
Dissociation of HeH+ in the electronic ground state using shaped mid-IR laser pulses
Kasper L.
Effersø
and
Niels E.
Henriksen

Department of Chemistry, Technical University of Denmark, Building 207, DK-2800 Kongens Lyngby, Denmark. E-mail: neh@kemi.dtu.dk
First published on 19th April 2024
Abstract
Inspired by recent experimental work, we study the control over the laser-driven dissociation of the HeH+ ion in the electronic ground state. Shaped pulses with peak intensities below 1012 W cm−2 are obtained by phase modulation of high-intensity transform-limited femtosecond pulses. We investigate the performance of pulse shaping for a number of shaping parameters targeting both vibrational and rotational excitation pathways. The numerical results show that pulse shaping is most effective at low pulse energies and broad spectral bandwidths, while intense transform-limited pulses with narrow spectral bandwidths maximize dissociation. We show that the control achieved with a quadratic chirped pulse optimized for vibrational ladder climbing, a cascade excitation process of adjacent vibrational levels, is hindered by rotational motion leading to significantly reduced dissociation. Moreover, pulse shaping using higher-order polynomial phase functions is found to provide only a marginal increase in dissociation yields. Our results provide additional insights into the coherent control of bond breaking in diatomic molecules, and demonstrate the efficacy of pulse shaping for a range of pulse energies.
1 Introduction
Over the past few decades, there has been considerable interest in exploiting the phase coherence properties of laser light to control quantum systems. Research on coherent control of photochemical reactions has largely focused on spectral ranges where electronic transitions predominate.1–9 Comparatively few studies have explored the possibility of controlling the femtosecond dynamics directly in the electronic ground state.10–14 Intense laser light can, e.g., induce conformational changes and chemical reactions in the electronic ground state of molecules. In order to avoid high intensities leading to ionization, pulse shaping is essential. Pulse shaping is achieved by manipulating the phase of the spectral components of ultrashort laser pulses;4,5 giving rise to coherent control of the molecular dynamics.Experimental demonstration of useful coherent control of chemical reactions under non-ionizing conditions continues to be a major challenge. To that end, we return to the mechanisms and optimal conditions for the simplest possible process related to chemical reactions, that is, breaking a chemical bond in the electronic ground state. Here, pulse shaping techniques in the relevant mid-infrared region (400–4000 cm−1) are under constant development.15–17
Recently, Wustelt et al. reported on the ground state photodissociation of the helium hydride ion (HeH+) in a combined experimental and theoretical study.18 They were able to demonstrate multiphoton dissociation of HeH+via vibrational excitation using transform-limited femtosecond pulses of high intensity (∼1014 W cm−2). Despite the intense laser pulses, ionization was found to be negligible due to the high ionization threshold of HeH+. However, at such high intensities, the competing process of ionization is expected to hinder selective bond dissociation for the majority of photochemical reactions.
Ultrashort laser pulses can enhance nonlinear light–matter interactions. Silberberg et al. demonstrated that a transform-limited pulse maximizes non-resonant two-photon absorption in atomic transitions, whereas a shaped pulse was optimal for resonant multiphoton transitions.19 Resonant multiphoton excitation in molecules is typically denoted as a vibrational ladder climbing (VLC) mechanism whenever higher vibrational states are successively populated during the interaction with the laser pulse. VLC has been proposed as an efficient means of controlling vibrational excitation in the electronic ground state. This was demonstrated in early theoretical work by Chelkowski et al.20 using a negatively chirped laser pulse with time-dependent frequencies matching the decrease in energy-spacing of excited vibrational states. For a one-dimensional Morse oscillator, genuine ladder climbing has been demonstrated, leading to a synchronous, successively complete population and depopulation of excited vibrational states.21,22 An additional advantage of VLC is that phase modulation elongates the pulse duration, which leads to a decrease in peak intensity and consequently, reduced ionization. However, the majority of theoretical studies on dissociation via VLC have been conducted in one dimension20–22 (for an exception, see ref. 23). Experimental reports on successful dissociation remain scarce, although controlled vibrational ladder climbing has been demonstrated.24–26
In this paper, we focus on coherent control of the molecular dynamics in the electronic ground state and demonstrate its ability to induce bond breaking using the helium hydride ion as a prototype system. The quantum dynamics is simulated in full dimension and the optimization of transform-limited femtosecond pulses is implemented in a closed-loop control scheme to ensure that the resultant pulse shapes are accessible to an experimental pulse shaper.27 We identify the genuine ladder climbing regime and investigate the extent to which this mechanism can be transferred to the full-dimensional case where rotational motion is included. To that end, we evaluate the efficacy of shaped pulses in two ways, comparing them to laser pulses with the same energy and no phase modulation, respectively: (i) transform-limited pulses, and (ii) a laser pulse with the same pulse envelope as the shaped pulse. All comparisons are done for different energies and bandwidths of the transform-limited pulse. We also demonstrate centrifugal fragmentation, i.e. dissociation via pure rotational excitation, using tailored pulses in the 500–900 cm−1 spectral region.
2 Theory
We consider a diatomic molecule in the electronic ground state subjected to a laser field![[E with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0045_20d1.gif) (t) and evolving in time according to the time-dependent Schrödinger equation (TDSE)
(t) and evolving in time according to the time-dependent Schrödinger equation (TDSE)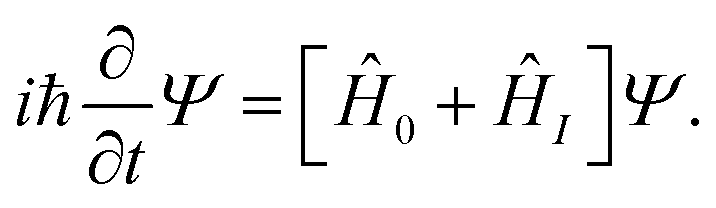 | (1) |
The three-dimensional field-free Hamiltonian can be expressed as
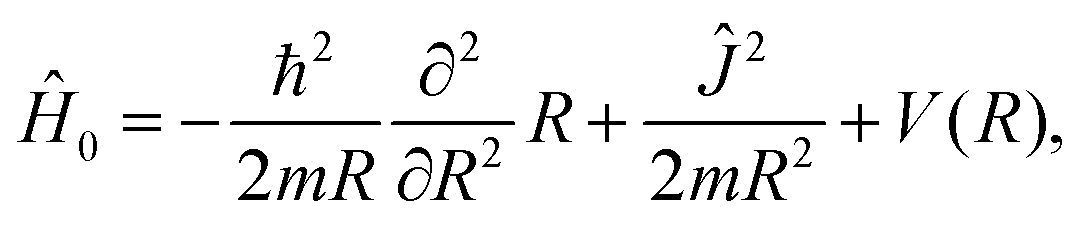 | (2) |
The electric field produced by the incident laser is treated classically and assumed to be linearly polarized along the laboratory frame z axis, (t) = E(t)ẑ. Within the electric-dipole approximation, the coupling between the molecule and field is described by the interaction Hamiltonian, ĤI(t) = −![[small mu, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b3.gif) ·(t). The molecular dipole moment, = 0 +
·(t). The molecular dipole moment, = 0 + ![[small alpha, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e101.gif) ·(t), contains contributions from the permanent dipole moment 0 and the first-order correction to the field-induced dipole moment . In this work, we consider fields with carrier frequencies far from electronic resonance and take to be the static polarizability.28 Under these conditions, the time-dependent field-molecule interaction is described by
·(t), contains contributions from the permanent dipole moment 0 and the first-order correction to the field-induced dipole moment . In this work, we consider fields with carrier frequencies far from electronic resonance and take to be the static polarizability.28 Under these conditions, the time-dependent field-molecule interaction is described by
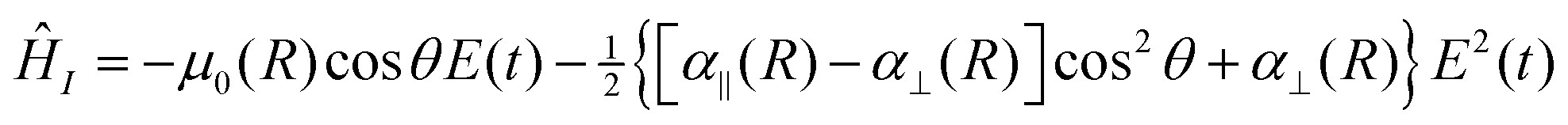 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, the polarizabilities have a relatively small magnitude, and the effect of including the quadratic Stark term is expected to be small at moderate field amplitudes.
:2, the polarizabilities have a relatively small magnitude, and the effect of including the quadratic Stark term is expected to be small at moderate field amplitudes.
The bound states |vJM〉 of the field-free Hamiltonian are computed by relaxation of a Gaussian trial wave packet, where v and J are the vibrational and rotational quantum numbers, respectively. In a linearly polarized field, the projection of the nuclear orbital angular momentum M along the polarization axis is conserved. Here, we will consider only states with M = 0, and the label is omitted in the following. Initially, the molecule is assumed to be in the ground vibrational and rotational state of the electronic ground state. The energy separation in the Franck–Condon region of the HeH+ ion, ranges from 13 to 39 eV between the electronic ground state and the first excited state.29 Thus, for the laser parameters considered in this study, the nuclear motion of the HeH+ ion is confined to the electronic ground state, which is described by a Morse potential, V(R) = De[e−β(R−Re) − 1]2, with well-depth, De = 2.04 eV, width parameter, β = 2.7407 Å−1, and equilibrium distance, Re = 0.7743 Å.31 For J = 0, the anharmonic potential supports a total of 10 bound vibrational states with a bond-dissociation energy of D0 = 1.85 eV.
The nuclear wave function Ψ(R,θ,t) corresponding to TDSE in eqn (1) is obtained by propagating the initial wave packet using the Heidelberg implementation32 of the multi-configuration time-dependent Hartree (MCTDH) method.33 The numerical grids used in the wave packet simulations are comprised of a fast Fourier transform collocation grid for the dissociative coordinate (R) with 256 grid points in the [0.65,14]a0 range and an associated-Legendre DVR-grid composed of 56 grid points in the [0,π] range for the rotational coordinate (θ). The specific grid ranges were chosen to ensure that all bound states are accommodated on the grid. To prevent wave packet reflection off the grid boundary, a complex absorbing potential (CAP) is placed in the dissociation channel to eliminate the part of the wave packet entering the region, R ≥ RCAP = 12a0.
The optimal pulse shape is determined through the spectral representation of a transform-limited Gaussian input pulse optimized by phase-only modulation in a closed-loop control scheme described in the following.
2.1 Simulation of pulse shaping
To obtain realistically shaped control fields, we simulate a phase-only pulse shaper equipped with a spatial light modulator (SLM). Pulse shaping using an SLM occurs in the Fourier domain, where the frequency components of the input pulse are spatially dispersed by the pulse shaper. Spectral phase control is obtained by applying a high-resolution phase mask to the frequency-dispersed input pulse defined by| Ẽ(ω) = M(ω)Ẽin(ω), | (4) |
![[small phi, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e2.gif) (ω). The phase modulation induced by the pixelated mask is described by
(ω). The phase modulation induced by the pixelated mask is described by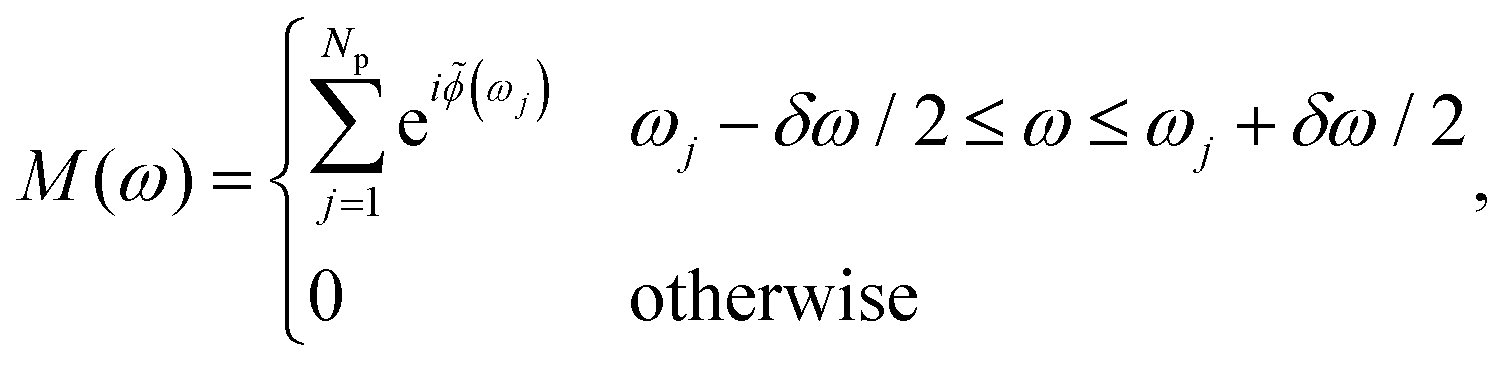 | (5) |
In order to obtain an analytic form of eqn (4) in the time domain, we assume that the spectral amplitude and phase of the input pulse are uniform over a given pixel. To that end, we consider a transform-limited input pulse with a Gaussian envelope and temporal center, tc = 0. In the spectral domain, the Gaussian input pulse is written as
Ẽin(ω) = Ẽ0e−![[small alpha, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0dc.gif) 2(ω−ω0)2/2, 2(ω−ω0)2/2, | (6) |
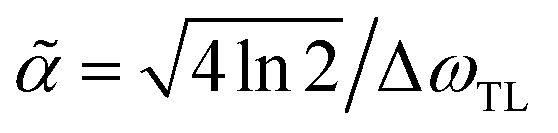 , with ΔωTL = FWHM[Ẽin2(ω)] being the full width at half maximum of the intensity. By taking the inverse Fourier transform of eqn (4), it can be shown that the analytic form of the temporal shaped pulse is34
, with ΔωTL = FWHM[Ẽin2(ω)] being the full width at half maximum of the intensity. By taking the inverse Fourier transform of eqn (4), it can be shown that the analytic form of the temporal shaped pulse is34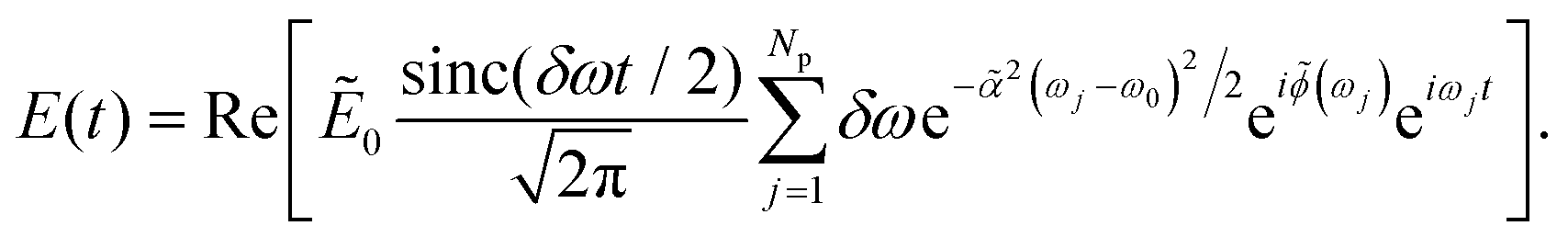 | (7) |
The pulse shaper is simulated with a spectral bandwidth corresponding to 6 standard deviations of the Gaussian input pulse, Δω = 6![[small sigma, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e10d.gif) in (in = −1), and a phase mask composed of Np = 10240 pixels.
in (in = −1), and a phase mask composed of Np = 10240 pixels.
The spectral phase induced by the SLM is expanded as a 4th-order Taylor series
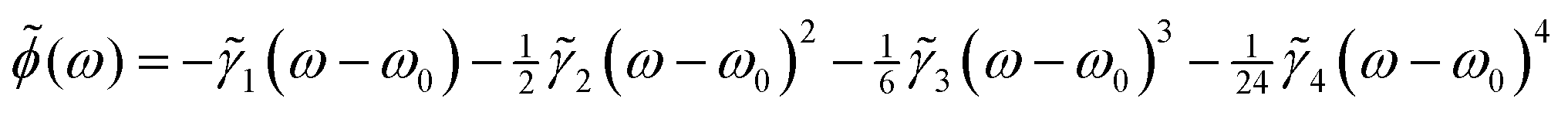 | (8) |
![[small gamma, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0dd.gif) 1 term introduces temporal translation, 2 denotes the group delay dispersion term and 3 and 4 are the 3rd and 4th-order dispersion terms, respectively. The four phase coefficients {i} determine the shape of the temporal pulse and are optimized as described below.
1 term introduces temporal translation, 2 denotes the group delay dispersion term and 3 and 4 are the 3rd and 4th-order dispersion terms, respectively. The four phase coefficients {i} determine the shape of the temporal pulse and are optimized as described below.
2.2 Optimization of shaped pulses
The optimization of the shaped pulses is treated as a blackbox, where the laser parameters are updated based on the results obtained by the wave packet simulation. The optimization parameters are collectively described by the vector, S = (Ẽ0,ω0,1,…,n), where {i} are the coefficients of the phase function (ω), and Ẽ0 and ω0 are the spectral amplitude and the carrier frequency of the Gaussian input pulse. The S parameters are updated using Bayesian optimization (BO) employing the upper confidence bound (UCB) as the acquisition function and the number of iterations was set to N = 500 for each optimization run. We used the open-source Bayesian Optimization library in Python35 for the implementation of BO. The library adopts the Gaussian process regression with the Matern kernel as the predictive model for the parameter space.
The optimization is subject to constraints on peak intensity I0 ≤ 1 TW cm−2, energy density F ≤ 2643 mJ cm−2 and pulse duration T ≤ 20 ps. This choice of constraints is motivated by the requirement that ionization is minimized and that the resulting pulse shapes are experimentally feasible. Since the maximal pulse shaping capability is dependent on the type of the SLM device utilized, the maximum pulse duration was limited to 20 ps. The pulse energies produced in the experimental study by Wustelt,18 were used as a reference for the upper limit on the energy density defined by
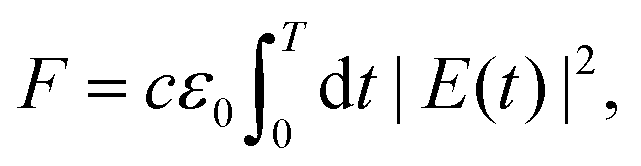 | (9) |
Under these conditions, the optimal combination of S parameters is determined so as to maximize the dissociation probability Pd. To that end, we define the objective function
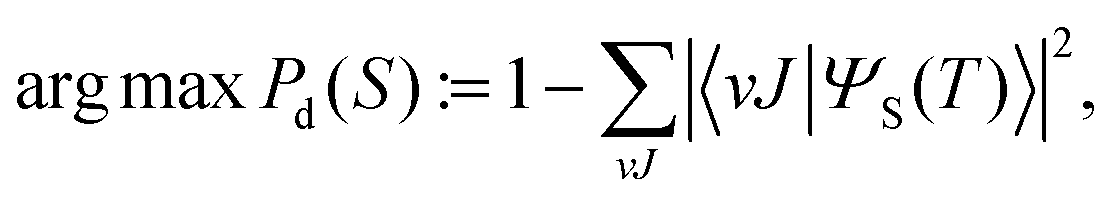 | (10) |
Reducing the parameter space is crucial for the efficient optimization of the laser pulses. In the one-dimensional case, a pulse with a linear temporal (quadratic spectral) chirp can efficiently excite vibrational states via ladder climbing and is therefore a natural starting point. The range of laser parameters in which VLC is the dominant excitation mechanism is estimated using the parameters proposed by Barth et al.21Fig. 1 provides a rough estimate of the VLC regime for the HeH+ ion indicated by the red region. The blue box represents the parameter range used to guide the optimization toward ladder climbing and corresponds to shaped pulses with 1.5–20.0 ps duration and a peak intensity of 0.1–1.0 TW cm−2.
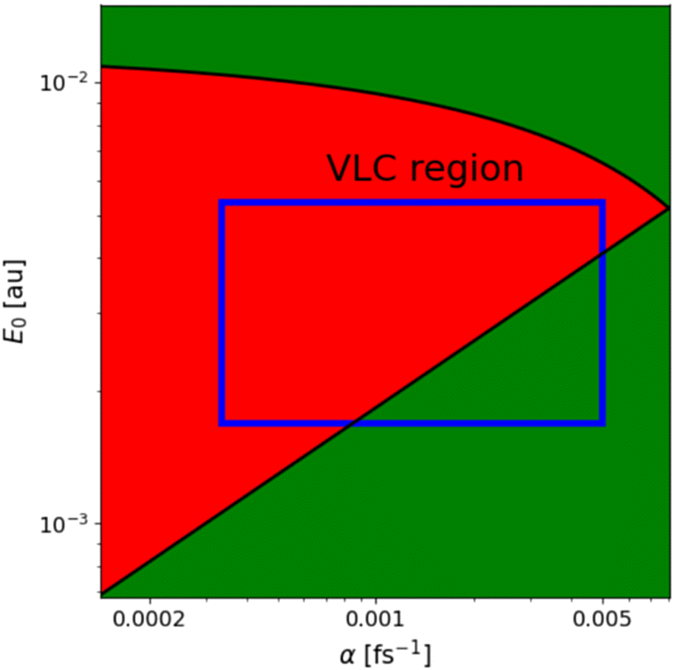 | ||
| Fig. 1 Estimate of the vibrational ladder climbing regime (red region) for the HeH+ ion in the one-dimensional case. The parameters E0 and α refer to a shaped pulse with a linear temporal chirp as described by eqn (12). The blue square represents the range of parameters, E0 = 1.7 × 10−3–5.3 × 10−3 a.u. and α = 3.3 × 10−4–5.0 × 10−3 fs−1, used in the optimization of the quadratic chirped pulses. | ||
The relationship between the spectral and temporal parameters is determined from the analytic expression in eqn (7) using a quadratic phase function, (ω) = −(ω − ω0)2/2. In the limit δω → 0, such that Np → ∞, the limiting value of the sinc function is unity and the sum is replaced by an integral
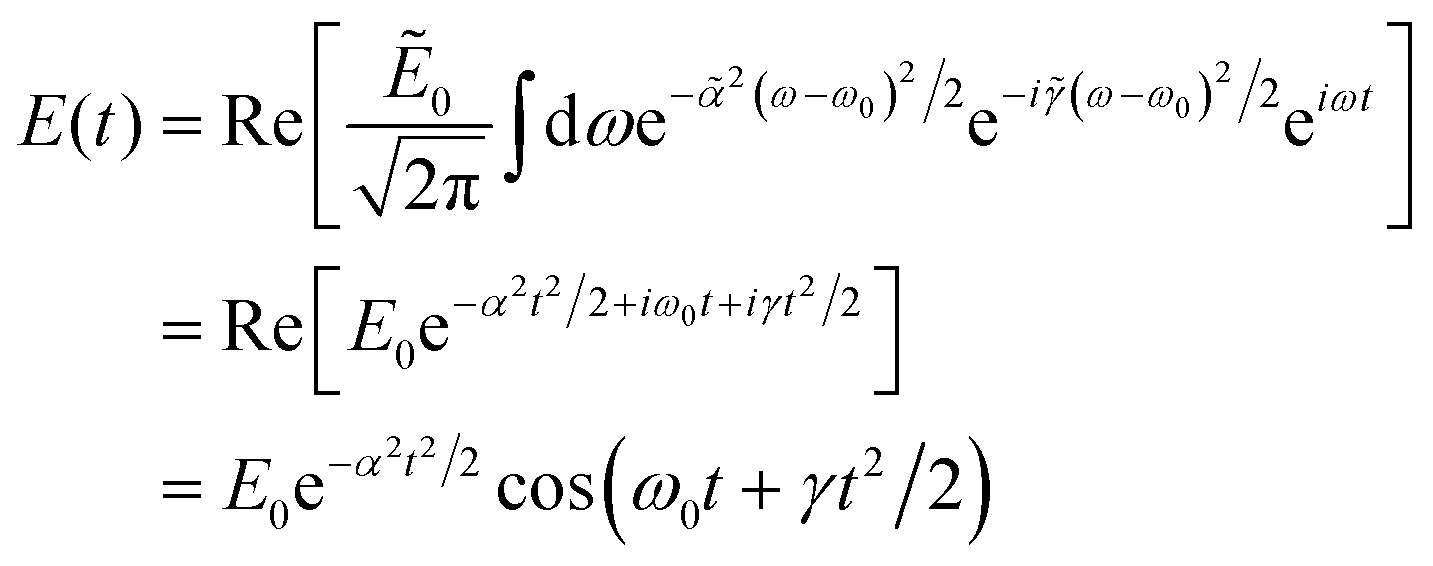 | (11) |
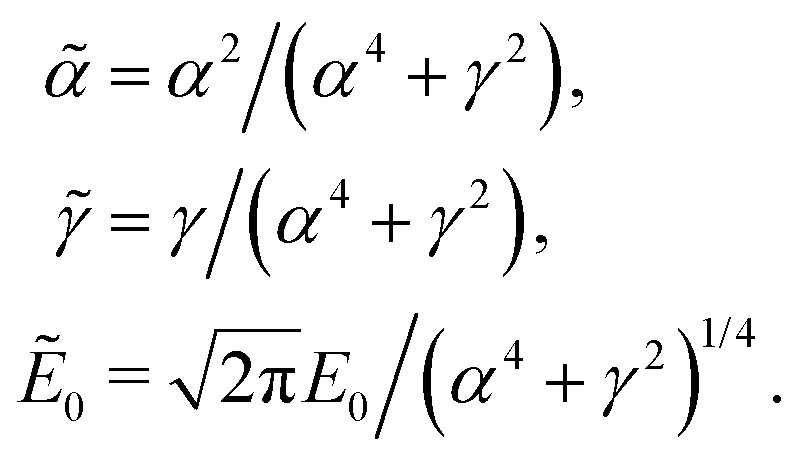 | (12) |
The spectral amplitude Ẽ0 and chirp rate corresponding to the VLC regime in Fig. 1 is determined using eqn (12) for a given spectral bandwidth of the input pulse defined by . The duration of the quadratic chirped pulse is defined by T = 8/α corresponding to 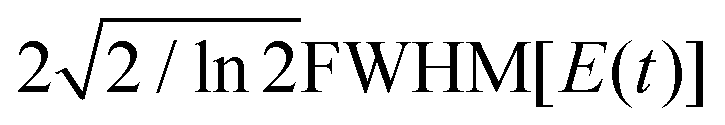 .
.
3 Results
This study is carried out in four parts in order to examine: (i) the validity of the VLC regime identified in Fig. 1 in one dimension using a quadratic chirped pulse, (ii) the extent to which genuine ladder climbing can be transferred to the three-dimensional case, (iii) the efficacy of pulse shaping using higher-order phase functions to enhance the dissociation beyond that obtained via the VLC mechanism and (iv) the effectiveness of centrifugal fragmentation by targeting rotational excitation as an alternative mechanism of inducing dissociation. To compare the effect of spectral bandwidth on the dissociation yield, two transform-limited input pulses, a narrowband and a broadband Gaussian, are employed in the pulse shaping. The narrowband pulse corresponds to the pulse utilized in the experimental study by Wustelt et al.18Table 1 summarizes the parameters of the two Gaussian input pulses.| Input pulse Ẽin | Peak intensity I0 (TW cm−2) | FWHM[Ein(t)] (fs) | FWHM[Ẽin2(ω)] (cm−1) |
(fs rad−1) |
|---|---|---|---|---|
| Narrowband | 70 | 50 | 415 | 21.3 |
| Broadband | 206 | 17 | 1225 | 7.22 |
The results of the wave packet simulations are evaluated by examining the population dynamics and the transition matrix elements depicted in Fig. 2. It is worth mentioning that the well-known selection rules, Δv = ±1 and ΔJ = 0,±1, are not strictly valid due to the anharmonicity and rovibrational coupling present in the system which allows for, e.g. vibrational overtone transitions. Furthermore, since the field-free Hamiltonian Ĥ0 is perturbed during the interaction with the laser pulse, identifying vibrational ladder climbing by projecting the wave packet onto the bound rovibrational states |vJ〉 is only meaningful as long as this perturbation remains small. In the full-dimensional case, the maximal instantaneous interaction energy is of the order of, |〈Ψ|·|Ψ〉| = 0.012 eV (97 cm−1), for a shaped pulse with a peak intensity of 1 TW cm−2. Thus, the eigenstates |vJ〉 of the field-free Hamiltonian are not significantly perturbed during the laser interaction and the magnitude of the dynamic Stark effect is small compared to the energy-spacing of |vJ〉.
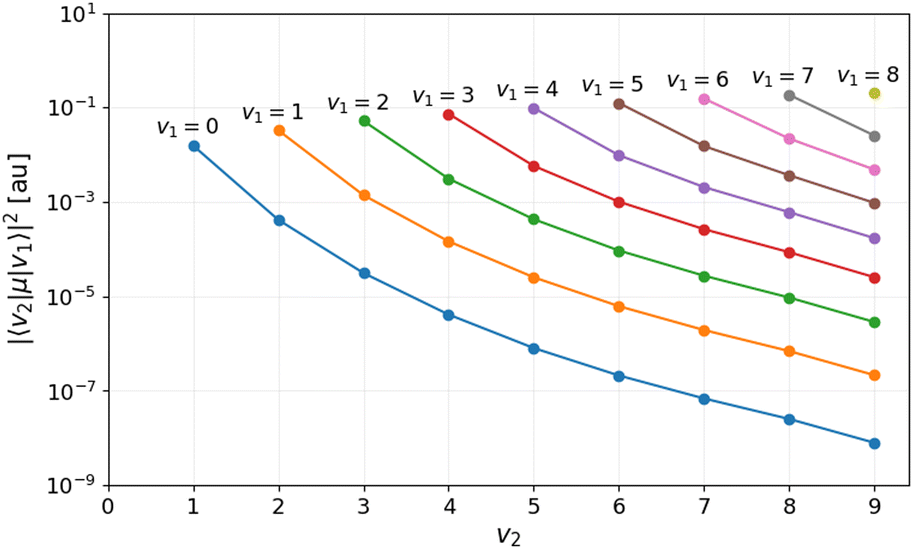 | ||
| Fig. 2 Transition matrix elements (1D) for the transition between bound states v1 → v2. | ||
3.1 Dissociation via vibrational ladder climbing
Fig. 3 shows the results of the wave packet simulations in one dimension for the two quadratic chirped pulses obtained by optimizing the laser parameters within the VLC region (blue box in Fig. 1). The optimal set of laser parameters and the resulting dissociation probabilities are shown in Table 2. It can be seen that the chirp rates for the two shaped pulses are negative corresponding to a linear decrease in the instantaneous frequency. For genuine ladder climbing to occur, the decrease in instantaneous frequency should match the resonance frequency between adjacent vibrational states and be temporally synchronized in order for successive population and depopulation to take place. Examining the population dynamics for the narrowband pulse in Fig. 3a, it is apparent that these conditions are not met. The population transfer is dominated by Rabi-like oscillations with the majority of the population residing in the v = 4 excited state after the laser interaction has subsided. The modest dissociation probability obtained, Pd = 6.2%, is attributable to the frequency content of the narrowband pulse lacking sufficiently low frequency components for resonant excitation between adjacent higher-lying vibrational states, Δv = ±1. In fact, ladder climbing is entirely absent at this energy density (2643 mJ cm−2) of the narrowband pulse, reflecting that the VLC regime identified in Fig. 1 is a rough estimate.
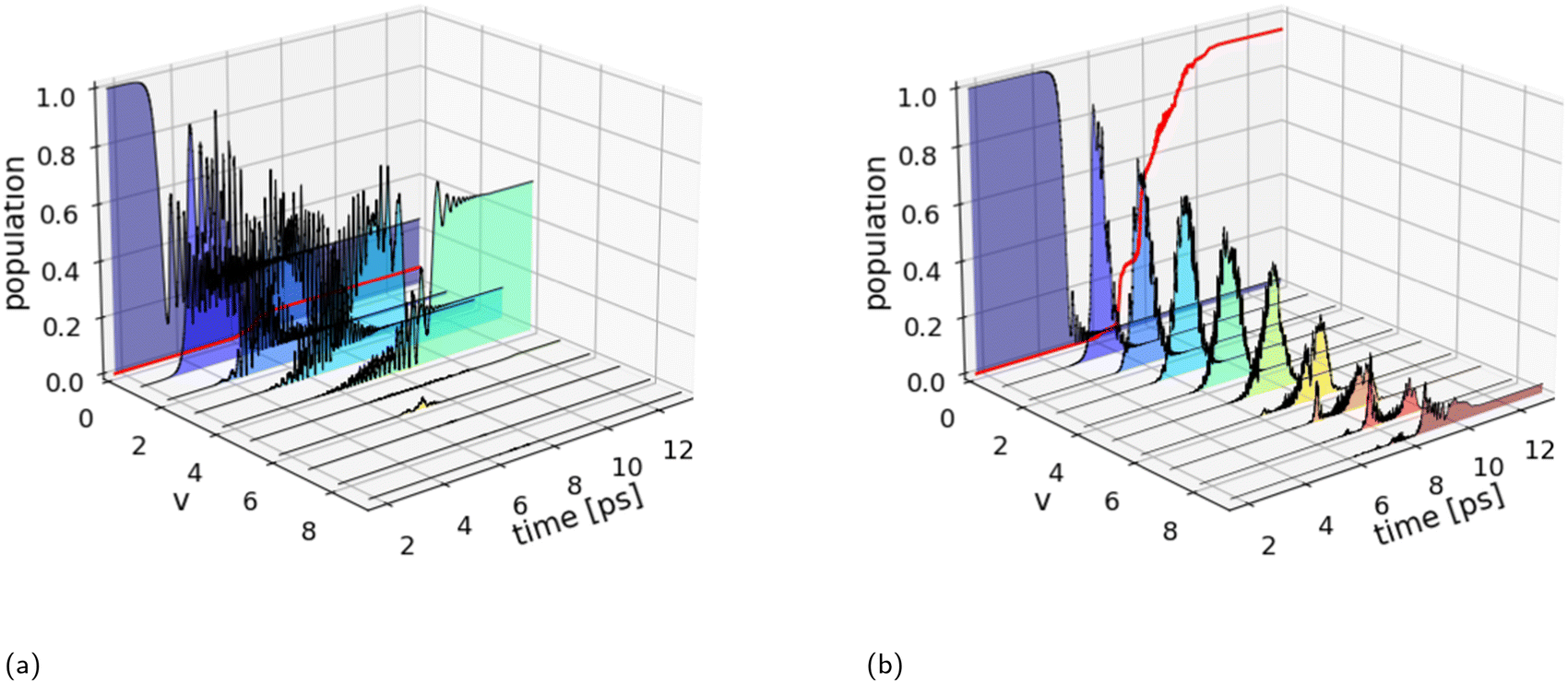 | ||
| Fig. 3 Population dynamics (1D) for the optimal quadratically chirped pulses shown in Table 2. (a) The excited state populations for the shaped narrowband pulse display Rabi-like oscillations with higher-lying vibrational states remaining unpopulated and a dissociation probability (red curve) of Pd = 6.2%. (b) The shaped broadband pulse shows close to genuine vibrational ladder climbing for the excited vibrational states v = 1–5, whereas the population transfer becomes desynchronized for the higher-lying excited states v = 6–9. Only the highest excited vibrational state v = 9 remains populated after the laser interaction resulting in a dissociation probability of Pd = 93.4%. | ||
| Shaped pulse E(t) | Peak intensity I0 (TW cm−2) | Duration T (ps) | Center freq. ω0 (cm−1) | Chirp rate (fs2 rad−2) |
Dissociation Pd | |
|---|---|---|---|---|---|---|
| 1D | Narrowband | 0.96 | 12.5 | 2436 | −33187.3 | 0.062 |
| Broadband | 0.95 | 12.6 | 2100 | −11323.8 | 0.934 | |
| 3D | Narrowband | 0.97 | 12.3 | 2553 | −32618.9 | 0.024 |
| Broadband | 0.75 | 15.9 | 2100 | −14379.6 | 0.116 |
In contrast, successive population and depopulation of the v = 1–5 vibrational states are observed for the broadband pulses in Fig. 3b, which is a characteristic of genuine VLC. This concerted population transfer is highly effective leading to near-complete dissociation with a dissociation probability of Pd = 93.4%. The magnitudes of the transition matrix elements in Fig. 2, suggest the possibility of overtone transitions, Δv ≥ ±2. However, overtone transitions to v ≤ 4 vibrational states require photon energies greater than 4200 cm−1, well above the frequency content of the input pulses. While both overtone transitions and direct excitation into the continuum are possible for the v = 6–9 excited states. Indeed, the magnitude of the transition dipole moment for the excitation to unbound states,18i.e. v6–9 → E, and the photon energy in the pulse for t > 6 ps is sufficiently large for direct excitation to the continuum to occur. As a result, the concerted population transfer becomes less pronounced for the higher-lying excited states.
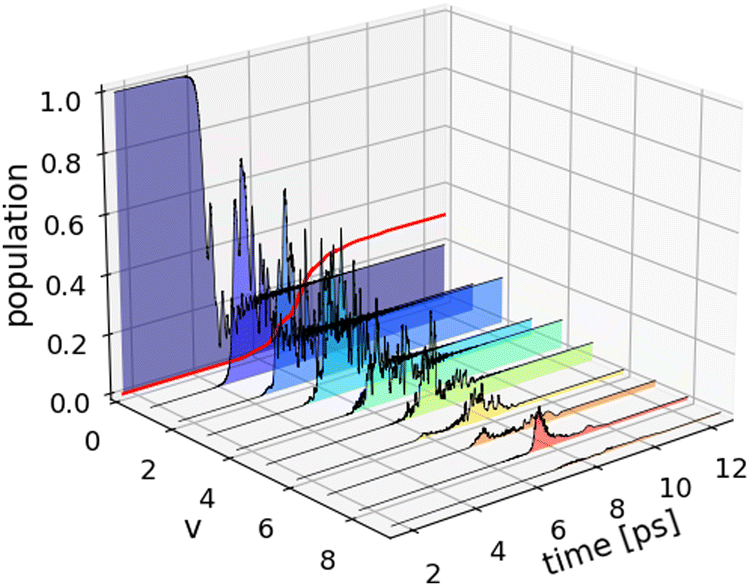
To examine ladder climbing in three dimensions, we adopt the optimization strategy in the previous discussion using the optimized parameters obtained from the one-dimensional simulations as the initial guess. Table 2 (3D) summarizes the optimized laser parameters and the corresponding dissociation probabilities. Fig. 4 shows the results of the wave packet simulations with the two optimal quadratic chirped pulses. In three dimensions, transitions involving Δv = ±m and ΔJ = ±n are allowed for m,n ≥ 1, with Δv = ±1 and ΔJ = ±1 being the dominant transitions. This increase in the number of allowed transitions significantly complicates the concerted population transfer between adjacent vibrational states. This is particularly apparent in the population dynamics for the shaped broadband pulse in Fig. 4b, where the fluctuating state occupations hinder the vibrational ladder climbing mechanism. As a consequence, the dissociation probabilities obtained with the shaped narrowband pulse Pd = 2.4% and the shaped broadband pulse Pd = 11.6% are significantly reduced compared to the one-dimensional results.
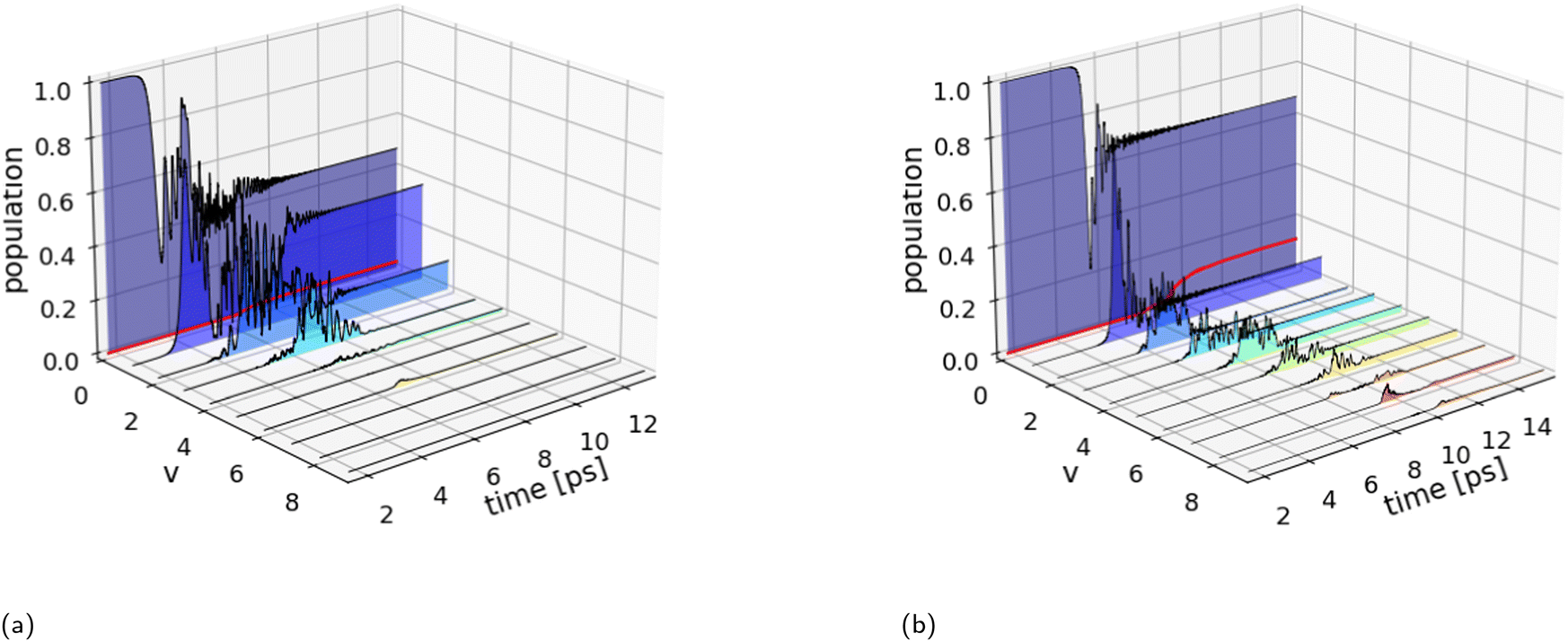 | ||
Fig. 4 Population dynamics (3D) for the optimal quadratically chirped pulses shown in Table 2. The evolution of the excited state populations is summed over all rotational states 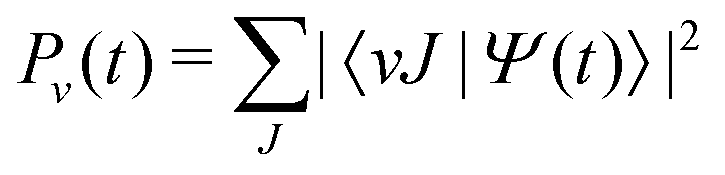 . (a) The excited state populations for the shaped narrowband pulse display Rabi-like oscillations with higher-lying vibrational states remaining unpopulated leading to a dissociation probability (red curve) of Pd = 2.4%. (b) The shaped broadband pulse shows less efficient and less smooth ladder climbing compared to the one-dimensional case (Fig. 3b). Consequently, leading to a significantly reduced dissociation probability of Pd = 11.6%. . (a) The excited state populations for the shaped narrowband pulse display Rabi-like oscillations with higher-lying vibrational states remaining unpopulated leading to a dissociation probability (red curve) of Pd = 2.4%. (b) The shaped broadband pulse shows less efficient and less smooth ladder climbing compared to the one-dimensional case (Fig. 3b). Consequently, leading to a significantly reduced dissociation probability of Pd = 11.6%. | ||
The results of the previous discussion raise the question of whether pulse shaping is always the best strategy for maximizing dissociation. In general, for the vast majority of molecular systems, ionization becomes appreciable above peak intensities of 1–10 TW cm−2 depending on frequency, and thus pulse shaping may be the only viable option in order to avoid ionization. However, for molecules with high ionization thresholds, as is the case for the HeH+ ion, pulse shaping may not always be the optimal choice. To investigate this aspect further, we compared the dissociation probability obtained with the two transform-limited pulses at different energy densities to that obtained with the corresponding shaped pulses. We also compared the performance of two zero-phase Gaussian pulses with identical envelope and peak intensity matching those of the shaped pulses. To that end, the chirp rates of the quadratic chirped pulses are re-optimized at each evaluated energy density, while the center frequencies are kept fixed at 2553 cm−1 and 2100 cm−1 corresponding to the optimal frequencies for the shaped narrowband and broadband pulses, respectively.
Fig. 5a shows the dissociation probability as a function of energy density for the narrowband pulses. It can be seen that the performance of the transform-limited pulse (orange line) exceeds that of the corresponding shaped pulse (blue line) by an order of magnitude. The dissociation probabilities obtained by the two pulses at the maximum energy density of 2643 mJ cm−2 are Pd = 30.4% and Pd = 2.4%, respectively. This observed difference cannot be attributed to the inclusion of the static polarizability terms in the interaction Hamiltonian, as the influence of the quadratic Stark effect on the dissociation probability is less than 0.1% for the transform-limited pulse with a peak intensity of 206 TW cm−2. At such high intensities, excitation occurs via both linear and non-linear transitions, whereas for peak intensities below 1 TW cm−2 excitation proceeds predominantly via linear transitions. In comparison, Fig. 5b shows that shaping of the broadband pulse significantly enhances dissociation. In particular, at lower pulse energies, where the dissociation probability is increased by a factor of 90 compared to that of the transform-limited pulse. However, it must be emphasized that the observed effect of pulse shaping is achieved at the optimal center frequency of the quadratic chirped pulse, ω0 = 2100 cm−1. While the frequency dependence of the transform-limited pulse is shifted toward higher center frequencies, e.g. the optimal center frequency was found to be ω0 = 2514 cm−1 at the maximal energy density F = 2643 TW cm−2, resulting in a dissociation probability of Pd = 14.1%. The results demonstrate that a transform-limited pulse with high peak intensity is more effective than a quadratic chirped pulse with peak intensity I0 < 1 TW cm−2 in inducing dissociation of the HeH+ ion, provided that the center frequency can be freely chosen.
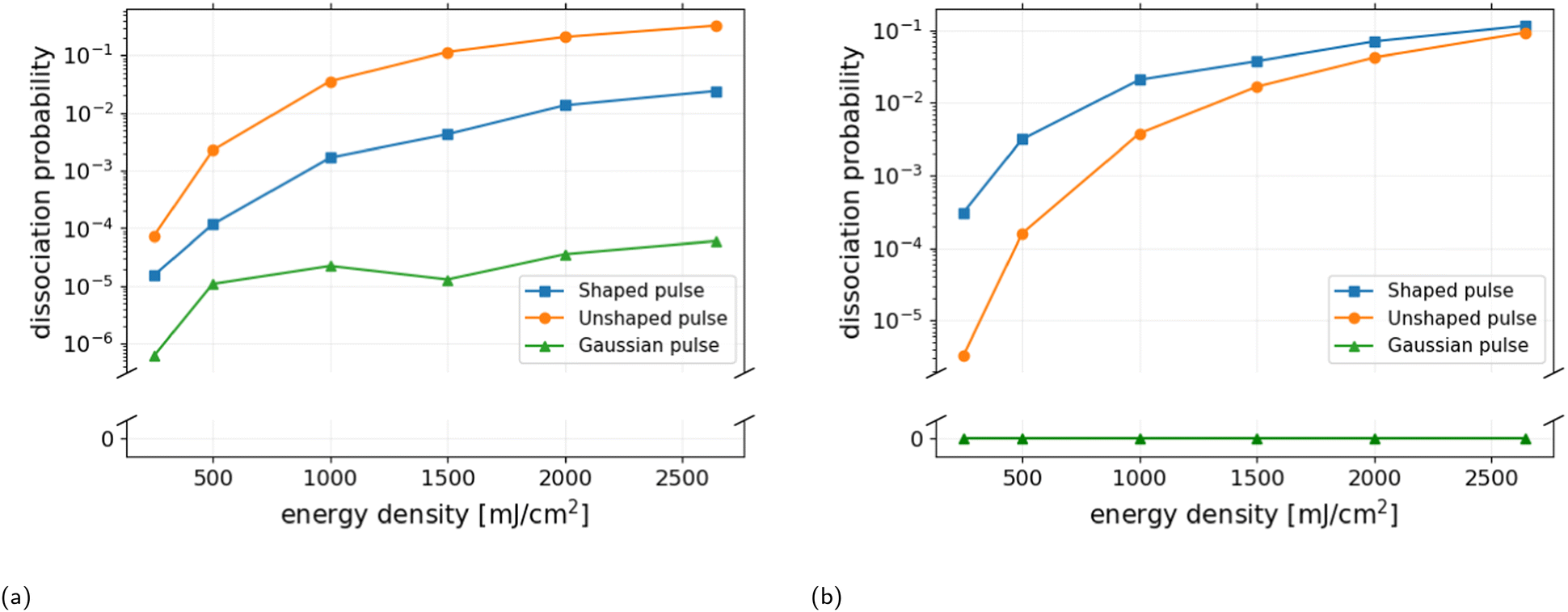 | ||
| Fig. 5 Dissociation probabilities (3D) as a function of energy density for (a) the transform-limited narrowband input pulse (orange curve) with a center frequency of 2553 cm−1, the corresponding quadratic chirped pulse (blue curve), and the zero-phase Gaussian pulse with envelope and peak intensity identical to that of the shaped pulse (green curve). The unshaped pulse achieves significantly higher dissociation probabilities at all energy densities reaching Pd = 30.4%. (b) The broadband input pulse with center frequency 2100 cm−1, the corresponding quadratic chirped pulse, and zero-phase Gaussian. At low energy densities, the shaped pulse enhances the dissociation probability by a factor of ∼90. | ||
The zero-phase Gaussian pulses (green line) shown in Fig. 5 have identical temporal envelope and peak intensity as the shaped pulses, and thus are transform-limited with a reduced spectral bandwidth given by = 1/α. It is clear that the negative quadratic chirp and the greater frequency content of the shaped pulses are responsible for the increase in dissociation probability observed for the entire range of energy densities considered. Thus, the performance of pulse shaping critically relies on the center frequency, spectral bandwidth and energy of the input pulse.
The wave packet simulations in the previous discussions have all been performed with |00〉 as the initial state. Since the magnitude of the molecule-laser interaction depends on the alignment of the molecular axis with the polarization axis, described by 〈Ψ|cos2θ|Ψ〉, it is expected that pre-alignment will lead to enhanced dissociation. This can be accomplished straightforwardly by choosing |01〉 as the initial state, which increases the initial alignment from 〈cos2θ〉00 = 1/3 to 〈cos2θ〉01 = 3/5. Fig. 6 shows the population dynamics with the optimized quadratic chirped broadband pulse in Table 3. The dissociation probability of Pd = 29.9% is enhanced by a factor of ∼3 compared to the result obtained with J = 0 in Fig. 4b. It should be noted that greater pre-alignment cannot be achieved starting from a single initial state with |0,J > 1〉, to obtain perfect alignment a superposition of |0,J〉 states is required.
 | ||
| Fig. 6 Population dynamics (3D) for the quadratic chirped pulse with the initial state |Ψ(0)〉 = |0,1〉. A single quantum in the initial rotational motion increases the initial alignment of the molecular axis with the polarization axis, 〈Ψ(0)|cos2θ|Ψ(0)〉. As a result, the population of the vibrational ground state is more efficiently transferred to excited states leading to a dissociation probability of Pd = 29.9%. | ||
| Shaped pulse E(t) | Peak intensity I0 (TW cm−2) | Duration T (ps) | Center freq. ω0 (cm−1) | Chirp rate (fs2 rad−2) |
Dissociation Pd |
|---|---|---|---|---|---|
| Broadband | 0.995 | 12 | 2100 | −10813.9 | 0.299 |
3.2 Beyond quadratic spectral chirp
The absence of a genuine concerted population transfer in the three-dimensional simulations, suggests that vibrational ladder climbing may not be the most efficient mechanism for controlling dissociation of the HeH+ ion. The fourth-order polynomial phase function in eqn (8), provides a greater degree of optical phase modulation during the optimization of the shaped pulses. This, in principle, enables greater coherent control of the complex dissociation dynamics resulting from the inclusion of rotational motion.To that end, we limit the discussion to shaping of the broadband input pulse, as the previous results achieved the highest dissociation with this bandwidth. We determined the results of the wave packet simulations with the optimized laser parameters: ω0 = 2126 cm−1, I0 = 0.7686 TW cm−2, 1 = 0.0, 2 = −14013.5, 3 = 6134.6 and 4 = −48942.7. The population dynamics is similar to that shown in Fig. 4b with slightly higher occupation in the v = 9 excited state and with a marginal increase in the dissociation probability, Pd = 11.9%. We considered additional phase modulation but were not able to identify phase functions leading to higher dissociation probabilities.
3.3 Dissociation via centrifugal fragmentation
As an alternative dissociation pathway, we consider centrifugal fragmentation via rotational excitation. To that end, we employ the transform-limited input pulse summarized in Table 4. Pulse shaping was performed with a positive quadratic chirp to accommodate the approximate linearly increasing energy-gap between adjacent rotational states. The dissociation probabilities are computed via, Pd = 1 − 〈Ψ|Θ(Rc − R)|Ψ〉, where Θ denotes the Heaviside function and the part of the wave packet beyond the cut-off, Rc = 10a0, represents unbound density, i.e. the dissociated part of the wave packet. This approach was adopted due to difficulties in computing high-lying rotational excited states, i.e. J > 23, via relaxation as the eigenenergy of these bound rovibrational states are comparable to that of unbound continuum states. Fig. 7 shows the results of the wave packet simulations using the optimized shaped pulse with a center frequency of 604 cm−1, peak intensity I0 = 0.9884 TW cm−2 and positive chirp rate = 42334.0 fs2. The population dynamics are highly oscillatory but with features indicating rotational ladder climbing leading to a dissociation probability of Pd = 25.5%. Thus, centrifugal fragmentation represents a highly effective means of inducing dissociation of the HeH+ ion.
| Peak intensity I0 (TW cm−2) | FWHM[Ein(t)] (fs) | FWHM[Ẽin2(ω)] (cm−1) |
(fs rad−1) |
|---|---|---|---|
| 53 | 66 | 315 | 28.1 |
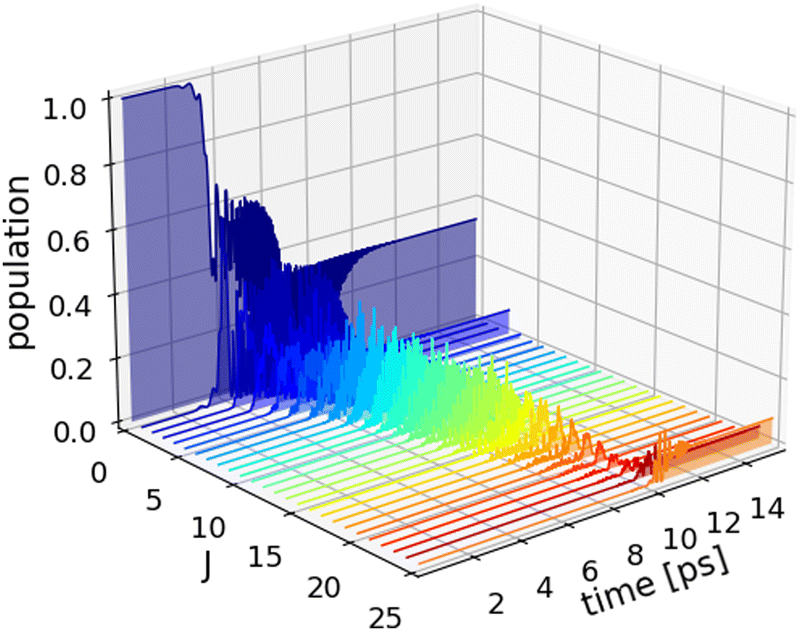 | ||
| Fig. 7 Population dynamics (3D) of the rovibrational states |0,J〉 for the optimal shaped pulse with positive quadratic chirps targeting rotational excitation. Rotational ladder climbing is observed leading to a dissociation probability of Pd = 25.5%. | ||
4 Conclusions
We have performed simulations of the photodissociation dynamics in the electronic ground state of the HeH+ ion. Dissociation was induced by shaped laser pulses targeting both vibrational and rotational excitation pathways. Pulse shaping was performed by phase modulation of transform-limited laser pulses and relied on constrained optimization of the laser parameters to generate shaped pulses with peak intensities below 1 TW cm−2. A number of experimental pulse shaping parameters and their effects on dissociation probabilities were investigated in both one and three dimensions. The parameters that were varied are (i) the pulse energy, (ii) the center frequency, and (iii) the coefficients of phase functions. Optimization was carried out for two transform-limited pulses with different spectral bandwidths.Initially, we identified the vibrational ladder climbing regime in one dimension and demonstrated that shaped pulses with a negative quadratic spectral chirp are capable of a high degree of selective vibrational excitation. Near-complete dissociation was achieved at the maximal pulse energy considered with a dissociation probability of 93.4%. This was extended to three dimensions by taking into account rotational motion. Our results show that selective population and depopulation of adjacent vibrational states is hindered by the additional transition pathways made accessible by rotational transitions. Consequently, oscillatory population dynamics is observed and the dissociation probability is significantly reduced. Under the constraints imposed on the pulse shaper, the efficacy of pulse shaping was found to be highly dependent on the frequency content of the shaped pulses.
At peak intensities below 1 TW cm−2, dissociation occurs predominantly via linear transitions and a shaped pulse with a broad spectral bandwidth is required for appreciable dissociation to occur via vibrational ladder climbing. The overtone and non-linear transitions contribute significantly to the dissociation dynamics at the high peak intensities of the transform-limited pulses considered here, which are up to 220 times greater than the corresponding shaped pulses. As a result, the transform-limited pulse was found to be optimal at narrow spectral bandwidths achieving a dissociation probability of 30.4%, whereas 2.4% dissociation was observed for the shaped pulse with a quadratic spectral chirp. By contrast, at the maximal pulse energy, a dissociation probability of 11.5% was obtained by the shaped pulse with a broad spectral bandwidth compared to 9.3% for the corresponding transform-limited pulse. The effectiveness of shaping broadband pulses is particularly noticeable at low pulse energies, where pulse shaping increases dissociation up to 90 times relative to the transform-limited pulse. Moreover, increasing the initial alignment of the molecule with respect to the polarization axis led to a further increase in dissociation, resulting in a dissociation probability of 29.9%. While phase modulation using a 4th order polynomial phase function resulted in a marginal increase of only 0.4%.
In addition, we investigated dissociation via centrifugal fragmentation using tailored pulses with a positive quadratic spectral chirp. Rotational ladder climbing was observed and found to be an effective method for inducing dissociation, resulting in a dissociation probability of 25.5%.
The HeH+ ion is an extreme example of a diatomic polar molecule with a high ionization threshold. In molecular systems that can withstand high-intensity laser pulses with minimal ionization, pulse shaping may not be the most effective method to induce dissociation. Effective vibrational control of bond cleavage in the HeH+ ion was achieved without stringent demands on the phase-coherence characteristics of the laser pulse. However, most molecules cannot tolerate high-intensity laser pulses, necessitating the use of pulse shaping. Furthermore, the objective of laser control is typically more subtle than simply breaking a bond in a diatomic molecule, e.g. state-to-state control or control of product branching ratios in polyatomic molecules, where the benefits of pulse shaping are more prominent.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has received funding from the Danish Council for Independent Research, grant no. 0135-00133B.Notes and references
- P. Brumer and M. Shapiro, Chem. Phys. Lett., 1986, 126, 541–546 CrossRef CAS.
- A. Giusti-Suzor, X. He, O. Atabek and F. H. Mies, Phys. Rev. Lett., 1990, 64, 515 CrossRef CAS PubMed.
- S. A. Rice and M. Zhao, Optical control of molecular dynamics, John Wiley, 2000 Search PubMed.
- A. M. Weiner, Rev. Sci. Instrum., 2000, 71, 1929–1960 CrossRef CAS.
- M. Wollenhaupt, A. Assion and T. Baumert, Springer Handbook of Lasers and Optics, 2007, pp. 937–983 Search PubMed.
- D. J. Tannor, Introduction to quantum mechanics: A time-dependent perspective, University Science Books, 2007 Search PubMed.
- C. Brif, R. Chakrabarti and H. Rabitz, New J. Phys., 2010, 12, 075008 CrossRef.
- M. Shapiro and P. Brumer, Quantum Control of Molecular Processes, Wiley-VCH, 2012 Search PubMed.
- A. S. Alnaser, M. Kübel, R. Siemering, B. Bergues, N. G. Kling, K. J. Betsch, Y. Deng, J. Schmidt, Z. A. Alahmed, A. M. Azzeer, J. Ullrich, I. Ben-Itzhak, R. Moshammer, U. Kleineberg, F. Krausz, R. D. Vivie-Riedle and M. F. Kling, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- A. Guldberg and G. D. Billing, Chem. Phys. Lett., 1991, 186, 229–237 CrossRef CAS.
- R. Schanz, V. Boţan and P. Hamm, J. Chem. Phys., 2005, 122, 044509 CrossRef PubMed.
- M. Artamonov, T. S. Ho and H. Rabitz, Chem. Phys., 2006, 328, 147–155 CrossRef CAS.
- C. C. Shu, E. F. Thomas and N. E. Henriksen, Chem. Phys. Lett., 2017, 683, 234–239 CrossRef CAS.
- K. Heyne and O. Kühn, J. Am. Chem. Soc., 2019, 141, 11730–11738 CrossRef CAS PubMed.
- A. Cartella, C. Manzoni, S. Bonora, M. Först, G. Cerullo and A. Cavalleri, Opt. Lett., 2014, 39, 1485–1488 CrossRef PubMed.
- M. A. Jakob, M. Namboodiri, M. J. Prandolini and T. Laarmann, Opt. Express, 2019, 27, 26979–26988 CrossRef CAS PubMed.
- F. Nicolai, N. Müller, T. Buckup, C. Manzoni and G. Cerullo, Opt. Express, 2021, 29, 20970–20980 CrossRef CAS PubMed.
- P. Wustelt, F. Oppermann, S. Mhatre, M. Kübel, A. M. Sayler, M. Lein, S. Gräfe and G. G. Paulus, Phys. Rev. Lett., 2021, 127, 043202 CrossRef CAS PubMed.
- N. Dudovich, B. Dayan, S. M. G. Faeder and Y. Silberberg, Phys. Rev. Lett., 2001, 86, 47 CrossRef CAS PubMed.
- S. Chelkowski, A. D. Bandrauk and P. B. Corkum, Phys. Rev. Lett., 1990, 65, 2355 CrossRef CAS PubMed.
- I. Barth, L. Friedland, O. Gat and A. G. Shagalov, Phys. Rev. A: At., Mol., Opt. Phys., 2011, 84, 013837 CrossRef.
- T. Horiba, S. Shirai and H. Hirai, Phys. Rev. A, 2022, 105, 013117 CrossRef CAS.
- J. T. Lin, D. S. Chuu and T. F. Jiang, Phys. Rev. A: At., Mol., Opt. Phys., 1998, 58, 2337 CrossRef CAS.
- T. Witte, T. Hornung, L. Windhorn, D. Proch, R. D. Vivie-Riedle, M. Motzkus and K. L. Kompa, J. Chem. Phys., 2003, 118, 2021–2024 CrossRef CAS.
- C. Ventalon, J. M. Fraser, M. H. Vos, A. Alexandrou, J. L. Martin and M. Joffre, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13216–13220 CrossRef CAS PubMed.
- D. B. Strasfeld, S. H. Shim and M. T. Zanni, Phys. Rev. Lett., 2007, 99, 038102 CrossRef PubMed.
- D. S. Seo, A. M. Weiner, Z. Jiang and D. E. Leaird, Opt. Lett., 2004, 29(16), 1864–1866 CrossRef CAS PubMed.
- E. F. Thomas and N. E. Henriksen, J. Chem. Phys., 2016, 144, 244307 CrossRef PubMed.
- Z. Qin, T. Bai, L. Liu, S.-D. Zhang, C. Liu, J. Loreau, J. Liévin, P. Palmeri, P. Quinet and N. Vaeck, J. Phys. B: At., Mol. Opt. Phys., 2010, 43, 065101 CrossRef.
- R. F. Stewart, D. K. Watson and A. Dalgarno, J. Chem. Phys., 1976, 65, 2104–2111 CrossRef CAS.
- P. Tran, Phys. Rev. A: At., Mol., Opt. Phys., 1999, 59, 1444 CrossRef CAS.
- G. A. Worth, M. H. Beck, A. Jäckle, O. Vendrell and H.-D. Meyer, The MCTDH Package, Version 8.2, (2000). H.-D. Meyer, Version 8.3 (2002), Version 8.4 (2007). O. Vendrell and H.-D. Meyer Version 8.5 (2013). Versions 8.5 and 8.6 contains the ML-MCTDH algorithm. Used version: 8.6.5.1 (July 2023). See https://mctdh.uni-hd.de/.
- M. H. Beck, A. Jäckle, G. A. Worth and H. D. Meyer, Phys. Rep., 2000, 324, 1–105 CrossRef CAS.
- R. R. Zaari and A. Brown, J. Chem. Phys., 2012, 137, 104306 CrossRef PubMed.
- F. Nogueira, Bayesian Optimization: Open source constrained global optimization tool for Python, https://github.com/fmfn/BayesianOptimization, 2014.
- A. K. Tiwari, K. B. Møller and N. E. Henriksen, Chem. Phys. Lett., 2007, 450, 6–11 CrossRef CAS.
| This journal is © the Owner Societies 2024 |