to ensure that the newly developed parameters are transferable to different systems. Similar to [C4NH3]+[OLE]−, the decomposed energy comparison for [EMIM]+[PALM]− demonstrates excellent agreement for total, electrostatics and vdW energies. Interestingly the correlation between MM and QM polarization energies for [EMIM]+[PALM]− improved significantly. This could be due to the fact that the electron density in [EMIM]+ is de-localized compared to [C4NH3]+.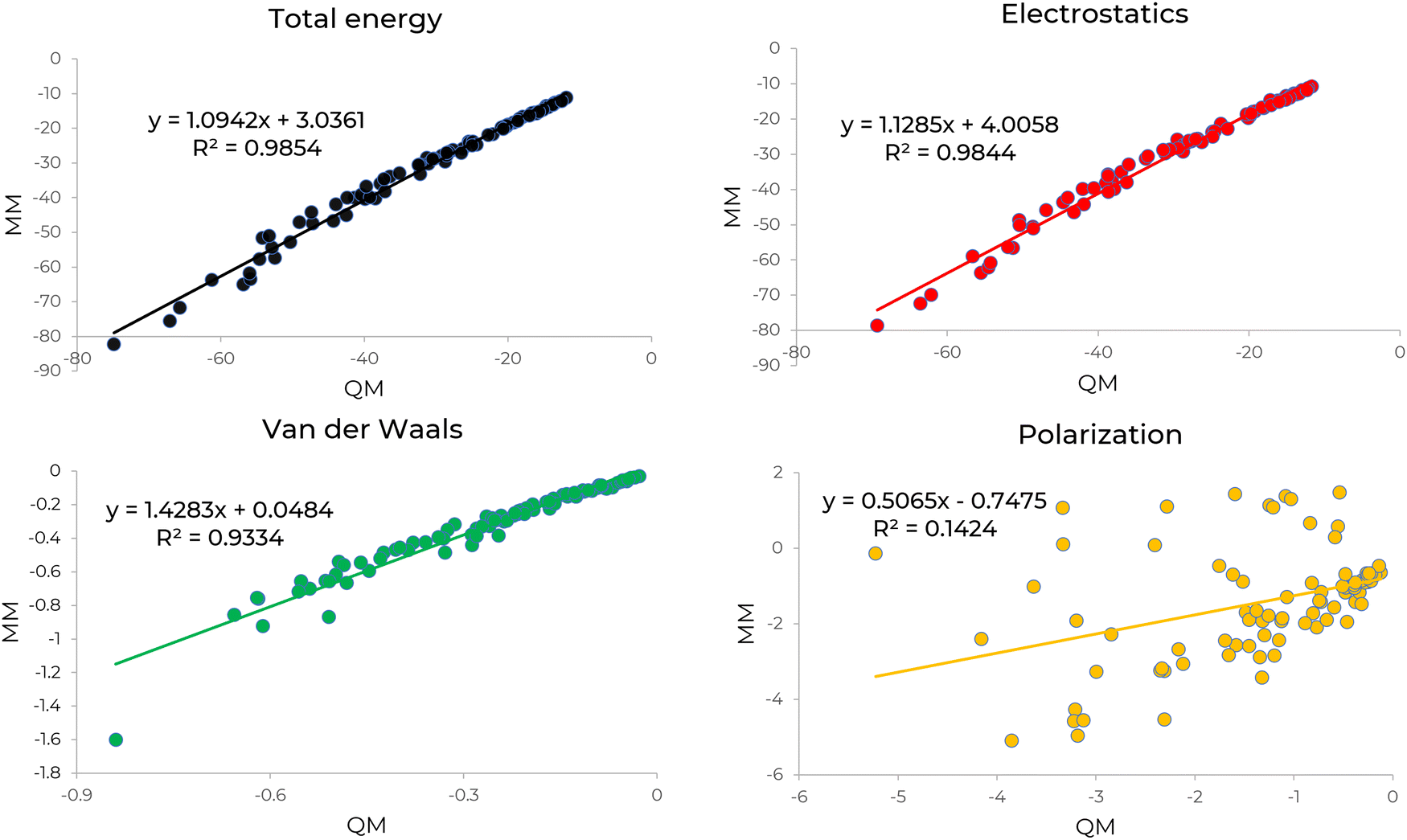 | ||
| Fig. 2 Comparison of intermolecular interaction energies for the [C4NH3]+[OLE]− ion pair with randomly varied distances between the anion and cation, using SAPT (QM) and the newly developed parameters (MM). | ||
3.2 Simulation results
| Ionic liquid | Temperature (K) | Calculated density (g cm−3) | Experimental density (g cm−3) | % Error |
|---|---|---|---|---|
| [C4NH3]+[OLE]− | 288.15 | 0.8950 ± 0.0025 | 0.8925 | 0.28 |
| [C4NH3]+[OLE]− | 293.15 | 0.8910 ± 0.0021 | 0.8893 | 0.19 |
| [C4NH3]+[OLE]− | 298.15 | 0.8876 ± 0.0026 | 0.8861 | 0.17 |
| [C4NH3]+[OLE]− | 303.15 | 0.8853 ± 0.0026 | 0.8829 | 0.27 |
| [C4NH3]+[OLE]− | 308.15 | 0.8815 ± 0.0030 | 0.9796 | 10.01 |
| [NH4]+[PALM]− | 298 | 0.9061 ± 0.0029 | 0.9109 | 0.52 |
| [EMIM]+[PALM]− | 315 | 0.9311 ± 0.0026 | — | — |
| [EMIM]+[PALM]− | 320 | 0.9298 ± 0.0028 | — | — |
To validate the transferability of our parameters to different FA-ILs, we opted for [NH4]+[PALM]− IL. Experimental density data for this compound is available at 298 K.66 Our model demonstrates an excellent agreement with the experiment, showing an error of approximately 0.5% (Table 1). Additionally, we used the same parameters to model [EMIM]+[PALM]− and predict their densities at 315 K and 320 K. These temperatures were selected to ensure that the system remains in its liquid phase throughout the simulation.29 These density comparisons confirm the reliability of our newly developed parameters to calculate other bulk and structural properties of different FA-ILs.
| ΔHvap = Egas − Eliq + nRT | (4) |
The potential energy in the gas phase was determined by conducting stochastic MD simulations for an isolated optimized cation–anion pair at the specified temperature. The potential energies of the liquid phase at different temperatures were extracted from the last five ns of the NPT simulations. The calculated ΔHvap values are provided in Table 2. While there is no experimental data to compare with our calculated ΔHvap, there is an expected trend in the results. For the [C4NH3]+[OLE]− system, ΔHvap decreased with increasing temperature. The [EMIM]+[PALM]− system also follows the trend, exhibiting a decrease in ΔHvap with increasing temperature.
| Ionic liquid | Temperature (K) | ΔHvap (kcal mol−1) |
|---|---|---|
| [C4NH3]+[OLE]− | 288.15 | 45.3959 |
| [C4NH3]+[OLE]− | 293.15 | 44.3247 |
| [C4NH3]+[OLE]− | 298.15 | 43.2353 |
| [C4NH3]+[OLE]− | 303.15 | 43.1013 |
| [C4NH3]+[OLE]− | 308.15 | 42.3501 |
| [NH4]+[PALM]− | 298 | 40.1900 |
| [EMIM]+[PALM]− | 315 | 50.2718 |
| [EMIM]+[PALM]− | 320 | 40.8265 |
 | (5) |
| Ionic liquid | Temperature (K) | D ± × 10−8 (cm2 s−1) |
|---|---|---|
| [C4NH3]+[OLE]− | 288.15 | 0.5 |
| [C4NH3]+[OLE]− | 293.15 | 0.6 |
| [C4NH3]+[OLE]− | 298.15 | 0.8 |
| [C4NH3]+[OLE]− | 303.15 | 1.2 |
| [C4NH3]+[OLE]− | 308.15 | 1.7 |
| [NH4]+[PALM]− | 298 | 0.5 |
| [EMIM]+[PALM]− | 315 | 0.5 |
| [EMIM]+[PALM]− | 320 | 0.5 |
According to Del Pópolo and Voth,68D± should be calculated from the MD trajectories at the diffusive regime (where the slope of the log–log plot of MSD-time equals one). We plotted log MSD over log time for all systems at different temperatures to find the diffusive regime (〈x2〉 ≈ t).69 Fig. S3 (ESI†) shows the MSD-time plots for all systems, and Table S1 (ESI†) shows the trajectory ranges from which diffusion is calculated. The average diffusion coefficients over the diffusive region were then calculated and reported in Table 3. The values are within the range of calculated diffusion coefficients for similar FA systems.70,71 At 353 K, diffusion coefficient values for myristic acid and lauric acid in FA-based deep eutectic solvents were reported as 0.1 × 10−8 cm2 s−1 and 6.4 × 10−8 cm2 s−1, respectively, which aligns with the range of our calculated D± for FA-based ILs.70,71 Moreover, D− for 2-hydroxyethylammonium oleate was experimentally determined at 353 K.72 The reported diffusion coefficient for oleate ranges from 32.6 × 10−8 cm2 s−1 to 14.4 × 10−8 cm2 s−1, aligning with the diffusion range we calculated. Although these values are higher than our calculated diffusion coefficient for [C4NH3]+[OLE]− at 308 K (1.7 × 10−8 cm2 s−1), this difference is expected due to the increase in diffusion with temperature and differences in structures. Additionally, self-diffusion coefficient for oleic acid was reported as 50.0 × 10−8 cm2 s−1 at 303.15 K,73 which is higher than that of [C4NH3]+[OLE]− at the same temperature (1.2 × 10−8 cm2 s−1). This difference is reasonable, as the interaction between the cation and anion in [C4NH3]+[OLE]− reduces diffusion. Furthermore, for [C4NH3]+[OLE]−, for which we calculated the diffusion coefficient at five different temperatures, the results are consistent with the fact that liquids diffuse more readily with increasing temperature.
 | ||
| Fig. 3 Calculated site–site radial distribution function (g(r)) and coordination number (n(r)) for [C4NH3]+[OLE]− at 298.15 K, [NH4]+[PALM]− at 298 K and [EMIM]+[PALM]− at 315 K with respect to the distance between the O atom of FA and N atom of cation. | ||
A complementary approach to studying the structure of ILs is calculating the structure factor (S(q)) from the RDF obtained through MD simulations.76 The S(q), represents local correlations between particles, indicating the likelihood of finding one particle at a certain distance from another.77 We calculated the total S(q) using viewSq78 module in VMD with X-ray form factor. Fig. S4 (ESI†) indicates the total S(q) for [C4NH3]+[OLE]−, [NH4]+[PALM]− and [EMIM]+[PALM]− at 298.15 K, 298 K and 315 K, respectively. The x axis is q (wave number) which is related to real-space distance by q = 2π/r. ViewSq calculates the S(q) from q = 0.5 Å−1 because theoretical data are unreliable below this q value due to the finite size of the simulation cell.79 The peaks at low q regions in Fig. S4 (ESI†) indicate the intermolecular correlations, for example, the peak at around 2.46 Å−1 correlates well with the O–N distance of 2.55 Å in RDF. A double-peaked pattern in S(q), with peaks near q = 0.8 Å−1 and 1.5 Å−1, is a characteristic feature of many ILs.80–84 The first peak is linked to charge alternation, reflecting periodic anion–anion and cation–cation ordering, while the second corresponds to charge adjacency, indicating near-neighbor ion interactions.85 This pattern has been consistently observed in neutron and X-ray scattering studies of various ionic liquids, including imidazolium86,87 and ammonium88–90-based systems. Therefore, although we lack experimental data for direct comparison, the calculated S(q) suggests similar and ordered structures across all three systems, indicating that the overall short-range structural shape of the liquids is almost the same.
4 Conclusions
This work presents an approach for FF parameterization of large systems, with a particular focus on modeling LCFA-ILs. We address the complexities associated with developing a multipolar-polarizable FF for large molecules by employing a fragment-based strategy. By segmenting the molecules into smaller, transferable fragments, we simplify the parameterization process without sacrificing accuracy. AMOEBA enables the effective incorporation of polarization effects, thereby enhancing the precision of modeling complex, charged systems such as LCFA-ILs. Furthermore, conducting MD simulations with our FF allows for predicting bulk and structural properties of diverse LCFA-ILs. The consistency between our computational results and available experimental data validates the reliability and accuracy of our approach, which holds implications for future research on even more intricate systems such as lipids, micelles, polymers and membrane proteins.Author contributions
H. T. designed and supervised the project; S. H. performed the computational studies and analyses; H. T. and S. H. analyzed the data and wrote the manuscript together.Data availability
Input files for the molecular dynamics simulations including parameters and initial coordinate files, are available at https://doi.org/10.5281/zenodo.13362550. The parameterization was conducted using Gaussian 16, while molecular dynamics simulations and subsequent analyses were performed using the Tinker software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the donors of ACS Petroleum Research Fund under Doctoral New Investigator Grant 66715-DNI6. H. T. served as Principal Investigator on ACS PRF 66715-DNI6 that provided support for S. H. We also acknowledge the Texas Advanced Computing Center (TACC) at the University of Texas at Austin, the Office of Information Technology and Cyber Infrastructure Research Computing (CIRC) department at the University of Texas at Dallas for providing HPC resources as well as Dr Dineli Ranathunga for providing the parameters of [EMIM]+.Notes and references
- I. Bahadur and N. Deenadayalu, Thermochim. Acta, 2013, 566, 77–83 CrossRef CAS.
- A. Shariati and C. J. Peters, J. Supercrit. Fluids, 2005, 34, 171–176 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- J. S. Wilkes, J. A. Levisky, R. A. Wilson and C. L. Hussey, Inorg. Chem., 1982, 21, 1263–1264 CrossRef CAS.
- F. U. Shah, R. An and N. Muhammad, Front. Chem., 2020, 8, 627213 CrossRef PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- B. Garcia, S. Lavallée, G. Perron, C. Michot and M. Armand, Electrochim. Acta, 2004, 49, 4583–4588 CrossRef CAS.
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS PubMed.
- D. Z. Troter, Z. B. Todorović, D. R. okić-Stojanović, O. S. Stamenković and V. B. Veljković, Renewable Sustainable Energy Rev., 2016, 61, 473–500 CrossRef CAS.
- C. Pretti, C. Chiappe, D. Pieraccini, M. Gregori, F. Abramo, G. Monni and L. Intorre, Green Chem., 2006, 8, 238–240 RSC.
- A. Foulet, O. B. Ghanem, M. El-Harbawi, J.-M. Lévêque, M. A. Mutalib and C.-Y. Yin, J. Mol. Liq., 2016, 221, 133–138 CrossRef CAS.
- R. Santiago, I. Diaz, M. González-Miquel, P. Navarro and J. Palomar, Fluid Phase Equilib., 2022, 560, 113495 CrossRef CAS.
- A. Tzani, M.-A. Karadendrou, S. Kalafateli, V. Kakokefalou and A. Detsi, Crystals, 2022, 12, 1776 CrossRef CAS.
- J. Hulsbosch, D. E. De Vos, K. Binnemans and R. Ameloot, ACS Sustainable Chem. Eng., 2016, 4, 2917–2931 CrossRef CAS.
- N. Muhammad, M. I. Hossain, Z. Man, M. El-Harbawi, M. A. Bustam, Y. A. Noaman, N. B. Mohamed Alitheen, M. K. Ng, G. Hefter and C.-Y. Yin, J. Chem. Eng. Data, 2012, 57, 2191–2196 CrossRef CAS.
- S. Kirchhecker and D. Esposito, Curr. Opin. Green Sustainable Chem., 2016, 2, 28–33 CrossRef.
- L. Poletti, C. Chiappe, L. Lay, D. Pieraccini, L. Polito and G. Russo, Green Chem., 2007, 9, 337–341 RSC.
- A. A. T. Hijo, H. D. Barros, G. J. Maximo, C. B. Cazarin, L. B. da Costa, J. F. Pereira, M. R. M. Junior and A. J. Meirelles, Food Res. Int., 2020, 134, 109125 CrossRef PubMed.
- N. V. Verssimo, F. A. Vicente, R. C. de Oliveira, B. Likozar, R. P. de Souza Oliveira and J. F. B. Pereira, Biotechnol. Adv., 2022, 61, 108055 CrossRef PubMed.
- S. M. Saadeh, Z. Yasseen, F. A. Sharif and H. M. A. Shawish, Ecotoxicol. Environ. Saf., 2009, 72, 1805–1809 CrossRef CAS PubMed.
- R. Gusain and O. P. Khatri, RSC Adv., 2016, 6, 3462–3469 RSC.
- Q. Yang, D. Xu, J. Zhang, Y. Zhu, Z. Zhang, C. Qian, Q. Ren and H. Xing, ACS Sustainable Chem. Eng., 2015, 3, 309–316 CrossRef CAS.
- M. Biswas, M. Dule, P. N. Samanta, S. Ghosh and T. K. Mandal, Phys. Chem. Chem. Phys., 2014, 16, 16255–16263 RSC.
- D. Parmentier, S. J. Metz and M. C. Kroon, Green Chem., 2013, 15, 205–209 RSC.
- D. Parmentier, T. Vander Hoogerstraete, S. J. Metz, K. Binnemans and M. C. Kroon, Ind. Eng. Chem. Res., 2015, 54, 5149–5158 CrossRef CAS.
- O. N. Starovoytov, H. Torabifard and G. A. Cisneros, J. Phys. Chem. B, 2014, 118, 7156–7166 CrossRef CAS PubMed.
- A. Mezzetta, L. Guazzelli, M. Seggiani, C. S. Pomelli, M. Puccini and C. Chiappe, Green Chem., 2017, 19, 3103–3111 RSC.
- J. N. Canongia Lopes, J. Deschamps and A. A. Pádua, J. Phys. Chem. B, 2004, 108, 2038–2047 CrossRef.
- B. Bhargava, S. Balasubramanian and M. L. Klein, Chem. Commun., 2008, 3339–3351 RSC.
- F. Dommert, K. Wendler, R. Berger, L. Delle Site and C. Holm, ChemPhysChem, 2012, 13, 1625–1637 CrossRef CAS PubMed.
- A. J. Stone, Chem. Phys. Lett., 1981, 83, 233–239 CrossRef CAS.
- P. Ren and J. W. Ponder, J. Phys. Chem. B, 2003, 107, 5933–5947 CrossRef CAS.
- Z. Jing, C. Liu, S. Y. Cheng, R. Qi, B. D. Walker, J.-P. Piquemal and P. Ren, Annu. Rev. Biophys., 2019, 48, 371–394 CrossRef CAS PubMed.
- J. C. Wu, G. Chattree and P. Ren, Theor. Chem. Acc., 2012, 131, 1–11 Search PubMed.
- T. Yan, Y. Wang and C. Knox, J. Phys. Chem. B, 2010, 114, 6905–6921 CrossRef CAS PubMed.
- G. A. Cisneros, K. T. Wikfeldt, L. Ojamae, J. Lu, Y. Xu, H. Torabifard, A. P. Bartók, G. Csányi, V. Molinero and F. Paesani, Chem. Rev., 2016, 116, 7501–7528 CrossRef CAS PubMed.
- D. Bedrov, O. Borodin, Z. Li and G. D. Smith, J. Phys. Chem. B, 2010, 114, 4984–4997 CrossRef CAS PubMed.
- E. A. Vázquez-Montelongo, J. E. Vázquez-Cervantes and G. A. Cisneros, Int. J. Mol. Sci., 2020, 21, 697 CrossRef PubMed.
- O. Borodin, J. Phys. Chem. B, 2009, 113, 11463–11478 CrossRef CAS PubMed.
- D. Bedrov, J.-P. Piquemal, O. Borodin, A. D. MacKerell Jr, B. Roux and C. Schroder, Chem. Rev., 2019, 119, 7940–7995 CrossRef CAS PubMed.
- J. W. Ponder, C. Wu, P. Ren, V. S. Pande, J. D. Chodera, M. J. Schnieders, I. Haque, D. L. Mobley, D. S. Lambrecht and R. A. DiStasio Jr, et al. , J. Phys. Chem. B, 2010, 114, 2549–2564 CrossRef CAS PubMed.
- P. Ren, C. Wu and J. W. Ponder, J. Chem. Theory Comput., 2011, 7, 3143–3161 CrossRef CAS PubMed.
- G. V. Olivieri, C. S. da Cunha, L. dos Santos Martins, P. A. M. Paegle, S. D. Nuncio, A. de Araújo Morandim-Giannetti and R. B. Torres, J. Therm. Anal. Calorim., 2018, 131, 2925–2942 CrossRef CAS.
- T. A. Halgren, J. Am. Chem. Soc., 1992, 114, 7827–7843 CrossRef CAS.
- P. Ren and J. W. Ponder, J. Comput. Chem., 2002, 23, 1497–1506 CrossRef CAS PubMed.
- H. Chu, X. Peng, Y. Li, Y. Zhang, H. Min and G. Li, Mol. Phys., 2018, 116, 1037–1050 CrossRef CAS.
- Y. Shi, M. L. Laury, Z. Wang and J. W. Ponder, J. Comput. Aided Mol. Des., 2021, 35, 79–93 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- J. A. Rackers, Z. Wang, C. Lu, M. L. Laury, L. Lagardère, M. J. Schnieders, J.-P. Piquemal, P. Ren and J. W. Ponder, J. Chem. Theory Comput., 2018, 14, 5273–5289 CrossRef CAS PubMed.
- Y. Shi, Z. Xia, J. Zhang, R. Best, C. Wu, J. W. Ponder and P. Ren, J. Chem. Theory Comput., 2013, 9, 4046–4063 CrossRef CAS PubMed.
- B. T. Thole, Chem. Phys., 1981, 59, 341–350 CrossRef CAS.
- P. T. Van Duijnen and M. Swart, J. Phys. Chem. A, 1998, 102, 2399–2407 CrossRef CAS.
- K. Szalewicz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 254–272 CAS.
- A. J. Misquitta, J. Chem. Theory Comput., 2013, 9, 5313–5326 CrossRef CAS PubMed.
- K. U. Lao and J. M. Herbert, J. Chem. Theory Comput., 2016, 12, 2569–2582 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- R. M. Parrish, L. A. Burns, D. G. Smith, A. C. Simmonett, A. E. DePrince III, E. G. Hohenstein, U. Bozkaya, A. Y. Sokolov, R. Di Remigio and R. M. Richard, J. Chem. Theory Comput., 2017, 13, 3185–3197 CrossRef CAS PubMed.
- L. Martnez, R. Andrade, E. G. Birgin and J. M. Martnez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- L. Lagardère, L.-H. Jolly, F. Lipparini, F. Aviat, B. Stamm, Z. F. Jing, M. Harger, H. Torabifard, G. A. Cisneros and M. J. Schnieders, Chem. Sci., 2018, 9, 956–972 RSC.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. Berendsen, J. v Postma, W. F. Van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- AMMONIUMPALMITATE.chemicalbook.com, https://m.chemicalbook.com/ProductChemicalPropertiesCB7506349_EN.htm.
- P. Berton, S. Manouchehr, K. Wong, Z. Ahmadi, E. Abdelfatah, R. D. Rogers and S. L. Bryant, ACS Sustainable Chem. Eng., 2019, 8, 632–641 CrossRef.
- M. G. Del Pópolo and G. A. Voth, J. Phys. Chem. B, 2004, 108, 1744–1752 CrossRef.
- E. Flenner, J. Das, M. C. Rheinstädter and I. Kosztin, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 79, 011907 CrossRef PubMed.
- S. Barani Pour, J. Jahanbin Sardroodi, A. Rastkar Ebrahimzadeh and G. Pazuki, Sci. Rep., 2023, 13, 7433 CrossRef CAS PubMed.
- S. Barani Pour, J. Jahanbin Sardroodi, A. Rastkar Ebrahimzadeh, G. Pazuki and V. Hadigheh Rezvan, Sci. Rep., 2024, 14, 1763 CrossRef CAS PubMed.
- V. H. Álvarez, S. Mattedi, M. Martin-Pastor, M. Aznar and M. Iglesias, Fluid Phase Equilib., 2010, 299, 42–50 CrossRef.
- M. Iwahashi, A. Umehara, K. Wakisaka, Y. Kasahara, H. Minami, H. Matsuzawa, H. Shinzawa, Y. Ozaki and M. Suzuki, J. Phys. Chem. B, 2007, 111, 740–747 CrossRef CAS PubMed.
- T. Mendez-Morales, J. Carrete, S. Bouzon-Capelo, M. Perez-Rodriguez, O. Cabeza, L. J. Gallego and L. M. Varela, J. Phys. Chem. B, 2013, 117, 3207–3220 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- K. Shimizu, C. E. Bernardes, A. Triolo and J. N. C. Lopes, Phys. Chem. Chem. Phys., 2013, 15, 16256–16262 RSC.
- C. Wu, D. Y. Chan and R. F. Tabor, J. Colloid Interface Sci., 2014, 426, 80–82 CrossRef CAS PubMed.
- T. Mackoy, B. Kale, M. E. Papka and R. A. Wheeler, Comput. Phys. Commun., 2021, 264, 107881 CrossRef CAS.
- M. Campetella, A. Le Donne, M. Daniele, L. Gontrani, S. Lupi, E. Bodo and F. Leonelli, J. Phys. Chem. B, 2018, 122, 2635–2645 CrossRef CAS PubMed.
- T. Murphy, R. Atkin and G. G. Warr, Curr. Opin. Colloid Interface Sci., 2015, 20, 282–292 CrossRef CAS.
- A. A. Freitas, K. Shimizu and J. N. Canongia Lopes, J. Chem. Eng. Data, 2014, 59, 3120–3129 CrossRef CAS.
- K. Shimizu, C. E. Bernardes and J. N. Canongia Lopes, J. Phys. Chem. B, 2014, 118, 567–576 CrossRef CAS PubMed.
- D. Edson, C. Pueblo, M. Blodgett, K. Kelton and N. Mauro, J. Mol. Liq., 2017, 242, 807–811 CrossRef CAS.
- L. J. Siqueira and M. C. Ribeiro, J. Chem. Phys., 2011, 135, 204506 CrossRef PubMed.
- J. C. Araque, J. J. Hettige and C. J. Margulis, J. Phys. Chem. B, 2015, 119, 12727–12740 CrossRef CAS PubMed.
- A. Triolo, A. Mandanici, O. Russina, V. Rodriguez-Mora, M. Cutroni, C. Hardacre, M. Nieuwenhuyzen, H.-J. Bleif, L. Keller and M. A. Ramos, J. Phys. Chem. B, 2006, 110, 21357–21364 CrossRef CAS PubMed.
- M. Deetlefs, C. Hardacre, M. Nieuwenhuyzen, A. A. Padua, O. Sheppard and A. K. Soper, J. Phys. Chem. B, 2006, 110, 12055–12061 CrossRef CAS PubMed.
- C. S. Santos, H. V. Annapureddy, N. S. Murthy, H. K. Kashyap, E. W. Castner and C. J. Margulis, J. Chem. Phys., 2011, 134, 064501 CrossRef PubMed.
- T. Pott and P. Méléard, Phys. Chem. Chem. Phys., 2009, 11, 5469–5475 RSC.
- T. L. Greaves, D. F. Kennedy, S. T. Mudie and C. J. Drummond, J. Phys. Chem. B, 2010, 114, 10022–10031 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S3 and Table S1. See DOI: https://doi.org/10.1039/d4cp01809e |
| This journal is © the Owner Societies 2024 |