
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile, green and scalable approach to fabricate hierarchical ZnAl-LDH for efficient removal of hexavalent chromium†
Tingting
Liu
a,
Meiqi
Zheng
a,
Kaiyue
Ji
d,
Xiaomeng
Xue
a,
Jiangrong
Yang
a,
Mingfei
Shao
a,
Haohong
Duan
 *bd and
Xianggui
Kong
*ac
*bd and
Xianggui
Kong
*ac
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing, 100029, China. E-mail: kongxg@mail.buct.edu.cn
bQingyuan Innovation Laboratory, Quanzhou, 362801, China
cQuzhou Institute for Innovation in Resource Chemical Engineering, Zhejiang, China
dDepartment of Chemistry, Tsinghua University, Beijing, China. E-mail: hhduan@mail.tsinghua.edu.cn
First published on 16th May 2023
Abstract
A convenient, green, and scalable production approach to cost-effective adsorbents with large adsorption capacity is exceedingly desirable yet challenging. Herein, a unique hierarchical zinc–aluminum layered double hydroxide (ZnAl-LDH-D) is obtained through a solid–liquid reaction with only zinc oxide (ZnO) and aluminum nitrate [Al(NO3)3] as the feedstocks. The synergistic activation involving hydrolysis of Al3+, etching of ZnO, and reconfiguration of LDH results in a fast-kinetic behavior (less than 1 min), and it is quite easy to produce at least 200 g in one batch in lab-scale. In the removal of CrVI, ZnAl-LDH-D can function steadily under different pH ranges (4–10) with a high adsorption capacity of 73.4 mg g−1 and a strong removal ability (from 10 mg L−1 to 3.3 μg L−1). Using a deuterated experiment and a series of characterization technologies, we clarified the removal mechanism of CrVI and revealed that the hydroxyl groups play a critical role in the removal process, including complexation, replacement, and reduction. Therefore, our work develops a new strategy for convenient scale-up production of ZnAl-LDH-D and provides guidance for exploring efficient adsorbents.
Introduction
The growing issue of water resource pollution by heavy metal contaminants has caused global concerns regarding severe adverse effects on environmental ecosystems and human health.1 While some of these elements are essential for biological functions, owing to persistent bioaccumulation in the body, they can induce life-threatening cancers after reaching an unacceptable level.2,3 Among them, hexavalent chromium (CrVI) is very toxic to biological systems and can induce lung and kidney cancer and bronchogenic carcinoma once present in the body at high doses.4,5 Given that, the World Health Organization (WHO) has established a maximum level of CrVI in drinking water at 50 μg L−1. Unfortunately, because of its intensive production (about 37.5 Mt per year in the world),6 widespread applications (e.g., electroplating, metal smelting, pigment production, etc.),7 and huge industrial effluent disposal without proper treatment,8 maintaining low levels of CrVI pollutants in water is a major challenge. For example, in Tamil Nadu, India, the CrVI concentration in surface water has exceeded 2000 μg L−1;2 and chromium is the second most abundant inorganic pollutant in the United States.2,9 In China, millions of tons of Cr-containing waste are discharged every year, resulting in serious environmental issues.10 Therefore, it is an urgent and compulsory task to remediate Cr(VI)-contaminated effluents to meet the stringent environmental discharge standards and achieve drinking-water quality.Adsorption technology has emerged as a strong candidate for removing heavy metals from solutions due to its flexibility in operation and low energy and maintenance costs.11–13 The greatest challenge inherent to adsorption is developing low-cost adsorbents with high efficiency. Natural adsorbents have the significant advantage of economic efficiency, including lignite coke, bentonite, vermiculite, and zeolites.14–16 However, low selectivity and capacity limit their widespread application. To overcome the drawbacks of poor adsorption capacity, various chemosynthetic adsorbents have been synthesized to remove contaminants from water,17,18 among which the metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) have emerged as the most common promising adsorbents,19,20 demonstrating high sorption capacity and outstanding specific selectivity for heavy metal ions owing to their large surface area, abundant porous structure and high surface reaction affinity. Despite these advantages, the synthesis strategies for most chemosynthetic adsorbents are not designed for affordable application at scale because of costly raw materials and sophisticated preparations. To tackle the bottleneck, it is critical to explore a simple and green synthetic approach that can produce cost-effective adsorbents at scale.
As a representative of two-dimensional anionic clays, layered double hydroxides (LDHs) consist of positively charged brucite-like layers and anions intercalated in interlayer galleries.21,22 Owing to their unique layered structure and tunable chemical composition, LDHs have shown great application potential in catalysis, optics, energy storage, and biological medicine.23–25 In the field of environmental remediation, LDHs have been recognized as ideal candidates for removing heavy metal ions from wastewater through surface adsorption, interlayer exchange, precipitation, and isomorphic substitution.26–28 For instance, Ma et al. successfully prepared MoS42−-LDH as an excellent adsorbent for Cu2+, Pb2+, Hg2+, UO22+, HAsO32−, and CrO42− through forming anionic complexes and/or intercalation into the gallery of LDHs.29,30 Our previous work demonstrated that ZnFe-LDH displays good immobilization performance toward HAsO32−, and that Cd2+ can be quickly transformed into CdAl-LDH using CaAl-LDH, exhibiting excellent remediation performance for heavy metal contaminated soil.31,32 Nevertheless, despite these advantages, it is still a big challenge to produce LDHs using a simple, efficient and green approach. For example, co-precipitation is the most common method to produce LDHs with metal salts and sodium hydroxide as raw materials, the significant disadvantage of this technique is the great demand for deionized water, which is used as an eluent to remove the by-products such as NaCl, Na2CO3, and Na2SO4. Besides, aging at a certain temperature and time is needed for LDHs synthesized by co-precipitation, resulting in high costs and inefficiency for large-scale production.
To overcome the aforementioned challenges, in this work, for the first time, we designed a convenient, green, and scalable synthetic strategy for constructing a zinc–aluminum layered double hydroxide with a 3D structure (denoted as ZnAl-LDH-D) with fast kinetics. Here, the raw materials are simply zinc oxide (ZnO) and aluminum nitrate [Al(NO3)3], preventing abundant by-products in the synthesis process. Benefiting from the convenience and simplicity of this strategy, we have completed the synthesis of ZnAl-LDH-D at about 200 g for one batch in lab-scale. The growth mechanism mainly depends on the synergistic effects of hydrolysis, dissolution, and reconfiguration. Moreover, as an adsorbent, the as-synthesized ZnAl-LDH-D exhibits excellent fast adsorption kinetics as well as high adsorption capacity for Cr(VI) with a broad pH range of 4–10, which is an indispensable advantage in comparison with other sorbents in practical applications. Furthermore, the effect of hydroxyl groups on the adsorption performance was thoroughly investigated through a series of experiments and characterization.
Results and discussion
Structural characterization
A schematic diagram of ZnAl-LDH-D is shown in Fig. 1a. Here, a suspension of commercial ZnO and solution of Al(NO3)3 were simultaneously pumped into the colloid mill reactor, rotating at 2000 rpm at room temperature. Subsequently, the mixture slurry was aged at 25 °C for 2 hours, followed by washing with deionized water and drying in an oven, after which ZnAl-LDH-D was finally obtained. In contrast to the conventional coprecipitation and hydrothermal synthesis, as shown in Video S1 in the ESI,† this solid–liquid reaction is very convenient and promising for large-scale production. X-ray diffraction (XRD) measurements were first employed to investigate the crystal structure of the obtained samples to certify the feasibility of this new construction strategy. As shown in the XRD patterns (Fig. 1b), the main diffraction peaks at 9.9, 19.9, 34.2, and 60.6° can be well indexed as the characteristic (003), (006), (009), and (110) facets of LDHs, respectively. The interplanar spacing in the thickness direction is 0.88 nm according to the facet of (003), indicating nitrate-intercalated LDHs. The presence of nitrate as interlayer anions can also be confirmed by Fourier transform infrared (FTIR) spectroscopy analysis (Fig. S1, ESI†). The broad band appearing at 3467 cm−1 is attributed to the O–H stretching vibration of adsorbed water and surface hydroxyl groups. The sharp and intense band at 1377 cm−1 corresponds to the N–O stretching vibration of NO3− species intercalated into the LDHs. Amorphous aluminum hydroxide is often erroneously assigned to the paramagnetic LDH phase. Therefore, solid-state 27Al magic angle spinning nuclear magnetic resonance spectroscopy (27Al MAS NMR) was adopted to investigate the local environment of Al species in ZnAl-LDH-D. As shown in Fig. 1c, the 27Al MAS NMR signal of ZnAl-LDH-D displayed only one symmetric resonance centered at 8.7 ppm, demonstrating that all of the Al in the layer has octahedral geometry corresponding to AlO6 sites. The morphology and structure of ZnAl-LDH-D were identified using Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM). Interestingly, unlike normal LDHs with independent plate-like structures, the SEM images show that the synthesized ZnAl-LDH-D exhibits a three-dimensional structure consisting of numerous nanoflakes intercrossed with each other (Fig. 1d, e and Fig. S2, ESI†). Furthermore, as displayed in the magnified high-resolution TEM (HRTEM) images (Fig. 1f), it is clear that the interconnected nanoflakes have a transparent nature, indicating an ultrathin thickness of the nanoflakes. The HRTEM images further revealed that ZnAl-LDH-D has a hierarchical structure with a solid core and a loose-density shell (Fig. 1f). The corresponding energy-dispersive X-ray (EDX) mapping of the Al and N elements demonstrated that Al and N were uniformly distributed on the edge and surface of ZnAl-LDH-D, and the Zn element was uniformly distributed throughout ZnAl-LDH-D (Fig. 1g). Fig. S3 (ESI†) shows the N2 adsorption–desorption isotherm and the pore size distribution of ZnAl-LDH-D. The isotherm exhibits a typical type IV shape with a H2-type hysteresis loop (P/P0 > 0.4),33 which suggested the existence of a mesoporous structure with relatively highly uniform channel-like pores. The Brunauer–Emmett–Teller (BET) specific surface area of ZnAl-LDH-D was calculated to be 66.8 m2 g−1, and the pore size distribution of ZnAl-LDH-D displays a single mode centered at 7.9 nm in the range of 2–10 nm. The corresponding pore volume is 0.13 cm3 g−1.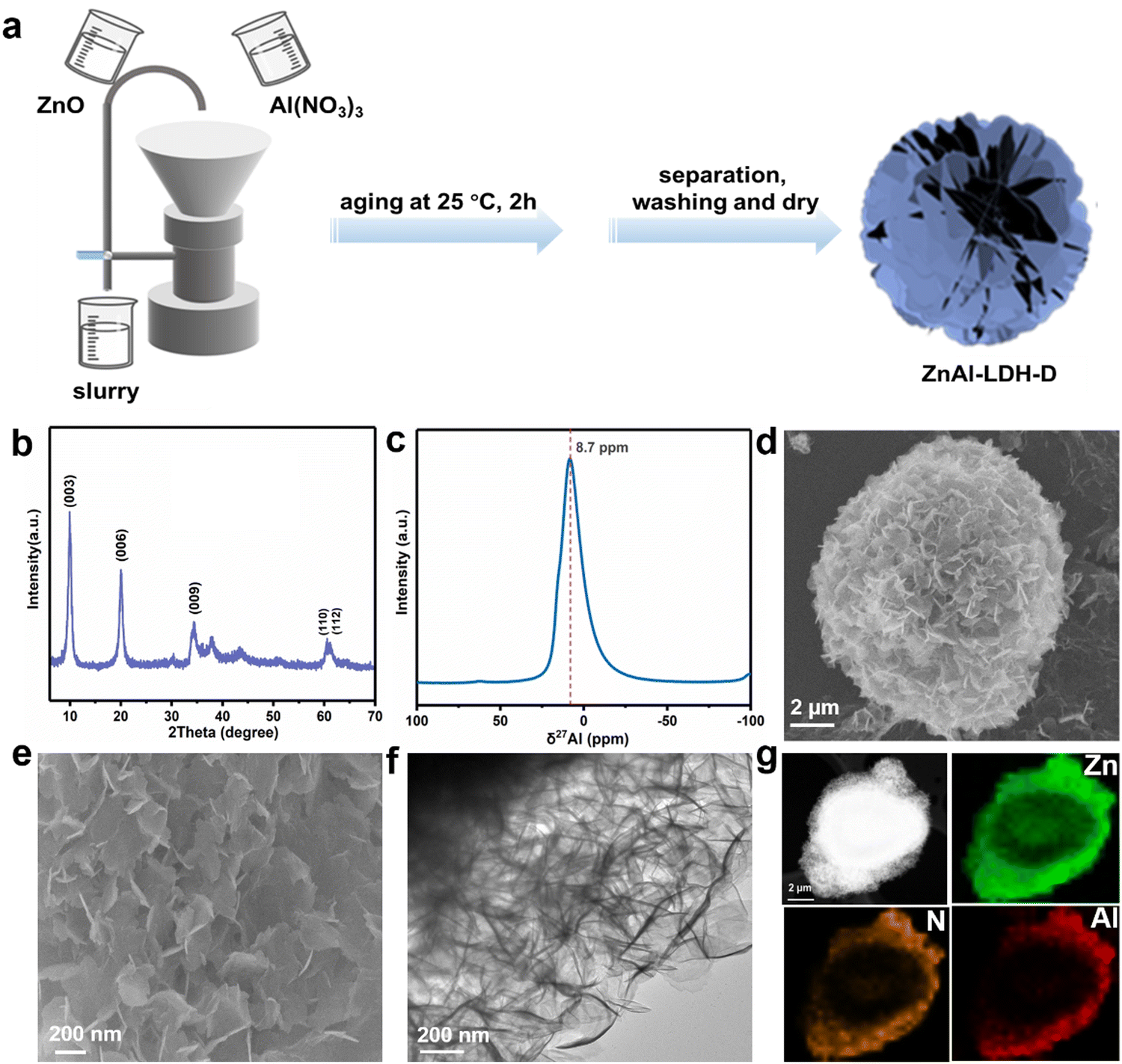 | ||
| Fig. 1 (a) Schematic illustration for the production of ZnAl-LDH-D; (b) XRD pattern of ZnAl-LDH-D; (c) 27Al MAS NMR spectrum of ZnAl-LDH-D; (d) and (e) SEM images of ZnAl-LDH-D; (f) and (g) TEM image and corresponding EDX mappings of ZnAl-LDH-D. | ||
To investigate the formation process of ZnAl-LDH-D, samples obtained at different reaction times were studied using XRD. As shown in Fig. 2a, despite the coexistence of unreacted ZnO, it is unambiguously clear that the crystalline ZnAl-LDH-D was successfully produced within 1 min, exhibiting an extremely fast synthesis rate. When the reaction time is extended to 5 min, the ZnO phase disappears. Instead, a pure crystal structure of ZnAl-LDH-D is formed. When the reaction time is prolonged to 2 h, no noticeable change in diffraction peaks occurs except for a higher crystallinity.
 | ||
| Fig. 2 (a) XRD patterns of samples obtained at different reaction times; (b) SEM image of the ZnO precursor; (c–f) SEM images of the samples obtained at different reaction times (t = 1 min, 5 min, 30 min, and 2 hours, respectively). | ||
Meanwhile, the morphological evolution of ZnAl-LDH-D at different stages was further examined using SEM. Fig. 2b and Fig. S4 (ESI†) show the SEM images of the original ZnO, revealing aggregated nanoparticles with approximate sizes of 30–300 nm. After being reacted with the Al(NO3)3 solution for 1 min (Fig. 2c), flower-like clusters built from nanosheets cover the surface of ZnO. When the reaction time is increased from 5 min to 2 h (Fig. 2d–f), the interconnected nanosheets become larger, forming an architectural network structure composed of ultrathin nanoflakes.
Due to the tremendous reaction rate, defining the exact formation mechanism is challenging. Therefore, we propose a plausible formation mechanism that is illustrated in Fig. 3. To verify the hypothesis, the intermediate sample was instantly separated from the reaction system in the early reaction stage (reaction time of less than 10 seconds) and dispersed into extensive anhydrous ethanol. The samples were then collected by centrifugation. As shown in the XRD pattern (Fig. S5, ESI†), besides the typical diffraction peaks of LDHs and ZnO, the broad diffraction peaks present at 18.2° and 22.2° can be attributed to Al(OH)3 (PDF#33-0018), indicating the generation of Al(OH)3 in the early reaction stage. The formation of Al(OH)3 can be explained by the hydrolysis reaction of Al3+ in the reaction system with a pH of 5.6, which results from the mixture of the ZnO suspension (pH = 7.9) and Al(NO3)3 solution (pH = 2.7). Under this condition, the Al3+ easily hydrolyzes to generate Al(OH)3 colloid and H+ followed the chemical reaction of Al3+ + 3H2O → Al(OH)3 + 3H+. This reaction preferentially occurs around the ZnO nanoparticles, and the generated Al(OH)3 colloid covers the surface of the ZnO substrate, resulting in ZnO@Al(OH)3. At the same time, Zn2+ is formed through the etching reaction of ZnO + 2H+ → Zn2+ + H2O. Because of the ultralow solubility product constants of ZnAl-LDH, it is thermodynamically favorable to generate ZnAl-LDH via the reaction of Zn2+ + Al(OH)3 + NO3− + OH− → ZnAl(OH)3NO3, which is similar to previous works on the synthesis of LDHs with metal ions and hydroxide as raw materials.34,35 The consumption of H+ and Zn2+ accelerates the hydrolysis reaction of Al3+ and the etching reaction of ZnO. As a result of combining hydrolysis, dissolution, and reconfiguration, ZnAl-LDH with a 3D structure can finally be formed.
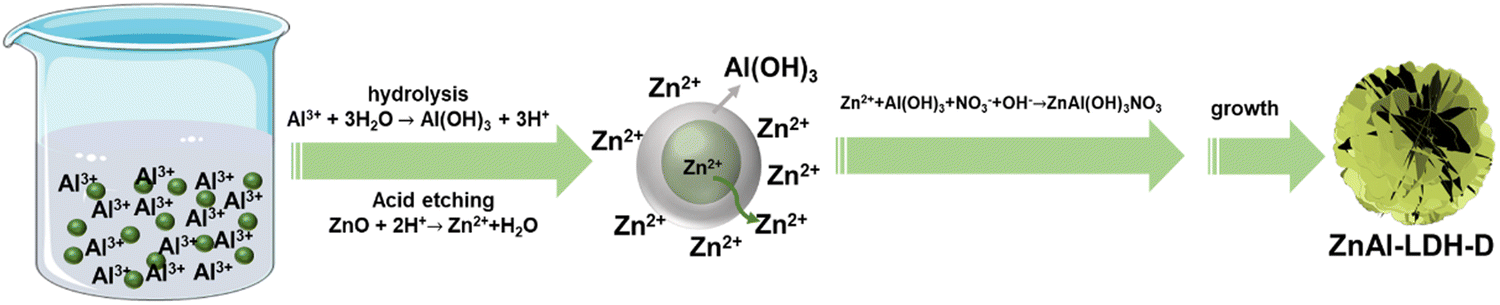 | ||
| Fig. 3 Growth mechanism of ZnAl-LDH-D. | ||
Adsorption performance
To evaluate the removal performance of ZnAl-LDH-D toward CrVI from water, sorption kinetics was first used to estimate the sorption efficiency. As shown in Fig. 4a, when the initial concentration of CrVI is 50 mg L−1, an adsorption equilibrium of ZnAl-LDH-D with CrVI with a dosage of 1 g L−1 can be reached within 60 min with 99% removal efficiency. This reveals a remarkable advantage in adsorption rate compared to numerous adsorbents (Table S1, ESI†), which ordinarily range from several hours to days. Besides adsorption efficiency, the adsorption capacity is another important metric for adsorbents. Fig. 4b shows the adsorption capacity of ZnAl-LDH-D toward CrVI at different initial concentrations. When the initial concentration of CrVI is 200 mg L−1, the saturated adsorption capacity is as high as 73.4 mg g−1. Furthermore, the classic Langmuir and Freundlich models were adopted to evaluate the adsorption behavior. As shown in Fig. S6 and Table S2 (ESI†), in comparison with the Freundlich model (R2 = 0.6506), the adsorption behavior of ZnAl-LDH-D toward CrVI fits the Langmuir model better (R2 = 0.9998), suggesting that the sorption behavior follows monolayer adsorption with a calculated maximum sorption capacity of 69.5 mg g−1. This is consistent with experimental data. The adsorption capacity of ZnAl-LDH-D toward CrVI is much higher than that of other adsorbents reported previously, including MOFs, resins, and silica-based materials (Table S1, ESI†).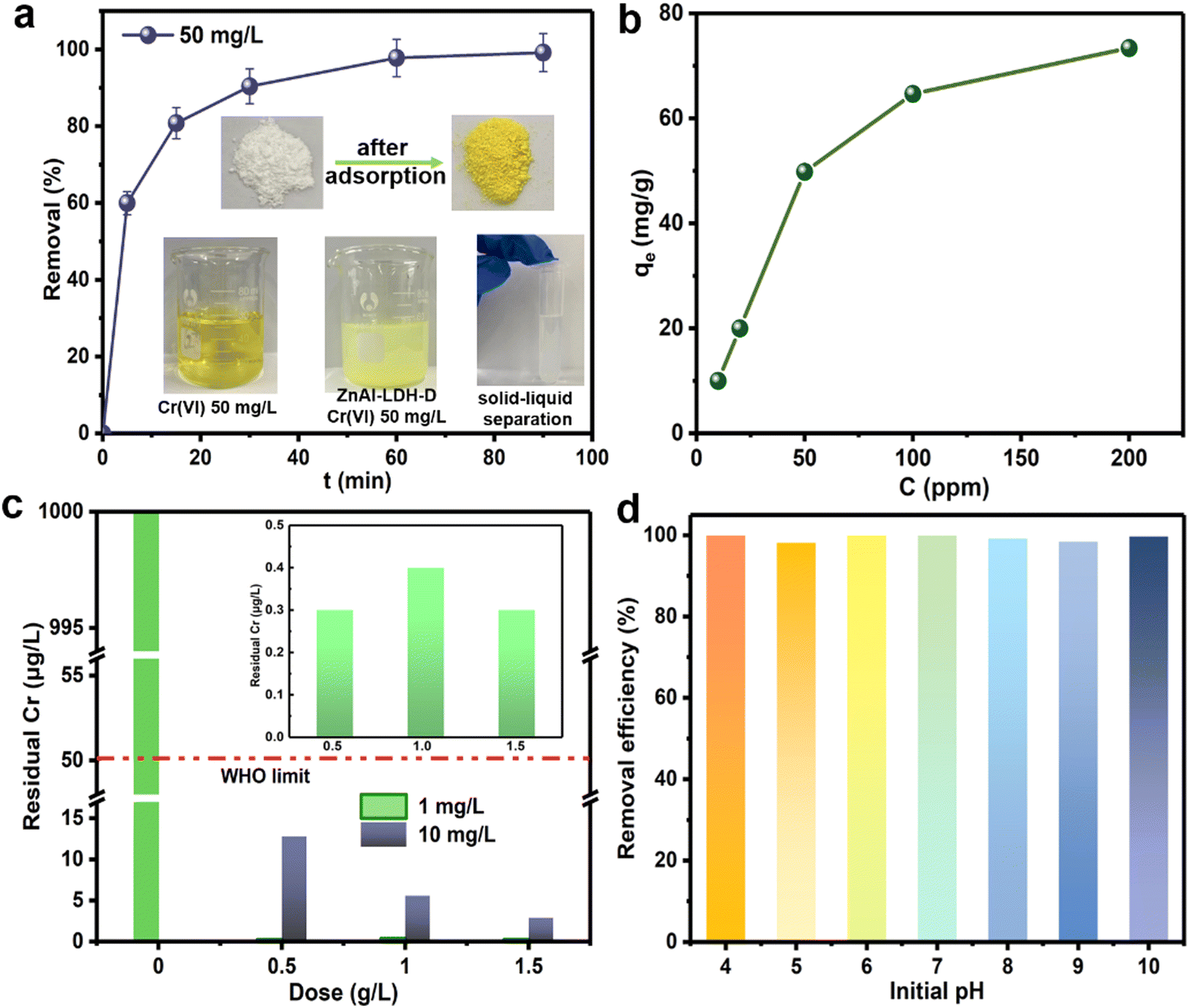 | ||
| Fig. 4 (a) Sorption kinetics of CrVI by ZnAl-LDH-D (dose = 0.5 g L−1, C0 = 50 mg L−1); (b) sorption isotherms of CrVI by ZnAl-LDH-D (dose = 0.5 g L−1, C0 =10–200 mg L−1, t = 2 h); (c) deep adsorption performance of ZnAl-LDH-D toward CrVI with a low concentration (t = 2 h); (d) influence of the initial pH on the adsorption performance (dose = 0.5 g L−1, C0 = 50 mg L−1, t = 2 h). | ||
Although enormous amounts of adsorbents have been reported for the removal of CrVI from wastewater, reducing the moderate or trace concentrations of total Cr below the maximum allowable standard of 50 μg L−1 still poses a big challenge. Excitingly, when an initial CrVI concentration of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μg L−1 is used as a target, the residual concentration of total Cr is significantly reduced to 2.9 μg L−1 after adsorption by ZnAl-LDH-D with a dosage of 1.5 g L−1 (Fig. 4c). This is well below the upper level recommended by the WHO (50 μg L−1). Even if the dosage of ZnAl-LDH-D decreases to 0.5 g L−1, a residual total Cr concentration of 12.8 μg L−1 is obtained. Furthermore, when the initial concentration of CrVI further decreases to 1000 μg L−1, treatment with ZnAl-LDH-D with a dosage of 0.5 g L−1 leads to a residual total Cr concentration of 0.3 μg L−1, suggesting an excellent deep adsorption ability toward CrVI.
000 μg L−1 is used as a target, the residual concentration of total Cr is significantly reduced to 2.9 μg L−1 after adsorption by ZnAl-LDH-D with a dosage of 1.5 g L−1 (Fig. 4c). This is well below the upper level recommended by the WHO (50 μg L−1). Even if the dosage of ZnAl-LDH-D decreases to 0.5 g L−1, a residual total Cr concentration of 12.8 μg L−1 is obtained. Furthermore, when the initial concentration of CrVI further decreases to 1000 μg L−1, treatment with ZnAl-LDH-D with a dosage of 0.5 g L−1 leads to a residual total Cr concentration of 0.3 μg L−1, suggesting an excellent deep adsorption ability toward CrVI.
It has been demonstrated that the presence speciation of CrVI ions in solution mainly depends on the pH value (e.g., H2CrO4, HCrO4−, Cr2O72−, and CrO42−).36,37 Given that, the optimum adsorption pH of many adsorbents usually ranges from 3 to 5.38 By comparison, Fig. 4d shows the influence of the initial pH on the adsorption performance of ZnAl-LDH-D toward CrVI. No difference in removal efficiency over a wide pH range (4–10) was observed, providing an apparent superiority compared to other adsorbents in practical applications. The possible reason for this can be explained as follows: according to the FTIR result (Fig. S7, ESI†), ZnAl-LDH-D possesses many more hydroxyl groups than that of ZnAl-LDH synthesized using the coprecipitation method, which play a significant role in the removal of Cr through complexation, replacement, and so on (detailed discussion in the next section). Furthermore, the abundant hydroxyl groups help to prolong the buffering effect of LDHs in a wide pH range. On the other hand, as shown in Fig. S8 (ESI†), the zeta potential of ZnAl-LDH-D was measured to be a positive value in the pH range of 4–10, and the value decreased slowly as pH increased. Therefore, with the combined action of the abundant hydroxyl groups and the relatively stable zeta potential, ZnAl-LDH-D exhibited a high adsorption capacity in a wide pH range.
In practical applications, owing to the coexistence of competing anions in wastewater, it is essential to evaluate the selectivity of ZnAl-LDH-D toward CrVI. As displayed in Fig. S9a (ESI†), the adsorption efficiency of ZnAl-LDH-D toward CrVI was still higher than 95% in the presence of CO32−, SO42−, NO3−, and Cl−. Furthermore, the adsorption performance of ZnAl-LDH-D toward CrVI in the presence of K+, Na+, Mg2+, Cd2+ and Pb2+ was also evaluated, in which the mass ratios of Cr to coexisting ions was set to 1:5. The experimental results displayed in Fig. S9b (ESI†) show that the influence of K+, Mg2+, Cd2+ and Pb2+ on the CrVI adsorption capacity of ZnAl-LDH-D was negligible, but the presence of Na+ reduced the sorption capacity of ZnAl-LDH-D toward CrVI, decreasing the removal efficiency from 99.6% to 95.1%. The recyclability of ZnAl-LDH-D was further investigated with Na2CO3 solution as eluent (0.5 mol L−1), in which the initial concentration of CrVI was 0.02 mg L−1. However, due to the strong binding ability between the adsorbed Cr anions and ZnAl-LDH-D, the complete desorption of Cr anions could not be achieved in the regeneration process. After the 4th cycle of reuse, the adsorption efficiency of ZnAl-LDH-D toward CrVI decreased to 53.7% (Fig. S10, ESI†). The investigation of the reusability of ZnAl-LDH-D is ongoing.
Adsorption mechanism analysis
To better understand the adsorption mechanisms leading to CrVI capture in ZnAl-LDH-D, firstly, two classic kinetic models, pseudo-second-order and pseudo-first-order model, were adopted to investigate the uptake behavior. The kinetic parameters and corresponding fitting results are displayed in Table S3 and Fig. S11 (ESI†). The pseudo-second-order kinetic equation model exhibits an excellent fit coefficient (R2 > 0.999), indicating that chemisorption is the predominant sorption behavior in the adsorption process of CrVI by ZnAl-LDH-D.39 To clarify the adsorption sites, we recorded the XRD patterns of samples after the adsorption of CrVI, as shown in Fig. 5a. All the diffraction peaks reveal a characteristic LDH structure. However, the interplanar spacing decreases from 0.88 nm to 0.75 nm, derived from (003), which is identical to the carbonate-intercalated LDHs.40 Additionally, in the Fourier transform infrared (FTIR) spectra, the characteristic band at 1367 cm−1 can be attributed to the stretching vibrations of CO32−, confirming the formation of carbonate-intercalated LDHs after adsorption (Fig. S12, ESI†). Furthermore, according to the results of previous works,41–43d003 should increase to > 0.9 nm after intercalation of CrVI species into the gallery of LDHs. Therefore, it can be concluded that the surface and edge of ZnAl-LDH-D are the main adsorption sites for CrVI species. Fig. 5b and c confirm that the 3D structure of ZnAl-LDH-D is maintained after the adsorption of CrVI. The EDS mapping images further confirm the presence and uniform distribution of the Cr element throughout the ZnAl-LDH-D nanosheets (Fig. 5d).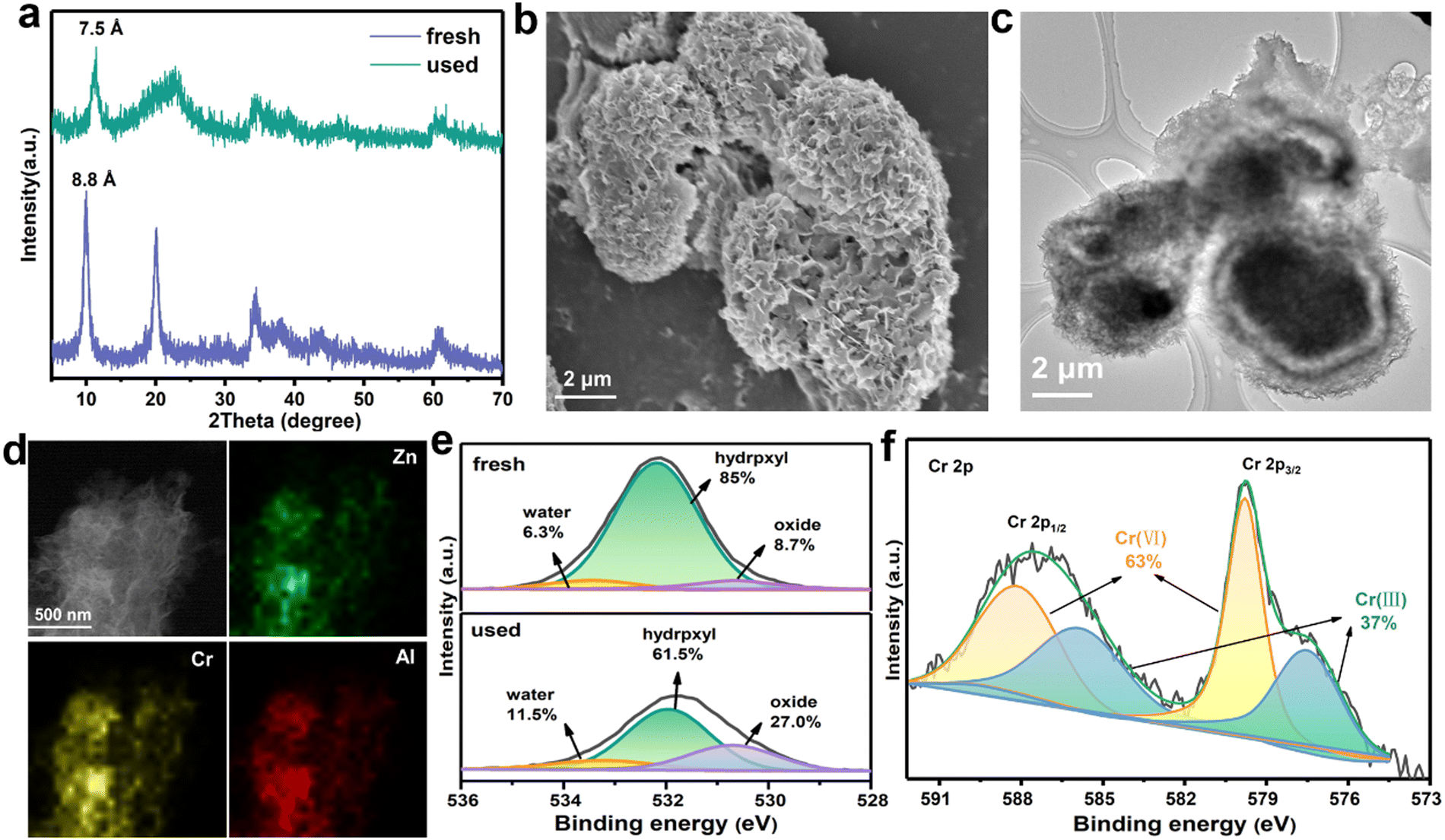 | ||
| Fig. 5 (a) XRD patterns of ZnAl-LDH-D before and after adsorption of CrVI; (b) SEM image of ZnAl-LDH-D after adsorption of CrVI; (c) and (d) TEM image and the EDS mapping of ZnAl-LDH-D after adsorption of CrVI; (e) high-resolution XPS O 1s spectra of ZnAl-LDH-D before and after adsorption of CrVI; (f) high-resolution XPS Cr 2p spectra of ZnAl-LDH-D after adsorption of CrVI. | ||
To obtain more detailed information on the adsorption behavior, surface-sensitive X-ray photoelectron spectroscopy (XPS) was introduced to elucidate the surface chemical status of ZnAl-LDH-D before and after the adsorption of CrVI. As a result, for the individual XPS spectra of O 1s (Fig. 5e), the peak can be deconvoluted into three components, where peaks at 530.6, 532.2, and 533.3 eV correspond to the lattice oxygen (O22−), hydroxyl groups at the surface of LDHs (OH−) and water molecules, respectively.44 Compared with the initial sample of ZnAl-LDH-D, the relative area ratios of hydroxyl groups are drastically reduced from 85% to 61.5%. In comparison, the area ratio of the lattice oxygen drastically increases from 8.7 to 27% after adsorption. Furthermore, the band energy of O 1s negatively shifts by 0.25eV, indicating a chemical bond formation between Cr, O, and the metals in the layer of ZnAl-LDH-D. All the above results suggested that a proportion of hydroxyl groups located on the surface of ZnAl-LDH-D is substituted by Cr species during the adsorption process. This phenomenon is consistent with the FTIR results (Fig. S12, ESI†), in which a new vibration of the Cr–O band can be found at 880 cm−1 after adsorption,45 and the bands for –OH at 1635 and 3500 cm−1 are clearly weakened.
To further certify the above suggestion, we synthesized deuterated ZnAl-LDH-D with D2O instead of H2O in the synthesis process. As shown in Fig. S13 (ESI†), all reflections of deuterated ZnAl-LDH-D are well-indexed to the typical hydrotalcite-like structure and are very similar to that of ZnAl-LDH-D. If the surface –OD groups are replaced by Cr species in the adsorption process, the free state of the –OD groups in solution transforms into –OH groups, resulting in a decrease of hydrogen content in solution, which can be checked by 1H NMR. Fig. S14 (ESI†) displays the 1H NMR signal of the Cr-containing solution before and after adsorption with deuterated ZnAl-LDH-D as the adsorbent. It is evident that the integrated area of hydrogen decreases from 1071.53 to 435.51. However, no apparent change in the integrated area of hydrogen in the 1H NMR spectra can be seen if the deuterated ZnAl-LDH-D is dispersed in deionized water under the same conditions without the presence of CrVI. Combining the above results, it is rational to assume that a portion of the surface –OH groups is replaced by CrVI species in the sorption process.
Furthermore, as shown in the XPS survey spectrum of ZnAl-LDH-D obtained after adsorption (Fig. S15, ESI†), new peaks that can be attributed to the presence of the element Cr appear at ∼580 and ∼48 eV, indicating the adsorption of Cr species over the surface of ZnAl-LDH-D. Based on the high-resolution XPS spectrum of Cr 2p (Fig. 5f), the peaks of Cr 2p3/2 (579.9 eV) and Cr 2p1/2 (588.3 eV) represent the typical features of CrVI.46 Amazingly, the visible signals at 585.9 and 577.5 eV are in good agreement with the characteristic peaks of CrIII with a percentage of 37%,47 suggesting that CrVI has been partially reduced to CrIII during the adsorption process. The presence of CrIII can also be verified by UV-vis spectral technology; as shown in Fig. S16 (ESI†), adsorption peaks appear at ∼430 nm and ∼640 nm for the used ZnAl-LDH-D, which can be attributed to the typical features of CrIII.48 According to previous reports, the CrVI species could be effectively reduced into CrIII, CrIV, and CrV species by hydroxyl groups.49,50 Therefore, we propose that the CrVI is reduced to CrIII by the plentiful hydroxyl groups on ZnAl-LDH-D, in which the O atom with more electronegativity donates electrons to CrVI. The XPS O 1s spectrum further certified the decrease of hydroxyl groups and increase of surface water molecules after the adsorption process (Fig. 5e). A similar result can also be found in previously reported work.51 In addition, with the exception of C 1s, the peaks of O 1s, Zn 2p, Al 2s, and Al 2p shift to lower binding energies after the adsorption of CrVI (Fig. 5c and Fig. S17, ESI†), suggesting that the local chemical environment of O, Zn, and Al in ZnAl-LDH-D has changed. Furthermore, the XPS analyses of used ZnAl-LDH-D displayed the Cr 2p2/3 and Cr 2p1/2 peaks at 577.2 and 586.3 eV, respectively (Fig. 5d). This is consistent with CrIII-based LDHs.52,53 Therefore, an isomorphic substitution of partial Al3+ by reduced CrIII in the sorption process is possible because of the very close ionic radius of Al3+ (0.054 nm) and CrIII (0.052 nm). Additionally, the replaced Al3+ may be transformed into amorphous Al(OH)3 at a pH of 5.1 in the adsorption system. This can be confirmed by the XRD pattern (Fig. 5a), in which the broad peak appearing at 21° represents the characteristic feature of amorphous Al(OH)3.54,55 Therefore, based on the above results, it is reasonable to propose that the hydroxyl groups play a critical role in the sorption performance of ZnAl-LDH-D toward CrVIvia complexation, replacement, and reduction.
Conclusions
In summary, we demonstrated a convenient yet highly efficient synthesis strategy for ZnAl-LDH with a hierarchical structure. According to the analysis, we can conclude that the surface hydroxyl groups of ZnAl-LDH-D play an essential role in the sorption of CrVI ions through replacement, complexation, and reduction. Due to its environmentally friendly and scalable features, the lab-scale synthesis of ZnAl-LDH-D can be effortlessly achieved with 200 g obtained in one batch. As an adsorbent for CrVI-contaminated water, the resulting ZnAl-LDH-D can effectively reduce moderate and trace concentrations of CrVI ions to drinkable levels with high adsorption capacity and fast kinetics, which are comparable with most state-of-the-art adsorbents. We expect that this result will provide a practicable strategy to scalably produce ZnAl-LDH and thus lead to broad applications, not merely as an effective adsorbent enabling the regeneration of CrVI-contaminated water.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Tingting Liu: methodology, investigation, writing–original draft. Meiqi Zheng: methodology, validation. Kaiyue Ji: investigation. Xiaomeng Xue: investigation. Jiangrong Yang: investigation. Mingfei Shao: conceptualization. Haohong Duan: conceptualization, supervision. Xianggui Kong: conceptualization, resources, supervision, writing–review & editing, funding acquisition.Conflicts of interest
The authors declare no competing financial or non-financial interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21978023, 22288102, 21838007, 21991102); Major Program of Qingyuan Innovation Laboratory (001220005). The authors appreciate Prof. Xue Duan for his constructive suggestions.References
- Y. Fei and Y. Hu, J. Mater. Chem. A, 2022, 10, 1047–1085 RSC
.
- C. M. Stern, T. O. Jegede, V. A. Hulse and N. Elgrishi, Chem. Soc. Rev., 2021, 50, 1642–1667 RSC
- L. Wang, C. Shi, L. Wang, L. Pan, X. Zhang and J. Zou, Nanoscale, 2020, 12, 4790–4815 RSC
- X. Zhang, F. Tian, L. Qiu, M. Gao, W. Yang, Y. Liu and Y. Yu, J. Mater. Chem. A, 2021, 9, 10297–10303 RSC
- K. Tan, S. Morikawa, K. R. Phillips, N. Ozbek and T. A. Hatton, ACS Appl. Mater. Interfaces, 2022, 14, 8974–8983 CrossRef CAS PubMed
- D. Hou, D. O’Connor, A. D. Igalavithana, D. S. Alessi, J. Luo, D. C. W. Tsang, D. L. Sparks, Y. Yamauchi, J. Rinklebe and Y. S. Ok, Nat. Rev. Earth Env., 2020, 1, 366–381, DOI:10.1038/s43017-020-0061-y
- S. Dutta, S. K. Srivastava and A. K. Gupta, Mater. Adv., 2021, 2, 2431–2443 RSC
- W. Yuan, W. Xu, Z. Zhang, X. Wang, Q. Zhang, J. Bai and J. Wang, Chemosphere, 2019, 227, 657–661 CrossRef CAS PubMed
-
National Research Council, Environmental Epidemiology, Volume 1: Public Health and Hazardous Wastes, National Academies Press, Washington, D. C, 1991, DOI:10.17226/1802
- X. Mo, X. Liu, J. Chen, S. Zhu, W. Xu, K. Tan, Q. Wang, Z. Lin and W. Liu, Environ. Sci.: Nano, 2022, 9, 1617–1626 RSC
- I. Ali, Chem. Rev., 2012, 112, 5073–5091 CrossRef CAS PubMed
- P. Zhao, M. Jian, Q. Zhang, R. Xu, R. Liu, X. Zhang and H. Liu, J. Mater. Chem. A, 2019, 7, 16598–16621 RSC
- L. Wang, C. Shi, L. Wang, L. Pan, X. Zhang and J. J. Zou, Nanoscale, 2020, 12, 4790–4815 RSC
- S. S. Gupta and K. G. Bhattacharyy, Phys. Chem. Chem. Phys., 2012, 14, 6698–6723 RSC
- X. Fang, W. Yuan, Y. Xiong and X. Qiu, Colloids Surf., A, 2022, 641, 128524 CrossRef CAS
- S. S. Guptaa and K. G. Bhattacharyya, RSC Adv., 2014, 4, 28537–28586 RSC
- X. Liu, R. Ma, X. Wang, Y. Ma, Y. Yang, L. Zhuang, S. Zhang, R. Jehan, J. Chen and X. Wang, Environ. Pollut., 2019, 252, 62–73 CrossRef CAS PubMed
- J. Li, X. Wang, G. Zhao, C. Chen, Z. Chai, A. Alsaedi, T. Hayat and X. Wang, Chem. Soc. Rev., 2018, 47, 2322–2356 RSC
- Y. Li, T. Huang, X. Liu, Z. Chen, H. Yang and X. Wang, Sep. Purif. Technol., 2023, 314, 123615 CrossRef CAS
- H. Gu, X. Liu, S. Wang, Z. Chen, H. Yang, B. Hu, C. Shen and X. Wang, Rev. Environ. Contam. Toxicol., 2022, 260, 23 CrossRef
- R. Gao, J. Zhu and D. Yan, Nanoscale, 2021, 13, 13593–13603 RSC
- M. Shao, R. Zhang, Z. Li, M. Wei, D. G. Evans and X. Duan, Chem. Commun., 2015, 51, 15880–15893 RSC
- W. Ye, X. Fang, X. Chen and D. Yan, Nanoscale, 2018, 10, 19484–19491 RSC
- S. Yang, W. Ye, D. Zhang, X. Fang and D. Yan, Inorg. Chem. Front., 2021, 8, 1762–1770 RSC
- R. Gao, D. Yan and X. Duan, Cell Rep. Phys. Sci., 2021, 2, 100536 CrossRef CAS
- S. Song, Q. Huang, G. Cheng, W. Wang, Z. Lu, R. Zhang, T. Wen, Y. Zhang, J. Wang and X. Wang, ACS Sustainable Chem. Eng., 2019, 7, 3475–3486 CrossRef CAS
- M. Zheng, F. Mao, X. Kong and X. Duan, Chem. J. Chin. Univ., 2022, 43, 20220456 Search PubMed
- F. Mao, P. Hao, X. Kong, X. Lei and X. Duan, Sci. Sin.: Chim., 2021, 51, 493–508 Search PubMed
- L. Ma, Q. Wang, S. M. Islam, Y. Liu, S. Ma and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 2858–2866 CrossRef CAS PubMed
- L. Ma, S. M. Islam, H. Liu, J. Zhao, G. Sun, H. Li, S. Ma and M. G. Kanatzidis, Chem. Mater., 2017, 29, 3274–3284 CrossRef CAS
- X. Kong, R. Ge, T. Liu, S. Xu, P. Hao, X. Zhao, Z. Li, X. Lei and H. Duan, Chem. Eng. J., 2021, 407, 127178 CrossRef CAS
- T. Liu, M. Zheng, P. Hao, K. Ji, M. Shao, H. Duan and X. Kong, J. Environ. Chem. Eng., 2023, 11, 109233 CrossRef CAS
- J. Zhang, J. Han, M. Wang and R. Guo, J. Mater. Chem. A, 2017, 5, 4058–4066 RSC
- S. S. C. Pushparaj, N. D. Jensen, C. Forano, G. J. Rees, J. Prevot, J. V. Hanna, D. B. Ravnsbæk, M. Bjerring and U. G. Nielsen, Inorg. Chem., 2016, 55, 9306–9315 CrossRef CAS PubMed
- S. Britto and P. V. Kamath, Inorg. Chem., 2010, 49, 11370–11377 CrossRef CAS PubMed
- M. K. Dinker and P. S. Kulkarni, J. Chem. Eng. Data, 2015, 60, 2521–2540 CrossRef CAS
- X. Y. Huang, G. Dognani, P. Hadi, M. Y. Yang, A. E. Job and B. S. Hsiao, ACS Sustainable Chem. Eng., 2020, 8, 4734–4744 CrossRef CAS
- K. Zhu, Y. Gao, X. Tan and C. Chen, ACS Sustainable Chem. Eng., 2016, 4, 4361–4369 CrossRef CAS
- R. S. Bangari, A. K. Singh, S. Namsani, J. K. Singh and N. Sinha, ACS Appl. Mater. Interfaces, 2019, 11, 19017–19028 CrossRef CAS PubMed
- R. M. M. Santos, J. Tronto, V. Brioisc and C. V. Santilli, J. Mater. Chem. A, 2017, 5, 9998–10009 RSC
- L. Yan, K. Yang, R. Shan, H. Yu and B. Du, RSC Adv., 2015, 5, 96495–96503 RSC
- X. Yu, T. Luo, Y. Jia, R. Xu, C. Gao, Y. Zhang, J. Liu and X. Huang, Nanoscale, 2012, 4, 3466–3474 RSC
- J. Li, H. Cui, X. Song, G. Zhang, X. Wang, Q. Song, N. Wei and J. Tian, RSC Adv., 2016, 6, 92402–92410 RSC
- T. Zhang, J. Wang, W. Zhang, C. Yang, L. Zhang, W. Zhu, J. Sun, G. Li, T. Li and J. Wang, J. Mater. Chem. A, 2019, 7, 2845–2854 RSC
- L. Yao, Y. Hu, Y. Zou, Z. Ji, S. Hu, C. Wang, P. Zhang, H. Yang, Z. Shen, D. Tang, S. Zhang, G. Zhao and X. Wang, Environ. Sci. Technol., 2022, 56, 14030–14037 CrossRef CAS PubMed
- L. Aboutorabi, A. Morsali, E. Tahmasebi and O. Büyükgüngor, Inorg. Chem., 2016, 55, 5507–5513 CrossRef CAS PubMed
- T. Wang, L. Zhang, C. Li, W. Yang, T. Song, C. Tang, Y. Meng, S. Dai, H. Wang, L. Chai and J. Luo, Environ. Sci. Technol., 2015, 49, 5654–5662 CrossRef CAS PubMed
- J. Ding, J. Ming, D. Lu, W. Wu, M. Liu, X. Zhao, C. Li, M. Yang and P. Fang, Catal. Sci. Technol., 2017, 7, 2283–2297 RSC
- L. Yao, Z. Shen, Z. Ji, Y. Hu, D. Tang, G. Zhao and X. Wang, Sci. Bull., 2022, 67, 2154–2157 CrossRef CAS PubMed
- L. Yao, Y. Hu, Y. Zou, Z. Ji, S. Hu, C. Wang, P. Zhang, H. Yang, Z. Shen, D. Tang, S. Zhang, G. Zhao and X. Wang, Environ. Sci. Technol., 2022, 56, 14030–14037 CrossRef CAS PubMed
- B. Zeng, W. Xu, S. Khan, Y. Wang, J. Ji, K. Yang, X. Su and Z. Lin, Chemosphere, 2021, 285, 131439 CrossRef CAS PubMed
- M. Shirotori, S. Nishimura and K. Ebitani, Catal. Sci. Technol., 2016, 6, 8200–8211 RSC
- S. Mansingh, D. P. Sahoo, L. Paramanik, M. Sahoo and K. Parida, Inorg. Chem. Front., 2022, 9, 559–576 RSC
- M. Takemoto, Y. Tokudome, D. Noguchi, R. Ueoka, K. Kanamori, K. Okada, H. Murata, A. Nakahira and M. Takahashi, Langmuir, 2020, 36, 9436–9442 CrossRef CAS PubMed
- X. Li, S. Hufnagel, H. Xu, S. A. Valdes, S. G. Thakkar, Z. R. Cui and H. Celio, ACS Appl. Mater. Interfaces, 2017, 9, 22893–22901 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00140g |
| This journal is © The Royal Society of Chemistry 2023 |