
Atomically dispersed copper catalysts for highly selective CO2 reduction†
Ruirui
Yun
 *a,
Beibei
Zhang
a,
Changsong
Shi
a,
Ruiming
Xu
a,
Junjie
Mao
*a and
Zhaoxu
Wang
*b
*a,
Beibei
Zhang
a,
Changsong
Shi
a,
Ruiming
Xu
a,
Junjie
Mao
*a and
Zhaoxu
Wang
*b
aThe Key Laboratory of Functional Molecular Solids, Ministry of Education, College of Chemistry and Materials Science, Anhui Normal University, Wuhu 241000, China. E-mail: ruirui58@ahnu.edu.cn; maochem@ahnu.edu.cn
bKey Laboratory of Theoretical Chemistry and Molecular Simulation of Ministry of Education, School of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan 411201, China. E-mail: hnust_chem@163.com
First published on 28th November 2022
Abstract
Support substrates play important roles in the catalysis process. Herein, atomically dispersed CuN3 catalysts supported by two different types of zirconia (denoted as CuN3/NC/T-ZrO2 and CuN3/NC/M-ZrO2) have been rationally fabricated to uncover the influence of the support. CuN3/NC/T-ZrO2 exhibits outstanding performance for electrochemical CO2 reduction towards CO at a wide range of potentials (∼96%, 0.6–0.8 V vs. RHE) owing to the acidic uncoordinated Zr4+ sites of T-ZrO2, which facilitate CO2 accumulation, and N-doped carbon (NC), which enhances the conductivity of the catalyst. Moreover, density functional theory calculations prove that T-ZrO2 effectively decreases the Gibbs free energy for CO2 to CO conversion. Significantly, this study reports the effects of the substrate on the electrocatalytic CO2RR and provides a promising strategy for tuning catalytic activity and selectivity during the process of converting CO2 into high-value products by controlling the phase of the support for the first time.
Introduction
As the consumption of fossil fuels increases, climate change has become a major global issue owing to sea-level rise and global warming.1 In recent years, many efforts have been made to convert CO2 into useful chemical feedstocks by designing and tuning appropriate catalytic active sites.2 These strategies are key to reducing fossil fuel dependence. Among them, the electrochemical CO2 reduction reaction (EC-CO2RR) has been considered a promising route to obtain value-added products such as methane, formate, and CO.3 However, the major competitive process of the H2 evolution reaction (HER) during the EC-CO2RR must be suppressed.4 Hence, it is necessary to develop robust electrocatalysts to selectively produce target products during the EC-CO2RR process.5Atomically dispersed single metal site catalysts are a new research topic that has attracted increasing attention. These catalysts are a promising technology with tunable active sites and substrates, aiming at higher activity and selectivity during heterogeneous catalytic reactions.6 Additionally, Cu species, as an abundant non-noble metal, have excellent performance in catalytic systems. Some examples include electrochemical CO2 reduction by Cu-based MOFs and Cu particles,7 Cu clusters for CO2 methanation,8 and single Cu atoms applied to the O2 evolution reaction.9 Additionally, the interaction between the catalyst and support is an important factor in the activity of catalysts.10 Zirconia is a commonly used substrate with many advantages, such as high thermal stability, good mechanical performance, and the presence of various phases with different chemical and physical characteristics.11 Some of the most widely used Cu/ZrO2 catalysts for the conversion of CO2 include Cu and Cu oxides combined with zirconia present in monoclinic or tetragonal phases.12
To explore the influence of different substrates on catalytic performance, we designed two catalysts with atomically dispersed CuN3 sites supported on zirconia with monoclinic (CuN3/NC/M-ZrO2) and tetragonal (CuN3/NC/T-ZrO2) phases. The synthetic approach has been simply divided into three steps, as illustrated in Scheme 1. There are equal Lewis and Brønsted acid centers in M-ZrO2, and water is a Lewis base that can be concentrated by M-ZrO2, while only the Brønsted acid sites of T-ZrO2 suppress the HER during the EC-CO2RR. As expected, the atomically dispersed CuN3 sites favoured the reduction of CO2, while the formation of strong metal–support interactions contributed to the enrichment of different reactants, resulting in the diverse catalytic activity of catalysts for the EC-CO2RR. CuN3/NC/T-ZrO2 exhibits high activity for the EC-CO2RR to produce CO with high faradaic efficiency of up to 96%, while CuN3/NC/M-ZrO2 exhibits negligible CO2RR activity with H2 as the main product.
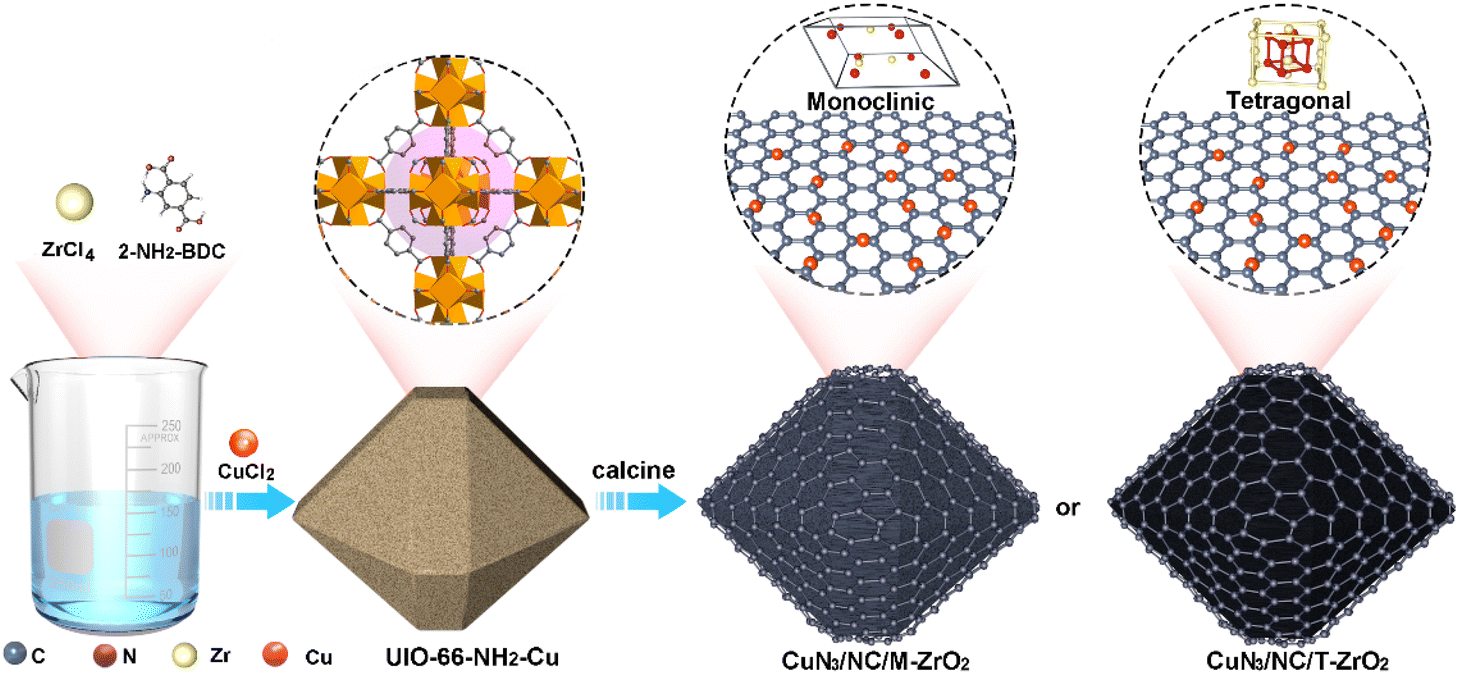 | ||
| Scheme 1 Schematic illustration of the synthesis of atomically dispersed Cu sites supported on zirconia. | ||
Results and discussion
Singly dispersed CuN3 sites were formed by pyrolyzing UIO-66-NH2-Cu, which prevents metal from aggregating owing to coordination bonds between the Cu and –NH2 groups (ESI, Fig. S1†). The powder X-ray diffraction (PXRD) patterns of CuN3/NC/T-ZrO2, CuN3/NC/M-ZrO2, and CuN3/NC possess a broad peak around 26° assigned to the (002) peaks of defective graphitic stacking, which allows the Cu atom to be isolated on the substrate support. The absence of significant Cu peaks in the PXRD profiles also agrees with the structural interpretation (Fig. S2†). The N2 and CO2 adsorption isotherms reveal that CuN3/NC/T-ZrO2 exhibits better BET surface area and CO2 uptake than CuN3/NC/M-ZrO2, indicating that CuN3/NC/T-ZrO2 possesses high CO2 affinity that facilitates CO2 conversion (Fig. S4 and S5†). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images show no clear evidence of particles (Fig. 1b–d). However, high-resolution scanning transmission electron microscopy (STEM) does not exhibit particles, indicating atomically dispersed Cu sites in the structure (Fig. 1f). As is well known, singly dispersed metal sites cannot exist solely on zirconia or carbon support owing to their higher surface energy. Hence, the N-doped carbon matrix obtained via the pyrolysis of UIO-66-NH2 plays an efficient role in fixing singly dispersed Cu sites through the formation of Cu–N bonds. As elemental mapping analysis shows, the homogeneous distribution of Cu, N, C, Zr, and O verifies that the Cu sites are dispersed uniformly over the substrate (Fig. 1g). | ||
| Fig. 1 (a) SEM image of Cu-UIO-66-NH2. (b and c) SEM and TEM images of CuN3/NC/T-ZrO2. (d–f) TEM, selected area electron diffraction (SAED), and STEM images of CuN3/NC/T-ZrO2. (g) EDS elemental mapping of CuN3/NC/T-ZrO2. | ||
Thereafter, X-ray photoelectron spectroscopy (XPS) was performed to verify the chemical formation of CuN3/NC/T-ZrO2. The XPS spectra confirm the existence of Cu, Zr, N, O, and C. The N 1s spectrum of the catalyst exhibits three deconvoluted peaks of 398.0 eV (pyridinic-N), 399.4 eV (Cu–N), and 400.3 eV (graphitic-N). This N distribution corresponds to previous reports on singly dispersed metal site catalysts, and the pyridinic-N forms a Cu–N bond. The two main peaks in the O 1s spectrum at 529.9 and 531.3 eV are attributed to the contribution of O bonded to Zr, which is consistent with the Zr 3d spectrum (Fig. 2c). The full width at half maximum of the peak in the Zr 3d spectra is approximately 1.5 eV, which verifies the existence of the tetragonal phase in the sample. The Zr 3d5/2 peak is located at 181.8 eV with a deviated value of 2.4 eV, which indicates the full oxidation of Zr4+ in accordance with the PXRD results. Furthermore, there is no evidence of Cu nanoparticles or clusters from the Zr 3d and O 1s spectra. The Cu 2p3/2 spectrum at 933.0 eV with a spread shape and spin–obit deviation of 19.0 eV indicates that Cu largely differs from the metallic form and oxidation state.13
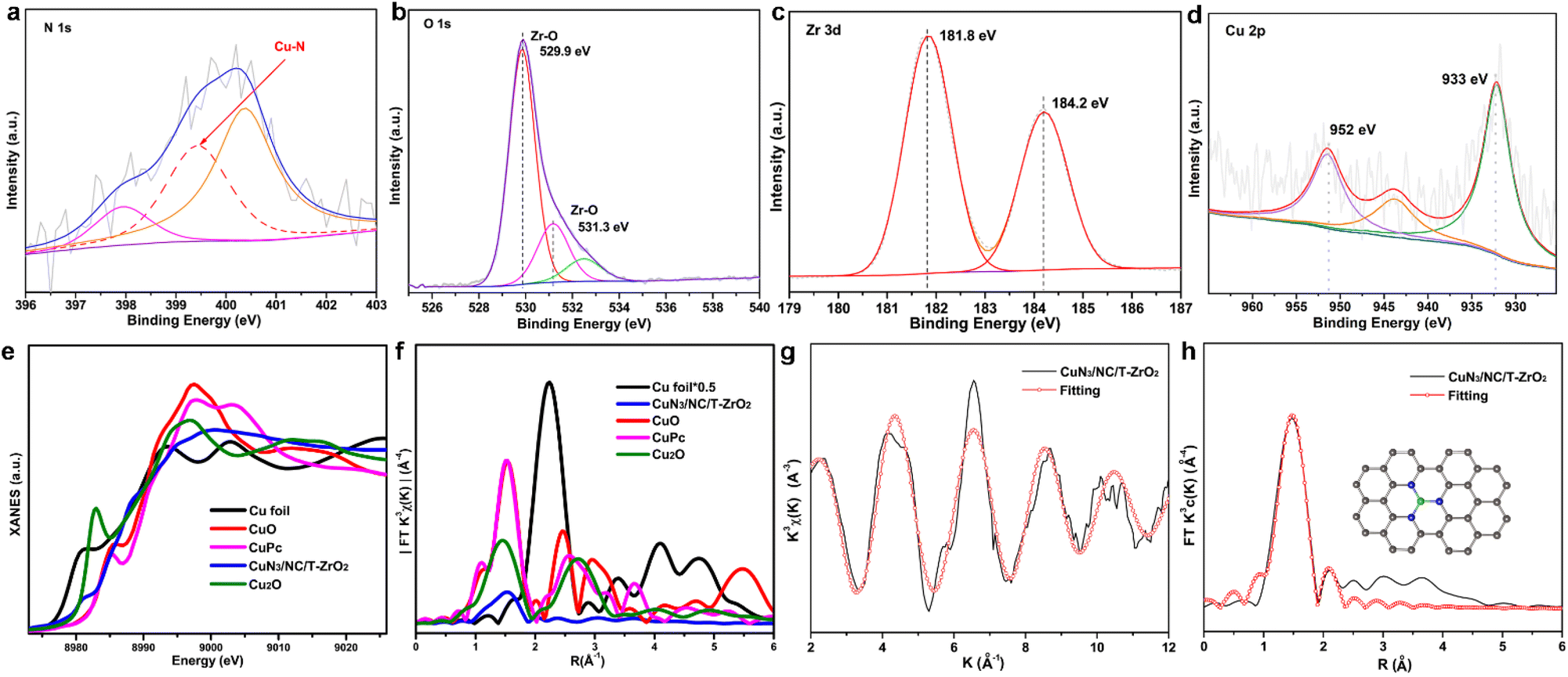 | ||
| Fig. 2 (a–d) XPS spectra of CuN3/NC/T-ZrO2: N 2p, O 1s, Zr 3d, and Cu 2p, respectively. (e) Cu K-edge data of Cu foil, CuO, CuPc, Cu2O, and CuN3/NC/T-ZrO2. (f) Fourier transform (FT) k3-weighted EXAFS profiles at the Cu k-edge. (g) Fitting curves. (h) EXAFS R space fitting curves of CuN3/NC/T-ZrO2. | ||
X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were further employed to investigate the local atomic structure of CuN3/NC/T-ZrO2. As shown in Fig. 2e, the absorption edge of CuN3/NC/T-ZrO2 was located between that of Cu foil and CuO, indicating that the atomically dispersed Cu atoms are positively charged in CuN3/NC/T-ZrO2. This result coincided with the aforementioned XPS results. Fig. 2f shows the Fourier transform (FT) k3-weighted extended EXAFS spectrum of CuN3/NC/T-ZrO2. Only one dominant peak is observed at about 1.5 Å, which is attributed to the Cu–N first coordination shell. No apparent Cu–Cu coordination peaks in the FT-EXAFS spectrum of CuN3/NC/T-ZrO2 are observed, demonstrating that the Cu atoms are atomically dispersed in the support. The quantitative structural parameters of Cu in CuN3/NC/T-ZrO2 were obtained by EXAFS fitting, and the fitting curves are shown in Fig. 2g and h. The fitting results indicate that the coordination number of Cu is about 3 (Cu–N3), and the corresponding Cu–N mean bond length is 1.95 Å. The optimized atomic structure model of the sample is shown in Fig. 2h.
The electrochemical activity towards the CO2RR of the as-synthesized catalysts was evaluated in a three-electrode system in a high-purity CO2-saturated 0.5 M KHCO3 (pH 6.8) aqueous solution. The obtained gaseous products (CO and H2) were monitored using gas chromatography (GC), and no liquid products were detected using 1H nuclear magnetic resonance (NMR) spectroscopy (Fig. S6†). As revealed using linear sweep voltammetry (Fig. 3a), CuN3/NC/T-ZrO2 and CuN3/NC/M-ZrO2 exhibit lower total current density than CuN3/NC as ZrO2 is an insulator. However, CuN3/NC/T-ZrO2 exhibits larger current densities of 5 mA cm−2 at −0.7 V and 12 mA cm−2 at −1.0 V (vs. RHE, Fig. 3a), indicating the importance of the N-doped carbon substrate towards facilitating electron transfer. Interestingly, the catalytic conversion of CO2 to CO is considerably affected by the phase of the ZrO2 substrate. In the case of T-ZrO2, the FECO is almost 94% (higher than other Cu-based catalysts, Table S2†) at wide potential ranges of −0.6 VRHE to 0.8 VRHE, while M-ZrO2 shows no activity for the CO2RR and is more selective to the HER through the entire potential range. In contrast, when zirconia was etched by hydrofluoric acid, the catalyst retained moderate activity towards CO2 to CO and with increasing overpotential, the HER is enhanced (Fig. 3b). The higher CO2 to CO activity indicates the key role of the tetragonal ZrO2 phase substrate. Compared with T-ZrO2, in the M-ZrO2 phase, acidic uncoordinated Zr4+ sites change to basic uncoordinated O2− sites, which reject the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond to reduce the collision between CO and the catalyst and decrease CO2 activity. Additionally, as shown in Fig. 3d, CuN3/NC/T-ZrO2 has a lower Tafel slope, which indicates that electrons are transferred from the surface of CuN3/NC/T-ZrO2 to CO2 molecules more easily for further reduction.14 In contrast, the significantly reduced Tafel slope for the singly dispersed CuN3 supported by T-ZrO2 indicates that the kinetics of electron transfer is greatly enhanced. Moreover, to evaluate the electrochemically active surface area of the three catalysts, the values of the electrochemical double-layer capacitance (Cdl) were obtained using cyclic voltammetry (Fig. 3e). The Cdl values suggest that CuN3/NC/T-ZrO2 (27 mF cm−2) possesses more active sites than CuN3/NC/M-ZrO2 (6 mF cm−2), increasing the reaction speed of the electrocatalytic process. CuN3/NC exhibits lower selectivity, which may be attributed to the lower CO2 uptake for subsequent reduction, which is further proof of the essentialness of the T-ZrO2 substrate during the EC-CO2RR to CO. More importantly, CuN3/NC/T-ZrO2 exhibits excellent stability for the EC-CO2RR, retaining approximately 95% of the initial faradaic efficiency for the CO product after 10 hours of continuous electrolysis. This result indicates that as a substrate, zirconia can support the active site well owing to its outstanding mechanical performance (Fig. 3f).
O bond to reduce the collision between CO and the catalyst and decrease CO2 activity. Additionally, as shown in Fig. 3d, CuN3/NC/T-ZrO2 has a lower Tafel slope, which indicates that electrons are transferred from the surface of CuN3/NC/T-ZrO2 to CO2 molecules more easily for further reduction.14 In contrast, the significantly reduced Tafel slope for the singly dispersed CuN3 supported by T-ZrO2 indicates that the kinetics of electron transfer is greatly enhanced. Moreover, to evaluate the electrochemically active surface area of the three catalysts, the values of the electrochemical double-layer capacitance (Cdl) were obtained using cyclic voltammetry (Fig. 3e). The Cdl values suggest that CuN3/NC/T-ZrO2 (27 mF cm−2) possesses more active sites than CuN3/NC/M-ZrO2 (6 mF cm−2), increasing the reaction speed of the electrocatalytic process. CuN3/NC exhibits lower selectivity, which may be attributed to the lower CO2 uptake for subsequent reduction, which is further proof of the essentialness of the T-ZrO2 substrate during the EC-CO2RR to CO. More importantly, CuN3/NC/T-ZrO2 exhibits excellent stability for the EC-CO2RR, retaining approximately 95% of the initial faradaic efficiency for the CO product after 10 hours of continuous electrolysis. This result indicates that as a substrate, zirconia can support the active site well owing to its outstanding mechanical performance (Fig. 3f).
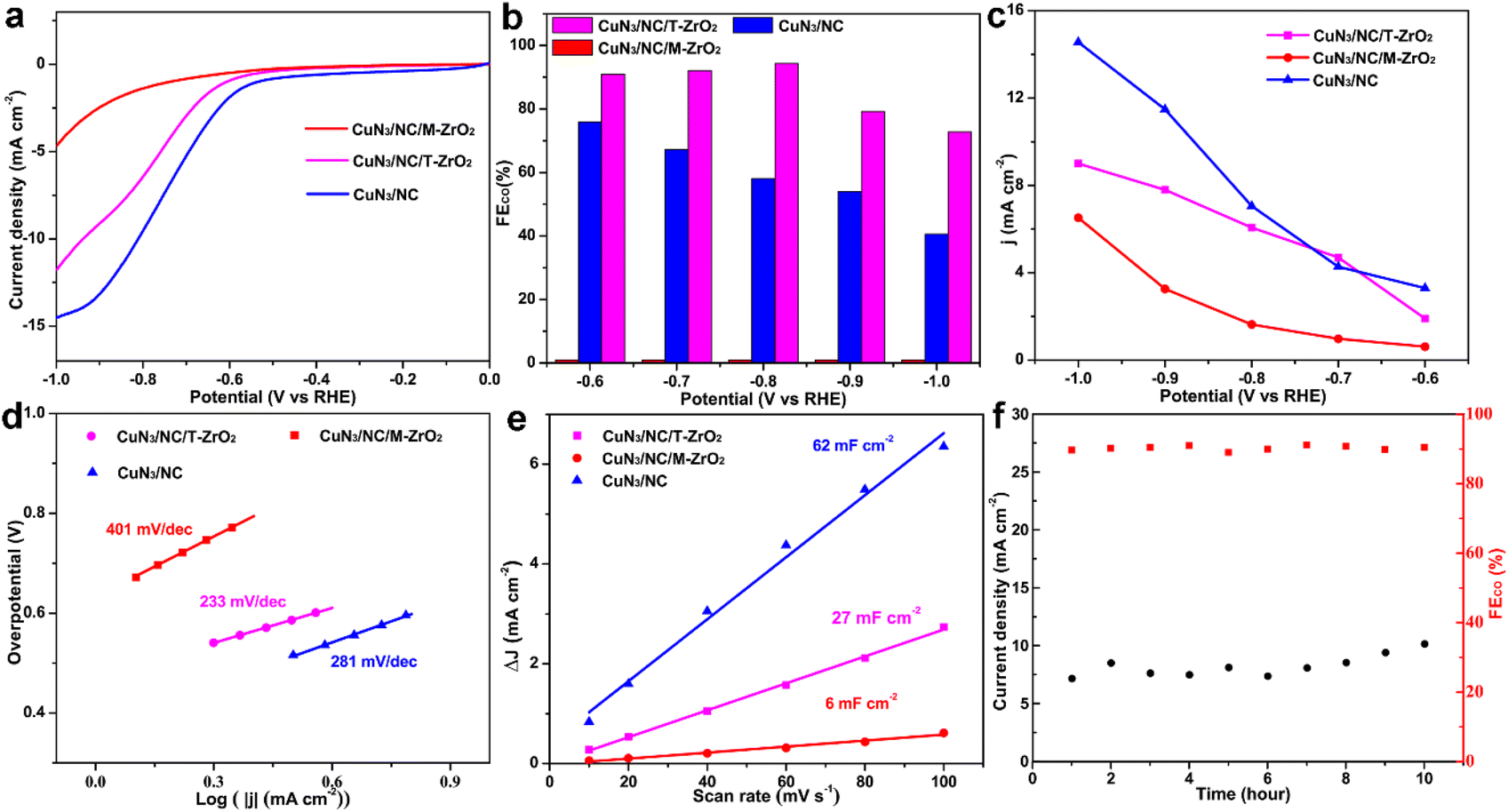 | ||
| Fig. 3 Electrochemical catalytic performance of the catalysts in CO2-saturated 0.5 M KHCO3. (a) LSVs of CuN3/NC/T-ZrO2, CuN3/NC/M-ZrO2, CuN3/NC. (b) Dependence of FECO for the catalysts. (c) Current densities at different potentials. (d) Tafel slopes of the catalysts. (e) Capacitances of the electrodes calculated from linear regression of the double-layer current as a function of scan rate. (f) Long-term stability performed at −1.0 vs. RHE. | ||
To further evaluate the effects of CuN3/NC/T-ZrO2 and CuN3/NC/M-ZrO2 catalysts on the activity and selectivity of the CO2RR and HER, we applied density functional theory (DFT) to calculate the free energy and binding energy at each reaction intermediate step of the CO2RR and HER. The most stable surfaces—the (101) face of tetragonal ZrO2 (T-ZrO2(101)) and (−111) of monoclinic ZrO2 (M-ZrO2(−111))—were employed to calculate the mechanism of the catalytic reaction (Fig. S9†).15Fig. 4a and b show the CO2RR to CO reaction processes on CuN3/NC/T-ZrO2 and CuN3/NC/M-ZrO2, in which the protonation of CO2 to form *COOH could be identified as a potential limiting step.16 Comparing Fig. 4a and b, the adsorption sites of the CO2RR intermediates were different in various phases. The reaction intermediates *COOH and *CO were bound at the Cu site for CuN3/NC/T-ZrO2, while the reaction intermediates were bound at the Zr site for CuN3/NC/M-ZrO2. Noticeably, the binding energies of the intermediates are significantly stronger on CuN3/NC/M-ZrO2 than on CuN3/NC/T-ZrO2 (Fig. 4c). Although the relatively strong *COOH adsorption on the catalytic sites should improve the electrocatalytic activity, the strong binding of *CO on CuN3/NC/M-ZrO2 (BE = −2.68 eV) makes its desorption difficult. Furthermore, DFT calculations demonstrated that the energy barriers of CuN3/NC/M-ZrO2 are higher than those of CuN3/NC/T-ZrO2, suggesting poor activity of CuN3/NC/M-ZrO2 for the CO2RR. Thus, CuN3/NC/T-ZrO2 exhibits the best selectivity for the CO2RR.
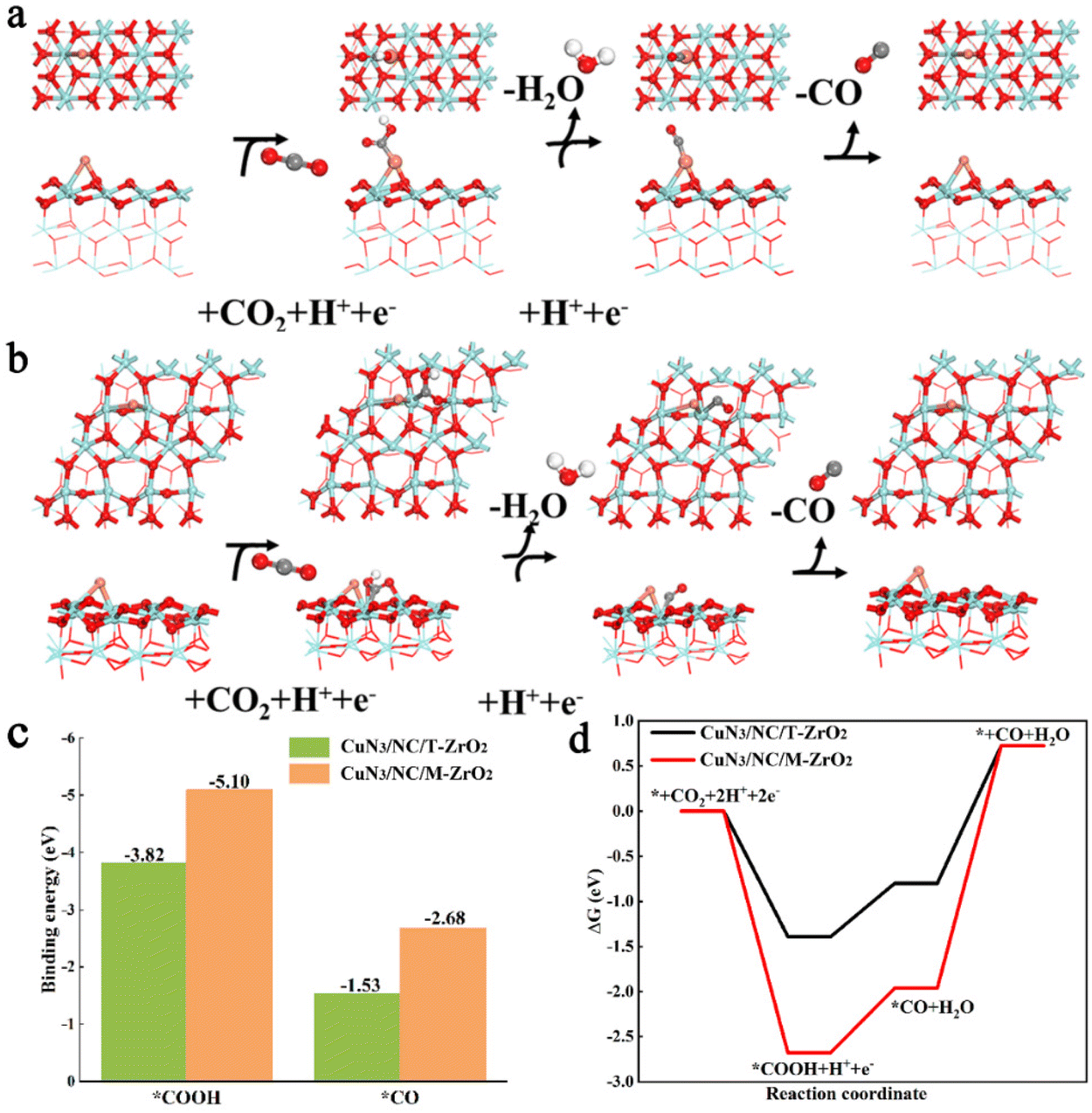 | ||
| Fig. 4 (a) CO2RR processes on CuN3/NC/T-ZrO2(101) and (b) CuN3/NC/M-ZrO2(−111). (c) Calculated binding energies (in eV) on CuN3/NC/T-ZrO2(101) and CuN3/NC/M-ZrO2(−111). (d) Calculated free energy diagrams of the CO2RR on CuN3/NC/T-ZrO2(101) and CuN3/NC/M-ZrO2(−111). | ||
The free energy of *H has been identified as a descriptor of the HER,17 a competing reaction in the CO2RR. Fig. 4d and5 show that the free energy of *H on CuN3/NC/M-ZrO2 is below the free energy of *COOH, suggesting that the HER should be more favorable on CuN3/NC/M-ZrO2. Additionally, CuN3/NC/T-ZrO2 is expected to possess excellent CO selectivity owing to the weaker binding of *H compared to that of COOH*. The DFT results further demonstrate that CuN3/NC/T-ZrO2 favors CO formation while CuN3/NC/M-ZrO2 is active for H2 evolution, which is consistent with our previously described experimental results.
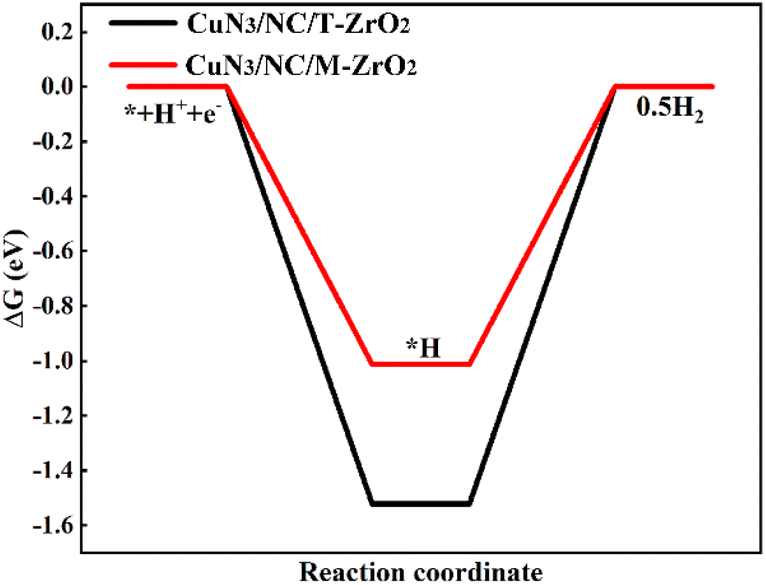 | ||
| Fig. 5 DFT-calculated free energy diagrams of the HER on CuN3/NC/T-ZrO2(101) and CuN3/NC/M-ZrO2(−111) at a potential (U) = 0 V. | ||
Conclusions
Herein, three EC-CO2RR catalysts with singly dispersed Cu–N sites supported by ZrO2 with different phases to tune the catalytic environment and enhance the selectivity for CO2 conversion to CO have been designed and synthesized. The tetragonal phase of zirconia with acidic uncoordinated Zr4+ sites enhances CO reduction, and N-doped carbon enhances the electrical conductivity of the catalysts. Together, the properties of the catalysts afford the excellent ability of CO2 to CO conversion of CuN3/NC/T-ZrO2. Systematic studies of CuN3/NC/T-ZrO2, CuN3/NC/M-ZrO2, and CuN3/NC via experimentation and DFT calculation reveal that the acidic catalytic centre of the substrate plays a crucial factor in the generation of CO owing to its ability to concentrate CO2 and prevent the HER. This work not only provides an in-depth understanding of the influence of the substrate during the electrocatalytic process but also provides a fundamental strategy to design promising catalysts for CO2 to CO conversion with high selectivity and durability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the NSFC (Grant No. 21401004), Natural Science Foundation of Anhui Province (Grant No. 1508085QB36, 1908085QB58, 2008085MB52), and Open Foundation of Anhui Laboratory of Molecule-based Materials (fzj19005).References
- (a) J. E. Kay, Early climate models successfully predicted global warming, Nature, 2020, 578, 45–46 CrossRef CAS PubMed; (b) C. A. Logan, J. P. Dunne, J. S. Ryan, M. L. Baskett and S. D. Donner, Quantifying global potential for coral evolutionary response to climate change, Nat. Clim. Change, 2021, 11, 537–542 CrossRef; (c) L. A. Parsons, D. Shindell, M. Tigchelaar, Y. Q. Zhang and J. T. Spector, Increased labor losses and decreased adaptation potential in a warmer world, Nat. Commun., 2021, 12, 7286 CrossRef CAS PubMed; (d) M. Klower, M. R. Allen, D. S. Lee, S. R. Proud, L. Gallagher and A. Skowron, Quantifying aviation's contribution to global warming, Environ. Res. Lett., 2021, 16, 104027 CrossRef.
- (a) I. Merino-Garcia, J. Albo, J. Solla-Gullón, V. Montiel and A. Irabien, Cu oxide/ZnO-based surfaces for a selective ethylene production from gasphase CO2 electroconversion, J. CO2 Util., 2019, 31, 135–142 CrossRef CAS; (b) M. Loipersberger, D. G. A. Cabral, D. B. K. Chu and M. Head-Gordon, Mechanistic Insights into Co and Fe Quaterpyridine-Based CO2 Reduction Catalysts: Metal-Ligand Orbital Interaction as the Key Driving Force for Distinct Pathways, J. Am. Chem. Soc., 2021, 143, 744–763 CrossRef CAS PubMed; (c) K. Kosugi, M. Kondo and S. Masaoka, Quick and Easy Method to Dramatically Improve the Electrochemical CO2 Reduction Activity of an Iron Porphyrin Complex, Angew. Chem., Int. Ed., 2021, 60, 22070–22074 CrossRef CAS.
- (a) Y. T. Liu, D. H. Deng and X. H. Bao, Catalysis for Selected C1 Chemistry, Chem, 2020, 6, 2497–2514 CrossRef CAS; (b) Y. J. Zhao, L. L. Zheng, D. Jiang, W. Xia, X. T. Xu, Y. Yamauchi, J. P. Ge and J. Tang, Nanoengineering Metal-Organic Framework-Based Materials for Use in Electrochemical CO2 Reduction Reactions, Small, 2021, 17, 2006590 CrossRef CAS PubMed; (c) J. B. Jakobsen, M. H. Ronne, K. Daasbjerg and T. Skrydstrup, Are Amines the Holy Grail for Facilitating CO2 Reduction, Angew. Chem., Int. Ed., 2021, 60, 9174–9179 CrossRef CAS.
- (a) S. R. Foit, I. C. Vinke, L. G. J. de Haart and R. A. Eichel, Power-to-Syngas: An Enabling Technology for the Transition of the Energy System, Angew. Chem., Int. Ed., 2017, 56, 5402–5411 CrossRef CAS PubMed; (b) Q. He, D. B. Liu, J. H. Lee, Y. M. Liu, Z. H. Xie, S. Hwang, S. Kattel, L. Song and J. G. G. Chen, Electrochemical Conversion of CO2 to Syngas with Controllable CO/H2 Ratios over Co and Ni Single-Atom Catalysts, Angew. Chem., Int. Ed., 2020, 59, 3033–3037 CrossRef CAS PubMed; (c) M. Zhang, Z. Hu, L. Gu, Q. H. Zhang, L. H. Zhang, Q. Song, W. Zhou and S. Hu, Electrochemical conversion of CO2 to syngas with a wide range of CO/H2 ratio over Ni/Fe binary single-atom catalysts, Nano Res., 2020, 13, 3206–3211 CrossRef CAS.
- (a) S. Z. Xu and E. A. Carter, Theoretical Insights into Heterogeneous (Photo)electrochemical CO2 Reduction, Chem. Rev., 2019, 119, 6631–6669 CrossRef CAS PubMed; (b) J. Y. Chen and L. Wang, Effects of the Catalyst Dynamic Changes and Influence of the Reaction Environment on the Performance of Electrochemical CO2 Reduction, Adv. Mater., 2021, 2103900 Search PubMed; (c) S. Y. Liang, L. Huang, Y. S. Gao, Q. Wang and B. Liu, Electrochemical Reduction of CO2 to CO over Transition Metal/N-Doped Carbon Catalysts: The Active Sites and Reaction Mechanism, Adv. Sci., 2021, 8, 2102886 CrossRef CAS PubMed.
- (a) Y. L. Jia, Z. Q. Xue, J. Yang, Q. L. Liu, J. H. Xian, Y. C. Zhong, Y. M. Sun, X. X. Zhang, Q. H. Liu, D. X. Yao and G. Q. Li, Tailoring the Electronic Structure of an Atomically Dispersed Zinc Electrocatalyst: Coordination Environment Regulation for High Selectivity Oxygen Reduction, Angew. Chem., Int. Ed., 2022, 61, e202110838 CAS; (b) R. R. Yun, T. H. Li, B. B. Zhang, L. He, S. J. Liu, C. Yu, Z. Chen and S. Z. Luo, Amino induced high-loading atomically dispersed Co sites on N-doped hollow carbon for efficient CO2 transformation, Chem. Commun., 2022, 58, 6602–6605 RSC; (c) L. L. Wan, X. L. Zhang, J. S. Cheng, R. Chen, L. X. Wu, J. W. Shi and J. S. Luo, Bimetallic Cu-Zn Catalysts for Electrochemical CO2 Reduction: Phase-Separated versus Core-Shell Distribution, ACS Catal., 2022, 12, 2741–2748 CrossRef CAS.
- (a) J. Albo, D. Vallejo, G. Beobide, O. Castillo, P. CastaÇo and A. Irabien, Copper-Based Metal–Organic Porous Materials for CO2 Electrocatalytic Reduction to Alcohols, ChemSusChem, 2017, 10, 1100–1109 CrossRef CAS; (b) L. Y. Ouyang, V. Noel, A. Courty, J. M. Campagne, A. Ouali and E. Vrancken, Copper Nanoparticles with a Tunable Size: Implications for Plasmonic Catalysis, ACS Appl. Nano Mater., 2022, 5(2), 2839–2847 CrossRef CAS.
- (a) M. R. Han, Y. N. Zhou, X. Zhou and W. Chu, Tunable Reactivity of MNi12 (M = Fe, Co, Cu, Zn) Nanoparticles Supported on Graphitic Carbon Nitride in Methanation, Acta Phys. – Chim. Sin., 2019, 35, 850–857 CAS; (b) A. Halder, C. Lenardi, J. Timoshenko, A. Mravak, B. Yang, L. K. Kolipaka, C. Piazzoni, S. Seifert, V. Bonacic-Koutecky, A. I. Frenkel, P. Milani and S. Vajda, CO2 Methanation on Cu-Cluster Decorated Zirconia Supports with Different Morphology: A Combined Experimental In Situ GIXANES/GISAXS, Ex Situ XPS and Theoretical DFT Study, ACS Catal., 2021, 11, 6210–6224 CrossRef CAS.
- (a) G. P. Gao, S. Bottle and A. J. Du, Understanding the activity and selectivity of single atom catalysts for hydrogen and oxygen evolution via ab initial study, Catal. Sci. Technol., 2018, 8, 996–1001 RSC; (b) Y. F. Wang, M. Jin, X. Zhang, C. J. Zhao, H. J. Wang, S. H. Li and Z. Y. Liu, Direct Conversion of Biomass into Compact Air Electrode with Atomically Dispersed Oxygen and Nitrogen Coordinated Copper Species for Flexible Zinc-Air Batteries, ACS Appl. Energy Mater., 2019, 2, 8659–8666 CrossRef CAS.
- (a) J. F. de Souza, M. S. Gularte, R. F. N. Quadrado, A. F. P. Biajoli and A. R. Fajardo, Copper species supported in polysaccharide-based materials: from preparation to application in catalysis, Catal. Rev., 2021, 1904543 Search PubMed; (b) I. Merino-Garciaa, J. Alboa, P. Krzywdab, G. Mulb and A. Irabien, Bimetallic Cu-based hollow fibre electrodes for CO2 electroreduction, Catal. Today, 2020, 346, 34–39 CrossRef.
- Y. Hisai, Q. B. Ma, T. Qureishy, T. Watanabe, T. Higo, T. Norby and Y. Sekine, Enhanced activity of catalysts on substrates with surface protonic current in an electrical field - a review, Chem. Commun., 2021, 57, 5737–5749 RSC.
- (a) M. Yang, J. F. Yu, X. Tong, X. T. Sun, H. Y. Xu and J. Sun, Flame-made Cu/ZrO2 catalysts with metastable phase and strengthened interactions for CO2 hydrogenation to methanol, Chem. Commun., 2021, 57, 7509–7512 RSC; (b) J. Lu, Y. Zhang, S. Wang and Z. Li, Preparation of highly dispersed CuO-ZnO-ZrO2 catalysts and their improved catalytic performance for hydrogenation of CO2, Can. J. Chem., 2022, 100, 387–395 CrossRef CAS.
- (a) T. Ghodselahi, M. A. Vesaghi, A. Shafiekhani, A. Baghizadeh and M. Lameii, XPS study of the Cu@Cu2O core-shell nanoparticles, Appl. Surf. Sci., 2008, 255, 2730–2734 CrossRef CAS; (b) G. U. Kulkarni and C. N. R. Rao, EXAFS and XPS investigations of Cu/ZnO catalysts and their interaction with CO and methanol, Top. Catal., 2003, 22, 183–189 CrossRef CAS.
- R. R. Yun, F. Y. Zhan, X. J. Wang, B. B. Zhang, T. Sheng, Z. F. Xin, J. J. Mao, S. J. Liu and B. S. Zheng, Design of Binary Cu-Fe Sites Coordinated with Nitrogen Dispersed in the Porous Carbon for Synergistic CO2 Electroreduction, Small, 2021, 17, 2006951 CrossRef CAS PubMed.
- A. Christensen and E. A. Carter, First-Principles Study of the Surfaces of Zirconia, Matter Mater. Phys., 1998, 58, 8050–8064 CrossRef CAS.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, Understanding Selectivity for the Electrochemical Reduction of Carbon Dioxide to Formic Acid and Carbon Monoxide on Metal Electrodes, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qi02288e |
| This journal is © the Partner Organisations 2023 |