
A trinuclear metallasilsesquioxane of uranium(III)†
Maxime
Tricoire‡
a,
Nadir
Jori‡
 a,
Farzaneh
Fadaei Tirani
b,
Rosario
Scopelliti
b,
Ivica
a,
Farzaneh
Fadaei Tirani
b,
Rosario
Scopelliti
b,
Ivica
![[Z with combining breve]](https://www.rsc.org/images/entities/char_005a_0306.gif) ivković
c,
Louise S.
Natrajan
d and
Marinella
Mazzanti
*a
ivković
c,
Louise S.
Natrajan
d and
Marinella
Mazzanti
*a
aGroup of Coordination Chemistry, Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne CH-1015, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bX-ray Diffraction and Surface Analytics Platform, Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne CH-1015, Switzerland
cLaboratory for Quantum Magnetism, Institute of Physics, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne CH-1015, Switzerland
dCentre for Radiochemistry Resesarch, Department of Chemistry, School of Natural Sciences and Photon Science Institute, The University of Manchester, Manchester M13 9PL, UK
First published on 23rd November 2023
Abstract
The silsesquioxane ligand (iBu)7Si7O9(OH)3 (iBuPOSSH3) is revealed as an attractive system for the assembly of robust polynuclear complexes of uranium(III) and allowed the isolation of the first example of a trinuclear U(III) complex ([U3(iBuPOSS)3]) that exhibits magnetic communication and promotes dinitrogen reduction in the presence of reducing agent.
The design of polymetallic assemblies is attracting high interest in uranium chemistry because of their ability to promote small molecule activation,1–10 and their interesting magnetic properties,11–15 yet synthetic routes to such assemblies remain elusive. Moreover, the recent identification of the first example of actinide metal–metal bonding interactions in a trinuclear thorium complex16,17 suggests that trinuclear assemblies may be used to promote metal–metal interactions in uranium compounds. Nevertheless, examples of trinuclear uranium complexes18–25 remain rare with only two examples reported so far for the +III oxidation state.26,27
Siloxides were shown to act as versatile supporting ligands in low-valent uranium chemistry which have led to unprecedented reactivity including dinitrogen reduction and cleavage.28–30 However, the use of the polydentate analogue silsesquioxane (POSS) in uranium chemistry remains limited to two reports describing the synthesis of mononuclear complexes [U(Cy7Si7O12)2]n− (Cy = cyclohexyl) with uranium in the oxidation states +IV, +V and +VI.31,32 In the most recent study by Hayton and coworkers,32 the formation of a trimeric assembly of uranyl(VI) was also described suggesting, together with several reports of POSS based di- and tetrametallic clusters of lanthanides,33,34 that POSS may provide a suitable ligand for the assembly of trinuclear U(III) complexes through silanolate bridges. Here we report the synthesis, structure, reactivity and magnetic properties of the first example of a trinuclear complex of uranium(III) which is assembled by using the silsesquioxane ligand (iBu)7Si7O9(OH)3, iBuPOSSH3.
The addition of one equiv. of iBuPOSSH3 to equimolar [U(N(SiMe3)2)3] in hexane at −40 °C led to a colour change from purple to dark brown (Scheme 1). The 1H NMR spectrum of the reaction mixture measured at 25 °C in cyclohexane shows a complex pattern suggesting the presence of several solution species. Storage of the resultant reaction mixture at −40 °C allowed to obtain midnight blue XRD-suitable crystals of the complex [U3(iBuPOSS)3], 1 (Fig. 1a). Due to the high solubility of 1, the careful removal of the HN(SiMe3)2 formed during the reaction was challenging but crucial to isolate analytically pure complex 1. After drying carefully the reaction mixture under dynamic vacuum for 16 h, complex 1 was isolated analytically pure in 19% yield, as a midnight blue powder, by recrystallization from cold (−40 °C) hexane (Scheme 1). Isolation of the trimeric complex in higher yields was prevented by its solubility and by the presence of species of higher nuclearity in the reaction mixture. Notably a few crystals of the tetranuclear complex [U4(iBuPOSS)4] (Fig. 1c) were isolated alongside the trinuclear complex when further concentrating the reaction mixture. The conditions required for the isolation of reasonable amounts of analytically pure [U4(iBuPOSS)4] could not so far be identified.
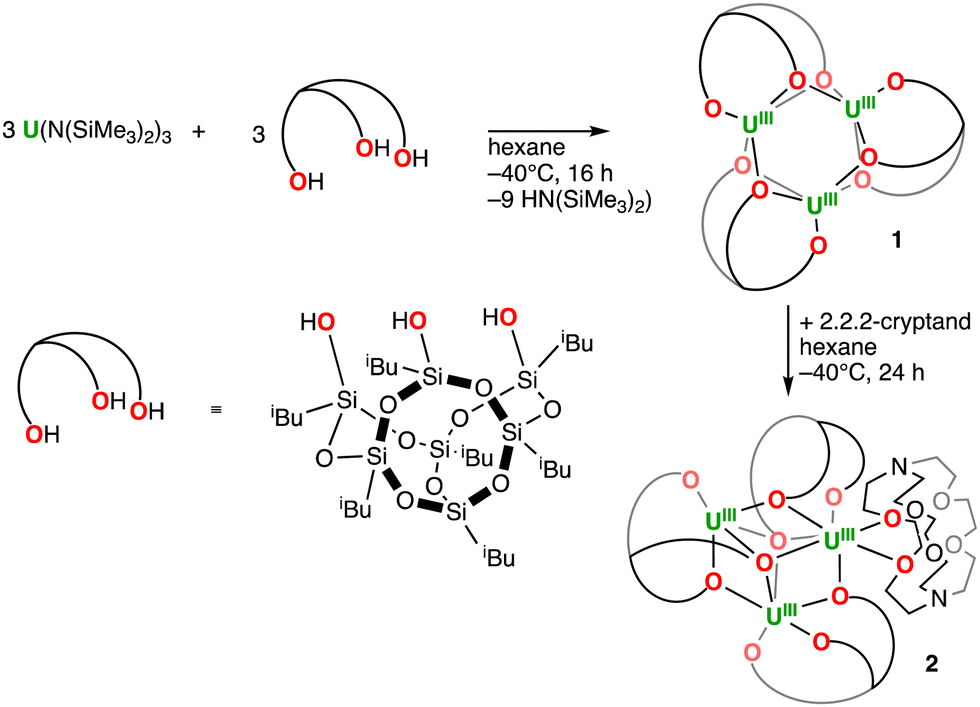 | ||
| Scheme 1 Synthesis of complexes 1 and 2. | ||
 | ||
| Fig. 1 ORTEP of complexes 1 (a), 2 (b) and [U4(iBuPOSS)4] (c) with thermal ellipsoids drawn at 50% probability level, close-up view of their core structure are shown on the right. The iso-butyl groups and the minor part of the disorder have been omitted for clarity. Selected bonds distances (Å): 1: U(1)–U(2) 3.6347(13), U(1)–U(3) 3.6493(12), U(2)–U(3) 3.6426(13). 2: U(1)–U(2) 3.8163(5), U(1)–U(3) 3.8124(5), U(2)–U(3) 3.7139(6). | ||
Complex 1 crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and shows a triangular uranium core that is disordered over two positions in a 77
space group and shows a triangular uranium core that is disordered over two positions in a 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :23 ratio. In the solid state, each uranium centre is coordinated by one iBuPOSS ligand.
:23 ratio. In the solid state, each uranium centre is coordinated by one iBuPOSS ligand.
The three U(iBuPOSS) fragments are assembled in a trinuclear architecture with a U3(μ-O)6 core, with three oxygen atoms in a triangular arrangement located above the plane formed by the three uranium centres and three oxygen atoms below the plane. A similar An3(μ-X)6 topology has only been observed twice in actinide chemistry with X = Cl.16,35 Although a few additional examples of trinuclear oxo- or nitride centred complexes of uranium with a U3(μ-X)6(μ3-Y) (Y = O, N) core21,22,25 have been reported, none of them contain only U(III) centres. The structure of complex 1 (Fig. 1a) shows that in each U(iBuPOSS) moiety, the uranium ion is coordinated by three anionic siloxide oxygen atoms and two oxygens of the silyl ether backbone of the iBuPOSS ligand. Each uranium ion is also bound by two additional bridging anionic siloxide oxygens from an adjacent U(iBuPOSS) unit. The U–Osiloxide distances range from 2.125(16) Å to 2.568(17) Å (average value of 2.36(2) Å), which lie within the range of distances found in previously reported U(III) siloxide complexes28,36,37 (2.182(3)–2.549(3) Å). The U3(μ-O)6 core in 1 displays alternating U–O bond lengths in which the shortest bonds are observed for an anionic oxygen from the coordinated iBuPOSS (U1–O1, U2–O13 and U3–O25) and for two bridging anionic oxygens from an adjacent U(iBuPOSS) fragment (U1–O35, U1–O36, U2–O11, U2–O12, U3–O23, U3–O24), thus ensuring the trimeric architecture's rigidity (Fig. 1). The U–U distances range from 3.6347(13) Å to 3.6493(12) Å, and are significantly shorter than the reported distance in the [UIII(OSi(OtBu)3)3]2 dimer (3.9862(2) Å).36 These values lie between the sum of the covalent radii of two uranium atoms (3.40 Å) and the sum of their van der Waals radii (3.72 Å).38 The observed arrangement of the U atoms in the U3(μ-O)6 core and the U–U short distance are similar to what observed in the tri-thorium Th3(μ-Cl)6 cluster displaying a σ-bonding Th–Th interaction with Th–Th distances in the 3.9896(4)–3.9947(5) Å range.16
The 1H NMR spectrum of isolated 1 recorded at 25 °C in cyclohexane-d12 shows the twenty-one peaks expected for the seven iso-butyl groups of the iBuPOSS ligands in a C3 symmetric coordination environment (Fig. S1, ESI†). Switching the solvent to thf-d8 led to a well resolved 1H NMR spectrum displaying 69 and 76 resonances at 25 and at −40 °C respectively (Fig. S2 and S3, ESI†), which indicates a loss of the C3 symmetry that is likely induced by thf coordination to the uranium centres. The fact that more than 63 peaks are observed suggests that not every iso-butyl is freely rotating at low temperature. Complex 1 did not show any sign of decomposition after 4 days at 25 °C in cyclohexane-d12 and is stable in thf-d8 at −40 °C up to a week but it slowly decomposes at 25 °C in thf-d8 over a week. The values of the Stokes radii obtained by 1H DOSY NMR spectroscopy (Table S1, ESI†) in cyclohexane-d12 (12.1 Å) and in thf-d8 (11.4 Å) correlate well with the 10.3 Å value estimated from the solid-state structure confirming that the trinuclear assembly of 1 is retained both in cyclohexane and in thf solution.
Attempts to reduce 1 by adding KC8 (up to 10 equiv.) and 2.2.2-cryptand (crypt) (1 equiv.) under Ar only led to the isolation of dark red crystals of a 2.2.2-cryptand adduct of 1, [U3(iBuPOSS)3(crypt-κ2-O,O′)] complex 2 (Fig. 1b). Complex 2 can be prepared in 48% yield by adding 1.0 equiv. of cryptand to 1.0 equiv. of complex 1 in hexane at −40 °C (Scheme 1).
Cryptand was previously found to encapsulate both low-valent lanthanides39,40 and actinides,41–43 but complex 2 is the first example of a bidentate cryptand coordinated to an actinide ion. Two examples of a similar bidentate binding mode were previously reported for rare-earth metals.44 Complex 2 crystallizes in the P space group and shows an interesting reorganization of the iBuPOSS ligands, leading to a modified core topology U3(μ-O)3(μ3-O)2 that allows the coordination of the cryptand. The number of the bridging oxygens (five) is different with respect to complex 1 (six) with the five bridging oxygen atoms now forming a distorted trigonal bipyramidal coordination polyhedron. The cryptand binding results in a lengthening of the U–U distances (3.7139(6) Å to 3.8163(5) Å) compared to 1.
The 1H NMR spectrum of complex 2, recorded at 25 °C in cyclohexane-d12, showed 63 peaks which is consistent with the loss of symmetry observed in the solid state upon cryptand coordination. Dissolving complex 2 in thf-d8 showed unbound cryptand signals and the proton resonances corresponding to complex 1 in thf-d8 indicating that cryptand is readily displaced in coordinating solvent (Fig. S14, ESI†).
To probe the impact of the structural differences observed in 1 and 2 on their magnetic properties, SQUID magnetic measurements were carried out in the 2–250 K range under an applied DC field of 1 T (Fig. 2).
 | ||
| Fig. 2 Solid-state μeff/complex vs. T data measured under an applied field of 1 T for complexes 1 and 2 and low temperature χ vs. T plot for 1 (inset). | ||
At 250 K, complex 1 displays an effective magnetic moment of 4.16 μB (2.40 μB per uranium centre) while a value of 5.12 μB (2.95 μB per uranium centre) was recorded for 2. Both values are in reasonable agreement with the presence of uranium centres being in the +III oxidation state.45 Upon cooling, the two complexes show different behaviours. Complex 2 displays a moderate and monotonic decrease of its magnetic moment to reach a value of 4.35 μB (2.51 μB per uranium centre) at 15 K before dropping to 3.58 μB (2.07 μB per uranium centre) at 2 K. The observed behaviour is similar to those measured for mononuclear complexes of U(III) suggesting that the three uranium centres in 2 are essentially magnetically independent.37,45,46
A different low temperature behaviour of the magnetic moment was observed for complex 1. Upon lowering the temperature, the magnetic moment decreases almost monotonically until 6 K (1.15 μB per complex, 0.67 μB per uranium centre), and then with a steeper curve to reach a value of 0.54 μB (0.31 μB per uranium centre) at 2 K. The observed low value of the magnetic moment with a χT value approaching 0 at low temperature indicates the presence of a non-magnetic ground state (Fig. S22, ESI†). Such behaviour is rare in systems comprising an odd number of unpaired electrons and was previously only observed for a triangular pentavalent uranium [UO2L]3 (L= 2-(4-tolyl)-1,3-bis(quinolyl)malondiiminate) system23,24 and for several Dy3 architectures.47,48 The analysis the χ vs. T plot for 1 revealed a sharp maximum of the susceptibility at 6 K which indicates the presence of an unambiguous antiferromagnetic coupling at low temperature (Fig. 2). The observed magnetic exchange is probably due to superexchange through the bridging oxygen atoms although a contribution from direct exchange cannot be ruled out considering the short U–U distances observed in the solid-state structure of complex 1. Only a few examples of complexes showing unambiguous magnetic coupling between U(III) centres have previously been reported.3,9,49,50 These results provide a rare example of structure-magnetic properties relation in uranium chemistry51 showing that structural changes lead to dramatic differences in the magnetism of polynuclear U(III) complexes.
Besides its interesting magnetic properties, the isolation of 1 also represented an excellent opportunity to study the reactivity of a trinuclear U(III) system. Exposing cyclohexane-d12 or thf-d8 solutions of complex 1 to N2 (1 atm) at 25 °C and −40 °C respectively did not lead to any observable changes in the 1H NMR spectrum. However, THF solutions of 1 were found to react with N2 at −40 °C in presence of excess KC8 (10 equiv.) leading to a slow color change from dark to pale brown over several days. The 1H NMR spectrum of a thf-d8 reaction mixture after 3 days at −40 °C showed only the complete disappearance of the resonances of 1. Crystallization attempts did not allow us to identify the reaction product(s). However, treating the reaction mixture after 5 days with a 2 M HCl solution in Et2O at −80 °C resulted in the formation of NH4Cl that was quantified to 2.0 equiv. per trimeric complex by 1H NMR spectroscopy in DMSO-d6. To further confirm that N2 activation had occurred the reaction was also performed with labelled 15N2 (Fig. S19, ESI†) yielding the distinctive signals of 15NH4Cl after quenching. The observed reduction of N2 by the complex 1 in the presence of KC8 suggests that N2 binding to the uranium centres must occur to some extent also in thf solution, but could not be detected by 1H NMR spectroscopy. The observed stoichiometry of 2 NH3 formed per complex suggested binding of one N2 molecule by each U(III) trimer.
In conclusion the iBuPOSS ligand revealed an attractive system for the assembly of robust polynuclear complexes of uranium(III) and allowed the isolation of the first example of a trinuclear U(III) complex showing magnetic exchange. Notably, the triangular U3(μ-O)6 core of complex 1 results in unusually short U–U distances and an antiferromagnetic coupling between the U(III) centres leading to a non-magnetic ground state. Coordination of 2.2.2-cryptand to one U(III) centre leads to a rearrangement to the core geometry and suppression of magnetic communication. Preliminary reactivity studies show that complex 1 promotes the reduction of one molecule of dinitrogen in the presence of an external reducing agent. These studies demonstrate the versatility of the silsesquioxane scaffold for assembling polymetallic complexes of low valent uranium that possess unusual properties.
We acknowledge support from the Swiss National Science Foundation grant number 212723 and 217133 and the Ecole Polytechnique Fédérale de Lausanne (EPFL).
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. L. Arnold, C. J. Stevens, N. L. Bell, R. M. Lord, J. M. Goldberg, G. S. Nichol and J. B. Love, Chem. Sci., 2017, 8, 3609–3617 RSC.
- M. Falcone, L. Chatelain, R. Scopelliti, I. Zivkovic and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS.
- M. Falcone, L. Barluzzi, J. Andrez, F. F. Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS.
- P. L. Arnold, T. Ochiai, F. Y. T. Lam, R. P. Kelly, M. L. Seymour and L. Maron, Nat. Chem., 2020, 12, 654–659 CrossRef CAS.
- X. Q. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Q. Zhu, J. Am. Chem. Soc., 2020, 142, 15004–15011 CrossRef CAS.
- D. R. Hartline and K. Meyer, Jacs Au, 2021, 1, 698–709 CrossRef CAS PubMed.
- D. K. Modder, C. T. Palumbo, I. Douair, F. Fadaei-Tirani, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2021, 60, 3737–3744 CrossRef CAS PubMed.
- W. Fang, Q. Zhu and C. Q. Zhu, Chem. Soc. Rev., 2022, 51, 8434–8449 RSC.
- N. Jori, T. Rajeshkumar, R. Scopelliti, I. Zivkovic, A. Sienkiewicz, L. Maron and M. Mazzanti, Chem. Sci., 2022, 13, 9232–9242 RSC.
- X. Q. Xin, I. Douair, Y. Zhao, S. O. Wang, L. Maron and C. Q. Zhu, Natl. Sci. Rev., 2023, 10, nwac144 CrossRef CAS PubMed.
- D. P. Mills, F. Moro, J. McMaster, J. van Slageren, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2011, 3, 454–460 CrossRef CAS.
- V. Mougel, L. Chatelain, J. Pecaut, R. Caciuffo, E. Colineau, J. C. Griveau and M. Mazzanti, Nat. Chem., 2012, 4, 1011–1017 CrossRef CAS PubMed.
- L. Chatelain, J. P. S. Walsh, J. Pecaut, F. Tuna and M. Mazzanti, Angew. Chem., Int. Ed., 2014, 53, 13434–13438 CrossRef CAS PubMed.
- L. Chatelain, F. Tuna, J. Pecaut and M. Mazzanti, Chem. Commun., 2015, 51, 11309–11312 RSC.
- D. K. Modder, M. S. Batov, T. Rajeshkumar, A. Sienkiewicz, I. Zivkovic, R. Scopelliti, L. Maron and M. Mazzanti, Chem. Sci., 2022, 13, 11294–11303 RSC.
- J. T. Boronski, J. A. Seed, D. Hunger, A. W. Woodward, J. van Slageren, A. J. Wooles, L. S. Natrajan, N. Kaltsoyannis and S. T. Liddle, Nature, 2021, 598, 72–75 CrossRef CAS PubMed.
- X. H. Lin and Y. R. Mo, Angew. Chem., Int. Ed., 2022, 61 DOI:10.1002/anie.202209658.
- D. L. Clark, J. C. Gordon, J. C. Huffman, J. G. Watkin and B. D. Zwick, New J. Chem., 1995, 19, 495–502 CAS.
- L. Karmazin, M. Mazzanti and J. Pecaut, Inorg. Chem., 2003, 42, 5900–5908 CrossRef CAS PubMed.
- J. L. Kiplinger, J. A. Pool, E. J. Schelter, J. D. Thompson, B. L. Scott and D. E. Morris, Angew. Chem., Int. Ed., 2006, 45, 2036–2041 CrossRef CAS.
- W. J. Evans, K. A. Miller, J. W. Ziller and J. Greaves, Inorg. Chem., 2007, 46, 8008–8018 CrossRef CAS PubMed.
- C. P. Larch, F. G. N. Cloke and P. B. Hitchcock, Chem. Commun., 2008, 82–84 RSC.
- L. Chatelain, V. Mougel, J. Pécaut and M. Mazzanti, Chem. Sci., 2012, 3, 1075–1079 RSC.
- S. Carretta, G. Amoretti, P. Santini, V. Mougel, M. Mazzanti, S. Gambarelli, E. Colineau and R. Caciuffo, J. Phys.: Condens. Matter, 2013, 25, 486001 CrossRef CAS PubMed.
- J. T. Boronski, L. R. Doyle, A. J. Wooles, J. A. Seed and S. T. Liddle, Organometallics, 2020, 39, 1824–1831 CrossRef CAS.
- J. M. Manriquez, P. J. Fagan, T. J. Marks, S. H. Vollmer, C. S. Day and V. W. Day, J. Am. Chem. Soc., 1979, 101, 5075–5078 CrossRef CAS.
- F. A. Cotton, W. Schwotzer and C. Q. Simpson, Angew. Chem., Int. Ed. Engl., 1986, 25, 637–639 CrossRef.
- L. Barluzzi, M. Falcone and M. Mazzanti, Chem. Commun., 2019, 55, 13031–13047 RSC.
- M. Falcone, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2019, 141, 9570–9577 CrossRef CAS PubMed.
- N. Jori, L. Barluzzi, I. Douair, L. Maron, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2021, 143, 11225–11234 CrossRef CAS PubMed.
- S. Giessmann, V. Lorenz, P. Liebing, L. Hilfert, A. Fischer and F. T. Edelmann, Dalton Trans., 2017, 46, 2415–2419 RSC.
- M. K. Assefa, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2020, 142, 8738–8747 CrossRef CAS.
- V. Lorenz, A. Edelmann, S. Giessmann, C. G. Hrib, S. Blaurock and F. T. Edelmann, Z. Anorg. Allg. Chem., 2010, 636, 2172–2191 CrossRef CAS.
- A. R. Willauer, A. M. Dabrowska, R. Scopelliti and M. Mazzanti, Chem. Commun., 2020, 56, 8936–8939 RSC.
- J. Old, A. A. Danopoulos and S. Winston, New J. Chem., 2003, 27, 672–674 RSC.
- V. Mougel, C. Camp, J. Pecaut, C. Coperet, L. Maron, C. E. Kefalidis and M. Mazzanti, Angew. Chem., Int. Ed., 2012, 51, 12280–12284 CrossRef CAS.
- M. Keener, R. A. K. Shivaraam, T. Rajeshkumar, M. Tricoire, R. Scopelliti, I. Zivkovic, A. S. Chauvin, L. Maron and M. Mazzanti, J. Am. Chem. Soc., 2023, 145, 16271–16283 CrossRef CAS.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef.
- D. N. Huh, J. W. Ziller and W. J. Evans, Inorg. Chem., 2019, 58, 9613–9617 CrossRef CAS PubMed.
- D. N. Huh, S. R. Ciccone, S. Bekoe, S. Roy, J. W. Ziller, F. Furche and W. J. Evans, Angew. Chem., Int. Ed., 2020, 59, 16141–16146 CrossRef CAS PubMed.
- D. N. Huh, C. J. Windorff, J. W. Ziller and W. J. Evans, Chem. Commun., 2018, 54, 10272–10275 RSC.
- D. N. Huh, J. M. Barlow, S. R. Ciccone, J. W. Ziller, J. Y. Yang and W. J. Evans, Inorg. Chem., 2020, 59, 17077–17083 CrossRef CAS PubMed.
- C. A. P. Goodwin, S. R. Ciccone, S. Bekoe, S. Majumdar, B. L. Scott, J. W. Ziller, A. J. Gaunt, F. Furche and W. J. Evans, Chem. Commun., 2022, 58, 997–1000 RSC.
- A. B. Chung, D. N. Huh, J. W. Ziller and W. J. Evans, Inorg. Chem. Front., 2020, 7, 4445–4451 RSC.
- D. R. Kindra and W. J. Evans, Chem. Rev., 2014, 114, 8865–8882 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS.
- J. K. Tang, I. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729–1733 CrossRef CAS PubMed.
- L. Ungur, S. Y. Lin, J. K. Tang and L. F. Chibotaru, Chem. Soc. Rev., 2014, 43, 6894–6905 RSC.
- C. Camp, V. Mougel, J. Pecaut, L. Maron and M. Mazzanti, Chem. – Eur. J., 2013, 19, 17528–17540 CrossRef CAS PubMed.
- B. Vlaisayljevich, P. L. Diaconescu, W. L. Lukens Jr, L. Gagliardi and C. C. Cummins, Organometallics, 2013, 32, 1341–1352 CrossRef.
- G. Nocton, P. Horeglad, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2008, 130, 16633–16645 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2240989, 2304632 and 2304633. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05390c |
| ‡ M. T. and N. J. contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |