
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization & catalysis of ITQ 2D metal–organic frameworks and spectroscopic & photodynamic properties of their composites with organic dyes
Mario
Gutiérrez
 a,
Urbano
Díaz
*b,
Boiko
Cohen
*a and
Abderrazzak
Douhal
*a
a,
Urbano
Díaz
*b,
Boiko
Cohen
*a and
Abderrazzak
Douhal
*a
aDepartamento de Química Física, Facultad de Ciencias Ambientales y Bioquímica, and Instituto de Nanociencia, Nanotecnología y Materiales Moleculares (INAMOL), Universidad de Castilla-La Mancha, Avenida Carlos III, S/N, 45071 Toledo, Spain. E-mail: boyko.koen@uclm.es; abderrazzak.douhal@uclm.es
bInstituto de Tecnología Química, Universitat Politécnica de Valéncia-Consejo Superior de Investigaciones Científicas (UPV-CSIC), Av. de los Naranjos, s/n, 46022 Valencia, Spain. E-mail: udiaz@itq.upv.es
First published on 25th September 2023
Abstract
Metal–organic frameworks (MOFs) have gained significant attention for their broad range of applications in a variety of scientific and technological fields. These materials are crystalline in nature and are formed through the self-assembly of metal ions or clusters with organic linkers via coordination bonds. Typically, MOFs adopt three-dimensional (3D) structures by assembling the subunits in all three spatial directions forming a relatively rigid network. However, in recent years, low-dimensional 2D-MOFs have garnered considerable interest due to their distinctive properties, particularly in gas adsorption/separation, sensing, catalysis, and chemical release applications. 2D-MOFs are created through the assembly of a single layer, resulting in sheet-like structures that provide larger active surface areas, thereby enhancing the diffusion of guest molecules. A notable example of this is the ITQ family of 2D-MOFs, which are formed through the assembly of 1D organic–inorganic structural subunits. The resulting mesoscopic lamellar structure of these MOFs favors interactions with organic molecules and hence improves the catalytic performance. Here, we provide a comprehensive overview of the available synthesis, structural characterization, physico-chemical properties, and specific catalytic performance of ITQ 2D MOF hybrid materials. We examine available strategies to obtain tuneable Lewis and Brønsted acid active sites from the 1D sub-domains as well as provide examples for the application of these MOFs in specific reactions. We also review studies on the effect of the nature of organic spacers as well as that of the metal clusters on the spectroscopic and photodynamical properties of composites formed by these MOFs as hosts with well-known organic fluorescent dyes (guests).
 Mario Gutiérrez | Mario Gutiérrez obtained his degrees in chemistry (2012), MSc (2014), and PhD in nanoscience and nanotechnology (2017) from the University of Castilla-La Mancha (UCLM). Then, he joined the University of Oxford (MMC Laboratory) as a postdoctoral researcher (2019–2020). From 2021, he is an Assistant Professor at UCLM (Faculty of Environmental Sciences and Biochemistry). His research interests are focused on the development and photophysical characterization of novel luminescent porous materials (MOFs, COFs, and HOFs) for applications in luminescence sensing and LED devices. |
 Urbano Díaz | Urbano Díaz is a permanent Scientific Researcher of CSIC at the Instituto de Tecnología Química (ITQ) from 2004. Born in Valencia, he studied chemistry at the University of Valencia, Spain, gaining his PhD in 2002 on the synthesis and characterization of novel delaminated zeolites (Universidad Politécnica de Valencia). He was awarded with a post-doctoral European Marie Curie fellowship at EniTecnologie in Italy on the catalytic applications of organic–inorganic hybrid materials from functional silsesquioxanes. In 2004, he returned to the ITQ where he has since worked in the field of multisite hybrid materials, layered solids, nanocomposites and multi-component materials with a particular focus on the effective combination between organic and inorganic structural builders. |
 Boiko Cohen | Boiko Cohen studied chemistry at Sofia University, Bulgaria, and obtained his diploma in 1997. He got his PhD in 2003 from Tel Aviv University, Israel under the supervision of Prof. Dan Huppert and Prof. Noam Agmon. After postdoctoral research at the Ohio State University and Northwestern University, he joined the University of Castilla – La Mancha in 2007 as Ramon y Cajal Fellow. He has been an Associate Professor at the University of Castilla – La Mancha since 2012. His main research interest is in the fast and ultrafast dynamics of photoinduced processes in solution and in confining chemical and biological media. |
 Abderrazzak Douhal | Abderrazzak Douhal (Femtochemistry and Microscopy (uclm.es)), a full Professor of physical chemistry at the University of Castilla La Mancha (UCLM, Toledo), has received his MSc and PhD degrees in chemistry from the University of Kadi Ayyad (Marrakech) under the supervision of Prof. J. L. Abboud and A. U. Acuña. From 1986 to 1990, he worked at the Institute of Physical Chemistry of CSIC (Madrid). During 1990–1992, he was a post-doctoral student at the Institute for Molecular Science (Okazaki, Japan) working in the group of Prof. K. Yoshihara. In 1992, he worked as a researcher associate to Prof. Françoise Lahmani at Laboratoire de Photophysique Moléculaire (University of Paris-Sud/CNRS, France). In 1993, he joined the UCLM as an assistant professor and in 1998 as an associate professor. He was a visiting researcher at the California Institute of Technology in several periods of 1995–2000, working in the group of Prof. Ahmed H. Zewail. Since 1998, he is heading the Femtoscience and Microscopy research group at the UCLM, focusing his research on the study of photoevents in condensed phase and advanced materials (silica-based materials, MOFs, COFs, HOFs and perovskites). In 2018, he was awarded the Elsevier Lecture Award from the Japanese Photochemistry Association. |
1. Introduction
Nowadays, nanoscience has become one of the most important branches of science and technology, providing high impact solutions to fulfil the demands of modern society, with organic–inorganic hybrid materials being at the forefront of these advances. The high chemical versatility that these materials offer has woken the interest of multidisciplinary researchers, since it is possible to rationally design and synthesize novel structures with controlled physico-chemical properties.1,2 In fact, these hybrid materials are being successfully implemented in a wide range of applications, from the most conventional to more nanotechnological ones, such as those related to photoresponsive applications.3–5 In this review, we focus on the development of novel 2D metal–organic frameworks (MOFs), structured from 1D builder sub-units, showing their more relevant properties and characteristics and evidencing their suitable properties to be used as catalysts and photoactive materials depending on their structure, morphology and composition.6Traditionally, it is possible to use different organic and inorganic structural units for the synthesis of 3D ordered porous frameworks, which through a self-assembly process generate specific metal–organic structures, MOF-type, with different properties and levels of structuration.7 Following this approach, a large number of this class of hybrid materials were described in the literature due to the possibility of using a great variety of inorganic units and organic ligands in the synthesis process.8–10 Furthermore, taking advantage of the versatility and reactivity of certain organic units, it is possible to modify and introduce new functionalities in the metal–organic structures through different post-synthesis treatments.11,12 At the borderline of the classical MOFs, in the last few years, new families of hybrid materials based on in situ generated 1D builder sub-units were prepared through solvothermal synthesis processes, using different types of metallic inorganic precursors and monotopic organic spacers usually not used up to that point for the preparation of conventional MOFs.13 Furthermore, post-synthesis modification was considered in some of these obtained materials for providing further functionalities to the hybrid network.14 In this way, MOFs with specific catalytic and photoactive properties were successfully generated.
This review is organized into several sections, with a summary of the main topics depicted in Scheme 1. After the Introduction section, we briefly comment on the novelty, advantages, and use of monotopic organic linkers for the synthesis of new 2D MOFs (Section 2). Section 3 focuses on the synthesis, structural characterization, physico-chemical properties and specific catalytic performance of MOF hybrid materials based on Al nodes and monotopic organic spacers. Section 4 examines the preparation procedures and characterization of 2- and 3D MOFs with tuneable Lewis acidity from 1D sub-domains. Section 5 is dedicated to MOFs based on 1D structural sub-domains with Brønsted acid and redox active sites as effective bifunctional catalysts. Section 6 examines the spectroscopic and photodynamical properties of composites formed by these 2D MOFs as hosts and two well-known organic fluorescent dyes (guests). The review ends with the Challenges and perspectives and Conclusions sections.
 | ||
| Scheme 1 Schematic representation of the topics and most important concepts covered in this review. | ||
2. Monotopic organic linkers for the synthesis of new metal–organic structures
Here we will describe the characteristics of the monomeric organic ligands that act as builder units of MOFs, as well as their ability to connect different oxy-metallic inorganic nodes with each other and their spatial arrangement, determine the order, regularity and structuring achieved by the resulting hybrid materials.15 The use of specific organic ligands with different coordination properties together with the employed solvothermal synthesis conditions is decisive to establish the organization and morphology of the obtained metal–organic hybrid materials,16–18 particularly when monotopic organic spacers are used.Traditionally, the formation of MOFs is based on the use of at least two types of structural building units with different characteristics and properties: one with inorganic nature, providing high thermal and mechanical stability, and the other with organic nature, providing associated flexibility and extra-functionalities. Normally, for the formation of these MOF-type materials, the organic building unit contains at least two reactive points capable of coordinatively interacting with the metal ions or clusters, although sometimes they may contain three or four connection points, which facilitate the formation of a crystalline, highly stable and, in some cases, rigid metal–organic structures after suitable solvothermal assembly processes.19
As a recent novelty, in the present review, a series of MOFs are described where monotopic organic spacers with only one point of interaction (in general carboxylic groups) combined with alkyl chains located in para positions of aryl groups are employed. This type of builder unit exerts a double function: as a structural agent and as a growth modulating assistant. Therefore, with the use of these monotopic ligands, the inhibition of the growth of the structure in the three directions of space is favoured, preventing the expansion during the synthesis process of the MOFs and favouring the generation of hybrid structures based on the in situ formation of 1D organic–inorganic sub-domains. The sub-domains are then organized and assembled along different dimensionalities and morphologies.13,20
These metal–organic structures are formed by inorganic chains composed of the association of metal octahedra, being the most frequent AlO4(OH)2. These are coordinated with monotopic organic spacers, generating 1D organic–inorganic structural units whose long-range associations form further families of MOF-type materials, depending on the nature of the metallic nodes used and the characteristics of the organic spacers involved in the solvothermal synthesis process. Specifically, aluminium octahedra are linked together through hydroxyl groups located in trans positions, typically present in inorganic materials based on trivalent nodes,21,22 such as those described in conventional aluminium MOFs similar to MIL-53 (Al).23–25Fig. 1 represents 1D sub-domains that are expected to form with greater probability during the synthesis process in the presence of monotopic organic ligands, being the fundamental builder units for the preparation of these new families of MOFs.
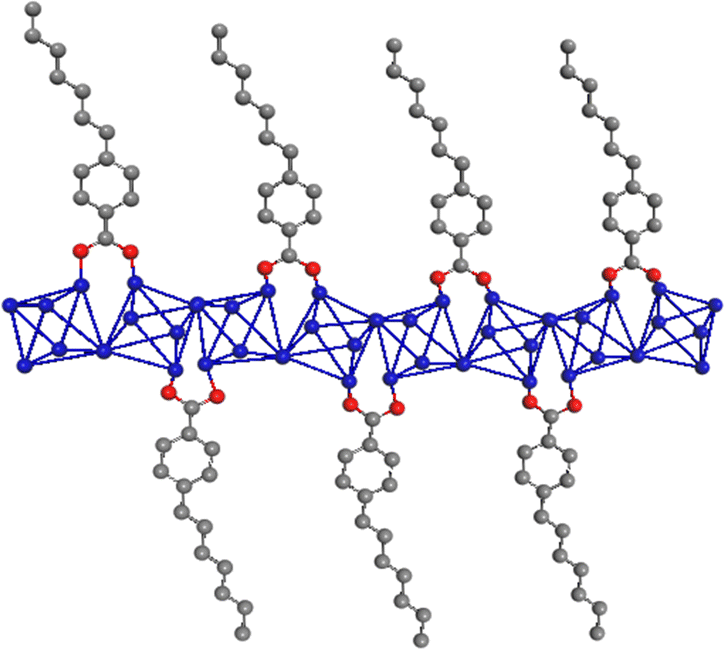 | ||
| Fig. 1 Schematic presentation of 1D subdomains of metalorganic structures with an organic spacer number/Al ratio of 1. In blue, a chain based on associated aluminium octahedra following a Kagomé conformation; in grey, hepthylbenzoate monodentate spacers; in red, oxygen atoms from carboxylate groups. | ||
The influence of different synthesis conditions on the properties and morphological characteristics finally achieved in these families of metalorganic structures should be considered, such as pressure, temperature, time and, especially, the nature of solvents.26 These experimental conditions can direct towards the formation of one or another type of hybrid structure depending on the associations that are favoured between the organic and inorganic low-dimensional building units formed in the solvothermal synthesis medium.27
3. Metal–Organic hybrid materials based on aluminium metal nodes and monotopic organic spacers
3.1 Structuration level and physico-chemical properties
The obtained results showed the importance of the solvothermal synthesis medium to reach different structuration levels in the metal–organic networks formed by 1D building units favoured by the use of monotopic organic linkers. In particular, it was confirmed that when only water was used as the synthesis medium, more compacted metal–organic structures were promoted, while if DMF was used as a solvent, the reagents achieved a higher degree of dispersion, generating less crystalline mesoscopic networks.28 Interestingly, when a mixture of both solvents (DMF and water) was employed, each builder unit was completely dissolved in one of the solvents separately, generating an interface in which associated 1D sub-domains in the form of organic–inorganic sheets were formed (Fig. 1).Specifically, the first family of MOFs obtained was based on the use of water as the only solvent. In this case, X-ray diffraction patterns of the obtained materials resembled those of the conventional 3D MIL-53 (Al)-type materials,29 regardless of the length of the aliphatic chain present in the monotopic organic ligand (ethylbenzoic acid, EB, or heptylbenzoic acid, HB) (Fig. 2a). These results support the hypothesis that structurally more compact 3D materials were formed, similar to the standard MOFs. This confirmed that the hybrid materials are isoreticular structures similar to MIL-53 (Al)-type materials (MIL-53(Al)-EB/HB).30
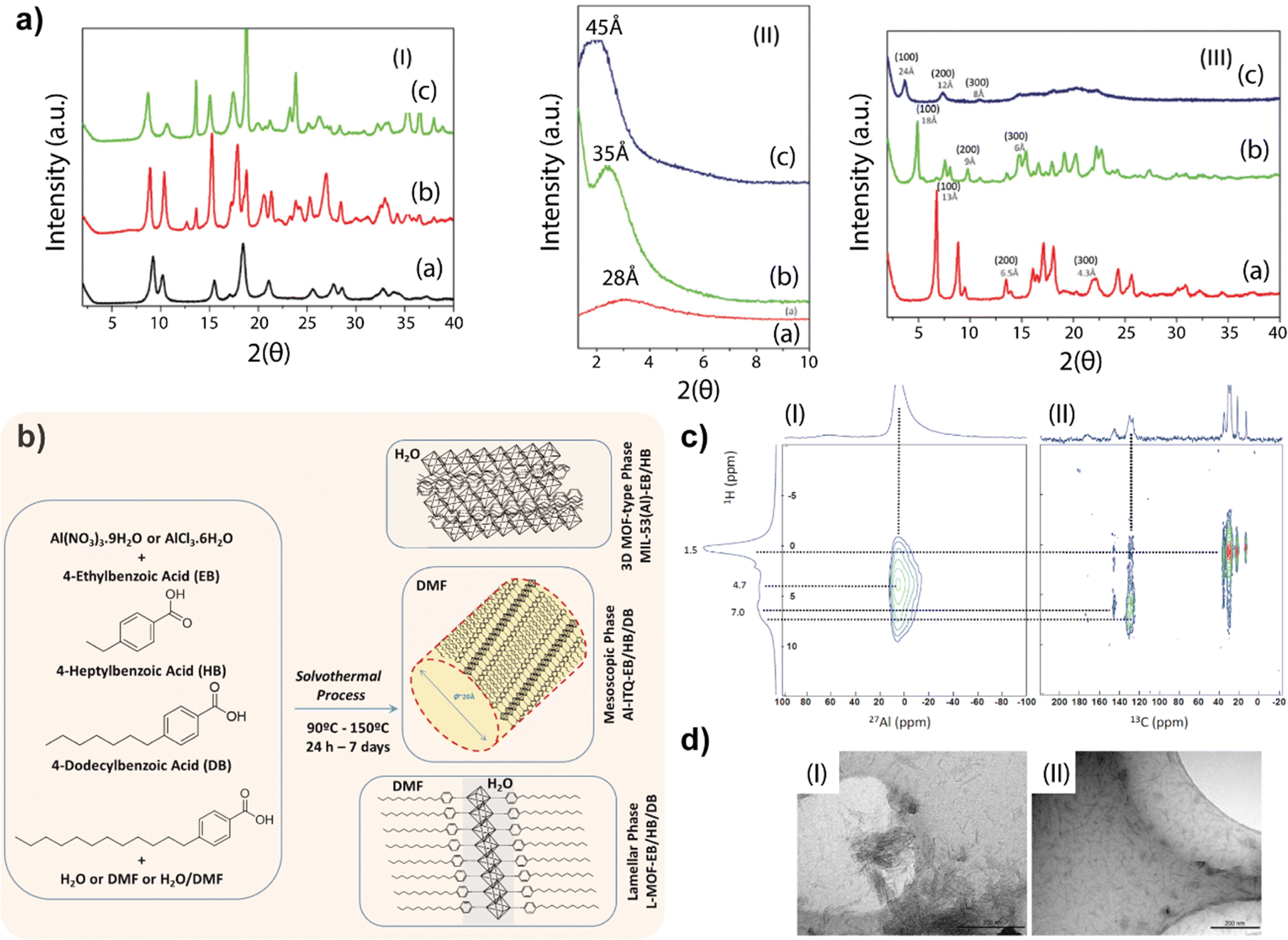 | ||
| Fig. 2 (a) (I) PXRD patterns of isoreticular MIL-53(Al)-type MOFs: (a) standard 3D MIL-53(Al), (b) MIL-53(Al)-EB and (c) MIL-53(Al)-HB. (II) PXRD patterns of mesoscopic samples: (a) Al-ITQ-EB, (b) Al-ITQ-HB and (c) Al-ITQ-DB. (III) PXRD patterns of lamellar MOFs: (a) L-MOF-EB, (b) L-MOF-HB and (c) L-MOF-DB. (b) Synthesis routes for obtaining aluminium-MOF-type materials with different morphologies based on the specific structural assembly of 1D nanoribbons as a function of solvothermal conditions. (c) 2D NMR spectra: (I) 1H and 27Al and (II) 1H and 13C (HETCOR) CP-MAS NMR for the Al-ITQ-HB material. (d) TEM images of the exfoliated MOFs with dichloromethane: (I) Al-ITQ-EB and (II) Al-ITQ-HB materials. Reproduced from ref. 28 with permission from the Royal Society of Chemistry. | ||
A second family of hybrid materials was obtained in the presence of monotopic ligands with alkyl groups in the para position and using only DMF as a solvent in the synthesis process.28 In this case, the X-ray diffraction patterns showed a single band (100) at low 2θ angles positions when specific organic spacers such as EB, HB and DB, respectively, were used (Al-ITQ-EB/HB/DB). The obtained results evidenced that the existence of a mesoscopic type of organization is possible. As it was also observed in the standard M41S materials,31 the mesoporous channels formed exhibited larger internal diameters when the length of the hydrocarbon chain of the incorporated organic unit was longer. This fact was corroborated by the position of the diffraction bands (100), which moved to lower diffraction angles 2θ as the length of the alkyl chain was greater. These results indicate that the use of DMF as the only solvent resulted in the complete dissolution of both organic and inorganic building units in the same medium. Due to the high affinity between the organic molecules and the DMF used as a solvent, a high dispersion of the solved structural sub-domains occurs, favouring the micellar-type self-assembly.
The last family of materials was obtained using a mixture of solvents, like DMF/MiliQ water, giving rise to a lamellar-type structure based on the association of 1D organic–inorganic units assembled along the ab plane and separated by the monotopic organic spacers (EB, HB or DB), which were perpendicularly positioned with respect to the aluminium nodes (L-MOF-EB/HB/DB).28 The obtained basal space was different for each sample (Fig. 2b), indicating that the variation in the separation between the individual sheets depended on the length of the organic spacer used during the synthesis process, which acted as a modulator of the growth of the materials, inhibiting 3D structuring and favouring the formation of lamellar metal–organic structures. In addition, the presence of the diffraction band (100) together with the bands assigned to the diffraction orders (200) and (300), confirmed the high regularity obtained in the distance between the successive individual layers. This mixture of solvents allowed the complete dissolution of both structural units separately, the organic spacer preferably being dissolved in DMF and the inorganic unit in the aqueous solvent. In this way, an interface was generated where the two types of construction units came into close contact, allowing a lamellar-type ordering.
Fig. 2b shows the different families of MOFs obtained using various solvents by the solvothermal process, roughly illustrating the most probable morphology obtained. It is remarkable that in all cases the same 1D organic–inorganic sub-domains, shown in Fig. 1, were forming the hybrid material network.
From IR and NMR spectroscopies, it was corroborated that the groups coming from the different organic units were coordinatively anchored to the aluminium octahedra, remaining intact as well as retaining their initial chemical configuration and preserving their composition. Additionally, 27Al-NMR spectra of the solids evidence that practically all aluminium atoms were incorporated into the structural network in octahedral positions. These formed aluminium octahedra were connected to form inorganic chains present in the 1D sub-domains. Furthermore, NMR measurements in two dimensions of the spectra of 1H with 27Al and 1H with 13C were carried out, confirming the proximity between the aluminium nodes and the carbon atoms of the aromatic rings and of the aliphatic chains of the used organic spacers (Fig. 2c). Specifically, this experiment was carried out for the Al-ITQ-HB material, showing a correlation between the protons of the aromatic ring and the alkyl chain of the 4-heptylbenzoate ligand at ∼7 ppm and ∼1.5 ppm, respectively, with the ∼6.5 ppm signal of the octahedral aluminium nodes. This result implies the effective formation of the 1D organic–inorganic units as structural sub-units, which are associated with each other to form the different MOF-type materials obtained depending on the synthesis conditions used.
On the other hand, the presence of low-dimensional structural sub-domains in the different networks that formed the hybrid materials was confirmed through post-synthesis treatments with solvents of different polarity (methanol, dichloromethane (DCM), diethyl ether (DEE) or toluene). For this, the obtained solutions were analyzed by TEM (Fig. 2d), confirming the presence of small isolated 1D nanostructures, generated after the expansion and exfoliation of the initial metal–organic structures. The possibility of isolating these 1D sub-domains through an exfoliation process allows their use to form further types of nanomaterials when they are combined with other sub-structures.
3.2 Al-MOFs like active and stable catalysts for the synthesis of fine chemicals. Synthesis of benzodiazepines and asymmetric transformations
The use of MOFs that present defects (CUSs, coordinatively unsaturated sites) in their structural network has been of great relevance in recent years to carry out different catalytic processes of interest.32 This phenomenon is based on the existence of free or vacant coordination positions in the inorganic building units. In the case of MOFs, the inorganic nodes may be weakly coordinated with their ligands, with some of them being easily removed without collapsing the crystal structure of which they are a part.33 In the materials reviewed here and formed by low-dimensional structural units, the use of specific monocarboxylic organic spacers favours the formation of free specific positions in the inorganic structure unit. For all these reasons, it was possible to observe the presence of sites with Lewis acidity in these hybrid materials thanks to the presence of CUS in their structure.34 Therefore, taking into account the textural and structural characteristics of the hybrid materials discussed here, their capacity as catalysts for the production of benzodiazepines was studied. This class of products is widely used in the pharmaceutical industry having therapeutic applications, being used as anticonvulsants, analgesics, sedatives, antidepressants and hypnotic agents. Furthermore, derivatives of these compounds are useful as dyes in acrylic fibres and are a valuable intermediate in the synthesis of triazolo-, oxadiazole-, oxazino- and furanobenzo-diazepines.35Generally, benzodiazepines are synthesized through condensation between 1,2-phenylenediamine and carbonyl compounds in the presence of catalysts that exhibit Lewis acidity.36 For this, the catalytic capacity of the hybrid materials, MIL-53(Al)-HB, Al-ITQ-HB and L-MOF-HB, was studied (Fig. 3a) to verify the effect of the different structuration and morphology of each of them in the cyclic condensation of 1,2-phenylenediamine with acetone to obtain 2,3-dihydro-2,2,4-trimethyl-1H-1,5-benzodiazepine, also known as 1,5-benzodiazepine. The reaction kinetics showed a similar catalytic behaviour for MIL-53(Al)-HB and Al-ITQ-HB materials. The reduced reaction rate obtained for the L-MOF-HB catalyst was attributed to the high organic content included in its structure, which is positioned in the interlayer space, causing diffusional problems, and preventing the arrival of the reactants to the active sites of the hybrid catalysts. Furthermore, the leaching phenomenon was not observed during the process and the high catalytic activity presented by the hybrid material MIL-53(Al)-HB to produce 1,5-benzodiazepine was exclusively assigned to the presence of unsaturated aluminium coordination sites (Fig. 3b and c). Finally, the stability and recyclability of the MIL-53(Al)-HB material were studied during several consecutive reaction cycles, evidencing how this hybrid material maintained its high catalytic activity during all uses. This fact would open the door to employ this type of material as an efficient solid acid catalyst in other fine chemical processes and, in particular, in those in which intramolecular cyclizations take place.37,38
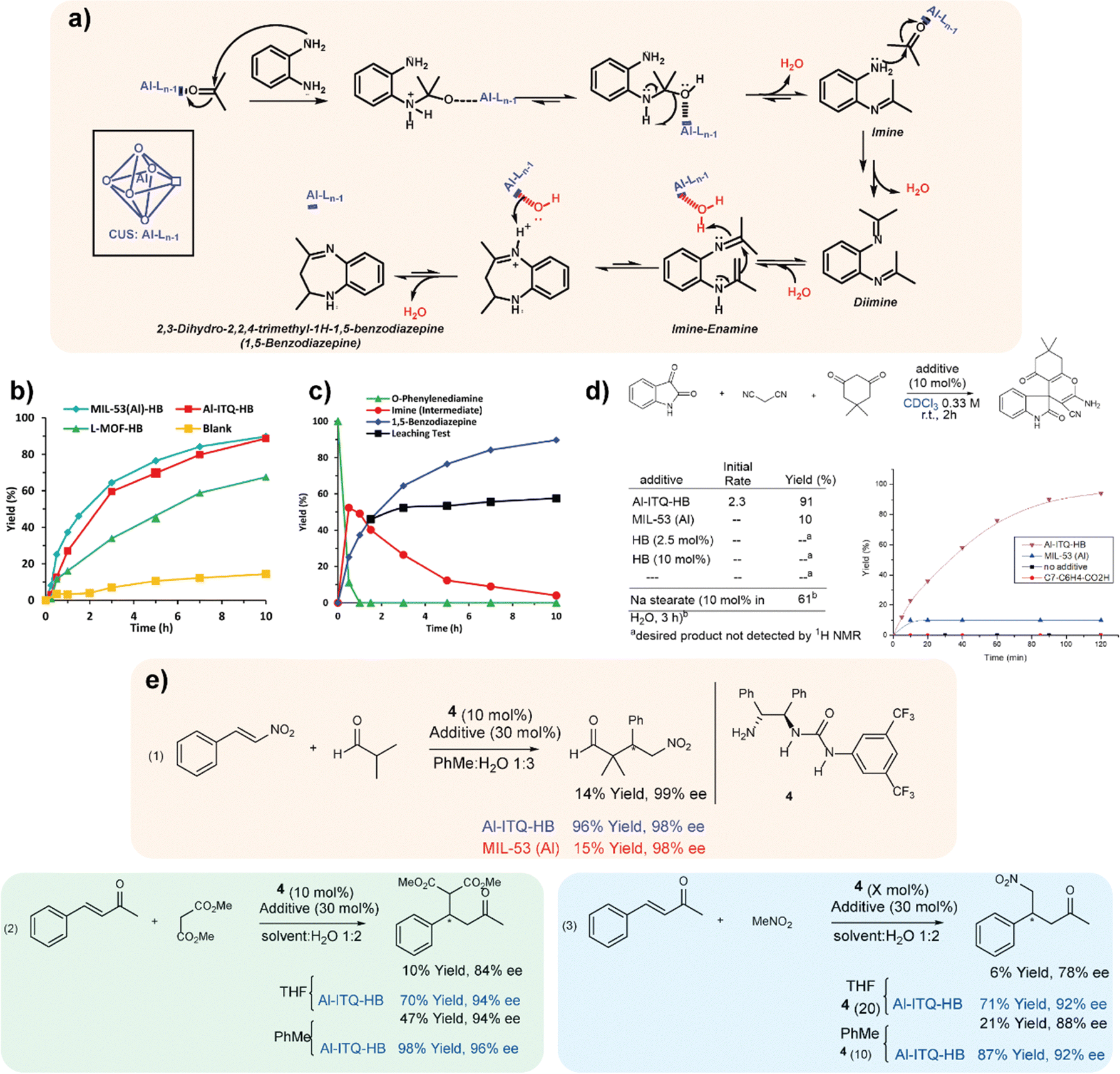 | ||
| Fig. 3 (a) Mechanism for the formation of 1,5-benzodiazepine. (b) Catalytic activity for the production of 1,5-benzodiazepine with the materials: MIL-53(Al)-HB (◊), Al-ITQ-HB (■) and L-MOF-HB (▲). Blank (●). (c) Catalytic activity with the MIL-53(Al)-HB material, observing the disappearance of 1,2-phenylenediamine (▲), the appearance and consumption of the reaction intermediate (●) and the production of 1,5-benzodiazepine (◊). Production of 1,5-benzodiazepine after removal of the catalyst (■).28 (d) Al-ITQ-HB catalyzed multicomponent reaction for the synthesis of spirooxindole product. (e) Enantioselective organocatalytic Michael reactions promoted by Al-ITQ-HB. Organocatalyst 4 was used in the amount of 10 mol% in all cases, except noted. Additives used were Al-ITQ-HB (results highlighted in blue) or MIL-53 (Al) (results highlighted in red) in the amount of 30 mol%. Reproduced from ref. 39 with permission from Springer Nature. | ||
The reactivity exhibited by this type of MOFs based on low-dimensional building units was extended to another relevant reaction processes, such as condensation reactions in water in the absence of acidic or basic catalysts, as well as organocatalytic Michael-type reactions that also showed superior enantioselectivity when comparing with the host-free transformation.39 Specifically, the mesoscopic Al-ITQ-HB catalyst was effective for the Knoevenagel reaction under mild conditions, being efficient for the condensation of a variety of aromatic aldehydes. Comparative experiments employing conventional MOFs, such as 3D MIL-53 (Al), showed no relevant conversion, remarking the important role of the hydrophobic sub-domains present in Al-ITQ-HB materials and confirming that substrate activation was not a simple consequence of Lewis acid activation by the network or the influence of π-stacking interaction of the aromatic aldehydes with the organic–inorganic network of the catalyst. The reaction process proceeded better with aromatic aldehydes containing electron donating groups, although rate acceleration was detected in all cases.
The reactivity of the mesoscopic hybrid material Al-ITQ-HB was also evaluated in the multicomponent reaction between isatin, dimedone and malononitrile for the synthesis of the spirooxindole product (Fig. 3d), through micellar catalysis in water by means of sodium stearate.40 One more time, in the presence of water in the reaction medium, an improved rate was observed for the Al-ITQ-HB catalyst compared to the MOF-free reaction under similar conditions. These excellent catalytic results were extended to asymmetric organocatalytic Michael-type reactions of isobutyraldehyde to nitrostyrene41 and even for Michael's addition of nitromethane to 4-phenyl-3-buten-2-one.42,43 This fact allowed establishing as general that our host system boosted the organic reactions in aqueous media (Fig. 3e).
4. 2D and 3D MOFs with tuneable Lewis acidity from preformed 1D hybrid sub-domains
4.1 Preparation and characterization
Considering the previous results, the interest in using specific monotopic organic spacers with different functional groups in their initial composition has elevated, being possible to influence the type of structuration achieved and the final reactivity of the hybrid materials obtained. In all cases, monotopic organic ligands were used, being of special relevance the introduction of functions in their composition located in the para position with respect to the benzylic ring. Likewise, the presence of electron-donor or electron-withdrawing substituent groups determines both the textural and catalytic properties of the synthesized MOFs.44–46Specifically, four new hybrid solids were prepared through the presence of different functional groups (nitro, bromine, ethyl and amino), which were found in the used monotopic organic spacers.47 In the selection of the aforementioned groups, two of them were chosen that exhibited the particularity of electron-attracting character (EWG, electron-withdrawing groups), nitro and bromine, which removed electron density from the conjugated system, making it more electrophilic. Thanks to the incorporation of these two types of substituents, a lower negative charge density in the carboxylic groups of the organic ligands was favoured. Therefore, the non-coordinated metallic centres (CUS, Coordinative Unsaturated Sites) included in the metallic nodes of the MOFs exhibited higher Lewis acidity.48,49 Furthermore, due to their ability to attract electrons, these EWG groups showed a high charge density. Therefore, electronic fields and dispersion forces were generated between the organic spacers themselves, inducing a certain disorder in the formation of the structural sub-domains, which facilitated the generation of 3D MOFs with a more compact structure. This was also favoured by the ease of self-assembly of the organic ligands and the aluminium octahedral units due to the highly polar nature of the organic units, which were easily stabilized in the synthesis medium (DMF/water). In consonance, the diffractograms obtained for these hybrid materials (Al-ITQ-Br and Al-ITQ-NO2) were more similar to those of the conventional 3D material MIL-53(Al) (Fig. 4). Similar to these materials and taking into account the possible use in different catalytic applications, the preparation of MOFs that contain more than one type of metallic nodes in their structure is of great interest, due to the possibility of carrying out different catalytic processes, in which each metal centre exhibits a singular role or can be favoured a cooperative effect, increasing the initial reactivity. For these reasons, bimetallic MOFs with a 3D structure were prepared, called Al/Fe-ITQ-NO2, in which the organic spacer with nitro groups in the para position of the aromatic rings and two different inorganic sources were used, one of aluminium and other of iron.47 Additionally, an EDS-STEM analysis confirmed a homogeneous distribution, along the metal–organic structure, of the two types of metallic nodes, iron and aluminium, present in the hybrid material Al/Fe-ITQ-NO2 (Fig. 5).
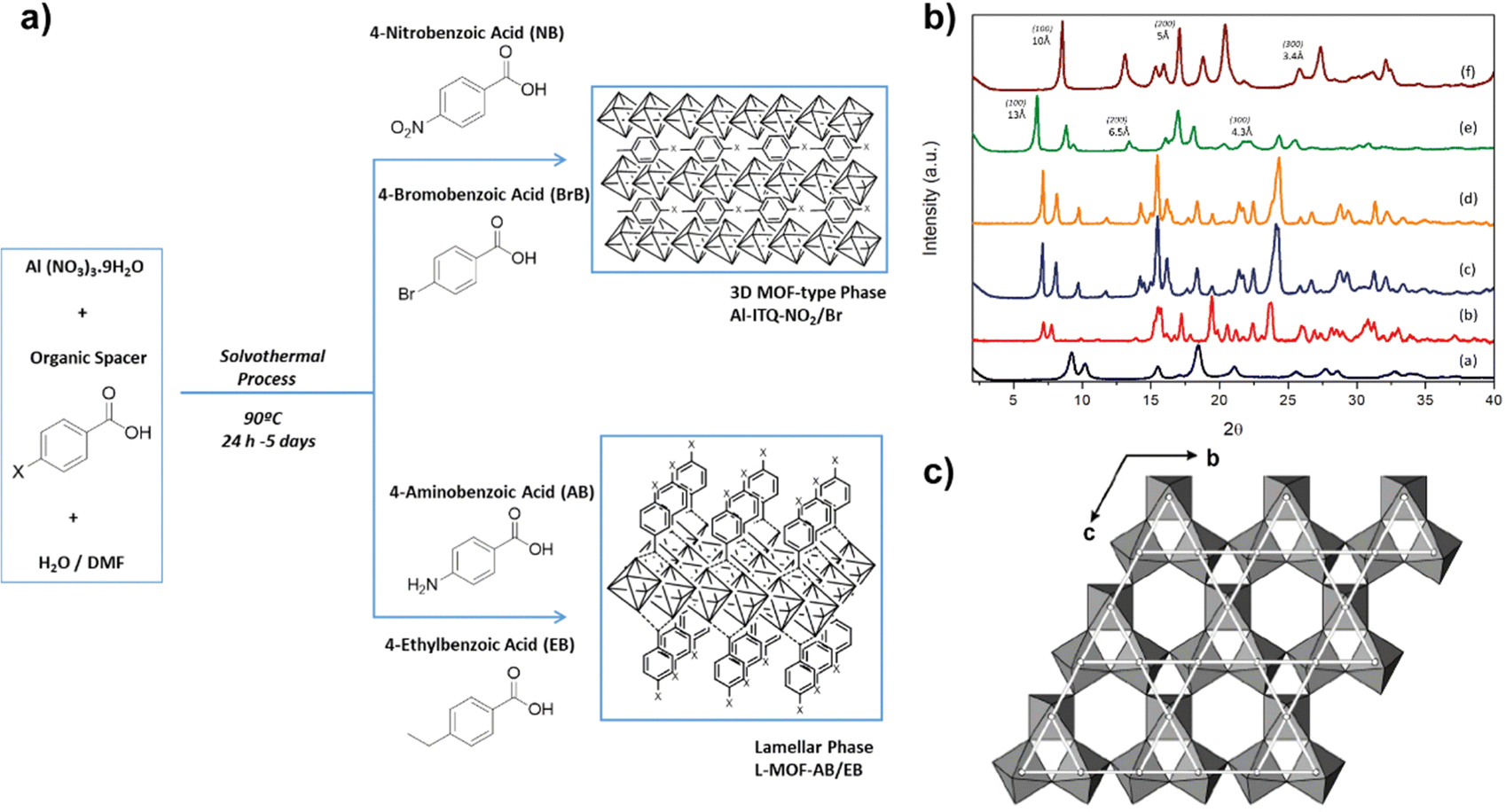 | ||
| Fig. 4 (a) Synthesis routes used to obtain MOFs with different morphologies using monotopic ligands with different substituent groups in the para position of the aromatic ring. (b) X-Ray diffractograms of the synthesized hybrid materials: (a) MIL-53(Al), (b) Al-ITQ-Br, (c) Al-ITQ-NO2, (d) Al/Fe-ITQ-NO2, (e) L-MOF-EB and (f) L-MOF-AB. (c) Representation of the layers present in the solids L-MOF-AB and L-MOF-EB, based on the association of aluminium octahedral in a Kagomé system. Spacers were omitted for clarity. Reproduced from ref. 47 with permission from the Royal Society of Chemistry. | ||
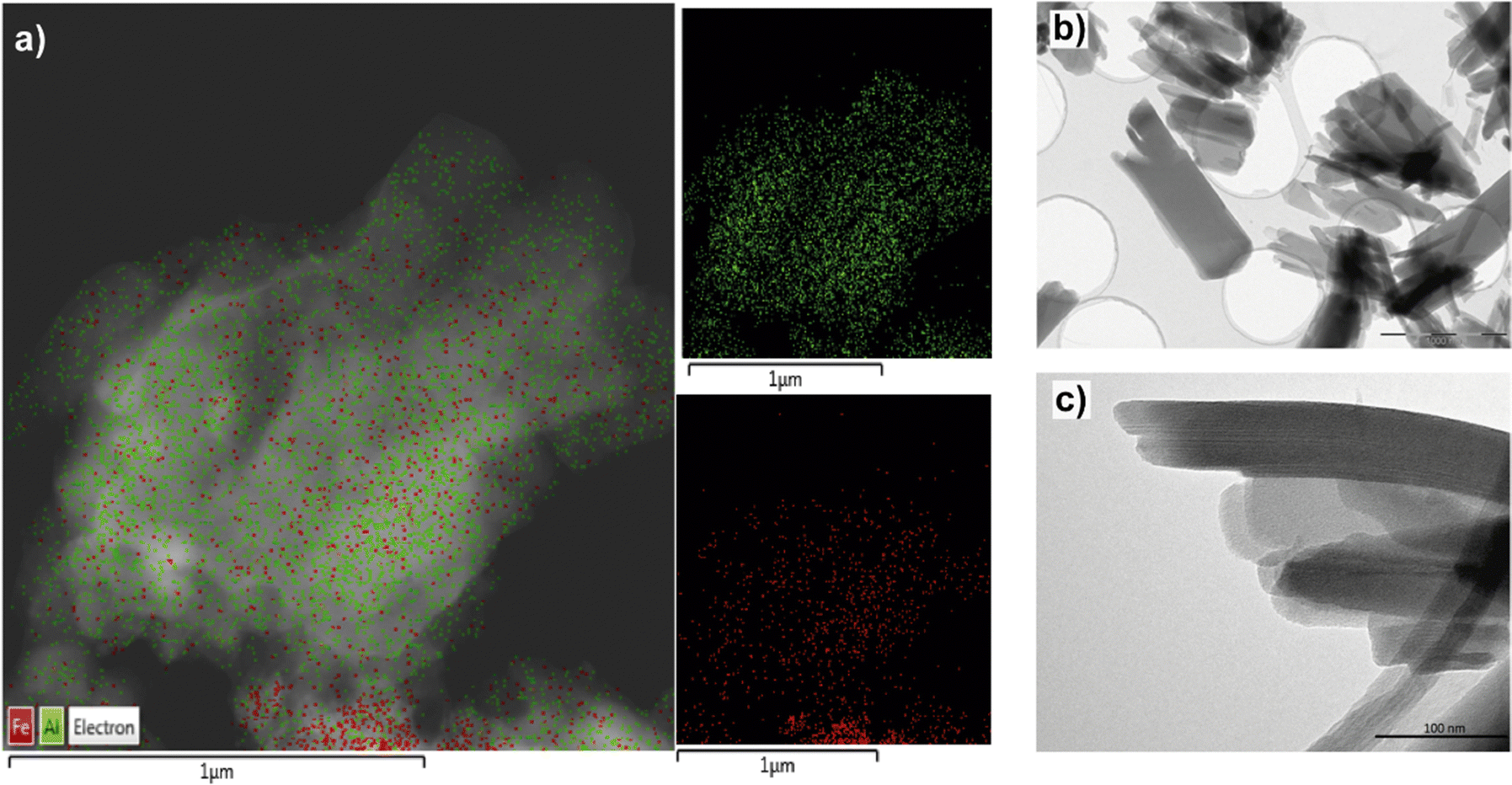 | ||
| Fig. 5 (a) EDS-STEM analysis for the two metallic elements, Al and Fe, incorporated in the 3D material Al/Fe-ITQ-NO2. (b) and (c) TEM images of the 2D material L-MOF-AB. Reproduced from ref. 47 with permission from the Royal Society of Chemistry. | ||
In contrast, the other two selected substituent groups, amino and ethyl, showed the ability to donate electrons (EDG, Electron-Donating Groups), providing electron density to the conjugated system and making the aromatic ligands more nucleophilic.47 Due to the incorporation of these two groups, an increase in the negative charge density of the carboxylic groups would be expected. Therefore, the uncoordinated metal centers present in the inorganic building units of MOFs exhibit lower Lewis acidity. From a structural point of view, it should be noted that although these two functional groups exhibit different chemical properties, a similar lamellar morphology was achieved. This morphology was favoured by the presence of amino groups, which could establish hydrogen bonds between them, facilitating a regular lamellar organization between these organic spacers during the solvothermal synthesis process. In the case of ethyl groups, they showed the tendency to remain ordered among themselves, because of the van der Waals forces that were established. Thus, in both cases, 2D materials with a lamellar morphology were obtained (L-MOF-AB and L-MOF-EB) (Fig. 4). Specifically, these individual sheets are formed by the association between the 1D organic–inorganic units, which favour a 2D order, with separated layers by the organic ligands that are located in the interlayer space. In this case, the aluminium octahedral is organized in the bc plane, following a Kagomé-type structural system, characteristic of lamellar materials formed by trivalent metal octahedra, forming sheets composed of hexagonal rings delimited by six triangular rings.50 TEM images of the samples obtained when electron donor groups (–NH2 and –C2H5) were incorporated into the final metalorganic network confirmed the lamellar morphology of the L-MOF-EB and L-MOF-AB hybrid materials (Fig. 5).
4.2 Synthesis of cyanohydrins and aerobic oxidation of thiols to disulfides
These hybrid materials were successfully used as heterogeneous catalysts due to the presence of uncoordinated metal centers (CUS), which can act as Lewis-type acids.51 The different hybrid materials exhibited variable Lewis acid strength in the function of substituent groups present in the organic building units. Electron donor groups (–NH2 or –C2H5) generated Lewis sites with a lower acid strength, while the incorporation in the metalorganic networks of electron-attracting groups (–NO2 and –Br) produced Lewis sites with higher acidity. In addition, it was important to consider the difference in the morphology of the synthesized hybrid materials, since it modified their textural properties, hydrophobic/hydrophilic character, porosity and accessibility to the active sites. Considering these reasons, the importance of the functionalized monotopic organic spacers was analyzed by comparing the catalytic behaviour of the different obtained hybrid materials in the production of cyanohydrins through the cyanosilylation of carbonyl compounds, which constitutes an important route to produce products containing the CN group in its composition through C–C bond formation reactions. The generated compounds are of great interest as intermediates in the synthesis of α-hydroxy acids and α-amino acids.52,53In the case of benzaldehyde cyanosilylation with cyanotrimethylsilane (TMSCN), without using solvents in the reaction medium, the catalytic results were better when the reaction was carried out in the presence of more compact 3D materials (Al-ITQ-NO2 and Al-ITQ-Br), which contained electron-withdrawing groups (Fig. 6a and b). The results showed that the organic ligand with the greatest capacity to attract electrons contributed to the formation of more active Lewis sites, and the catalyst Al-ITQ-NO2 presented higher activity (∼99%). An illustrative way of seeing the influence of the different substituents on the obtained kinetic results was achieved by plotting the relative initial velocities against Hammet's constant (σpara) in each case (Fig. 6c).
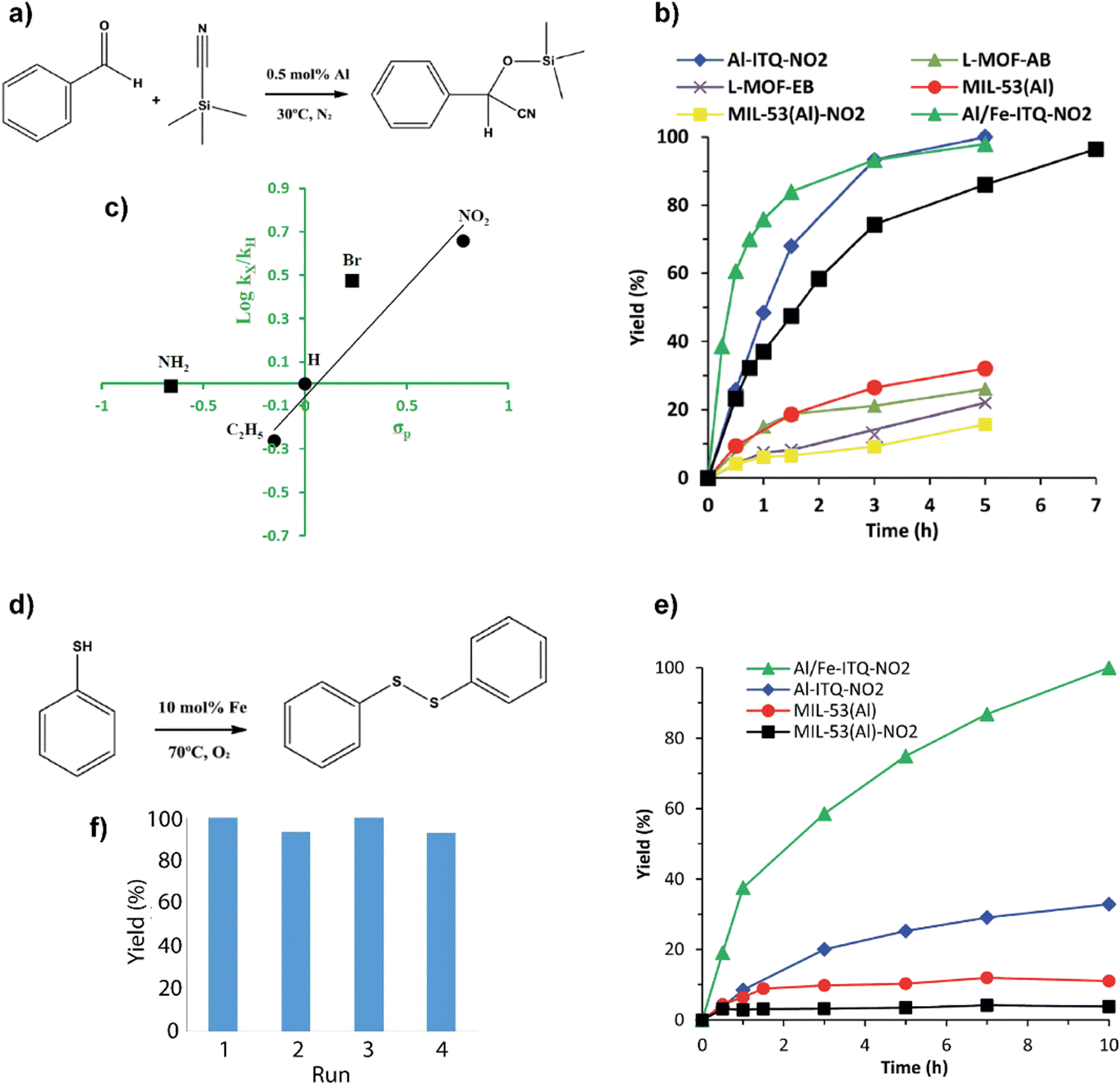 | ||
| Fig. 6 (a) Benzaldehyde cyanosilylation reaction. (b) Kinetics data obtained for the production of cyanohydrins with the different metalorganic materials. The reaction blank reached a 24% yield after 5 hours of reaction. (c) Plot of Hammet's constant against relative initial rates obtained for benzaldehyde cyanosilation with the different Al-ITQ-X catalysts (–X: –NO2, –Br, –NH2 and –C2H5). (d) Oxidation of thiophenol to diphenyl disulfide. (e) Kinetic curves for the production of diphenyl disulfide using Al/Fe-ITQ-NO2, Al-ITQ-NO2, MIL-53(Al) and MIL-53(Al)-NO2 as heterogeneous catalysts under O2 atmosphere. The reaction blank reached 3% conversion after 10 h of reaction. (f) Yields for four consecutive uses in the thiophenol oxidation reaction using Al/Fe-ITQ-NO2 material as a catalyst under an O2 atmosphere. Reproduced from ref. 47 with permission from the Royal Society of Chemistry. | ||
In addition, catalytic results showed that the incorporation of iron provided additional advantages (Al/Fe-ITQ-NO2), being capable of carrying out different catalytic applications, and even positively modifying the catalytic properties of the aluminium sites.54,55 Specifically, the aerobic oxidation of thiols to disulfides is an important process to obtain interesting chemical compounds in different biological applications, in which the use of molecular oxygen as an oxidizing agent presents a sustainable and environmentally favourable way of carrying out this type of processes.56,57 For all these reasons, the activity of the synthesized bimetallic hybrid material (Al/Fe-ITQ-NO2) was explored by using it as a catalyst in the aerobic oxidation of thiophenol to diphenyl disulfide, using acetonitrile as a reaction solvent (Fig. 6d and e). The results showed high yields (100%) to the desired product in the presence of the Al/Fe-ITQ-NO2 material, while much more reduced were reached when solid Al-ITQ-NO2 or conventional MIL-53(Al) MOFs were used in the same reaction time. Heterogenization of active functions through lixiviated tests and recyclability was clearly confirmed for the used hybrid catalysts. Furthermore, when iron was successfully incorporated into the metal–organic structure, exhibited higher catalytic activity in the benzaldehyde cyanosilylation, presenting a positive effect combined with the structural aluminium, modifying and increasing the final activity of the Lewis acid sites (Fig. 6f).
5. MOFs based on 1D structural sub-domains with Brønsted acid and redox active sites as effective bi-functional catalysts
5.1 Preparation and characterization
With the objective to increase the reactivity of the metal–organic solids based on low-dimensional building units, a monotopic organic spacer with thiol groups was used in the para position with respect to the aromatic ring, whose subsequent oxidation generated sulfonic groups are active in acid-demanding reactions. In addition, thiol groups were employed as stabilizers of palladium nanoparticles, with the aim of generating bi-functional materials capable of carrying out catalytic processes based on several consecutive steps (tandem reactions) that were catalyzed by a single solid and recoverable metalorganic catalyst,58–60 with the consequent energy savings and the possibility of eliminating stages of separation and/or purification (Fig. 7).61 In general, these materials exhibited a lamellar morphology, as evidenced from X-ray diffractograms obtained for the metalorganic solids prepared when the organic spacers containing –SH groups (Al-ITQ-SH) and after oxidation (Al-ITQ-SO3H) or incorporation of Pd nanoparticles (Al-ITQ-SH/Pd and Al-ITQ-SO3H/Pd) during the synthesis process. Fig. 8 shows TEM images of the Al-ITQ-SH/Pd hybrid materials, in which it was observed that the lamellar morphology persisted even with the inclusion of palladium nanoparticles with diameters ranging between 5 and 13 nm, approximately. The images obtained for the oxidized sample Al-ITQ-SO3H/Pd evidenced that the lamellar morphology was maintained, observing a decrease in the size of incorporated palladium nanoparticles, this fact being favoured by the oxidation process (2–4 nm). Fig. 8 shows the analysis performed by EDS-STEM, identifying the different metallic species existing in the solid, and observing a homogeneous distribution with the incorporation of both aluminium and palladium species.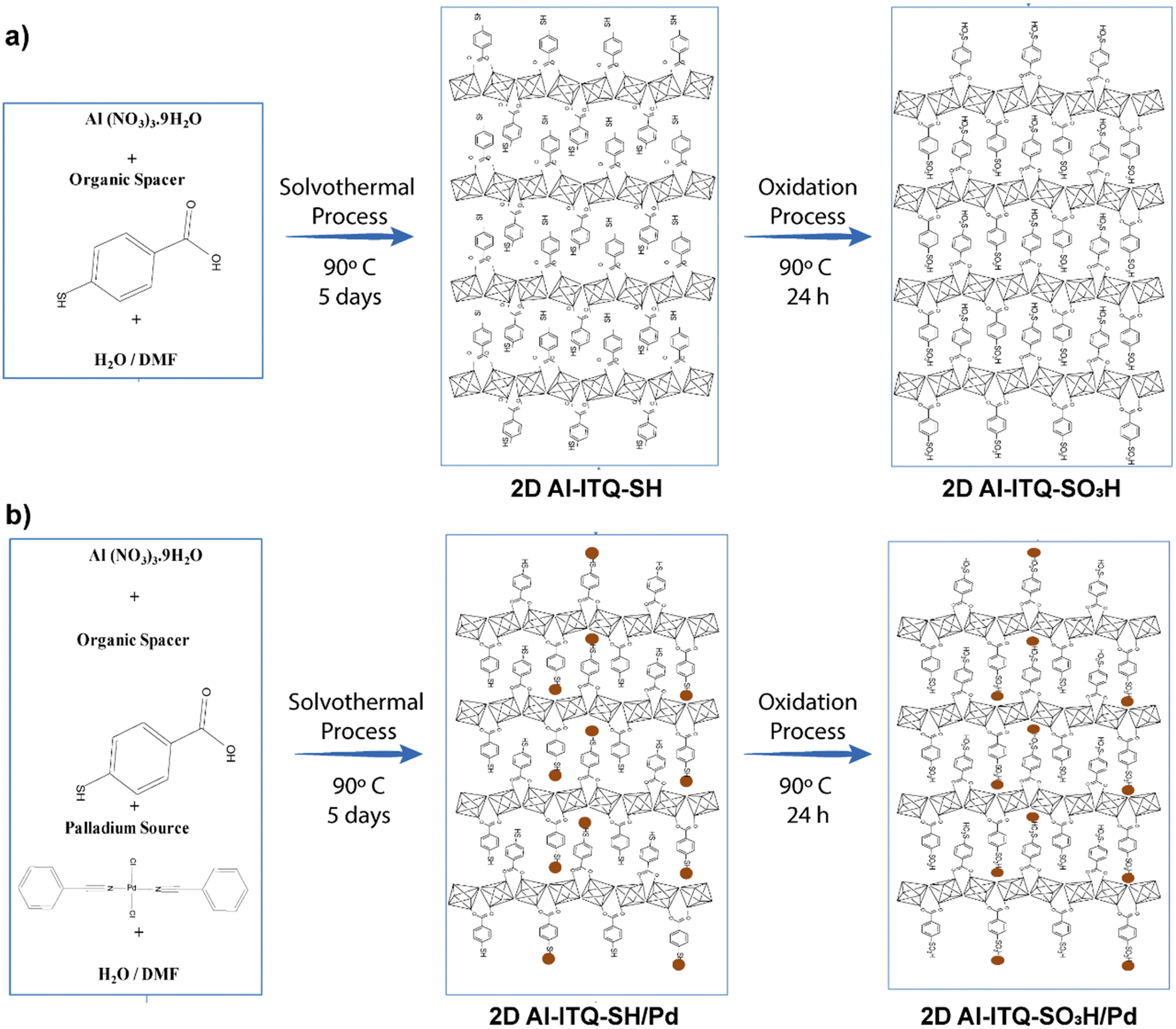 | ||
| Fig. 7 Representation of metalorganic materials synthesis: (a) Al-ITQ-SH and Al-ITQ-SO3H and (b) Al-ITQ-SH/Pd and Al-ITQ-SO3H/Pd. The dark spheres represent the palladium nanoparticles. Reproduced from ref. 61 with permission from the Royal Society of Chemistry. | ||
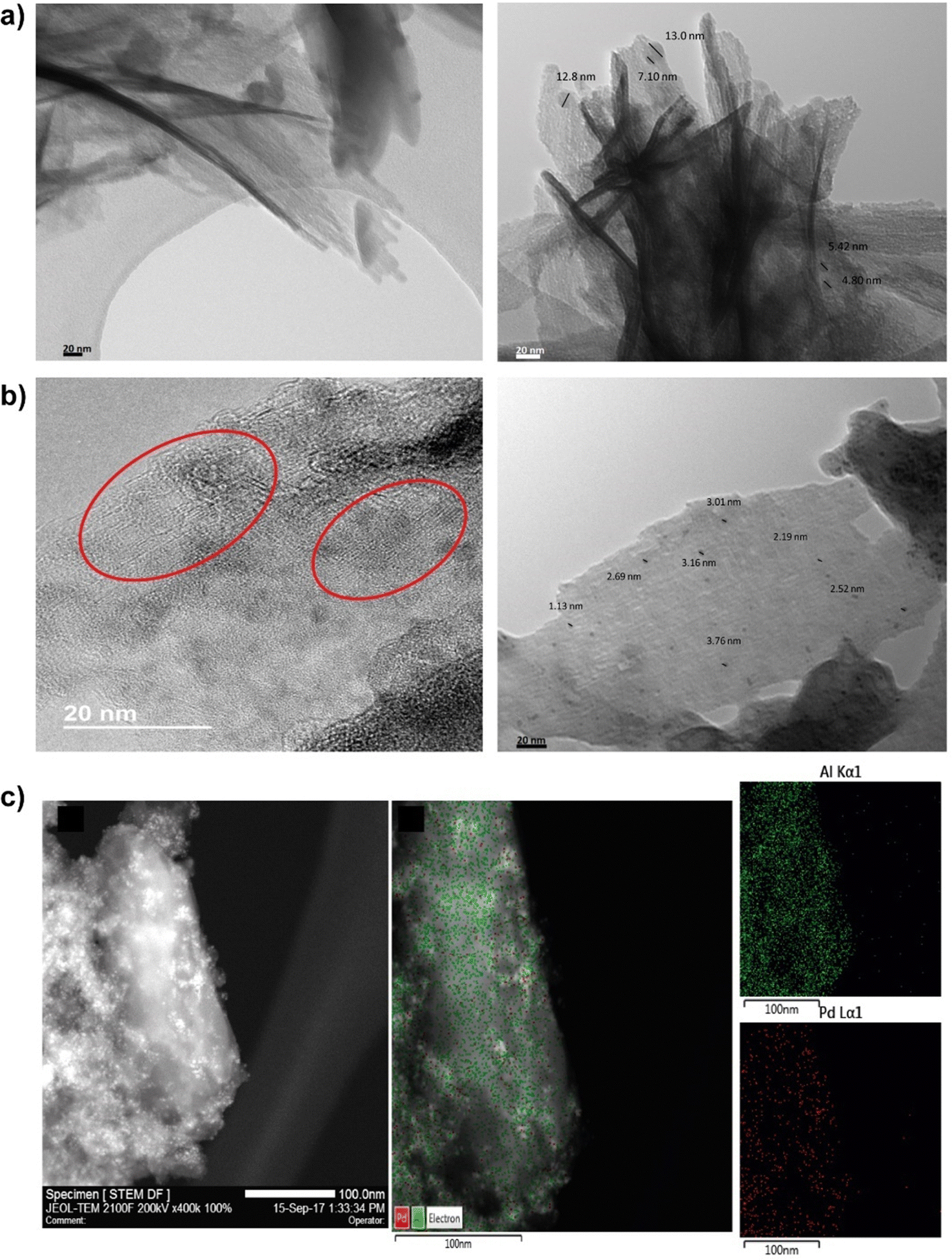 | ||
| Fig. 8 (a) TEM images of the Al-ITQ-SH/Pd laminar hybrid materials. (b) HRTEM images of the Al-ITQSO3H/Pd lamellar hybrid materials. The red circles highlight an area where the lamellar morphology is better appreciated. (c) STEM image and EDS-STEM analysis for the two metallic elements incorporated in the Al-ITQ-SO3H/Pd laminar material. Reproduced from ref. 61 with permission from the Royal Society of Chemistry. | ||
5.2 Acid reactions: acetalization, esterification and ring opening of epoxides
Acetalization of carbonyl compounds for the formation of an acetal is one of the most used methods in organic synthesis for the protection of compounds, being necessary to react aldehydes or ketones with an alcohol or diol using acid catalysts.62,63 The production of acetals has been widely studied due to their great relevance as reaction intermediates and final products in different fields of application, such as in the pharmaceutical industry, in the production of fragrances and perfumes, and in the food industry. Taking into account the importance of this process, the acid capacity of the Al-ITQ-SO3H lamellar hybrid materials was studied for the acetalization of benzaldehyde with methanol, obtaining yields higher than 95%. Furthermore, the production of cyclic acetals was studied through the acetalization reaction between benzaldehyde or vanillin in the presence of 1,2-propanediol. After optimization of the reaction conditions, analyzing the influence of concentration of active sites, alcohol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aldehyde ratios and temperature reaction, high yields were achieved in short reaction times, achieving complete conversion at short reaction times (Fig. 9a).
:aldehyde ratios and temperature reaction, high yields were achieved in short reaction times, achieving complete conversion at short reaction times (Fig. 9a).
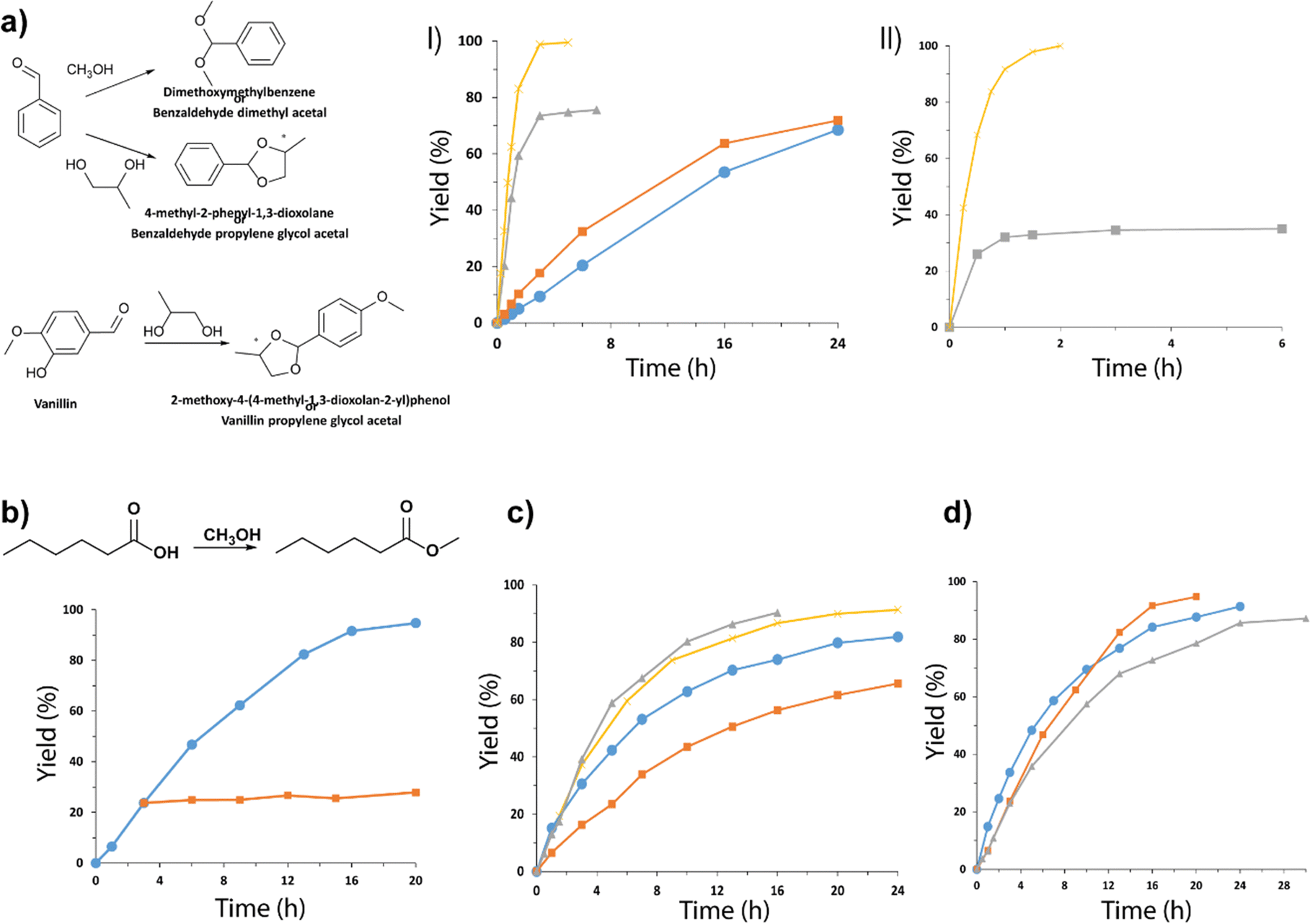 | ||
Fig. 9 (a) Kinetic curves obtained from the acetalization of 1,2-propanediol with: (I) benzaldehyde, (●) ratio 1:2, 10 mol% SO3H at 90 °C, (■) ratio 1:2, 20 mol% SO3H at 90 °C, (▲) ratio 1:6, 20 mol% SO3H at 90 °C and (×) ratio 1:6, 20 mol% SO3H under solvent reflux (toluene), and (II) vanillin, (■) ratio 1:6, 20 mol% SO3H at 90 °C and (×) ratio 1:6, 20 mol% SO3H under solvent reflux, using the hybrid material Al-ITQ-SO3H as the catalyst. (b) Kinetic curves obtained for the esterification of hexanoic acid with methanol at 60 °C, using 10 mol% SO3H of the Al-ITQ-SO3H material as the hybrid catalyst. The separation of the solid at 3 h and the kinetics obtained by leaching generated under the same reaction conditions are represented. (c) The yield of different esters was plotted versus time when esterification was performed in the presence of Al-ITQ-SO3H: ethyl hexanoate ( ) using 10 mol% SO3H at 90 °C and ethanol as the solvent; ( ) using 10 mol% SO3H at 90 °C and ethanol as the solvent; ( ) butyl hexanoate using 10 mol% SO3H at 90 °C and butanol as the solvent; ( ) butyl hexanoate using 10 mol% SO3H at 90 °C and butanol as the solvent; ( ) hexyl hexanoate using 10 mol% SO3H at 90 °C and hexanol as the solvent; ( ) hexyl hexanoate using 10 mol% SO3H at 90 °C and hexanol as the solvent; ( ) hexyl hexanoate using 10 mol% SO3H at 120 °C and hexanol as the solvent. (d) The yield of different esters was plotted versus time when esterification was performed in the presence of Al-ITQ-SO3H: ( ) hexyl hexanoate using 10 mol% SO3H at 120 °C and hexanol as the solvent. (d) The yield of different esters was plotted versus time when esterification was performed in the presence of Al-ITQ-SO3H: ( ) methyl hexanoate, ( ) methyl hexanoate, ( ) methyl octanate and ( ) methyl octanate and ( ) methyl laurate. Reaction conditions: 10 mol% SO3H at 60 °C, using methanol as the solvent. Reproduced from ref. 61 with permission from the Royal Society of Chemistry. ) methyl laurate. Reaction conditions: 10 mol% SO3H at 60 °C, using methanol as the solvent. Reproduced from ref. 61 with permission from the Royal Society of Chemistry. | ||
The esterification of an alcohol with a carboxylic acid to form the corresponding ester is a widely known reaction.64,65 Currently, there is great interest in this catalytic process since it is of relevance in different application fields, because organic esters are commonly used as plastic derivatives, solvents, and fragrances as well as in the food and agrochemical industries. Following this scope, the lamellar hybrid material Al-ITQ-SO3H was used as the acid catalyst in the esterification of hexanoic acid with methanol. Fig. 9b shows the high catalytic activity (95%) that the hybrid catalyst reached in short reaction times. In addition, subsequent analysis of leaching showed how the performance remained practically unchanged, confirming the stability and heterogeneous nature of the hybrid catalyst under the reaction conditions, since the release of active species from it to the reaction medium did not occur.
Considering the excellent results obtained, the acid capacity of the Al-ITQ-SO3H hybrid catalyst was studied for the production of other organic esters of great interest in the food industry, as described by FEMA. Fig. 9c shows the kinetics obtained for the different esters produced, using hexanoic acid in all cases and increasing the number of carbon atoms of the alcohol used at the same time as a reaction reagent and a solvent. The results confirmed that by increasing the length of the aliphatic chain of the used alcohol, lower yields were achieved, even requiring more severe reaction conditions to overcome this effect. Similarly, by increasing the length of the alkyl chain of the acid, lower yields were obtained (Fig. 9d). In both cases, the observed effect could be due to the greater impediment exhibited by the reagents by increasing the length of their hydrocarbon chains to react with the active sites of the catalyst and produce the corresponding ester.
On the other hand, epoxides are highly important intermediates in organic synthesis, being used as starting materials for products with high added value on an industrial scale. One of the most relevant processes is the opening of the epoxide ring with an alcohol. This process is catalyzed by an acid to produce β-alkoxyalcohols, which are of interest to the pharmaceutical industry.66,67 For this, the acid capacity of the metal–organic lamellar material Al-ITQ-SO3H was evaluated for the opening reaction of the styrene oxide ring with methanol, using mild reaction conditions during the process. The obtained results showed that the catalytic process was successfully carried out, obtaining 98% yield, being reused during several consecutive reaction cycles without losing the reactivity and maintaining the heterogeneity of the process. This confirmed the validity of the MOFs based on low-dimensional builder units as acid active heterogeneous catalysts for fine chemical processes with industrial interest.
5.3 One-pot two-step processes: oxidation–acetalization reactions
The direct synthesis of acetals from alcohols has not been widely studied yet, being a great challenge to be able to carry out the two catalytic transformations using a single catalyst in a single reactor, avoiding the subsequent stages of separation and purification of intermediate products with the consequent energy savings, and helping to develop a more environmentally sustainable process.68,69 Specifically, different catalysts have been used to carry out this process: inorganic acids combined with palladium species, palladium metalloporphyrins, MOFs (UiO-66-NH2-Pd), etc.70–72 Due to the importance of obtaining new active materials that carry out this catalytic process, the bi-functional metal–organic material, Al-ITQ-SO3H/Pd, is an excellent candidate for the aforementioned process. This consecutive process (one-pot) consists of two steps: the first is based on the oxidation of benzyl alcohol (1) under an oxygen atmosphere catalyzed through the palladium incorporated in the hybrid material, obtaining benzaldehyde (2), and the second step being the acetalization of the benzaldehyde produced with methanol to obtain the corresponding acetal (3) due to the presence of acid sites, that is, sulfonic groups, in the catalyst (Fig. 10). The catalytic study was carried out with three different percentages of active sites, considering in this case palladium as the active phase (1, 3 and 5 mol% Pd). The results showed that the optimal amount required was 3 mol% Pd, since high conversion was again achieved in the first step of the reaction (97%) and the final production of acetal was improved by up to 90%. To corroborate the efficacy of the synthesized bi-functional material, the oxidation reaction was carried out with previously commented metalorganic materials Al-ITQ-SO3H, Al-ITQ-SH and MIL-53(Al), all of them showing low activity, which confirmed the need for the incorporation of palladium in them. In addition, TON values were calculated for a series of Pd@MOFs described in the literature, obtaining values comparable to those shown by the catalyst discussed in this study. The recyclability of the bi-functional hybrid material in the consecutive oxidation-acetalization reaction was studied, keeping the catalyst active in the two reaction steps for several consecutive uses in the same reaction time (Fig. 10). The obtained diffractograms of the hybrid catalyst, after each use, showed practically the same diffraction bands, only a slight decrease in crystallinity being observed and, therefore, maintained the lamellar morphology throughout the reactive processes (Table 1 and Fig. 10c).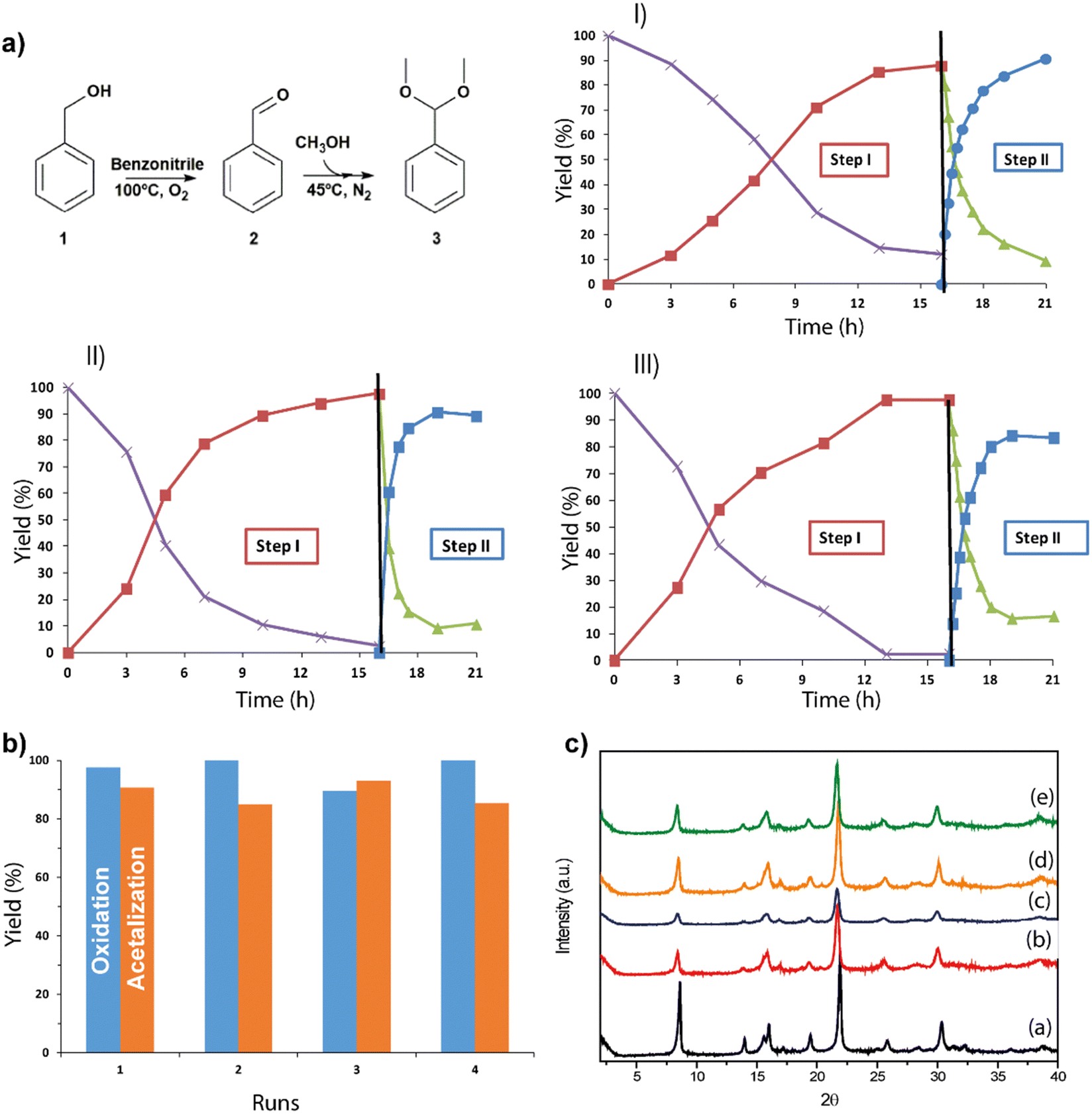 | ||
| Fig. 10 (a) Kinetic curves obtained for the consecutive reaction of oxidation–acetalization in the presence of different percentages of the Al-ITQ-SO3H/Pd catalyst: (I) 1 mol% Pd, (II) 3 mol% Pd and (III) 5 mol% Pd, where step I was carried out at 100 °C under an oxygen atmosphere and step II at 45 °C under nitrogen atmosphere. (b) Yields obtained during four consecutive cycles in the oxidation–acetalization reaction, using 3 mol% Pd of the Al-ITQ-SO3H/Pd catalyst. (c) Diffractograms for the Al-ITQ-SO3H/Pd catalyst: (a) as-synthesized, (b) first use, (c) second use, (d) third use and (e) fourth use, from bottom to top. Reproduced from ref. 61 with permission from the Royal Society of Chemistry. | ||
| Section | Example | Ref. | Catalyst |
|---|---|---|---|
| Section 2 | Synthesis of benzodiazepines and asymmetric transformations such as Michael and Knoevenagel condensations, evidencing the influence of defect structural sites together with Lewis acidity in the reactivity of the 2D metalorganic materials | 28 and 39 | Al-ITQ-HB |
| Section 3 | Synthesis of cyanohydrins and aerobic oxidation of thiols to disulfides, showing the capacity of tuning the Lewis acid character of the solids through the structural incorporation of electron-donor or electron-attracting organic functions. | 47 | Al-ITQ-X (–X: –NO2, –Br, –NH2 and –C2H5); Al/Fe-ITQ-NO2 |
| Section 4 | Acid reactions such as acetalization, esterification and ring opening of epoxides, showing the reactivity of the solids in different reactions which are habitual for organic synthesis of high added value products and also an industrial scale. | 61 | Al-ITQ-SO3H |
| Section 5 | Tandem or consecutive processes such as oxidation–acetalization reactions, confirming the possibilities to use these materials like efficient bi-functional catalysts by the incorporation of acid and redox sites, acting concomitantly, for one-pot two-step processes. | 61 | Al-ITQ-SO3H/Pd |
6. Photodynamical properties of guests interacting with ITQ-MOFs
6.1 Fluorescent molecular probes: special case of NR and DCM dyes
Fluorescent molecules have been used as probes for the local environment over the past few decades due to the combination of their outstanding fluorescent properties (e.g., high photoluminescence quantum yield and large Stokes shift) and the fact that their luminescence characteristics strongly depend on the surrounding environment, turning these molecules into ideal candidates for developing luminescence-based sensors.73–76 Nile red (NR, Fig. 11a) and trans-4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran (DCM, Fig. 11a) are among the most explored molecular probes not only from a spectroscopic and photophysics point of view, but also for developing novel materials. Indeed, many researchers have focused their efforts to synthesize different NR and DCM derivatives anchoring a great variety of functional groups or spacers, aiming to improve the spectroscopic properties of these fluorescent molecular probes.77–81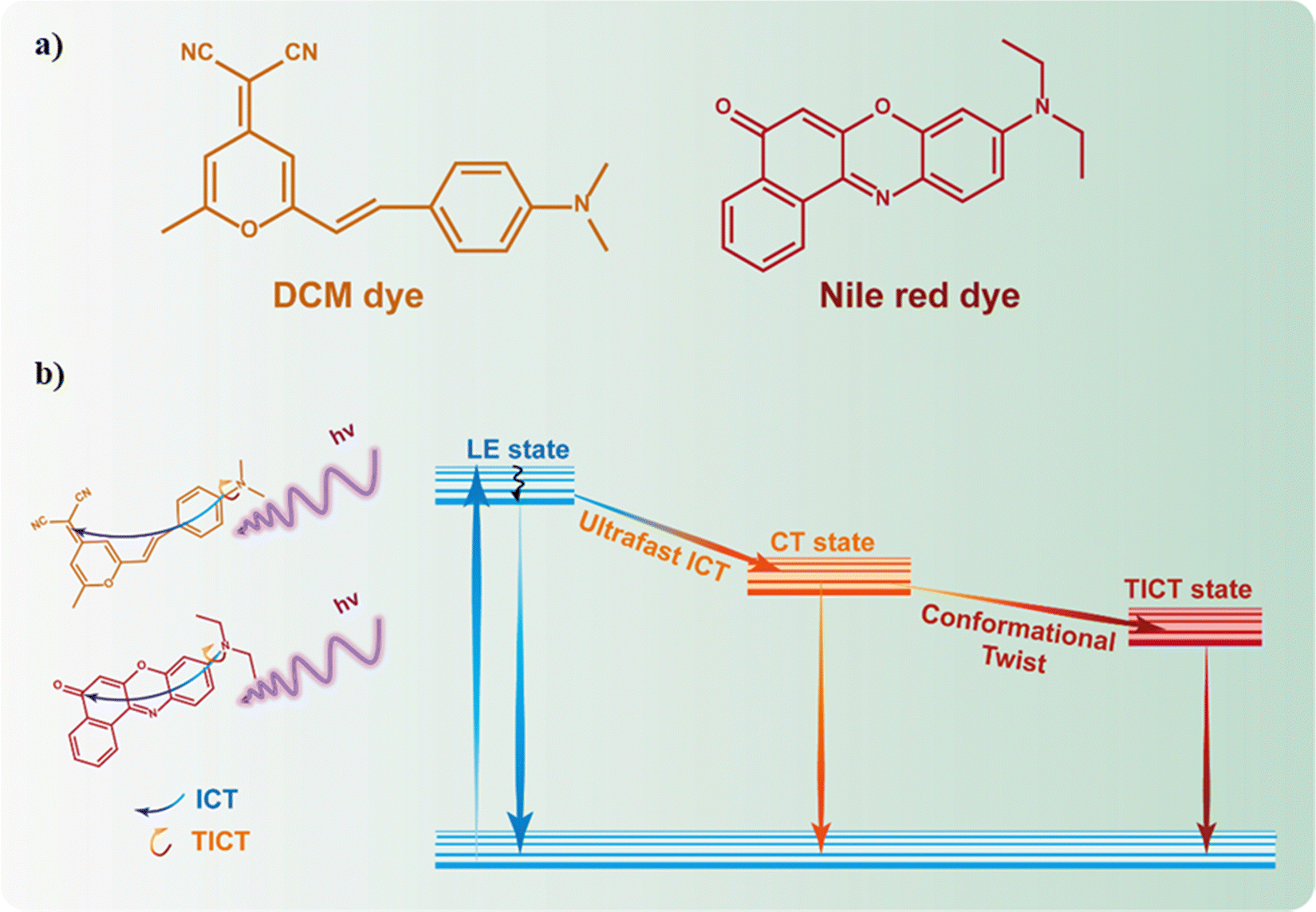 | ||
| Fig. 11 (a) Representation of the chemical structures of trans-4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran (DCM) and Nile red (NR) dyes. (b) Schematic representation of the photophysical scheme of NR and DCM dyes. Upon photoexcitation, both dyes can be deactivated from the LE state (in non-polar aprotic solvents) or might undergo ultrafast ICT or TICT processes emitting at longer wavelengths. | ||
The photophysical behavior of NR and DCM dyes has been a subject of intense debate. For instance, their photophysical properties have been explored in a multitude of different media, trying to reach a consensus about their steady-state and time-resolved photobehavior.82–86 In general, the fluorescence properties of both dyes are similar, showing a well-resolved structured band in non-polar aprotic solvents, which becomes a broader, unstructured band, shifted towards longer wavelengths, in polar and protic solvents.82 This spectral behavior is typically associated with an excited state intramolecular charge transfer (ICT).87 In fact, NR and DCM can be considered as push–pull fluorescent molecules, with an electron donor moiety (N,N-dimethylaniline for DCM and N,N-diethylaniline for NR) connected through a π-conjugated spacer to an electron acceptor group (dicyanomethylene for DCM, and keto group for NR). Moreover, both dyes might present a rotation of the electron donor group, inducing a twist in the conformation known as twisted intramolecular charge transfer (TICT) state.84,88–91 Hence, after photoexcitation, the emission of these dyes might originate from the locally excited (LE), the ICT, or the TICT states depending on the surrounding environment. Based on this, it has been proposed that when these fluorescent probes are in non-polar aprotic dyes, the well-structured fluorescence band is a consequence of the emissive deactivation from their LE state.82,84,90 On the other hand, in polar protic media, the emission might originate from the ICT or the TICT states, being the latter more stabilized and therefore, shifted towards longer wavelengths.82,84,90 These photoproperties have been also investigated by employing time-resolved laser-based spectroscopic techniques. For instance, in solution, NR undergoes an ultrafast (∼1 ps) polarity dependent TICT in its first singlet excited state (S1). Following the TICT, the photoproduced state relaxes radiatively to the ground state in ∼4.4 ns.88 Similarly, DCM can undergo an ICT to produce an emissive charge transfer (CT) state that can be followed by a twisting motion, thus populating an excited TICT state.92–94 A summary of the general photophysics scheme is depicted in Fig. 11b.
Even though these fluorescent molecular probes exhibit great potential to be implemented in the construction of luminescent-based chemical sensors, they suffer from an intrinsic limitation, which is the quenching of their fluorescence in the solid state, a phenomenon known as aggregation-caused quenching (AQC).95 Since many of the sensing detectors require solid-state active materials, the use of NR and DCM dyes as powders constitutes a limitation for their exploitation in real sensing devices. A possible solution to overcome this issue is to encapsulate these dyes within porous materials, in which the porous structure allows the isolation of the fluorophores, minimizing or preventing the aggregation of the dyes, and therefore, enhancing their fluorescence intensity in the solid state. A number of materials have been proposed to encapsulate these fluorescent dyes, including but not limited, to polymers,96,97 micelles,98–100 mesoporous silica based materials,101–103 zeolites,104–106 and MOFs.107–110 When a molecular probe, such as NR and DCM interacts with the MOF support either within the available pores or on its surface, several photoinduced processes can occur. Some examples include electron transfer to the metal cluster part (ligand-to-cluster charge transfer, LCCT) generating a charge separated state (electrons and holes), excited state proton transfer (ESPT), CT and Förster resonance energy transfer (FRET) phenomena. The efficiency of these depends on different intrinsic and external factors, such as the dye concentration, the polarity of the local environment, the presence of electron-donating or accepting groups, etc. A variety of spectroscopic techniques, such as time-correlated single-photon counting, femtosecond–ns Raman and IR, transient absorption and fluorescence up-conversion, can be employed to obtain a detailed information for the related parameters. These photoinduced processes and the available techniques for their characterization in MOFs have been reviewed in detail recently.111 Additionally, the photophysical properties of these dyes upon encapsulation are significantly altered with the concentration of the encapsulated guests.102–104,112,113 This effect has been well explained considering the formation of J- and H-aggregates. In an H-aggregate, the molecules stack predominantly face-to-face, while J-aggregates form when the molecules primarily stack in a head-to-tail arrangement. The formation of such aggregates has important consequences for the energies of the excited states and the oscillator strengths of the transitions to these states from the ground state. Consequently, H- and J-aggregation can strongly modify the optical absorption and photoluminescence spectra.114–116 Following the classical excitonic theory, S1 → S0 is forbidden for the H-aggregates, while for the J-aggregates it will be less energetic than for the monomers. A recent review provides a more detailed overview of the most recent theories for H- and J-aggregates.117 Several strategies can be explored to control the formation of these molecular aggregates. These include, but are not limited to, precise control of the dye concentration, the introduction of bulkier substituents in the molecular structure of the probe or by applying alternative approaches for the inclusion of the dye in the MOF structure – either by diffusion or in a one-pot synthesis. The sensitivity of the photophysical properties of NR and DCM to subtle changes in the local environment was exploited to characterize different mesoporous materials. For example, when NR is encapsulated within Zr- and Ti-doped MCM-41 materials, its photoexcitation leads to an ultrafast ICT phenomenon of <200 fs.113 The photodynamics of NR encapsulated within these materials shows a multiexponential behavior with time constants of τ1 = 0.18–0.34 ns, τ2 = 0.7–1.5 ns and τ3 =1.2–4.0 ns, attributed to the emission lifetimes of H-aggregates, J-aggregates and monomers, respectively.113 This multiexponential behavior was also observed when these molecules are encapsulated within different silica-based materials. Moreover, it has also been reported that NR dye possesses the ability to interact with Lewis and Brønsted acid sites in metal-doped mesoporous materials and zeolites, with key implications for the improvement of catalytic systems.103,104,113 In another example, NR and DCM dyes have been also encapsulated within a Zr-MOF to combine the emission of these dyes with that of the MOF in order to obtain high quality white light emitting materials.107 These and others are examples of how these dyes can be implemented for the development of hybrid emissive materials having great potential for their application in different photonic devices.
In the following subsections, we will focus on the photophysical properties of NR and DCM dyes upon their interaction with the ITQ 2D-MOF family, since these 2D-MOFs exhibit a higher surface area than conventional 3D materials as the ones described above. Hence, these materials are excellent candidates for the development of chemical sensors, but prior to that, it is primordial to deeply understand their photobehavior.
6.2 NR encapsulated within Al-ITQ-HB 2D-MOF
Encapsulation of NR by Al-ITQ-4-heptylbenzoic acid (HB) 2D-MOF gives rise to different dye populations and affects its photobehavior.118 NR/Al-ITQ-HB composites with three initial dye concentrations (1 × 10−4 M, 1 × 10−3 M and 1 × 10−2 M) were prepared and studied by steady-state and time-resolved spectroscopic techniques. To prepare the composites, dichloromethane solutions of NR with the selected molar concentrations were prepared. Then, 50 mg of MOF was added to 1 mL of the dye solution, and the mixture was stirred for 24 h at room temperature. The material was then washed several times to remove excess dye adsorbed on the surface of the MOF until no trace of NR was observed in the UV-visible absorption spectra of the supernatant. As a result, the estimated loadings were 1.8 × 1018, 2.3 × 1017, and 3.2 × 1016 NR molecules per gMOF for 1 × 10−2, 1 × 10−3, and 1 × 10−4 M initial dye concentrations, respectively. Steady-state UV-visible experiments have shown that upon interaction with the MOF, several species are formed, which was evidenced by the broad absorption spectra of the composites (Fig. 12a). Upon increasing the initial dye concentration, the spectra became broader concomitant with a red shift (from 595 nm at 1 × 10−4 M to 618 nm at 1 × 10−2 M) of the absorption maximum. This behavior suggests that since the host material consists of a layered structure where the dye can be located and interact with the units that form the framework, several possible orientations and interaction sites of NR molecules in the MOF, as well as intermolecular interactions might take place. The significantly narrower emission spectra of these populations were also affected by the initial dye concentrations in the composites consistent with formation of different types of aggregates. This was further confirmed by the excitation spectra, recorded at the emission maxima, that showed an identical shape to the absorption ones but with notably reduced full width at half maxima, which indicated the presence of weak- or non-emissive species. Time-resolved emission experiments (Fig. 12b) have shown the contribution of two monomer populations, experiencing different local environments along with J- and H-type aggregates, showing that the photobehavior of NR is affected by its location and organization within the MOF (Fig. 12c). The lifetime of the monomer in the LE state was reported as 0.71 ns, while the one that emits from the photoproduced CT state was 5.1 ns. Similar values have been reported for NR encapsulated in different host materials, such as zeolites, micelles and cyclodextrins.88,103,104,113,119,120 The presence of two monomer species was explained in terms of the different environment experienced by the encapsulated NR molecules (Fig. 12c): (1) linker domain, where the heptyl chains provide a nonpolar environment giving rise to the monomer emitting from the LE state (0.71 ns), and (2) domain involving the aromatic and carboxylic groups and the metal clusters, where hydroxyl or oxy groups might be present, resulting in a higher polarity of the region and associated with the CT monomer emission (5.1 ns). A third component of ∼2.6 ns, assigned to the emission lifetime of J-aggregates, was also reported. All lifetimes decreased with the increase in the concentration of the encapsulated NR (Fig. 12b). This decrease is associated with formation of weakly- or non-emissive H-aggregates because of stronger intermolecular interactions due to the increased NR concentration.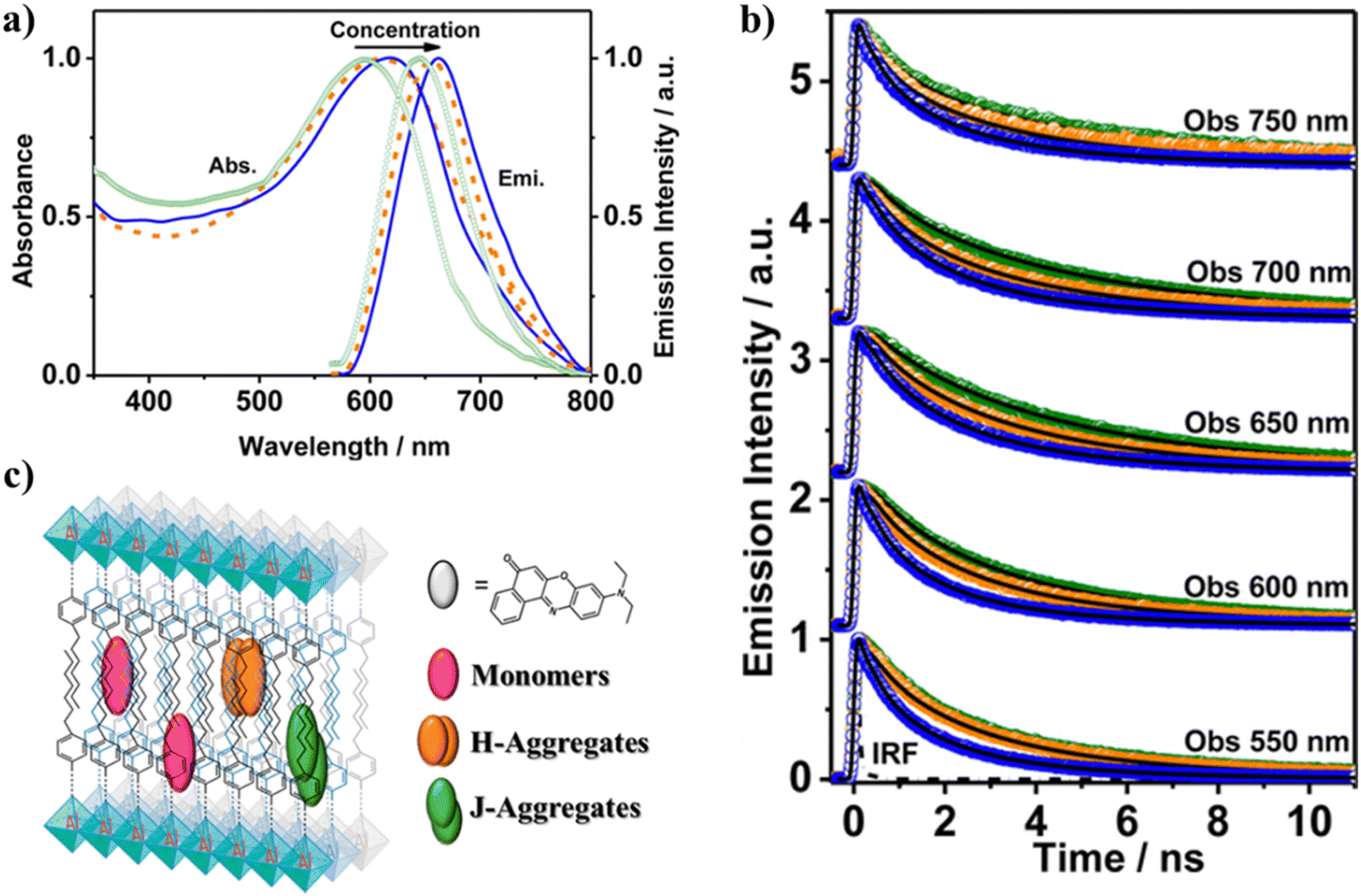 | ||
| Fig. 12 (a) UV-visible absorption and emission spectra of NR/Al–ITQ-HB at different initial dye concentrations: 1 × 10−4 M (green lines), 1 × 10−3 M (orange lines), and 1 × 10−2 M (blue lines) after thoroughly washing the samples. The emission spectra were recorded upon excitation at 550 nm. (b) Normalized emission decays of NR/Al–ITQ-HB at different initial dye concentrations: 1 × 10−2 M (blue circles), 1 × 10−3 M (orange circles), and 1 × 10−4 M (green circles) upon excitation at 470 nm and recorded at the indicated wavelengths. (c) Schematic illustration of the distribution of NR molecules within the Al-ITQ-HB framework in the form of monomers, and J- and H-aggregates. Adapted with permission from ref. 118 (ACS Omega, 2018, 3, 1600–1608). Copyright 2018 American Chemical Society. | ||
Femtosecond time-resolved emission experiments have characterized the ultrafast intramolecular charge transfer to produce the CT state (∼1 ps), which is then followed by a vibrational cooling (VC) in ∼8 ps, while the homogeneous distribution of the dye within the MOF was confirmed by scanning confocal fluorescence microscopy of single crystals and agglomerates of the composites. Both time constants are slower than those reported for the ultrafast dynamics of NR in silica-based materials.103,104,113 The slowing down of the ultrafast dynamics was explained in terms of the different types of interactions that occur between the dye and the host materials. In silica-based materials, the hydroxyl groups of the framework can interact with NR via H-bonds, favoring vibrational energy transfer from excited NR molecules to the surrounding environment, while for the NR/Al–ITQ-HB composites, the coupling between the dye and the MOF host is weaker, and occurs through nonspecific interactions thus slowing down the related energy dissipation processes.
6.3 NR Adsorbed on Al-ITQ-HB 2D-MOF
To understand the behavior of excited NR adsorbed on Al-ITQ-HB surfaces, three different initial NR concentrations (10−5, 10−4, and 10−3 M) were used to make NR@Al-ITQ-HB composites.121 As opposed to the NR/Al-ITQ-HB composites,118 where the formed composites were washed several times to remove the excess NR resulting in the dye being predominantly encapsulated within the layered MOF, for NR@Al-ITQ-HB the solvent was eliminated by evaporation, resulting in NR being mainly deposited on the surface of the MOF. Fig. 13a shows the difference in colors of both composites (blue for the encapsulated NR and purple for the surface-adsorbed one) made using the different synthesis procedures. The distinction of the colors suggests different types of interactions between the adsorbed NR molecules on one hand and within the MOF framework on the other. The steady state UV-visible absorption and emission spectral behavior resembles the one observed for the encapsulated NR. While the absorption spectra are broad and the fwhm increases with the increase in the initial concentration, the emission ones are narrower, suggesting the presence of both emitting (monomer and J-aggregates) and weakly- or non-emitting species (H-aggregates). Due to the strong overlap between the absorption and emission spectra, for the NR@Al-ITQ-HB composites, the possibility of homo-energy transfer (ET) and its concentration dependence on the surface-adsorbed NR molecules were explored. In similarity with the composites where NR is encapsulated within the MOF structure,118 the photodynamics of the NR@Al-ITQ-HB ones (Fig. 13b) has shown emission from NR populations that relax from the LE state and H-aggregates (∼0.5 ns) and from the CT state (2–4 ns). Additionally, a new component of 2.5 ns for the lowest NR concentration (10−5 M), which was not observed for the emission transients of the encapsulated NR, was present as a decay and a rise at the blue and red sides of the emission spectra, respectively. This component was assigned to an ET process between adsorbed NR molecules on the surface of Al-ITQ-HB. The photoproduced NR species as a result of the ET process then relax in ∼4.8 ns. This component was also detected upon excitation at 635 nm (lower excess energy at S1) and was assigned to the emission lifetime of the directly excited species (Fig. 13c). Upon increasing the concentration of the adsorbed NR molecules (initial concentrations of 10−4 and 10−3 M), an additional, faster ET process (∼0.2–0.3 ns) was also observed. The presence of this faster ET process was explained in terms of the decreased distances between the NR molecules due to the higher concentration of adsorbed dyes, thus allowing stronger dipole–dipole interactions.122,123 The other lifetimes of the monomer species also become shorter upon increasing dye content, which is explained in terms of homo-quenching of the NR emission due to the adsorbed molecules forming aggregates. The Al-ITQ-HB surface also affected the ultrafast dynamics of the adsorbed NR, where a decrease in the value of the ICT process was observed (Fig. 13c). While this process occurs in 1 ps when NR is encapsulated within the layered 2D Al-ITQ-HB, it takes only ∼ 400 fs when the dye is adsorbed on the MOF surface. This speeding up of the ICT, along with the occurrence of ET in the NR@Al-ITQ-HB composites suggests the presence of defects, typically found on the surface of solid structures, that can provide specific interactions that condition the dye photodynamics and facilitate both processes.124 This in turn reflects how the singular local environment created due to the different probe locations when interacting with Al-ITQ-HB (either adsorbed on its surface or entrapped within the interior) might provide different outcomes for the catalytic reactions using this MOF.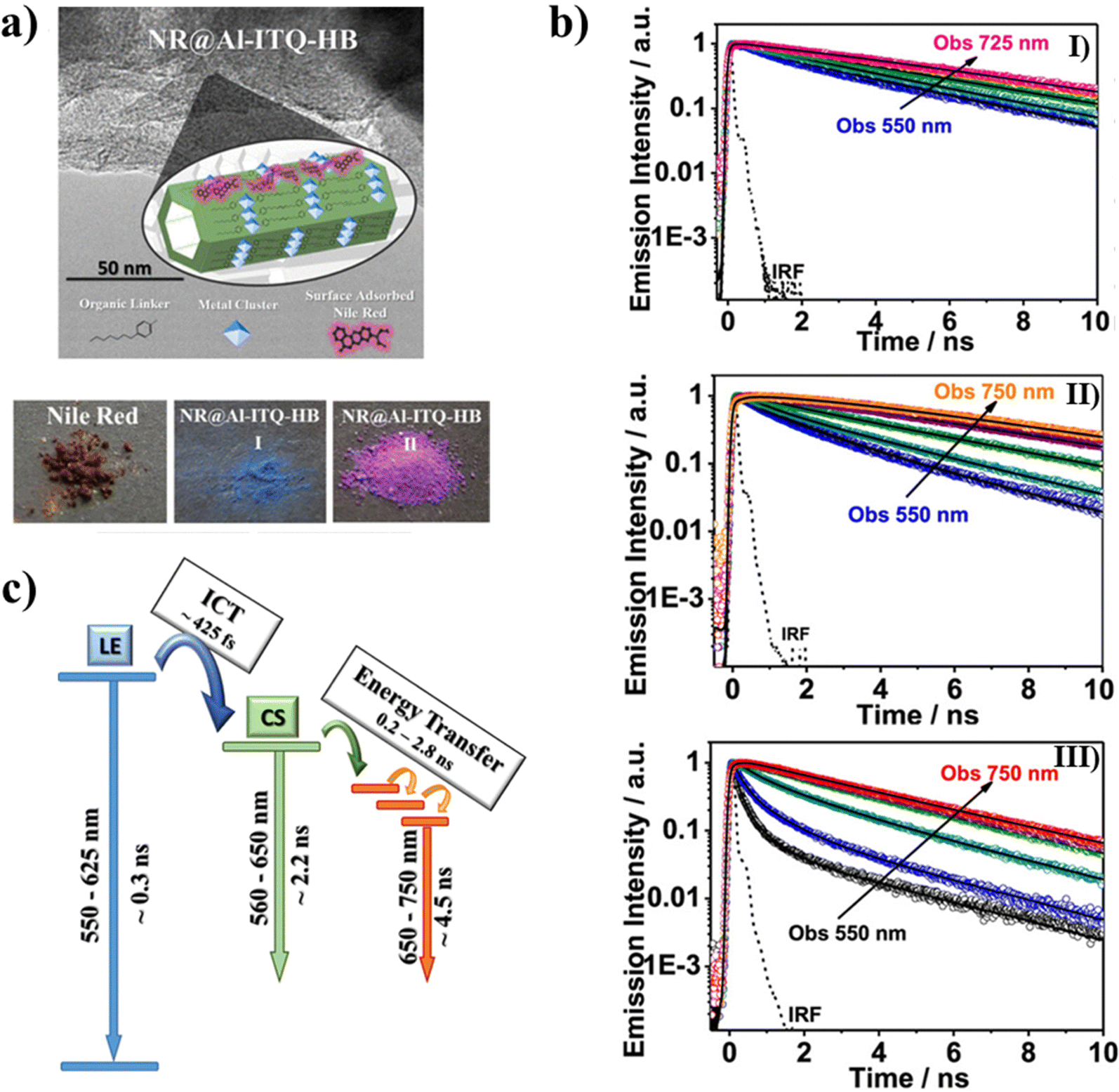 | ||
| Fig. 13 (a) The top image is a schematic illustration of the adsorption of NR molecules on the surface of the Al-ITQ-HB MOF. The bottom images are real photos of NR, thoroughly washed NR@Al-ITQ-HB, and unwashed NR@Al-ITQ-HB powders. (b) Normalized emission decays of unwashed NR@Al–ITQ-HB powder at different initial NR concentrations: 1 × 10−5 M (I), 1 × 10−4 M (II), and 1 × 10−3 M (III) upon excitation at 470 nm, and observation at different wavelengths. (c) Schematic representation of the photoprocesses occurring after the photoexcitation of the unwashed NR@Al–ITQ-HB composites. Adapted with permission from ref. 121 (ACS Appl. Mater. Interfaces, 2018, 10, 20159–20169). Copyright 2018 American Chemical Society. | ||
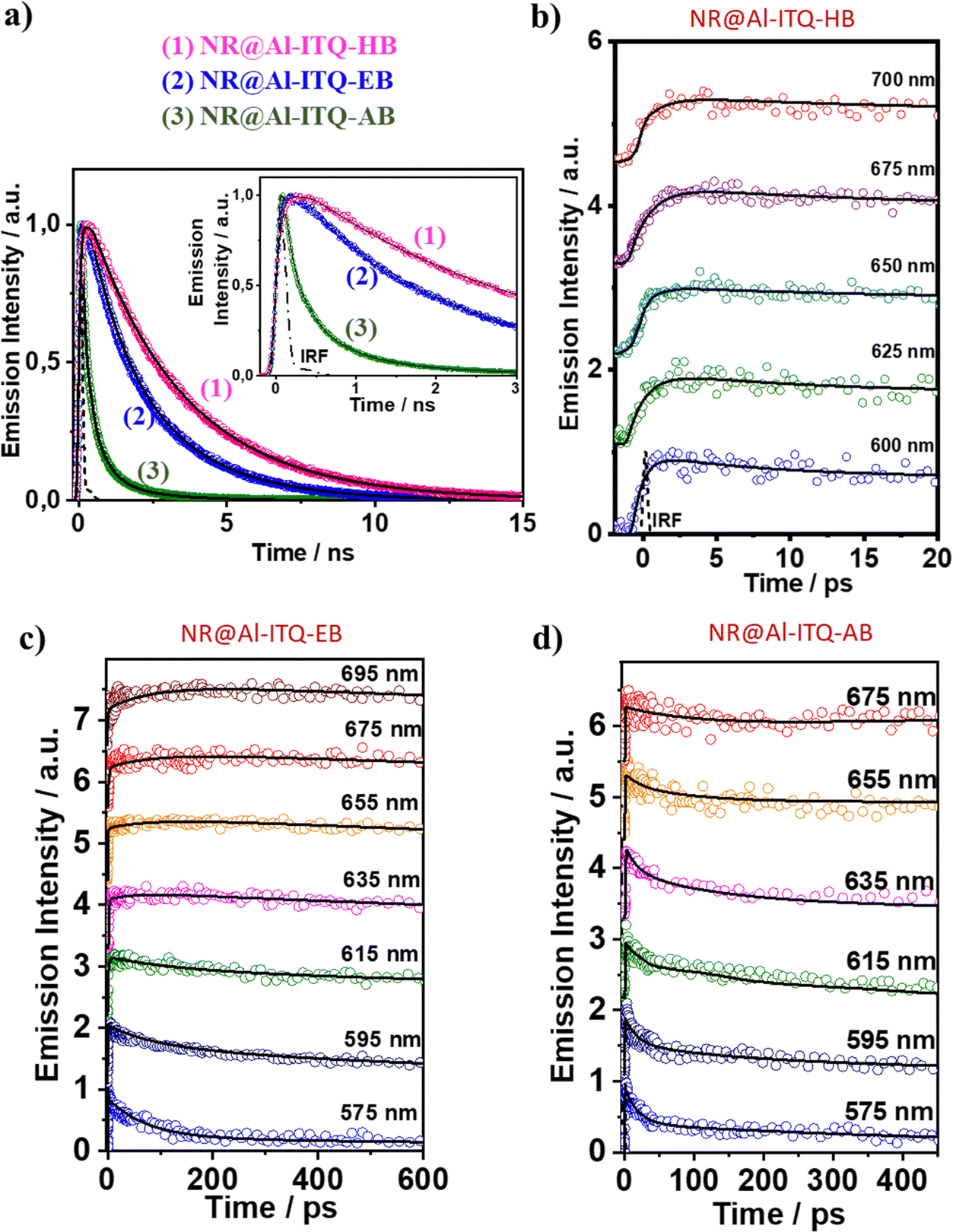 | ||
| Fig. 14 (a) Comparison of the ps–ns emission decays of 1: NR@Al-ITQ-HB; 2: NR@Al-ITQ-EB; and 3: NR@Al-ITQ-AB upon photoexcitation at 470 nm and recording the signals at 725 nm. (b)–(d) fs-emission decays of (b) NR@Al-ITQ-HB; (c) NR@Al-ITQ-EB; and (d) NR@Al-ITQ-AB, upon their photoexcitation with 470 nm light and gating the signal at the indicated wavelengths. Adapted with permission from ref. 125 (ACS Appl. Mater. Interfaces, 2018, 10, 32885–32894). Copyright 2018 American Chemical Society. | ||
Significantly different behavior was reported for NR interacting with Al-ITQ-4-aminobenzoic acid (AB) 2D-MOF, in which the HB acid linker was replaced with 4-aminobenzoic acid.125 Although the absorption spectrum of the resulting composite is comparable to those of the other MOF composites from this family, its emission spectrum is distinct, showing a much broader band in the 550–650 nm range concomitant with a shift in the emission intensity maximum towards lower wavelengths (from 660 nm to 645 nm). This behavior suggests the presence of additional emissive NR populations and that different photoinduced processes are acting in this NR@Al-ITQ-AB composite. This was further supported by the results obtained from the time-resolved emission experiments, where the decays of NR@Al-ITQ-AB were well-fitted by a three-exponential function giving time constants of τ1 = 0.13 ns, τ2 = 0.54 ns and τ3 = 2.10 ns, and an average lifetime value much shorter than those obtained for its counterparts, as represented in Fig. 14a. The contribution of the longest τ3-component – attributed to NR molecules formed as a result of the homo-ET reaction – decreases from 70 to 5%, suggesting that the ET deactivation channel is not as efficient as in the other MOFs. Moreover, the shortest τ1-component is decaying in the whole spectral region, and therefore, the lack of short rising component indicates that either the homo-ET is not occurring in this MOF or it is too fast to be detected by the system. Interestingly, the amino group of the AB linkers of this MOF can interact with the NR molecules via H-bonds, favoring alternative and more efficient relaxation pathways such as electron transfer from the organic linker to the NR dye molecules. These new deactivation channels could explain the inhibition of the ET phenomenon observed for the other NR composites (Fig. 15a). To further corroborate this mechanism, time-resolved experiments of NR@Al-ITQ-EB in the presence of vapor of aniline were performed. When this material was exposed to aniline, the obtained time constants (τ1 = 0.11 ns, τ2 = 0.55 ns and τ3 = 2.31 ns) were very similar to those reported for NR@Al-ITQ-AB. In contrast to the pristine NR@Al-ITQ-EB, the shortest component was observed as decay in the whole spectral range, providing a clear proof that in the presence of aniline or other electron donating amine moieties (4-aminobenzoic acid – AB), there exists a faster and more efficient deactivation pathway involving an electron transfer from the amine derivative to the adsorbed NR molecules, which hampers the homo-ET reaction (Fig. 15b).
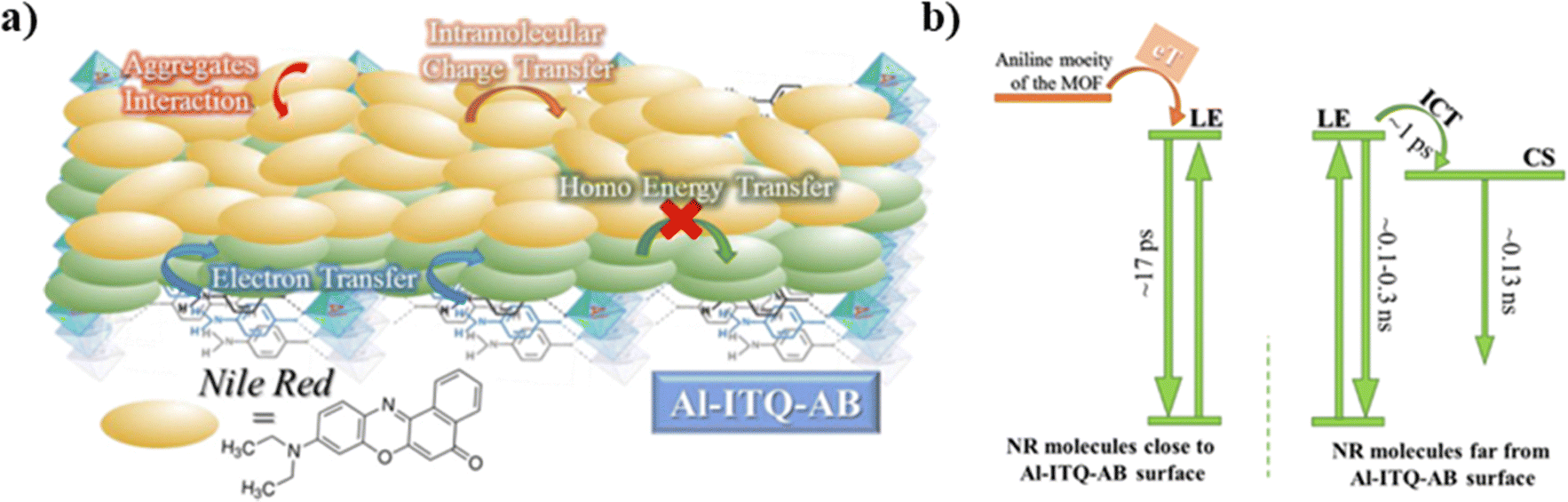 | ||
| Fig. 15 (a) Illustration of the proposed scenario observed when NR molecules are distributed over the Al-ITQ-AB MOF, while (b) is a scheme displaying the photophysical processes and their corresponding times happening in the excited NR@Al-ITQ-AB composite. Adapted with permission from ref. 125 (ACS Appl. Mater. Interfaces, 2018, 10, 32885–32894). Copyright 2018 American Chemical Society. | ||
The ultrafast dynamics of NR@Al-ITQ-EB and NR@Al-ITQ-AB was also characterized and compared with the NR@Al-ITQ-HB hybrid material (Fig. 14b and c). The emission decays of NR@Al-ITQ-EB were similar to those described for NR@Al-ITQ-HB, with three time components of 1.2 ps, 87 ps and 2.59 ns. The two shortest components appear as decays in the bluest region and as rising components in the reddest one, reflecting the occurrence of two photoinduced processes. In agreement with the data described above, the intermediate (87 ps) component corresponds to the homo-ET process, while 1.2 ps is the ICT taking place within the NR after its photoexcitation. However, this ICT reaction is much slower (1.2 ps) than that observed for the NR@Al-ITQ-HB composite (0.5 ps) due to the interaction of the adsorbed NR molecules with the more polar regions (Al clusters) of the 2D-MOF because of the shorter linker in Al-ITQ-EB. This increased polarity stabilizes the locally excited state of the NR and increases the energy barrier of the ICT reaction. On the other hand, the ultrafast behavior of NR@Al-ITQ-AB is different from the other materials, with 3 time components of (1 ps, 17 ps and 115 ps, Fig. 14d). The longest component corresponds to the lifetime of NR aggregates, while the shortest reflects the ICT reaction within the NR molecules. However, the intermediate component, not detected for the other composites, reveals the presence of a new deactivation pathway, and in agreement with the mechanism described above, is evidence of the emission quenching of a population of NR species interacting with the AB linker and undergoing an electron transfer reaction (Fig. 15b).
The photodynamics of this material is rich and complex, with the emission decays fitted by a sum of 4 exponential components, giving time constants of τ1 = 0.15 ns, τ2 = 0.34 ns, τ3 = 1.18 ns, and τ4 = 3.31 ns.126 The shortest τ1-component is the emission of NR molecules from their LE state, while the second τ2-component, which appears as a decay in the shorter wavelengths and as a rise in the longer ones, is related to the homo-ET process, similar to what was found for the NR@Al-ITQ-HB composite. However, this ET reaction is much slower (from 0.22 to 0.34 ns) because of the change in the polarity of the MOF material due to the replacement of the Al clusters by the Zr ones. Finally, the 1.18 and 3.31 ns-components are assigned to the emission of NR aggregates and NR molecules formed through the ET reaction, respectively. To further understand this mechanism, the photodynamics of NR@Zr-ITQ-HB was investigated by measuring its time-resolved emission spectra (TRES, Fig. 16a). Clearly, at short times (<200 ps), it is possible to observe an emission band in the region ranging between 600 and 650 nm, which promptly diminishes its intensity until its complete disappearance at 1 ns. Hence, this band corresponds to the emission of NR molecules from their LE state. In addition, at shorter times there is a band at 650 nm, whose intensity decreases, with a concomitant increase in the intensity of the band located at 700 nm (Fig. 16a). This clearly correlates with the ET process where the NR molecules (emission at 650 nm) suffer from a homo-ET process generating the population of NR species emitting at the reddest spectral region (700 nm) and having the longest lifetimes (3.31 ns), therefore further corroborating the proposed photodynamical mechanism.
 | ||
| Fig. 16 (a) Normalized time-resolved emission spectra (TRES) of NR@Zr-ITQ-HB upon its photoexcitation at 470 nm. (b) fs-emission transient decays of NR@Zr-ITQ-HB in powder form upon its photoexcitation at 470 nm and gated at the indicated wavelengths. Adapted from ref. 126; J. Photochem. Photobiol., A, 2021, 404, 112887, Copyright (2021), with permission from Elsevier. | ||
The ultrafast dynamics of NR@Zr-ITQ-HB is not very different from that described for NR@Al-ITQ-HB.126 The analysis of the fs-emission decays gives a sub-picosecond component of 0.6 ps, which is decaying at shorter wavelengths while rising at longer ones, and a second one of 10 ps, observed only at shorter wavelengths (Fig. 16b). The first component is assigned to the ICT reaction happening within the photoexcited NR, while the 10 ps component is the VC of the molecule. The only difference between the ultrafast photodynamics of those 2 materials is the slightly slower ICT reaction of NR molecules adsorbed on the Zr-based MOF and explained in terms of the different polarity of the local environment in the two materials having different metal clusters.
6.4 DCM dye adsorbed on the Al-ITQ-HB MOF
Once the spectroscopic and photodynamical properties of NR@ITQ-MOF materials were unveiled, the need for novel materials with different properties that could expand the range of actuation of these luminescent materials led to the fabrication of DCM@Al-ITQ-HB composites.127 In these materials, different amounts of initial DCM dye were deposited on the surface of the Al-ITQ-HB MOF. As shown in Fig. 17a, the absorption spectra of these composites become more intense and much broader with the increase in the concentration of DCM, while the emission spectra exhibit a red shift with the increase of DCM molecules deposited on the MOF. Both facts agree with the formation of DCM aggregates upon increasing the concentration of the dye. Interestingly, a deconvolution analysis of the broad absorption band of these samples showed a contribution of 3 different absorbing species: (i) one with a maximum at 395 nm, assigned to the absorption of H-aggregates; (ii) a second with a maximum at 470 nm, which corresponds to the absorption by DCM monomers and (iii) a third red-shifted one with a maximum at 528–538 nm, which is the absorption of the J-aggregates. Based on this, it is possible to envisage that when the concentration of DCM on the MOF surface increases, the concentration of both H- and J-aggregates should also increase. Hence, this explains the red shift observed in the emission spectra with the DCM concentration, since the emission of the H-aggregates is very weak (or non-existent), while the J-aggregates emit at lower energies than the monomer.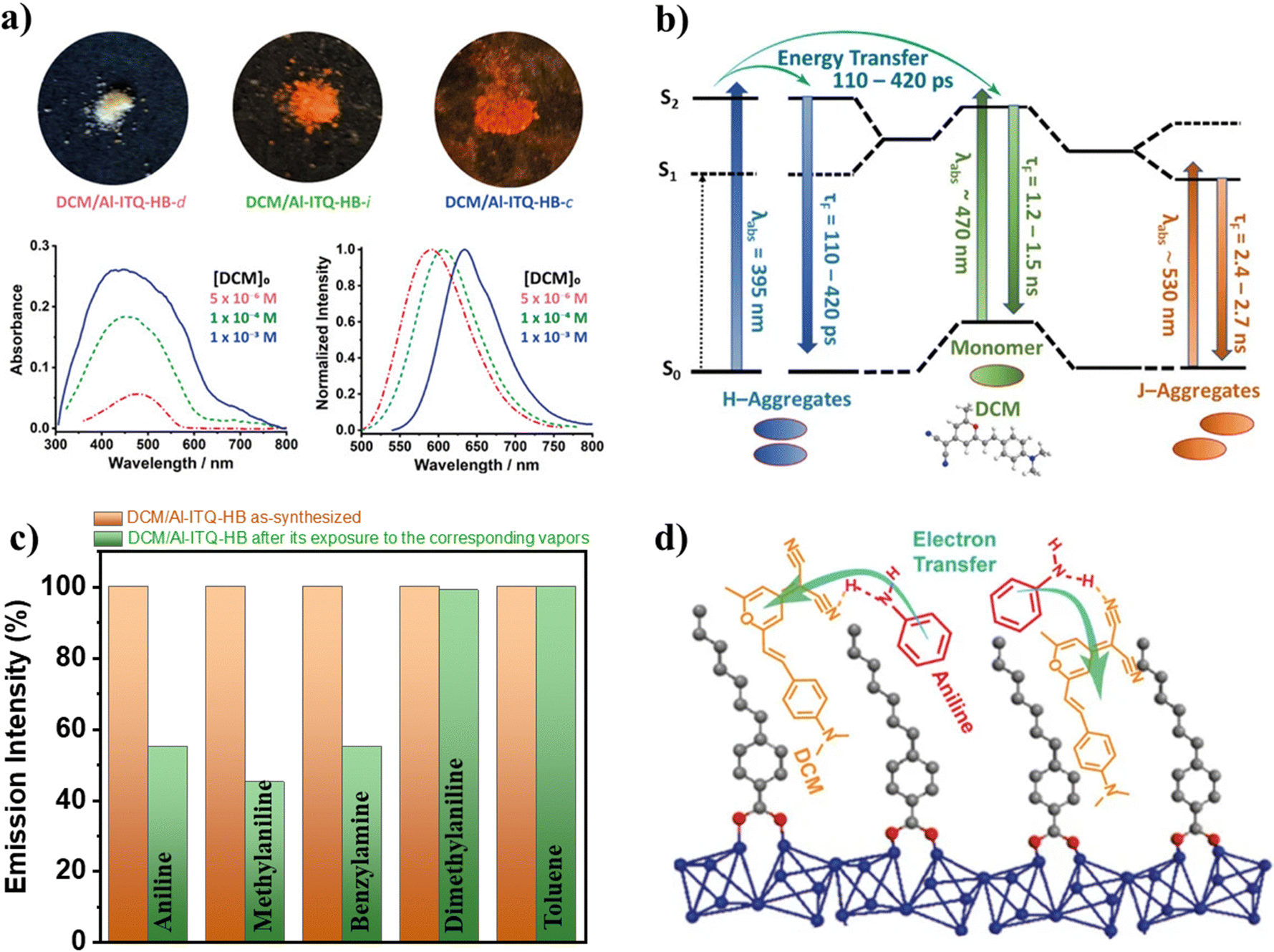 | ||
| Fig. 17 (a) The top sections are real photos of the DCM/Al-ITQ-HB (under daylight) containing increasing amounts of DCM, while the top graphs correspond to the UV-visible absorption (left) and emission (right) spectra of these materials. The different dye concentrations are 1 × 10−3 M (blue solid line), 1 × 10−4 M (green dashed line), and 5 × 10−6 M (pink dashed-dotted line). (b) Illustration of the photoprocesses and related times taking place upon photoexcitation of DCM/Al-ITQ-HB materials. (c) Representation of the emission intensity (in %) of the DCM/Al-ITQ-HB material before and after its exposure to vapors of the indicated aniline or aniline derivatives. (d) Schematic artwork of the emission quenching mechanism of the DCM/Al-ITQ-HB in the presence of aniline, involving an electron transfer process favored by H-bond interactions. Adapted from ref. 127 with permission from MDPI. | ||
In similarity with the NR composites, the time-resolved emission decays of these materials also showed multiexponential behavior with time constants of τ1 = 180–420 ps, τ2 = 1.4–1.5 ns, and τ3 = 2.5–2.7 ns. The intermediate component (τ2) is attributed to the emission lifetime of DCM monomers, as it resembles the emission lifetime of DCM in different organic solvent solutions, whereas the longest component (τ3), whose contribution is higher at longer wavelengths, corresponds to the emission lifetime of J-aggregates (Fig. 17b). The shortest component (τ1), which is decaying at shorter wavelengths but rising at longer ones is attributed to two deactivation pathways. On the one side, this component contributes more to the signal at shorter wavelengths, so it reflects the emission lifetime of DCM H-aggregates. On the other, as it is rising in the reddest spectral region, it also evidences the occurrence of a photophysical phenomenon at S1. In accordance with the mechanism described for the NR@Al-ITQ-HB, this is assigned to a homo-ET reaction happening between neighboring adsorbed DCM molecules (Fig. 17b). Moreover, it was proved that this ET occurs from the H-aggregates, since the rising component disappeared when the material was excited with longer wavelengths (510 nm), where the H-aggregates are not photoexcited.
The luminescent properties of the DCM@Al-ITQ-HB (the initial concentration of DCM was 1 × 10−4 M) were leveraged to detect vapors of harmful compounds such as aniline and some amino derivatives.127 When this material was exposed to a saturated atmosphere of aniline, methylaniline, or benzylamine, it experienced a quenching of its emission intensity (up to 45% of the initial emission intensity, Fig. 17c). At first instance, this quenching could be explained in terms of an electron transfer from the amine derivatives to the photoexcited DCM dye; however, the energy levels of benzylamine are not suitable for such an electron transfer to occur, and the dimethylamine molecule, with a HOMO level much higher than the aniline itself, did not produce any quenching in the emission intensity of DCM@Al-ITQ-HB (Fig. 17c). Hence, the quenching mechanism was explained by attending to the molecular structure of the analytes. Aniline, methylaniline and benzylamine have a H-atom in their structure, which can interact with the DCM dye through H-bonds, thus favoring the electron transfer reaction and quenching the emission of the composite (Fig. 17d). Therefore, despite that the dimethylaniline has a higher energetic HOMO, it cannot undergo this electron transfer, as it does not contain any H-atom in its structure to interact with the DCM molecules adsorbed on the MOF. These results open a novel avenue for the development of luminescent guest@2D-MOF materials for their implementation in sensing devices of toxic pollutants in the vapor phase.
Finally, to summarize Section 6, Table 2 displays the values of the time constants obtained for the related photoprocesses and the fluorescence lifetimes recorded for the different composites formed by NR and DCM interacting with the different MOFs of the ITQ family: Al-ITQ-HB, Al-ITQ-EB, Al-ITQ-AB and Zr-ITQ-HB, where HB = heptylbenzoate, EB = ethylbenzoate and AB = aminobenzoate.
| Composite | Photoprocesses | Fluorescence lifetimes | |||||
|---|---|---|---|---|---|---|---|
| ICT/ps | VC/ps | ET/ns | eT/ps | CSS/ns | ETS/ns | Aggregates/ns | |
| The terms ICT, VC, ET, eT, CSS and ETS stand for intramolecular charge transfer, vibrational cooling, energy transfer, electron transfer, electron injection, charge-separated state and energy transfer state, respectively.a For DCM@Al-ITQ-HB, the 0.18 ns component is for H-aggregates while the 2.5 ns one is for J-aggregates. | |||||||
| NR@Al-ITQ-HB121 | 0.42 | 9.0 | 0.22 | — | 0.22 | 3.61 | 1.33 |
| NR@Al-ITQ-EB125 | 1.2 | — | 0.09 | — | 0.09 | 2.60 | 0.93 |
| NR@Al-ITQ-AB125 | 1.0 | — | — | 17 | 0.13 | — | 0.54 |
| NR@Zr-ITQ-HB126 | 0.60 | 10.0 | 0.34 | — | 0.34 | 3.31 | 1.18 |
| DCM@Al-ITQ-HB127 | — | — | — | — | — | 1.4–1.5 | 0.18/2.5a |
| NR/dichloromethane113 | 1.0 | 1.0 | — | — | 4.4 | — | — |
| DCM/dichloromethane102 | — | — | — | — | 1.1 | — | — |
7. Challenges and perspectives
Of particular interest for the future development of the field will be the use of 2D MOFs or containing 1D or 2D sub-domains for the generation of organic–inorganic nanofibers or even nanosheets,14 which could be the starting point for new hybrid materials with applications in catalysis (photo and electro),128 separation–adsorption,129 storage of active principles or gases,130 sensing,111,131 or molecular photodynamic therapies.132 In addition, modified MOFs based on low-dimensional building units would be highly effective for combining intimately and cooperatively with other components, such as graphene, chalcogenides or metal oxides. This will enable the preparation of new nanocomposites or multi-component materials through hybridization methodologies with a myriad of uses.133 For instance, 2D MOFs might present many advantages over their 3D counterparts, such as a higher surface area, unprecedent electronic properties (with higher carrier mobility and electrical conductivity), high thermal conductivity and excellent optical transparency.134,135 Consequently, 2D MOFs are expected to surpass the performance of their 3D counterparts in a wide variety of technological applications. This is an incipient field that will provide exciting results in the near future.For the synthesis of more complex hybrid organic–inorganic materials by assembling preformed 1D nanofibers, several methodologies can be explored in the future. The layer-by-layer assembly could be used to build complex multilayer structures by alternately depositing oppositely charged materials. Through this method, preformed 1D metalorganic nanofibers would serve as building blocks. The nanofibers could be surface modified with charged functional groups or even polyelectrolytes to impart electrostatic interactions and immersed in a positively charged solution followed by rinsing to remove excess material and then dipped in a negatively charged solution. This process could be repeated to create multiple layers, allowing control over the overall thickness and composition of the hybrid materials. Another method is the template-assisted assembly based on using a sacrificial template to guide the assembly of 1D nanofibers. One valid approach would be to utilize porous templates, such as membranes or nanoporous materials, with controlled pore sizes and shapes. The preformed 1D nanofibers could be dispersed or impregnated within the template. Post-treatment steps, such as washing or annealing, might be required to enhance the interfacial interactions and stability. Another recently proposed approach is an eco-friendly salt-templated synthesis of luminescent guests incarcerated within 2D MOF nanosheets (MONs) for tunable emissive materials.135 This is a promising methodology for synthesizing 2D guest@MON materials, but more information is still required about how to control the entrapment of guests and whether it is possible to incarcerate guests of larger sizes within the 2D sheets. Finally, a recently reported strategy for fine-tuning the MOF color that could be applied also to 2D ITQ MOFs combines postsynthetic modification (PSM) of the chromophoric linkers of an amino-functionalized UiO-68 MOF with computationally guided selection of specific linker modifiers to rationally tune the chromophore band gap.136
On the other hand, advanced theoretical methods that describe the complex structure of the family of 2D ITQ MOFs and the behaviour of the excited guests interacting with the frameworks are important to get a precise picture of the atomic and orbital levels. Time-dependent density functional theory (TD-DFT) and time-dependent density functional perturbation theory (TD-DFPT) methods with refined functionals and basis sets can be used to calculate the optical properties of the hybrid materials.136 With the advancement and further development of machine learning and artificial intelligence algorithms, there is a great opportunity to apply these prediction tools to design, select and fine-tune the structure and properties of the desired MOFs.137–139 The results of such high-level predictions and calculations must be also experimentally verified. However, the ultrafast spectroscopic techniques still present experimental limitations and technical issues. These limitations are mostly related to the heterogeneous nature of the samples, which affects the quality of the obtained experimental data. In that sense, advanced ultrafast vibrational, electronic and X-ray spectroscopies, and 4D electron microscopy are some of the modern tools to explore the intimate interactions in the formed composites at atomic and short-time scales.140–143 Another step towards relating the structure of the 2D ITQ MOFs with the localization and photodynamics of the guests would be the combination of techniques with enough spectral and spatial resolution that can monitor the weakly bonded composites. In this direction, recent developments have demonstrated the feasibility of scattering-type scanning near-field optical microscopy (s-SNOM) and nano-Fourier transform infrared (nanoFTIR) spectroscopy to characterize MOF–guest interactions at the nanoscale.135,144 This review has demonstrated the possibility to confer luminescence properties to 2D ITQ MOFs by employing the more classical strategy of encapsulating fluorescent dyes within the MOF structures.111 This approach has been successfully applied in both 3D and other 2D MOF composites giving rise to materials with tunable optoelectronic properties, such as generating long-lived charge separated states or long-lived photoluminescence.145–152 However, a more challenging aspect that needs to be addressed would be the fabrication of composites with intrinsic luminescence arising from the organic ligands and the interactions with the metal clusters. Thus, with the improvement of our fundamental knowledge of both the synthesis, structure and photoinduced processes within these MOFs, we should be able to address further challenges in photocatalysis, such as selective control and better yield of photoinduced reaction, and even expand the applicability to the field of photonics to produce sensors with better sensitivity and selectivity or more efficient light absorbing and converting LEDs.
8. Conclusions
Throughout this review, we have shown the possibility of using different organic and inorganic structural units like effective low-dimensional building units, which through an effective self-assembly process can generate specific functional MOFs with different properties, morphologies, and structuration. Taking advantage of the versatility and reactivity of the used monotopic organic builders, it is possible to modify and introduce new functionalities in the novel metal–organic structures through different synthesis and post-synthetic treatments to introduce additional active sites into the hybrid networks, controlling the crystalline growth of the obtained hybrid materials. In this way, advanced low-dimensional metal–organic solids with specific catalytic and photo-responsive properties could be generated, making it possible to tune their physico-chemical characteristics according to applications in which they will be finally used. We have further shown how specific and directed changes in the structure of these 2D systems change the properties of the resulting MOF and how these changes affect the photophysical behaviour of composites formed with these 2D MOFs and organic fluorescent dyes.Author contributions
All the authors have contributed to the writing and revision of this manuscript. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is dedicated to Prof. Avelino Corma for his 72nd birthday. This research was supported by grants: PID2020-116519RB-I00 and TED2021–131650B–I00 funded by MCIN/AEI/10.13039/501100011033 and by the European Union (EU); SBPLY/19/180501/000212 and SBPLY/21/180501/000108 funded by JCCM and by the EU through “Fondo Europeo de Desarollo Regional” (FEDER). M. G. also thanks the EU for financial support through Fondo Social Europeo Plus (FSE+). U. D. acknowledges support from Grant PID2020-112590GB-C21 and from Severo Ochoa excellence program CEX2021-001230-S, both funded by MCIN/AEI/10.13039/501100011033. This study forms part of the Advanced Materials program and was supported by MICIN with funding from European Union NextGeneration (PRTR-C17.I1) and by Generalitat Valenciana (MFA/2022/003).References
- A. K. Cheetham, G. Férey and T. Loiseau, Angew. Chem., Int. Ed., 1999, 38, 3268–3292 CrossRef CAS PubMed
.
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC
- A. Corma, J. Catal., 2003, 216, 298–312 CrossRef CAS
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC
- U. Díaz and A. Corma, Coord. Chem. Rev., 2016, 311, 85–124 CrossRef
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC
- M. O’Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef PubMed
- A. Schneemann, R. Dong, F. Schwotzer, H. Zhong, I. Senkovska, X. Feng and S. Kaskel, Chem. Sci., 2021, 12, 1600–1619 RSC
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC
- C. N. R. Rao, H. S. S. Ramakrishna Matte and U. Maitra, Angew. Chem., Int. Ed., 2013, 52, 13162–13185 CrossRef CAS PubMed
- J. M. Moreno, I. Navarro, U. Díaz, J. Primo and A. Corma, Angew. Chem., Int. Ed., 2016, 55, 11026–11030 CrossRef CAS PubMed
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed
- C. G. Carson, K. Hardcastle, J. Schwartz, X. Liu, C. Hoffmann, R. A. Gerhardt and R. Tannenbaum, Eur. J. Inorg. Chem., 2009, 2338–2343 CrossRef CAS
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O’Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed
- J. M. Moreno, A. Velty and U. Diaz, Materials, 2019, 12, 1953 CrossRef CAS PubMed
- I. Syôzi, Prog. Theor. Phys., 1951, 6, 306–308 CrossRef
- C. Volkringer, M. Meddouri, T. Loiseau, N. Guillou, J. Marrot, G. Férey, M. Haouas, F. Taulelle, N. Audebrand and M. Latroche, Inorg. Chem., 2008, 47, 11892–11901 CrossRef CAS PubMed
- G. Férey, J. Solid State Chem., 2000, 152, 37–48 CrossRef
- S. Surblé, F. Millange, C. Serre, G. Férey and R. I. Walton, Chem. Commun., 2006, 1518–1520, 10.1039/B600709K
- C. Serre, F. Millange, S. Surblé and G. Férey, Angew. Chem., Int. Ed., 2004, 43, 6285–6289 CrossRef PubMed
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed
- J. M. Moreno, A. Velty, J. A. Vidal-Moya, U. Díaz and A. Corma, Dalton Trans., 2018, 47, 5492–5502 RSC
- G. Cai and H.-L. Jiang, Angew. Chem., Int. Ed., 2017, 56, 563–567 CrossRef CAS PubMed
- Q. Yang, S. Vaesen, M. Vishnuvarthan, F. Ragon, C. Serre, A. Vimont, M. Daturi, G. De Weireld and G. Maurin, J. Mater. Chem., 2012, 22, 10210–10220 RSC
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS PubMed
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS
- P. García-García, M. Müller and A. Corma, Chem. Sci., 2014, 5, 2979–3007 RSC
- R. Fazaeli and H. Aliyan, Appl. Catal., A, 2007, 331, 78–83 CrossRef CAS
- V. R. Jumde, E. Cini, A. Porcheddu and M. Taddei, Eur. J. Org. Chem., 2015, 1068–1074 CrossRef CAS
- G. Huang, Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 7379–7383 CrossRef CAS PubMed
- M. Jeganathan and K. Pitchumani, ACS Sustainable Chem. Eng., 2014, 2, 1169–1176 CrossRef CAS
- P. García-García, J. M. Moreno, U. Díaz, M. Bruix and A. Corma, Nat. Commun., 2016, 7, 10835 CrossRef PubMed
- M. Zhang, Q.-Y. Fu, G. Gao, H.-Y. He, Y. Zhang, Y.-S. Wu and Z.-H. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 6175–6182 CrossRef CAS
-
B. List, Science of Synthesis: Asymmetric Organocatalysis 1, Lewis Base and Acid Catalysts, Georg Thieme Verlag, 2012 Search PubMed
- T. He, Q. Gu and X.-Y. Wu, Tetrahedron, 2010, 66, 3195–3198 CrossRef CAS
- A. Avila, R. Chinchilla, B. Fiser, E. Gómez-Bengoa and C. Nájera, Tetrahedron: Asymmetry, 2014, 25, 462–467 CrossRef CAS
- S. Biswas, T. Ahnfeldt and N. Stock, Inorg. Chem., 2011, 50, 9518–9526 CrossRef CAS PubMed
- F. Vermoortele, M. Vandichel, B. Van de Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Angew. Chem., Int. Ed., 2012, 51, 4887–4890 CrossRef CAS PubMed
- A. Santiago-Portillo, S. Navalón, P. Concepción, M. Álvaro and H. García, ChemCatChem, 2017, 9, 2506–2511 CrossRef CAS
- J. M. Moreno, A. Velty, U. Díaz and A. Corma, Chem. Sci., 2019, 10, 2053–2066 RSC
- U. Ravon, M. Savonnet, S. Aguado, M. E. Domine, E. Janneau and D. Farrusseng, Microporous Mesoporous Mater., 2010, 129, 319–329 CrossRef CAS
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed
- J. Frunzke, T. Hansen, A. Harrison, J. S. Lord, G. S. Oakley, D. Visser and A. S. Wills, J. Mater. Chem., 2001, 11, 179–185 RSC
- Z. Fang, J. P. Dürholt, M. Kauer, W. Zhang, C. Lochenie, B. Jee, B. Albada, N. Metzler-Nolte, A. Pöppl, B. Weber, M. Muhler, Y. Wang, R. Schmid and R. A. Fischer, J. Am. Chem. Soc., 2014, 136, 9627–9636 CrossRef CAS PubMed
- L. Alaerts, E. Séguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem. – Eur. J., 2006, 12, 7353–7363 CrossRef CAS PubMed
- N. B. Pathan, A. M. Rahatgaonkar and M. S. Chorghade, Catal. Commun., 2011, 12, 1170–1176 CrossRef CAS
- S. Zhang, B. Zhang, H. Liang, Y. Liu, Y. Qiao and Y. Qin, Angew. Chem., Int. Ed., 2018, 57, 1091–1095 CrossRef CAS PubMed
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2010, 46, 6476–6478 RSC
- A. Rapeyko, M. J. Climent, A. Corma, P. Concepción and S. Iborra, ChemSusChem, 2015, 8, 3270–3282 CrossRef CAS PubMed
- M. Oba, K. Tanaka, K. Nishiyama and W. Ando, J. Org. Chem., 2011, 76, 4173–4177 CrossRef CAS PubMed
- P. T. Anastas, Chem. Rev., 2007, 107, 2167–2168 CrossRef
- D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS
- I. T. Horváth and P. T. Anastas, Chem. Rev., 2007, 107, 2169–2173 CrossRef PubMed
- J. M. Moreno, A. Velty and U. Díaz, Catal. Sci. Technol., 2020, 10, 3572–3585 RSC
-
H. Surburg and J. Panten, Common Fragrance and Flavor Materials, Wiley-VCH Verlag GmbH & Co., 2006, pp. 7–175 Search PubMed
- M. N. Timofeeva, V. N. Panchenko, J. W. Jun, Z. Hasan, M. M. Matrosova and S. H. Jhung, Appl. Catal., A, 2014, 471, 91–97 CrossRef CAS
-
J. Otera, Esterification: Methods, Reactions and Applications, Wiley VCH, Weinheim, 2003 Search PubMed
- B. R. Jermy and A. Pandurangan, Appl. Catal., A, 2005, 288, 25–33 CrossRef CAS
- J. G. Smith, Synthesis, 1984, 629–656 CrossRef CAS
- I. Vilotijevic and T. F. Jamison, Angew. Chem., Int. Ed., 2009, 48, 5250–5281 CrossRef CAS PubMed
- X. Li, Z. Guo, C. Xiao, T. W. Goh, D. Tesfagaber and W. Huang, ACS Catal., 2014, 4, 3490–3497 CrossRef CAS
- M. I. Vohra, D.-J. Li, Z.-G. Gu and J. Zhang, Nanoscale, 2017, 9, 7734–7738 RSC
- L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990–12993 CrossRef CAS PubMed
- A. Steinegger, Y. Moritz and S. M. Borisov, J. Photochem. Photobiol., A, 2021, 407, 113046 CrossRef CAS
- L. Shen, W. Wu, R. Liang, R. Lin and L. Wu, Nanoscale, 2013, 5, 9374–9382 RSC
- J. Krämer, R. Kang, L. M. Grimm, L. De Cola, P. Picchetti and F. Biedermann, Chem. Rev., 2022, 122, 3459–3636 CrossRef PubMed
- X. Lu, Y. Zhan and W. He, J. Photochem. Photobiol., B, 2022, 234, 112528 CrossRef CAS PubMed
- Y. Suzuki and K. Yokoyama, Biosensors, 2015, 5, 337–363 CrossRef PubMed
- J. L. Kolanowski, F. Liu and E. J. New, Chem. Soc. Rev., 2018, 47, 195–208 RSC
- B. J. Jung, C. B. Yoon, H. K. Shim, L. M. Do and T. Zyung, Adv. Funct. Mater., 2001, 11, 430–434 CrossRef CAS
- X. Wang, Z. Guo, S. Zhu, Y. Liu, P. Shi, H. Tian and W.-H. Zhu, J. Mater. Chem. B, 2016, 4, 4683–4689 RSC
- M. T. Sturm, H. Horn and K. Schuhen, Anal. Bioanal. Chem., 2021, 413, 1059–1071 CrossRef CAS PubMed
- M. W. Mulberg, M. Hornum, P. Reinholdt, B. B. Jensen, M. Szomek, J. R. Brewer, D. Wüstner, J. Kongsted and P. Nielsen, Eur. J. Org. Chem., 2023, e202300238 CrossRef CAS
- D. Madea, M. Martínek, L. Muchová, J. Váňa, L. Vítek and P. Klán, J. Org. Chem., 2020, 85, 3473–3489 CrossRef CAS PubMed
- B. Boldrini, E. Cavalli, A. Painelli and F. Terenziani, J. Phys. Chem. A, 2002, 106, 6286–6294 CrossRef CAS
- M. Meyer and J. C. Mialocq, Opt. Commun., 1987, 64, 264–268 CrossRef CAS
- A. K. Dutta, K. Kamada and K. Ohta, J. Photochem. Photobiol., A, 1996, 93, 57–64 CrossRef CAS
- J. C. Mialocq and M. Meyer, Laser Chem., 1990, 10, 277–296 CrossRef
- C.-W. Chang, Y.-T. Kao and E. W.-G. Diau, Chem. Phys. Lett., 2003, 374, 110–118 CrossRef CAS
-
R. B. Misra and S. P. Bhattacharyya, Intramolecular Charge Transfer: Theory and Applications, Wiley-VCH, Weinheim, 2018, ch. 4, pp. 115–148 DOI:10.1002/9783527801916
- N. Sarkar, K. Das, D. N. Nath and K. Bhattacharyya, Langmuir, 1994, 10, 326–329 CrossRef CAS
- A. Ya. Freidzon, A. A. Safonov, A. A. Bagaturyants and M. V. Alfimov, Int. J. Quantum Chem., 2012, 112, 3059–3067 CrossRef
- S. Lee, M. Jen and Y. Pang, Int. J. Mol. Sci., 2020, 21 CrossRef PubMed
-
K. Ravi Kumar, S. Satyen, S. Manjeev and M. Akhila, in Photophysics, Photochemical and Substitution Reactions, ed. S. Satyen, K. Ravi Kumar and V. S. Tanja, IntechOpen, Rijeka, 2020, ch. 1 DOI:10.5772/intechopen.93149
- S. Marguet, J. C. Mialocq, P. Millie, G. Berthier and F. Momicchioli, Chem. Phys., 1992, 160, 265–279 CrossRef CAS
- W. Rettig and W. Majenz, Chem. Phys. Lett., 1989, 154, 335–341 CrossRef CAS
- Z. Hsing-Kang, M. Ren-Lan, N. Er-pin and G. Chu, J. Photochem., 1985, 29, 397–404 CrossRef CAS
- J. Qi, X. Hu, X. Dong, Y. Lu, H. Lu, W. Zhao and W. Wu, Adv. Drug Delivery Rev., 2019, 143, 206–225 CrossRef CAS PubMed
- M. Bartel, B. Wysocka, P. Krug, D. Kępińska, K. Kijewska, G. J. Blanchard, K. Kaczyńska, K. Lubelska, K. Wiktorska, P. Głowala, M. Wilczek, M. Pisarek, J. Szczytko, A. Twardowski and M. Mazur, Spectrochim. Acta, Part A, 2018, 195, 148–156 CrossRef CAS PubMed
- C. Martin, S. Bhattacharyya, A. Patra and A. Douhal, Photochem. Photobiol., 2014, 13, 1241–1252 CrossRef CAS PubMed
- M. M. G. Krishna, J. Phys. Chem. A, 1999, 103, 3589–3595 CrossRef CAS
- N. C. Maiti, M. M. G. Krishna, P. J. Britto and N. Periasamy, J. Phys. Chem. B, 1997, 101, 11051–11060 CrossRef CAS
- S. K. Pal, D. Sukul, D. Mandal, S. Sen and K. Bhattacharyya, Chem. Phys. Lett., 2000, 327, 91–96 CrossRef CAS
- H. Guo, X. Zhang, M. Aydin, W. Xu, H.-R. Zhu and D. L. Akins, J. Mol. Struct., 2004, 689, 153–158 CrossRef CAS
- M. R. di Nunzio, G. Perenlei and A. Douhal, Int. J. Mol. Sci., 2019, 20, 1316 CrossRef CAS PubMed
- C. Martin, B. Cohen, M. T. Navarro, A. Corma and A. Douhal, Phys. Chem. Chem. Phys., 2016, 18, 2152–2163 RSC
- M. R. di Nunzio, E. Caballero-Mancebo, C. Martín, B. Cohen, M. T. Navarro, A. Corma and A. Douhal, Microporous Mesoporous Mater., 2018, 256, 214–226 CrossRef CAS
- J. L. Meinershagen and T. Bein, J. Am. Chem. Soc., 1999, 121, 448–449 CrossRef CAS
- S. Uppili, K. J. Thomas, E. M. Crompton and V. Ramamurthy, Langmuir, 2000, 16, 265–274 CrossRef CAS
- M. Gutiérrez, F. Sánchez and A. Douhal, J. Mater. Chem. C, 2015, 3, 11300–11310 RSC
- D. Yan, Y. Tang, H. Lin and D. Wang, Sci. Rep., 2014, 4, 4337 CrossRef PubMed
- H.-Y. Chen and C.-W. Chang, J. Phys. Chem. C, 2020, 124, 8854–8860 CrossRef CAS
- Z. Xu, J. Zhang, T. Pan, H. Li, F. Huo, B. Zheng and W. Zhang, Chem. Mater., 2020, 32, 3553–3560 CrossRef CAS
- M. Gutiérrez, Y. Zhang and J.-C. Tan, Chem. Rev., 2022, 122, 10438–10483 CrossRef PubMed
- A. Synak, P. Bojarski, B. Grobelna, L. Kułak, E. Szczepańska and M. Mońka, J. Mol. Liq., 2020, 306, 112899 CrossRef CAS
- C. Martín, P. Piatkowski, B. Cohen, M. Gil, M. T. Navarro, A. Corma and A. Douhal, J. Phys. Chem. C, 2015, 119, 13283–13296 CrossRef
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS
- K. Michael, Radiat. Res., 2012, 178 Search PubMed
- M. Más-Montoya and R. A. J. Janssen, Adv. Funct. Mater., 2017, 27, 1605779 CrossRef
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed
- E. Caballero-Mancebo, B. Cohen, J. M. Moreno, A. Corma, U. Díaz and A. Douhal, ACS Omega, 2018, 3, 1600–1608 CrossRef CAS PubMed
- A. Datta, D. Mandal, S. K. Pal and K. Bhattacharyya, J. Phys. Chem. B, 1997, 101, 10221–10225 CrossRef CAS
- P. Hazra, D. Chakrabarty, A. Chakraborty and N. Sarkar, Chem. Phys. Lett., 2004, 388, 150–157 CrossRef CAS
- E. Caballero-Mancebo, J. M. Moreno, A. Corma, U. Díaz, B. Cohen and A. Douhal, ACS Appl. Mater. Interfaces, 2018, 10, 20159–20169 CrossRef CAS PubMed
- R. F. Chen and J. R. Knutson, Anal. Biochem., 1988, 172, 61–77 CrossRef CAS PubMed
- Y. Kawamura, J. Brooks, J. J. Brown, H. Sasabe and C. Adachi, Phys. Rev. Lett., 2006, 96, 017404 CrossRef PubMed
-
O. Halbherr and R. A. Fischer, The Chemistry of Metal–Organic Frameworks, 2016, pp. 795–822 DOI:10.1002/9783527693078.ch26
- E. Caballero-Mancebo, J. M. Moreno, B. Cohen, U. Díaz, A. Corma and A. Douhal, ACS Appl. Mater. Interfaces, 2018, 10, 32885–32894 CrossRef CAS PubMed
- E. Caballero-Mancebo, J. M. Moreno, A. Corma, U. Díaz, B. Cohen and A. Douhal, J. Photochem. Photobiol., A, 2021, 404, 112887 CrossRef CAS
- M. R. di Nunzio, M. Gutiérrez, J. M. Moreno, A. Corma, U. Díaz and A. Douhal, Int. J. Mol. Sci., 2022, 23, 330 CrossRef CAS PubMed
- J. Gao, Q. Huang, Y. Wu, Y.-Q. Lan and B. Chen, Adv. Energy Sustainability Res., 2021, 2, 2100033 CrossRef CAS
- M. Chang, F. Wang, Y. Wei, Q. Yang, J.-X. Wang, D. Liu and J.-F. Chen, Open Med. Chem. J., 2022, 68, e17794 CAS
- W. Fan, X. Zhang, Z. Kang, X. Liu and D. Sun, Coord. Chem. Rev., 2021, 443, 213968 CrossRef CAS
- J. Kaur, M. Kaur, S. K. Kansal, A. Umar and H. Algadi, Chemosphere, 2023, 311, 136832 CrossRef CAS PubMed
- H. Wang, T. Chen, H. Ren, W. Liu, F. Nan, J. Ge and P. Wang, ACS Appl. Bio Mater., 2023, 6, 3376–3386 CrossRef CAS PubMed
- C. Guo, F. Duan, S. Zhang, L. He, M. Wang, J. Chen, J. Zhang, Q. Jia, Z. Zhang and M. Du, J. Mater. Chem. A, 2022, 10, 475–507 RSC
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed
- D. A. Sherman, M. Gutiérrez, I. Griffiths, S. Mollick, N. Amin, A. Douhal and J.-C. Tan, Adv. Funct. Mater., 2023, 2214307, DOI:10.1002/adfm.202214307
- Z. M. Soilis, T. H. Choi, J. Brennan, R. R. Frontiera, J. K. Johnson and N. L. Rosi, Chem. Mater., 2023, 35, 7741–7749 CrossRef CAS
- Y. Luo, S. Bag, O. Zaremba, A. Cierpka, J. Andreo, S. Wuttke, P. Friederich and M. Tsotsalas, Angew. Chem., Int. Ed., 2022, 61, e202200242 CrossRef CAS PubMed
- A. S. Rosen, S. M. Iyer, D. Ray, Z. Yao, A. Aspuru-Guzik, L. Gagliardi, J. M. Notestein and R. Q. Snurr, Matter, 2021, 4, 1578–1597 CrossRef CAS
- S. Chong, S. Lee, B. Kim and J. Kim, Coord. Chem. Rev., 2020, 423, 213487 CrossRef CAS
- B.-K. Yoo, Z. Su, J. M. Thomas and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 503–508 CrossRef CAS PubMed
- A. M. Lindenberg, S. L. Johnson and D. A. Reis, Annu. Rev. Mater. Res., 2017, 47, 425–449 CrossRef CAS
- F. Perakis, L. De Marco, A. Shalit, F. Tang, Z. R. Kann, T. D. Kühne, R. Torre, M. Bonn and Y. Nagata, Chem. Rev., 2016, 116, 7590–7607 CrossRef CAS PubMed
- J. P. Kraack and P. Hamm, Chem. Rev., 2017, 117, 10623–10664 CrossRef CAS PubMed
- A. F. Möslein, M. Gutiérrez, B. Cohen and J.-C. Tan, Nano Lett., 2020, 20, 7446–7454 CrossRef PubMed
- X. Yang, X. Lin, Y. S. Zhao and D. Yan, Chem. – Eur. J., 2018, 24, 6484–6493 CrossRef CAS PubMed
- B. Zhou, Z. Qi and D. Yan, Angew. Chem., Int. Ed., 2022, 61, e202208735 CrossRef CAS PubMed
- Y.-J. Ma, X. Fang, G. Xiao and D. Yan, Angew. Chem., Int. Ed., 2022, 61, e202114100 CrossRef CAS PubMed
- X. Yang and D. Yan, Chem. Sci., 2016, 7, 4519–4526 RSC
- X.-G. Yang, J.-H. Qin, Y.-D. Huang, Z.-M. Zhai, L.-F. Ma and D. Yan, J. Mater. Chem. C, 2020, 8, 17169–17175 RSC
- D. Yan, G. O. Lloyd, A. Delori, W. Jones and X. Duan, ChemPlusChem, 2012, 77, 1112–1118 CrossRef CAS
- M. Gutiérrez, C. Martín, J. Hofkens and J.-C. Tan, J. Mater. Chem. C, 2021, 9, 15463–15469 RSC
- F. Sánchez, M. Gutiérrez and A. Douhal, ACS Appl. Mater. Interfaces, 2022, 14, 42656–42670 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2023 |