
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA perspective of COx conversion to aromatics
Guo
Tian†
,
Xiaoyu
Liang†
,
Hao
Xiong
 ,
Chenxi
Zhang
* and
Fei
Wei
*
,
Chenxi
Zhang
* and
Fei
Wei
*
Beijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing, China 100084. E-mail: wf-dce@tsinghua.edu.cn; cxzhang@tsinghua.edu.cn
First published on 30th June 2023
Abstract
The sustainable production of chemicals through COx hydrogenation is a growing area of interest, with thermal catalytic conversion showing the most promise. Selective hydrogenation to high carbon number products (C8+) remains a challenge, and this perspective focuses on recent advancements in heterogeneous catalytic COx hydrogenation to aromatics. Efficient conversion has been achieved using tandem catalysts composed of metal oxides and nano-porous zeolites, particularly H-ZSM-5, which activate COx and dissociate H2 while promoting precise C–C coupling and cyclization. Such behavior facilitated the system towards a simple biological system. However, understanding the reaction mechanisms, including product selectivity and catalyst activity regulation is still a challenge. This perspective reviews recent progress and integrates quantitative activity descriptors for metal-dependent speciation within the biological metabolic system. H* adsorption energy in the presence of C1 oxygenate intermediates is identified as a speciation-sensitive activity descriptor, while zeolite topologies serve as product selectivity descriptors. These findings establish robust structure–performance relationships and guide the rational design of high-performance COx hydrogenation to aromatic catalysts.
Broader contextIn the burgeoning field of COx hydrogenation for sustainable chemical production, thermal catalytic conversion stands out as the most promising avenue. This perspective emphasizes recent strides made in selective hydrogenation to higher carbon number products (C8+) via heterogeneous catalytic COx hydrogenation to aromatics, a challenge that remains at the forefront of the field. Notably, substantial conversion efficiency has been realized through the employment of tandem catalysts, specifically metal oxides, and nano-porous zeolites like H-ZSM-5. These catalysts activate COx and dissociate H2, simultaneously fostering precise C–C coupling and cyclization, and thereby drawing parallels to simple biological systems. Nevertheless, fully deciphering the reaction mechanisms, including aspects of product selectivity and catalyst activity regulation, remains a daunting task. This perspective integrates the recent breakthroughs and introduces quantitative activity descriptors for metal-dependent speciation in relation to the biological metabolic system. It pinpoints the H* adsorption energy in the presence of formyl as a key activity descriptor sensitive to speciation, while zeolite topologies emerge as significant product selectivity descriptors. These insights solidify structure–performance relationships and illuminate the path towards rational design of high-performance catalysts for COx hydrogenation to aromatics. |
Introduction
In the realm of sustainable chemical production, the hydrogenation of CO2 or CO (collectively known as COx) to generate value-added compounds has attracted considerable interest.1–3 Numerous reviews have delved into the heterogeneous catalytic hydrogenation of COx, examining techniques such as thermal, electrochemical, and photochemical processes.4,5 While each approach possesses promise, thermal catalytic conversion exhibits superior yields and scalability. Recent advancements have facilitated the synthesis of oxygenates (e.g., CH3OH and C2H5OH), light olefins (C=2–4), gasoline (C5+), and heavy aromatics (C8–12) using COx as a feedstock.6,7 Nevertheless, the selective hydrogenation of COx to high carbon number products (C8+) remains a formidable challenge due to unpredictable C–C coupling and a multitude of elementary reactions.Aromatic compounds characterized by a low H/C ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–1:1.5), similar to biomass-derived feedstocks, may represent an ideal outcome from renewable pathways (Fig. 1(a)). Furthermore, the limited H2 consumption capacity could yield substantial economic value (Fig. 1(b)). In comparison to conventional hydrocarbons and oxygenates, the favorable thermodynamic driving force at low temperatures offers superior energy conservation and lays the groundwork for rational catalyst design (Fig. 1(c)). Generally, the entropy contribution arising from mixed products results in more favorable Gibbs free energy than that of most individual products (Fig. 1(d)). This observation offers a unique perspective on the production of high-quality synthetic fuels rather than single fine chemicals. The escalating global demand for aromatic compounds necessitates alternative production routes beyond petroleum-based methods (Fig. 1(e)). Based on the aforementioned acknowledgements, our perspective focuses on the heterogeneous catalytic hydrogenation of COx to aromatics, highlighting the reaction mechanisms responsible for generating these valuable materials. Our efforts aim to bridge the knowledge gap and guide researchers in this field towards the development of more efficient and selective catalysts.
:1–1:1.5), similar to biomass-derived feedstocks, may represent an ideal outcome from renewable pathways (Fig. 1(a)). Furthermore, the limited H2 consumption capacity could yield substantial economic value (Fig. 1(b)). In comparison to conventional hydrocarbons and oxygenates, the favorable thermodynamic driving force at low temperatures offers superior energy conservation and lays the groundwork for rational catalyst design (Fig. 1(c)). Generally, the entropy contribution arising from mixed products results in more favorable Gibbs free energy than that of most individual products (Fig. 1(d)). This observation offers a unique perspective on the production of high-quality synthetic fuels rather than single fine chemicals. The escalating global demand for aromatic compounds necessitates alternative production routes beyond petroleum-based methods (Fig. 1(e)). Based on the aforementioned acknowledgements, our perspective focuses on the heterogeneous catalytic hydrogenation of COx to aromatics, highlighting the reaction mechanisms responsible for generating these valuable materials. Our efforts aim to bridge the knowledge gap and guide researchers in this field towards the development of more efficient and selective catalysts.
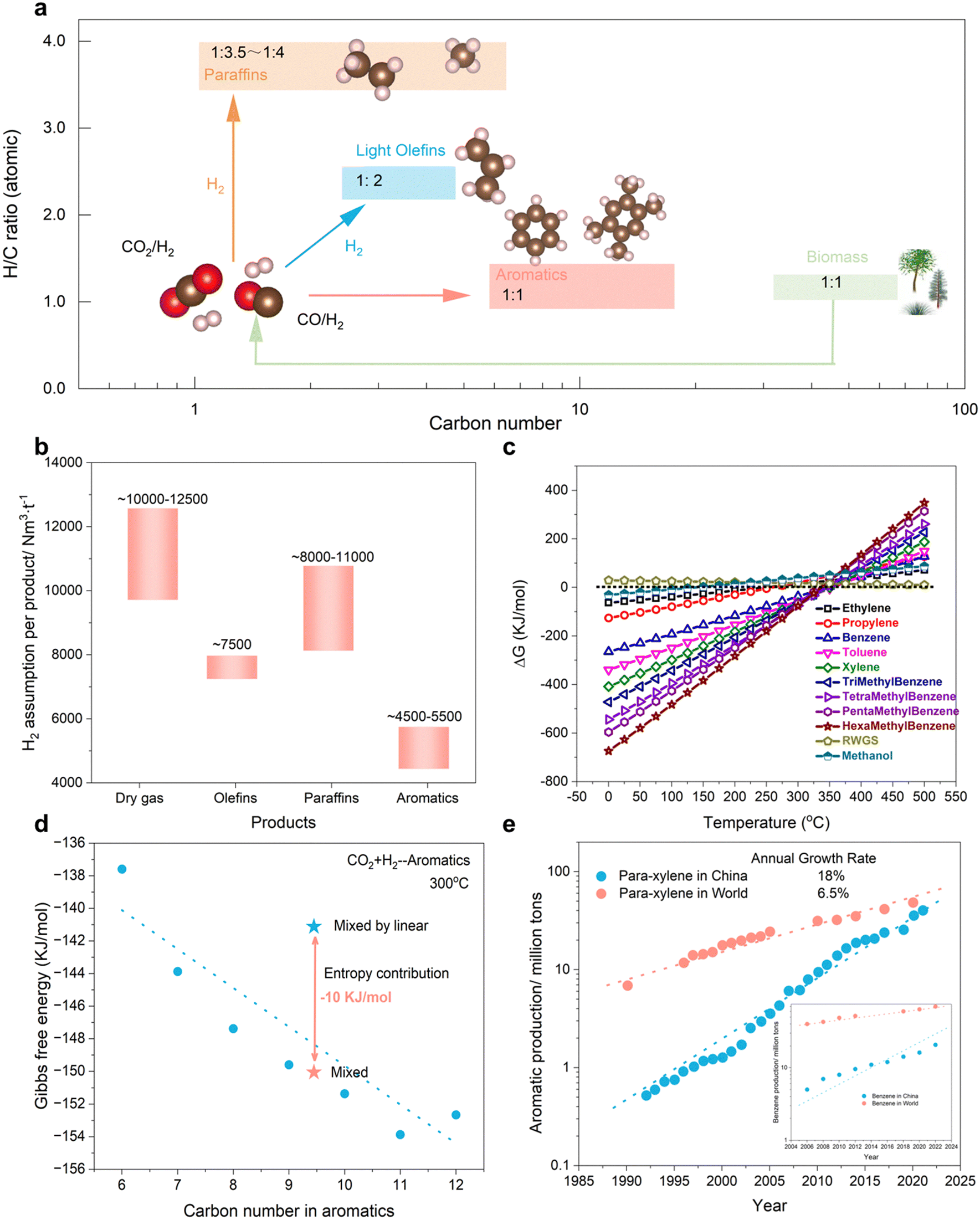 | ||
| Fig. 1 Economic and atomic analysis of COx conversion to aromatics versus other hydrocarbons. (a) Atomic and economic analysis of COx conversion to paraffins, light olefins and aromatics. (b) H2 assumption profile over typical hydrocarbons. (c) Thermodynamical equilibrium of CO conversion to hydrocarbons and oxygenates. (d) Gibbs free energy of mixed aromatics versus single aromatic products. (e) Some typical aromatic productivity in the world versus China. | ||
The proficient transformation of COx to heavy aromatics has been accomplished via the utilization of tandem catalysts, specifically comprising metal oxides and nano-porous zeolites, with H-ZSM-5 being particularly noteworthy.8 In this arrangement, the metal oxides function to activate COx and dissociate H2, while the confined acidic pores of H-ZSM-5 facilitate accurate C–C coupling and cyclization.9 Intriguingly, the tailored diameter pores of H-ZSM-5, possessing a dynamic diameter compatible with benzene, toluene, and xylene (BTX), result in heightened aromatic selectivity.10 Following this, surface methylation of BTX transpires at the exterior surface of H-ZSM-5, forming heavy aromatics (C8+), with CH3O* generated on the metal oxide surface, a mechanism widely acknowledged by researchers.11,12 Moreover, an acid–base heterostructure formed by the close proximity between metal oxides and H-ZSM-5 ensures the rapid diffusion of key intermediates originating from the metal oxides.13,14 This catalytic process bears similarity to a rudimentary one,15 wherein the transfer of matter (C–C coupling) and energy (thermodynamic driving force) is optimized. Nonetheless, despite its potential, a significant challenge lies in developing a comprehensive understanding of the metabolic system and establishing an activity and selectivity descriptor to accurately delineate its catalytic behavior.
A multitude of unresolved questions persist regarding the diverse reaction mechanisms implicated in catalysts for COx hydrogenation to aromatics, encompassing aspects such as product selectivity, catalyst activity regulation, and the enhancement of catalytic performance. The fundamental challenge associated with these catalysts remains a crucial research focus. Despite employing an array of methods, including in situ IR, NMR, and synchrotron-based photoionization mass spectrometry, the actual mechanism remains highly contentious.16 The intricacy of coexistent species, coupled with the elevated reaction temperatures and pressures necessitated for certain steps, frequently results in active and low-concentration intermediates, further exacerbating the issue and rendering most techniques inadequate for detecting these elusive species. Such disputes surrounding the mechanism have significantly impacted the advancement and comprehension of next-generation high-performance catalysts.
In this perspective, we comprehensively review recent advancements in COx hydrogenation to aromatics, integrating these developments to derive quantitative activity descriptors that account for metal-dependent speciation within this biological metabolic system. Moreover, through in-depth multivariate analysis, we investigate the chemistry involved in different zeolite topologies when combined with highly active oxide-based catalysts for COx hydrogenation to olefins, aromatics, and paraffins. By combining kinetic, transient, and chemisorption analyses with modelling, we identify H* adsorption energy in the presence of C1 oxygenate intermediates as a speciation-sensitive activity descriptor, while zeolite topologies serve as product selectivity descriptors. These findings are crucial in establishing robust structure-performance relationships and guiding the rational design of next-generation, high-performance COx hydrogenation to aromatic catalysts.
Results and discussion
Activity side on the metal oxides
In general, the thermodynamic driving force of mixed aromatic products is more favorable than that of single aromatics. Additionally, mixed aromatic products with carbon numbers ranging between C8–12 may also serve as aviation fuel additives.17,18 Consequently, recent studies have primarily concentrated on synthesizing mixed aromatic products. Within the domain of thermocatalytic hydrogenation of COx to aromatics, metal oxides are pivotal in activating COx and dissociating H2.19,20 Two categories of metal oxides have been investigated for this process: Fischer–Tropsch-based oxides, such as Fe, Co, and Mo, and spinel-type metal oxides rich in oxygen vacancies.21 The Fischer–Tropsch categories combining the Fischer–Tropsch-based oxides with the zeolite can achieve a high CO single pass conversion with moderate aromatics selectivity. Ma et al.6 exploited a combination of Na–Zn–Fe5C2 and hierarchical HZSM-5 showed a high selectivity of aromatics of 51% under the stable stage with CO conversion over 85%. Although the addition of zeolite can break the Anderson–Schulz–Flory (ASF) distribution of Fisher–Tropsch synthesis to a certain extent, the formation of C–C bonds at the metal oxide is still not well controlled, resulting in limited selectivity of aromatics.In contrast, the spinel-type metal oxides rich in oxygen vacancies are particularly fascinating, as they circumvent the Anderson–Schulz–Flory rule limitation and exhibit enhanced selectivity towards target aromatics when paired with H-ZSM-5, which exerts significant constraint effects.22 Therefore, numerous researchers have concentrated on developing highly effective metal oxide + H-ZSM-5 composite catalysts. Examples of these composite catalysts include ZnAl2O4, ZnCr2O4, and ZnZrOx, which can generate C1 oxygenates from COx hydrogenation and subsequently transform them into aromatics utilizing the H-ZSM-5 component.23 Various studies have revealed that, in this biological metabolic system, catalytic activity is predominantly constrained by the metal oxide aspect. Without compromising aromatic selectivity, considerable efforts have been dedicated to enhancing the capacity of COx conversion over traditional spinel-based metal oxides.
In a recent study, Wang and colleagues shed light on the advantageous effects of heteroatom doping, including Al, Ce, Zn, Mo, and Pt, into ZrO2 catalysts on H2 adsorption and dissociation.24 Their research showcased the significant impact of these modifications on the catalytic performance of syngas to aromatics when combined with H-ZSM-5, as depicted in Fig. 2(a). These findings suggested that the d-band electrons of metals could be tuned while preserving the catalyst structure, resulting in effective regulation of reaction activity primarily controlled by the metal oxide side. A similar approach was adopted by Wei et al.,25 introducing appropriate amounts of F–T metals, such as Fe and Co, into spinel-type ZnCr2O4 to significantly improve CO conversion without sacrificing total aromatic selectivity, as depicted in Fig. 2(b). To evaluate the ability of doping metals for COx and H2 activation, Wang et al. conducted H2–D2 exchange studies for M-ZrO2 catalysts, as well as pristine ZrO2, as depicted in Fig. 2(c). They deduced that metals with moderate H2 activation energy, such as Zn and Mo, are an ideal choice to balance the trade-off between activity and target product selectivity. Additionally, Wei et al.25 carried out density functional theory calculations (DFT) to quantitatively evaluate CO adsorption and H2 dissociation energy. As shown in Fig. 2(d), matched CO adsorption energy with H2 dissociation is speculated to achieve high performance COx to aromatics. Overall, the results and analysis suggest that measuring the COx adsorption and H2 dissociation ability is a feasible method for evaluating metal oxides, further enabling high-throughput screening for suitable metals.
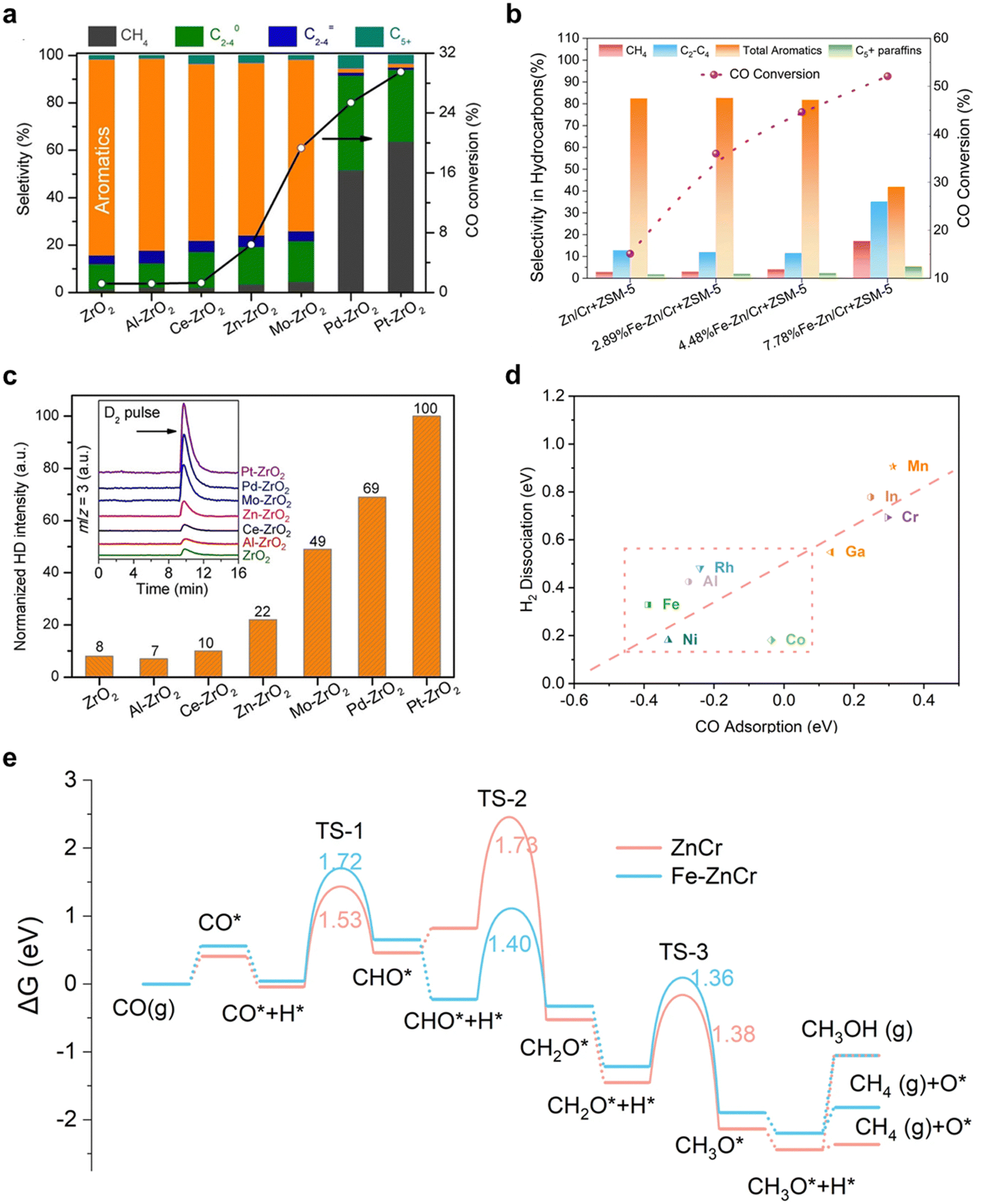 | ||
| Fig. 2 The modulation on metal oxides in syngas to aromatics. (a) Effect of metal doping on the catalytic performance of M-ZrO2/H-ZSM-5 (M denotes metal oxides). Reprinted with permission. Copyright 2019 Chemistry Europe. (b) Catalyst evaluation of pristine ZnCr2O4 and different Fe–Zn/Cr with H-ZSM-5. Reprinted with permission. Copyright The Author(s) 2022. (c) Normalized HD formation rate over M-ZrO2. Reprinted with permission. Copyright 2019 Chemistry Europe. (d) H2 dissociation energy versus CO adsorption over different M-ZnCr catalysts. (e) Gibbs free energy diagrams of syngas conversion to CH3OH/CH4 on pristine and Fe/ZnCr(111) surfaces with 1/4 ML Ov. Reprinted with permission. Copyright The Author(s) 2022. | ||
In a recent study, Wei et al.25 employed density functional theory (DFT) calculations to examine the reaction pathway of CO hydrogenation, as depicted in Fig. 2(e). CO adsorption takes place at Ov, succeeded by H2 dissociation near the surface Ov and H* adsorption on an O situated in proximity to 16d site metals. The CO* then undergoes progressive hydrogenation, forming CHO*, CH2O*, CH3O*, and ultimately CH3OH/CH4. The authors discovered that on pristine ZnCr2O4 (111) with Ov, H* formation is impeded due to the presence of CHO* and the robust adsorption of CH3O*. Conversely, the integration of isolated Fe stabilizes the state of CHO* + H*, rendering the hydrogenation step to produce H2CO* energetically exothermic while destabilizing the state of CH2O* + H*, thereby promoting CH3O* formation. As illustrated in Fig. 2(e), the generation of CH3OH and CH4 diverges from CH3O* on the oxide catalysts, both being endothermic with reaction energies as high as 1.14 and 0.38 eV, respectively, and less thermodynamically viable than the preceding endothermic steps to form H2CO*/CH3O* from CO. Furthermore, earlier theoretical investigations propose that the hydrogenation of CH3O* to CH3OH or CH4 presents the highest kinetic barrier.26,27 Consequently, Fe doping can enhance CO conversion, resulting in the production of significant quantities of CH4, suggesting that Fe aids in the transformation of syngas to an intermediate capable of rapid conversion to CH4. H-ZSM-5 is subsequently necessary for the further alteration of the intermediate. Wei et al. posited that the intermediate is CH3O*/CH2O*, which has recently been successfully identified in an experiment by Hou et al.28
In consideration of recent investigations on metal oxides in tandem biological metabolism systems, we have assembled evidence signifying that the activity of COx hydrogenation to aromatics is predominantly constrained by metal oxide properties, specifically, the activation of COx and H2 dissociation capability. Furthermore, we have ascertained that the formation rate of CH2O* represents the rate-limiting step within the extensive reaction pathway. To tackle this, we put forward a quantitative activity descriptor of H* adsorption energy in the presence of C1 oxygenate intermediates. By modulating the d-band center of metal oxides, we can effectively regulate the adsorption energy of key intermediates and accelerate the reaction towards desired products.
Surface diffusion of key intermediates
It is widely acknowledged that C1 oxygenates predominantly form on the surface of metal oxides. The diffusion of C1 oxygenates into the acidic pores of H-ZSM-5 may establish a bridge connecting two distinct reactions.29 The crucial role of key intermediate diffusion between catalyst A and catalyst B in tandem catalytic systems is well-established, necessitating a comprehensive examination of the system's configuration to understand its catalytic behavior. Up to this point, active site proximity between catalyst A and catalyst B has primarily been dictated by particle size or catalyst arrangement.30,31 Guided by these insights, numerous investigations have been undertaken to explore the surface diffusion propensity within this framework.As illustrated in Fig. 3(a), differing catalyst placement orientations yield distinct catalytic performances and product distributions in syngas-to-aromatics conversion. Physically mixed composite catalysts exhibit notably improved aromatics selectivity and CO conversion in comparison to separated/layered catalyst orientations. Analogous observations are documented in Xu et al.'s32 research utilizing a Ce–Zr/ZSM-5 catalyst and Wang et al.'s19 investigation employing a Zn–ZrO2/H-ZSM-5 catalyst, both suggesting that decreased catalyst particle size, resulting in closer proximity, leads to heightened aromatics selectivity. These findings collectively emphasize the vital role of intermediate diffusion between the bifunctional catalyst's two active components in tandem reaction product distribution. Fig. 3(b) portrays the influence of surface diffusion on the tandem reaction, where intermediates formed on the metal oxide side transfer to the zeolite side for further conversion to aromatics, and H species from aromatics formation return to the metal oxide side for CO hydrogenation. In addition to the diffusion of C1 intermediates and H species, the diffusion of aromatics bears considerable significance. As depicted in Fig. 3(c), Wei et al.'s study33 discloses that short b-axis nano ZSM-5 surfaces with ZnCr2O4 attachment display superior desorption capabilities compared to those lacking ZnCr2O4, while the desorption ability of phenol exhibits an opposite trend. This asymmetric desorption in bifunctional catalysts contributes to heightened tandem reaction catalytic performance. Such behaviors indicate the existence of dual transfer in this tandem system, which profoundly affects the catalytic performance.
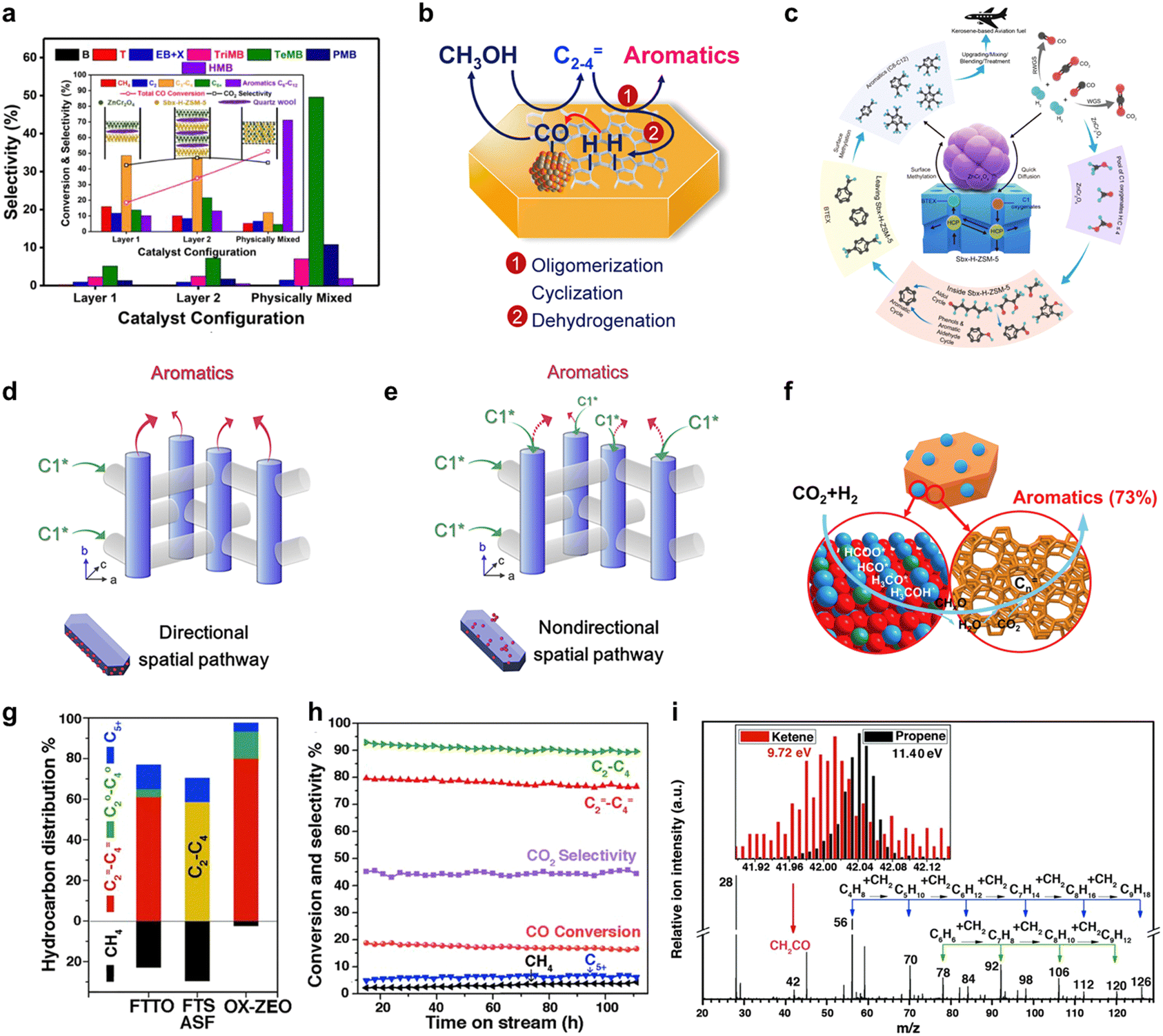 | ||
| Fig. 3 The identification of key intermediates and their diffusion ability over bifunctional catalysts. (a) Product distribution and catalytic activity in different catalyst placement orientations. Reprinted with permission. Copyright 2020 American Chemical Society. (b) Schematic of the proposed reaction mechanism of CO2 hydrogenation on the tandem catalyst. Reprinted with permission. Copyright 2017 Elsevier Inc. (c) Functioning mechanism of diffusion ability of CH3OH into H-ZSM-5. Reprinted with permission. Copyright 2022 American Chemical Society (d) and (e) comparison between the directional and nondirectional spatial pathway for tandem reaction of syngas to aromatics; (d) directional spatial pathway and (e) nondirectional spatial pathway, C1* represents C1 intermediate. Reprinted with permission. Copyright 2021 Elsevier Inc. (f) Proposed reaction mechanism for the highly selective conversion of CO2 or syngas into aviation fuel precursors over the nano-sized bifunctional catalyst ZnCr2O4/H-ZSM-5. Reprinted with permission. Copyright 2018 Elsevier Inc. (g) and (h) The catalyst performance in syngas to light olefins. Reprinted with permission. Copyright 2016, American Association for the Advancement of Science. (i) In situ study of syngas conversion over ZnCrOx with CH2CO as a key intermediate by SVUV-PIMS. Reprinted with permission. Copyright 2016, American Association for the Advancement of Science. | ||
Intermediates and product diffusion can be directionally modulated by designing the proximity of bifunctional catalysts in an oriented manner. As depicted in Fig. 3(d) and (e), Xie et al.34 introduced a directional-diffusion catalyst featuring a highly exposed (010) ZSM-5 surface and preferentially attached Cr2O3 to (100) and (101) surfaces via a recrystallization method. This arrangement facilitates C1 intermediates, generated on metal oxide sites, to likely diffuse into adjacent sinusoidal channels of ZSM-5, while enabling aromatic products to easily diffuse out through straight channels. The directional pathway resulted in a striking enhancement in catalytic performance. Regarding CO2 conversion to aromatics, surface diffusion of key intermediates also plays a pivotal role. Analogous to the CO-to-aromatics conversion, CHxO species generated on the metal oxide side through CO2 hydrogenation are transferred to the zeolite side for further conversion to aromatics.35 Notably, catalyst performance exhibits pronounced differences depending on the proximity between the active components. Beyond COx-to-aromatics conversion, intermediate diffusion assumes a significant role in this tandem system, such as the transformation of COx to light olefins. As illustrated in Fig. 3(g)–(i), CH2CO, generated from CO hydrogenation, serves as the key intermediate in the tandem reaction, as detected by highly sensitive synchrotron-based vacuum ultraviolet photoionization mass spectrometry (SVUV-PIMS).7,36 Through surface diffusion, intermediates reach the MSAPO side and subsequently convert to light olefins under the selective catalysis of zeolite.
Considering recent advancements in zeolite research within tandem biological metabolism systems, it is evident that surface transfer of key intermediates establishes a crucial connection between the COx active reaction on the metal oxide side and the selective production of target products on the zeolite side. By modulating the proximity of active components and designing the diffusion pathway, both COx conversion and target product selectivity can be significantly enhanced.
Selectivity side on the zeolites
While the H* adsorption energy with the existence of C1 oxygenate intermediates is a speciation-sensitive activity descriptor, zeolite topologies act as a product selectivity descriptor.30,37 Several studies have demonstrated that in this biological metabolic system, the product distribution mainly depends on the side of the zeolite. Through the surface diffusion, the C1 oxide intermediates, generated from COx catalyzed by metal oxide, enter the zeolite pore, which acts as a selective reaction vessel and are further transformed into target products through carbon chain growth, dehydrogenation, cyclization, etc.38 Various pore zeolite topology structures with different pore sizes and cage structures, show unique domain limiting effects leading to diverse product selectivity. So that when metal oxides are coupled with different zeolites, the restriction of thermodynamics can be broken and different target products can be obtained with high selectivity.SAPO-34, featuring a CHA framework type, possesses an ellipsoidal cage (1.1 × 0.65 nm) and forms a three-dimensional pore structure through six eight-membered rings, with a pore size of 3.8 Å.39 This narrow pore allows passage of only small molecules and hydrocarbons, while restricting diffusion of long-chain hydrocarbons and larger aromatic molecules.40 As a result, SAPO-34 serves as a common catalyst for the targeted synthesis of light olefins (C=2–4). Bao et al.7 utilized OX-ZEO and combined the ZnCrOx with MSAPO to achieve the conversion of syngas to C2−4 with the selectivity up to 94% at a CO conversion of 17%. Recently, Li et al.41 developed a ZnZrO/SAPO-34 tandem catalyst that combines a ZnO–ZrO2 solid solution with a Zn-modified SAPO-34 zeolite, as depicted in Fig. 4(a) and (b). By leveraging the constraining effect of SAPO-34, the selectivity for lower olefins reaches 80–90% among hydrocarbon products under conditions of 380 °C, 2 MPa, and 3600 ml gcat−1 h−1. Furthermore, this catalytic performance can be sustained over 100 h under the same conditions, laying the groundwork for its industrialization. Similar results can be achieved under the catalysis of In2O3/ZrO2/SAPO-3442 or In2O3–ZnZrOx/SAPO–34.43
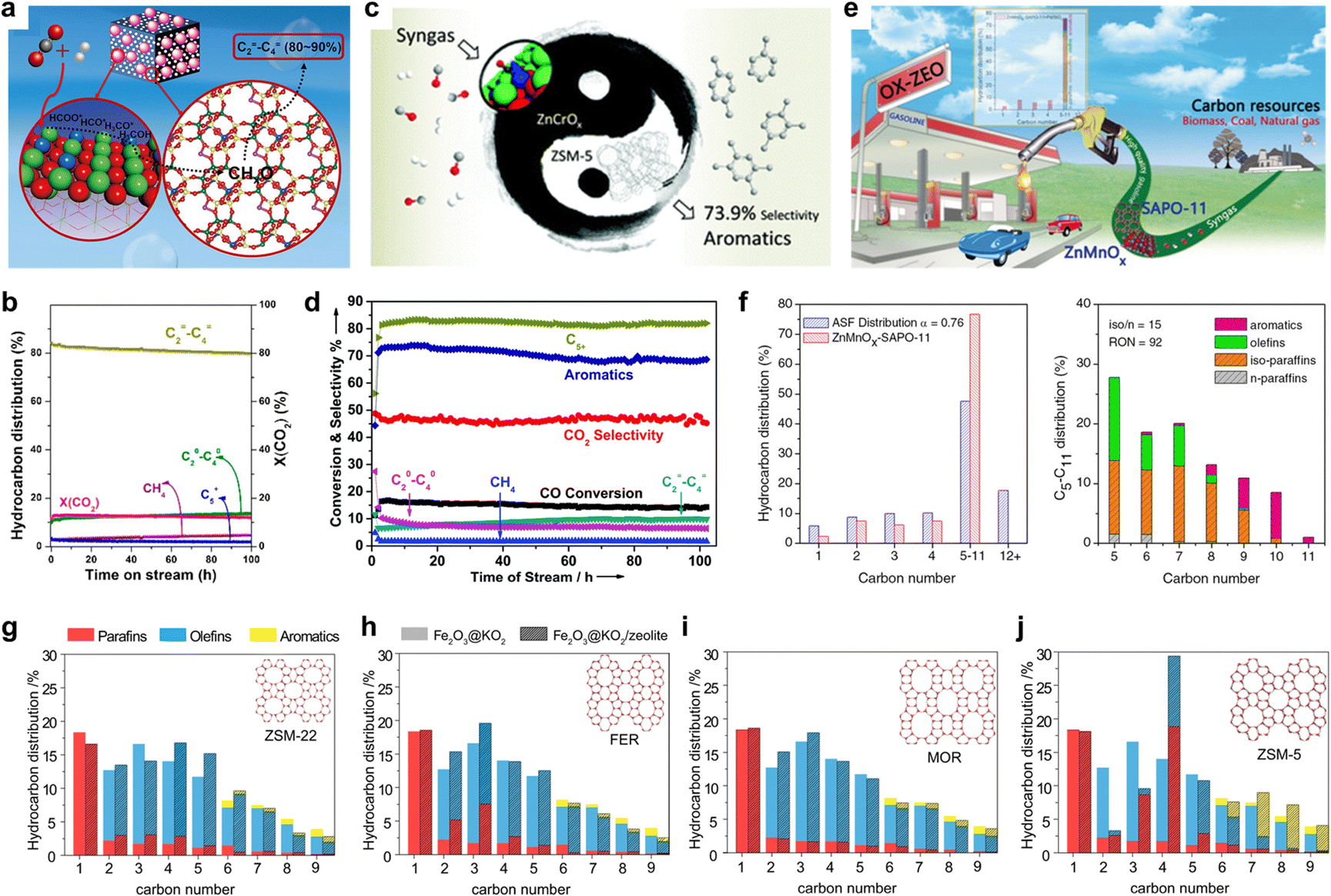 | ||
| Fig. 4 Different catalytic performances over bifunctional catalysts. (a) Schematic of the proposed reaction mechanism of CO2 hydrogenation to light olefins. Reprinted with permission. Copyright 2017 American Chemical Society. (b) Catalytic performance over ZnO–ZrO2/SAPO-34. Reprinted with permission. Copyright 2017 American Chemical Society. (c) Schematic of the proposed reaction mechanism of CO2/CO hydrogenation to aromatics. Reprinted with permission. Copyright The Royal Society of Chemistry 2017. (d) Catalytic performance over ZnO–ZrO2/H-ZSM-5. Reprinted with permission. Copyright The Royal Society of Chemistry 2017. (e) Schematic of the proposed reaction mechanism of CO hydrogenation to gasoline. Reprinted with permission. Copyright 2019 Wiley-VCH VerlagGmbH & Co. KGaA, Weinheim. (f) Catalytic performance over ZnMnOx/SAPO-11. Reprinted with permission. Copyright 2019 Wiley-VCH VerlagGmbH & Co. KGaA, Weinheim. (g)–(j) Detailed hydrocarbon distribution for selected zeolite frameworks in a dual-bed configuration during Fe2O3@KO2/zeolite bifunctional material catalyzed hydrogenation of CO2. Reprinted with permission. Copyright The Author(s) 2021. | ||
Owing to the well-matched straight channel of H-ZSM-5 (5.6 × 5.3 Å) with the dynamic diameter of benzene, toluene, and xylene (BTX), employing H-ZSM-5 with an MFI structure as the selectivity component enables the direct conversion of COx to aromatics.44 As illustrated in Fig. 4(c) and (d), Wang et al.19,45 achieved an aromatics selectivity of ∼80% at a single-pass CO conversion of 16.0%. It should be noted that, due to the strong external acidic properties of H-ZSM-5, BTX undergoes alkylation reactions at the (010) surface of H-ZSM-5 to form heavier aromatics such as tri- or tetra-methylbenzene. Recently, Wei et al.33 accomplished the conversion of CO2 to high-energy-density aviation fuel, polymethyl benzene (C10+), with high selectivity by employing short b-axis ZSM-5 with superior specific surface area and diffusion properties. Tsubaki et al. achieved highly selective preparation of PX by modifying ZSM-5 with silica46 or employing tailor-made ZSM-547 to reduce the acidic sites on the external surface of the zeolite.
Furthermore, similar strategies for designing product-oriented catalysis have also been adopted by researchers in this field. Bao et al.48 explored zeolites with 1D ten-membered-ring (10-MR) channel structures, such as SAPO-11 and ZSM-22, combined with ZnaMnbOx. This approach facilitated the direct conversion of syngas into C5+ gasoline, achieving a selectivity of 76.7% among hydrocarbons at a 20.3% CO conversion, as depicted in Fig. 4(e) and (f). Generally, this catalytic process exhibits a longer lifetime compared to traditional methanol conversion to gasoline, light olefins, or aromatics, owing to the facile diffusion properties of the final products facilitated by the synergistic effect between metal oxides and zeolites.49
The substantial influence of zeolite topology on reaction product distribution has garnered the attention of several researchers. Abhishek Dutta Chowdhury et al.37 investigated zeolite as a descriptor governing ultimate product selectivity. In their study, eight zeolite catalysts, including MOR, SAPO-34, ZSM-58, BETA, Y, FER, ZSM-22, and ZSM-5, were tested for CO2 conversion, with Fe2O3@KO2 remaining constant as the metal oxide on the activity side, as depicted in Fig. 4(g)–(j). The results reveal that different zeolites yield vastly distinct product distributions, while CO2 conversion remains nearly constant. MOR exhibits exceptional diffusion performance, resulting in high selectivity for C=2–4. Conversely, ZSM-22 with 1D, 10MR and FER with 2D, 10MR facilitate the targeted synthesis of C=5–9 and C2–9, respectively, due to the linear expansion effect. Moreover, ZSM-5 demonstrates outstanding aromatic selectivity. The combined outcomes of experimental and computational analyses highlight the vital role played by the hybrid nature of the active zeolite catalyst, its preferred CO2-derived reaction intermediates, and zeolite topology in determining the ultimate product selectivity. Recent studies on zeolites in tandem biological metabolism systems have revealed that product distribution is predominantly constrained by the zeolite. By judiciously selecting zeolites and further refining their topological structures and acid sites, it is possible to achieve higher selectivity for the desired product.
Conclusion
Based on recent research on zeolites in tandem biological metabolism systems, we propose that the tandem reaction proceeds via the following steps (Fig. 5): (1) activation of COx on the metal oxide side, generating CHxO intermediates through hydrogenation; (2) surface diffusion of key intermediates; and (3) selective catalysis on the zeolites.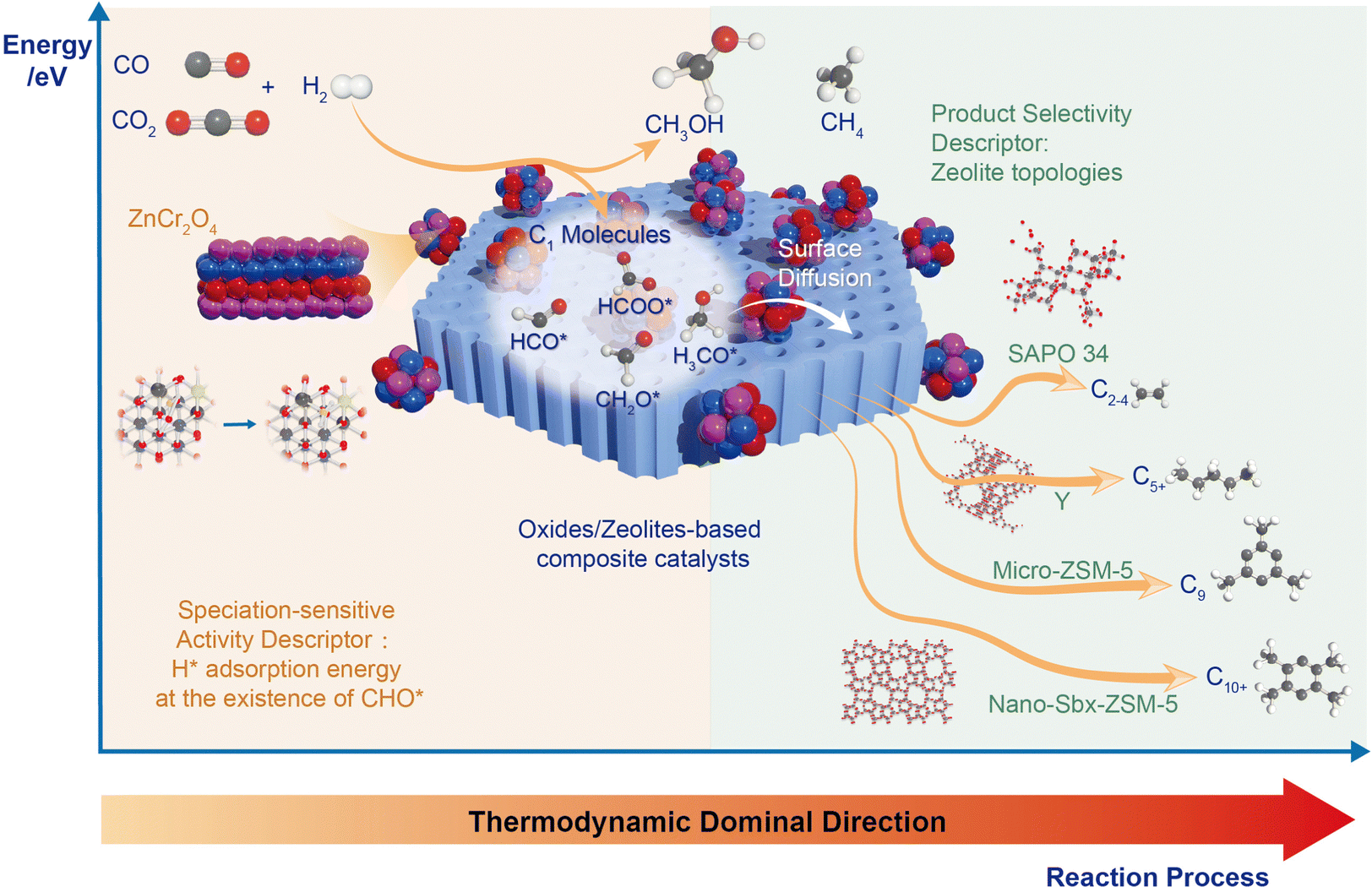 | ||
| Fig. 5 Schematical diagram of bifunctional catalysts and corresponding activity and selectivity descriptor. | ||
In the first step of this tandem biological metabolism system, CO or CO2 undergoes activation via stepwise hydrogenation. CO is adsorbed at Ov, followed by H2 dissociation near the surface Ov, on an appropriate metal oxide with suitable CO adsorption and H2 dissociation energy. The CO* then undergoes gradual hydrogenation along the pathway: CO* → CHO* → CH2O* → CH3O* → CH3OH/CH4.50 Similarly, CO2 activation follows a stepwise hydrogenation path with two possible pathways: the formate pathway and the carboxyl pathway.51 The formation of the key intermediate H2CO*, which has the highest formation energy and is easily desorbed into the gas phase, is the decisive step in the activation of both CO2 and CO. Hence, the H* adsorption energy in the presence of C1 oxygenate intermediates serves as the quantitative active descriptor in this step.
With the presence of zeolite, H2CO* intermediates effectively desorb into the zeolite pore for further reaction, whereas in the absence of zeolite, these intermediates must continue hydrogenation to produce methanol or methane, which have higher reaction energy barriers. The introduction of zeolites facilitates the desorption and surface diffusion of critical intermediates, allowing the modulation of proximity between the two active components to effectively regulate diffusion, leading to dramatic changes in conversion and selectivity.
Within the acidic zeolite pore, key intermediates transferred from the metal oxide side undergo further transformation into hydrocarbons. An aldol-aromatic mechanism, supported by in situ spectroscopic and microscopic characterization methods and theoretical calculations, explains the reactions on the zeolite side.33,52 This mechanism involves C–C coupling of reaction intermediates via aldol condensation and Prins reaction, generating crucial hydrocarbon pool species under the spatial constraints of the zeolite. Oxygenate intermediates are then converted to single-ring aromatics through hydrogenation/dehydrogenation and dehydration. The product distribution is primarily limited by the topology structures of the zeolite, with different zeolites exhibiting various pore sizes, cage structures, and acid sites. The match between the zeolite pore size and the molecular dynamics size of the product is the fundamental factor for the differing product selectivity. In these tandem biological metabolism systems, we can store the energy of high-energy gaseous phase reactants in target hydrocarbons via thermochemical storage. Analyzing this system allows for directed performance improvement through appropriate control of key descriptors at each step, offering guidance for designing the next generation of COx transformation reactions.
Challenges and perspective
In this review, we present an in-depth analysis of tandem catalysis systems for the conversion of COx into aromatics and other value-added products, with a particular emphasis on heavy aromatics. While these novel tandem catalyst systems hold promise as alternatives to traditional crude oil-based production methods for aromatics, significant advancements in performance are still required. A critical aspect of this endeavor involves elucidating the underlying mechanisms within the “black box” where the actual catalysis occurs. Specifically, identifying descriptors that connect micro-scale characteristics such as adsorption and activation energy with macro-scale performance indicators like reactant conversion and product selectivity is crucial. As we navigate this dynamic landscape, we are met with both immense opportunities and formidable challenges in the pursuit of sustainable and efficient catalytic systems.First of all, in the context of COx to aromatics conversion processes, thermodynamical constraints present a significant challenge. Mixed aromatics, in particular, are less thermodynamically restricted at lower temperatures. Nevertheless, the extensive chain-growth and aromatization reaction pathway required for the reduction of inert CO2 demands a considerable energy input, necessitating high temperatures. As a result, the development of high-activity catalysts that can operate effectively at low temperatures is crucial for the advancement of this field. One viable approach is to introduce high activity metals such as Fe, Mo, Pt, etc. into the metal oxides while avoiding the C–C coupling. By addressing these thermodynamic hurdles, we can tap into the full potential of these processes and foster more efficient and sustainable production strategies for aromatics.
Secondly, the unique attributes of aromatics as self-catalyzed ring-terminated carbons with fewer than 12 carbon atoms position them as essential high-carbon building blocks in modern chemical and material industries. These inherent properties enable the circumvention of challenges associated with product selectivity and hydrogen consumption, which commonly arise from the Anderson–Schulz–Flory (ASF) distribution. By concentrating research efforts on these high-carbon components, the scientific community can propel the development and optimization of conversion processes. Nonetheless, most catalysts in this system are prone to carbon deposition, leading to a decline in catalytic performance. Consequently, extending the lifetime of this innovative catalysis system from hundreds to thousands of hours is imperative for enhancing its practicality and viability. This advancement, in turn, will establish more efficient and accurate production methods for valuable aromatic compounds, further solidifying their significance and applications across various sectors.
Thirdly, the intricate structure of metal oxides and zeolites presents an intriguing area of study. Under a reduction atmosphere (CO, CO2, H2), the physical mixture of metal oxides and zeolites may undergo dynamic structural evolution, leading to the formation of heterostructures. Unraveling the interface between metal oxides and zeolites and its capacity for key intermediate diffusion remains a challenge. It is anticipated that density functional theory (DFT) studies, in conjunction with highly sensitive in situ/operando spectroscopic techniques, can offer more profound insights. Deciphering the high efficiency of this catalytic performance, conferred by the complex catalytic structure, may guide the design and creation of numerous active sites, ultimately enhancing overall activity.
Taking into account these challenges, the fundamental investigation of reaction mechanisms and the elucidation of active sites may be an ongoing pursuit. Concurrently, persistent efforts in optimizing catalyst compositions and structures, especially the exploration of emerging novel catalytic materials, could accelerate the development of more active and selective catalysts. This progress would potentially enable novel processes for synthesizing value-added chemicals and further advance the field.
Author contributions
Conceptualization and supervision: C. Z. and F. W. Investigation, resources, and visualization: G. T., X. L., and H. X. Writing – original draft: G. T., and X. L. Writing – review and editing: G.T., C. Z., and F. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (grant no. 22278238, C. Z.), the Key Research and Development Program of Inner Mongolia and Ordos (20211140095, C. Z.), and CNPC Innovation Funds (2020990028, C. Z.).References
- J. Li, Y. He, L. Tan, P. Zhang, X. Peng, A. Oruganti, G. Yang, H. Abe, Y. Wang and N. Tsubaki, Nat. Catal., 2018, 1, 787–793 CrossRef CAS.
- J. Hu, L. Yu, J. Deng, Y. Wang, K. Cheng, C. Ma, Q. Zhang, W. Wen, S. Yu and Y. Pan, Nat. Catal., 2021, 4, 242–250 CrossRef CAS.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef PubMed.
- H.-J. Peng, M. T. Tang, J. Halldin Stenlid, X. Liu and F. Abild-Pedersen, Nat. Commun., 2022, 13, 1399 CrossRef CAS PubMed.
- Y. Wang, X. Liu, X. Han, R. Godin, J. Chen, W. Zhou, C. Jiang, J. F. Thompson, K. B. Mustafa and S. A. Shevlin, Nat. Commun., 2020, 11, 2531 CrossRef CAS PubMed.
- B. Zhao, P. Zhai, P. Wang, J. Li, T. Li, M. Peng, M. Zhao, G. Hu, Y. Yang and Y.-W. Li, Chem, 2017, 3, 323–333 CAS.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou and M. Li, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Nat. Commun., 2018, 9, 3457 CrossRef PubMed.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell and Q. Li, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- H. Xiong, Z. Liu, X. Chen, H. Wang, W. Qian, C. Zhang, A. Zheng and F. Wei, Science, 2022, 376, 491–496 CrossRef CAS PubMed.
- M. T. Arslan, B. Ali, S. Z. A. Gilani, Y. Hou, Q. Wang, D. Cai, Y. Wang and F. Wei, ACS Catal., 2020, 10, 2477–2488 CrossRef CAS.
- M. T. Arslan, B. A. Qureshi, S. A. Gilani, D. Cai, Y. Ma, M. Usman, X. Chen, Y. Wang and F. Wei, ACS Catal., 2019, 9, 2203–2212 CrossRef CAS.
- S. Z. A. Gilani, L. Lu, M. T. Arslan, B. Ali, Q. Wang and F. Wei, Catal. Sci. Technol., 2020, 10, 3366–3375 RSC.
- G. Tian, C. Zhang and F. Wei, Nanoscale Horiz., 2022, 7, 1478–1487 RSC.
- L. A. Wessjohann, W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625–1648 CrossRef CAS PubMed.
- X. Pan, F. Jiao, D. Miao and X. Bao, Chem. Rev., 2021, 121, 6588–6609 CrossRef CAS PubMed.
- L. Zhang, Y. Dang, X. Zhou, P. Gao, A. P. van Bavel, H. Wang, S. Li, L. Shi, Y. Yang and E. I. Vovk, Innovation, 2021, 2, 100170 CAS.
- B. Yao, T. Xiao, O. A. Makgae, X. Jie, S. Gonzalez-Cortes, S. Guan, A. I. Kirkland, J. R. Dilworth, H. A. Al-Megren and S. M. Alshihri, Nat. Commun., 2020, 11, 6395 CrossRef CAS PubMed.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS.
- W. Zhou, C. Zhou, H. Yin, J. Shi, G. Zhang, X. Zheng, X. Min, Z. Zhang, K. Cheng and J. Kang, Chem. Commun., 2020, 56, 5239–5242 RSC.
- B. Bai, C. Guo, F. Jiao, J. Xiao, Y. Ding, S. Qu, Y. Pan, X. Pan and X. Bao, Angew. Chem., Int. Ed., 2023, e202217701 CAS.
- S. Wang, L. Zhang, P. Wang, W. Jiao, Z. Qin, M. Dong, J. Wang, U. Olsbye and W. Fan, Nat. Catal., 2022, 1–13 Search PubMed.
- Q. Han, P. Gao, K. Chen, L. Liang, Z. Zhao, X. Yao, D. Xiao, X. Han and G. Hou, Chem, 2023, 9, 721–738 CAS.
- W. Zhou, S. Shi, Y. Wang, L. Zhang, Y. Wang, G. Zhang, X. Min, K. Cheng, Q. Zhang and J. Kang, ChemCatChem, 2019, 11, 1681–1688 CrossRef CAS.
- G. Tian, X. Liu, C. Zhang, X. Fan, H. Xiong, X. Chen, Z. Li, B. Yan, L. Zhang and N. Wang, Nat. Commun., 2022, 13, 5567 CrossRef CAS PubMed.
- S. Ma, S.-D. Huang and Z.-P. Liu, Nat. Catal., 2019, 2, 671–677 CrossRef CAS.
- Z. Zhang, X. Chen, J. Kang, Z. Yu, J. Tian, Z. Gong, A. Jia, R. You, K. Qian and S. He, Nat. Commun., 2021, 12, 4331 CrossRef CAS PubMed.
- Y. Ji, P. Gao, Z. Zhao, D. Xiao, Q. Han, H. Chen, K. Gong, K. Chen, X. Han and X. Bao, Nat. Catal., 2022, 5, 594–604 CrossRef CAS.
- C. Liu, J. Su, S. Liu, H. Zhou, X. Yuan, Y. Ye, Y. Wang, W. Jiao, L. Zhang and Y. Lu, ACS Catal., 2020, 10, 15227–15237 CrossRef CAS.
- C. C. Amoo, C. Xing, N. Tsubaki and J. Sun, ACS Cent. Sci., 2022, 8, 1047–1062 CrossRef CAS PubMed.
- Y. Wang, W. Gao, K. Wang, X. Gao, B. Zhang, H. Zhao, Q. Ma, P. Zhang, G. Yang and M. Wu, Chem. Sci., 2021, 12, 7786–7792 RSC.
- Z. Huang, S. Wang, F. Qin, L. Huang, Y. Yue, W. Hua, M. Qiao, H. He, W. Shen and H. Xu, ChemCatChem, 2018, 10, 4519–4524 CrossRef CAS.
- M. T. Arslan, G. Tian, B. Ali, C. Zhang, H. Xiong, Z. Li, L. Luo, X. Chen and F. Wei, ACS Catal., 2022, 12, 2023–2033 CrossRef CAS.
- C. Liu, J. Su, Y. Xiao, J. Zhou, S. Liu, H. Zhou, Y. Ye, Y. Lu, Y. Zhang and W. Jiao, Chem Catal., 2021, 1, 896–907 CrossRef CAS.
- Z. Li, Y. Qu, J. Wang, H. Liu, M. Li, S. Miao and C. Li, Joule, 2019, 3, 570–583 CrossRef CAS.
- N. Li, Y. Zhu, F. Jiao, X. Pan, Q. Jiang, J. Cai, Y. Li, W. Tong, C. Xu and S. Qu, Nat. Commun., 2022, 13, 2742 CrossRef CAS PubMed.
- A. Ramirez, X. Gong, M. Caglayan, S.-A. F. Nastase, E. Abou-Hamad, L. Gevers, L. Cavallo, A. Dutta Chowdhury and J. Gascon, Nat. Commun., 2021, 12, 5914 CrossRef CAS PubMed.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- L. Yang, C. Wang, L. Zhang, W. Dai, Y. Chu, J. Xu, G. Wu, M. Gao, W. Liu and Z. Xu, Nat. Commun., 2021, 12, 4661 CrossRef CAS PubMed.
- J. Zhou, M. Gao, J. Zhang, W. Liu, T. Zhang, H. Li, Z. Xu, M. Ye and Z. Liu, Nat. Commun., 2021, 12, 17 CrossRef CAS PubMed.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS.
- J. Gao, C. Jia and B. Liu, Catal. Sci. Technol., 2017, 7, 5602–5607 RSC.
- S. Dang, S. Li, C. Yang, X. Chen, X. Li, L. Zhong, P. Gao and Y. Sun, ChemSusChem, 2019, 12, 3582–3591 CrossRef CAS PubMed.
- G. Qi, T. E. Davies, A. Nasrallah, M. A. Sainna, A. G. Howe, R. J. Lewis, M. Quesne, C. R. A. Catlow, D. J. Willock and Q. He, Nat. Catal., 2022, 5, 45–54 CrossRef CAS.
- C. Zhou, J. Shi, W. Zhou, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ACS Catal., 2019, 10, 302–310 CrossRef.
- Y. Wang, W. Gao, S. Kazumi, H. Li, G. Yang and N. Tsubaki, Chem. – Eur. J., 2019, 25, 5149–5153 CrossRef CAS PubMed.
- W. Gao, L. Guo, Y. Cui, G. Yang, Y. He, C. Zeng, A. Taguchi, T. Abe, Q. Ma and Y. Yoneyama, ChemSusChem, 2020, 13, 6541–6545 CrossRef CAS PubMed.
- N. Li, F. Jiao, X. Pan, Y. Chen, J. Feng, G. Li and X. Bao, Angew. Chem., Int. Ed., 2019, 58, 7400–7404 CrossRef CAS PubMed.
- S. Müller, Y. Liu, M. Vishnuvarthan, X. Sun, A. C. van Veen, G. L. Haller, M. Sanchez-Sanchez and J. A. Lercher, J. Catal., 2015, 325, 48–59 CrossRef.
- D. Laudenschleger, H. Ruland and M. Muhler, Nat. Commun., 2020, 11, 3898 CrossRef PubMed.
- H. Zhao, R. Yu, S. Ma, K. Xu, Y. Chen, K. Jiang, Y. Fang, C. Zhu, X. Liu and Y. Tang, Nat. Catal., 2022, 5, 818–831 CrossRef CAS.
- Y. Liu, F. M. Kirchberger, S. Müller, M. Eder, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2019, 10, 1462 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |