
Unraveling the ultrafast dynamics of thermal-energy chemical reactions
Matthew S.
Robinson
 ab and
Jochen
Küpper
*abc
ab and
Jochen
Küpper
*abc
aCenter for Free-Electron Laser Science CFEL, Deutsches Elektronen-Synchrotron DESY, Notkestr. 85, 22607 Hamburg, Germany. E-mail: jochen.kuepper@cfel.de; Web: https://www.controlled-molecule-imaging.org
bCenter for Ultrafast Imaging, Universität Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany
cDepartment of Physics, Universität Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany
First published on 4th November 2023
Abstract
In this perspective, we discuss how one can initiate, image, and disentangle the ultrafast elementary steps of thermal-energy chemical dynamics, building upon advances in technology and scientific insight. We propose that combinations of ultrashort mid-infrared laser pulses, controlled molecular species in the gas phase, and forefront imaging techniques allow to unravel the elementary steps of general-chemistry reaction processes in real time. We detail, for prototypical first reaction systems, experimental methods enabling these investigations, how to sufficiently prepare and promote gas-phase samples to thermal-energy reactive states with contemporary ultrashort mid-infrared laser systems, and how to image the initiated ultrafast chemical dynamics. The results of such experiments will clearly further our understanding of general-chemistry reaction dynamics.
I. Introduction
For decades, femtochemistry has given us glimpses into the ultrafast world of chemical dynamics.1,2 With the tried and tested pump–probe technique, a vast array of probes were used to track and interpret the photo-induced excited-state dynamics of countless molecules using transient-absorption,3–5 photoelectron spectroscopy,6–8 and diffraction9–14 techniques. To date, the majority of these experiments initiate the dynamics of interest through electronically-excited states, using high-energy visible,12,15 ultraviolet,6,16 or X-ray photons17 or the strong fields of intense ultrashort ionizing laser pulses.18–20However, most chemical processes that are key to everyday life, from biology to materials, occur at lower, thermal energies. As an example, the final, heat-induced decarboxylation step in the Reissert indole synthesis reaction21 is depicted in Fig. 1. We propose to mimic such thermal-energy processes by triggering cold molecules with low-energy photons, i.e., using light in the mid-infrared (mid-IR) region of the electromagnetic spectrum. To time-resolve these processes they must be initiated and probed with pulses of suitably short duration, typically from ultrafast-laser sources.
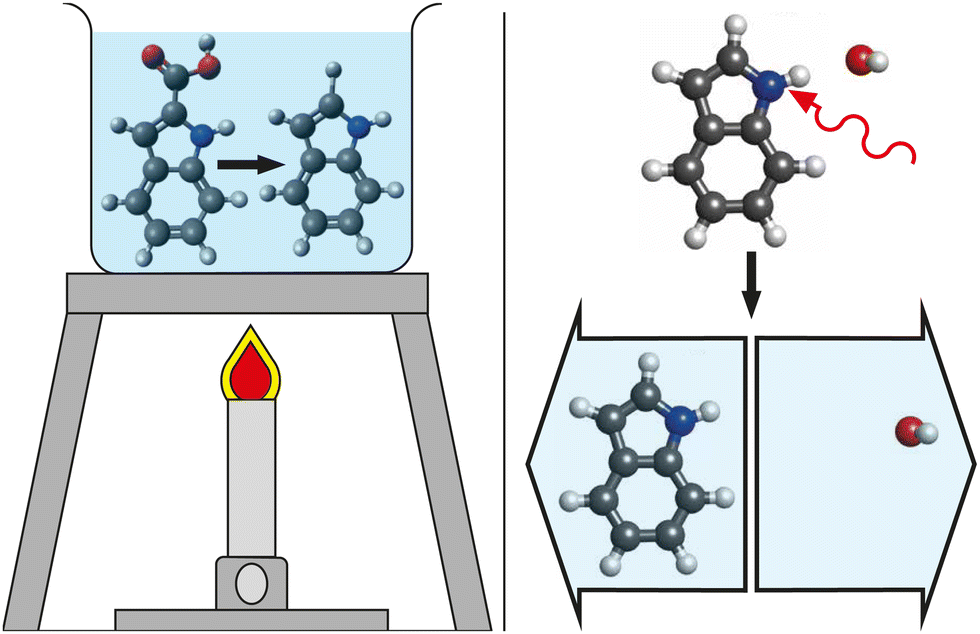 | ||
| Fig. 1 Cartoon depictions of thermal-energy chemical reactions. (left) The heating by a Bunsen burner induces the final decarboxylation step of the Reissert indole synthesis in solution. (right) The mode-specific excitation of the N–H bond of a solvated indole system using a single photon of mid-IR light (red arrow) leads to bond breaking and the dissociation of the molecular system. | ||
The time-resolved dynamics of thermal-energy-excited condensed-phase systems were investigated, for example, through 2D-IR spectroscopy in solution.22,23 This allowed for the study of the secondary structures of proteins24 and for biomolecular recognition.25 Femtosecond infrared pulses enabled the triggering and time-resolved spectroscopic observation of the isomerization of HONO in cryogenic solid matrices26 as well as the acceleration of thermal (poly)urethane formation in room-temperature solution.27 Furthermore, the evaporative dissociation of iron-pentacarbonyl clusters due to infrared-radiation-induced heating was tracked on a (sub)nanosecond timescale.28 However, the imaging of such dynamics at thermal energies in the gas phase, free from solvent backgrounds and with femtosecond temporal resolution, is a milestone that has yet to be reached, but one that we step closer to with progressing technologies.
Advances in laser technology, especially the development of mid-IR optical parametric amplifiers (OPAs), now allow for simultaneously suitably bright and ultrashort mid-IR pump pulses for initiating reactions.29,30 Molecular beams combined with skimming and deflection techniques allow us to cool, control and choose specific species31,32 that are suitable for initiating and observing reactions with thermal-energy pump pulses. The high-repetition rates at free-electron laser (FEL) facilities provide us with the opportunity to probe the initiated thermal-energy dynamics using techniques ranging from Coulomb-explosion ion imaging33–35 to coherent X-ray diffraction9,36 in a reasonable time frame. Table-top-laser-based methods like photoion37,38 and photoelectron imaging39–41 as well as laser-induced electron diffraction42–47 provide the opportunity to observe such dynamics with low-density targets in the laboratory.
We propose that combining these techniques allows for thermal-energy reactions to be investigated using advanced femtochemistry techniques. Prototypical reactions include the dynamic interaction between water and (bio)molecules in microsolvated systems,16 isomerizations of organic molecules,48–50 or folding pathways of larger biological systems.18,51 Disentangling these prototypical reactions will provide time-resolved rationales to the generally-used statistical models of everyday textbook chemistry and it will allow us to derive intrinsic key reaction modes for a dynamical basis of chemistry.
In this perspective, we detail experimental approaches to be used for studying ultrafast thermal-energy reaction dynamics in gas-phase systems and how these mimic the initial steps of thermal-energy processes. This includes details on how suitably cooled samples can be prepared and selected, how contemporary mid-IR OPAs can be used to trigger the reactions of interest, and how the ongoing reactions can be observed in real time. We provide quantitative estimates of excitation and detection efficiencies, which demonstrate that these experiments are now possible.
II. Proposed initial prototypical reaction systems
A fundamental process in “everyday chemistry” is the dissociation of a species into parts. This is a process that can be induced by mid-IR excitation for both small clusters52 and molecular ions53 alike. One system that we propose as a suitable starting point for time-resolving, and thus better understanding, mid-IR induced dissociation dynamics is the prototypical microsolvated (bio)molecular cluster indole–water, which has a well-defined structure54 depicted in Fig. 1 (top right). Indole is the chromophore of tryptophan, one of the strongest near-UV-absorbing amino acids in nature, whose photochemistry was shown to strongly depend on its environment.55 With indole, we take a bottom-up approach to understanding how this chromophore and building block of proteins behaves under thermal-excitation conditions. Investigating its hydrated cluster, we provide insight into the dynamics of these systems in aqueous environments. Ultimately, this will allow for a better understanding of the elementary steps of such effects of the molecular environment, which in turn can be related back to studies of aqueous systems previously studied using 2D-IR spectroscopy.56,57With this wider context in mind, several key properties of the cluster make indole–water a prime candidate for benchmarking techniques to study ultrafast thermal-energy-reaction dynamics. The location of the water molecule, which is localized at the N–H bond of indole through a hydrogen bond,54 yields a well-defined reactant system that can be purified in the gas phase58 and is even of relevance in aqueous solution.59 Following solvation, the cross section of the N–H stretch of the indole moiety at ∼2.9 μm (3425 cm−1, 0.425 eV) becomes significantly enhanced with respect to all other vibrational modes in the system, by approximately an order of magnitude in comparison to the unsolvated system.60 This strong contrast of oscillator strengths between the N–H stretch and other stretching modes means that one can expect the N–H-stretch vibration to be the dominant absorption of the system in this spectral range, even considering the broad spectral bandwidth of the ultrashort mid-IR pulses necessary to excite the system with high temporal resolution. As the excitation energy of this transition is considerably larger than the binding energy of the hydrogen-bound cluster (∼0.210 eV61) the system could directly dissociate after the vibrational excitation as depicted in Fig. 2, pathways 1. However, intramolecular vibrational redistribution (IVR) could also lead to more involved threshold-like reaction dynamics, such as roaming,62 as depicted in Fig. 2, pathways 2.
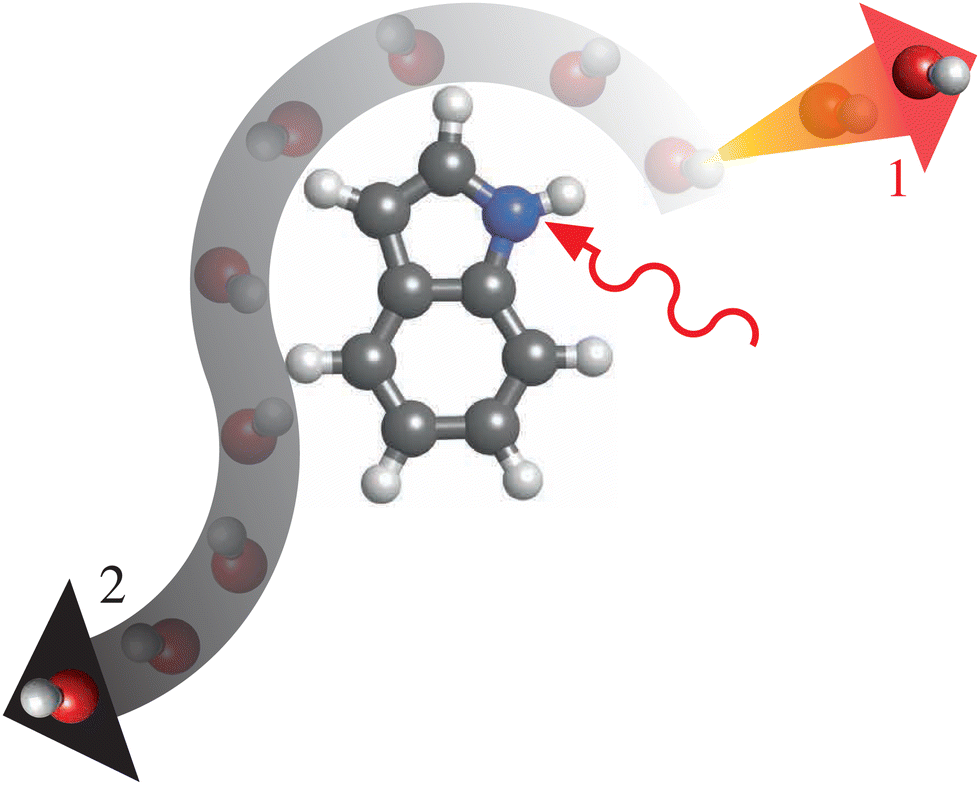 | ||
| Fig. 2 Artistic interpretation of two possible dissociation pathways for indole–water following excitation of the molecule by a mid-IR photon (solid red arrow), including (pathway 1, orange-red arrow) the direct ejection of the water molecule and (pathway 2, white-gray arrow) a longer, roaming-like process before eventual dissociation. | ||
Further prototypical experiments to consider would be the mid-IR induced conformational changes of organic or biological molecules – processes that are key to folding mechanisms of larger biological systems like proteins.63,64 Initial investigations could focus on single-bond rotations in small molecules, like that of the dihedral rotation around the C–C bond of cyclopropane carboxaldehyde (CPCA),50,65i.e., ϕHCCO, as depicted in Fig. 3. Previous work utilized a mid-IR laser, centered around 2750 cm−1, to induce a reversible anti ↔ syn-conformational change that was observed on a timescale on the order of a few 100 ps, 16 times slower than what was predicted by statistical methods.50 This highlights the need for ultrafast-dynamics techniques, including atomic-resolution imaging, to disentangle these fundamental processes.
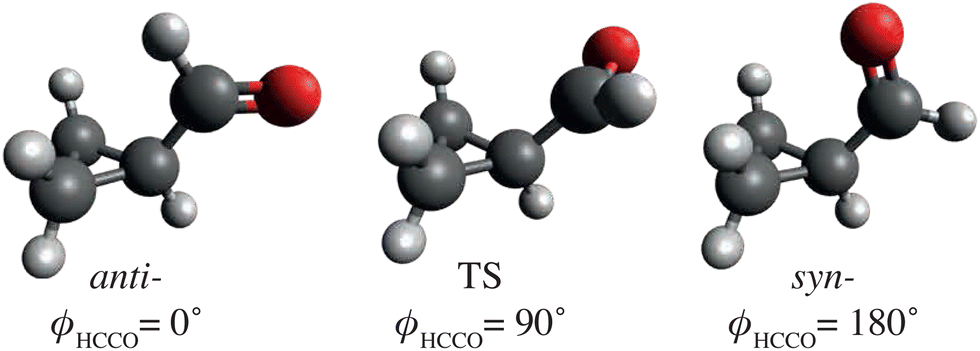 | ||
| Fig. 3 Sketch of anti- and syn-cyclopropane carboxaldehyde (CPCA) and a possible transition state (TS) structure, including ϕHCCO dihedral angles. These structures are based on computational results from ref. 65. | ||
Larger molecular species that showed interesting thermal-energy induced conformational changes include tryptamine, for which different isomerization pathways became accessible as certain thermal-energy barriers were overcome when scanning the IR excitation over the range 7–13 μm (∼1500–800 cm−1),49 see Fig. 4. Similarly, tunable mid-IR lasers were used to induce conformer-specific isomerization processes in gas-phase samples of N-acetyl-tryptophan methyl amide (NATMA) via single-mode vibrational excitation of the N–H stretch at ∼2.9 μm.48
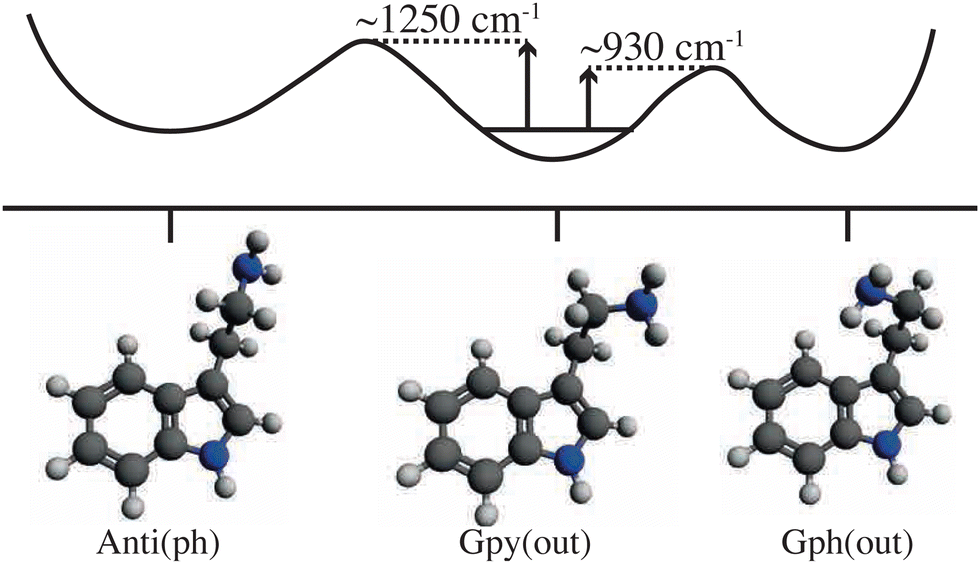 | ||
| Fig. 4 Diagrammatic representation of selected ground-state structures of tryptamine and a schematic potential energy curve that connects them. Transition-state energy barriers are represented by the vertical arrows and are based on experimental results.49 | ||
Infrared multiple-photon dissociation (IRMPD) experiments were used to induce unfolding and dissociation processes of protein(-like) structures,66,67 including short sequences that match DNA telomeres,51 following excitation with mid-IR light in the 6–11 μm region.
Investigating these thermal-energy-induced intramolecular structural changes, and wider (un)folding mechanisms of protein(-like) systems, in an ultrafast-time-resolved manner, will allow us to develop a better understanding of these low-energy vibrational and conformational changes. This should also enable comparisons to statistical methods that were typically used to describe these processes so far, as well as to coherent-wavepacket-dynamics simulations, and it should trigger further theoretical work on these processes to eventually unravel the actual physics underlying these elementary steps of thermal-energy chemistry.
Further work could be guided by discussions of non-statistical reactivity, e.g., due to phase-space bottlenecks.68,69 Moreover, even reactions with seemingly well-behaving kinetics could show intriguing ultrafast dynamics, including roaming- or trapping-like processes due to long-range interactions of the reactants and products.70,71
III. Sample preparation
To be able to effectively monitor the gas-phase thermal-energy dynamics one must prepare samples so that the induced changes can be observed above background signals. Preferably, one would prepare a molecular ensemble of a single well-defined reactant. For small molecular species, supersonic expansion of a seeded gas into a vacuum, combined with skimmers, is common practice to produce vibrationally- and rotationally-cold molecules in the few/sub-Kelvin temperature regime.72,73 These beams can be further controlled or species selected for better purification. For example, the electric deflector allows for the dispersion and separation of individual species in a molecular beam according to their dipole-moment-to-mass ratios.31 This allows for the production of pure samples, clear from the seed gas, as well as the selection of specific conformers or clusters,31 including, for instance, pure samples of indole–water16,32 or similar hydrated-molecule dimers.74,75 It also allowed for the separation of individual conformers.76–78 Experimentally, these deflected beams have typical thicknesses of ∼2 mm,32 with sample densities of 107–109 molecules per cm3.9,71,78For larger systems, alternative methods are available to bring samples into the gas phase. For example, laser desorption techniques, such as MALDI (matrix-assisted laser desorption/ionization),79 were used to transfer neutral and charged systems into the gas phase, ranging from single molecules80–82 to kilo-Dalton-sized structures.83 Electrospray-ionization methods84 can be used to bring solvated systems into the gas-phase in various charge states. High densities of uncharged species can also be obtained when utilizing neutralizers.85 Once these larger molecules are in the gas-phase, contemporary apparatus, such as drift cells and ion-mobility techniques, can be used to separate these larger species based on the their size and shape.86,87 Recently, control techniques developed for small molecules were applied to larger species, including the use of inhomogeneous electric fields to separate artificial and biological nanoparticles as a function of their mass-to-charge ratio.85,88 Proposals have also suggested that this can be applied to neutral large molecules, separating out different protein conformers as a function of their dipole-moment-to-mass ratios.85
IV. Ultrashort mid-IR pulses for inducing thermal-energy excitations
We propose to initiate the thermal-energy dynamics in a temporally well-defined fashion though the absorption of a single, or a few, mid-IR photon(s) provided by an ultrashort laser pulse. However, this range of 2.5–25 μm (4000–400 cm−1, ∼0.5–0.05 eV, ∼120–12 THz) is on the cusp of the so-called “THz gap”, a wavelength region where it was notoriously difficult to develop compact and intense light sources,89 which until recently limited our ability to use mid-IR light in ultrafast experiments. Modern OPAs are now able to produce mid-IR pulses with sub-picosecond durations and sufficient brightness to effectively induce excitation of vibrational modes in a controlled manner.30,90 A full review of OPAs is outside the scope of this perspective and provided elsewhere,29,30 however, as reference, a current state-of-the-art mid-IR OPA centered at ∼2.8 μm can provide 520 μJ per pulse at a repetition rate of 1 kHz, with a spectral bandwidth of ∼500 nm, corresponding to a Fourier-limited pulse duration of ∼25 fs.91 Typical commercial OPAs have similar bandwidth and pulse duration properties, albeit with a reduced output power for higher wavelength tunability. To reach sufficient intensities for performing the envisioned pump–probe experiments, the mid-IR light from an OPA needs to be focused using lenses made from CaF2 or similar materials possessing high transmission and low dispersion for mid-IR light. Assuming a 30 cm focal-length lens,92 1 cm diameter beams will be focused to a spot size of ∼110 μm.Alternatively, one could use free-electron laser sources, such as specific IR and THz facilities93,94 or THz-undulator sources at X-ray free-electron lasers.95 This could be especially interesting to extend the proposed studies to lower-energy-photon excitations, at the cost of limited accessibility to beamtime at facilities.
V. Scientific feasibility
With both the sample preparation and mid-IR lasers for inducing thermal-energy dynamics clarified, the remaining experimental details depend on the exact probing technique of interest. We do not expect this to differ widely from established experimental set-ups used in other pump–probe experiments.16,32 To quantify the probability of exciting the desired vibrations to trigger a chemical reaction, we return to the prototypical indole–water dimer, i.e., the breaking of its hydrogen bond. Our calculation takes into account the mid-IR absorption properties of indole–water as well as the experimental parameters defined in the previous sections. It is similar to models used in other IR experiments.28,96 Advanced experimental techniques, such as pulse shaping, which could increase the excitation probability by several orders of magnitude,97 were not included, but could eventually be used to yield higher reaction probabilities as well as to control reaction pathways.The N–H stretch of indole–water at ∼2.9 μm (∼3450 cm−1) has a recorded absorption width of ∼10 cm−1.60 No absolute absorption cross section for indole–water were found in the literature, though typical absorption cross sections for comparable systems are generally on the order of 10−18–10−20 cm2 per molecule.98 The two peaks in the indole–water spectrum at ∼3650 and ∼3750 cm−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 60 correspond to water stretching bands, which have known absorption coefficients of ∼2 × 10−19 cm2.99 Normalizing the spectrum to these peaks suggests that the N–H stretch of indole–water is a “strong” transition with an absorption coefficient of ≳1 × 10−18 cm2.
60 correspond to water stretching bands, which have known absorption coefficients of ∼2 × 10−19 cm2.99 Normalizing the spectrum to these peaks suggests that the N–H stretch of indole–water is a “strong” transition with an absorption coefficient of ≳1 × 10−18 cm2.
We assume the use of an OPA with its output central wavelength matching the center of the N–H-stretch band and with a pulse energy at the sample of ∼70 μJ, i.e., ∼1015 photons per pulse. This pulse energy is representative of commercial OPAs, while taking into account transport losses between the OPA and the sample. The use of sub-50 fs laser pulses corresponds to a ∼500 cm−1 bandwidth. This is significantly wider than the width of the N–H-stretch band and hence only 2% of the photons will have the correct wavelength to be absorbed, i.e., ∼2 × 1013 photons. A typical focal spot size of 110 μm equates to an area of ∼1 × 10−4 cm−2. Together, the above parameters yield an excitation probability for an indole–water molecule of ∼20%.
The excitation probability seems very adequate. Standard femtochemistry experiments typically work with a 2–10% excitation probability as a balance between significant single-photon excitation signals and the minimization of undesirable multi-photon processes. In this light, the predicted value for the mid-IR excitation probability may be considered to be too high. However, it is generally easier to reduce laser intensity than to increase it and thus, importantly, these experiments can be considered clearly feasible with current technology.
Nonetheless, certain experimental features could move this excitation probability to one side or the other. For example, a ∼100 fs laser pulse would allow for a much narrower bandwidth of ∼200 cm−1, yielding a better spectral overlap with the molecular absorption and hence a higher effective number of usable photons. Other molecules or vibrational bands will have smaller absorption cross sections than what was used in the estimate above, corresponding to smaller excitation probabilities. While our calculations suggest that these experiments are feasible, careful planning is still needed to execute them. Properties such as the absorption strengths and the temporal resolution required to observe the dynamics of the system of interest will need to be considered on a case-by-case basis.
Returning to indole–water, appropriate techniques for studying the thermal-energy dynamics need to be carefully considered. Assuming the aforementioned molecular beam density and laser spot size, we expect ∼40 of the ∼200 molecules in our laser-interaction region to be excited and undergo dynamics per shot. Suitably sensitive probes are necessary.
VI. Time-resolved imaging of ultrafast thermal-energy dynamics
When investigating the properties of highly-diluted gases, photoion and photoelectron imaging techniques are at the forefront of experimental techniques for elucidating ultrafast dynamics. We will, therefore, discuss these methods, as well as advanced atomic-resolution imaging, utilizing laser-induced electron diffraction, coherent X-ray diffraction, and electron diffraction, for obtaining time-resolved information of the ultrafast thermal-energy dynamics.A. Ion and electron imaging
Photoion-detection techniques are a powerful tool for identifying energetic and structural changes in gas-phase systems after photoexcitation, with photoion mass spectrometry acting as a valuable first-principle tool. Achievable with relatively simple setups,100 one typically monitors the changes in signal strength of the parent and fragment ions of a system produced in the ionization process as a function of the pump–probe delay. Careful analysis, often guided by computational chemistry, allows one to unravel how the individual atoms or functional groups within a molecule rearrange. This was used to deduce the dynamics of many small molecules7,37,38,101 and protein(-like) structures.18,51 Time-resolved photoion mass spectrometry was also used for studying the mid-IR-induced dissociation of [Fe(CO)5]n by monitoring the time-dependent signal of the Fe(CO)5+ ion.28 Similar tools were utilized to track the dissociation of the indole–water dimer following UV excitation.16Tunable lasers were used to selectively excite and ionize structural isomers.102 This allowed for the determination of the composition of a molecular beam of the dipeptide Ac-Phe-Cys-NH2103 and for monitoring the degree of separation of different conformers after the beam passed through a Stark deflector.77,78 In principle, similar spectroscopic approaches could track the appearance or loss of conformers after mid-IR excitation. However, for ultrafast-dynamics studies the necessarily broad bandwidth of the laser pulses would reduce specificity, as typically the resonances of conformers are spectrally close.
Coulomb-explosion imaging (CEI),33 leading to the complete35 or partial stripping of the electrons of a molecule19,34,104–106 following multiple ionization, by X-ray35,106 or intense-ultrashort-laser-pulse ionization,15,45,104,105,107–109 provided structural information of gas-phase systems. This includes the identification of structural isomers106,110 and individual enantiomers,34,111 and it was used to image nuclear wavepackets in pump–probe experiments.15,45,104,105,107–109,112,113 A combination of mid-IR pump pulses and CEI observations would allow one to track the structural changes in a system undergoing ultrafast thermal-energy dynamics.
Photoelectrons provide information about the intrinsic and dynamic electronic properties of gas-phase molecules. The kinetic energy of electrons emitted by photoionization provides information about the electronic states of molecules.39,114–116 Changes in the photoelectron spectra also allowed for the tracking of structural changes.40,117 Similarly, high-resolution techniques, like zero-kinetic-energy (ZEKE) photoelectron spectroscopy,118 were able to distinguish different conformers.119,120
Photoelectron-momentum-distribution (PEMD) techniques, including velocity-map-imaging spectrometers or so-called reaction microscopes, map the momentum of the emitted electrons and could provide information on the orbital from which the electron is ejected.121,122 Photoelectron circular dichroism (PECD) was used to identify populations of enantiomers and conformers in the gas phase and how this may change over time after photoexcitation.123,124
All the techniques listed above could provide important information on ultrafast structural changes in molecular systems following thermal-energy excitation. However, for a significant number of the techniques discussed in this section, the structural information is largely indirectly inferred through the comparison of experimental results to theoretical models.119,123 CEI conceptionally provides direct structural information. Small-charge CEI can provides information on certain bonds within the axial recoil approximation. High-charge CEI, with practically all atoms individually charged, could conceptually provide direct information on the full molecular structure. It can also provide high-order correlations of nuclear positions. However, for all but the simplest molecules one has to retreat to machine-learning or similar big-data approaches to invert the experimental data to molecular structure.35 Moreover, the approach is limited in molecule size by charge-trapping effects leading to incomplete explosion.125
With these considerations in mind, we turn to diffractive-imaging techniques for the recording of thermal-energy ultrafast dynamics with atomic resolution.
B. Atomic-resolution diffractive imaging
Coherent-diffractive-imaging (CDI) techniques allow one to follow the ultrafast atomic-scale structural changes of a reacting molecule, with ultrafast electron and X-ray diffraction being at the forefront of these methods. Ultrafast electron diffraction (UED),11,14,126,127 performed both with table-top128,129 and accelerator-facility-based sources,10,130,131 proved to be a powerful tool for observing the ultrafast structural dynamics of gas-phase molecules. However, the temporal resolution was limited to ∼100 fs due to charge repulsion between electrons,132 although shorter electron pulses were demonstrated.133 While these temporal limitations currently prevent us from observing the fastest molecular dynamics using UED, the temporal resolution is still sufficient for many important chemical processes at thermal energies which will include significant structural dynamics at picosecond timescales.16X-ray CDI, using ultrashort pulses from XFELs, on the other hand, can already image molecules with sub-100 fs pulses,134 but suffers from significantly smaller scattering cross sections compared to electrons.135 Serial femtosecond crystallography successfully demonstrated the recording of ultrafast dynamics of proteins in nanocrystals.134,136–138 CDI was also demonstrated for small gas-phase molecules,9,139,140 based on access to the molecular frame through the imaging of laser-aligned species.9,141–143 Assuming that analyzable gas-phase diffraction patterns can be captured in hours, if not minutes,36,144 and the recent drive to develop and integrate THz/mid-IR sources at FEL experimental endstations to deliver thermal-energy-scale pump pulses,95 coupled with Stark deflectors for separating out specific molecular species,9 the coherent-diffractive imaging of thermal-energy induced reactions at FELs is closer than ever.
Most gas-phase diffraction experiments worked with sample densities on the order of 1014–1017 molecules per cm3. However, as noted above, the densities provided by controlled molecular beams after passing through the deflector are lower. While a few CDI experiments were performed with these very diluted samples, these were so far limited in signal-to-noise levels.
Laser-induced electron diffraction (LIED) is a very promising table-top-laser-based, sensitive technique42,145–147 for the imaging of ultrafast thermal-energy dynamics. LIED is an extension of PEMD to the rescattering regime, as sketched in Fig. 5. Using intense ultrashort IR laser pulses, molecules can be strong-field ionized. The ejected electron is subsequently accelerated in the laser field, resulting in velocities proportional to the ponderomotive energy, Up of the ionizing laser field.148 However, as the electric field of the laser reverses with respect to the interaction region, a fraction of the electrons are driven back towards the molecular ion to rescatter at energies up to 3.2·Up. The elastically back-scattered electrons yield kinetic energies of up to 10Up149 and ultimately contain diffraction information that can be extracted to provide structural information on the system.42
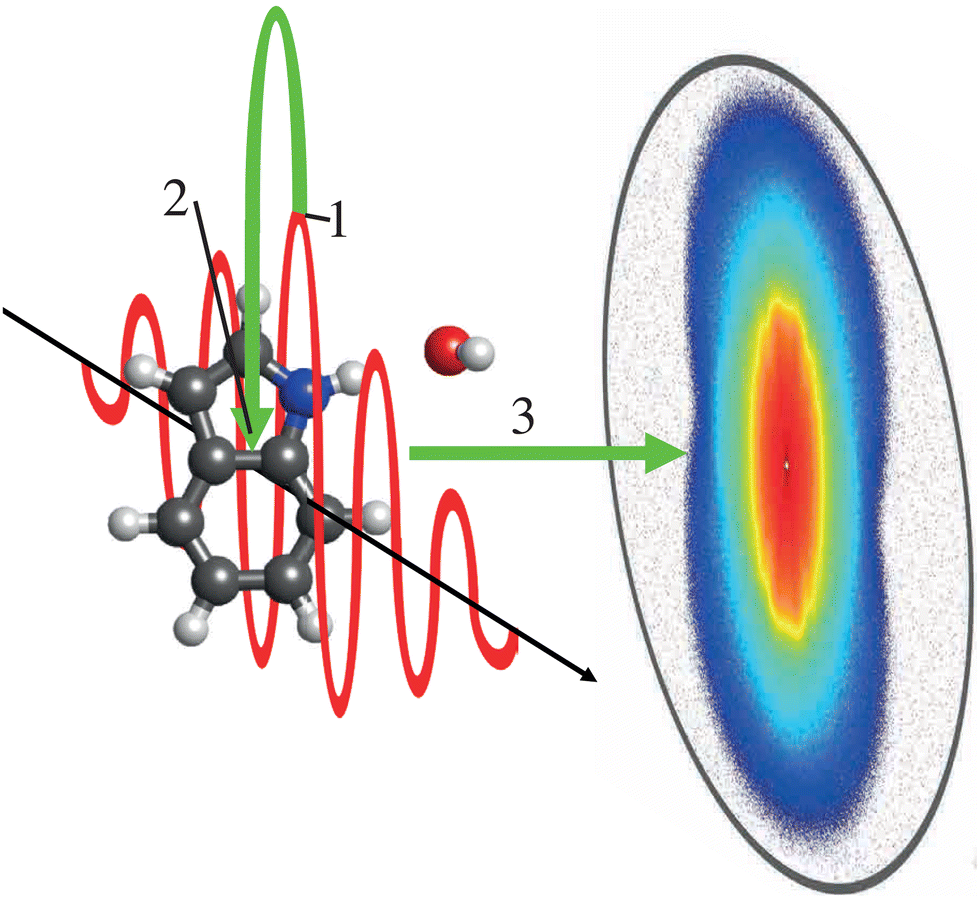 | ||
| Fig. 5 Schematic of the LIED process in indole–water. The strong electric field of the laser (red) initiates ionization of the system and ejection of an electron (green) at times around peak intensity (1). As the electric field changes direction, (2) the electron can return to its ion and scatter. Using a velocity-map imaging (VMI) apparatus, the electron momenta can (3) be imaged to produce an photoelectron diffraction pattern such as the shown preliminary experimental data. | ||
On the scale of diffractive-imaging experiments, the returning electrons have relatively low energies, typically on the order of a few 100 eV, yielding large scattering cross sections – making it a powerful technique for looking at low-density gas targets – but still have a small enough de Broglie wavelength to allow for few-picometer-resolved structure retrieval.44 The technique was already used to image a number of small molecules.42,44,46,47,150 As the electron typically returns within a single cycle of the ionizing laser, the technique has the potential to image molecular structure on the few-femtosecond timescale42 or even electronic dynamics.43,147
However, while LIED is, in principle, single-molecule sensitive, the signal of the backscattered electrons is significantly weaker, by several orders of magnitude, compared to the so-called “direct” electrons.151 This leads to experiments needing typically 104–108 laser pulses to produce a single diffraction pattern,44,145,152i.e., a data collection time between ∼10 s and ∼1 day using a 1 kHz laser. Another consideration is the ionization energy of the molecular system of interest: when the photon energy of the ionizing laser is on the same order as, or greater than, the ionization energy of the molecule, single-photon or few-photon ionization processes will be favored over strong-field-tunneling processes. This ultimately limits the effectiveness of the technique, especially as one moves to image larger systems, which typically posses smaller ionization energies. However, this can be mitigated by advanced analysis techniques153 or longer-wavelength laser fields.
Moreover, the application of LIED to pump–probe “femtochemistry”2 experiments is hindered by the strongly varying ionization energies while the system traverses different electronic states.147 With thermal-energy dynamics, one can expect small changes in the ionization energy of the system of interest, suggesting that the efficiency of the tunnel-ionization process, which is intrinsically linked to the Keldysh parameter,154,155 will remain relatively constant. Conversely, as near-/mid-IR light is typically used to perform the strong-field ionization step, careful consideration of the wavelength used for LIED must be made to ensure that the probe does not inadvertently induce or alter the dynamics in the system itself.
To demonstrate the power of diffraction techniques for monitoring thermal-energy-induced ultrafast structural dynamics, we simulated electron-diffraction signals for the thermal-energy anti–syn-conformer conversion of CPCA, cf.Fig. 3, shown in Fig. 6. The simulation was performed for experimental parameters, specified in Fig. 6, corresponding to a typical UED apparatus156 and an isotropic-gas sample. The predicted diffraction patterns were radially averaged about the center of the detector. One can expect similar results if the dynamics were studied using X-ray diffraction or LIED.
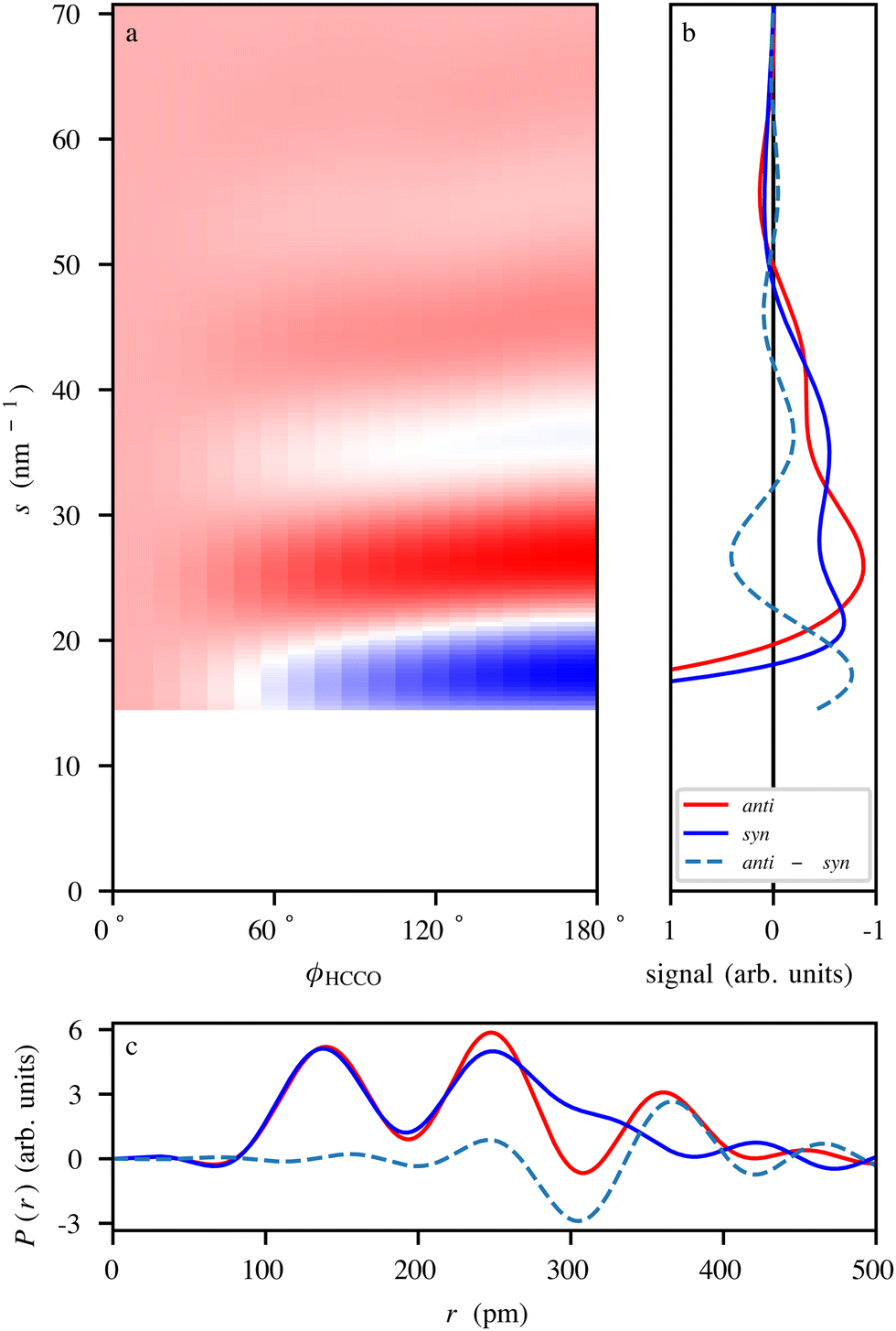 | ||
| Fig. 6 (a) Simulated electron-diffraction-difference pattern of CPCA as it undergoes the anti–syn-conformer change, presented as function of the ϕHCCO dihedral angle. The difference signal was obtained by subtracting the anti-CPCA signal from the individual ϕHCCO angle-dependent signals. The simulation assumed the use of a UED setup operating at an electron energy of 3.7 MeV, with a 3 m sample-to-detector distance, and a 2 mm radius hole in the center of the detector.156 (b) Simulated scattering curves for anti- and syn-CPCA as well as the difference signal. (c) Simulated radial distribution curves of anti and syn-CPCA obtained through the Fourier transform of the data in (b). | ||
Fig. 6a shows the differences in the radial scattering signal with respect to the anti-conformer as a function of the ϕHCCO dihedral angle. Fig. 6b shows the individual radial scattering signals for the anti- and syn-conformers, shown in Fig. 3, as well as the difference signal between the two. The data in both panels display clear signal changes between the conformational structures that are well within the experimentally demonstrated resolution limits.10,131 Changes in atomic distances are clearly visible in the structural data in Fig. 6c obtained by Fourier transforming the diffraction data. For example, the peak at ∼360 pm for anti-CPCA moves to become a shoulder at ∼330 pm for syn-CPCA. This relates to the shortening of the C–O distance as the CO group moves atop the cyclopropane ring. Given that the rotation is expected to occur on a timescale of ∼100 ps,50 similar to the actual indole–water bond-breaking timescale,16vide supra, all CDI methods discussed here should be able to capture such structural dynamics.
VII. Mimicking thermal chemistry
We note that the dynamics of isolated gas-phase systems is not identical to thermal reactions in gases or the condensed phase. However, we point out that there is sufficient similarity that the insights gained from the experiments proposed here will strongly benefit our understanding of truly thermal chemistry. Moreover, in any case the benchmarking of computational chemistry approaches with precision data from the isolated systems will directly benefit predictions for condensed-phase reactions.Regarding condensed-phase chemistry, e.g., in aqueous solution, one surely has to consider the influence of the solvent. In the isolated-molecule approach, this can be partly built up using microsolvation, i.e., investigations of the reaction while adding water molecules one by one.16 Furthermore, even in solution there are hierarchies of interactions, with solvation effects often being weak and thus slow couplings. The indole(H2O) system discussed above is in fact a good example for indole in aqueous solution,59 highlighting the hierarchy in that case. Therefore, especially the ultrafast first steps of a chemical reaction, in solution, might often not be strongly influenced by the weak interactions due to solvation and thus can be mimicked by the dynamics of the, possibly microsolvated, isolated system.
Furthermore, it is intriguing to consider the presence and the effect of coherence in these processes. For thermal chemistry, one would possibly consider the system, at any given time, to be in an eigenstate, whereas the excitation with an ultrashort laser pulse would routinely be ascribed to trigger wavepacket dynamics. However, the difference could be quite small. As an example, we consider the excitation of the N–H stretch vibration in indole(H2O) in a molecular beam at ∼3400 cm−1.60 One could describe this as an excitation of a single vibrational eigenstate, as no other bright modes exist in the wavenumber range ∼3200–3650 cm−1. However, the shift of the excitation line compared to bare indole60 highlights that this N–H-stretch vibration is significantly coupled to other vibrations, i.e., combination modes likely including various of the low-wavenumber intermolecular vibrations. Similar couplings would also exist in the modes leading to conformational rearrangements discussed in Section II. Ascribing this to the excitation of a wavepacket of a set of coupled modes with intensity-borrowing from the N–H stretch is essentially identical to the excitation of an eigenstate of the NH-stretch vibration with subsequent ultrafast energy flow into coupled dark modes.157 The same holds for the corresponding excitations due to statistical fluctuations in the thermal solution-phase system.
The coherence times in the condensed phase will be shorter than in the isolated gas-phase system, as the solvation bath will always provide additional couplings. Considering coherence times as defined by the time-dependent autocorrelation function, for instance, of the excited N–H-stretch mode, indicates significant correspondence between the microsolvated gas-phase system and the thermal condensed-phase system.
These arguments are not valid in the case of broad wavepackets produced by simultaneous coherent excitation of multiple quanta of bright modes, e.g., v = 1, 2,… of the N–H-stretch, which would be a different approach and possibly also open up control of chemical reactivity.
VIII. Conclusion
The imaging of ultrafast elementary steps of thermal-energy chemical dynamics with atomic resolution is within reach, thanks to advances in both techniques for preparing pure, controlled samples and mid-infrared laser technology. Over the next few years, we expect to see experiments that utilize ultrashort mid-IR laser pulses to initiate ground-state thermal-energy dynamics within gas-phase systems and, in first instances, to be probed with ion- and electron-imaging techniques. Moreover, lab- and facility-based diffraction techniques will provide opportunities to monitor these thermal-energy dynamics with atomic resolution. While early work will likely focus on smaller and highly-controllable systems, like indole–water or CPCA, the techniques and methods detailed here are general and will be extended to the imaging of larger molecular systems.Beyond imaging the structural dynamics of individual elementary steps of thermal-energy chemistry, datasets of a variety of molecular systems and reactions will enable application of these time-resolved discoveries to further disentangle general chemistry and everyday processes. This includes the benchmarking of computational models and enabling their advance as well as building up correlations regarding decoherence times and statistical descriptions. It might eventually allow us to extract the key reaction coordinates of chemical processes at, for example, transition states, thus furthering our conceptional understanding of general chemical-reaction dynamics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We kindly thank Carl Trindle, Zikri Altun, and Erdi Ata Bleda for providing the structural coordinates of CPCA from ref. 65. We thank Joss Wiese, Jolijn Onvlee, and Sebastian Trippel for their permission to use the unpublished experimental photoelectron pattern of indole–water used in Fig. 5. This work was supported by Deutsches Elektronen-Synchrotron DESY, a member of the Helmholtz Association (HGF), and the Cluster of Excellence “Advanced Imaging of Matter” of the Deutsche Forschungsgemeinschaft (DFG) (AIM, EXC 2056, ID 390715994).References
- A. H. Zewail, Laser femtochemistry, Science, 1988, 242, 1645–1653, DOI:10.1126/science.242.4886.1645.
- A. H. Zewail, Femtochemistry: Atomic-scale dynamics of the chemical bond, J. Phys. Chem. A, 2000, 104, 5660–5694, DOI:10.1021/jp001460h.
- K. F. Chang, M. Reduzzi, H. Wang, S. M. Poullain, Y. Kobayashi, L. Barreau, D. Prendergast, D. M. Neumark and S. R. Leone, Revealing electronic state-switching at conical intersections in alkyl iodides by ultrafast XUV transient absorption spectroscopy, Nat. Commun., 2020, 11, 4042, DOI:10.1038/s41467-020-17745-w , https://arxiv.org/abs/2005.00668.
- Y. Pertot, C. Schmidt, M. Matthews, A. Chauvet, M. Huppert, V. Svoboda, A. von Conta, A. Tehlar, D. Baykusheva, J.-P. Wolf and H. J. Wörner, Time-resolved X-ray absorption spectroscopy with a water window high-harmonic source, Science, 2017, 355, 264–267, DOI:10.1126/science.aah6114.
- Z.-H. Loh and S. R. Leone, Ultrafast strong-field dissociative ionization dynamics of CH2Br2 probed by femtosecond soft X-ray transient absorption spectroscopy, J. Chem. Phys., 2008, 128, 204302, DOI:10.1063/1.2925268.
- T. J. A. Wolf, R. M. Parrish, R. H. Myhre, T. J. Martínez, H. Koch and M. Gühr, Observation of ultrafast intersystem crossing in thymine by extreme ultraviolet time-resolved photoelectron spectroscopy, J. Phys. Chem. A, 2019, 123, 6897–6903, DOI:10.1021/acs.jpca.9b05573.
- T. Baumert, B. Bühler, R. Thalweiser and G. Gerber, Femtosecond spectroscopy of molecular autoionization and fragmentation, Phys. Rev. Lett., 1990, 64, 733–736, DOI:10.1103/physrevlett.64.733.
- D. H. Paik, T. M. Bernhardt, N. J. Kim and A. H. Zewail, Femtochemistry of mass-selected negative-ion clusters of dioxygen: Charge-transfer and solvation dynamics, J. Chem. Phys., 2001, 115, 612–616, DOI:10.1063/1.1384549.
- J. Küpper, S. Stern, L. Holmegaard, F. Filsinger, A. Rouzée, A. Rudenko, P. Johnsson, A. V. Martin, M. Adolph, A. Aquila, S. Bajt, A. Barty, C. Bostedt, J. Bozek, C. Caleman, R. Coffee, N. Coppola, T. Delmas, S. Epp, B. Erk, L. Foucar, T. Gorkhover, L. Gumprecht, A. Hartmann, R. Hartmann, G. Hauser, P. Holl, A. Hömke, N. Kimmel, F. Krasniqi, K.-U. Kühnel, J. Maurer, M. Messerschmidt, R. Moshammer, C. Reich, B. Rudek, R. Santra, I. Schlichting, C. Schmidt, S. Schorb, J. Schulz, H. Soltau, J. C. H. Spence, D. Starodub, L. Strüder, J. Thøgersen, M. J. J. Vrakking, G. Weidenspointner, T. A. White, C. Wunderer, G. Meijer, J. Ullrich, H. Stapelfeldt, D. Rolles and H. N. Chapman, X-ray diffraction from isolated and strongly aligned gas-phase molecules with a free-electron laser, Phys. Rev. Lett., 2014, 112, 083002, DOI:10.1103/PhysRevLett.112.083002 , https://arxiv.org/abs/1307.4577.
- J. Yang, X. Zhu, T. J. A. Wolf, Z. Li, J. P. F. Nunes, R. Coffee, J. P. Cryan, M. Gühr, K. Hegazy, T. F. Heinz, K. Jobe, R. Li, X. Shen, T. Veccione, S. Weathersby, K. J. Wilkin, C. Yoneda, Q. Zheng, T. J. Martínez, M. Centurion and X. Wang, Imaging CF3I conical intersection and photodissociation dynamics with ultrafast electron diffraction, Science, 2018, 361, 64–67, DOI:10.1126/science.aat0049.
- G. Sciaini and R. J. D. Miller, Femtosecond electron diffraction: Heralding the era of atomically resolved dynamics, Rep. Prog. Phys., 2011, 74, 096101, DOI:10.1088/0034-4885/74/9/096101.
- A. Barty, S. Boutet, M. J. Bogan, S. Hau-Riege, S. Marchesini, K. Sokolowski-Tinten, N. Stojanovic, R. Tobey, H. Ehrke, A. Cavalleri, S. Düsterer, M. Frank, S. Bajt, B. W. Woods, M. M. Seibert, J. Hajdu, R. Treusch and H. N. Chapman, Ultrafast single-shot diffraction imaging of nanoscale dynamics, Nat. Photonics, 2008, 2, 415–419, DOI:10.1038/nphoton.2008.128.
- R. J. D. Miller, R. Ernstorfer, M. Harb, M. Gao, C. T. Hebeisen, H. Jean-Ruel, C. Lu, G. Moriena and G. Sciaini, ‘Making the molecular movie’: First frames, Acta Crystallogr., 2010, 66, 137–156, DOI:10.1107/S0108767309053926.
- J. C. Williamson, J. M. Cao, H. Ihee, H. Frey and A. H. Zewail, Clocking transient chemical changes by ultrafast electron diffraction, Nature, 1997, 386, 159–162, DOI:10.1038/386159a0.
- H. Stapelfeldt, E. Constant and P. B. Corkum, Wave-packet structure and dynamics measured by Coulomb explosion, Phys. Rev. Lett., 1995, 74, 3780–3783, DOI:10.1103/PhysRevLett.74.3780.
- J. Onvlee, S. Trippel and J. Küpper, Ultrafast light-induced dynamics in solvated biomolecules: The indole chromophore with water, Nat. Commun., 2022, 13, 7462, DOI:10.1038/s41467-022-33901-w , https://arxiv.org/abs/2103.07171.
- C. S. Lehmann, A. Picón, C. Bostedt, A. Rudenko, A. Marinelli, D. Moonshiram, T. Osipov, D. Rolles, N. Berrah, C. Bomme, M. Bucher, G. Doumy, B. Erk, K. R. Ferguson, T. Gorkhover, P. J. Ho, E. P. Kanter, B. Krässig, J. Krzywinski, A. A. Lutman, A. M. March, D. Ray, L. Young, S. T. Pratt and S. H. Southworth, Ultrafast X-ray-induced nuclear dynamics in diatomic molecules using femtosecond X-ray-pump-X-ray-probe spectroscopy, Phys. Rev. A, 2016, 94, 013426, DOI:10.1103/PhysRevA.94.013426.
- W. Li, O. Kavatsyuk, W. Douma, X. Wang, R. Hoekstra, D. Mayer, M. S. Robinson, M. Gühr, M. Lalande, M. Abdelmouleh, M. Ryszka, J.-C. Poully and T. Schlathölter, Multiple valence electron detachment following Auger decay of inner-shell vacancies in gas-phase DNA, Chem. Sci., 2021, 12, 13177–13186, 10.1039/D1SC02885E.
- M. Johny, C. A. Schouder, A. Al-Refaie, L. He, J. Wiese, H. Stapelfeldt, S. Trippel and J. Küpper, Molecular sunscreen: water protects pyrrole from radiation damage, 2020, submitted, https://arxiv.org/abs/2010.00453.
- A. Rudenko, V. Makhija, A. Vajdi, T. Ergler, M. Schürholz, R. K. Kushawaha, J. Ullrich, R. Moshammer and V. Kumarappan, Strong-field-induced wave packet dynamics in carbon dioxide molecule, Faraday Discuss., 2016, 194, 463–478, 10.1039/c6fd00152a.
- A. Reissert, Einwirkung von Oxalester und Natriumäthylat auf Nitrotoluole. Synthese nitrirter Phenylbrenztraubensäuren, Ber. Dtsch. Chem. Ges., 1897, 30, 1030–1053, DOI:10.1002/cber.189703001200.
- P. Hamm and M. Zanni, Concepts and Methods of 2D Infrared Spectroscopy, Cambridge University Press, 2011 DOI:10.1017/CBO9780511675935.
- R. Fritzsch, S. Hume, L. Minnes, M. J. Baker, G. A. Burley and N. T. Hunt, Two-dimensional infrared spectroscopy: An emerging analytical tool? Analyst, 2020, 145, 2014–2024 10.1039/C9AN02035G.
- L. Minnes, D. J. Shaw, B. P. Cossins, P. M. Donaldson, G. M. Greetham, M. Towrie, A. W. Parker, M. J. Baker, A. J. Henry, R. J. Taylor and N. T. Hunt, Quantifying secondary structure changes in calmodulin using 2D-IR spectroscopy, Anal. Chem., 2017, 89, 10898–10906, DOI:10.1021/acs.analchem.7b02610.
- P. J. M. Johnson, K. L. Koziol and P. Hamm, Quantifying biomolecular recognition with site-specific 2D infrared probes, J. Phys. Chem. Lett., 2017, 8, 2280–2284, DOI:10.1021/acs.jpclett.7b00742.
- R. Schanz, V. Boţan and P. Hamm, A femtosecond study of the infrared-driven cis-trans isomerization of nitrous acid (HONO), J. Chem. Phys., 2005, 122, 044509, DOI:10.1063/1.1834567.
- T. Stensitzki, Y. Yang, V. Kozich, A. A. Ahmed, F. Kössl, O. Köhn and K. Heyne, Acceleration of a ground-state reaction by selective femtosecond-infrared-laser-pulse excitation, Nat. Chem., 2018, 10, 126–131, DOI:10.1038/nchem.2909.
- D. G. Poydashev, V. N. Lokhman, V. O. Kompanets, S. V. Chekalin and E. A. Ryabov, Ultrafast dissociation dynamics of [Fe(CO)5]n clusters induced by femtosecond IR radiation, J. Phys. Chem. A, 2014, 118, 11177–11184, DOI:10.1021/jp510130x.
- G. Cerullo and S. De Silvestri, Ultrafast optical parametric amplifiers, Rev. Sci. Instrum., 2003, 74, 1–18, DOI:10.1063/1.1523642.
- A. G. Ciriolo, M. Negro, M. Devetta, E. Cinquanta, D. Faccialà, A. Pusala, S. De Silvestri, S. Stagira and C. Vozzi, Optical parametric amplification techniques for the generation of high-energy few-optical-cycles IR pulses for strong field applications, Appl. Sci., 2017, 7, 265, DOI:10.3390/app7030265.
- Y.-P. Chang, D. A. Horke, S. Trippel and J. Küpper, Spatially-controlled complex molecules and their applications, Int. Rev. Phys. Chem., 2015, 34, 557–590, DOI:10.1080/0144235X.2015.1077838 , https://arxiv.org/abs/1505.05632.
- S. Trippel, M. Johny, T. Kierspel, J. Onvlee, H. Bieker, H. Ye, T. Mullins, L. Gumprecht, K. Długołȩcki and J. Küpper, Knife edge skimming for improved separation of molecular species by the deflector, Rev. Sci. Instrum., 2018, 89, 096110, DOI:10.1063/1.5026145 , https://arxiv.org/abs/1802.04053.
- Z. Vager, R. Naaman and E. P. Kanter, Coulomb explosion imaging of small molecules, Science, 1989, 244, 426–431, DOI:10.1126/science.244.4903.426.
- M. Pitzer, M. Kunitski, A. S. Johnson, T. Jahnke, H. Sann, F. Sturm, L. P. H. Schmidt, H. Schmidt-Böcking, R. Dörner, J. Stohner, J. Kiedrowski, M. Reggelin, S. Marquardt, A. Schießer, R. Berger and M. S. Schöffler, Direct determination of absolute molecular stereochemistry in gas phase by Coulomb explosion imaging, Science, 2013, 341, 1096–1100, DOI:10.1126/science.1240362.
- R. Boll, J. M. Schäfer, B. Richard, K. Fehre, G. Kastirke, Z. Jurek, M. S. Schöffler, M. M. Abdullah, N. Anders, T. M. Baumann, S. Eckart, B. Erk, A. De Fanis, R. Dörner, S. Grundmann, P. Grychtol, A. Hartung, M. Hofmann, M. Ilchen, L. Inhester, C. Janke, R. Jin, M. Kircher, K. Kubicek, M. Kunitski, X. Li, T. Mazza, S. Meister, N. Melzer, J. Montano, V. Music, G. Nalin, Y. Ovcharenko, C. Passow, A. Pier, N. Rennhack, J. Rist, D. E. Rivas, D. Rolles, I. Schlichting, L. P. H. Schmidt, P. Schmidt, J. Siebert, N. Strenger, D. Trabert, F. Trinter, I. Vela-Perez, R. Wagner, P. Walter, M. Weller, P. Ziolkowski, S.-K. Son, A. Rudenko, M. Meyer, R. Santra and T. Jahnke, X-ray multiphoton-induced Coulomb explosion images complex single molecules, Nat. Phys., 2022, 18, 423–428, DOI:10.1038/s41567-022-01507-0.
- A. Barty, J. Küpper and H. N. Chapman, Molecular imaging using X-ray free-electron lasers, Annu. Rev. Phys. Chem., 2013, 64, 415–435, DOI:10.1146/annurev-physchem-032511-143708.
- M. Dantus, M. H. M. Janssen and A. H. Zewail, Femtosecond probing of molecular dynamics by mass-spectrometry in a molecular beam, Chem. Phys. Lett., 1991, 181, 281–287, DOI:10.1016/0009-2614(91)80071-5.
- D. Zhong and A. H. Zewail, Femtosecond real-time probing of reactions. 23. Studies of temporal, velocity, angular, and state dynamics from transition states to final products by femtosecond-resolved mass spectrometry, J. Phys. Chem. A, 1998, 102, 4031–4058, DOI:10.1021/jp9805196.
- A. Stolow, A. E. Bragg and D. M. Neumark, Femtosecond time-resolved photoelectron spectroscopy, Chem. Rev., 2004, 104, 1719–1757, DOI:10.1021/cr020683w.
- T. Baumert, T. Frohnmeyer, B. Kiefer, P. Niklaus, M. Strehle, G. Gerber and A. H. Zewail, Femtosecond transition state dynamics of cis-stilbene, Appl. Phys. B: Lasers Opt., 2001, 72, 105–108, DOI:10.1007/s003400000497.
- S. G. Walt, N. B. Ram, M. Atala, N. I. Shvetsov-Shilovski, A. von Conta, D. Baykusheva, M. Lein and H. J. Wörner, Dynamics of valence-shell electrons and nuclei probed by strong-field holography and rescattering, Nat. Commun., 2017, 8, 15651, DOI:10.1038/ncomms15651.
- C. I. Blaga, J. Xu, A. D. DiChiara, E. Sistrunk, K. Zhang, P. Agostini, T. A. Miller, L. F. DiMauro and C. D. Lin, Imaging ultrafast molecular dynamics with laser-induced electron diffraction, Nature, 2012, 483, 194–197, DOI:10.1038/nature10820.
- A. Trabattoni, J. Wiese, U. De Giovannini, J.-F. Olivieri, T. Mullins, J. Onvlee, S.-K. Son, B. Frusteri, A. Rubio, S. Trippel and J. Küpper, Setting the photoelectron clock through molecular alignment, Nat. Commun., 2020, 11, 2546, DOI:10.1038/s41467-020-16270-0 , https://arxiv.org/abs/1802.06622.
- E. T. Karamatskos, G. Goldsztejn, S. Raabe, P. Stammer, T. Mullins, A. Trabattoni, R. R. Johansen, H. Stapelfeldt, S. Trippel, M. J. J. Vrakking, J. Küpper and A. Rouzée, Atomic-resolution imaging of carbonyl sulfide by laser-induced electron diffraction, J. Chem. Phys., 2019, 150, 244301, DOI:10.1063/1.5093959 , https://arxiv.org/abs/1905.03541.
- E. T. Karamatskos, S. Raabe, T. Mullins, A. Trabattoni, P. Stammer, G. Goldsztejn, R. R. Johansen, K. Długołȩcki, H. Stapelfeldt, M. J. J. Vrakking, S. Trippel, A. Rouzée and J. Küpper, Molecular movie of ultrafast coherent rotational dynamics of OCS, Nat. Commun., 2019, 10, 3364, DOI:10.1038/s41467-019-11122-y , https://arxiv.org/abs/1807.01034.
- B. Wolter, M. G. Pullen, A.-T. Le, M. Baudisch, K. Doblhoff-Dier, A. Senftleben, M. Hemmer, C. D. Schroter, J. Ullrich, T. Pfeifer, R. Moshammer, S. Gräfe, O. Vendrell, C. D. Lin and J. Biegert, Ultrafast electron diffraction imaging of bond breaking in di-ionized acetylene, Science, 2016, 354, 308–312, DOI:10.1126/science.aah3429.
- Y. Ito, C. Wang, A.-T. Le, M. Okunishi, D. Ding, C. D. Lin and K. Ueda, Extracting conformational structure information of benzene molecules via laser-induced electron diffraction, Struct. Dyn., 2016, 3, 034303, DOI:10.1063/1.4952602.
- B. C. Dian, A. Longarte and T. S. Zwier, Conformational dynamics in a dipeptide after single-mode vibrational excitation, Science, 2002, 296, 2369–2373, DOI:10.1126/science.1071563.
- B. C. Dian, J. R. Clarkson and T. S. Zwier, Direct measurement of energy thresholds to conformational isomerization in tryptamine, Science, 2004, 303, 1169–1173, DOI:10.1126/science.1093731.
- B. C. Dian, G. G. Brown, K. O. Douglass and B. H. Pate, Measuring picosecond isomerization kinetics via broadband microwave spectroscopy, Science, 2008, 320, 924–928, DOI:10.1126/science.1155736.
- V. Gabelica, F. Rosu, E. De Pauw, J. Lemaire, J.-C. Gillet, J.-C. Poully, F. Lecomte, G. Grégoire, J.-P. Schermann and C. Desfrançois, Infrared signature of DNA G-quadruplexes in the gas phase, J. Am. Chem. Soc., 2008, 130, 1810–1811, DOI:10.1021/ja077321w.
- L. Oudejans and R. E. Miller, Photofragment translational spectroscopy of weakly bound complexes: Probing the interfragment correlated final state distributions, Annu. Rev. Phys. Chem., 2001, 52, 607–637, DOI:10.1146/annurev.physchem.52.1.607.
- L. I. Yeh, M. Okumura, J. D. Myers, J. M. Price and Y. T. Lee, Vibrational spectroscopy of the hydrated hydronium cluster ions H3O+.(H2O)n (n = 1, 2, 3), J. Chem. Phys., 1989, 91, 7319–7330, DOI:10.1063/1.457305.
- T. M. Korter, D. W. Pratt and J. Küpper, Indole-H2O in the gas phase. Structures, barriers to internal motion, and S1 ← S0 transition moment orientation. Solvent reorganization in the electronically excited state, J. Phys. Chem. A, 1998, 102, 7211–7216, DOI:10.1021/jp982456x.
- J. T. Vivian and P. R. Callis, Mechanisms of tryptophan fluorescence shifts in proteins, Biophys. J., 2001, 80, 2093–2109, DOI:10.1016/S0006-3495(01)76183-8.
- V. Volkov and P. Hamm, A two-dimensional infrared study of localization, structure, and dynamics of a dipeptide in membrane environment, Biophys. J., 2004, 87, 4213–4225, DOI:10.1529/biophysj.104.045435.
- S. Bagchi, Y. S. Kim, A. K. Charnley, A. B. Smith III and R. M. Hochstrasser, Two-dimensional infrared investigation of N-acetyl tryptophan methyl amide in solution, J. Phys. Chem. B, 2007, 111, 3010–3018, DOI:10.1021/jp067348m.
- S. Trippel, Y.-P. Chang, S. Stern, T. Mullins, L. Holmegaard and J. Küpper, Spatial separation of state- and size-selected neutral clusters, Phys. Rev. A: At., Mol., Opt. Phys., 2012, 86, 033202, DOI:10.1103/PhysRevA.86.033202 , https://arxiv.org/abs/1208.4935.
- L. He, S. Malerz, F. Trinter, S. Trippel, L. Tomaník, M. Belina, P. Slavíček, B. Winter and J. Küpper, Specific versus non-specific solvent interactions of a biomolecule in water, J. Phys. Chem. Lett., 2023, 14, 10499–10508, DOI:10.1021/acs.jpclett.3c01763 , https://arxiv.org/abs/2205.08217.
- J. R. Carney and T. S. Zwier, Infrared and ultraviolet spectroscopy of water-containing clusters of indole, 1-methylindole, and 3-methylindole, J. Phys. Chem. A, 1999, 103, 9943–9957, DOI:10.1021/jp992222t.
- M. Mons, I. Dimicoli, B. Tardivel, F. Piuzzi, V. Brenner and P. Millié, Site dependence of the binding energy of water to indole: Microscopic approach to the side chain hydration of tryptophan, J. Phys. Chem. A, 1999, 103, 9958–9965, DOI:10.1021/jp992285b.
- A. G. Suits, Roaming reactions and dynamics in the van der Waals region, Annu. Rev. Phys. Chem., 2020, 71, 77–100, DOI:10.1146/annurev-physchem-050317-020929.
- T. Dartigalongue and F. Hache, Ultrafast conformational changes in carboxymyoglobin studied by time-resolved circular dichroism, in Ultrafast Phenomena XV, ed. P. Corkum, D. M. Jonas, R. J. D. Miller and A. M. Weiner, Springer Verlag, Berlin, Heidelberg, 2007, pp. 495–497 DOI:10.1007/978-3-540-68781-8_160.
- O. A. Sytina, D. J. Heyes, C. N. Hunter, M. T. Alexandre, I. H. M. Van Stokkum, R. Van Grondelle and M. L. Groot, Conformational changes in an ultrafast light-driven enzyme determine catalytic activity, Nature, 2008, 456, 1001–1004, DOI:10.1038/nature07354.
- C. Trindle, E. A. Bleda and Z. Altun, Structure and energetics of cyclopropane carboxaldehyde, Int. J. Quantum Chem., 2013, 113, 1155–1161, DOI:10.1002/qua.24201.
- J. Oomens, N. Polfer, D. T. Moore, L. van der Meer, A. G. Marshall, J. R. Eyler, G. Meijer and G. von Helden, Charge-state resolved mid-infrared spectroscopy of a gas-phase protein, Phys. Chem. Chem. Phys., 2005, 7, 1345–1348, 10.1039/B502322J.
- N. C. Polfer, Infrared multiple photon dissociation spectroscopy of trapped ions, Chem. Soc. Rev., 2011, 40, 2211–2221, 10.1039/c0cs00171f.
- B. K. Carpenter, Nonstatistical dynamics in thermal reactions of polyatomic molecules, Annu. Rev. Phys. Chem., 2005, 56, 57–89, DOI:10.1146/annurev.physchem.56.092503.141240.
- B. Jayee and W. L. Hase, Nonstatistical reaction dynamics, Annu. Rev. Phys. Chem., 2020, 71, 289–313, DOI:10.1146/annurev-physchem-112519-110208.
- D. Townsend, S. A. Lahankar, S. K. Lee, S. D. Chambreau, A. G. Suits, X. Zhang, J. Rheinecker, L. B. Harding and J. M. Bowman, The roaming atom: Straying from the reaction path in formaldehyde decomposition, Science, 2004, 306, 1158–1161, DOI:10.1126/science.1104386.
- Y.-P. Chang, K. Długołȩcki, J. Küpper, D. Rösch, D. Wild and S. Willitsch, Specific chemical reactivities of spatially separated 3-aminophenol conformers with cold Ca+ ions, Science, 2013, 342, 98–101, DOI:10.1126/science.1242271 , https://arxiv.org/abs/1308.6538.
- D. H. Levy, The spectroscopy of very cold gases, Science, 1981, 214, 263–269, DOI:10.1126/science.214.4518.263.
- Atomic and molecular beam methods, ed. G. Scoles, Oxford University Press, New York, NY, USA, vol. 1 & 2, 1988 & 1992 Search PubMed.
- M. Johny, J. Onvlee, T. Kierspel, H. Bieker, S. Trippel and J. Küpper, Spatial separation of pyrrole and pyrrole-water clusters, Chem. Phys. Lett., 2019, 721, 149–152, DOI:10.1016/j.cplett.2019.01.052 , https://arxiv.org/abs/1901.05267.
- H. Bieker, J. Onvlee, M. Johny, L. He, T. Kierspel, S. Trippel, D. A. Horke and J. Küpper, Pure molecular beam of water dimer, J. Phys. Chem. A, 2019, 123, 7486–7490, DOI:10.1021/acs.jpca.9b06460 , https://arxiv.org/abs/1904.08716.
- F. Filsinger, U. Erlekam, G. von Helden, J. Küpper and G. Meijer, Selector for structural isomers of neutral molecules, Phys. Rev. Lett., 2008, 100, 133003, DOI:10.1103/PhysRevLett.100.133003 , https://arxiv.org/abs/0802.2795.
- F. Filsinger, J. Küpper, G. Meijer, J. L. Hansen, J. Maurer, J. H. Nielsen, L. Holmegaard and H. Stapelfeldt, Pure samples of individual conformers: The separation of stereo-isomers of complex molecules using electric fields, Angew. Chem., Int. Ed., 2009, 48, 6900–6902, DOI:10.1002/anie.200902650.
- N. Teschmit, D. A. Horke and J. Küpper, Spatially separating the conformers of a dipeptide, Angew. Chem., Int. Ed., 2018, 57, 13775–13779, DOI:10.1002/anie.201807646 , https://arxiv.org/abs/1805.12396.
- F. Hillenkamp, M. Karas, R. C. Beavis and B. T. Chait, Matrix-assisted laser desorption/ionization mass spectrometry of biopolymers, Anal. Chem., 1991, 63, 1193A–1203A, DOI:10.1021/ac00024a716.
- J. R. Cable, M. J. Tubergen and D. H. Levy, Laser desorption molecular beam spectroscopy: The electronic spectra of tryptophan peptides in the gas phase, J. Am. Chem. Soc., 1987, 109, 6198–6199, DOI:10.1021/ja00254a057.
- G. Meijer, M. S. de Vries, H. E. Hunziker and H. R. Wendt, Laser desorption jet-cooling of organic molecules - cooling characteristics and detection sensitivity, Appl. Phys. B: Photophys. Laser Chem., 1990, 51, 395–403, DOI:10.1007/BF00329101.
- J. W. Elam and D. H. Levy, Ultraviolet laser desorption of indole, J. Chem. Phys., 1997, 106, 10368–10378, DOI:10.1063/1.474071.
- M. Karas and F. Hillenkamp, Laser desorption ionization of proteins with molecular masses exceeding 10000 daltons, Anal. Chem., 1988, 60, 2299–2301, DOI:10.1021/ac00171a028.
- J. B. Fenn, M. Mann, C. K. Meng, S. F. Wong and C. M. Whitehouse, Electrospray ionization for mass-spectrometry of large biomolecules, Science, 1989, 246, 64–71, DOI:10.1126/science.2675315.
- J. Lübke, Control of Bionanoparticles with Electric Fields, Dissertation, Universität Hamburg, Hamburg, Germany, 2022, submitted, https://ediss.sub.uni-hamburg.de/handle/ediss/10012 Search PubMed.
- K. J. Pacholarz, R. A. Garlish, R. J. Taylor and P. E. Barran, Mass spectrometry based tools to investigate protein-ligand interactions for drug discovery, Chem. Soc. Rev., 2012, 41, 4335–4355, 10.1039/c2cs35035a.
- J. A. McLean, The mass-mobility correlation redux: The conformational landscape of anhydrous biomolecules, J. Am. Soc. Mass Spectrom., 2009, 20, 1775–1781, DOI:10.1016/j.jasms.2009.06.016.
- J. Lübke, N. Roth, L. Worbs, D. A. Horke, A. D. Estillore, A. K. Samanta and J. Küpper, Charge-state distribution of aerosolized nanoparticles, J. Phys. Chem. C, 2021, 125, 25794–25798, DOI:10.1021/acs.jpcc.1c06912 , https://arxiv.org/abs/2108.04710.
- S. S. Dhillon, M. S. Vitiello, E. H. Linfield, A. G. Davies, M. C. Hoffmann, J. Booske, C. Paoloni, M. Gensch, P. Weightman, G. P. Williams, E. Castro-Camus, D. R. S. Cumming, F. Simoens, I. Escorcia-Carranza, J. Grant, S. Lucyszyn, M. Kuwata-Gonokami, K. Konishi, M. Koch, C. A. Schmuttenmaer, T. L. Cocker, R. Huber, A. G. Markelz, Z. D. Taylor, V. P. Wallace, J. A. Zeitler, J. Sibik, T. M. Korter, B. Ellison, S. Rea, P. Goldsmith, K. B. Cooper, R. Appleby, D. Pardo, P. G. Huggard, V. Krozer, H. Shams, M. Fice, C. Renaud, A. Seeds, A. Stöhr, M. Naftaly, N. Ridler, R. Clarke, J. E. Cunningham and M. B. Johnston, The 2017 terahertz science and technology roadmap, J. Phys. D: Appl. Phys., 2017, 50, 043001, DOI:10.1088/1361-6463/50/4/043001.
- M. Musheghyan, P. P. Geetha, D. Faccial, A. Pusala, G. Crippa, A. Campolo, A. G. Ciriolo, M. Devetta, A. Assion, C. Manzoni, C. Vozzi and S. Stagira, Tunable, few-cycle, CEP-stable mid-IR optical parametric amplifier for strong field applications, J. Phys. B: At., Mol. Opt. Phys., 2020, 53, 185402, DOI:10.1088/1361-6455/aba127.
- H. He, Z. Wang, C. Hu, J. Jiang, S. Qin, P. He, N. Zhang, P. Yang, Z. Li and Z. Wei, 520 μJ mid-infrared femtosecond laser at 2.8 μm by 1 kHz KTA optical parametric amplifier, Appl. Phys. B: Lasers Opt., 2018, 124, 31, DOI:10.1007/s00340-018-6896-y.
- S. Trippel, T. G. Mullins, N. L. M. Müller, J. S. Kienitz, K. Długołȩcki and J. Küpper, Strongly aligned and oriented molecular samples at a kHz repetition rate, Mol. Phys., 2013, 111, 1738, DOI:10.1080/00268976.2013.780334 , https://arxiv.org/abs/1301.1826.
- D. Oepts, A. van der Meer and P. van Amersfoort, The free-electron-laser user facility FELIX, Infrared Phys. Technol., 1995, 36, 297–308, DOI:10.1016/1350-4495(94)00074-u.
- S. Winnerl, D. Stehr, O. Drachenko, H. Schneider, M. Helm, W. Seidel, P. Michel, S. Schneider, J. Seidel, S. Grafstrom, L.-M. Eng, T. Roch, G. Strasser, T. Maier, and M. Walther, FELBE Free-Electron Laser: Status and application for time resolved spectroscopy experiments, in Joint 31st International Conference on Infrared Millimeter Waves and 14th International Conference on Teraherz Electronics (2006) pp. 159-159 DOI:10.1109/ICIMW.2006.368367.
- M. Beye, M. Gühr, I. Hartl, E. Plönjes, L. Schaper, S. Schreiber, K. Tiedtke and R. Treusch, FLASH and the FLASH2020+ project - current status and upgrades for the free-electron laser in Hamburg at DESY, Eur. Phys. J. Plus, 2023, 138, 193, DOI:10.1140/epjp/s13360-023-03814-8.
- L. Windhorn, T. Witte, J. S. Yeston, D. Proch, M. Motzkus, K. L. Kompa and W. Fuß, Molecular dissociation by mid-IR femtosecond pulses, Chem. Phys. Lett., 2002, 357, 85–90, DOI:10.1016/S0009-2614(02)00444-X.
- S. Chelkowski, A. D. Bandrauk and P. B. Corkum, Efficient molecular dissociation by a chirped ultrashort infrared-laser pulse, Phys. Rev. Lett., 1990, 65, 2355–2358, DOI:10.1103/PhysRevLett.65.2355.
- S. W. Sharpe, T. J. Johnson, R. L. Sams, P. M. Chu, G. C. Rhoderick and P. A. Johnson, Gas-phase databases for quantitative infrared spectroscopy, Appl. Spectrosc., 2004, 58, 1452–1461, DOI:10.1366/0003702042641281.
- I. Gordon, L. Rothman, R. Hargreaves, R. Hashemi, E. Karlovets, F. Skinner, E. Conway, C. Hill, R. Kochanov, Y. Tan, P. Wcisło, A. Finenko, K. Nelson, P. Bernath, M. Birk, V. Boudon, A. Campargue, K. Chance, A. Coustenis, B. Drouin, J. Flaud, R. Gamache, J. Hodges, D. Jacquemart, E. Mlawer, A. Nikitin, V. Perevalov, M. Rotger, J. Tennyson, G. Toon, H. Tran, V. Tyuterev, E. Adkins, A. Baker, A. Barbe, E. Canè, A. Császár, A. Dudaryonok, O. Egorov, A. Fleisher, H. Fleurbaey, A. Foltynowicz, T. Furtenbacher, J. Harrison, J. Hartmann, V. Horneman, X. Huang, T. Karman, J. Karns, S. Kassi, I. Kleiner, V. Kofman, F. Kwabia-Tchana, N. Lavrentieva, T. Lee, D. Long, A. Lukashevskaya, O. Lyulin, V. Makhnev, W. Matt, S. Massie, M. Melosso, S. Mikhailenko, D. Mondelain, H. Müller, O. Naumenko, A. Perrin, O. Polyansky, E. Raddaoui, P. Raston, Z. Reed, M. Rey, C. Richard, R. Tóbiás, I. Sadiek, D. Schwenke, E. Starikova, K. Sung, F. Tamassia, S. Tashkun, J. Vander Auwera, I. Vasilenko, A. Vigasin, G. Villanueva, B. Vispoel, G. Wagner, A. Yachmenev and S. Yurchenko, The HITRAN2020 molecular spectroscopic database, J. Quant. Spectrosc. Radiat. Transfer, 2022, 277, 107949, DOI:10.1016/j.jqsrt.2021.107949.
- W. C. Wiley and I. H. Mclaren, Time-of-flight mass spectrometer with improved resolution, Rev. Sci. Instrum., 1955, 26, 1150–1157, DOI:10.1063/1.1715212.
- V. G. Stavros and J. R. Verlet, Gas-phase femtosecond particle spectroscopy: A bottom-up approach to nucleotide dynamics, Annu. Rev. Phys. Chem., 2016, 67, 211–232, DOI:10.1146/annurev-physchem-040215-112428.
- R. Zimmermann, U. Boesl, C. Weickhardt, D. Lenoir, K.-W. Schramm, A. Kettrup and E. W. Schlag, Isomer-selective ionization of chlorinated aromatics with lasers for analytical time-of-flight mass spectrometry: First results for polychlorinated dibenzo-p-dioxins (PCDD), biphenyls (PCB) and benzenes (PCBz), Chemosphere, 1994, 29, 1877–1888, DOI:10.1016/0045-6535(94)90353-0.
- B. Yan, S. Jaeqx, W. J. van der Zande and A. M. Rijs, A conformation-selective IR-UV study of the dipeptides Ac-Phe-Ser-NH2 and Ac-Phe-Cys-NH2: probing the SH⋯O and OH⋯O hydrogen bond interactions, Phys. Chem. Chem. Phys., 2014, 16, 10770–10778, 10.1039/C4CP00810C.
- C. B. Madsen, L. B. Madsen, S. S. Viftrup, M. P. Johansson, T. B. Poulsen, L. Holmegaard, V. Kumarappan, K. Jørgensen and H. Stapelfeldt, Manipulating the torsion of molecules by strong laser pulses, Phys. Rev. Lett., 2009, 102, 073007, DOI:10.1103/PhysRevLett.102.073007 , https://arxiv.org/abs/0809.2935.
- M. E. Corrales, J. González-Vázquez, G. Balerdi, I. R. Solá, R. D. Nalda and L. Bañares, Control of ultrafast molecular photodissociation by laser-field-induced potentials, Nat. Chem., 2014, 6, 785–790, DOI:10.1038/nchem.2006.
- S. Pathak, R. Obaid, S. Bhattacharyya, J. Bürger, X. Li, J. Tross, T. Severt, B. Davis, R. C. Bilodeau, C. A. Trallero-Herrero, A. Rudenko, N. Berrah and D. Rolles, Differentiating and quantifying gas-phase conformational isomers using Coulomb explosion imaging, J. Phys. Chem. Lett., 2020, 11, 10205–10211, DOI:10.1021/acs.jpclett.0c02959.
- M. Burt, R. Boll, J. W. L. Lee, K. Amini, H. Köckert, C. Vallance, A. S. Gentleman, S. R. Mackenzie, S. Bari, C. Bomme, S. Düsterer, B. Erk, B. Manschwetus, E. Müller, D. Rompotis, E. Savelyev, N. Schirmel, S. Techert, R. Treusch, J. Küpper, S. Trippel, J. Wiese, H. Stapelfeldt, B. Cunha de Miranda, R. Guillemin, I. Ismail, L. Journel, T. Marchenko, J. Palaudoux, F. Penent, M. N. Piancastelli, M. Simon, O. Travnikova, F. Brauße, G. Goldsztejn, A. Rouzée, M. Géléoc, R. Geneaux, T. Ruchon, J. Underwood, D. M. P. Holland, A. S. Mereshchenko, P. K. Olshin, P. Johnsson, S. Maclot, J. Lahl, A. Rudenko, F. Ziaee, M. Brouard and D. Rolles, Coulomb-explosion imaging of concurrent CH2BrI photodissociation dynamics, Phys. Rev. A, 2017, 96, 043415, DOI:10.1103/PhysRevA.96.043415 , https://arxiv.org/abs/1710.02372.
- H. Stapelfeldt, E. Constant, H. Sakai and P. B. Corkum, Time-resolved Coulomb explosion imaging: A method to measure structure and dynamics of molecular nuclear wave packets, Phys. Rev. A: At., Mol., Opt. Phys., 1998, 58, 426–433, DOI:10.1103/PhysRevA.58.426.
- M. E. Corrales, J. González-Vázquez, R. de Nalda and L. Bañares, Coulomb explosion imaging for the visualization of a conical intersection, J. Phys. Chem. Lett., 2019, 10, 138–143, DOI:10.1021/acs.jpclett.8b03726.
- M. Burt, K. Amini, J. W. L. Lee, L. Christiansen, R. R. Johansen, Y. Kobayashi, J. D. Pickering, C. Vallance, M. Brouard and H. Stapelfeldt, Communication: Gas-phase structural isomer identification by Coulomb explosion of aligned molecules, J. Chem. Phys., 2018, 148, 091102, DOI:10.1063/1.5023441 , https://arxiv.org/abs/1902.06974.
- C. Saribal, A. Owens, A. Yachmenev and J. Küpper, Detecting handedness of spatially oriented molecules by Coulomb explosion imaging, J. Chem. Phys., 2021, 154, 071101, DOI:10.1063/5.0029792 , https://arxiv.org/abs/2009.09783.
- J. Gagnon, K. F. Lee, D. M. Rayner, P. B. Corkum and V. R. Bhardwaj, Coincidence imaging of polyatomic molecules via laser-induced Coulomb explosion, J. Phys. B: At., Mol. Opt. Phys., 2008, 41, 215104, DOI:10.1088/0953-4075/41/21/215104.
- T. Havermeier, T. Jahnke, K. Kreidi, R. Wallauer, S. Voss, M. Schöffler, S. Schössler, L. Foucar, N. Neumann, J. Titze, H. Sann, M. Kühnel, J. Voigtsberger, J. H. Morilla, W. Schöllkopf, H. Schmidt-Böcking, R. E. Grisenti and R. Dörner, Interatomic coulombic decay following photoionization of the helium dimer: Observation of vibrational structure, Phys. Rev. Lett., 2010, 104, 133401, DOI:10.1103/physrevlett.104.133401 , https://arxiv.org/abs/1006.2664.
- A. Assion, M. Geisler, J. Helbing, V. Seyfried and T. Baumert, Femtosecond pump-probe photoelectron spectroscopy: Mapping of vibrational wave-packet motion, Phys. Rev. A: At., Mol., Opt. Phys., 1996, 54, R4605–R4608, DOI:10.1103/PhysRevA.54.R4605.
- A. Padva, P. R. LeBreton, R. J. Dinerstein and J. N. A. Ridyard, UV photoelectron studies of biological pyrimidines: The electronic structure of uracil, Biochem. Biophys. Res. Commun., 1974, 60, 1262–1268, DOI:10.1016/0006-291X(74)90334-9.
- R. Livingstone, O. Schalk, A. E. Boguslavskiy, G. Wu, L. T. Bergendahl, A. Stolow, M. J. Paterson and D. Townsend, Following the excited state relaxation dynamics of indole and 5-hydroxyindole using time-resolved photoelectron spectroscopy, J. Chem. Phys., 2011, 135, 194307, DOI:10.1063/1.3659231.
- S. Lochbrunner, T. Schultz, M. Schmitt, J. P. Shaffer, M. Z. Zgierski and A. Stolow, Dynamics of excited-state proton transfer systems via time-resolved photoelectron spectroscopy, J. Chem. Phys., 2001, 114, 2519–2522, DOI:10.1063/1.1345876.
- K. Müller-Dethlefs and E. W. Schlag, High-resolution zero kinetic energy (ZEKE) photoelectron spectroscopy of molecular systems, Annu. Rev. Phys. Chem., 1991, 42, 109–136, DOI:10.1146/annurev.pc.42.100191.000545.
- S. Ullrich, G. Tarczay, X. Tong and C. E. H. Dessent and K. müller-Dethlefs, A ZEKE photoelectron spectroscopy and ab initio study of the cis- and trans-isomers of formanilide: Characterizing the cationic amide bond? Phys. Chem. Chem. Phys., 2001, 3, 5450–5458 10.1039/b107700g.
- T. H. Gan, J. B. Peel and G. D. Willett, Photoelectron spectra of the gauche and trans conformers of 1,2-dichloroethane, J. Chem. Soc., Faraday Trans. 2, 1977,(73), 965–972, 10.1039/F29777300965.
- K. L. Reid, Photoelectron angular distributions, Annu. Rev. Phys. Chem., 2003, 54, 397–424, DOI:10.1146/annurev.physchem.54.011002.103814.
- J. Ullrich, R. Moshammer, A. Dorn, R. Dörner, L. P. H. Schmidt and H. Schmidt-Böcking, Recoil-ion and electron momentum spectroscopy: Reaction-microscopes, Rep. Prog. Phys., 2003, 66, 1463–1545, DOI:10.1088/0034-4885/66/9/203.
- S. Turchini, D. Catone, G. Contini, N. Zema, S. Irrera, M. Stener, D. Di Tommaso, P. Decleva and T. Prosperi, Conformational effects in photoelectron circular dichroism of alaninol, ChemPhysChem, 2009, 10, 1839–1846, DOI:10.1002/cphc.200800862.
- A. Comby, S. Beaulieu, M. Boggio-Pasqua, D. Descamps, F. Légaré, L. Nahon, S. Petit, B. Pons, B. Fabre, Y. Mairesse and V. Blanchet, Relaxation dynamics in photoexcited chiral molecules studied by time-resolved photoelectron circular dichroism: Toward chiral femtochemistry, J. Phys. Chem. Lett., 2016, 7, 4514–4519, DOI:10.1021/acs.jpclett.6b02065 , https://arxiv.org/abs/1611.06226.
- C. Bostedt, H. Thomas, M. Hoener, E. Eremina, T. Fennel, K. H. Meiwes-Broer, H. Wabnitz, M. Kuhlmann, E. Ploenjes, K. Tiedtke, R. Treusch, J. Feldhaus, A. R. B. D. Castro and T. Moeller, Multistep ionization of argon clusters in intense femtosecond extreme ultraviolet pulses, Phys. Rev. Lett., 2008, 100, 133401, DOI:10.1103/physrevlett.100.133401.
- B. J. Siwick, J. R. Dwyer, R. E. Jordan and R. J. D. Miller, An atomic-level view of melting using femtosecond electron diffraction, Science, 2003, 302, 1382–1385, DOI:10.1126/science.1090052.
- H. Ihee, V. A. Lobastov, U. M. Gomez, B. M. Goodson, R. Srinivasan, C.-Y. Ruan and A. H. Zewail, Direct imaging of transient molecular structures with ultrafast diffraction, Science, 2001, 291, 458–462, DOI:10.1126/science.291.5503.458.
- C. J. Hensley, J. Yang and M. Centurion, Imaging of isolated molecules with ultrafast electron pulses, Phys. Rev. Lett., 2012, 109, 133202, DOI:10.1103/PhysRevLett.109.133202.
- H. Jean-Ruel, M. Gao, M. A. Kochman, C. Lu, L. C. Liu, R. R. Cooney, C. A. Morrison and R. J. D. Miller, Ring-closing reaction in diarylethene captured by femtosecond electron crystallography, J. Phys. Chem. B, 2013, 117, 15894–15902, DOI:10.1021/jp409245h.
- S. Manz, A. Casandruc, D. Zhang, Y. Zhong, R. A. Loch, A. Marx, T. Hasegawa, L. C. Liu, S. Bayesteh, H. Delsim-Hashemi, M. Hoffmann, M. Felber, M. Hachmann, F. Mayet, J. Hirscht, S. Keskin, M. Hada, S. W. Epp, K. Flöttmann and R. J. D. Miller, Mapping atomic motions with ultrabright electrons: towards fundamental limits in space-time resolution, Faraday Discuss., 2015, 177, 467–491, 10.1039/c4fd00204k.
- T. J. A. Wolf, D. M. Sanchez, J. Yang, R. M. Parrish, J. P. F. Nunes, M. Centurion, R. Coffee, J. P. Cryan, M. Gühr, K. Hegazy, A. Kirrander, R. K. Li, J. Ruddock, X. Shen, T. Vecchione, S. P. Weathersby, P. M. Weber, K. Wilkin, H. Yong, Q. Zheng, X. J. Wang, M. P. Minitti and T. J. Martinez, The photochemical ring-opening of 1,3-cyclohexadiene imaged by ultrafast electron diffraction, Nat. Chem., 2019, 11, 504–509, DOI:10.1038/s41557-019-0252-7 , https://arxiv.org/abs/1810.02900.
- B. J. Siwick, J. R. Dwyer, R. E. Jordan and R. J. D. Miller, Ultrafast electron optics: Propagation dynamics of femtosecond electron packets, J. Appl. Phys., 2002, 92, 1643–1648, DOI:10.1063/1.1487437.
- Y. Morimoto and P. Baum, Diffraction and microscopy with attosecond electron pulse trains, Nat. Phys., 2018, 14, 252–256, DOI:10.1038/s41567-017-0007-6.
- H. N. Chapman, P. Fromme, A. Barty, T. A. White, R. A. Kirian, A. Aquila, M. S. Hunter, J. Schulz, D. P. Deponte, U. Weierstall, R. B. Doak, F. R. N. C. Maia, A. V. Martin, I. Schlichting, L. Lomb, N. Coppola, R. L. Shoeman, S. W. Epp, R. Hartmann, D. Rolles, A. Rudenko, L. Foucar, N. Kimmel, G. Weidenspointner, P. Holl, M. Liang, M. Barthelmess, C. Caleman, S. Boutet, M. J. Bogan, J. Krzywinski, C. Bostedt, S. Bajt, L. Gumprecht, B. Rudek, B. Erk, C. Schmidt, A. Hömke, C. Reich, D. Pietschner, L. Strüder, G. Hauser, H. Gorke, J. Ullrich, S. Herrmann, G. Schaller, F. Schopper, H. Soltau, K.-U. Kühnel, M. Messerschmidt, J. D. Bozek, S. P. Hau-Riege, M. Frank, C. Y. Hampton, R. G. Sierra, D. Starodub, G. J. Williams, J. Hajdu, N. Timneanu, M. M. Seibert, J. Andreasson, A. Rocker, O. Jönsson, M. Svenda, S. Stern, K. Nass, R. Andritschke, C.-D. Schröter, F. Krasniqi, M. Bott, K. E. Schmidt, X. Wang, I. Grotjohann, J. M. Holton, T. R. M. Barends, R. Neutze, S. Marchesini, R. Fromme, S. Schorb, D. Rupp, M. Adolph, T. Gorkhover, I. Andersson, H. Hirsemann, G. Potdevin, H. Graafsma, B. Nilsson and J. C. H. Spence, Femtosecond X-ray protein nanocrystallography, Nature, 2011, 470, 73–77, DOI:10.1038/nature09750.
- R. Henderson, The potential and limitations of neutrons, electrons and X-rays for atomic-resolution microscopy of unstained biological molecules, Q. Rev. Biophys., 1995, 28, 171–193, DOI:10.1017/S003358350000305X.
- M. M. Seibert, T. Ekeberg, F. R. N. C. Maia, M. Svenda, J. Andreasson, O. Jönsson, D. Odić, B. Iwan, A. Rocker, D. Westphal, M. Hantke, D. P. Deponte, A. Barty, J. Schulz, L. Gumprecht, N. Coppola, A. Aquila, M. Liang, T. A. White, A. Martin, C. Caleman, S. Stern, C. Abergel, V. Seltzer, J.-M. Claverie, C. Bostedt, J. D. Bozek, S. Boutet, A. A. Miahnahri, M. Messerschmidt, J. Krzywinski, G. Williams, K. O. Hodgson, M. J. Bogan, C. Y. Hampton, R. G. Sierra, D. Starodub, I. Andersson, S. Bajt, M. Barthelmess, J. C. H. Spence, P. Fromme, U. Weierstall, R. Kirian, M. Hunter, R. B. Doak, S. Marchesini, S. P. Hau-Riege, M. Frank, R. L. Shoeman, L. Lomb, S. W. Epp, R. Hartmann, D. Rolles, A. Rudenko, C. Schmidt, L. Foucar, N. Kimmel, P. Holl, B. Rudek, B. Erk, A. Hömke, C. Reich, D. Pietschner, G. Weidenspointner, L. Strüder, G. Hauser, H. Gorke, J. Ullrich, I. Schlichting, S. Herrmann, G. Schaller, F. Schopper, H. Soltau, K.-U. Kühnel, R. Andritschke, C.-D. Schröter, F. Krasniqi, M. Bott, S. Schorb, D. Rupp, M. Adolph, T. Gorkhover, H. Hirsemann, G. Potdevin, H. Graafsma, B. Nilsson, H. N. Chapman and J. Hajdu, Single mimivirus particles intercepted and imaged with an X-ray laser, Nature, 2011, 470, 78–81, DOI:10.1038/nature09748.
- J. Tenboer, S. Basu, N. Zatsepin, K. Pande, D. Milathianaki, M. Frank, M. Hunter, S. Boutet, G. J. Williams, J. E. Koglin, D. Oberthuer, M. Heymann, C. Kupitz, C. Conrad, J. Coe, S. Roy-Chowdhury, U. Weierstall, D. James, D. Wang, T. Grant, A. Barty, O. Yefanov, J. Scales, C. Gati, C. Seuring, V. Srajer, R. Henning, P. Schwander, R. Fromme, A. Ourmazd, K. Moffat, J. J. Van Thor, J. C. H. Spence, P. Fromme, H. N. Chapman and M. Schmidt, Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein, Science, 2014, 346, 1242–1246, DOI:10.1126/science.1259357.
- K. Pande, C. D. M. Hutchison, G. Groenhof, A. Aquila, J. S. Robinson, J. Tenboer, S. Basu, S. Boutet, D. P. DePonte, M. Liang, T. A. White, N. A. Zatsepin, O. Yefanov, D. Morozov, D. Oberthuer, C. Gati, G. Subramanian, D. James, Y. Zhao, J. Koralek, J. Brayshaw, C. Kupitz, C. Conrad, S. Roy-Chowdhury, J. D. Coe, M. Metz, P. L. Xavier, T. D. Grant, J. E. Koglin, G. Ketawala, R. Fromme, V. Šrajer, R. Henning, J. C. H. Spence, A. Ourmazd, P. Schwander, U. Weierstall, M. Frank, P. Fromme, A. Barty, H. N. Chapman, K. Moffat, J. J. van Thor and M. Schmidt, Femtosecond structural dynamics drives the trans/cis isomerization in photoactive yellow protein, Science, 2016, 352, 725–729, DOI:10.1126/science.aad5081.
- S. Stern, L. Holmegaard, F. Filsinger, A. Rouzée, A. Rudenko, P. Johnsson, A. V. Martin, A. Barty, C. Bostedt, J. D. Bozek, R. N. Coffee, S. Epp, B. Erk, L. Foucar, R. Hartmann, N. Kimmel, K.-U. Kühnel, J. Maurer, M. Messerschmidt, B. Rudek, D. G. Starodub, J. Thøgersen, G. Weidenspointner, T. A. White, H. Stapelfeldt, D. Rolles, H. N. Chapman and J. Küpper, Toward atomic resolution diffractive imaging of isolated molecules with X-ray free-electron lasers, Faraday Discuss., 2014, 171, 393, 10.1039/c4fd00028e , https://arxiv.org/abs/1403.2553.
- T. Kierspel, A. Morgan, J. Wiese, T. Mullins, A. Aquila, A. Barty, R. Bean, R. Boll, S. Boutet, P. Bucksbaum, H. N. Chapman, L. Christensen, A. Fry, M. Hunter, J. E. Koglin, M. Liang, V. Mariani, A. Natan, J. Robinson, D. Rolles, A. Rudenko, K. Schnorr, H. Stapelfeldt, S. Stern, J. Thøgersen, C. H. Yoon, F. Wang and J. Küpper, X-ray diffractive imaging of controlled gas-phase molecules: Toward imaging of dynamics in the molecular frame, J. Chem. Phys., 2020, 152, 084307, DOI:10.1063/1.5133963 , https://arxiv.org/abs/1910.13494.
- J. C. H. Spence and R. B. Doak, Single molecule diffraction, Phys. Rev. Lett., 2004, 92, 198102, DOI:10.1103/PhysRevLett.92.198102.
- J. C. H. Spence, K. Schmidt, J. S. Wu, G. Hembree, U. Weierstall, R. B. Doak and P. Fromme, Diffraction and imaging from a beam of laser-aligned proteins: resolution limits, Acta Crystallogr., Sect. A: Found. Crystallogr., 2005, 61, 237–245, DOI:10.1107/S0108767305002710.
- M. Amin, J.-M. Hartmann, A. K. Samanta, and J. Küpper, Laser-induced alignment of nanoparticles and macromolecules for imaging applications, 2023, https://arxiv.org/abs/2306.05870.
- F. Filsinger, G. Meijer, H. Stapelfeldt, H. N. Chapman and J. Küpper, State- and conformer-selected beams of aligned and oriented molecules for ultrafast diffraction studies, Phys. Chem. Chem. Phys., 2011, 13, 2076–2087, 10.1039/C0CP01585G , https://arxiv.org/abs/1009.0871.
- C. D. Lin, A.-T. Le, Z. Chen, T. Morishita and R. Lucchese, Strong-field rescattering physics - self-imaging of a molecule by its own electrons, J. Phys. B: At., Mol. Opt. Phys., 2010, 43, 122001, DOI:10.1088/0953-4075/43/12/122001.
- K. Amini and J. Biegert, Ultrafast electron diffraction imaging of gas-phase molecules, Adv. At., Mol., Opt. Phys., 2020, 69, 163–231, DOI:10.1016/bs.aamop.2020.04.001 , https://arxiv.org/abs/2003.02971.
- U. D. Giovannini, J. Küpper and A. Trabattoni, New perspectives in time-resolved laser-induced electron diffraction, J. Phys. B: At., Mol. Opt. Phys., 2023, 56, 054002, DOI:10.1088/1361-6455/acb872.
- P. B. Corkum, Plasma perspective on strong-field multiphoton ionization, Phys. Rev. Lett., 1993, 71, 1994–1997, DOI:10.1103/PhysRevLett.71.1994.
- M. Y. Ivanov, M. Spanner and O. Smirnova, Anatomy of strong field ionization, J. Mod. Opt., 2005, 52, 165–184, DOI:10.1080/0950034042000275360.
- M. G. Pullen, B. Wolter, A.-T. Le, M. Baudisch, M. Hemmer, A. Senftleben, C. D. Schroter, J. Ullrich, R. Moshammer, C. D. Lin and J. Biegert, Imaging an aligned polyatomic molecule with laser-induced electron diffraction, Nat. Commun., 2015, 6, 7262, DOI:10.1038/ncomms8262 , https://arxiv.org/abs/1503.03294.
- G. G. Paulus, W. Becker, W. Nicklich and H. Walther, Rescattering effects in above threshold ionization: a classical model, J. Phys. B: At., Mol. Opt. Phys., 1994, 27, L703–L708, DOI:10.1088/0953-4075/27/21/003.
- F. Schell, T. Bredtmann, C.-P. Schulz, S. Patchkovskii, M. J. J. Vrakking and J. Mikosch, Molecular orbital imprint in laser-driven electron recollision, Sci. Adv., 2018, 4, eaap8148, DOI:10.1126/sciadv.aap8148.
- B. Belsa, K. M. Ziems, A. Sanchez, K. Chirvi, X. Liu, S. Gräfe and J. Biegert, Laser-induced electron diffraction in the over-the-barrier-ionization regime, Phys. Rev. A, 2022, 106, 043105, DOI:10.1103/PhysRevA.106.043105.
- L. Keldysh, Ionization in the field of a strong electromagnetic wave, J. Exp. Theor. Phys, 1965, 20, 1307–1314 Search PubMed , https://jetp.ras.ru/cgi-bin/e/index/e/20/5/p1307?a=list.
- B. Wolter, M. G. Pullen, M. Baudisch, M. Sclafani, M. Hemmer, A. Senftleben, C. D. Schröter, J. Ullrich, R. Moshammer and J. Biegert, Strong-field physics with mid-IR fields, Phys. Rev. X, 2015, 5, 021034, DOI:10.1103/PhysRevX.5.021034 , https://arxiv.org/abs/1506.03636.
- J. Yang, M. Guehr, T. Vecchione, M. S. Robinson, R. Li, N. Hartmann, X. Shen, R. Coffee, J. Corbett, A. Fry, K. Gaffney, T. Gorkhover, C. Hast, K. Jobe, I. Makasyuk, A. Reid, J. Robinson, S. Vetter, F. Wang, S. Weathersby, C. Yoneda, M. Centurion and X. Wang, Diffractive imaging of a rotational wavepacket in nitrogen molecules with femtosecond megaelectronvolt electron pulses, Nat. Commun., 2016, 7, 11232, DOI:10.1038/ncomms11232 , https://arxiv.org/abs/1510.06426.
- E. J. Heller, The semiclassical way to molecular spectroscopy, Acc. Chem. Res., 1981, 14, 368–375, DOI:10.1021/ar00072a002.
| This journal is © the Owner Societies 2024 |