
A supported polymeric organic framework composed of dual electrocatalytically active sites for high-performance carbon dioxide electroreduction†
Shengsheng
Huang
a,
Qizhe
He
a,
Hongwei
Li
a,
Jinjie
Qian
 *b,
Wei
Xu
a and
Ting-Ting
Li
*a
*b,
Wei
Xu
a and
Ting-Ting
Li
*a
aSchool of Materials Science and Chemical Engineering, Ningbo University, Ningbo, 315211, China. E-mail: litingting@nbu.edu.cn
bCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, 325000, China. E-mail: jinjieqian@wzu.edu.cn
First published on 30th May 2023
Abstract
The electrocatalytic reduction of CO2 to CO and other sustainable fuels presents an attractive approach for alleviating the energy crisis and environmental problems. Carbon nanotube (CNT) hybridization and the incorporation of dual catalytic sites into a polymeric organic framework (POF) are promising strategies for boosting the electrocatalytic performance of POF-tailored electrocatalysts. Herein, we demonstrate a combined nanoscale and molecular-level strategy to construct an organic–inorganic hybrid material by the template-directed in situ polymerization of ultrathin POF layers with isolated Co(II) porphyrin (Por-) and Co(II) bipyridine sites on the CNT scaffold. As an electrocatalyst for CO2-to-CO conversion in an aqueous solution, the resultant POF@CNT shows higher electrocatalytic activity and selectivity compared to other comparative materials. Specifically, it exhibits the highest faradaic efficiency for CO production (FECO) of 97.5% at the overpotential of 600 mV, a superior turnover frequency (TOF) of 36.6 s−1 at −1.0 V vs. the reversible hydrogen electrode (RHE), and remarkable long-term stability. These metrics are superior to or at least comparable to most of its organic or inorganic competitors. The remarkable catalytic performance of POF@CNT can be attributed to the combined effects of CNT hybridization and the dual active sites in the POF structure, which facilitates interfacial electron transfer and enables a high degree of catalytically active site exposure.
1. Introduction
The undue use of fossil fuels leads to an increase in anthropogenic CO2 emission and results in serious climate change and other environmental problems, such as ocean acidification and land desertification. The above issues have inspired extensive research on sustainable technologies that enable efficient conversion of CO2 into some high-value-added carbon-based fuels and chemicals. Among them, the electrocatalytic CO2 reduction reaction (CO2RR), particularly when the electric energy is generated from renewable sources such as solar power and wind energy in a mode parallel to natural photosynthesis, is an attractive strategy to capture and utilize CO2.1–14 Nevertheless, owing to the high thermodynamic robustness of CO2 and the kinetically favorable hydrogen evolution reaction (HER), electrocatalytic CO2RR has poor activity and product selectivity, and the primary hurdle for CO2RR remains the shortage of high-performance electrocatalysts.15–19 Tremendous efforts have been made over the past several decades to develop different types of transition metal-based electrocatalysts such as transition-metal complexes,20,21 inorganic nanomaterials,22–24 and organic–inorganic hybrid nanomaterials.25–27 Among them, transition-metal complexes such as Por-, phthalocyanine, and other N4− macrocyclic compounds have drawn more attention due to the good establishment of mechanisms for transition-metal complexes, which offers well-defined blueprints for an efficient CO2RR.28–33 Additionally, the key catalytic performance such as activity and selectivity can be rationally modulated by changing metals and organic ligands via direct synthesis or post-synthetic modification. However, the poor durability and the difficulties in synthesis and recycling associated with these transition-metal complexe-based electrocatalysts limit their practical application. In this context, the heterogenization of transition-metal complexes by forming cross-linked frameworks has been proven as an effective strategy to solve the above issues.34–36 POFs including covalent organic frameworks (COFs) are classes of crystalline porous materials fabricated by covalent bonding with organic monomers in a periodic arrangement.37,38 Due to the large accessible surface area, programmable topological designability, and high structural and chemical stability, the rational design of COFs brings much more possibilities for developing different kinds of electrocatalysts with enhanced performance.34,39–42 Furthermore, the permanent porosity of COFs can also offer an increasing local CO2 concentration near the active centers and rich transport channels for carriers. In the past few years, several investigations have revealed the strengths of COF-based CO2RR electrocatalysts.29,34 Particularly, the high activity and selectivity of some Por-based COFs have been confirmed in aqueous electrolytes since the pioneering work from Yaghi and coworkers.5,43 Despite this encouraging progress, pristine COFs have relatively lower electrocatalytic activity when compared with traditional metallic electrocatalysts, and the fundamental reasons are as follows: (i) pristine COFs generally have poor electronic conductivity, which results in undesired electron transmission efficiency between COF layers; (ii) the high polarization of heteroatom-containing linkages including imine, imide, and hydrazone in Por-based COFs is inefficient in supporting π-electron delocalization between the organic monomers, thus leading to a low intramolecular electron migration rate.44,45To address the above challenges, rationally integrating Por-based COFs with a conductive matrix to construct COF-based hybrid electrocatalysts with an effective electron transfer ability represents a universal strategy to boost the electrocatalytic performance of COF-based electrocatalysts.29,39,46–48 Beyond that, the incorporation of metalloporphyrin sites with other metal nodes in COFs can afford dual active sites to enhance the electrocatalytic activity or induce the synergistic electrocatalysis functionality. Recently, some newly designed COFs with metallopyridine units have been reported as a new kind of electro-/photocatalytic platform for CO2RR.35,49–51 In this direction, COFs with dual distinct active sites can be prepared by connecting metalloporphyrin sites and metallopyridine units, which would be favorable for formulating design principles for the development of electroactive COFs. However, this strategy has not been systematically explored.
Based on the above consideration, herein, we demonstrate a combined nanoscale and molecular-level strategy for enhancing the intrinsic activity of Por-based POFs by constructing a POF@CNT hybrid electrocatalyst, which was prepared through a simple one-pot reaction of pyrrole and 2,2′-bipyridine-5,5′-diformaldehyde (Bpy) on the surface of CNTs. Post-synthetic modification with CoCl2 leads to the coordination of Co(II) ions into Por- and Bpy building blocks, forming a POF with dual electrocatalytic active sites. The in situ polymerization of pyrrole and Bpy provides an ultrathin POF nanolayer around CNTs, and the ultrathin thickness of the POF nanolayer not only greatly reduces the electron transfer distance from CNTs to active sites and maximizes the catalytically active surface area, but also enhances the mechanical and catalytic stability. Accordingly, the resultant hybrid electrocatalyst can selectively catalyze CO2RR with the highest CO2-to-CO conversion FECO of 97.5% at the overpotential of 600 mV, the highest TOF of 36.6 s−1 at −1.0 V vs. RHE, and excellent stability in a CO2-saturated 0.5 M KHCO3 solution.
2. Experimental section
2.1. Chemicals and materials
All the chemicals and solvents were commercially available and used directly without further purification. Pyrrole, benzene-1,4-dialdehyde (Bda), biphenyl-4,4′-dicarboxaldehyde (Bpda), nitrobenzene (NBZ), and Bpy were purchased from Macklin Co., Ltd.2.2. Preparation of CoPOF@CNT
CoPOF@CNT composites were prepared according to the reported methods.52–54 For the preparation of CoPOF-Bpy@CNT, CNTs (50 mg) were added into a round-bottom flask with 100 mL of acetic acid (AcOH) and sonicated for 30 min. And then, 100 μL of trifluoroacetic acid (TFA), 18.2 μL of pyrrole, 0.9 mL of NBZ, and Bpy (27.6 mg) were added into the above suspension and sonicated for another 30 min. The resulting suspension was stirred at 80 °C for 24 h. After the reaction cooled down to room temperature, the obtained black powder was filtered and washed three times with water and ethanol, respectively. Finally, it was dried under vacuum at 60 °C overnight. To introduce Co(II) ions inside the POF structure, 50 mg of the obtained black powder was dispersed in 15 mL of N,N-dimethylformamide (DMF) and heated at 100 °C. Then, 129.8 mg of CoCl2 was added into the suspension and heated at 160 °C for 6 h under stirring conditions. After cooling to room temperature, the product was filtered and washed with water and methanol, respectively. At last, the product was dried under vacuum at 60 °C overnight.The pure CoPOF-Bpy sample was prepared through the same method without the addition of CNTs.
The synthesis of CoPOF-Bda@CNT and CoPOF-Bpda@CNT followed the synthetic procedure of CoPOF-Bpy@CNT with Bda (17.4 mg) and Bpda (27.3 mg) as the starting materials, respectively.
2.3. Instrumentation and characterization
X-ray diffraction (XRD) patterns of the products were recorded using an X-ray diffractometer (Bruker D8 Advance) with a Cu Kα radiation source at 1600 W (40 kV voltage, 40 mA) power. Fourier transform infrared (FT-IR) spectroscopy was performed using an FT-IR spectrometer (Thermo Scientific Nicolet iS20) in a frequency range of 4000–450 cm−1. The Co content of CoPOF@CNT was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES, Thermo Fisher iCAP PRO). Scanning electron microscopy (SEM) was carried out using a Hitachi S-4800 II field emission scanning electron microscope with an accelerating voltage of 20 kV. Transmission electron microscopy (TEM) and high-resolution TEM (HR-TEM) were performed using an FEI Tecnai G2 F20 electron microscope equipped with an elemental mapping and energy dispersive spectroscopy (EDS) detector and operated at an accelerating voltage of 200 kV. CO2 adsorption isotherms were recorded at 298 K using an adsorption apparatus (ASAP 2020). X-ray photoelectron spectroscopy (XPS) measurement was recorded on a Thermo Scientific K-Alpha XPS system using C 1s (284.8 eV) as the reference binding energy. To analyze the liquid products, 1H nuclear magnetic resonance (NMR) spectra were obtained using a Bruker Ascend spectrometer operating at 500 MHz. The generated gaseous products were successively analyzed using a gas chromatography (GC) instrument equipped with a thermal conductivity detector (TCD) and a hydrogen flame ionization detector (FID).2.4. Electrochemical measurements
All the electrochemical measurements were carried out using an electrochemical workstation (CHI 660E) with a three-electrode configuration. A gas-tight two-compartment electrolytic cell with a piece of Nafion membrane as the separator was used. 2 mg of CoPOF@CNT was added into 2 mL of isopropanol with 10 μL of 5 wt% Nafion solution. Then, the suspension was sonicated for 30 min to obtain a homogeneous catalyst ink. The catalyst ink was dropped onto a glassy carbon electrode (GCE) and dried at room temperature to obtain the working electrode with a catalyst loading of 50 μg cm−2. An Ag/AgCl electrode and a Pt wire were utilized as the reference and the counter electrode, respectively. Unless otherwise noted, 0.5 M KHCO3 solution saturated with CO2 was used as the electrolyte. The reported potentials in this work were converted into the RHE scale based on the Nernst equation. Linear sweep voltammetric (LSV) measurement was conducted in a potential range from 0 to −1.1 V vs. RHE with a scan rate of 5 mV s−1, and all the LSV plots were displayed without iR correction. Electrochemical surface area (ECSA) was calculated from the electrical double layer capacitance (Cdl) data, and the cyclic voltammetric (CV) measurements were conducted by measuring Cdl with various scan rates in a non-faradaic potential range from −0.1 to −0.2 V vs. RHE with different scan rates. Electrochemical impedance spectroscopy (EIS) measurement was conducted in a frequency range from 100 kHz to 0.1 kHz with an AC amplitude of 5 mV.2.5. Calculation of faradaic efficiency and TOF
Faradaic efficiency was calculated as follows: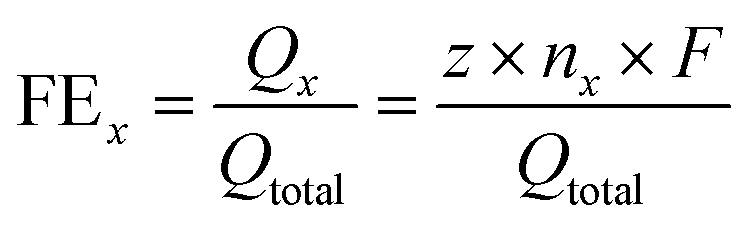 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1); x: CO or H2.
485 C mol−1); x: CO or H2.
The TOF for CO production was calculated as follows:
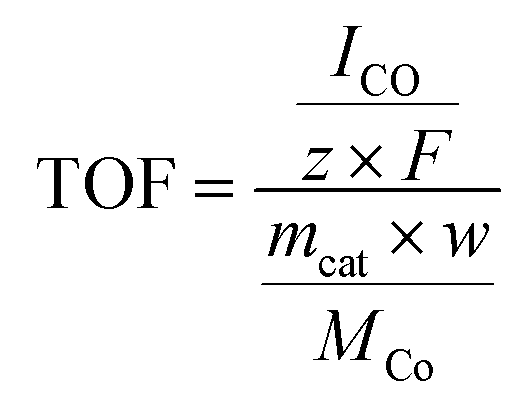 | (2) |
3. Results and discussion
3.1. Characterization of CoPOF-Bpy@CNT
Due to the high conductivity, to determine the electron transfer pathways and to strengthen ion transportation, a CNT was used as the scaffold. The synthesis process of CoPOF-Bpy@CNT is schematically illustrated in Scheme 1. Specifically, through an acid-catalyzed dehydration condensation reaction, POF-Bpy was constructed and in situ coaxially coated onto the CNT scaffold to obtain POF-Bpy@CNT, and then POF-Bpy@CNT was coordinated with Co(II) ions in DMF solution to afford CoPOF-Bpy@CNT. During the coordination reaction, both the Por- and Bpy moieties prefer to coordinate with Co(II) ions, giving rise to CoPOF-Bpy@CNT composed of both Co(II) Por- and Co(II)N2Cl2 sites. It has been reported that Por- and Bpy sites are the well-defined platform for confining metal sites, which would enable us to investigate the dual active catalytic sites for CO2RR.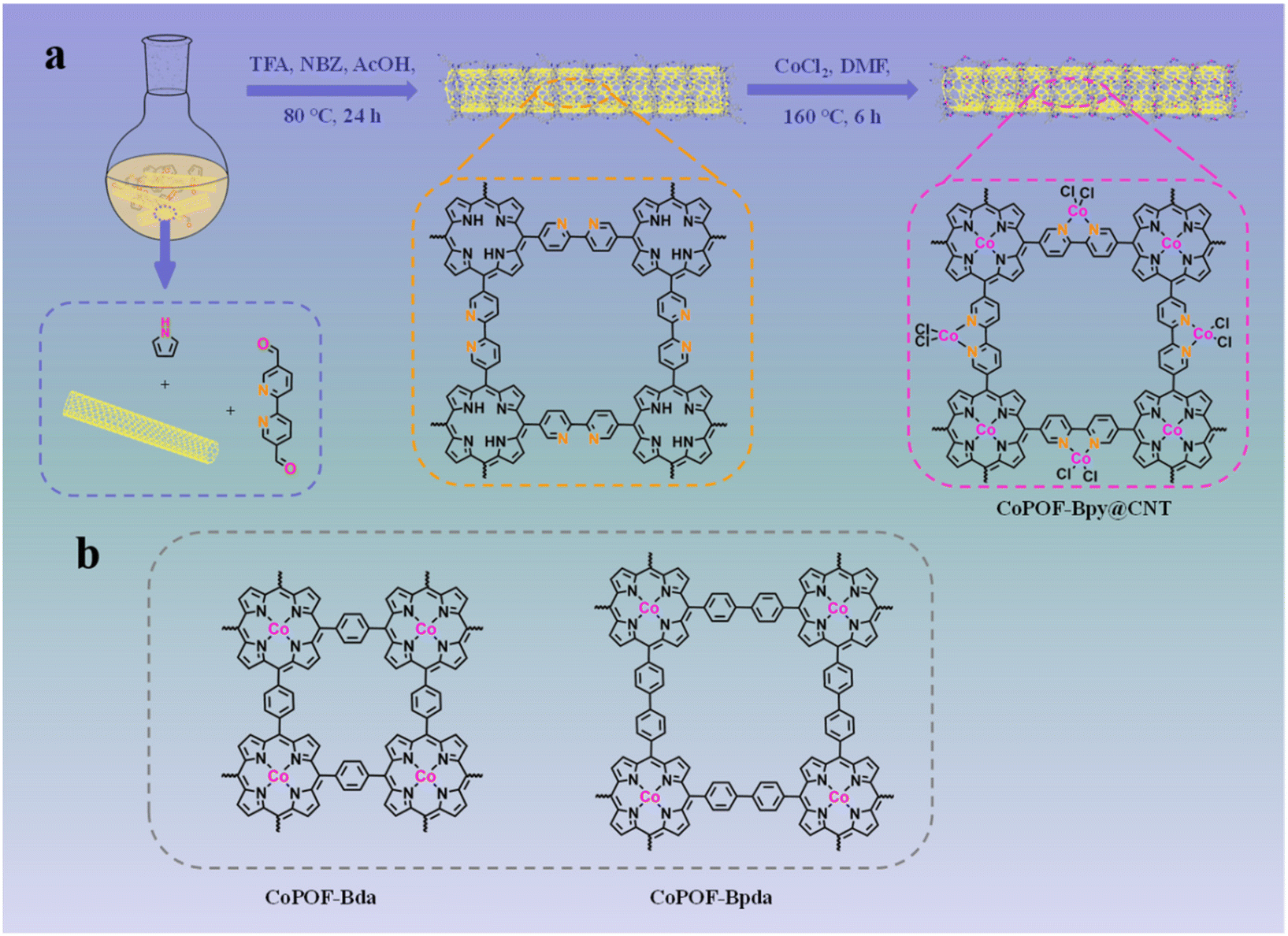 | ||
| Scheme 1 (a) Schematic illustration of the synthetic process of CoPOF-Bpy@CNT; (b) molecular structures of CoPOF-Bda and CoPOF-Bpda. | ||
The structural morphologies of CoPOF-Bpy@CNT and CoPOF-Bpy were investigated by SEM and TEM. In the SEM image of CoPOF-Bpy@CNT (Fig. S1a†), the hybrid sample retains the tubular morphology, in which the surfaces of these CNTs become roughened and the diameters are larger than those of the pure CNTs, suggesting the uniform dispersion of the CoPOF-Bpy film on the CNT scaffold. There was no sign of pure CoPOF-Bpy generation. In contrast, pure CoPOF-Bpy has a spherical morphology with a diameter of ca. 500 nm and aggregated together in the absence of the CNT scaffold (Fig. S1b†); the dense morphology with few exposed active sites of pure CoPOF-Bpy signifies it is not suitable as the CO2RR electrocatalyst alone. Due to the strong intermolecular π–π interactions between CNTs and CoPOF-Bpy, CoPOF-Bpy tends to uniformly coat on the CNT scaffold, which is verified by TEM. Compared with pure CNTs, the morphology of CoPOF-Bpy@CNT does not undergo great change after hybridization (Fig. 1a). The HR-TEM figure of CoPOF-Bpy@CNT shown in Fig. 1b and its inset display that the crystalline walls of the CNTs are coaxially wrapped by smooth CoPOF-Bpy layers with an average thickness of 2.6 nm, the thin conformal polymer coating layer of CoPOF-Bpy around the CNT scaffold guarantees the high conductivity of this hybrid material. Moreover, the EDS elemental mapping of CoPOF-Bpy@CNT shown in Fig. 1c discloses the corroborated cylindrical element distribution of C, N, and Co without aggregation, indicative of an even morphology of CoPOF-Bpy on the surface of CNTs. The aberration-corrected high-angle annular dark-field scanning TEM (AC HAADF-STEM) image shown in Fig. S2† exhibits that the supported CoPOF-Bpy layer has various individual bright spots, which are responsible for the atomically dispersed Co metal. The amount of Co in CoPOF-Bpy@CNT was determined to be 3.7 wt% based on the ICP-OES result (Table S1†), which is almost twice as high as that in CoPOF-Bpda@CNT (1.8 wt%), further indicating the generation of Co(II) Bpy sites in CoPOF-Bpy@CNT.
 | ||
| Fig. 1 (a) TEM image of CoPOF-Bpy@CNT; (b) HR-TEM image of CoPOF-Bpy@CNT, and the inset is the enlarged view; (c) the corresponding EDX mapping of CoPOF-Bpy@CNT; (d) FT-IR spectra and (e) XRD patterns of CoPOF-Bpy@CNT and CoPOF-Bpy. | ||
FT-IR spectroscopy was used to make a comprehensive diagnosis of the molecular structures. In the FT-IR spectra of CoPOF-Bpy@CNT and CoPOF-Bpy (Fig. 1d), no band from 1740 to 1700 cm−1 was observed, indicative of the thorough conversion of carbonyl groups in Bpy. The band at 1600 cm−1 can be attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, and the peaks at 1250 and 1100 cm−1 are related to the C–N and C–H stretching, respectively.52–54 Additionally, the absorption bands between 800 and 700 cm−1 are assigned to Por-deformation modes, implying that pyrrole is successfully combined with aromatic aldehydes to generate Por-structures.52 Furthermore, the structure of CoPOF-Bpy@CNT was studied using the XRD technique. As displayed in Fig. 1e, CNTs exhibit an intensive diffraction peak at 26° and a weak peak at 43°, which correspond to the (002) and (100) lattice planes, respectively.31,36 Except for the characteristic peaks originating from pure CNTs, the XRD pattern of CoPOF-Bpy@CNT shows another broad diffraction peak at 22°, suggesting the intrinsically ordered framework structure of CoPOF-Bpy.52,53
N bond, and the peaks at 1250 and 1100 cm−1 are related to the C–N and C–H stretching, respectively.52–54 Additionally, the absorption bands between 800 and 700 cm−1 are assigned to Por-deformation modes, implying that pyrrole is successfully combined with aromatic aldehydes to generate Por-structures.52 Furthermore, the structure of CoPOF-Bpy@CNT was studied using the XRD technique. As displayed in Fig. 1e, CNTs exhibit an intensive diffraction peak at 26° and a weak peak at 43°, which correspond to the (002) and (100) lattice planes, respectively.31,36 Except for the characteristic peaks originating from pure CNTs, the XRD pattern of CoPOF-Bpy@CNT shows another broad diffraction peak at 22°, suggesting the intrinsically ordered framework structure of CoPOF-Bpy.52,53
XPS measurements were conducted to reveal the coordination environment and the valence states of the cobalt sites in CoPOF-Bpy@CNT and CoPOF-Bpy. The XPS survey spectrum of CoPOF-Bpy@CNT shows that the hybrid material contains C, N, Co, and Cl elements, which is in accordance with the theoretical elemental composition of CoPOF-Bpy@CNT, and the additional O element can be assigned to the absorbed H2O molecules (Fig. S3†). The HR-XPS spectrum of Co 2p in CoPOF-Bpy@CNT is shown in Fig. 2a, the chemical shift signals at 780.9 and 796.8 eV are assigned to the Co 2p3/2 and Co 2p1/2 spin–orbit peaks, respectively, suggesting the Co2+ oxidation state in this hybrid material.35,55,56 It should be pointed out that the Co 2p peaks in CoPOF-Bpy@CNT shift to higher binding energies compared to those in CoPOF-Bpy, revealing that the electronic structure of Co(II) centers has been modulated due to the interfacial electron transfer from CNTs to Co(II) centers via strong π–π interactions between the CoPOF-Bpy layer and CNTs. Also, the N 1s XPS spectrum of CoPOF-Bpy@CNT is deconvoluted into two peaks, the peaks with binding energies of 399.8 and 398.7 eV belong to pyrrolic N (originating from the pyrrole moiety) and pyridinic N (originating from the Bpy moiety), respectively (Fig. 2b).35,57–59 Additionally, according to the XPS result, the molar ratio of nitrogen to cobalt in CoPOF-Bpy@CNT is about 3:1, matching well with the theoretical value. Based on the above characterization studies, it can be concluded that the pre-designed framework structure of CoPOF-Bpy@CNT with both Co(II) Por- and Co(II) Bpy sites has been precisely constructed. The Raman spectra of CNTs and CoPOF-Bpy@CNT were recorded for comparison. The ID/IG intensity ratios in CNTs (1.19) and CoPOF-Bpy@CNT (1.21) are almost the same, suggesting that CoPOF-Bpy@CNT can well retain the high conductivity of the CNT scaffold (Fig. S4†).
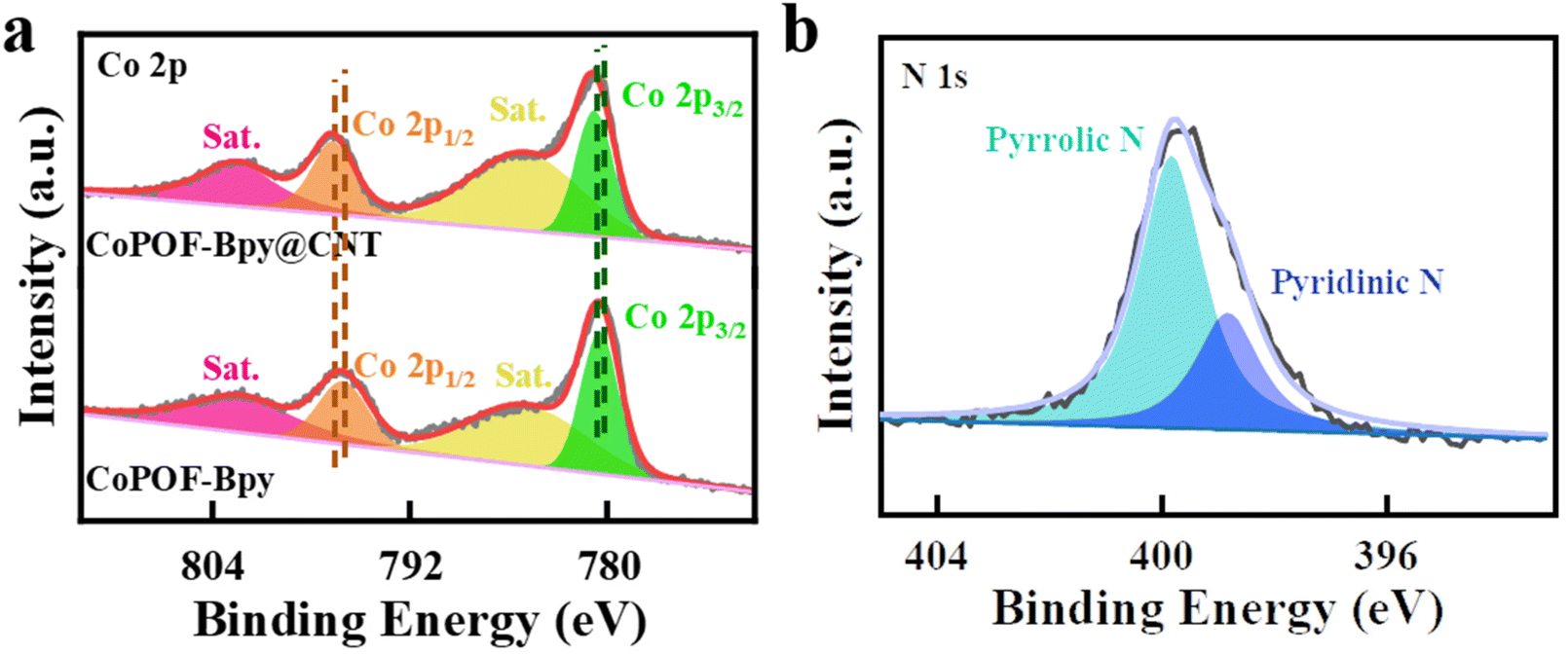 | ||
| Fig. 2 (a) Co 2p XPS spectra of CoPOF-Bpy@CNT and CoPOF-Bpy; (b) the N 1s XPS spectrum of CoPOF-Bpy@CNT. | ||
The CO2 uptake amounts of CNTs, CoPOF-Bpy@CNT, CoPOF-Bda@CNT, and CoPOF-Bpda@CNT were measured at 298 K. The CO2 uptake amount of pure CNTs is the lowest among the four samples, indicating that CO2 adsorption is mainly located on the CoPOF layers. With a larger linker between Por-moieties and the extended π-conjugate chain, the CO2 adsorption capacity of CoPOF-Bpda@CNT (52.8 cm3 g−1) is higher than that of CoPOF-Bda@CNT (26.8 cm3 g−1). As for CoPOF-Bpy@CNT, it shows the highest CO2 uptake amount (77.4 cm3 g−1), which might be attributed to the synergy between accessible Co(II) Por- and Co(II)N2Cl2 sites (Fig. S5†). The highest CO2 adsorption capacity of CoPOF-Bpy@CNT is beneficial for the electrocatalytic CO2RR.
3.2. Electrocatalytic performance of CoPOF-Bpy@CNT
Evidenced by the comprehensive characterization studies performed above, the in situ polymerization of pyrrole and Bpy provides a thin conformal polymeric layer around CNTs. CoPOF-Bpy@CNT reveals well-defined Co(II) Por- and Co(II)N2Cl2 sites to be under precise control, and it not only offers highly exposed active sites and abundant mass channels for electrocatalysis but also boosts the mechanical and chemical robustness attributed to the CNT scaffold and the covalently constructed CoPOF-Bpy. Considering these unique features, CoPOF-Bpy@CNT inspires subsequent application as a promising electrocatalyst for the CO2RR. To verify the conjecture, we assessed the electrocatalytic performance of CoPOF-Bpy@CNT. The powder of CoPOF-Bpy@CNT was coated on a GCE with the assistance of a Nafion binder to prepare the working electrode for testing the CO2RR activity in an H-type electrolytic cell with two compartments. LSV tests were performed in a 0.5 M KHCO3 aqueous solution saturated with CO2. To highlight the superiority of dual electrocatalytic active sites in CoPOF-Bpy@CNT, CoPOF-Bda@CNT and CoPOF-Bpda@CNT were selected as the comparative materials, and their electrocatalytic activities were assessed under the same conditions.Compared with CoPOF-Bpy@CNT, CoPOF-Bda@CNT and CoPOF-Bpda@CNT were synthesized according to a similar procedure and characterized by FT-IR spectroscopy, XRD, TEM, and XPS (Fig. S6–S14†). TEM images of CoPOF-Bda@CNT and CoPOF-Bpda@CNT indicate that there is no agglomeration, and further verify the successful wrapping of POF nanolayers on the CNTs (Fig. S10†). To quantify the CO2 reduction product, the evolved gaseous and liquid products were analyzed by GC and 1H NMR spectroscopy, respectively. For the sake of avoiding the measuring error, three parallel measurements were carried out. As shown in Fig. 3a, for CoPOF-Bpy@CNT, a negative sweep beyond −0.5 V vs. RHE triggers a significant increase in the reduction current density caused by the CO2RR. At −0.8 V vs. RHE, the cathodic current density reaches −20 mA cm−2. In comparison, the LSV plots of CoPOF-Bda@CNT and CoPOF-Bpda@CNT show smaller and cathodically shifted current densities. This result demonstrates that CoPOF-Bpy@CNT is more favorable for the CO2RR than other samples. Furthermore, we quantified the mass activities of these electrocatalysts by normalizing the current densities in Fig. 3a with respect to the Co contents. As shown in Fig. S15,† the mass activity of CoPOF-Bpy@CNT is larger than the other two beyond −0.85 V vs. RHE. Pure CNT shows negligible catalytic current density in the CO2-saturated electrolyte, excluding the possibility of catalytic activity originating from the CNT scaffold (Fig. S16†). Additionally, the control experiment shows metal-free POF-Bpy@CNT is non-CO2RR active (Fig. S17†), further confirming that the electrocatalytic activity of CoPOF-Bpy@CNT originates from Co centered active sites. We further optimized the CoPOF-Bpy loading on CNTs by preparing CoPOF-Bpy@CNT(X) with the addition of various amounts (X represents the mass of CNTs) of CNTs. The LSV plots manifest that the catalytic current density increases with the CNT percentage and starts to saturate when the mass of CNTs is 50 mg for synthesis, whereas a further increase in the mass of CNTs brings a negative effect on the electrocatalytic activity (Fig. S18†). We suspected that the incomplete coverage of CoPOF-Bpy on CNTs with a high mass of CNTs may provide limited active sites. However, the addition of a low mass of CNTs results in thicker layers of CoPOF-Bpy wrapped on CNTs and even the generation of CoPOF-Bpy aggregates, which is confirmed by SEM. The SEM image of CoPOF-Bpy@CNT(30) in Fig. S19† shows that the diameter of CNTs become thicker and some aggregates are observed, which is harmful to the conductivity of this hybrid material and leads to a decrease in the mass/charge transfer ability. Consequently, we focused on CoPOF-Bpy@CNT(50) (the Co content is 3.7 wt%) in the following studies. As a control, we loaded CoPOF-Bpy on CNTs by physical mixing to prepare CoPOF-Bpy/CNT with the same Co content, and the obtained CoPOF-Bpy/CNT exhibits an inferior catalytic current (Fig. S20†). It is noteworthy that the solution-phase polymerization strategy in our work distinguishes itself from the former strategy where molecular electrocatalysts are dip-coated or physically mixed with CNTs. Such a direct strategy may form less-conductive molecular aggregates, which limits the exposure of active sites.
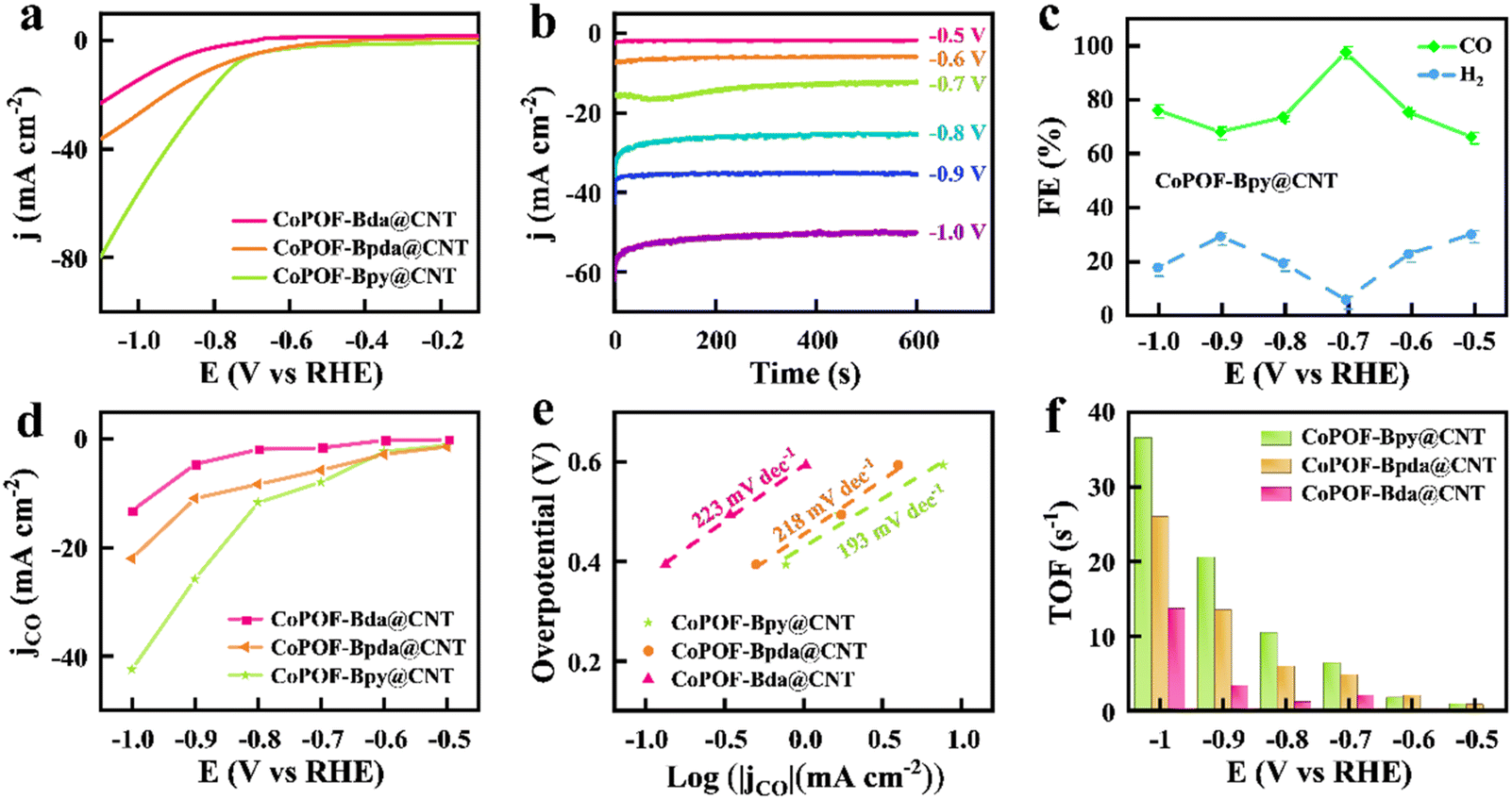 | ||
| Fig. 3 (a) LSV plots of CoPOF-Bpy@CNT, CoPOF-Bpda@CNT, and CoPOF-Bda@CNT in a CO2-saturated 0.5 M KHCO3 solution; (b) time-dependent total catalytic current densities (jtotal ∼ t) of CoPOF-Bpy@CNT in a wide potential range from −0.5 V to −1.0 V vs. RHE; (c) FECO and FEH2 of CoPOF-Bpy@CNT at different applied potentials; (d) partial current densities of CO production; (e) Tafel plots, and (f) TOF values of CoPOF-Bpy@CNT, CoPOF-Bpda@CNT, and CoPOF-Bda@CNT. | ||
Then, we performed chronoamperometry at different potentials to determine the FE values of the CO2RR. Fig. 3b displays the time-dependent total catalytic current densities (jtotal ∼ t) of CoPOF-Bpy@CNT in a wide potential range from −0.5 V to −1.0 V vs. RHE. All of the jtotal ∼ t plots remain stable for 600 s, which suggests a good short-term stability of CoPOF-Bpy@CNT. CoPOF-Bda@CNT and CoPOF-Bpda@CNT also show short-term stabilities, but the jtotal are lower than those of CoPOF-Bpy@CNT (Fig. S21 and S22†). FECO over the entire potential range was also determined. As shown in Fig. 3c, with an applied potential of −0.7 V vs. RHE, the FECO of CoPOF-Bpy@CNT is 97.5%, accompanied by a minor amount of H2 as the byproduct originating from water reduction, and no liquid product could be detected (Fig. S23†), which suggests the excellent selectivity of CoPOF-Bpy@CNT for electrocatalytic CO2-to-CO conversion. Comparatively, FECO of CoPOF-Bda@CNT and CoPOF-Bpda@CNT are inferior, particularly in the low potential region (Fig. S24†). These results indicate that in addition to the CO2RR activity, the electrocatalytic selectivity of CoPOF can likewise be effective by introducing dual active sites. Furthermore, to deeply understand the efficiency of each sample for electrocatalytic CO2RR, the partial catalytic current density for CO production (jCO) was calculated by multiplying the jtotal with the relevant FECO. As displayed in Fig. 3d, due to the larger jtotal and higher FECO value, the jCO of CoPOF-Bpy@CNT far surpasses those of CoPOF-Bda@CNT and CoPOF-Bpda@CNT over the entire potential range. Specifically, CoPOF-Bpy@CNT exhibits a jCO of −42.4 mA cm−2 at −1.0 V vs. RHE, much higher than those of CoPOF-Bda@CNT (−13.2 mA cm−2) and CoPOF-Bpda@CNT (−22.0 mA cm−2). Tafel analysis was further performed to reveal the reaction kinetic activity of these electrocatalysts for the CO2RR. Tafel slope was determined by plotting the applied potentials vs. the logarithm of jCO. As presented in Fig. 3e, the Tafel slope of CoPOF-Bpy@CNT was determined to be 193 mV dec−1, much smaller than those of CoPOF-Bda@CNT (223 mV dec−1) and CoPOF-Bpda@CNT (218 mV dec−1), suggesting the faster kinetics of electrocatalytic CO2-to-CO conversion over CoPOF-Bpy@CNT attributed to its higher activity of the sites. We assessed the specific activities of these electrocatalysts based on the TOF under the hypothesis that all the cobalt-centered sites participate in the CO2RR. Notably, CoPOF-Bpy@CNT shows a remarkable TOF value of 36.6 s−1 at −1.0 V vs. RHE (Fig. 3f), higher than those of CoPOF-Bda@CNT (13.7 s−1) and CoPOF-Bpda@CNT (26.0 s−1). Table S2† summarizes the dependence of the TOF values of CoPor-based COFs collected from recent literature. Remarkably, CoPOF-Bpy@CNT is superior to the majority of them in activity, which places it as one of the best candidates for electrocatalysts. Furthermore, the electrocatalytic activities of CoPOF-Bpy and CoPOF-Bpy/CNT in terms of FECO, jCO, Tafel slope, and TOF were determined. Specifically, CoPOF-Bpy@CNT exhibits a remarkable improvement in the formation of CO (Fig. S25†), which is consistent with its superior catalytic current density response (Fig. S26†).
The above electrocatalytic results reveal a high CO2RR activity and selectivity of CoPOF-Bpy@CNT. To deeply reveal the origin of its excellence, we first investigated the redox property of CoPOF-Bpy@CNT via CV. The CV plot displays a redox couple of E1/2 = −0.78 V vs. Ag/AgCl in DMF, and this redox event belongs to the Co(II/I) couple (Fig. S27†).60,61 The Co(II/I) redox potential of CoPOF-Bpy is identical to that of CoPOF-Bpy@CNT, which strongly supports the maintenance of the molecular structure after loading on the CNT scaffold. Importantly, compared with CoPOF-Bpy@CNT, the Co(II/I) redox potential of CoPOF-Bda@CNT negatively shifted by 50 mV, suggesting the tuning of the electronic structure in CoPOF-Bpy@CNT, due to the interaction between Co(II) Por- and Co(II)N2Cl2 sites (Fig. S28†). Additionally, the Co(II/I) redox event of CoPOF-Bpy@CNT at a more positive potential renders a higher fraction of Co(I) sites than in the CoPOF-Bda@CNT, indicating that dual active sites in CoPOF-Bpy@CNT can facilitate the generation of Co(I) species, and Co(I) species are generally considered as active sites for reducing CO2.62 And then, Cdl of each electrocatalyst was determined by analyzing the CV plots in the non-faradaic region with various scan rates (10–100 mV s−1) to calculate the ECSA (Fig. S29–S31†). As illustrated in Fig. 4a, CoPOF-Bpy@CNT shows a larger Cdl value (2.38 mF cm−2) than those of CoPOF-Bda@CNT (0.71 mF cm−2) and CoPOF-Bpda@CNT (0.77 mF cm−2). As ECSA is in proportion with Cdl, it reveals that CoPOF-Bpy@CNT offers a larger electrochemical area and more efficient active sites for CO2RR. EIS tests were performed to study the electrocatalytic kinetics of the electrode–electrolyte interface during the CO2RR. As revealed by the Nyquist plots in Fig. 4b and the corresponding equivalent circuit, CoPOF-Bpy@CNT has the smallest charge-transfer resistance compared to others, indicating more efficient electron transfer from the surface active sites of CoPOF-Bpy@CNT to the absorbed CO2 molecules, which is favorable for the CO2RR performance.
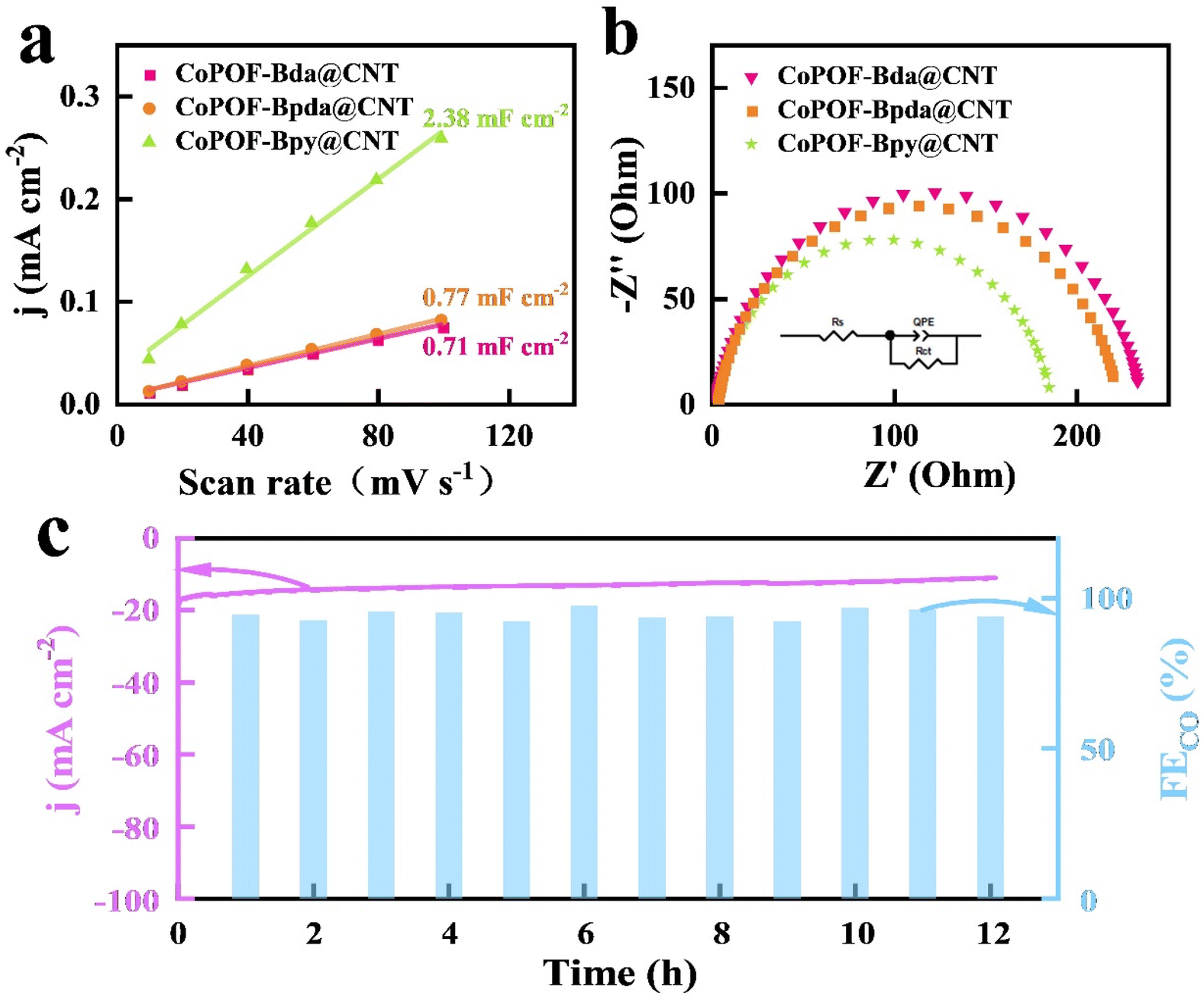 | ||
| Fig. 4 (a) Cdl values of CoPOF-Bpy@CNT, CoPOF-Bda@CNT, and CoPOF-Bpda@CNT in the non-faradaic region with various scan rates (10–100 mV s−1); (b) Nyquist plots by EIS; (c) measurements for assessing the long-term stability of CoPOF-Bpy@CNT at −0.7 V vs. RHE in a CO2-saturated 0.5 M KHCO3 solution for 12 h. | ||
To evaluate the long-term stability of CoPOF-Bpy@CNT, an electrocatalytic experiment was conducted at −0.7 V vs. RHE. The jtotal of −14 mA cm−2 was maintained for 12 h (Fig. 4c). Moreover, the gaseous product was periodically sampled and analyzed by GC every 1 h. Notably, the FECO was over 95% during the entire course of the measurement, which afforded an impressive turnover number of 2.8 × 104 for CO2 reduction to CO based on the amount of overall Co(II) sites. To ensure the component integrity of CoPOF-Bpy@CNT after long-term electrocatalysis, it was characterized by FT-IR, XRD, and XPS. A comparison of the sample before and after the reaction using FT-IR spectra shows that CoPOF-Bpy@CNT maintains the initial molecular structure (Fig. S32†). The recorded XRD pattern of CoPOF-Bpy@CNT is also in accordance with that of a fresh sample (Fig. S33†). As for the XPS measurement, the Co 2p XPS spectrum (Fig. S34†) shows that the binding energies of Co 2p1/2 and Co 2p3/2 remain unchanged after the reaction, indicating the molecular integrity of CoPOF-Bpy. The excellent long-term stability of CoPOF-Bpy is believed to be a consequence of the homogeneous polymerization of CoPOF-Bpy on the CNT scaffold, which enhances the mechanical and chemical stability of the electrochemically active species.
The exceptional catalytic performance including activity, selectivity, and long-term stability of CoPOF-Bpy@CNT can be ascribed to the following reasons: (1) the combined nanoscale and molecular strategy by hybridization of CNTs on the nanoscale and the introduction of dual active sites on the molecular-level allows the homogeneous coverage of CoPOF-Bpy composed of both Co(II) Por- and Co(II)N2Cl2 sites on the CNT scaffold, and thus enables a high degree of catalytically active site exposure; (2) strong interactions between the CoPOF-Bpy layer and CNTs can lead to the electron transfer from the electrode to the CoPOF-Bpy layer immobilized on CNTs, and also facilitate rapid repetitive cycling of Co(II) to Co(I) to promote CO2 reduction to CO; (3) the homogeneous coverage of CoPOF-Bpy on CNTs is beneficial for limiting the exposure of CNTs, which is kinetically favorable for hydrogen evolution.
4. Conclusions
In conclusion, we have demonstrated a combined nanoscale and molecular strategy for enhancing the intrinsic activity of POF-based electrocatalysts by constructing a CoPOF-Bpy@CNT composite material, which is synthesized by the template-directed in situ polymerization of ultrathin POF layers with isolated Co(II) Por- and Co(II)N2Cl2 sites on the CNT scaffold via π–π interactions. As a result, the resultant CoPOF-Bpy@CNT electrocatalyst displays remarkable electrocatalytic CO2-to-CO activity and selectivity, as well as excellent long-term stability in an aqueous electrolyte. At the nanoscale, Por-based POF layers were homogeneously anchored on the CNT scaffold to allow strengthened interfacial electron transfer from CNTs to Co(II) centers and enhanced long-term stability. At the molecular level, the electrocatalytic performance was boosted by incorporating dual catalytic sites into the molecular structure, which enables a high degree of catalytically active site exposure. The strategy reported herein introduces a facile and versatile approach for introducing dual active sites within a POF to construct an attractive type of electrocatalyst for converting CO2 to sustainable fuels.Author contributions
Shengsheng Huang: conception, investigation, and writing – review and editing. Qizhe He: formal analysis and data curation. Hongwei Li: formal analysis and data curation. Jinjie Qian: supervision. Wei Xu: data curation. Ting-Ting Li: supervision and project administration.Conflicts of interest
The authors declare that they have no known conflict of interest or personal relationships that could have appeared to influence the results reported in this work.Acknowledgements
This work was financially supported by the National Science Foundation of Zhejiang Province (LY20B030002, LY21B010003) and the Fundamental Research Funds for the Provincial Universities of Zhejiang (SJLY2021005). The authors also thank the Shiyanjia Lab (https://www.shiyanjia.com) for the XPS measurement analysis.References
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Defect and interface engineering for aqueous electrocatalytic CO2 reduction, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- N. Wang, R. K. Miao, G. Lee, A. Vomiero, D. Sinton, A. H. Ip, H. Liang and E. H. Sargent, Suppressing the liquid product crossover in electrochemical CO2 reduction, SmartMat, 2021, 2, 12–16 CrossRef CAS.
- S. Huang, K. Chen and T.-T. Li, Porphyrin and phthalocyanine based covalent organic frameworks for electrocatalysis, Coord. Chem. Rev., 2022, 464, 314563 CrossRef.
- D. Grammatico, A. J. Bagnall, L. Riccardi, M. Fontecave, B.-L. Su and L. Billon, Heterogenised molecular catalysts for sustainable electrochemical CO2 reduction, Angew. Chem., Int. Ed., 2022, 61, e202206399 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- J. Su, Y. Liu, Y. Song, L. Huang, W. Guo, X. Cao, Y. Dou, L. Cheng, G. Li, Q. Hu and R. Ye, Recent development of nanomaterials for carbon dioxide electroreduction, SmartMat, 2022, 3, 35–53 CrossRef CAS.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Sustainable conversion of carbon dioxide: an integrated review of catalysis and life cycle assessment, Chem. Soc. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- M. Hou, Y. Shi, J. Li, Z. Gao and Z. Zhang, Cu-based organic-inorganic composite materials for electrochemical CO2 reduction, Chem. – Asian J., 2022, 17, e202200624 CAS.
- N. Qiu, J. Li, H. Wang and Z. Zhang, Emerging dual-atomic-site catalysts for electrocatalytic CO2 reduction, Sci. China Mater., 2022, 65, 3302–3323 CrossRef CAS.
- C. Yang, Z. Gao, D. Wang, S. Li, J. Li, Y. Zhu, H. Wang, W. Yang, X. J. Gao, Z. Zhang and W. Hu, Bimetallic phthalocyanine heterostructure used for highly selective electrocatalytic CO2 reduction, Sci. China Mater., 2022, 65, 155–162 CrossRef CAS.
- S. Chu and A. Majumdar, Opportunities and challenges for a sustainable energy future, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- K. S. Song, P. W. Fritz and A. Coskun, Porous organic polymers for CO2 capture, separation and conversion, Chem. Soc. Rev., 2022, 51, 9831–9852 RSC.
- X.-Z. Wang, S.-L. Meng, J.-Y. Chen, H.-X. Wang, Y. Wang, S. Zhou, X.-B. Li, R.-Z. Liao, C.-H. Tung and L.-Z. Wu, Mechanistic insights into iron(II) bis(pyridyl)amine-bipyridine skeleton for selective CO2 photoreduction, Angew. Chem., Int. Ed., 2021, 60, 26072–26079 CrossRef CAS PubMed.
- J. F. Kurisingal, H. Kim, J. H. Choe and C. S. Hong, Covalent organic framework-based catalysts for efficient CO2 utilization reactions, Coord. Chem. Rev., 2022, 473, 214835 CrossRef.
- C. Yan, L. Lin, G. Wang and X. Bao, Transition metal-nitrogen sites for electrochemical carbon dioxide reduction reaction, Chin. J. Catal., 2019, 40, 23–37 CrossRef CAS.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, Fe-porphyrin-based metal-organic framework films as high-surface concentration, heterogeneous catalysts for electrochemical reduction of CO2, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS.
- Y. Yang, Z. Yang, C. Zhang, J. Zhou, S. Liu and Q. Cao, Single-atom catalysts supported on the graphene/graphdiyne heterostructure for effective CO2 electroreduction, Inorg. Chem., 2022, 61, 12012–12022 CrossRef CAS PubMed.
- S. Gu, A. N. Marianov, Y. Zhu and Y. Jiang, Cobalt porphyrin immobilized on the TiO2 nanotube electrode for CO2 electroreduction in aqueous solution, J. Energy Chem., 2021, 55, 219–227 CrossRef CAS.
- J. Han, N. Wang, X. Li, H. Lei, Y. Wang, H. Guo, X. Jin, Q. Zhang, X. Peng, X.-P. Zhang, W. Zhang, U.-P. Apfel and R. Cao, Bioinspired iron porphyrins with appended polypyridine/amine units for boosted electrocatalytic CO2 reduction reaction, eScience, 2022, 2, 623–631 CrossRef.
- H. Li, W. Xu, J. Qian and T.-T. Li, Construction of a polymeric cobalt phthalocyanine@mesoporous graphitic carbon nitride composite for efficient photocatalytic CO2 reduction, Chem. Commun., 2021, 57, 6987–6990 RSC.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Electrocatalytic reduction of carbon dioxide: opportunities with heterogeneous molecular catalysts, Energy Environ. Sci., 2020, 13, 374–403 RSC.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, Structural sensitivities in bimetallic catalysts for electrochemical CO2 reduction revealed by Ag-Cu nanodimers, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- X.-M. Liang, H.-J. Wang, C. Zhang, D.-C. Zhong and T.-B. Lu, Controlled synthesis of a Ni2 dual-atom catalyst for synergistic CO2 electroreduction, Appl. Catal., B, 2023, 322, 122073 CrossRef CAS.
- S. Liu, H. Yang, X. Huang, L. Liu, W. Cai, J. Gao, X. Li, T. Zhang, Y. Huang and B. Liu, Identifying active sites of nitrogen-doped carbon materials for the CO2 reduction reaction, Adv. Funct. Mater., 2018, 28, 1800499 CrossRef.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S.-T. Lee and Y. Li, Supported cobalt polyphthalocyanine for high-performance electrocatalytic CO2 reduction, Chem, 2017, 3, 652–664 CAS.
- D.-C. Zhong, Y.-N. Gong, C. Zhang and T.-B. Lu, Dinuclear metal synergistic catalysis for energy conversion, Chem. Soc. Rev., 2023, 52, 3170–3214 RSC.
- C. Yang, S. Li, Z. Zhang, H. Wang, H. Liu, F. Jiao, Z. Guo, X. Zhang and W. Hu, Organic–inorganic hybrid nanomaterials for electrocatalytic CO2 reduction, Small, 2020, 16, 2001847 CrossRef CAS PubMed.
- Z. Liang, H.-Y. Wang, H. Zheng, W. Zheng and R. Cao, Porphyrin-based frameworks for oxygen electrocatalysis and catalytic reduction of carbon dioxide, Chem. Soc. Rev., 2021, 50, 2540–2581 RSC.
- H. Dong, M. Lu, Y. Wang, H.-L. Tang, D. Wu, X. Sun and F.-M. Zhang, Covalently anchoring covalent organic framework on carbon nanotubes for highly efficient electrocatalytic CO2 reduction, Appl. Catal., B, 2022, 303, 120897 CrossRef CAS.
- H. Wang, H. Ding, X. Meng and C. Wang, Two-dimensional porphyrin- and phthalocyanine-based covalent organic frameworks, Chin. Chem. Lett., 2016, 27, 1376–1382 CrossRef CAS.
- S. Liu, H. B. Yang, S.-F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Elucidating the electrocatalytic CO2 reduction reaction over a model single-atom nickel catalyst, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- K. Chi, Y. Wu, X. Wang, Q. Zhang, W. Gao, L. Yang, X. Chen, D. Chang, Y. Zhang, T. Shen, X. Lu, Y. Zhao and Y. Liu, Single atom catalysts with out-of-plane coordination structure on conjugated covalent organic frameworks, Small, 2022, 18, 2203966 CrossRef CAS PubMed.
- H. Gu, G. Shi, L. Zhong, L. Liu, H. Zhang, C. Yang, K. Yu, C. Zhu, J. Li, S. Zhang, C. Chen, Y. Han, S. Li and L. Zhang, A two-dimensional van der Waals heterostructure with isolated electron-deficient cobalt sites toward high-efficiency CO2 electroreduction, J. Am. Chem. Soc., 2022, 144, 21502–21511 CrossRef CAS PubMed.
- H.-J. Zhu, M. Lu, Y.-R. Wang, S.-J. Yao, M. Zhang, Y.-H. Kan, J. Liu, Y. Chen, S.-L. Li and Y.-Q. Lan, Efficient electron transmission in covalent organic framework nanosheets for highly active electrocatalytic carbon dioxide reduction, Nat. Commun., 2020, 11, 497 CrossRef CAS PubMed.
- E. M. Johnson, R. Haiges and S. C. Marinescu, Covalent-organic framework composed of rhenium bipyridine and metal porphyrin: designing heterobimetallic frameworks with two distinct metal sites, ACS Appl. Mater. Interfaces, 2018, 10, 37919–37927 CrossRef CAS PubMed.
- T. Wang, L. Xu, Z. Chen, L. Guo, Y. Zhang, R. Li and T. Peng, Central site regulation of cobalt porphyrin conjugated polymer to give highly active and selective CO2 reduction to CO in aqueous solution, Appl. Catal., B, 2021, 291, 120128 CrossRef CAS.
- S. Li, R. Ma, S. Xu, T. Zheng, H. Wang, G. Fu, H. Yang, Y. Hou, Z. Liao, B. Wu, X. Feng, L.-Z. Wu, X.-B. Li and T. Zhang, Two-dimensional benzobisthiazole-vinylene-linked covalent organic frameworks outperform one-dimensional counterparts in photocatalysis, ACS Catal., 2023, 13, 1089–1096 CrossRef CAS.
- S. Li, R. Ma, S. Xu, T. Zheng, G. Fu, Y. Wu, Z. Liao, Y. Kuang, Y. Hou, D. Wang, P. S. Petkov, K. Simeonova, X. Feng, L.-Z. Wu, X.-B. Li and T. Zhang, Direct construction of isomeric benzobisoxazole-vinylene-linked covalent organic frameworks with distinct photocatalytic properties, J. Am. Chem. Soc., 2022, 144, 13953–13960 CrossRef CAS PubMed.
- Y. Liu, X. Yan, T. Li, W.-D. Zhang, Q.-T. Fu, H.-S. Lu, X. Wang and Z.-G. Gu, Three-dimensional porphyrin-based covalent organic frameworks with tetrahedral building blocks for single-site catalysis, New J. Chem., 2019, 16907–16914 RSC.
- M. Chen, H. Li, C. Liu, J. Liu, Y. Feng, A. G. H. Wee and B. Zhang, Porphyrin- and porphyrinoid-based covalent organic frameworks (COFs): from design, synthesis to applications, Coord. Chem. Rev., 2021, 435, 213778 CrossRef CAS.
- Q. Wu, M.-J. Mao, Q.-J. Wu, J. Liang, Y.-B. Huang and R. Cao, Construction of donor–acceptor heterojunctions in covalent organic framework for enhanced CO2 electroreduction, Small, 2021, 17, 2004933 CrossRef CAS PubMed.
- S. An, C. Lu, Q. Xu, C. Lian, C. Peng, J. Hu, X. Zhuang and H. Liu, Constructing catalytic crown ether-based covalent organic frameworks for electroreduction of CO2, ACS Energy Lett., 2021, 6, 3496–3502 CrossRef CAS.
- C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang and O. M. Yaghi, Reticular electronic tuning of porphyrin active sites in covalent organic frameworks for electrocatalytic carbon dioxide reduction, J. Am. Chem. Soc., 2018, 140, 1116–1122 CrossRef CAS PubMed.
- Y. Xiang, W. Dong, P. Wang, S. Wang, X. Ding, F. Ichihara, Z. Wang, Y. Wada, S. Jin, Y. Weng, H. Chen and J. Ye, Constructing electron delocalization channels in covalent organic frameworks powering CO2 photoreduction in water, Appl. Catal., B, 2020, 274, 119096 CrossRef CAS.
- M. R. Rao, Y. Fang, S. D. Feyter and D. F. Perepichka, Conjugated covalent organic frameworks via Michael addition-elimination, J. Am. Chem. Soc., 2017, 139, 2421–2427 CrossRef CAS PubMed.
- Y. Wang, D. Song, J. Li, Q. Shi, J. Zhao, Y. Hu, F. Zeng and N. Wang, Covalent metalloporphyrin polymer coated on carbon nanotubes as bifunctional electrocatalysts for water splitting, Inorg. Chem., 2022, 61, 10198–10204 CrossRef CAS PubMed.
- H. Jia, Z. Sun, D. Jiang and P. Du, Covalent cobalt porphyrin framework on multiwalled carbon nanotubes for efficient water oxidation at low overpotential, Chem. Mater., 2015, 27, 4586–4593 CrossRef CAS.
- Y. Lu, J. Zhang, W. Wei, D.-D. Ma, X.-T. Wu and Q.-L. Zhu, Efficient carbon dioxide electroreduction over ultrathin covalent organic framework nanolayers with isolated cobalt porphyrin units, ACS Appl. Mater. Interfaces, 2020, 12, 37986–37992 CrossRef CAS PubMed.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, A covalent organic framework bearing single Ni sites as a synergistic photocatalyst for selective photoreduction of CO2 to CO, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- Z. Liu, Y. Huang, S. Chang, X. Zhu, Y. Fu, R. Ma, X. Lu, F. Zhang, W. Zhu and M. Fan, Highly dispersed Ru nanoparticles on a bipyridine-linked covalent organic framework for efficient photocatalytic CO2 reduction, Sustainable Energy Fuels, 2021, 5, 2871–2876 RSC.
- X. Ding, B. Yu, B. Han, H. Wang, T. Zheng, B. Chen, J. Wang, Z. Yu, T. Sun, X. Fu, D. Qi and J. Jiang, Porphyrin coordination polymer with dual photocatalytic sites for efficient carbon dioxide reduction, ACS Appl. Mater. Interfaces, 2022, 14, 8048–8057 CrossRef CAS PubMed.
- X. Yao, K. Chen, L.-Q. Qiu, Z.-W. Yang and L.-N. He, Ferric porphyrin-based porous organic polymers for CO2 photocatalytic reduction to syngas with selectivity control, Chem. Mater., 2021, 33, 8863–8872 CrossRef CAS.
- B.-Q. Li, S.-Y. Zhang, X. Chen, C.-Y. Chen, Z.-J. Xia and Q. Zhang, One-pot synthesis of framework porphyrin materials and their applications in bifunctional oxygen electrocatalysis, Adv. Funct. Mater., 2019, 29, 1901301 CrossRef.
- B.-Q. Li, S.-Y. Zhang, B. Wang, Z.-J. Xia, C. Tang and Q. Zhang, A porphyrin covalent organic framework cathode for flexible Zn-air batteries, Energy Environ. Sci., 2018, 11, 1723–1729 RSC.
- M. Zhu, J. Chen, L. Huang, R. Ye, J. Xu and Y.-F. Han, Covalently grafting cobalt porphyrin onto carbon nanotubes for efficient CO2 electroreduction, Angew. Chem., Int. Ed., 2019, 58, 6595–6599 CrossRef CAS PubMed.
- M. Zhu, J. Chen, R. Guo, J. Xu, X. Fang and Y.-F. Han, Cobalt phthalocyanine coordinated to pyridine-functionalized carbon nanotubes with enhanced CO2 electroreduction, Appl. Catal., B, 2019, 251, 112–118 CrossRef CAS.
- J. Chen, X. Tao, C. Li, Y. Ma, L. Tao, D. Zheng, J. Zhu, H. Li, R. Li and Q. Yang, Synthesis of bipyridine-based covalent organic frameworks for visible-light-driven photocatalytic water oxidation, Appl. Catal., B, 2020, 262, 118271 CrossRef CAS.
- M. Abdinejad, C. Dao, B. Deng, F. Dinic, O. Voznyy, X.-A. Zhang and H.-B. Kraatz, Electrocatalytic reduction of CO2 to CH4 and CO in aqueous solution using pyridine-porphyrins immobilized onto carbon nanotubes, ACS Sustainable Chem. Eng., 2020, 8, 9549–9557 CrossRef CAS.
- L. Ma, W. Hu, B. Mei, H. Liu, B. Yuan, J. Zang, T. Chen, L. Zou, Z. Zou, B. Yang, Y. Yu, J. Ma, Z. Jiang, K. Wen and H. Yang, Covalent triazine framework confined copper catalysts for selective electrochemical CO2 reduction: operando diagnosis of active sites, ACS Catal., 2020, 10, 4534–4542 CrossRef CAS.
- A. N. Marianov, A. S. Kochubei, T. Roman, O. J. Conquest, C. Stampfl and Y. Jiang, Resolving deactivation pathways of Co porphyrin-based electrocatalysts for CO2 reduction in aqueous medium, ACS Catal., 2021, 11, 3715–3729 CrossRef CAS.
- Y. Wang, X.-P. Zhang, H. Lei, K. Guo, G. Xu, L. Xie, X. Li, W. Zhang, U.-P. Apfel and R. Cao, Tuning electronic structures of covalent Co porphyrin polymers for electrocatalytic CO2 reduction in aqueous solution, CCS Chem., 2022, 4, 2959–2967 CrossRef CAS.
- K. Leung, I. M. B. Nielsen, N. Sai, C. Medforth and J. A. Shelnutt, Cobalt porphyrin catalyzed electrochemical reduction of carbon dioxide in water. 2. Mechanism from first principles, J. Phys. Chem. A, 2010, 114, 10174–10184 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qi00589e |
| This journal is © the Partner Organisations 2023 |