
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular geometry and the photophysics of thermally activated delayed fluorescence: the strange case of DMAC-py-TRZ†‡
Ettore
Crovini§
a,
Rama
Dhali§
b,
Dianming
Sun
 *a,
Tomas
Matulaitis
a,
Thomas
Comerford
a,
Alexandra M. Z.
Slawin
a,
Cristina
Sissa
b,
Francesco
Azzolin
b,
Francesco
Di Maiolo
b,
Anna
Painelli
*b and
Eli
Zysman-Colman
*a
*a,
Tomas
Matulaitis
a,
Thomas
Comerford
a,
Alexandra M. Z.
Slawin
a,
Cristina
Sissa
b,
Francesco
Azzolin
b,
Francesco
Di Maiolo
b,
Anna
Painelli
*b and
Eli
Zysman-Colman
*a
aOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife, KY16 9ST, UK. E-mail: eli.zysman-colman@st-andrews.ac.uk; Web: https://www.zysman-colman.com Tel: +44 (0)1334 463826
bDepartment of Chemistry, Life Science and Environmental Sustainability, University of Parma, 43124 Parma, Italy
First published on 5th April 2023
Abstract
We present the synthesis, optoelectronic characterization, and a detailed theoretical study of DMAC-py-TRZ, a novel, efficient TADF emitter. This compound is a structural relative of the well-known TADF compound DMAC-TRZ, substituting the bridging phenylene for a pyridyl group. This marginal change has an enormous impact on the structure and hence on the photophysics as the steric interactions between the DMAC donor and the bridge that force DMAC-TRZ into an orthogonal conformation are attenuated and permit DMAC-py-TRZ to adopt a planar and slightly bent structure in the ground state. The large degree of conjugation in the bent DMAC-py-TRZ structure, demonstrated by the strong intensity of the lowest excitation with CT character, is responsible for a large singlet triplet gap, hence hindering TADF of this bent conformer. The computational analysis predicts that emission occurs, however, from a relaxed orthogonal excited-state geometry, as confirmed by the huge Stokes shift observed in non-polar solvents. In this relaxed orthogonal geometry TADF is indeed observed. Emission from the unrelaxed state is recovered in glassy frozen solvents, where the emission band is largely blue-shifted compared with measurements in liquid solvent, and TADF is quenched. In amorphous matrices, structural disorder leads to the coexistence of both conformers, even if, depending on the emitter concentration, dual fluorescence may disappear due to a fast energy transfer from the bent to the orthogonal conformers. We maintain that this efficient energy transfer is responsible for the good efficiency of DMAC-py-TRZ devices, because of the presence in the matrix of a sizable proportion of compounds that adopt the bent structure, favorable to act as the host for the orthogonal TADF conformer of DMAC-py-TRZ.
10th Anniversary StatementThe Journal of Materials Chemistry C has been one of our favourite journals for OLED materials research. The quality and breadth of the science covered in the journal particularly in the area of emitter development for electroluminescent devices has been excellent. It is always a pleasure to peruse each week's table of contents and to then read exciting and new science in optoelectronic materials. We look forward to the next 10 years and beyond and will continue to support JMCC. |
Introduction
In an organic light-emitting diode (OLED), electrons and holes, injected in the device from opposite electrodes, combine to form excitons. According to spin statistics, 25% of the electrically generated excitons are in a singlet state and 75% are in a triplet state. If the excitons form on a fluorescent molecule, emission will only occur from the singlet excitons, effectively limiting the device Internal Quantum Efficiency (IQE). Phosphorescent dyes harvest both singlet and triplet excitons to emit light from the triplet excited state resulting in an IQE of up to 100%. Most phosphorescent emitters, however, contain noble metals such as platinum or iridium, among the scarcest elements on Earth.1–3 Thermally activated delayed fluorescence (TADF) offers a different yet equally appealing strategy to triplet harvesting where 100% IQE is also possible in the device. In TADF emitters, dark triplet excitons are thermally upconverted into emissive singlets via reverse intersystem crossing (RISC). RISC is possible when the energy gap, ΔEST, between the lowest lying singlet and triplet excited states is of the order of the thermal energy (ca. <0.02 eV), provided that spin–orbit coupling (SOC) between the two states is non-negligible. To minimize ΔEST, the overlap between the HOMO and LUMO of the molecule must be reduced, localizing the two orbitals in separate electron-donating (for the HOMO) and electron-accepting (for the LUMO) parts of the molecule.4 This separation is most often obtained by enforcing a large dihedral angle between the electron donor and acceptor moieties.5 This strategy, however, leads to a reduction of the fluorescence efficiency and, according El Sayed rule,6 to a reduction of the spin–orbit coupling between the singlet and triplet states as well.Two of the most common moieties employed as donors and acceptors, respectively, are 9,9-dimethy-9,10-dihydroacridine (DMAC) and 2,4,6-triphenyl-1,3,5-triazine (TRZ), and the combination of the two produces the emitter DMAC-TRZ, first reported by Tsai et al.7DMAC-TRZ shows a very high photoluminescence quantum yield, ΦPL, of 90%, at λPL of 495 nm, as an 8 wt% doped film in mCPCN [9-(3-(9H-carbazol-9-yl)phenyl)-9H-carbazole-3-carbonitrile]. In DMAC-TRZ, the ΔEST amounts to a few tens of meV, depending on the host matrix, with a delayed fluorescence lifetime of 1.9 μs in mCBPBN, in line with an efficient RISC process.8 The OLED shows a high maximum external quantum yield, EQEmax, of 26.5% at λEL of 500 nm, while devices prepared from neat DMAC-TRZ films show a comparable EQEmax of 20.0%.7 The modest reduction of the EQEmax at high concentration can be understood in terms of the orthogonal conformation of the emitter, which effectively prevents aggregation. Both the doped and non-doped devices show a relatively small efficiency roll-off, with an EQE100 of 25.1% and 18.9%, respectively.
The emitters a-DMAC-TRZ9 and MA-TA10 are derivatives of DMAC-TRZ where adamantane groups are incorporated into the structure (Fig. 1). In a-DMAC-TRZ, the adamantane-functionalization of the donor moiety leads to a deformed structure that results in an increased optical gap and thus a bluer emission. Dual fluorescence is observed from two different conformers, but overall, the electroluminescence (EL) performance remains similar to the DMAC-TRZ OLED, with an EQEmax of 28.9%, yet with a λEL of 488 nm. In the report by Wada et al.,10 the replacement of the distal phenyl moieties on the TRZ with adamantyl groups results in a weaker acceptor, leading to a blue-shifted electroluminescence compared to DMAC-TRZ. The adamantyl substitution also reduces the non-radiative decay leading to a ΦPL of 99%. The blue solution-processed device (λEL of 475 nm) shows an EQEmax at 22.1%. Conversely, replacement of the distal phenyl rings in TRZ by electron-withdrawing pyrimidines (DMAC-bPmT) results in a red-shifted emission (λPL of 520 nm vs. 500 nm for DMAC-TRZ in toluene).11 The delayed emission lifetime of DMAC-bPmT is 3.3 μs in toluene, which is shorter than that of DMAC-TRZ at 8.8 μs in the same medium. The RISC rate constant, kRISC, of DMAC-bPmT is 8.8 × 105 s−1, is three times faster than that of DMAC-TRZ (2.9 × 105 s−1). However, its ΦPL of 70% in toluene is reduced compared to that of DMAC-TRZ (ΦPL = 93% in toluene). Rajamalli et al.,12 and Dos Santos et al.,13 showed that the introduction of a heteroaromatic bridge in sulfone-based D–A TADF emitters can prevent structural relaxation, enhance ΦPL and improve the color purity due to the narrower emission, and demonstrated an improvement in the efficiency of the devices compared to that with the reference emitter pDTCz-DPS. The materials in the study of Rajamalli et al.,12pDTCz-2DPyS, and pDTCz-3DPyS, both show good ΦPL of ca. 60%. The blue OLEDs, at λEL of 466 nm and 452 nm for the devices with pDTCz-2DPyS, and pDTCz-3Dpy, respectively, showed EQEmax ∼12–13%, which are considerably improved over the parent device with pDTCz-DPS (EQEmax of 4.7%). Dos Santos et al.,13 showed that the addition of second nitrogen atom within the bridging heterocycle in pDTCz-DPzS, and pDTCz-DPmS contributed to a further enhancement of the EQEmax to 18% and 14%, respectively, at λEL of 522 nm and 461 nm for the devices with pDTCz-DPzS, and pDTCz-DPmS, respectively.
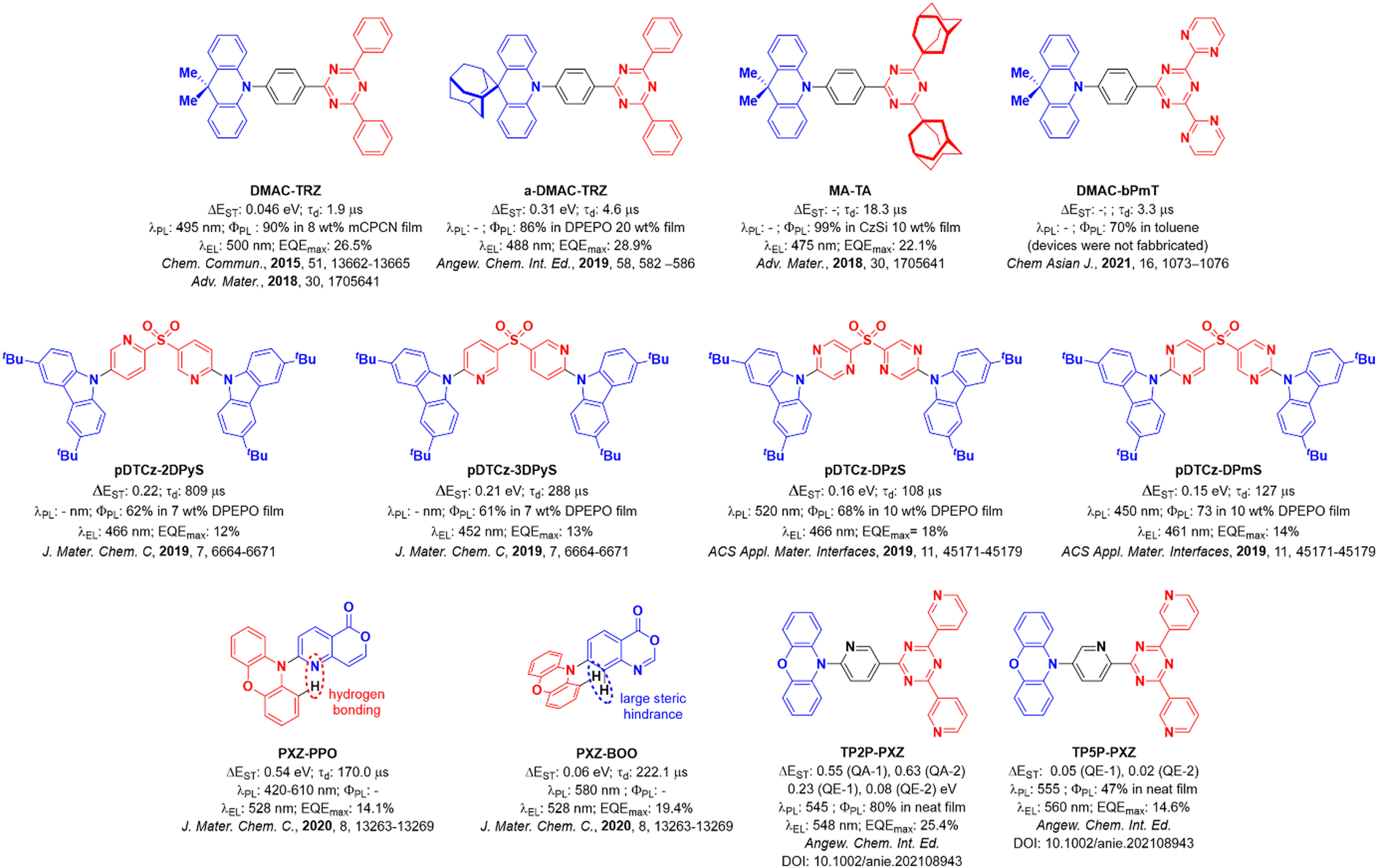 | ||
| Fig. 1 Molecular structures of selected emitters from the literature with selected photophysical and device performance data. | ||
The impact of intramolecular H-bonding on the photophysics of TADF emitters was also investigated by Chen et al. in two phenoxazine-based emitters PXZ-PPO and PXZ-BOO.14PXZ-BOO shows TADF, relevant devices having EQEmax of 19.4% at λEL of 528 nm. In THF solution, PXZ-PPO is present as a mixture of two conformers, a more planar structure with N–H interaction and a twisted structure, where hydrogen bonding is not present. The planar conformation emits in the deep blue at 420 nm but shows no TADF, while the twisted conformer shows green TADF (610 nm) but with a very short lifetime of 170 ns. The planar conformer is dominant in the crystalline phase, while in solution the twisted conformer is largely responsible for the observed photophysics, giving rise to TADF. The PXZ-PPO-based device, with EQEmax of 14.1% at λEL of 528 nm, is slightly inferior to the OLED based on PXZ-BOO, which has an EQEmax of 19.4% at λEL of 528 nm.
The presence of two conformers of a TADF emitter has also been documented by Shi et al. in the TRZ derivative compounds TP2P-PXZ and TP5P-PXZ.15 Phenoxazine and a central terpyridine bridge were used to promote the formation of an intramolecular hydrogen bond in TP2P-PXZ, while in the control compound TP5P-PXZ this interaction is absent. The presence of quasi-equatorial (QE) and quasi-axial (QA) conformations of TP2P-PXZ led to a self-doped system, where the QA conformer effectively acts as the host material. This led to an efficient OLED with an EQEmax of 25.4% at λEL of 548 nm while the performance of the device with TP5P-PXZ was somewhat attenuated with an EQEmax of 14.6% at a slightly red-shifted λEL of 560 nm. Similarly, two conformers have been found coexisting in the crystalline phase for the compounds Trz-Py-NCS and Trz-Py-SAC.16
These examples show that modification of the nature of the aromatic bridge in a TADF emitter can lead to significant changes in the molecular geometry, with the co-existence of different conformers that show distinctive photophysics. In this work, we introduce the emitter DMAC-py-TRZ, where the phenylene bridge in DMAC-TRZ7 is replaced by a 2-pyridyl bridge. DMAC-py-TRZ emits at λPL of 539 nm and has a ΦPL of 58% in toluene solution while as a 10 wt% doped mCP film the ΦPL is 496 nm and the ΦPL is 57%. Its crystal structure (Fig. 2) documents a small dihedral angle between the DMAC and pyridyl bridge of 19.7(2)° and a V-shaped or bent structure of the DMAC donor, with an associated bending angle (deviation from a planar conformation) of 45°. This behaviour is in line with that observed by Shi et al.15 We present an in-depth computational study and an extensive optoelectronic and photophysical characterization that showcases the impact that conformational changes in the excited state and not just in the ground state have on the photophysics of the compound.
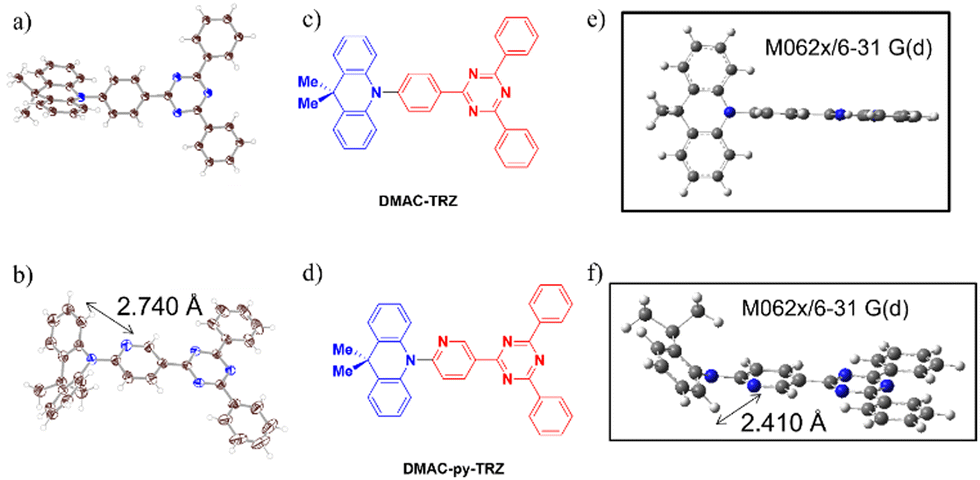 | ||
| Fig. 2 (a) and (b) The crystal structure shown as ORTEP at 50% ellipsoid probability; (c) and (d) the molecular structure; (e) and (f) the calculated ground state structure of DMAC-TRZ and DMAC-py-TRZ. | ||
Results and discussion
Computational analysis
The ground-state geometries of DMAC-TRZ and DMAC-py-TRZ were optimized in the gas phase using density functional theory (DFT) at the M062X/6-31G(d) level of theory.17–19 The excited-state energies were calculated using time-dependent density functional theory (TD-DFT) within the Tamm–Dancoff approximation20 at the same level of theory (TDA-DFT). We employ the term “orthogonal” to describe the structure with a dihedral angle between the DMAC and the bridge that is close to 90° and we dub as “bent” the structure with the small dihedral angle and V-shaped geometry of the DMAC.In a recent publication,21 an extensive computational analysis of DMAC-TRZ set the basis for a few-state model that was carefully validated against spectroscopic properties in solution. Then the same model was exploited to calculate ISC and RISC rate constants, also accounting for environmental effects including dielectric and conformational disorder.22,23 In the ground-state equilibrium geometry of DMAC-TRZ, the DMAC and TRZ moieties are mutually orthogonal (Fig. 2), in line with the crystal structure.21 In this orthogonal geometry, the S1 and T1 states each have a pure charge transfer (CT) character. In other terms, the HOMO and LUMO have negligible overlap so that the singlet–triplet gap is almost closed, with ΔEST = 0.01 eV. The close similarity between the orbitals involved in S1 and T1 states implies a vanishing SOC, according to El Sayed's rule,6 as to hinder direct RISC from T1 to S1.
As for the excited states of DMAC-TRZ, S1 (with a strong 1CT character) retains an orthogonal geometry, while T1 (3CT) undergoes a large conformational deformation where the dihedral becomes ∼60°. At this angle, ΔEST increases as does SOC. A rigid scan of the dihedral angle of DMAC-TRZ (Fig. S9c, ESI‡)21 is informative. Specifically, starting from the optimized ground-state geometry, we calculated the ground and excited state energies upon gradual rotation of the DMAC unit about the phenylene bridge without allowing for any additional molecular relaxation (the dihedral angle for the scan is defined in Fig. S9a, ESI‡). The resulting S0, S1, T2 and T3 potential energy surfaces (PES) all show a flat minimum for the orthogonal geometry, while T1 shows a double minimum around (90 ± 30)° angle.19 The molecular orbitals (MOs) and natural transition orbitals (NTOs) shown in Fig. S11 and S12 (ESI‡) reveal that in the orthogonal structure, the HOMO is localized on DMAC and the LUMO is on the TRZ.
In contrast with DMAC-TRZ, the ground-state optimized geometry of DMAC-py-TRZ has a bent structure (Fig. 2f), as also observed in the crystal structure (Fig. 2b). To better understand the structural differences between DMAC-TRZ and DMAC-py-TRZ, a rigid dihedral angle scan of DMAC-py-TRZ starting from an analogous orthogonal conformation to that of DMAC-TRZ has been performed. The rigid scan leads to a qualitatively similar picture for the two compounds (Fig. S9, ESI‡), with relevant MOs and NTOs in Fig. S11 and S12 (ESI‡), showing the HOMO and LUMO localized on the donor and acceptor moieties, respectively. The presence of the nitrogen atom in the pyridine bridge effectively increases the electron-withdrawing strength of the acceptor, resulting in a stabilized LUMO and a smaller HOMO–LUMO gap in DMAC-py-TRZ (EHOMO–LUMO = 4.78 eV for the orthogonal structure) vs.DMAC-TRZ (EHOMO–LUMO = 4.99 eV for the orthogonal structure). Accordingly, the S1 and T1 excitations occur at lower energy in DMAC-py-TRZ than in DMAC-TRZ and both the S1 and T1 states are stabilized compared to those of DMAC-TRZ (Fig. S9c and d, ESI‡). The rigid energy scan, however, points to a large increase of the ground-state energy when the dihedral angle deviates significantly from orthogonality, so that non-orthogonal conformations are hardly accessible. To address the bent conformer, we performed a relaxed scan of the dihedral angle, relevant results being shown in Fig. 3 (see also ESI,‡ Fig. S18).
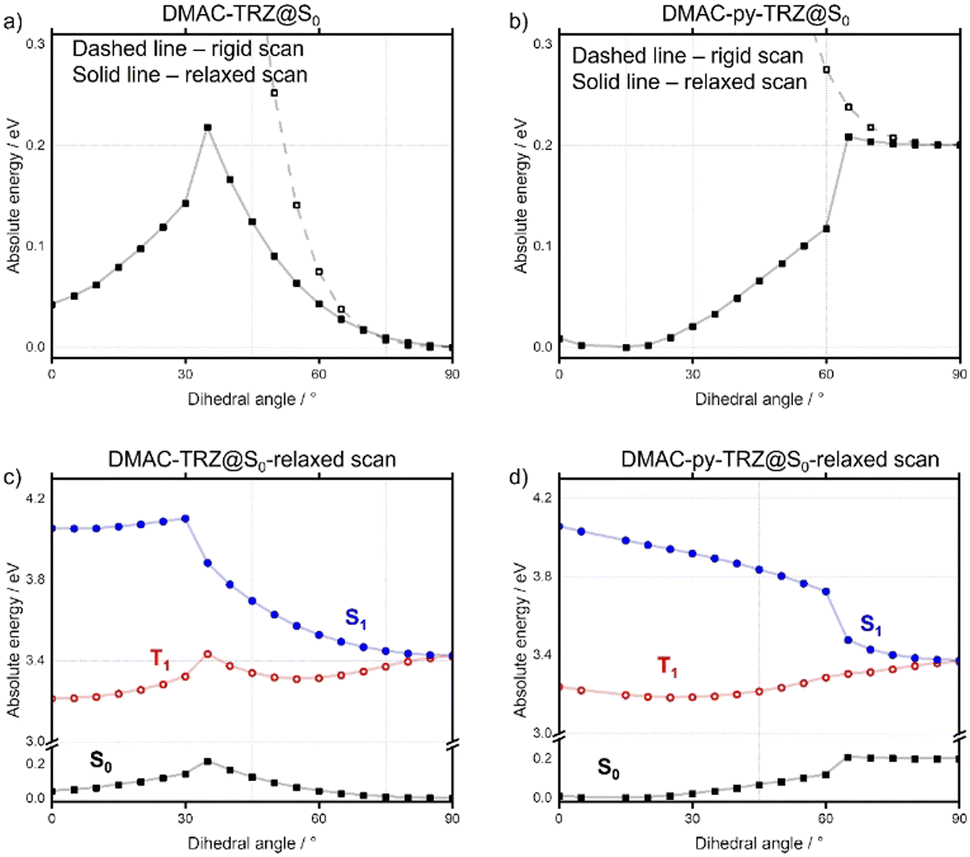 | ||
| Fig. 3 (a) and (b) compare the ground state (S0) energy calculated as a function of the dihedral angle in a rigid (dashed lines) and relaxed scan (continuous lines) for DMAC-TRZ and DMAC-py-TRZ, respectively; (c) and (d) show the energies of the S0, S1 and T1. | ||
The relaxed scans show that for each of the emitters, two minima are present, corresponding to the orthogonal and bent structures. For DMAC-TRZ, the energy difference between the two conformers amounts to 0.04 eV, slightly larger than thermal energy at room temperature. The energy barrier for the interconversion between the two conformers, 0.22 eV (21.2 kcal mol−1), is, however, much larger than thermal energy so that only the orthogonal geometry is expected to be significantly populated at room temperature. The situation is very different for DMAC-py-TRZ where the bent conformer (dihedral angle ∼10°) is lower in energy than the orthogonal conformer by 0.20 eV and the energy barrier for the interconversion between the bent and orthogonal conformers is 0.20 eV (19.3 kcal mol−1). Thus, at room temperature only the bent conformer is populated. The MOs and NTOs calculated for the bent structure (Fig. S11 and S12, ESI‡) show that in both molecules there is a partial delocalization of the HOMO onto the acceptor moiety and, for DMAC-py-TRZ also a partial delocalization of the LUMO onto the donor moiety.
Divergent results are obtained for the excited state energies at the geometries relevant to the rigid/relaxed scans of the dihedral angle (Fig. 3c and d) of the two compounds. For DMAC-TRZ, the rigid (Fig. S9c, ESI‡) and the relaxed scans (Fig. 3c) lead to the same picture: the S1 state maintains the same orthogonal conformation as the ground state, while the T1 state is stabilized and adopts a twisted structure (dihedral angle: ∼60°). Full optimisations of S1 and T1 confirm this result.21 In the orthogonal geometry, the S1 state is an almost pure CT state and, hence, has a negligible oscillator strength. The scenario is much more interesting for DMAC-py-TRZ. In the bent geometry (the energy minimum, Fig. 3b), the vertical excitation energy to S1 amounts to 4.034 eV and the ΔEST is 0.786 eV, which is far too large for TADF to be operational at ambient temperature. Moreover, the oscillator strength for the S0 → S1 is large (1.27, Fig. S12c, ESI‡) in this geometry due to the significant overlap of the orbitals involved in the transition (Fig. 4a). However, the bent geometry is not the equilibrium geometry for S1 (Fig. 4d) and a huge structural deformation is predicted in the S1 state from the bent to the orthogonal structure. In other terms, in DMAC-py-TRZ the absorption occurs from the bent geometry and the lowest energy transition is both high in energy and has a large oscillator strength. By contrast, fluorescence occurs from the orthogonal structure at a much lower energy (3.17 eV) and with negligible oscillator strength (as per the non-overlapping orbitals, in Fig. 4b). In this orthogonal geometry the ΔEST reduces to only 8.2 meV), making TADF possible. To better appreciate the differing photophysics of the two different conformers in either DMAC-TRZ or DMAC-py-TRZ, Fig. S19 (ESI‡) compares relevant calculated spectra and Table S29 (ESI‡) summarizes the calculated ΔEST.
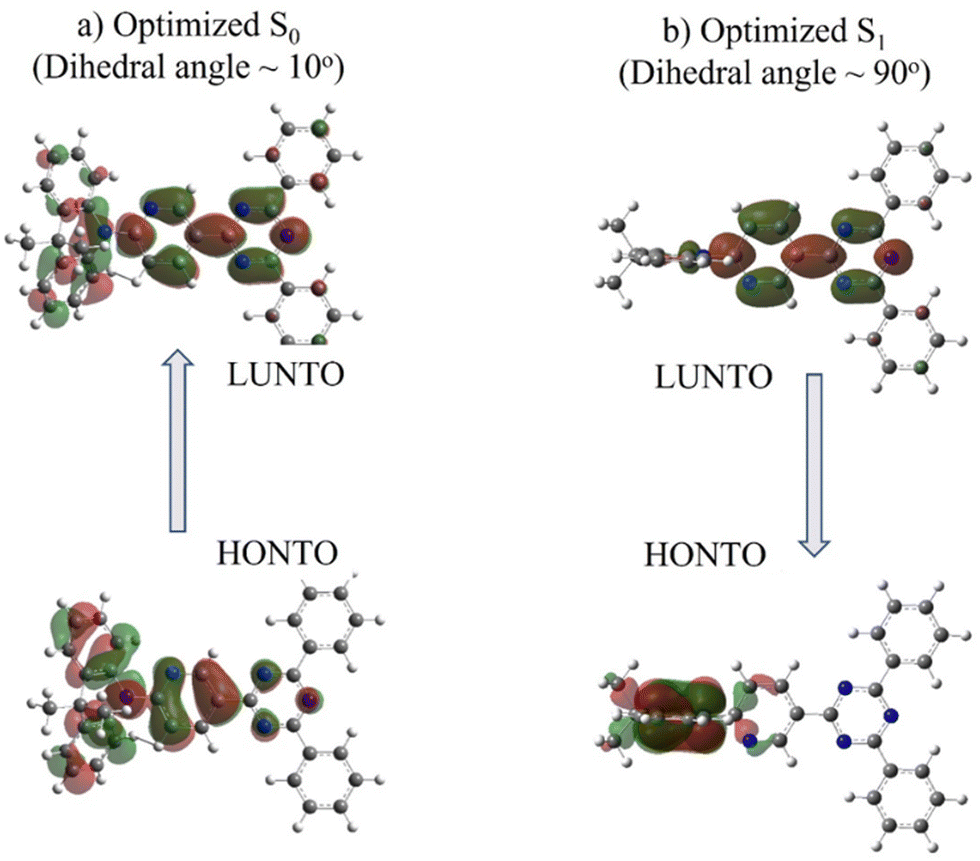 | ||
| Fig. 4 NTOs of DMAC-py-TRZ calculated for the S0–S1 transition (a) at the S0-optimized geometry, which is relevant to the absorption process; (b) at the S1-optimized geometry, which is relevant to the emission process. | ||
In DMAC-py-TRZ, the T1 state is predicted to exist as a twisted geometry in both the rigid (Fig. S9d, ESI‡) and relaxed scans (Fig. 3d). The full optimization of the excited state geometry, however, yields conflicting results. For S1 the situation is clear: taking either the bent or the orthogonal geometry as starting points for the excited state optimization, the S1 geometry (Fig. 3d) always converges to the orthogonal conformation, supporting the results from the relaxed scan analysis. For T1, instead, two different structures are reached depending on the starting geometry (Fig. S10, ESI‡), with slightly different ΔES1–T1 values (0.93 eV where the dihedral angle is 30° and 0.41 eV where the dihedral angle is 60°). The energy of the two triplet conformations is similar (ΔET1 60°–30° = 0.07 eV), so that a firm conclusion about the equilibrium geometry for T1 cannot be reached.
Optoelectronic properties
Cyclic Voltammetry (CV) and Differential Pulse Voltammetry (DPV) of DMAC-py-TRZ and DMAC-TRZ were measured in degassed DCM with tetra-n-butylammonium hexafluorophosphate as the electrolyte and Fc/Fc+ as the internal reference. The voltammograms in Fig. 5a are reported versus a Saturated Calomel Electrode (SCE). Both materials show pseudo-reversible reduction and oxidation waves. Both oxidation and reduction waves for DMAC-py-TRZ (Eox/Ered = 1.11/−1.64 V) are anodically shifted compared to those of DMAC-TRZ (0.97/−1.72 V). The corresponding HOMO and LUMO energies are −5.31/−2.62 eV and −5.45 eV/−2.70 eV for DMAC-TRZ and DMAC-py-TRZ, respectively. Comparing electrochemical redox gaps measured in solution with gas-phase DFT results is tricky.25,26 Experimentally, the HOMO–LUMO gap of DMAC-py-TRZ, ∼2.75 eV, is larger than the HOMO–LUMO gap of DMAC-TRZ ∼2.69 eV. Considering an orthogonal geometry for both dyes, DFT results for the HOMO–LUMO gaps are 4.78 eV and 4.99 eV for DMAC-py-TRZ and DMAC-TRZ, respectively, showing the opposite trend vs. experiment. The same is true if the bent geometry is considered for both dyes, with calculated HOMO–LUMO gaps of 5.73 eV and 5.77 eV for DMAC-py-TRZ and DMAC-TRZ, respectively. However, if we properly consider an orthogonal structure for DMAC-TRZ (EHOMO–LUMO = 4.99 eV for orthogonal geometry) and a bent structure for DMAC-py-TRZ (EHOMO–LUMO = 5.73 eV for bent geometry), calculated HOMO–LUMO gaps reflect the trend observed experimentally. A better estimate of the ionization potential (IP) and electron affinity (EA) of both dyes is obtained by comparing the energy of the radical cation and radical anion molecules in their relaxed geometry with the energy of the neutral dye: IP = Eradical cation − Eneutral and EA = Eradical![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) anion − Eneutral. In this case, the IP-EA gap (to be compared with the experimental data) are 7.59 eV and 7.24 eV for DMAC-py-TRZ and DMAC-TRZ, respectively, for the orthogonal geometries. For the bent geometries, the gaps are 7.63 eV and 7.25 eV for DMAC-py-TRZ and DMAC-TRZ, respectively. Also in this case, if we consider the orthogonal conformer for DMAC-TRZ and a bent conformer for DMAC-py-TRZ the calculated gaps reflect the trend observed experimentally. A perfect agreement between DFT estimates and experimental data is, however, not expected since our gas-phase calculations do not account for solvation effects.25,26 In any case, the HOMO stabilization in DMAC-py-TRZvs.DMAC-TRZ is ascribed to the presence of the electron-withdrawing pyridyl bridge, which reduces the electron density on the donor. Similarly, the electron density of the acceptor is reduced, resulting in a more stabilized LUMO level.
anion − Eneutral. In this case, the IP-EA gap (to be compared with the experimental data) are 7.59 eV and 7.24 eV for DMAC-py-TRZ and DMAC-TRZ, respectively, for the orthogonal geometries. For the bent geometries, the gaps are 7.63 eV and 7.25 eV for DMAC-py-TRZ and DMAC-TRZ, respectively. Also in this case, if we consider the orthogonal conformer for DMAC-TRZ and a bent conformer for DMAC-py-TRZ the calculated gaps reflect the trend observed experimentally. A perfect agreement between DFT estimates and experimental data is, however, not expected since our gas-phase calculations do not account for solvation effects.25,26 In any case, the HOMO stabilization in DMAC-py-TRZvs.DMAC-TRZ is ascribed to the presence of the electron-withdrawing pyridyl bridge, which reduces the electron density on the donor. Similarly, the electron density of the acceptor is reduced, resulting in a more stabilized LUMO level.
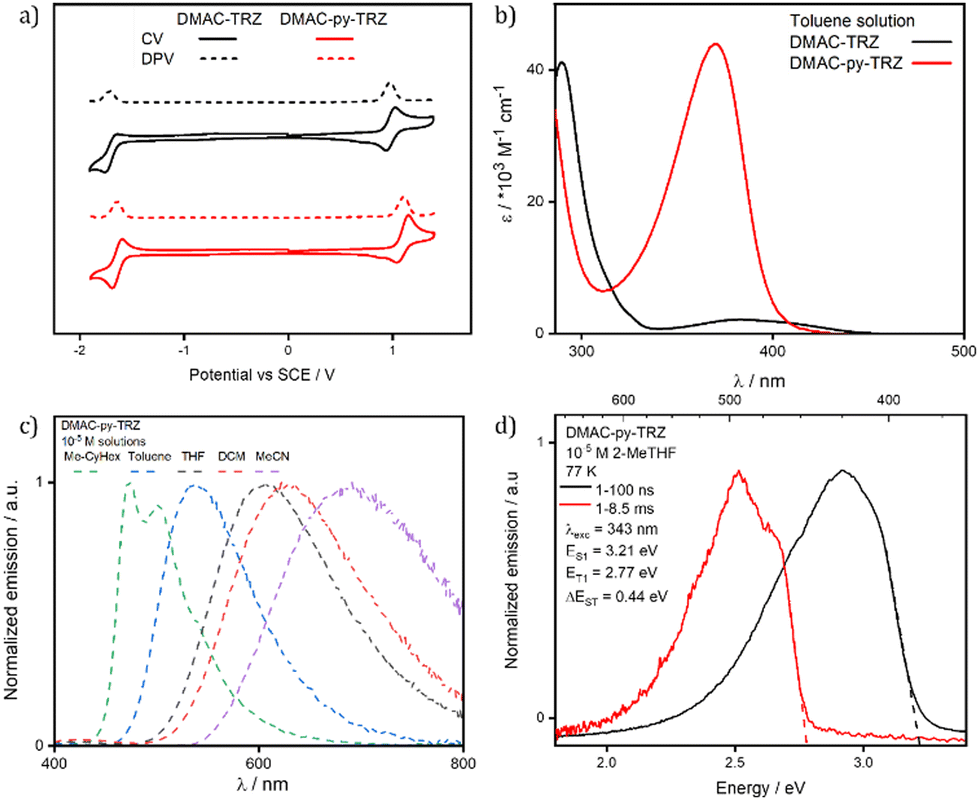 | ||
| Fig. 5 (a) Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) of DMAC-TRZ (black) and DMAC-py-TRZ (red) in DCM (scan rate = 100 mV s−1, tetrabutylammonium hexafluorophosphate as electrolyte, reported relative to a saturated calomel electrode (SCE) with a ferrocene/ferrocenium (Fc/Fc+) redox couple as the internal standard, 0.46 V vs. SCE for DCM24). (b) UV-vis absorption spectra of DMAC-TRZ (black) and DMAC-py-TRZ (red) in 10−5 M toluene. (c) Solvatochromism photoluminescence study of DMAC-py-TRZ (λexc, = 340 nm); (d) prompt fluorescence and phosphorescence spectra at 77 K in 10−5 M 2-methyltetrahydrofuran (2-MeTHF) glass (λexc = 343 nm, prompt and delayed fluorescence spectra were obtained in the 1–100 ns and 1–8.5 ms time range, respectively). | ||
Fig. 5b compares the absorption spectra of DMAC-TRZ and DMAC-py-TRZ in toluene. The CT band of DMAC-py-TRZ at 370 nm is slightly blue-shifted with respect to DMAC-TRZ at 382 nm. The most striking difference is recognized, however, in the much larger intensity of the band measured for DMAC-py-TRZ (ε = 43800 M−1 cm−1) vs.DMAC-TRZ (ε = 2100 M−1 cm−1). This is a direct consequence of the different conformations adopted by the two compounds in the ground state: the orthogonal conformation of DMAC-TRZ (observed in the crystal structure and predicted by DFT) hinders an effective conjugation and suppresses the intensity of the low-energy CT transition. On the other hand, the bent conformation of DMAC-py-TRZ (observed in the crystal structure and predicted by DFT) promotes an efficient conjugation of the two moieties, perfectly in line with the oscillator strength calculated with TD-DFT (oscillator strengths are reported in Fig. S12 (ESI‡), together with NTOs).
Neither the absorption spectra of DMAC-TRZ and DMAC-py-TRZ (Table S27, ESI‡) show solvatochromism, in line with a very small permanent dipole moment of the molecule in the ground state. Photoluminescence (PL) spectra or DMAC-py-TRZ in Fig. 5c (Table S27, ESI‡) show a strong positive solvatochromism akin to that observed for DMAC-TRZ (Fig. S13 and ref. 20, ESI‡) The large positive PL solvatochromism observed for both compounds suggests that the emissive excited state has a large permanent dipole moment, thus confirming the CT character of this state in both compounds.27,28 The progressive broadening of the emission band in the solvatochromism study is a result of polarity-induced inhomogeneous broadening.28,29 The well-resolved vibronic structure of the emission band in non-polar solvents is often considered an indication of a local nature of the relevant excitation. Therefore, to prove the CT nature of the lowest transition in DMAC-py-TRZ in all solvent, including non-polar ones, Fig. S14 (ESI‡) shows spectra collected for the two molecular fragments, DMAC and py-TRZ. For py-TRZ, only absorption spectra are shown since the species is not emissive. The spectroscopic features of both molecular fragments are located at higher energies than the lowest energy feature seen in DMAC-py-TRZ, confirming that this specific feature is related to a CT state. An important and unusual result is recognized in the large Stokes shift observed for DMAC-py-TRZ in non-polar solvents (Table S27, ESI‡): in methylcyclohexane, the absorption band is located at 370 nm, while the emission is seen at 472 nm, amounting to a Stokes shift of ∼0.7 eV. This large Stokes shift can only be explained in terms of a very large molecular relaxation upon photoexcitation, well in line with the TD-DFT results that predict the relaxation of the S1 state from the bent to the orthogonal geometry.
In degassed toluene, DMAC-TRZ and DMAC-py-TRZ have similar ΦPL of 67% and 58%, respectively, in line with emission originating in both compounds from a similar orthogonal geometry. The ΦPL decrease in air (ΦPL = 22% and 17%, respectively), indicating the presence of accessible triplet excited states. The prompt and delayed lifetimes, τp and τd, for DMAC-TRZ in degassed toluene are of 20.8 ns (1.1%) and 5.2 μs (98.9%), in line with those previously reported,7 while the τp and τd for DMAC-py-TRZ are 44.0 ns (8.1%) and 1.5 μs (91.9%), respectively (Fig. S15, ESI‡).
To summarize, theoretical and spectroscopic studies agree with the picture where DMAC-TRZ maintains the same orthogonal conformation in both the ground and the S1 states. However, DMAC-py-TRZ undergoes a significant geometric reorganization from the bent geometry in the ground state to the orthogonal geometry in the excited state.
Spectra collected in a glassy 2-MeTHF matrix at 77 K (Fig. 5d) shed further light on the geometrical relaxation of DMAC-py-TRZ upon photoexcitation. In the frozen matrix, the emission peaks at 404 nm, blue-shifted compared to that in liquid 2-MeTHF at ambient conditions (λPL = 596 nm, Table 1 and Fig. S16, ESI‡). Apparently, in the frozen matrix the excited compound cannot relax, so that emission occurs from the bent structure and hence peaks at much higher energy than in the (non-polar) liquid solvent. The gated signal collected in the glassy matrix (red line in Fig. 5d) is ascribed to phosphorescence, suggesting a large ΔEST for this conformer under these conditions, again in line with that calculated for the bent structure.
| Material | Environment | λ PL /nm | Φ PL N2 (air)b/% | τ p, τde/ns; μs | S1f/eV | T1g/eV | ΔESTh/eV |
|---|---|---|---|---|---|---|---|
| a Measured at room temperature. b λ exc = 340 nm. c Obtained via the optically dilute method32 (see ESI), quinine sulfate (0.5 M) in H2SO4 (aq) was used as the reference, ΦPL: 54.6%, λexc = 360 nm.33 d Obtained using an integrating sphere. e τ p (prompt lifetime) and τd (delayed lifetime) were obtained from the transient PL decay of degassed solution/doped film, λexc = 378 nm. f S1 was obtained from the onset of the prompt fluorescence measured at 77 K, obtained in the 1–100 ns time range. g T1 was obtained from the onset of the phosphorescence spectrum measured at 77 K, obtained in the 1–8.5 ms time range. h ΔEST = S1 − T1. | |||||||
| DMAC-TRZ | Toluene (10−5 M)31 | 499 | 67 (22)c | 20.8; 5.2 | 2.88 | 2.57 | 0.31 |
| mCP 10 wt% | 499 | 47 (45) | 22.9; 15.3 | — | — | — | |
| PMMA 10 wt% | 523 | 18 (15) | 41.6; 17.0 | — | — | — | |
| DMAC-py-TRZ | Toluene (10−5 M) | 539 | 58 (17)c | 44.0; 1.5 | 3.21 | 2.77 | 0.44 |
| mCP 10 wt% | 496 | 57 (54)d | 24.9; 5.3 | — | — | — | |
| PMMA 10 wt% | 516 | 64 (58)d | 26.0; 4.7 | — | — | — | |
The PL behavior of DMAC-py-TRZ was also characterized in polyTHF, a viscous solvent where conformational relaxation is hindered. Interestingly, two emission bands are observed in this viscous medium (Fig. S17, ESI‡). The first emission band at 410 nm is similar to the one observed in the glassy matrix at 77 K while the second emission at 600 nm is similar to the emission observed in DMSO at room temperature. At ambient temperature, the excited state relaxation, fully hindered in glassy matrices at low temperature, is only partially hindered in the viscous polyTHF. Accordingly, the presence of the two emission bands is evidence of the simultaneous presence of the unrelaxed (bent) emissive species (as in the glassy matrix) as well as of relaxed (orthogonal) species (as in the liquid solvent).
Having observed that the S1 relaxation of DMAC-py-TRZ in frozen glassy matrices at low temperature is fully hindered while it is only partially hindered in viscous solvents at ambient condition, we next transitioned to an investigation of the behavior of this compound in amorphous matrices where large geometric reorganization is also likely to be hindered (Fig. 6). Spin-coated thin films of DMAC-py-TRZ doped into PMMA at 10 wt% were first prepared (Fig. 6a). Emission at λPL of 516 nm was observed with a ΦPL of 63.8% under a N2 atmosphere, which decreased to 58.0% upon exposure to oxygen. Biexponential decay kinetics were observed in the time-resolved PL measurements, with τp of 26.0 ns and an average τd of 4.7 μs [τ1 = 1.0 μs (32.6%), τ2 = 7.4 μs (67.4%)], respectively. The presence of a delayed fluorescence suggests that at least some emitter molecules adopt an orthogonal conformation, as to allow for TADF. Compared to the data obtained in toluene (τp of 44.0 ns and τd of 1.5 μs), DMAC-py-TRZ possesses a shorter-lived prompt component and a slightly longer-lived delayed component. We then investigated the photophysics in mCP (1,3-bis(N-carbazolyl)benzene) as the host matrix, a suitable high triplet energy host for both compounds that would be relevant for OLEDs. Fig. 6d shows results at a 10 wt% doping concentration. The emission in mCP is blue-shifted at τPL of 496 nm, compared to that of the doped PMMA film. The ΦPL of the doped film in mCP is 57.4% under N2, which decreased to 53.5% in air. The τp = 24.9 ns and the average τd = 5.3 μs [τ1 = 1.4 μs (46.8%), τ2 = 7.7 μs (53.2%)]. In both PMMA and mCP matrices at 10% doping the delayed emission is thermally activated (Fig. 7). However, extracting detailed information from such highly doped matrices is dangerous because of spurious phenomena, including homo energy-transfer and inner filter effects (self-absorption). Specifically, the ΔEST could be largely underestimated in films at 10% dye concentration due to self-absorption that moves the apparent onset of the absorption peak to the red. However, extracting the ΔEST from rates is similarly dangerous in view of the large error bars associated with rates.30
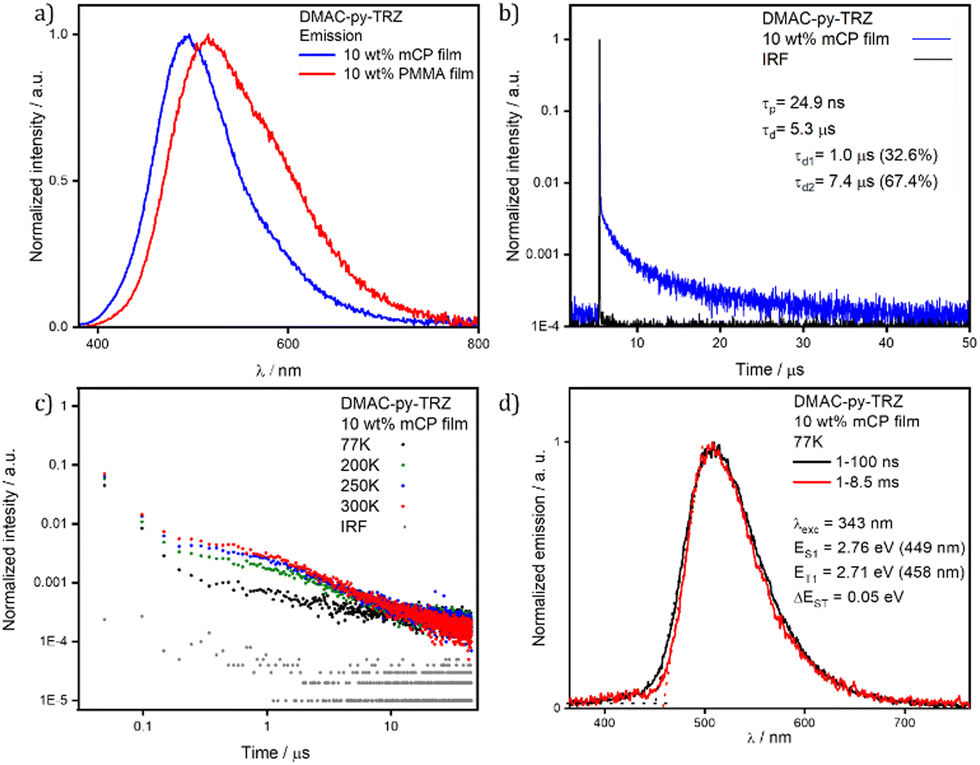 | ||
| Fig. 6 (a) PL spectra of DMAC-py-TRZ in spin-coated 10 wt% mCP film, spin-coated 10 wt% PMMA film (λexc = 340 nm); (b) time-resolved PL decay in spin-coated 10 wt% mCP film of DMAC-py-TRZ (λexc = 378 nm); (c) temperature-dependent delayed fluorescence decays in spin-coated 10 wt% mCP film of DMAC-py-TRZ (λexc = 378 nm); (d) prompt fluorescence and phosphorescence spectra at 77 K in drop-cast 10 wt% mCP film (λexc = 343 nm, prompt and delayed fluorescence spectra were obtained in the 1–100 ns and 1–8.5 ms time range, respectively). | ||
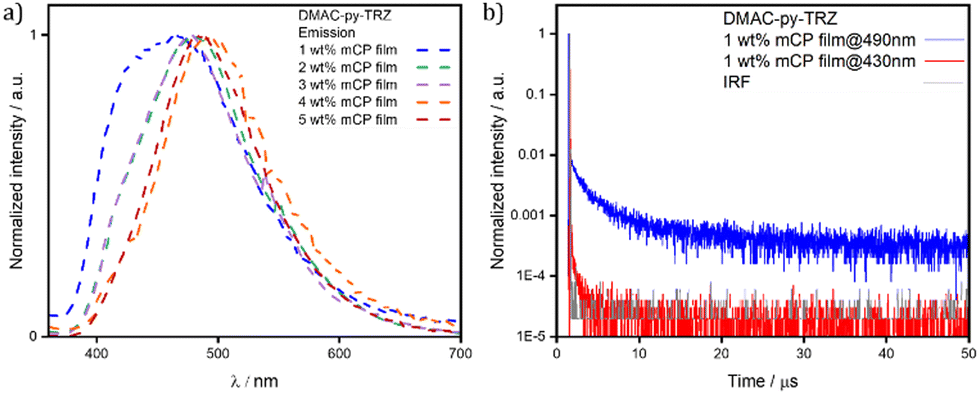 | ||
| Fig. 7 (a) Emission spectra of DMAC-py-TRZ in spin-coated 1–5 wt% mCP films (λexc = 340 nm); (b) time-resolved PL decay in spin-coated 1 wt% mCP film of DMAC-py-TRZ collected at λem = 490 nm and λem = 430 nm (λexc = 378 nm). | ||
To minimize spurious concentration effects, low-concentration (down to 1 wt%) films were fabricated, relevant spectra being shown in Fig. 7. In these films both the high frequency emission originating from the bent structure, and the low-frequency emission from the orthogonal structure, are present, suggesting that both conformers are present in all films. The high frequency emission decays much more rapidly (Fig. 7b), again confirming that it originates from the bent conformer. Upon increasing concentration, the high frequency emission progressively weakens and disappears for doping concentrations above 3 wt%.
Two phenomena may exist to explain this observation, both related to the large transition dipole moment (large oscillator strength) of the S0 → S1 transition in the bent geometry: (1) self-absorption; and (2) energy transfer from the bent to the orthogonal structure. Both phenomena are expected to become more efficient upon increasing the concentration of the emitter in the host matrix.
Conclusions
The synthesis of a new TADF emitter, DMAC-py-TRZ, is presented, together with an extensive computational analysis and experimental characterization. The chemical structure of DMAC-py-TRZ only marginally differs from that of the parent DMAC-TRZ compound. However, this minor change of the bridging moiety between the donor and acceptor has an enormous impact on the conformation and hence on the photophysics of the compound. Specifically, while DMAC-TRZ maintains the same orthogonal geometry in both the ground and the first excited singlet states, DMAC-py-TRZ assumes a bent geometry in the ground state, as highlighted by the large oscillator strength measured in solution for this dye. However, upon excitation to the S1 state, the system undergoes a large geometrical rearrangement to the orthogonal structure. This large relaxation is confirmed by the very large Stokes shift measured in non-polar solvents. In frozen 2-MeTHF glass at very dilute conditions, the relaxation is hindered and only a blue-shifted emission is seen from the unrelaxed bent conformer, without any hint of emission from the orthogonal structure. In mCP films a distribution of conformers exists and at low concentrations dual emission is observed, originating both from both the bent and orthogonal structures. However, upon increasing the doping concentration, the emission from the orthogonal conformer dominates. While self-absorption can be partly responsible for the phenomenon, we conclude that an efficient energy transfer from one conformer to the other also contributes to the spectral change. Indeed, TADF is not expected nor observed in the bent structure, due to a too large ΔEST. The good TADF efficiency of DMAC-py-TRZ in solution, similar as for DMAC-TRZ, is in line with the very fast molecular relaxation from the bent (TADF-silent) to the orthogonal geometry (TADF-active) in solution. The situation is more delicate in matrices where the host rigidity hinders a large molecular rearrangement. The observed good efficiency of TADF in matrices then suggests efficient energy transfer of excitons created on the bent (and TADF silent) structures towards molecules in the orthogonal (and TADF-active) structure as to retrieve all photogenerated singlet states for TADF activity. Most probably, efficient triplet-to-triplet energy transfer is also required to explain the good efficiency of DMAC-py-TRZ OLEDs, but this will be subject of a subsequent study.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 5 April 2023, in which Table 1 included values of S1 and T1 (as well as their difference, ΔEST) for the environments mCP 10 wt% and PMMA 10 wt%, for each of the materials DMAC-TRZ and DMAC-py-TRZ. These values have been removed in the current version because the authors no longer believe they can accurately measure them due to self-absorption, as is explained in the article. For completeness, the removed values of S1 and T1 have instead been included in the ESI,‡ where they can be found in Table S28.Acknowledgements
We thank EU Horizon 2020 Grant Agreement No. 812872 (TADFlife) for funding. The St Andrews team acknowledges support from the Engineering and Physical Sciences Research Council of the UK (grant EP/P010482/1). The authors from the University of Parma acknowledge the support from the HPC (High Performance Computing) facility of the University of Parma, Italy. Moreover, authors from University of Parma benefited from the equipment and support of the COMP-HUB Initiative, funded by the “Departments of Excellence” program of the Italian Ministry for Education, University and Research (MIUR, 2018-2022).Notes and references
- C. Adachi, Jpn. J. Appl. Phys., 2014, 53, 060101 CrossRef.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- F. M. Xie, J. X. Zhou, Y. Q. Li and J. X. Tang, J. Mater. Chem. C, 2020, 8, 9476–9494 RSC.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- B. Milián-Medina and J. Gierschner, Org. Electron., 2012, 13, 985–991 CrossRef.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- W. L. Tsai, M. H. Huang, W. K. Lee, Y. J. Hsu, K. C. Pan, Y. H. Huang, H. C. Ting, M. Sarma, Y. Y. Ho, H. C. Hu, C. C. Chen, M. T. Lee, K. T. Wong and C. C. Wu, Chem. Commun., 2015, 51, 13662–13665 RSC.
- K. Stavrou, L. G. Franca and A. P. Monkman, ACS Appl. Electron. Mater., 2020, 2, 2868–2881 CrossRef CAS PubMed.
- W. Li, X. Cai, B. Li, L. Gan, Y. He, K. Liu, D. Chen, Y. C. Wu and S. J. Su, Angew. Chem., Int. Ed., 2019, 58, 582–586 CrossRef CAS PubMed.
- Y. Wada, S. Kubo and H. Kaji, Adv. Mater., 2018, 30, 1705641 CrossRef PubMed.
- Y. Wada, H. Nakagawa and H. Kaji, Chem. – Asian J., 2021, 16, 1073–1076 CrossRef CAS PubMed.
- P. Rajamalli, D. Chen, W. Li, I. D. W. Samuel, D. B. Cordes, A. M. Z. Slawin and E. Zysman-Colman, J. Mater. Chem. C, 2019, 7, 6664–6671 RSC.
- P. L. Dos Santos, D. Chen, P. Rajamalli, T. Matulaitis, D. B. Cordes, A. M. Z. Slawin, D. Jacquemin, E. Zysman-Colman and I. D. W. Samuel, ACS Appl. Mater. Interfaces, 2019, 11, 45171–45179 CrossRef CAS PubMed.
- J. X. Chen, Y. F. Xiao, K. Wang, X. C. Fan, C. Cao, W. C. Chen, X. Zhang, Y. Z. Shi, J. Yu, F. X. Geng, X. H. Zhang and C. S. Lee, J. Mater. Chem. C, 2020, 8, 13263–13269 RSC.
- Y. Shi, K. Wang, S. Zhang, X. Fan, Y. Tsuchiya, Y. Lee, G. Dai, J. Chen, C. Zheng, S. Xiong, X. Ou, J. Yu, J. Jie, C. Lee, C. Adachi and X. Zhang, Angew. Chem., 2021, 133, 26082–26087 CrossRef.
- X. Fan, K. Wang, Y. Shi, D. Sun, J. Chen, F. Huang, H. Wang, J. Yu, C. Lee and X. Zhang, SmartMat, 2023, 4, e1122 CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1988, 94, 6081–6090 CrossRef.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- R. Dhali, D. K. A. Phan Huu, F. Bertocchi, C. Sissa, F. Terenziani and A. Painelli, Phys. Chem. Chem. Phys., 2021, 23, 378–387 RSC.
- D. K. A. Phan Huu, S. Saseendran and A. Painelli, J. Mater. Chem. C, 2022, 10, 4620–4628 RSC.
- D. K. A. Phan Huu, S. Saseendran, R. Dhali, L. G. Franca, K. Stavrou, A. Monkman and A. Painelli, J. Am. Chem. Soc., 2022, 144, 15211–15222 CrossRef CAS PubMed.
- W. E. Connelly and N. G. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef PubMed.
- R. Dhali, D. K. A. Phan Huu, F. Terenziani, C. Sissa and A. Painelli, J. Chem. Phys., 2021, 154, 134112–134119 CrossRef CAS PubMed.
- D. K. A. Phan Huu, R. Dhali, C. Pieroni, F. Di Maiolo, C. Sissa, F. Terenziani and A. Painelli, Phys. Rev. Lett., 2020, 124, 107401 CrossRef CAS PubMed.
- A. Painelli and F. Terenziani, Chem. Phys. Lett., 1999, 312, 211–220 CrossRef CAS.
- B. Boldrini, E. Cavalli, A. Painelli and F. Terenziani, J. Phys. Chem. A, 2002, 106, 6286–6294 CrossRef CAS.
- F. Terenziani, A. Painelli, A. Girlando and R. M. Metzger, J. Phys. Chem. B, 2004, 108, 10743–10750 CrossRef CAS.
- M. Zheng, Y. Li, Y. Wei, L. Chen, X. Zhou and S. Liu, J. Phys. Chem. Lett., 2022, 13, 2507–2515 CrossRef CAS PubMed.
- Z. Zhang, E. Crovini, P. L. dos Santos, B. A. Naqvi, D. B. Cordes, A. M. Z. Slawin, P. Sahay, W. Brütting, I. D. W. Samuel, S. Bräse and E. Zysman-Colman, Adv. Opt. Mater., 2020, 8, 2001354 CrossRef CAS.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- F. Song, Z. Xu, Q. Zhang, Z. Zhao, H. Zhang, W. Zhao, Z. Qiu, C. Qi, H. Zhang, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, Z. Zhao, A. Qin, D. Ma and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1800051 CrossRef.
Footnotes |
| † The research data supporting this publication can be accessed at https://doi.org/10.17630/2152973b-5584-40ec-ac34-3f30c0d7893d |
| ‡ Electronic supplementary information (ESI) available. CCDC 2224273 and 2224274. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc05213j |
| § Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |