
Functionalization of methane using molecular metal complexes as catalysts
Hiroto
Fujisaki
 and
Takahiko
Kojima
*
and
Takahiko
Kojima
*
Department of Chemistry, Faculty of Pure and Applied Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8571, Japan. E-mail: kojima@chem.tsukuba.ac.jp
First published on 25th May 2023
Abstract
Efficient and selective functionalization of methane is one of the most important tasks in chemistry in light of its utilization as a naturally abundant feedstock toward the development of a sustainable society. This article surveys recent progress in oxidative conversion and C–H activation of methane to obtain useful functionalized products using molecular metal complexes as catalysts. The reactions proceed through C–H bond cleavage by high-valent metal–oxo complexes and also through C–H bond activation at lower-valent metal centers to form a C–O or C–C bond as a result of oxygen-rebound, reductive elimination or insertion processes. We also mention the importance of hydrophobic environments around metal centers to capture the methane molecule to be ready for the oxidative conversion toward the improvement of efficiency and product selectivity of CH4 oxidation.
1. Introduction
Methane (CH4) is an abundant C1 feedstock as a major component of natural gas, which is approximately 90%, and has been mainly used as a fuel and also as a raw material for producing many useful compounds including methanol.1 Methane oxidation to afford methanol is quite important for further production of useful molecules including formaldehyde, acetic acid, dimethyl ether and so on.2 Due to the serious situation in Europe, the natural gas supply has been reduced and alternative energy supplies have been considered such as renewables.3 The reduced supply of natural gas causes inflation of the price and enforces us to develop more efficient methods to convert CH4 to useful chemicals.3 On the other hand, CH4 shows 25-times higher global warming potential than CO2;4 thus, its discharge into air is avoided by flaring CH4, which is an inevitable waste of the resource. The main strategy of large-scale CH4 utilization is highly energy-consuming steam reforming to form syngas as a mixture of carbon monoxide (CO) and dihydrogen (H2) in the presence of water and alumina-supported nickel catalysts at 1100 K.5 In order to achieve efficient and selective conversion of methane, tremendous efforts have been devoted to the development of heterogeneous catalysts, including zeolite-based metal catalysts; however, these heterogeneous catalysts have been operated under harsh conditions including high temperature and high pressure.6The difficulty of oxidative conversion of CH4 to other compounds mainly stems from the high bond dissociation energy (BDE) of C–H bonds of CH4, which is 105 kcal mol−1.7 The other difficulty is due to the fact that the BDE of C–H bonds of methanol (CH3OH), which is a 2e−-oxidized product of CH4, is 96 kcal mol−1, lower than that of CH4.7 Therefore, the selective oxidation of CH4 to obtain CH3OH is very difficult due to overoxidation of CH3OH to HCHO as a 4e−-oxidized product, which is much easier to oxidize to HCOOH as a 6e−-oxidized product.
As molecular and homogeneous catalysts that can oxidize CH4 efficiently and selectively to afford CH3OH, the most excellent performance can be found in the catalysis by methane monooxygenases (MMOs), which include metal ions such as Cu and Fe at the active sites to activate O2 for generating oxidative active species.8,9 These enzymes are Cu-containing particulate MMOs (pMMOs)8 and Fe-containing soluble MMOs (sMMOs).9 As for the crystal structure of sMMOs, a hydrophobic tunnel which is called the “W308 tunnel” is observed and the tunnel has been recognized to control the selectivity and accessibility of substrates, CH4 and O2, to let them react at the diiron centre through the formation of a reactive intermediate called “compound Q”, which is a bis(μ-oxo) diiron(IV) species.10 Any naturally occurring heme enzyme has never been reported to oxidize CH4.
A number of small metal complexes as functional models of MMOs have been prepared to perform catalytic functionalization of alkanes to shed some light on the structure–reactivity relationship and to gain mechanistic insights into the reactions including the detection of intermediates involved such as high-valent metal–oxo complexes.11 Although catalytic oxidation of gaseous alkanes including CH4 has yet to be successful by using molecular catalysts so far, it should be important to develop molecular catalysts for selective CH4 functionalization to attain fundamental knowledge to gain a deeper understanding about reaction mechanisms and requisites for the selectivity and efficiency of the catalysis.
In this article, we focus on homogeneous molecular catalysts for methane oxidation, and thus, oxidative methane conversion using heterogeneous catalysts is not covered. Some examples of molecular catalysts supported on solid surfaces are also mentioned. Molecular catalysis includes a biomimetic approach inspired by metalloenzymes mentioned above and an organometallic strategy such as C–H activation.
2. Methane functionalization through C–H activation
In 1950, Snyder and Grosse reported the functionalization of methane to generate methane sulfonic acid (CH3SO3H; MSA) and methyl bisulfate (CH3OSO3H; MBS).12 The process involved the use of HgSO4 as a catalyst and SO3 at 673 K and 93 bar of methane. The total conversion of methane to MSA/MBS was 44% in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio; however, mechanistic insights into the catalytic process were not provided.12 In 1993, Periana and colleagues reported a Hg-catalysed high-yield system in H2SO4.13 This system involved the formation of Hg(OSO3H)2 as a catalyst by the reaction between HgSO4 and H2SO4. The catalytic system involving Hg(OSO3H)2 achieved a turnover frequency (TOF) of 10−3 s−1 and 50% conversion of methane with 85% selectivity for MBS (CO2 as the major by-product) at 350 K.13 In the catalysis, H2SO4 acts as a reaction solvent, a reagent for esterification of methane, and an oxidant (Scheme 1).13
:2 ratio; however, mechanistic insights into the catalytic process were not provided.12 In 1993, Periana and colleagues reported a Hg-catalysed high-yield system in H2SO4.13 This system involved the formation of Hg(OSO3H)2 as a catalyst by the reaction between HgSO4 and H2SO4. The catalytic system involving Hg(OSO3H)2 achieved a turnover frequency (TOF) of 10−3 s−1 and 50% conversion of methane with 85% selectivity for MBS (CO2 as the major by-product) at 350 K.13 In the catalysis, H2SO4 acts as a reaction solvent, a reagent for esterification of methane, and an oxidant (Scheme 1).13
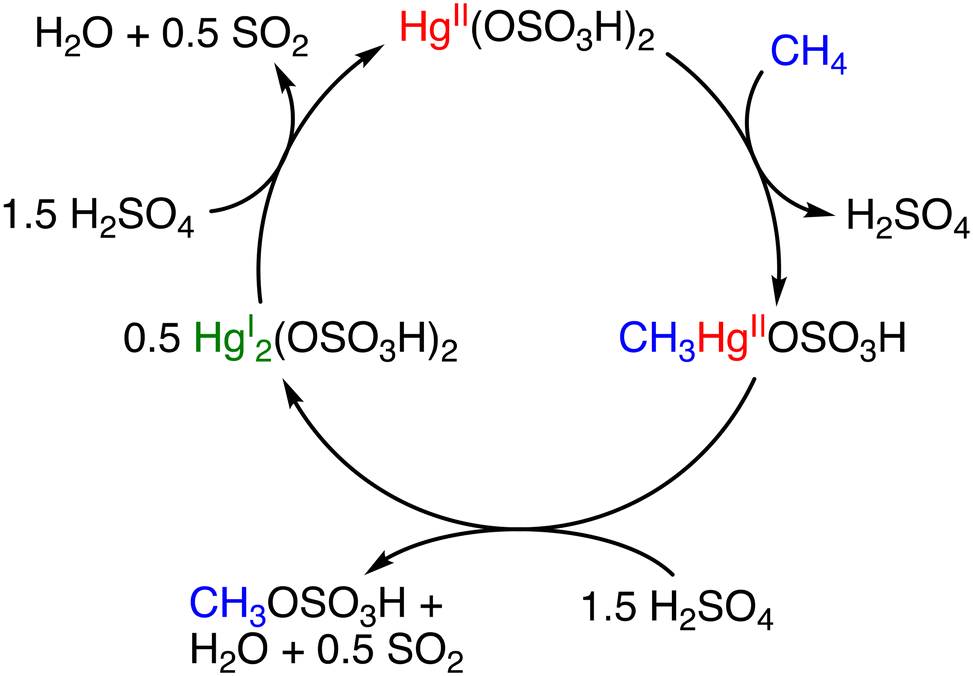 | ||
| Scheme 1 Catalytic cycle for the methane activation with Hg(OSO3H)2 to generate MBS.13 | ||
The first example of Pt-catalysed methane functionalization has been reported by Shilov.14 In this system, K2PtCl4 and K2PtCl6 catalyse the conversion of methane to methanol and chloromethane under 10 MPa of methane at 393 K in water. Under these conditions, a low TON of <20 has been obtained due to decomposition of the catalyst to form Pt black.14 The proposed mechanism involves three steps: (1) formation of a PtII–CH3 intermediate through C–H activation of methane by a PtII complex, (2) oxidation of the PtII–CH3 species with PtCl62− to form a PtIV–CH3 complex, and (3) reductive elimination of CH3OH or CH3Cl from the PtIV–CH3 species.14
In 1998, Periana and colleagues introduced a more effective Pt catalyst, dichloro(η2-{2,2′-bipyrimidyl}) platinum(II) ([(bpym)PtCl2]), known as the Periana catalyst.15 The Periana catalyst, based on the Shilov system mentioned above,14 converts methane to MBS with 90% conversion in oleum (fuming sulfuric acid; SO3/H2SO4) at 500 K and 34 bar of methane. Under the conditions, MBS has been produced with a TON of 500 and a TOF of 10−3 s−1 with 81% selectivity.15 The catalytic mechanism of the Periana-Catalytica system is depicted in Scheme 2.16 The Periana-Catalytica system was significantly better than the Shilov system using K2PtCl4 due to the good solubility of Pt complexes with organic ligands.17 More recently, a simplified Pt catalyst, (DMSO)2PtCl2, has been reported by Lee and co-workers.18 This catalyst in oleum shows a very high TON of close to 20000 and the highest MBS yield of 85% for 3 h under 35 bar of CH4 and at 453 K. The role of the DMSO ligand, instead of bpym, is to increase the catalyst's stability and prevent deactivation to form PtCl2 or PtO2.18 The Hg and Pt catalysts have a high reactivity in methane oxidation to MSA or MSB with high selectivity; however, these catalysts require prohibitively high Hg- and Pt-inventory costs. A practical metal-free process for producing MSA with 99% selectivity and 80% conversion of methane using only methane and SO3 as reactants in H2SO4 has been also reported;19 however, we do not go into detail about a metal-free system here.
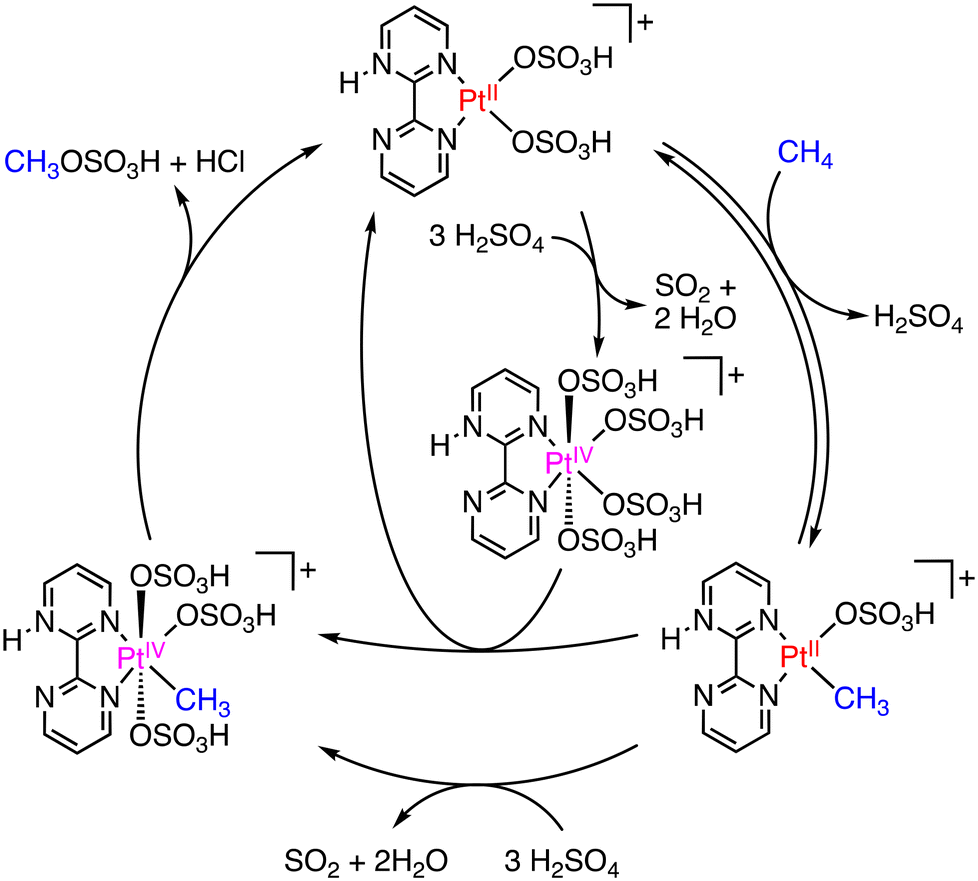 | ||
| Scheme 2 Plausible mechanism in the Periana-Catalytica system to produce MBS.16 | ||
The aforementioned Hg- and Pt-catalysed methane-to-MSA or MBS conversion processes exhibit high methane conversion and MSA or MBS selectivity; however, adding water is required to hydrolyse MSA or MBS for methanol production. Therefore, the catalytic system needs to be further improved toward direct conversion of methane to methanol.
Other than the examples reported by Periana and co-workers,15 C–H activation of CH4 by Pd complexes having an N-heterocyclic carbene (NHC) ligand has been investigated.20 PdII–NHC complexes catalysed the conversion of methane to CF3COOCH3 (MeTFA) with K2S2O8 as an oxidant at 363 K and 30 bar of methane in CF3COOH (HTFA) and (CF3CO)2O (TFAA) (Fig. 1a). Under optimized conditions, a TON of 30 was obtained using PdIIBr2L1 (L1: 1,1′-dimethyl-3,3′-methylenebisimidazolium dichloride) for 14 h as a catalyst (Fig. 1b).20a When an ethylene-bridged complex, PdIICl2L2 (L2: 1,1′-dimethyl-3,3′-(1,2-dimethylene)bisimidazolium dichloride), was used as a catalyst (Fig. 1b), the TON was improved to 33 for 17 h.20b In 2009, Strassner and co-workers reported the pyrimidine–NHC PdII complex [PdIICl(L3)2][PdIICl3(dmso)] (L3: 1-(2-pyrimidyl)-3-(methyl)imidazolium chloride) (Fig. 1b), which could functionalize methane to MeTFA with the highest TON of 41 for 17 h.20c These Pd–NHC complexes are suitable for C–H activation; the extraordinary thermal and chemical stability allows the reaction to proceed due to the suppression of the catalyst deactivation to form Pd black.20
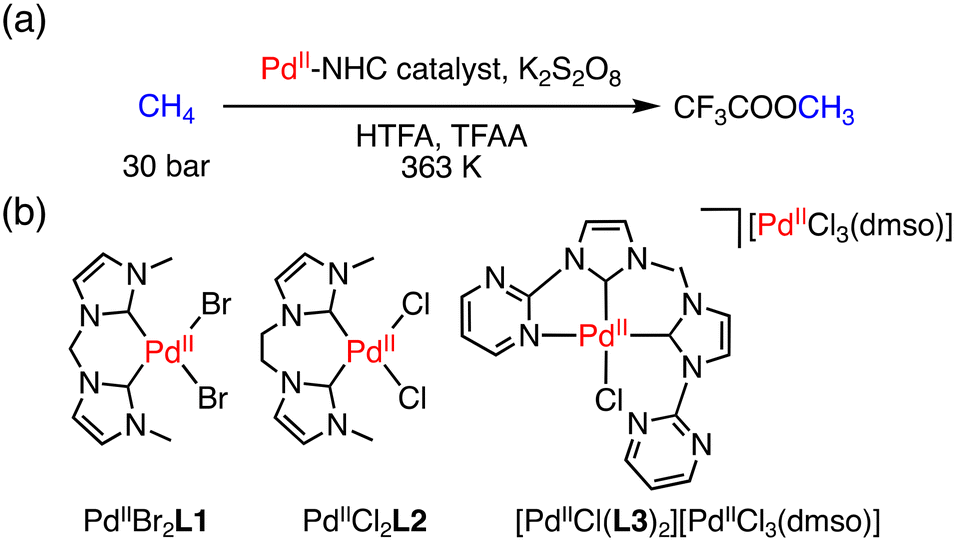 | ||
| Fig. 1 (a) A schematic representation of conversion of methane to MeTFA; (b) structures of PdIIBr2L1, PdIIBr2L1 and [PdIICl(L3)2][PdIICl3(dmso)] as catalysts.20 | ||
On the other hand, Pd-catalysed oxidative functionalization of CH4 has been achieved using O2 as a terminal oxidant in HTFA in the presence of NaNO2 as an NO source, although TONs are low (1–7) based on [PdII].21 In this reaction, CH4 is assumed to undergo C–H activation at the PdII centre of PdII(OAc)2 followed by ligand substitution to form a cis-[CF3COO–PdII–CH3] intermediate, which affords MeTFA as the product through reductive elimination. The Pd0 species formed via the reductive elimination is oxidized to a PdII species by quinone (Q). The quinone is reduced to hydroquinone (H2Q), which is re-oxidized by NO2 to regenerate PdII species. NO2 is reduced to NO in the course of oxidation of H2Q to Q and NO is oxidized by O2 to recover NO2 (Scheme 3).21
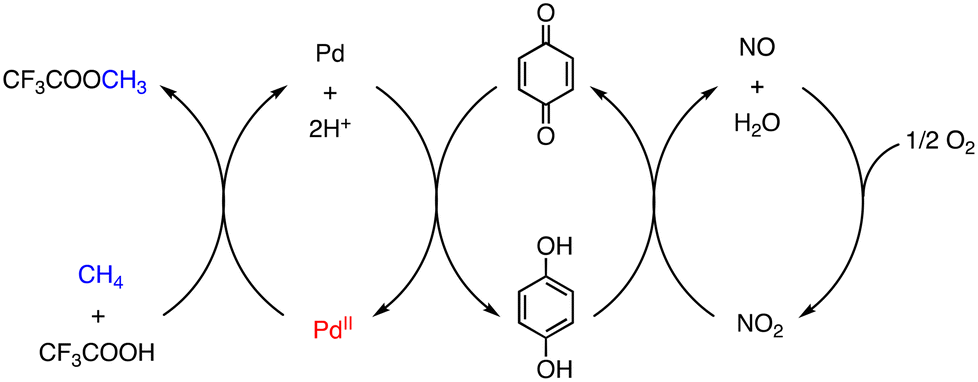 | ||
| Scheme 3 Proposed reaction mechanism of a PdII/Q/NO2/O2 system to oxidize CH4 in CF3COOH.21 | ||
As another example of metal-salt-catalysed direct conversion of methane into MeTFA, Zeller and co-workers have reported a Co-catalysed oxidation system.22 Yields of up to 50% based on methane were obtained using Co(OAc)2·4H2O as a pre-catalyst in HTFA/TFAA = 10:60 at 453 K under 20 bar of methane and 10 bar of O2 as a terminal oxidant (Scheme 4). In this system, TFAA acts as an inhibitor of the deactivation of the catalyst by preventing the precipitation of the Co catalysts.22
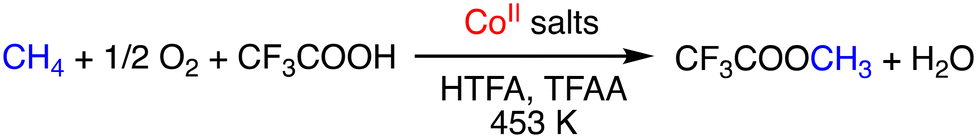 | ||
| Scheme 4 Schematic representation of Co salt catalysed direct conversion of methane to MeTFA.22 | ||
In 2022, Lee and co-workers reported that PdCl42− can act as a catalyst to effectively convert methane to CF3COOMe by using K2S2O8 as an oxidant in HTFA in the presence of TFAA to remove water generated.23 Under optimized conditions (catalyst: 0.01 mmol, [K2S2O8] = 10 mmol, [TFAA] = 24 mmol, HTFA: 30 g, under 20 bar of methane, 353 K, 15 h), [Me4N]2[PdCl4] shows the highest level of MeTFA production (TON 330, 33% selectivity) from methane oxidation.23 The main overoxidized by-product is CO2. The high catalytic reactivity of [Me4N]2[PdCl4] is derived from its ionic properties: a larger amount of PdCl42− can dissolve in the polar protic HTFA compared to the neutral PdII species, promoting the formation of a larger number of active catalytic species in the solution. During the catalytic reaction, PdCl42− is converted to Pd(TFA)42− as a pre-catalyst. Upon dissociation of one of the TFA ligands from Pd(TFA)42−, the resulting complex reacts with methane to form cis-[CF3COO–PdIV–CH3] as an intermediate, which then undergoes reductive elimination to afford MeTFA as the product (Scheme 5).23
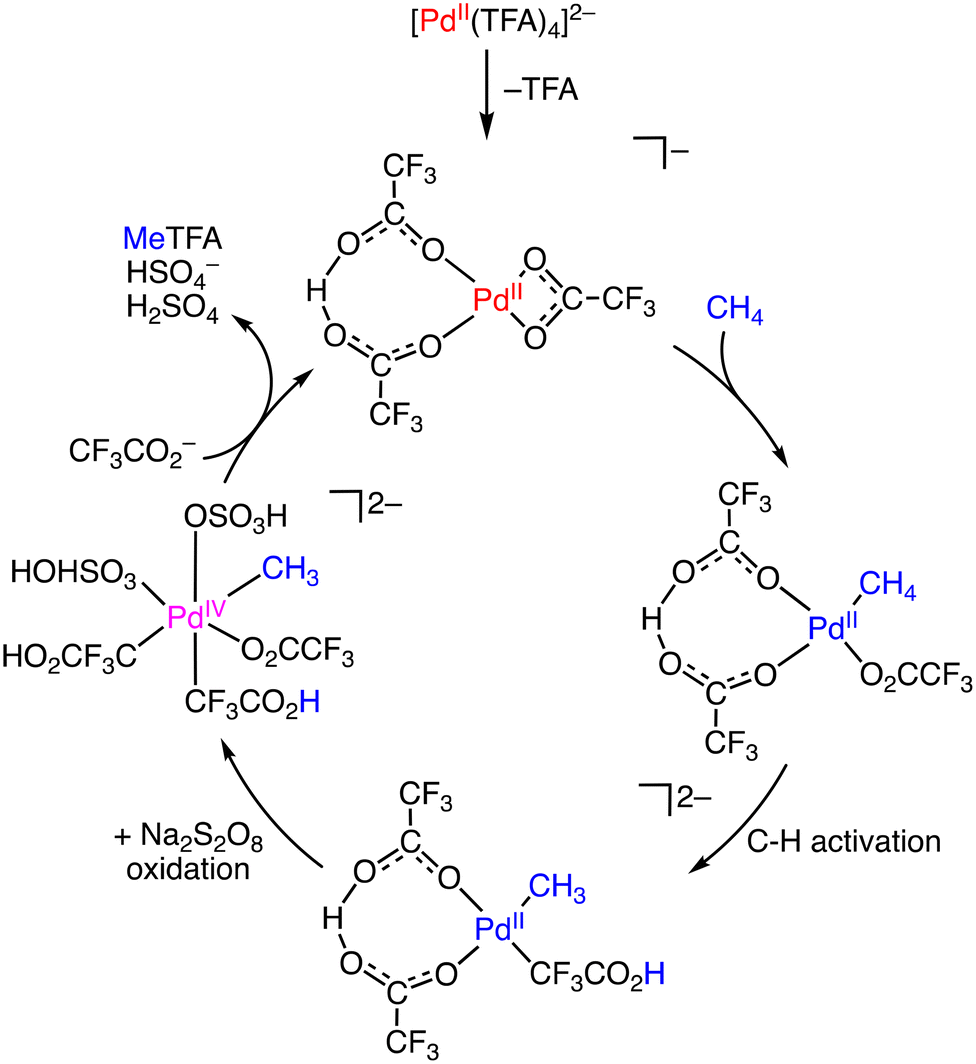 | ||
| Scheme 5 Plausible reaction mechanism for methane oxidation using Pd(TFA)42−.23 | ||
The direct conversion of methane to MeTFA depicted in Scheme 5 exhibits high selectivity with the use of metal salts or metal complexes as catalysts and S2O82− or O2 as an oxidant in HTFA/TFAA. In addition, van Bokhoven and co-workers reported the same conversion with a TON of 33 for 17 h.24 In this system, CuO has been employed as a pre-catalyst to form in situ an unknown homogeneous catalyst.24
Other than the conversion of methane to MeTFA, C–H activation of CH4 using transition metal complexes has been investigated using 4d- and 5d-transition metal complexes as catalysts. Two kinds of catalytic borylation of CH4 have been achieved independently.25 Sanford and co-workers have reported C–H borylation of CH4 (2800 to 3500 kPa) using Ir, Rh, and Ru complexes as catalysts with bis-pinacolborane (B2pin2) in cyclohexane at 423 K as described in Scheme 6.25 The products obtained are CH3Bpin, CH2(Bpin)2, c-C6H11Bpin, and HBpin. The product distribution can be controlled by the choice of catalysts. A dinuclear Ru(II) complex (Scheme 6), which acts as a pre-catalyst showing an induction period, afforded the ratio between CH3Bpin and CH2(Bpin)2 of 21:1 and the highest selectivity between CH3Bpin and c-C6H11Bpin (83:1).25 On the other hand, a Rh(I) complex (Scheme 6) exhibited the highest turnover number of 68 for 14 h with the ratio between CH3Bpin and c-C6H11Bpin of 46:1 and that between CH3Bpin and CH2(Bpin)2 of 18:1 at the concentration of 0.75 mol% of the catalyst. The competitive reactions have been examined using mixtures of CH4 (3500 kPa, 1.1 M) and CH3Bpin (0.13 M, 1 equivalent to (Bpin)2) as substrates, demonstrating that the reaction rate of CH3Bpin formation is faster than that of the borylation of CH3Bpin to afford CH2(Bpin)2 for the Ru catalyst; however, the Rh complex showed the opposite reactivity with a faster rate of borylation of CH3Bpin than that of CH4.25 Note that the catalysts exhibited higher reactivity in CH4 borylation than that of ethane.
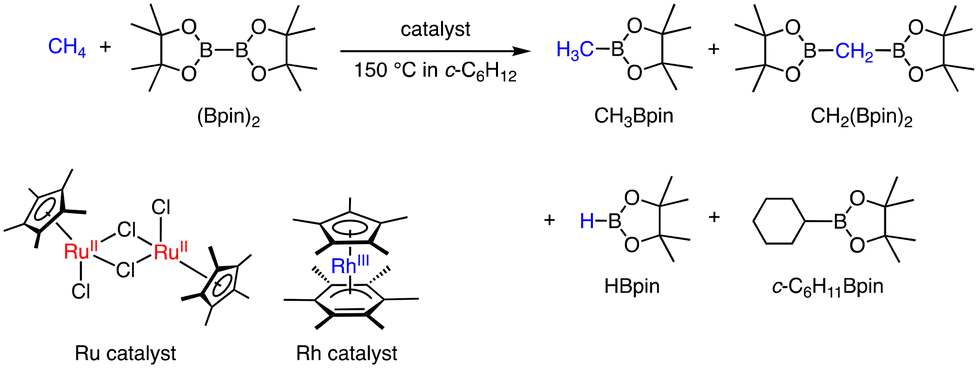 | ||
| Scheme 6 Catalytic borylation of CH4 by a Ru or Rh complex in c-C6H12 at 423 K.25 | ||
At the same time, Mindiola and co-workers have reported catalytic borylation of CH4 using Ir complexes bearing 1,10-phenathroline (phen) and its derivatives as ligands at 393 K in c-C6H12 or THF.26 They used dinuclear IrI complexes, [IrI(COD)(μ-X)]2 (COD = cyclooctadiene; X = Cl, OMe), as pre-catalysts. The phen ligands are added in a 2:1 molar ratio to the dimeric IrI complexes. In the case of the methoxo derivative, the products derived from CH4 are CH3Bpin, CH2(Bpin)2, and HBpin as in the case mentioned above.25 Concomitantly, CH3OBpin and ClBpin are also obtained as by-products derived from the bridging ligands, indicating that the dinuclear complexes turn to be mononuclear species during the reaction. The proposed reaction mechanism is depicted in Scheme 7.26 The dimeric precursor complex is converted to a five-coordinated square-pyramidal IrI(phen) complex through the elimination of the bridging ligand and coordination of phen. The five-coordinate intermediate, which has been rationalized by DFT calculations, reacts with CH4 to generate a cis-CH3, H–IrIII(phen) complex through oxidative addition of CH4. The intermediate is proposed to undergo isomerization, followed by reductive elimination of CH3–Bpin as a product. Mindiola and co-workers have also reported catalytic borylation of CH4 using [IrI(COD)(dmpe)] (dmpe = 1,2-bis(dimethylphosphino)-ethane) attached onto amorphous silica as a catalyst in c-C6H12 or c-C8H16 at 423 K.27 The TON of borylation of CH4 has been 12-fold improved by binding the molecular catalyst to the silica surface through the coordination of a Si–O− moiety to the Ir centre. A proposed reaction mechanism is similar to that depicted in Scheme 7.27
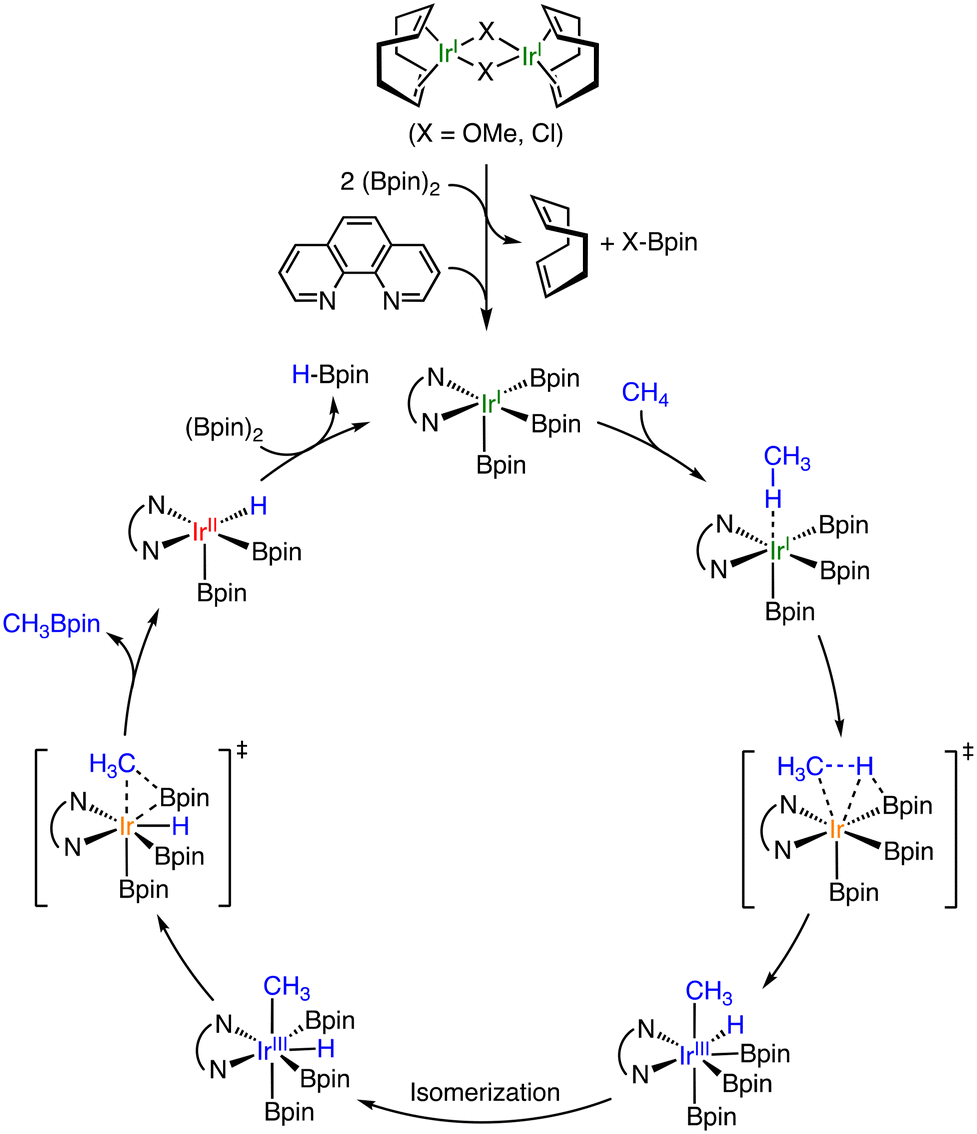 | ||
| Scheme 7 A proposed mechanism of catalytic borylation of CH4 by an Ir complex having phen as an auxiliary ligand.26 | ||
Pérez and co-workers have reported that Ag complexes bearing perfluorinated tris(indazolyl)borate ligands catalyse the reaction of methane with ethyl diazoacetate (N2CHCOOEt) to yield ethyl propionate (CH3CH2COOEt).28 F27–Tp4Bo,3CF2CF3Ag (Fig. 2(a)) as a catalyst has afforded a TON of 478 in supercritical CO2 (sc-CO2) at 313 K and PCH4:PCO2 = 160:90 (Fig. 2a). In this reaction, sc-CO2 was used as the solvent to dissolve the perfluorinated silver catalyst.28 A plausible mechanism for C–H functionalization of methane to ethyl propionate involves Ag-catalysed N2 elimination from ethyl diazoacetate followed by carbene transfer as depicted in Fig. 2(b).28
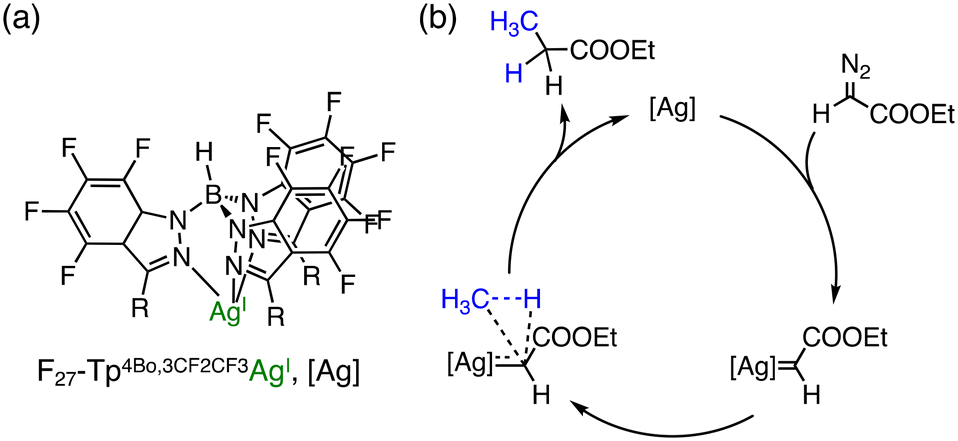 | ||
| Fig. 2 (a) Schematic representation of F27–Tp4Bo,3CF2CF3Ag; (b) a plausible mechanism of Ag-catalysed methane functionalization in supercritical CO2.28 | ||
3. Formation of acetic acid using methane and CO
The oxidative coupling of methane with CO to form acetic acid has significant implications for the large-scale application of methane. Lin and Sen have reported a catalytic system that uses RhCl3 as a catalyst and operates in an aqueous medium at 373 K (Scheme 8).29 RhCl3 acts as a catalyst in the production of acetic acid, methanol, and formic acid in the presence of methane (800 psi), CO (200 psi), and O2 (100 psi). Additives such as HI and KI serve as promoters of the conversion to acetic acid by accelerating the formation of Rh–CH3 species from methanol via methyl iodide formed in situ. Rh–CH3 is then carbonylated to form a Rh–C(O)CH3 species, which is subsequently hydrolyzed to form acetic acid. In the presence of KI (0.025 M), the yield of acetic acid has been improved more than two-fold from 34% to 80%.29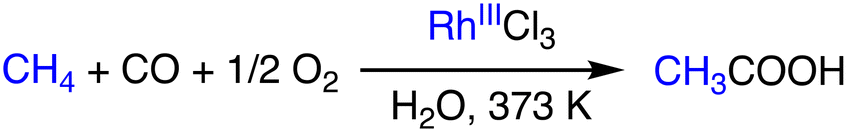 | ||
| Scheme 8 Schematic representation of methane to acetic acid conversion using RhCl3.29 | ||
On the other hand, Periana and co-workers reported a Pd/H2SO4 system to afford acetic acid from methane.30 The reaction is catalysed by Pd, and the results are consistent with a tandem catalysis mechanism, which involves methane C–H activation to generate Pd–CH3 species affording methanol. Methanol is also converted to CO to react with the Pd–CH3 intermediate to produce acetic acid. The origin of carbon sources of acetic acid has been confirmed by isotopic labelling experiments as shown in Scheme 9; with the use of a mixture of 12CH4 and 13CH3OH,12CH313COOH was obtained as the product without forming 13CH313COOH or 13CH312COOH.30
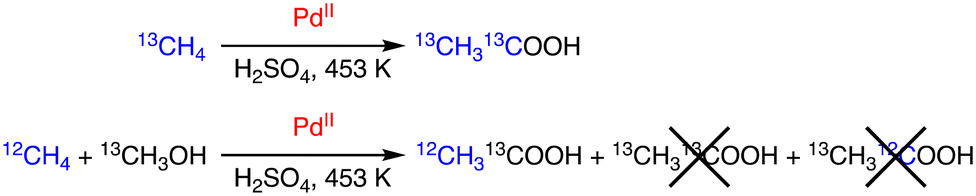 | ||
| Scheme 9 Isotopic labelling for clarifying the origin of carbon sources of acetic acid.30 | ||
In 2007, Pombeiro and co-workers reported that vanadium complexes with N,O- or O,O-ligands can efficiently convert methane into acetic acid in HTFA under ambient conditions, using a peroxodisulfate salt, K2S2O8, as an oxidant at 353 K.31 The most effective catalysts were found to be triethanolaminate or (hydroxyimino)dicarboxylates, which lead to CH3COOH production in 50% yield and high TONs up to 5.6 × 103.31 Carboxylation proceeds through free radical mechanisms involving the sequential formation of CH3˙, CH3CO˙, and CH3COO˙ upon H-abstraction. In this reaction, S2O82− has been proposed to act as a source of sulfate radical anions (SO4˙−) and their protonated form (HSO4˙) that may act as H-abstractors from methane, as an oxidizing agent for vanadium, and as an oxidizing and coupling agent for CH3CO˙. TFA is involved in the formation of CH3COOH by carbonylating CH3˙, acting as an H-source to CH3COO˙, and enhancing the oxidizing power of a peroxo-VV complex upon protonation.31
4. Photochemical oxidation of methane
In 2022, Groves and co-workers reported the photo-driven oxidation of methane to MeTFA using a commercial FeIII source as a catalyst and dioxygen as the terminal oxygen in a mixture of HTFA and TFAA at a 4:1 ratio.32 Fe(OTf)3 was found to exhibit the best catalytic activity, producing a 60% yield of MeTFA with a TON of 24 based on the amount of FeIII salt, without detectable overoxidation products. The reaction conditions involve a catalyst loading of 0.025 mmol, 15 mmol O2, 100 psig of methane, 278 K, 370 nm LED, and 24 h reaction time. TFAA plays a crucial role in trapping H2O as a by-product from O2, similarly as mentioned above, to prevent deactivation of the FeIII(TFA)3 cluster (Scheme 10).32 Note that the reaction mechanism of this system has yet to be clarified.
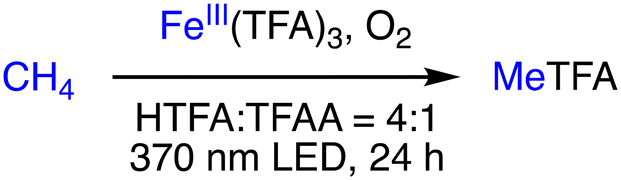 | ||
| Scheme 10 Schematic representation of photocatalytic oxidation of methane to MeTFA.32 | ||
Photochemical functionalization of methane has been reported using CeIV salts as catalysts in CH3CN under photoirradiation at 400 nm in the presence of 2,2,2-trichloroethanol (Cl3CCH2OH).33 In the catalytic system, methylation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of EtO(O)CC(H)C(C(O)OEt)2 was achieved to give the corresponding mono-methylated product in 58% yield (Scheme 11). Isoquinoline is methylated at the 1-position through Minisci-type nucleophilic alkylation in 19% yield. Di-tert-butyl azodicarboxylate (DBAD) is also methylated under the conditions to form an N-methyl hydralazine derivative through C–N bond formation. In those reactions, the initiation is LMCT in a CeIV–OCH2CCl3 complex to generate a CeIII species and an alkoxo radical, ˙OCH2CCl3, which can abstract a hydrogen atom from CH4 to form the methyl radical, ˙CH3, as a nucleophilic reactant to CC double bonds as well as aromatic rings.33
C double bond of EtO(O)CC(H)C(C(O)OEt)2 was achieved to give the corresponding mono-methylated product in 58% yield (Scheme 11). Isoquinoline is methylated at the 1-position through Minisci-type nucleophilic alkylation in 19% yield. Di-tert-butyl azodicarboxylate (DBAD) is also methylated under the conditions to form an N-methyl hydralazine derivative through C–N bond formation. In those reactions, the initiation is LMCT in a CeIV–OCH2CCl3 complex to generate a CeIII species and an alkoxo radical, ˙OCH2CCl3, which can abstract a hydrogen atom from CH4 to form the methyl radical, ˙CH3, as a nucleophilic reactant to CC double bonds as well as aromatic rings.33
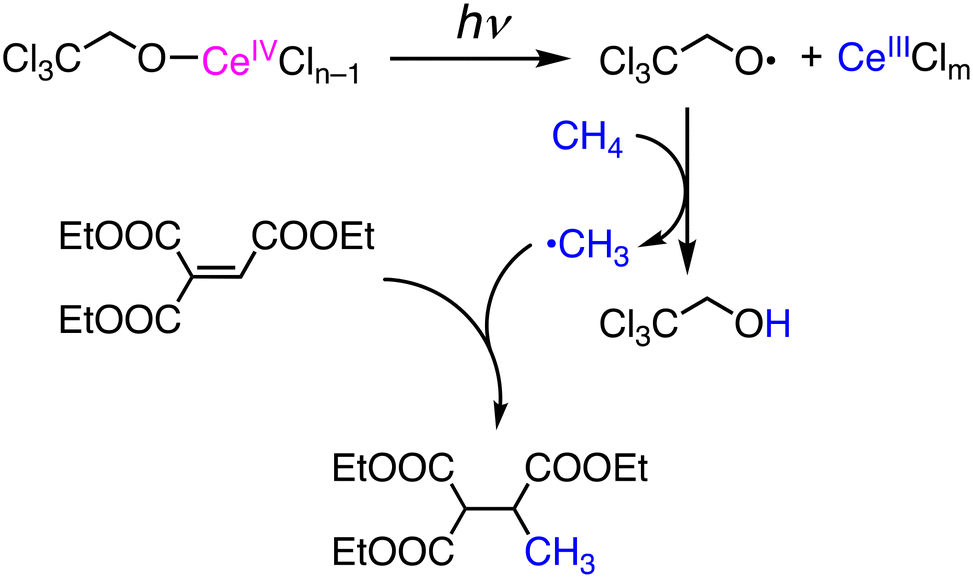 | ||
| Scheme 11 Photoinduced functionalization of CH4 based on LMCT in a CeIV–alkoxo complex to generate an alkoxy radical.33 | ||
Another proposal has been raised by Schelter and co-workers on the Ce-catalysed methane oxidation reaction against the mechanism depicted in Scheme 11.33,34 [NEt4]2[CeCl6] catalyses a coupling reaction of methane with diazo compounds to afford methylated hydrazine derivatives under photoirradiation at 390 nm at room temperature. The reaction is initiated by the formation of Cl˙ as the oxidant through the photoinduced homolysis of a CeIV–Cl− bond (Scheme 12). A yield of methane functionalization of 66% was obtained in the presence of [NEt4]Cl (25 mol%) and HOCH2CCl3 (20 mol%).34 The presence of Cl− in the reaction solution strongly affects the reactivity of the photocatalyst. This effect is due to the complexation of Cl˙ with Cl−, which can either stabilize or destabilize the radicals and affect the overall reaction pathway (Scheme 12).34 The participation of Cl˙ has been rationalized by the formation of chlorinated products in the presence of excess Cl−.34
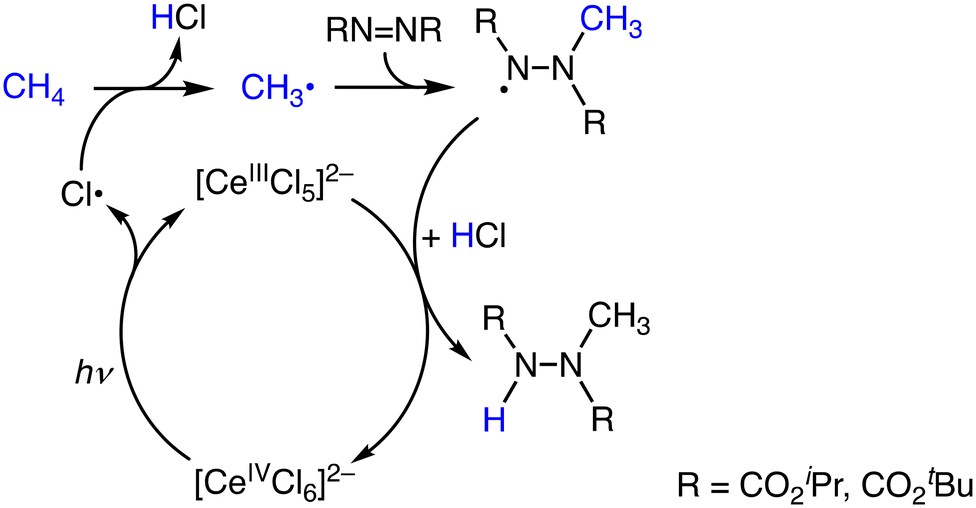 | ||
| Scheme 12 Plausible reaction mechanism of the catalytic amination of methane using [NEt4]2[CeCl6].34 | ||
More recently, Noël and co-workers reported a new strategy to methane functionalization through hydrogen atom transfer using inexpensive decatungstate (W10O324−) as a photocatalyst at room temperature (Fig. 3).35 W10O324− undergoes hydrogen atom transfer from methane (45 bar) under photoirradiation at 365 nm to generate the methyl radical (CH3˙), which reacts with alkenes to afford hydroalkylated adducts in good yields and high selectivity in CD3CN:H2O (7:1) at room temperature under flow conditions.35 The reaction mechanism involves an excited state of [W10O32]4− (*[W10O32]4−) to abstract a hydrogen atom from a C(sp3)–H bond of methane. After the formation of CH3˙, hydroalkylated adducts can be formed by trapping the radical with a variety of Michael acceptors (Fig. 3).35
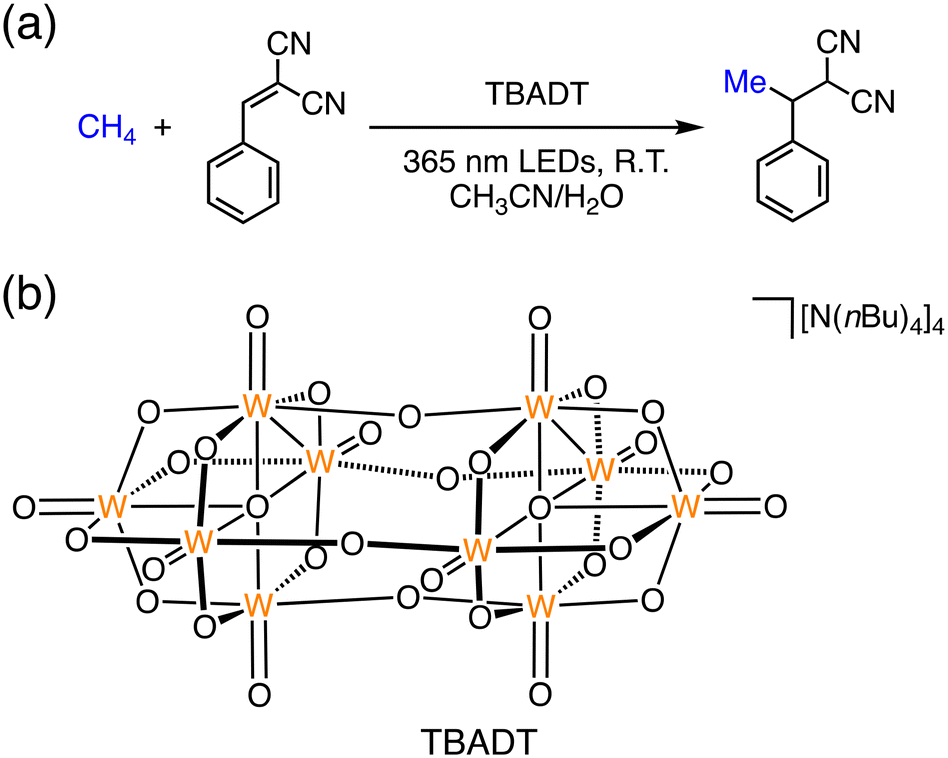 | ||
| Fig. 3 (a) Schematic representation of the C(sp3)–H functionalization of methane; (b) schematic description of the structure of [N(nBu)4]4[W10O32] (TBADT).35 | ||
5. Catalytic oxidation of methane using peroxides
The first example of a molecular catalyst that can convert methane to methanol directly was reported in 1991 by Drago and co-workers with the use of H2O2.36a The sterically crowded cis-[RuII(dmp)2(S)2]2+ (dmp = 2,9-dimethyl-1,10-phenanthroline) (S = MeCN or OH2) complex is capable of methane hydroxylation under mild conditions using H2O2 as an oxidant (catalyst: 0.1 μmol, 30% aqueous H2O2: 0.1 mmol, under 4 atm of methane, 348 K, 30 h) (Scheme 13).36a Methanol and formaldehyde were obtained as main oxidation products with a total TON of 125 per day and 75% methanol selectivity. The reactive species of the catalytic cycle is assumed to be cis-[RuVI(O)2(dmp)2]2+, which can be derived from a reaction of cis-[RuII(dmp)2(S)2]2+ with H2O2 as a reactive species of the oxidation.36b Hydrogen atom transfer from CH4 to the RuO moiety is followed by an oxygen-rebound process to form methanol bound to the Ru centre. Further ligand substitution of a bound methanol molecule with a solvent molecule (S) affords methanol and cis-[RuIV(O)(dmp)2(S)]2+.36b The cis-[RuVI(O)2(dmp)2]2+ oxidant can then be regenerated from cis-[RuIV(O)(dmp)2(S)]2+ in the presence of excess H2O2.36b
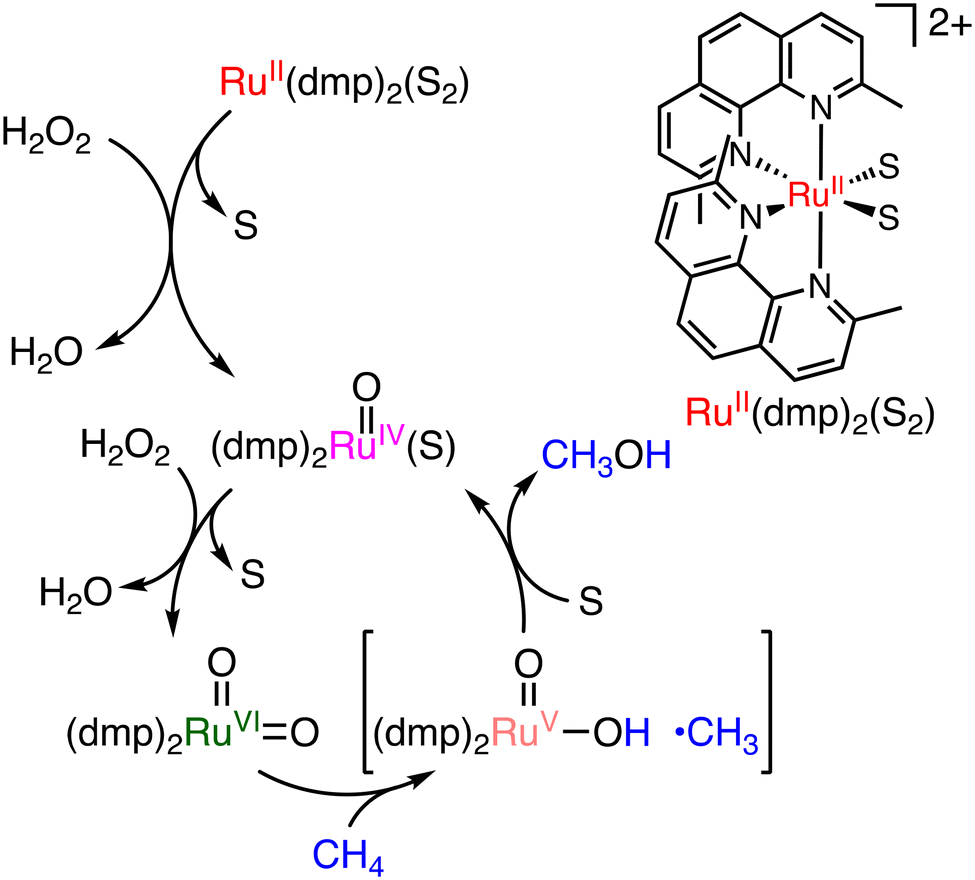 | ||
| Scheme 13 A plausible mechanism of methane oxidation using [RuII(dmp)2(S)2]2+ with H2O2 to form [RuVI(dmp)2(O)2]2+ as a reactive species (S = MeCN or H2O).36b | ||
A number of artificial catalysts have been developed to mimic the structures of pMMOs having multiple copper centres and sMMOs having a dinuclear iron site for catalysing methane oxidation under mild conditions.
As a functional model of pMMOs, in 2003, Chan and co-workers reported a tricopper cluster catalyst, [CuICuICuI(7-N-Etppz)]+ (7-N-Etppz: 3,3′-(1,4-diazepane-1,4-diyl)bis[1-(4-ethyl piperazine-1-yl)propan-2-ol]) (Fig. 4a), which converts methane to methanol in acetonitrile at room temperature using molecular oxygen and hydrogen peroxide.37,38 Under optimized conditions ([catalyst] = 8 mM, [H2O2] = 160 mM, [CH4] = 11 mM, under an O2 atmosphere, 1 h), the tricopper cluster catalyst exhibits a TON of 7 with 100% methanol selectivity.38 During the catalytic cycle, [CuICuICuI(7-N-Etppz)]+ reacts with molecular oxygen to form [CuIICuII(μ-O)2CuIII(7-N-Etppz)]+ as the active species, which is responsible for the methane oxidation to afford methanol. H2O2 has been proposed to be required as a reductant for regenerating [CuICuICuI(7-N-Etppz)]+ from [CuICuII(μ-O)CuII(7-N-Etppz)]+, which is an intermediate formed concomitantly with methanol (Fig. 4b).38,39 Furthermore, the TON of methane oxidation was improved to 18 under modified conditions, where 20 equivalents of H2O2 were used to initiate the reaction, followed by incremental dropwise additions of the same amount of H2O2 at 10 minute intervals.40 One of the advantages of this reaction is high methanol selectivity in methane oxidation. The disadvantages, however, include a low TON, the use of an organic solvent, and catalyst deactivation caused by hydrogen peroxide.41
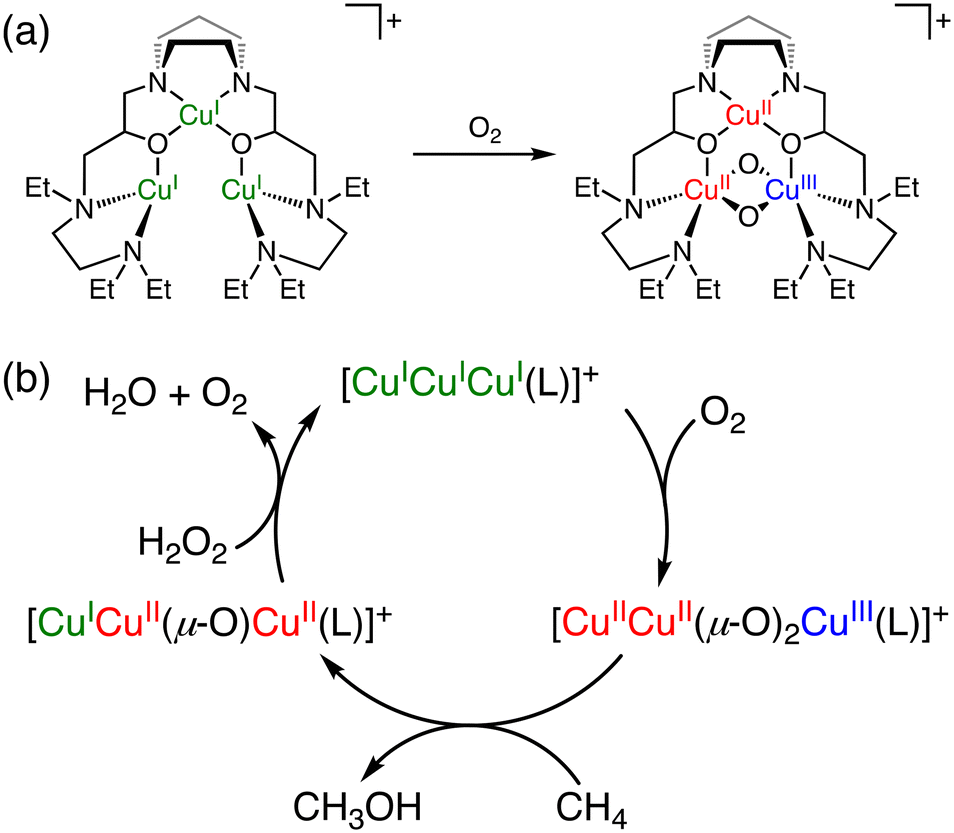 | ||
| Fig. 4 (a) The schematic representation of reaction of [CuICuICuI(7-N-Etppz)]+ with O2 to form [CuIICuII(μ-O)2CuIII(7-N-Etppz)]+.37,38 (b) A proposed mechanism of methane oxidation with [CuICuICuI(7-N-Etppz)]+ with the use of O2 and H2O2.38 | ||
More recently, Kodera and co-workers have reported a dinuclear Cu complex, [Cu2(μ-OH)(6-hpa)]3+ (hpa = 1,2-bis{2-[bis(2-pyridylmethyl)aminomethyl]pyridine-6-yl}ethane), which can oxidize methane to methanol and formaldehyde in MeCN/H2O (4:1) at 323 K using H2O2 as an oxidant.42 In this reaction, methyl hydroperoxide (CH3OOH) is obtained as the major product and formaldehyde as a minor product. The TONMeOH, TONHCHO and TONtotal were determined to be 43, 7.4 and 50.4 after the treatment of PPh3 to convert MeOOH to MeOH (Fig. 5a). In this catalytic system, [Cu2(O˙)(O2˙)(6-hpa)]2+ has been proposed to be formed as a reactive species through the reaction of [Cu2(μ-OH)(6-hpa)]3+ with H2O2 (Fig. 5b). Based on the DFT calculations, the CuII–O˙ moiety of the active species should have a high oxidation ability to cleave the strong C–H bond of methane.42
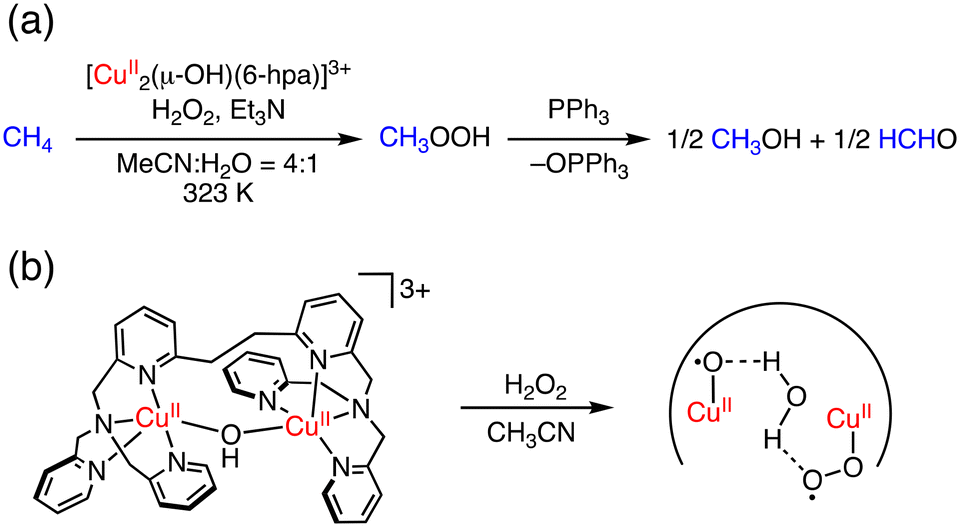 | ||
| Fig. 5 (a) Methane oxidation with H2O2 catalysed by [Cu2(μ-OH)(6-hpa)]3+. (b) Plausible active species of benzene oxidation by [Cu2(μ-OH)(6-hpa)]3+ with H2O2.42 | ||
Nitride-bridged dinuclear iron phthalocyanine complexes, (FePc)2N, are designed on the basis of the diiron active site of sMMOs.9 (FePc)2N contains 2 equivalent Fe centres with a +3.5-oxidation state linearly bridged by the nitride ligand (Scheme 14).43 In 2008, Bouchu and co-workers reported an iron μ-nitrido tetra-tert-butylphthalocyanine, (FePctBu4)2N complex, which can oxidize methane with H2O2 as an oxidant in water.44
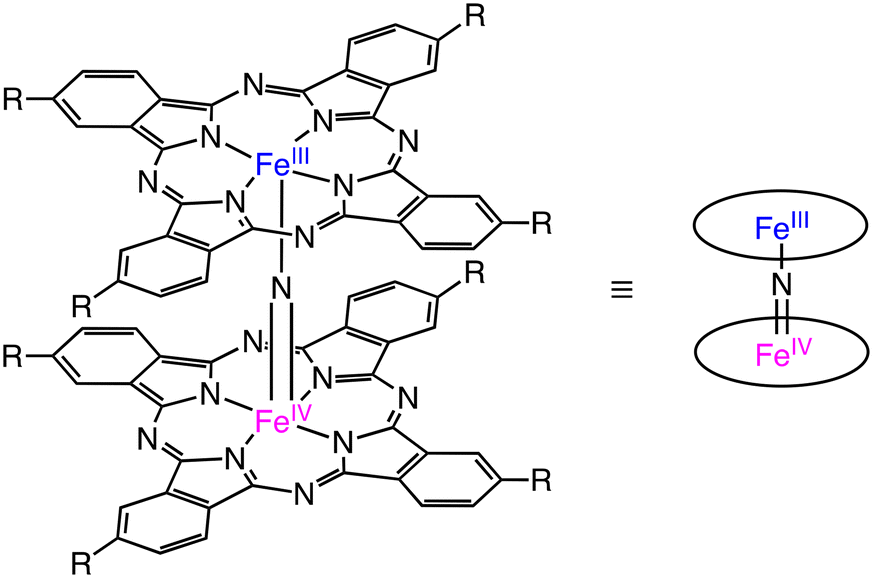 | ||
| Scheme 14 The schematic representation of (FePcR4)2N.43 | ||
Under optimized conditions (catalyst: 0.925 μmol, H2O2: 678 μmol, under 32 bar of CH4, 20 h), the maximum total TON of 84 was obtained at 323 K.44 The selectivity to methanol, formaldehyde, and formic acid was 0%, 32%, and 68%, respectively.44 The key step of the catalytic reaction is the formation of FeIVNFeVO by the reaction between (FePctBu4)2N and H2O2. FeIVNFeIVO or FeIVNFeVO was formed by the O–O bond cleavage in FeIVNFeIIIOOH. In the homolytic O–O bond cleavage, FeIVNFeIVO and the hydroxy radical (˙OH) would be formed. In this case, (FePctBu4)2N would be decomposed by the strongly oxidizing ˙OH. On the other hand, FeIVNFeVO and −OH were formed through the heterolytic O–O bond cleavage. In this catalytic system, the Fe–N–Fe unit of (FePctBu4)2N plays an important role in the catalytic reactivity by stabilizing the high-valent Fe–oxo intermediate with the strongly electron-donating μ-nitride ligand.44–46 Therefore, the proposed mechanism involves the heterolytic μ-nitrido tetra-tert-butylphthalocyanine cleavage of the O–O bond, leading to the formation of a highly oxidizing FeIVNFeVO species, which is generated by the release of an H2O molecule from the FeIVNFeIIIOOH complex in the presence of an acid (Scheme 15).44–46
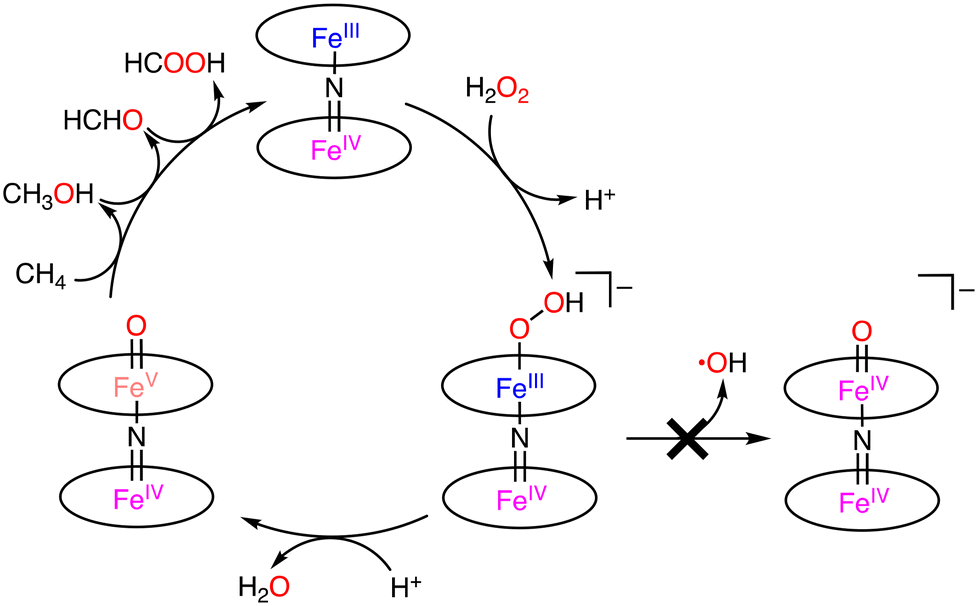 | ||
| Scheme 15 Proposed mechanism of the formation of active species in the system.44–46 | ||
In 2012, Sorokin and co-workers reported an oxo-diiron(IV) porphyrin π-radical complex, which was obtained from the reaction of a nitride-bridged diiron meso-tetraphenylporphyrin complex, [(TPP)FeIII(μ-N)FeIV(TPP)]0, with m-chloroperbenzoic acid (mCPBA) as an oxygen atom transfer reagent (Scheme 16).47 Catalytic methane oxidation was achieved using a silica-supported [(TPP)FeIII(μ-N)FeIV(TPP)]0 catalyst, [(TPP)FeIII(μ-N)FeIV(TPP)]0–SiO2, and 100 equivalents of mCPBA. The overoxidized product, formic acid, was obtained in a yield of 44% based on the oxidant. The result indicates that [(TPP)(μ-CBA)FeIV(μ-N)FeIV(O)(TPP˙+)]−, which is proposed as an active species of this catalytic system, should have a comparable oxidizing ability to that of putative FeIVNFeVO species derived from (FePctBu4)2N.44–47
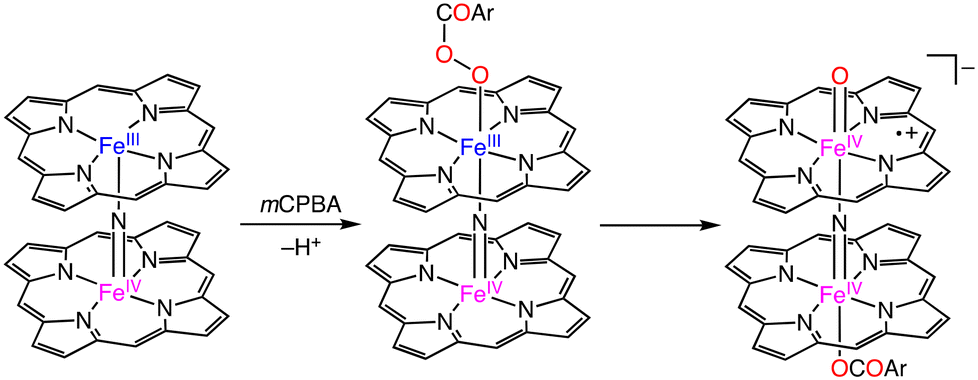 | ||
| Scheme 16 Proposed mechanism for the formation of the N-bridged high-valent diiron–oxo porphyrin cation radical complex.47 | ||
More recently, Tanaka and co-workers reported the utilization of supramolecular catalysts based on a nitride-bridged iron porphyrinoid dimer using a porphyrin–phthalocyanine heterodimer connected via a four-fold rotaxane structure (Scheme 17).48,49 The μ-nitride [(Por)Fe–N–Fe(Pc)]5+ complex with a terminal stopper and tetraanionic metalloporphyrin (M-TPPS4−, 5,10,15,20-tetrakis(4-sulfonatophenyl) porphyrin metal complex, M = Cu(II) or Ni(II)) resulted in the extension of the stacked structure to form [(Por)Fe–N–Fe(Pc)–M–TPPS]+ through π–π stacking and electrostatic interaction, as depicted in Scheme 17. Under optimized conditions ([silica supported catalyst] = 141 μM, [H2O2] = 160 mM, [HTFA] = 51 mM, under 1 MPa of methane, solvent: H2O, 333 K, 8 h), the reactivity of the supramolecular catalysts in methane oxidation was enhanced via electron donation through π–π stacking to afford a maximum total TON of ca. 50.50 Furthermore, Tanaka and co-workers reported (FePc(12-crown-4)4)2N and (FePcMe8)2N complexes bearing an electron-donating group on phthalocyanine moieties (Scheme 17).50,51 The catalytic activity of (FePc(12-crown-4)4)2N and H2O2 was lower than that of (FePctBu4)2N due to the decomposition of 12-crown-4 moieties during the reaction.50 In contrast, the total TON reached 100 using (FePcMe8)2N–SiO2 as a silica-supported catalyst and H2O2 as an oxidant.51 Sorokin and de Visser proposed that the introduction of electron-donating substituents is advantageous for H atom abstraction due to increasing the basicity of the oxo species.52 Based on the lack of the catalytic oxidation reaction in the presence of excess Na2SO3 as a radical scavenger, Tanaka proposed the possibility of a Fenton-type reaction,53–55 in which the ˙OH radical acts as a reactive species instead of FeIVNFeVO (Scheme 18).49–51 In addition, the bleaching of the colour of catalysts and overoxidation of oxidized products were observed.49–51
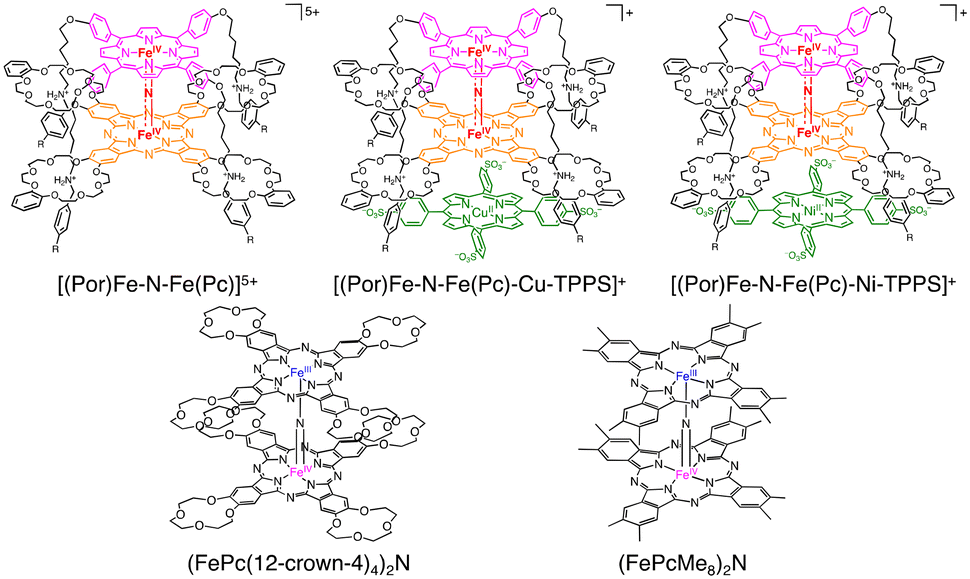 | ||
| Scheme 17 Schematic representations of supramolecular extension of a μ-nitrido-bridged dinuclear iron complex of a four-fold rotaxane heterodimer of a porphyrin and a phthalocyanine.49–51 | ||
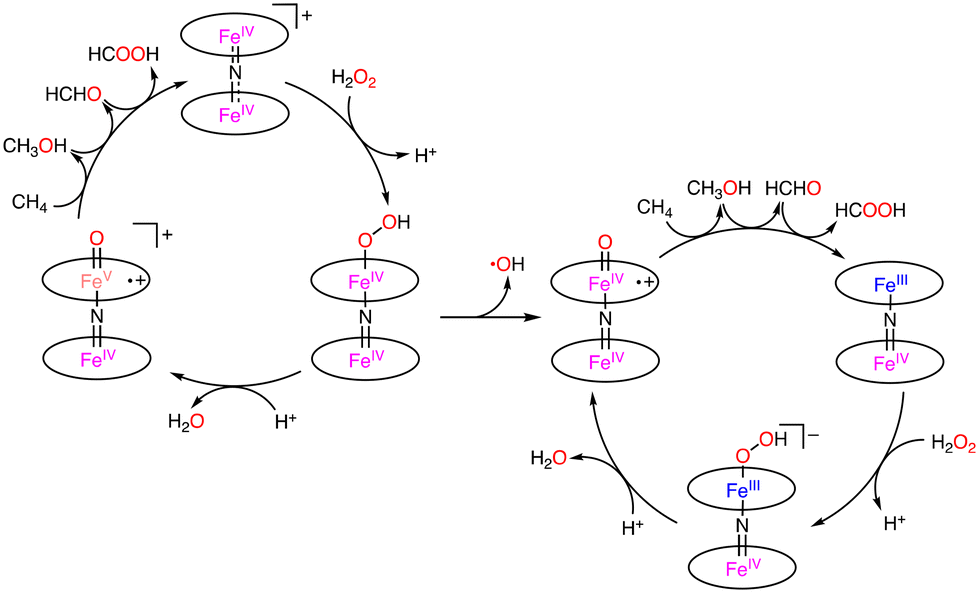 | ||
| Scheme 18 Plausible reaction mechanism for methane oxidation by [(Por)Fe–N–Fe(Pc)]5+.49–51 | ||
Therefore, strategies to suppress the overoxidation of the oxidized products and the decomposition of the catalyst need to be developed for efficient and selective oxidation of methane.
6. Molecular catalysts having hydrophobic moieties in the second coordination sphere for methane oxidation
To mimic the catalytic oxidation of organic substrates by natural enzymes, heme model complexes have been intensively studied and the high reactivity has been reproduced by the model complexes so far.56,57 In order to oxidize methane efficiently, a methane molecule should be trapped in the vicinity of an active metal centre, which can be converted to a reactive state as observed in enzymatic oxidation reactions.As for the introduction of a hydrophobic second coordination sphere (SCS) which can bind to a methane molecule in the close vicinity of a metal centre, Martinez and co-workers have reported the capture of a methane molecule by a hemicryptophane (Hm) moiety attached to metal–pyridylamine complexes as a hydrophobic cavity; the 1H NMR signal attributed to the methane molecule showed a significant upfield shift, indicating the interaction of the methane molecule with Hm.58
In methane oxidation reactions by using a silica-supported [CuII(Hm–TREN)]2+ (TREN: tris(2-aminoethyl)amine) catalyst and naked [CuII(TREN)]2+, total TONs of 2 and 1 have been obtained, respectively, indicating no significant changes in the reactivity (reaction conditions: silica supported catalyst: 1.0 μmol, [H2O2] = 0.33 M, [H2SO4] = 0.07 M, 30 bar of methane, 333 K, 20 h) (Scheme 19).58 Martinez and co-workers have reported oxido-vanadium(V) complexes that can act as catalysts for sulfoxidation of thioanisole with the use of alkyl hydroperoxides (tBuOOH and cumyl-OOH) as oxidants in CH2Cl2.59,60 The complexes have nitrilotriacetic-acid-based tripodal ligands and are referred to as VV(Hm–TKA), VV(Hm–BINOL–TKA), and VV(Bz–BINOL–TKA) bearing the respective hydrophobic SCS near the metal centre (Scheme 20).59,60
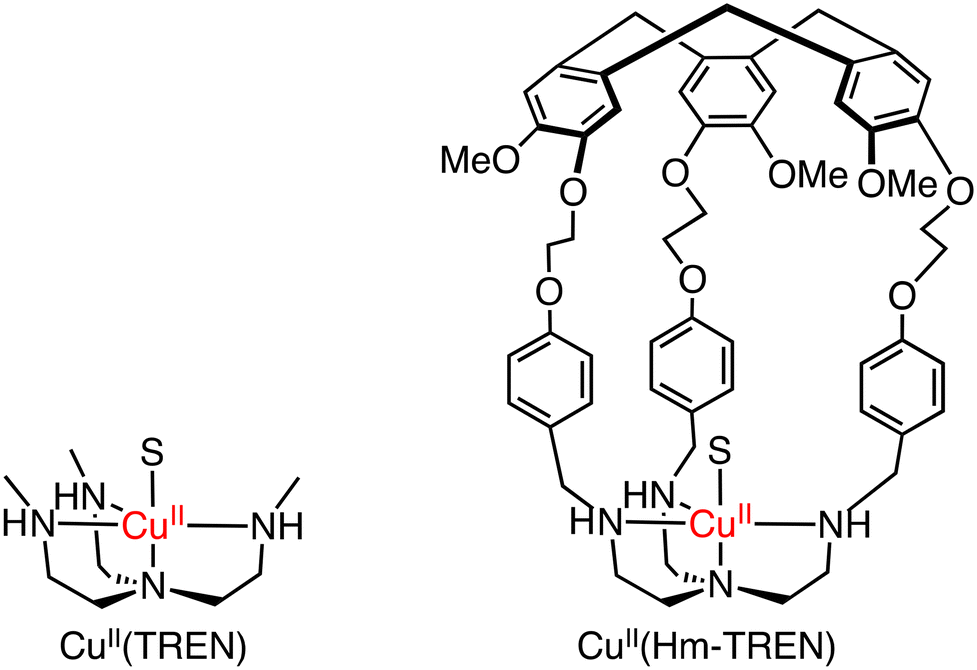 | ||
| Scheme 19 The schematic representations of Cu(TREN) and CuII(Hm–TREN). S: solvent.58 | ||
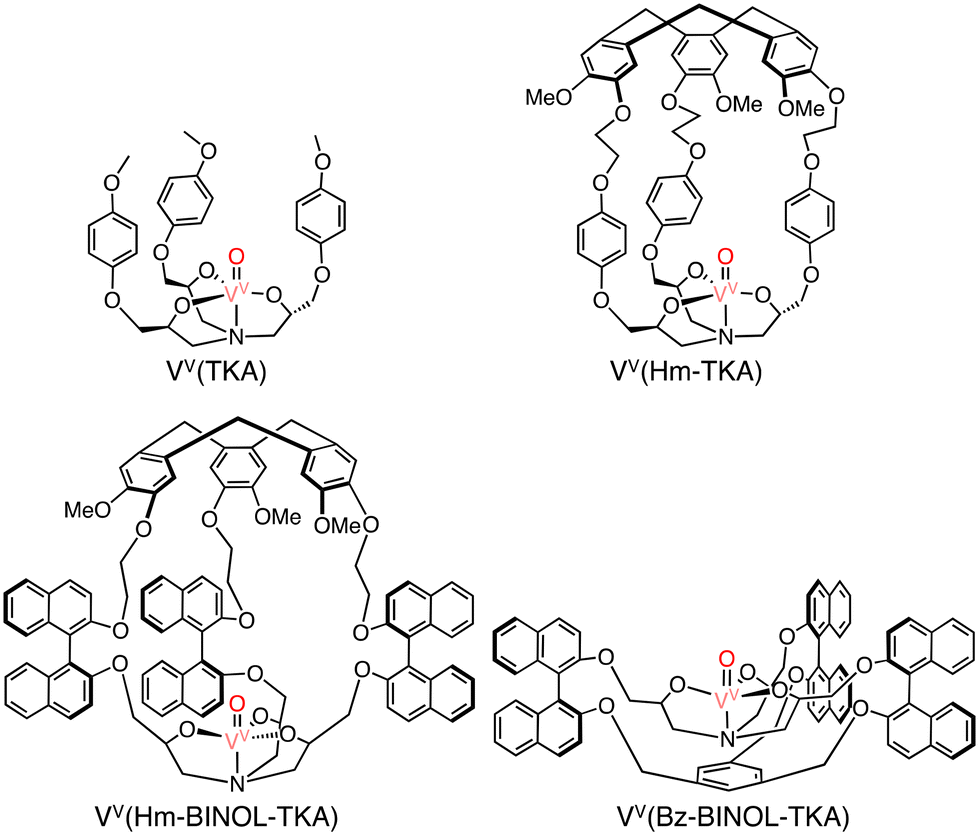 | ||
| Scheme 20 The schematic representations of VV(TKA), VV(Hm–TKA), VV(Hm–BINOL–TKA) and VV(Bz–BINOL–TKA).58 | ||
The same complexes have been applied as catalysts to methane oxidation. The introduction of a hydrophobic SCS led to an increase in the yield of oxidized products in oxidation of methane (30 bar) using H2O2 as an oxidant in H2O at 60 °C.58 VV(Bz–BINOL–TKA) was found to be the most effective catalyst with a TON of 18.3 (20 h), higher than those of VV(TKA), VV(Hm–TKA), and VV(Hm–BINOL–TKA), although the highest selectivity to 2-electron-oxidized products (CH3OH and CH3OOH) has been observed for VV(Hm–TKA) to be 15%.58 These results suggest that the size and shape of the hydrophobic SCS have a significant impact on the efficiency of methane oxidation. Methane oxidation was also achieved by using silica-supported [FeII(Hm–TPA)]2+ (TPA: tris(2-pyridyl-methyl)amine) having the Hm moiety at the TPA ligand as a catalyst (Scheme 21) and H2O2 as an oxidant. Introducing the Hm moiety to the Fe–TPA complex has improved the total TON from 4.7 to 9.7 and the selectivity to CH3OH and CH3OOH from 15% to 27%.58
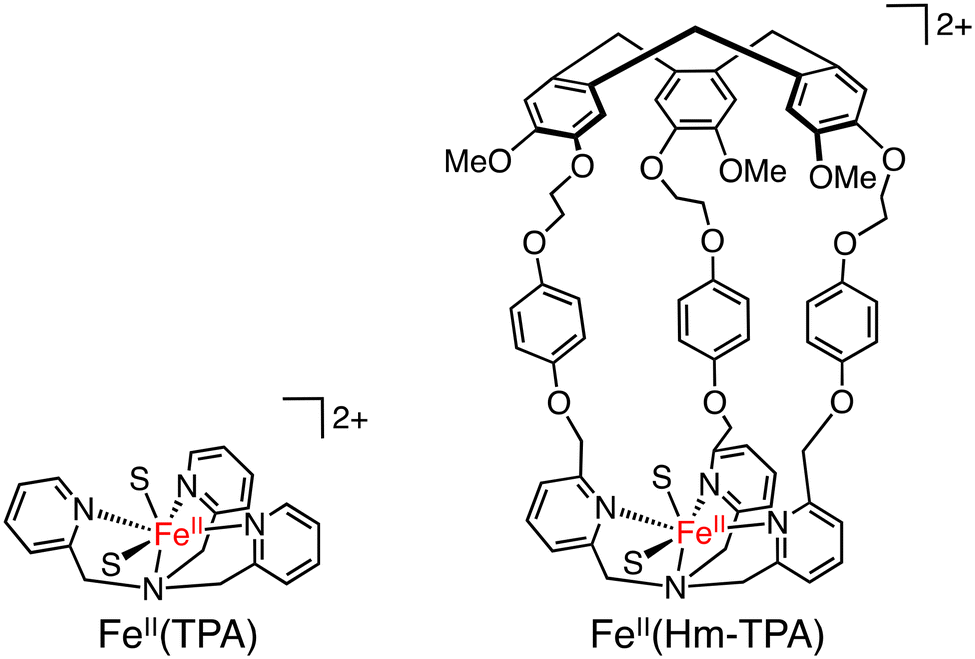 | ||
| Scheme 21 The schematic representations of FeII(TPA) and FeII(Hm–TPA). S: solvent.58 | ||
Although these results demonstrate the positive impact of the hydrophobic SCS on the efficiency in terms of TONs and the selectivity to 2-electron-oxidized products such as CH3OH and CH3OOH, very low TONs have been observed. Therefore, one of the most promising approaches to suppressing the overoxidation is the use of catalysts with a hydrophobic SCS close to the catalytically active metal centre (Schemes 19–21).
7. Fe–NHC complex having a hydrophobic cavity for “catch-and-release” oxidation of methane
Kojima and co-workers have synthesized FeII complexes bearing an N-heterocyclic carbene (NHC) ligand, [FeII(HPY4Cl2BIm)(OH2)]2+, which have been reported to exhibit the reactivity in C–H oxidation using Na2S2O8 as an ET oxidant in H2O, showing high selectivity to afford 2-electron-oxidized products.61 Based on this observation, as well as inspired by the arrangement of the hydrophobic cavity near the active iron centre in sMMOs, they have prepared FeII complexes bearing N-heterocyclic carbene ligands, [FeII(RPY4Cl2BIm)(NCMe)]2+ (R = mesityl (Mes), anthracenyl (Ant)).62 Among those FeII–NHC complexes prepared, [FeII(AntPY4Cl2BIm)(NCMe)]2+ has a hydrophobic SCS constructed from four anthracenyl moieties near a mononuclear iron centre (Fig. 6a).62 The iron catalyst can trap one methane molecule into the hydrophobic SCS in aqueous media. The association constant of methane with [FeII(AntPY4Cl2BIm)(OD2)]2+ at 298 K has been determined to be (2.1 ± 0.4) × 103 M−1, which is relatively high compared with the values reported so far for methane encapsulation.62–67 The iron catalyst having a densely surrounded and rigid hydrophobic SCS has allowed us to observe a total TON of 5.0 × 102 with 83% methanol selectivity in a 3 h methane oxidation reaction using sodium persulfate as an oxidant (reaction conditions: [catalyst] = 1.0 mM, [Na2S2O8] = 5.0 mM, P(CH4) = 0.98 MPa, T = 323 K, H2O:CH3CN = 95:5) (Fig. 6b). In this catalytic system, a hydrophobic CH4 molecule is captured in the hydrophobic SCS of [FeII(AntPY4Cl2BIm)(OH2)]2+, which is formed by ligand substitution of CH3CN in [FeII(AntPY4Cl2BIm)(NCMe)]2+ with H2O. [FeII(AntPY4Cl2BIm)(OH2)]2+ undergoes proton-coupled electron-transfer (PCET) oxidation to generate an FeIV–oxo complex ([FeIV(O)(AntPY4Cl2BIm)]2+), which hydroxylates the CH4 molecule (Fig. 6c). The hydroxylation affords a methanol-bound intermediate, FeII–O(H)CH3, as a result of the oxygen-rebound mechanism. The putative FeII–O(H)CH3 intermediate undergoes ligand substitution with H2O to release the hydrophilic methanol molecule into the aqueous media to accomplish the catalytic cycle.62 The high TON and methanol selectivity in catalytic methane oxidation have been achieved by trapping a hydrophobic methane molecule in the hydrophobic SCS and by releasing a hydrophilic methanol molecule from the hydrophobic SCS into the surrounding aqueous medium. Kojima and co-workers have proved the validity of the “catch and release” strategy mimicking the catalytic performance of sMMOs for efficient and selective conversion of methane to methanol in aqueous medium (Fig. 7).
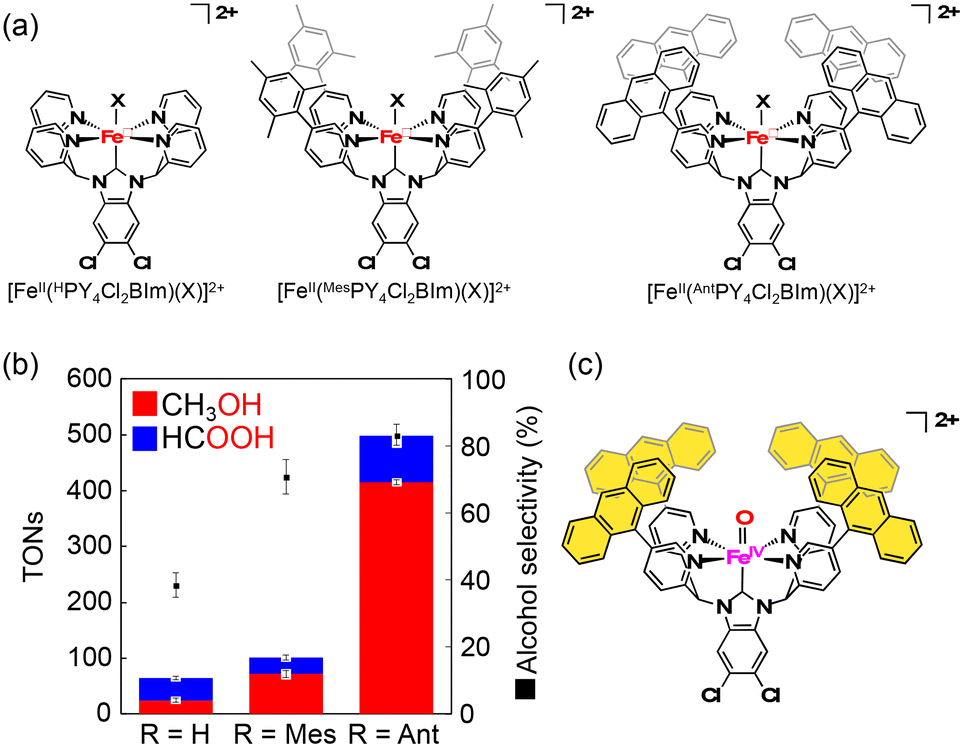 | ||
| Fig. 6 (a) Schematic representations of [FeII(RPY4Cl2BIm)(X)]2+ (R = H, Mes, Ant, X: OH2, OD2, NCCH3 or NCC6H5). (b) Comparison of TONs and alcohol selectivity among the three catalysts. (c) Schematic representation of active species for methane oxidation by [FeII(AntPY4Cl2BIm)(OH2)]2+ and Na2S2O8 in H2O:CH3CN = 95:5.62 | ||
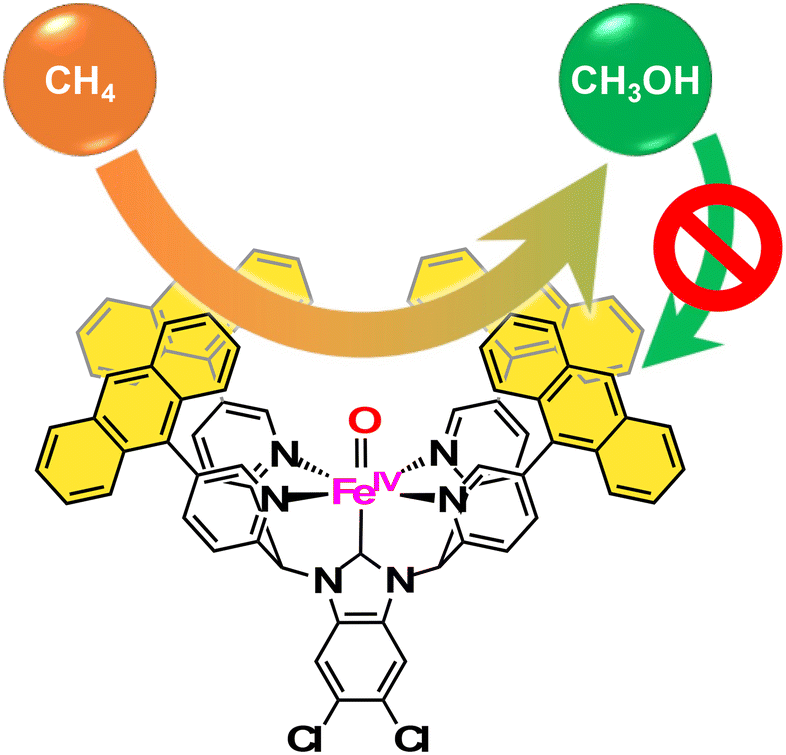 | ||
| Fig. 7 The concept of “catch and release” strategy to suppress the overoxidation for selective conversion of methane to methanol.62 | ||
8. Summary and outlook
In this minireview, we have surveyed homogeneous catalysts for functionalization of methane. Tables 1 and 2 summarize the state-of-the-art functionalization of methane using homogeneous molecular catalysts.| Catalysts and conditions | Solvent | Reaction time | Total TON | Product, selectivities | Ref. |
|---|---|---|---|---|---|
|
a Reaction conditions: SO3:CH4 = 6.9, 93.1 bar of methane, 673 K.
b Catalyst: 2.0 mmol, TFA: 2.0 mmol, 34.5 bar of methane, 453 K.
c [Catalyst] = 0.77 mM, 35 bar of methane, 453 K.
d [PdIICl(L3)2][PdIICl3(dmso)]: 0.084 mmol, K2S2O8: 8.4 mmol, HTFA: 32 mL, TFAA: 24 mL, 30 bar of methane, 363 K.
e Pd(OAc)2: 10 μmol, quinone: 20 μmol, NaNO2: 10 μmol, 1 atm of O2, 54 atm of methane, 353 K.
f Co(OAc)2·2H2O: 3.8 mol%, HTFA/TFAA = 10:60, 20 bar of methane, 10 bar of O2, 453 K.
g Catalyst: 0.01 mmol, [K2S2O8] = 10 mmol, [TFAA] = 24 mmol, HTFA: 30 g, under 20 bars of methane, 353 K.
h [CuO] = 9.4 mM, [K2S2O8] = 0.3 M, HTFA: 23 g, TFAA: 5 g, 5.2 bar of methane, 383 K.
i Catalyst: 3 mol%, [B2pin2] = 0.13 M, 3500 kPa of methane, 423 K.
j Catalyst: 0.5 mol%, dmpe: 1.0 mol%, B2pin2: 20 mol%, 3447 kPa of methane, 423 K.
k Catalyst: 0.035 mol%, B2pin2: 0.0512 mmol, 500 psi of methane, 423 K.
l F27–Tp4Bo,3CF2CF3Ag: 0.03 mmol, N2CHCOOEt: 3.0 mmol, 160 atm of methane, 313 K.
m [RhCl3] = 0.01 M, [HCl] = 0.13 M, methane (800 psi), CO (200 psi), and O2 (100 psi), 373 K.
n [PdSO4] = 20 mM, 27.2 atm of methane, 453 K.
o [VO{N(CH2CH2O)3}] = 0.04 μmol, K2S2O8: 4.2 mmol, 12 atm of methane, 15 atm of CO, 353 K.
p Fe(TFA)3: 0.025 mmol, O2: 15 mmol, under 100 psig of methane, 278 K, 370 nm LED.
q Ce(OTf)3: 0.01 mol%, CCl3CH2OH: 20 mol%, TBACl: 0.05 mol%, DBAD: 1 equivalent, 5000 kPa of methane, R.T. 400 nm LED.
r TBADT: 0.5 mol%, [alkene] = 0.02 M, 45 bar of methane, R.T., 365 nm LED.
|
|||||
| HgSO4a | H2SO4 | — | — | MSA, 38%; MBS, 62% | 12 |
| Hg(OSO3H)2b | H2SO4 | 3 h | 11 | MBS, 85% | 13 |
| K2PtCl4, K2PtCl6 | H2O | 0.25–5 h | <20 | MeOH, —; CHCl3, — | 14 |
| [(bpym)PtCl2]c | H2SO4 | 3 h | 500 | MBS, 84%; CO2, 16% | 15 and 18 |
| K2PtCl4c | H2SO4 | 3 h | 17000 |
MBS, 62%; CO2, 38% | 18 |
| (DMSO)2PtCl2c | H2SO4 | 3 h | 20000 |
MBS, 85%; CO2, 15% | 18 |
| [PdIICl(L3)2][PdIICl3(dmso)]d | HTFA, TFAA | 17 h | 41 | MeTFA, 100% | 20 |
| Pd(OAc)2, quinone, NaNO2, O2e | HTFA | 10 h | 7 | MeTFA, 100% | 21 |
| Co(OAc)2·4H2O, O2f | HTFA, TFAA | 24 h | 13 | MeTFA, 100% | 22 |
| [Me4N]2[PdCl4], K2S2O8g | HTFA, TFAA | 15 h | 330 | MeTFA, 45%; CO2, 55% | 23 |
| CuO, K2S2O8h | HTFA, TFAA | 17 h | 33 | MeTFA, 86% | 24 |
| (MesH)Ir(Bpin)3, B2pin2i | c-C6H12 | 14 h | 15 | CH3Bpin, 63%; CyBpin, 21%; CH2(Bpin)2, 16% | 25 |
| Cp*Rh, B2pin2i | c-C6H12 | 14 h | 33 | CH3Bpin, 89%; CyBpin, 1%; CH2(Bpin)2, 10% | 25 |
| Cp*RuCl(μ-Cl)2RuClCp*, B2pin2i | c-C6H12 | 14 h | 22 | CH3Bpin, 94%; CyBpin, 1%; CH2(Bpin)2, 5% | 25 |
| [Ir(COD)(μ-Cl)]2, dmpe, B2pin2j | c-C6H12 | 16 h | 104 | CH3Bpin, 75%; CH2(Bpin)2, 25% | 26 |
| [(dmpe)Ir(cod)CH3]–SiO2, B2pin2k | c-C8H16 | 16 h | 1857 | CH3Bpin, >99% | 27 |
| F27–Tp4Bo,3CF2CF3Ag, N2CHCOOEtl | Sc–CO2 | 14 h | 478 | CH3CH2COOEt, 100% | 28 |
| RhCl3, CO, O2, HCl, KIm | H2O | 352 h | 3.8 | CH3COOH, 71%; CH3OH, 2%; HCOOH, 27% | 29 |
| PdSO4n | H2SO4 | 7 h | 18 | CH3COOH, 72%; CH3OH, 17%; CO2, 11% | 30 |
| VO{N(CH2CH2O)3}, K2S2O8, COo | TFA | 20 h | 5.6 × 103 | CH3COOH, 100% | 31 |
| Fe(TFA)3, O2, hνp | HTFA, TFAA | 24 h | 24 | MeTFA, 56%; CO2, 44% | 32 |
| Ce(OTf)3, CCl3CH2OH, TBACl, DBAD, hνq | CH3CN | 18 h | 2900 | CH3N(Boc)NH(Boc), 100% | 33 and 34 |
| TBADT, alkenesr | CD3CN:H2O = 7:1 |
6 h | <200 | Corresponding hydroalkylated adducts, <90% | 35 |
| Catalysts | Oxidants | Solvent | Reaction time | Total TON | Selectivities | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| MeOOH | MeOH | HCHO | HCOOH | CO2 | ||||||
| a Reaction conditions: catalyst: 0.1 μmol, 30% aqueous H2O2: 0.1 mmol, under 4 atm of methane, 348 K. b [Catalyst] = 8 mM, [H2O2] = 160 mM, [CH4] = 11 mM, under an O2 atmosphere, 1 h, 278 K. c Catalyst: 0.03 μmol, H2O2: 300 μmol, Et3N: 0.3 μmol, 8 MPa of methane, 323 K. d Catalyst: 0.925 μmol, H2O2: 678 μmol, 32 bar of methane, 323 K. e Catalyst: 1.1 μmol, mCPBA: 105 μmol, 32 bar of methane, 333 K. f [Catalyst] = 55 μM, [H2O2] = 189 mM, [TFA] = 51 mM, 1 MPa of methane, 333 K. g Catalyst: 1 μmol, [H2O2] = 0.33 M, 30 bar of methane, 333 K. h [Catalyst] = 1 μM, [Na2S2O8] = 5 mM, 0.98 MPa of methane, 323 K. | ||||||||||
| cis-[Ru(dmp)2(OH2)2]2+a | H2O2 | H2O | 30 h | 125 | — | 75% | 25% | — | Trace | 36 |
| [CuICuICuI(7–N–Etppz)]+b | O2 | MeCN | 1 h | 18 | — | 100% | — | — | — | 40 |
| [Cu2(μ-OH)(6-hpa)]3+c | H2O2 | MeCN:H2O = 4:1 |
3 h | 50.4 | — | 85% | 15% | — | — | 42 |
| (FePctBu4)2Nd | H2O2 | H2O | 20 h | 84 | — | — | 75% | 25% | — | 43 |
| [(TPP)FeIII(μ-N)FeIV(TPP)]0–SiO2e | mCPBA | H2O | 3 h | 41 | — | — | — | 100% | — | 47 |
| [(Por)Fe–N–Fe(Pc)]5+–SiO2f | H2O2 | H2O | 8 h | 30 | — | 32% | 6% | 62% | — | 49 |
| [(Por)Fe–N–Fe(Pc)–Cu–TPPS]+–SiO2f | H2O2 | H2O | 8 h | 44 | — | — | — | — | — | 49 |
| [(Por)Fe–N–Fe(Pc)–Ni–TPPS]+–SiO2f | H2O2 | H2O | 8 h | 47 | — | — | — | — | — | 49 |
| (FePc(12-crown-4)4)2N–SiO2f | H2O2 | H2O | 8 h | 26 | — | 22% | 37% | 42% | — | 50 |
| (FePctMe8)2N–SiO2f | H2O2 | H2O | 16 h | 147 | — | 7% | 23% | 71% | — | 51 |
| [CuII(TREN)]2+–SiO2g | H2O2 | H2O | 20 h | 0.6 | 17% | — | — | 83% | — | 58 |
| [CuII(Hm–TREN)]2+–SiO2g | H2O2 | H2O | 20 h | 1.8 | — | 17% | 6% | 78% | — | 58 |
| V(TKA)–SiO2g | H2O2 | H2O | 20 h | 7.4 | 4% | 4% | 3% | 89% | — | 58 |
| V(Hm–TKA)–SiO2g | H2O2 | H2O | 20 h | 6.6 | — | 15% | 12% | 73% | — | 58 |
| V(Hm–BINOL–TKA)–SiO2g | H2O2 | H2O | 20 h | 13.2 | — | 5% | 18% | 77% | — | 58 |
| V(Bz–BINOL–TKA)–SiO2g | H2O2 | H2O | 20 h | 18.3 | — | 4% | 21% | 75% | — | 58 |
| [FeII(TPA)]2+–SiO2g | H2O2 | H2O | 20 h | 4.7 | 15% | — | 49% | 36% | — | 58 |
| [FeII(Hm–TPA)]2+–SiO2g | H2O2 | H2O | 20 h | 9.2 | 15% | 12% | 22% | 51% | — | 58 |
| [FeII(HPY4Cl2BIm)(NCMe)]2+h | Na2S2O8 | D2O:CD3CN = 95:5 |
3 h | 64 | — | 38% | — | 62% | — | 62 |
| [FeII(MesPY4Cl2BIm)(NCMe)]2+h | Na2S2O8 | D2O:CD3CN = 95:5 |
3 h | 101 | — | 71% | — | 29% | — | 62 |
| [FeII(AntPY4Cl2BIm)(NCMe)]2+h | Na2S2O8 | D2O:CD3CN = 95:5 |
3 h | 500 | — | 83% | — | 17% | — | 62 |
While Hg and Pt catalysts show high efficiency and selectivity in the conversion of methane to MBS or MSA, the cost of product manipulation is high due to the need for water addition for hydrolysis of the products to obtain methanol. A Pd(II) catalyst can perform the formation of MeTFA using quinone as an electron mediator and O2 as a terminal oxidant through the C–H activation in HTFA and reductive elimination of the product: this reaction system is reminiscent of the Wacker process to oxidize ethylene to produce acetaldehyde.68 C–H activation of methane has been made by using late-transition-metal complexes such as Ru, Rh, and Ir complexes as catalysts to achieve borylation of methane through C–H activation as summarized in Table 1. An Ag(I) catalyst can perform the conversion of N2CHCOOEt to CH3CH2COOEt in sc-CO2.
Photocatalytic methane oxidation has been achieved using CeIV–chloro complexes and a TFA salt of FeIII as catalysts under photoirradiation. In the case of CeIV salts, photoirradiation causes charge transfer from negatively charged ligands to the CeIV centre to generate radical species, which is responsible for hydrogen abstraction from methane to form oxidized products. Furthermore, photo-excited *[W10O32]4− enables hydrogen atom transfer from methane to form CH3˙, reacting with alkenes to afford hydroalkylated products. Photocatalytic methane functionalization will be developed through various approaches including emerging molecular catalysts.69
Dinuclear and trinuclear copper complexes as well as μ-nitride iron phthalocyanine catalysts, inspired by the active sites of pMMOs and sMMOs, respectively, can catalyse methane oxidation using peroxides as oxidants under ambient conditions. The tricopper cluster catalyst shows a moderate TON with high methanol selectivity. On the other hand, μ-nitride iron phthalocyanine catalysts exhibit a high TON but low methanol selectivity in water due to decomposition or overoxidation by ˙OH derived from the oxidants used. Furthermore, vanadium and iron catalysts with hydrophobic cavities constructed from hemicryptophane or BINOL moieties can improve the TON and methanol selectivity in water. Very recently, Kojima and co-workers have launched a “catch-and-release” strategy in aqueous medium, inspired by the functionality of the hydrophobic cavity near the diiron active site of sMMOs. They have developed a catalyst with a hydrophobic SCS constructed from four anthracenyl moieties attached to the NHC ligand. The catalyst can convert methane into methanol with a total TON of 500 for 3 h and 83% methanol selectivity. The “catch-and-release” strategy allows a hydrophobic methane molecule to be trapped in the hydrophobic SCS for oxidation, and the resulting hydrophilic methanol molecule is released into the surrounding aqueous solution.
Note that, in the arguments on methane oxidation using metal complexes having organic ligands, precaution should be taken to confirm that the origin of the carbon source of product(s) is exclusively methane by employing isotope-labelled methane, such as 13CH4 (ref. 13, 15, 21, 26, 29–32 and 42) or CD4.39,46,62
Toward the construction of a sustainable society, manipulation of methane is getting more important than ever in light of sustainable development goals (SDGs) to solve energy and environmental issues. By virtue of molecular catalysts, where we can fine-tune reactivity of metal centres by manipulating ligand sets, electronic properties and structures of SCSs, we can pave the way for effective and selective utilization of methane as a C1 resource under milder and sustainable conditions, as enzymes do so. The stability of molecular catalysts would be of concern; however, a certain range of robustness can be gained by introduction of suitable substituents at appropriate positions of a ligand. Molecular catalysts for methane functionalization should be developed in many ways in terms of not only technical improvement for the catalysis but also accumulation of fundamental scientific knowledge in our toolbox, which should be applicable to many kinds of reactions we need.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by CREST (JPMJCR16P1) from Japan Science and Technology Agency (JST).Notes and references
- (a) H. Arakawa, et al. , Chem. Rev., 2001, 101, 953 CrossRef CAS PubMed; (b) R. Horn and R. Schlögl, Catal. Lett., 2015, 145, 23 CrossRef CAS.
- X. Zhen and Y. Wang, Renewable Sustainable Energy Rev., 2015, 52, 477 CrossRef CAS.
- World Energy Outlook, International Energy Agency, 2022 Search PubMed.
- (a) P. Schwach, X. Pa and X. Bao, Chem. Rev., 2017, 117, 8497 CrossRef CAS PubMed; (b) AR6 WGI Report – List of corrigenda to be implemented, IPCC, 2021 Search PubMed; (c) see also ref. 3.
- (a) J. R. Rostrup-Nielsen, J. S. Sehested and J. K. Nørskov, Adv. Catal., 2002, 47, 65 CAS; (b) M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464 CrossRef CAS PubMed.
- Some representative references: TiO2-supported metal species: (a) J. Xie, R. Jin, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar, D. Ma and J. Tang, Nat. Catal., 2018, 1, 889 CrossRef CAS; (b) H. Song, X. Meng, S. Wang, W. Zhou, S. Song, T. Kako and J. Ye, ACS Catal., 2020, 10, 14318 CrossRef CAS; Au-Pd colloids: (c) M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock, D. M. Murphy, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 52, 1280 CrossRef PubMed; (d) N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Science, 2017, 358, 223 CrossRef CAS PubMed; (e) R. Serra-Maia, F. M. Michel, T. A. Douglas, Y. Kang and E. A. Stach, ACS Catal., 2021, 11, 2837 CrossRef CAS; Metal oxide: (f) S. Park, J. M. Vohs and R. J. Gorte, Nature, 2000, 406, 265 CrossRef PubMed; (g) C. Okolie, Y. F. Belhseine, Y. Lyu, M. M. Yung, M. H. Engelhard, L. Kovarik, E. Stavitski and C. Sievers, Angew. Chem., Int. Ed., 2017, 56, 13876 CrossRef CAS PubMed; (h) H. Song, X. Meng, S. Wang, W. Zhou, X. Wang, T. Kako and J. Ye, J. Am. Chem. Soc., 2019, 141, 20507 CrossRef CAS PubMed; MOF: (i) M. C. Simons, S. D. Prinslow, M. Babucci, A. S. Hoffman, J. Hong, J. G. Vitillo, S. R. Bare, B. C. Gates, C. C. Lu, L. Gagliardi and A. Bhan, J. Am. Chem. Soc., 2021, 143, 12165 CrossRef CAS PubMed; (j) N. Antil, M. Chauhan, N. Akhtar, R. Kalita and K. Manna, J. Am. Chem. Soc., 2023, 145, 6156 CrossRef CAS PubMed; (k) B. An, Z. Li, Z. Wang, X. Zeng, X. Han, Y. Cheng, A. M. Sheveleva, Z. Zhang, F. Tuna, E. J. L. McInnes, M. D. Frogley, A. J. Ramirez-Cuesta, L. S. Natrajan, C. Wang, W. Lin, S. Yang and M. Schröder, Nat. Mater., 2022, 21, 932 CrossRef CAS PubMed; P4O10: (l) N. Dietl, M. Engeser and H. Schwarz, Angew. Chem., Int. Ed., 2009, 48, 4861 CrossRef CAS PubMed; Pd/C: (m) M. Lin and A. Sen, J. Am. Chem. Soc., 1992, 114, 7308 CrossRef; (n) M. Lin, T. Hogan and A. Sen, J. Am. Chem. Soc., 1997, 119, 6048 CrossRef CASZeolite: (o) C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129 CrossRef CAS PubMed; (p) C. Hammond, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, M. H. Ab Rahim, M. M. Forde, A. Thetford, D. M. Murphy, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, Chem. – Eur. J., 2012, 18, 15735 CrossRef CAS PubMed; (q) W. Huang, S. Zhang, Y. Tang, Y. Li, L. Nguyen, Y. Li, J. Shan, D. Xiao, R. Gagne, A. I. Frenkel and F. F. Tao, Angew. Chem., Int. Ed., 2016, 55, 13441 CrossRef CAS PubMed; (r) B. Ipek and R. F. Lobo, Chem. Commun., 2016, 52, 13401 RSC; (s) J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605 CrossRef CAS PubMed; (t) V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523 CrossRef CAS PubMed; (u) E. Tabor, M. Lemishka, Z. Sobalik, K. Mlekodaj, P. C. Andrikopoulos, J. Dedecek and S. Sklenak, Commun. Chem., 2019, 2, 71 CrossRef; (v) L. Sun, Y. Wang, C. Wang, Z. Xie, N. Guan and L. Li, Chem, 2021, 7, 1557 CrossRef CAS.
- (a) Y.-R. Luo, Handbook of bond dissociation energies in organic compounds, CRC Press LLC, Boca Raton, FL, 1st edn, 2003 Search PubMed; (b) S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255 CrossRef CAS PubMed.
- For pMMO: (a) M. A. Culpepper and A. C. Rosenzweig, Crit. Rev. Biochem. Mol. Biol., 2012, 47, 483 CrossRef CAS PubMed; (b) C. W. Koo and A. C. Rosenzweig, Chem. Soc. Rev., 2021, 50, 3424 RSC; (c) R. L. Lieberman and A. C. Rosenzweig, Nature, 2005, 434, 171 CrossRef PubMed; (d) V. C.-C. Wang, S. Maji, P. P.-Y. Chen, H. K. Lee, S. S.-F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8754 CrossRef PubMed; (e) W. Peng, X. Qu, S. Shaik and B. Wang, Nat. Catal., 2021, 4, 266 CrossRef CAS.
- For sMMO: (a) V. Srinivas, et al. , J. Am. Chem. Soc., 2020, 142, 14249 CrossRef CAS PubMed; (b) A. C. Rosenzweig, C. A. Frederick, S. J. Lippard and P. Nordlund, Nature, 1993, 366, 537 CrossRef CAS PubMed; (c) M.-H. Baik, M. Newcomb, R. A. Friesner and S. J. Lippard, Chem. Rev., 2003, 103, 2385 CrossRef CAS PubMed.
- (a) R. Banerjee and J. D. Lipscomb, Acc. Chem. Res., 2021, 54, 2185 CrossRef CAS PubMed; (b) R. G. Castillo, R. Banerjee, C. J. Allpress, G. T. Rohde, E. Bill, L. Que Jr., J. D. Lipscomb and S. DeBeer, J. Am. Chem. Soc., 2017, 139, 18024 CrossRef CAS PubMed; (c) S.-K. Lee, B. G. Fox, W. A. Froland, J. D. Lipscomb and E. Münck, J. Am. Chem. Soc., 1993, 115, 6450 CrossRef CAS.
- (a) L. Que Jr. and W. B. Tolman, Nature, 2008, 455, 333 CrossRef PubMed; (b) A. R. McDonald and L. Que Jr., Coord. Chem. Rev., 2013, 257, 414 CrossRef CAS; (c) M. Guo, Y.-M. Lee, S. Fukuzumi and W. Nam, Coord. Chem. Rev., 2021, 435, 213807 CrossRef CAS; (d) W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2018, 51, 2014 CrossRef CAS PubMed; (e) W. Zhu, A. Kumar, J. Xiong, M. J. Abernathy, X.-X. Li, M. S. Seo, Y.-M. Lee, R. Sarangi, Y. Guo and W. Nam, J. Am. Chem. Soc., 2023, 145, 4389 CrossRef CAS PubMed; (f) V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Nat. Rev. Chem., 2020, 4, 404 CrossRef CAS PubMed; (g) G. L. Tripodi, M. M. J. Dekker, J. Roithová and L. Que Jr., Angew. Chem., 2021, 60, 7126 CrossRef CAS PubMed.
- J. C. Snyder and A. V. Grosse, US Pat., 2493038, 1950 Search PubMed.
- R. A. Periana, D. J. Taube, E. R. Evitt, D. J. Löffler, P. R. Wentrcek, G. Voss and T. Masuda, Science, 1993, 259, 340 CrossRef CAS PubMed.
- (a) A. E. Shilov, Activation of saturated hydrocarbons by transition metal complexes, D. Reidel Publishing Co.: Dordrecht, The Netherlands, 1984 Search PubMed; (b) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS PubMed; (c) A. E. Shilov and G. B. Shul'pin, Russ. Chem. Rev., 1987, 56, 442 CrossRef; (d) G. A. Luinstra, L. Wang, S. S. Stahl, L. A. Labinger and J. E. Bercaw, J. Org. Chem., 1995, 504, 75 CrossRef CAS.
- R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560 CrossRef CAS PubMed.
- O. A. Mironov, S. M. Bischof, M. M. Konnick, B. G. Hashiguchi, V. R. Ziatdinov, W. A. Goddard III, M. Ahlquist and R. A. Periana, J. Am. Chem. Soc., 2013, 135, 14644 CrossRef CAS PubMed.
- (a) T. Zimmermann, M. Soorholtz, M. Bilke and F. Schüth, J. Am. Chem. Soc., 2016, 138, 12395 CrossRef CAS PubMed; (b) T. Zimmermann, M. Bilke, M. Soorholtz and F. Schüth, ACS Catal., 2018, 8, 9262 CrossRef CAS.
- H. T. Dang, H. W. Lee, J. Lee, H. Choo, S. H. Hong, M. Cheong and H. Lee, ACS Catal., 2018, 8, 11854 CrossRef CAS.
- C. Díaz-Urrutia and T. Otto, Science, 2019, 363, 1326 CrossRef PubMed.
- (a) M. Muehlhofer, T. Strassner and W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1745 CrossRef CAS; (b) S. Ahrens, A. Zeller, M. Taige and T. Strassner, Organometallics, 2006, 25, 5409 CrossRef CAS; (c) D. Meyer, M. A. Taige, A. Zeller, K. Hohlfeld, S. Ahrens and T. Strassner, Organometallics, 2009, 28, 2142 CrossRef CAS.
- Z. An, X. Pan, X. Liu, X. Han and X. Bao, J. Am. Chem. Soc., 2006, 128, 16028 CrossRef CAS PubMed.
- T. Strassner, S. Ahrens, M. Muehlhofer, D. Munz and A. Zeller, Eur. J. Inorg. Chem., 2013, 3659 CrossRef CAS.
- S.-H. Cheong, D. Kim, H. T. Dang, D. Kim, B. Seo, M. Cheong, S. H. Hong and H. Lee, J. Catal., 2022, 413, 803 CrossRef CAS.
- M. Ravi and J. A. van Bokhoven, ChemCatChem, 2018, 10, 2383 CrossRef CAS.
- A. K. Cook, S. D. Schimler, A. J. Matzger and M. S. Sanford, Science, 2016, 351, 1421 CrossRef CAS PubMed.
- K. T. Smith, S. Berritt, M. González-Moreiras, S. Ahn, M. R. Smith III, M.-H. Baik and D. J. Mindiola, Science, 2016, 351, 1424 CrossRef CAS PubMed.
- O. Staples, M. S. Ferrandon, G. P. Laurent, U. Kanbur, A. J. Kropf, M. R. Gau, P. J. Carroll, K. McCullough, D. Sorsche, F. A. Perras, M. Delferro, D. M. Kaphan and D. J. Mindiola, J. Am. Chem. Soc., 2023, 145, 7992 CrossRef CAS PubMed.
- A. Caballero, E. Despagnet-Ayoub, M. M. Díaz-Requejo, A. Díaz-Rodríguez, M. E. González-Núñez, R. Mello, B. K. Muñoz, W.-S. Ojo, G. Asensio, M. Etienne and P. J. Pérez, Science, 2011, 332, 835 CrossRef CAS PubMed.
- M. Lin and A. Sen, Nature, 1994, 368, 613 CrossRef CAS.
- R. A. Periana, O. Mironov, D. Taube, G. Bhalla and C. Jones, Science, 2003, 301, 814 CrossRef CAS PubMed.
- M. V. Kirillova, M. L. Kuznetsov, P. M. Reis, J. A. L. da Silva, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, J. Am. Chem. Soc., 2007, 129, 10531 CrossRef CAS PubMed.
- N. Coutard, J. M. Goldberg, H. U. Valle, Y. Cao, X. Jia, P. D. Jeffrey, T. B. Gunnoe and J. T. Groves, Inorg. Chem., 2021, 61, 759 CrossRef PubMed.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668 CrossRef CAS PubMed.
- Q. Yang, Y.-H. Wang, Y. Qiao, M. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Science, 2021, 372, 847 CrossRef CAS PubMed.
- G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92 CrossRef CAS PubMed.
- (a) A. S. Goldstein and R. S. Drago, J. Chem. Soc., Chem. Commun., 1991, 21 RSC; (b) A. S. Goldstein, R. H. Beer and R. S. Drago, J. Am. Chem. Soc., 1994, 116, 2424 CrossRef CAS.
- P. P.-Y. Chen, R. B.-G. Yang, J. C.-M. Lee and S. I. Chan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14571 Search PubMed.
- S. I. Chan, Y.-J. Lu, P. Naganobu, S. Maji, M.-C. Hung, M. M. Lee, I.-J. Hsu, P. D. Minh, J. C.-H. Lai, K. Y. Ng, S. Ramalingam, S. S.-F. Yu and M. K. Chan, Angew. Chem., Int. Ed., 2013, 52, 3731 CrossRef CAS PubMed.
- P. P.-Y. Chen, P. Naganobu, S. S.-F. Yu and S. I. Chan, ChemCatChem, 2014, 6, 429 CrossRef CAS.
- P. Naganobu, S. S.-F. Yu, R. Ramu and S. I. Chan, Catal. Sci. Technol., 2014, 4, 930 RSC.
- Y.-H. Chen, C.-Q. Wu, P.-H. Sung, S. I. Chan and P. P.-Y. Chen, ChemCatChem, 2020, 12, 3088 CrossRef CAS.
- H. Takahashi, K. Wada, K. Tanaka, K. Fujikawa, Y. Hitomi, T. Endo and M. Kodera, Bull. Chem. Soc. Jpn., 2022, 95, 1148 CrossRef CAS.
- L. A. Bottomley, J.-N. Gorce, V. L. Goedken and C. Ercolani, Inorg. Chem., 1985, 24, 3733 CrossRef CAS.
- A. B. Solokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008, 2562 RSC.
- P. Afanasiev, E. V. Kudrik, J.-M. M. Millet, D. Bouchu and A. B. Solokin, Dalton Trans., 2011, 40, 701 RSC.
- M. G. Quesne, D. Senthilnathan, D. Singh, D. Kumar, P. Maldivi, A. B. Solokin and S. P. de Visser, ACS Catal., 2016, 6, 2230 CrossRef CAS.
- E. V. Kudrik, P. Afanasiev, L. X. Alvarez, P. Dubourdeaux, M. Clémancey, J.-M. Latour, G. Blondin, D. Bouchu, F. Albrieux, S. E. Nefedov and A. B. Sorokin, Nat. Chem., 2012, 4, 1024 CrossRef CAS PubMed.
- N. Mihara, Y. Yamada, H. Takuya, Y. Kitagawa, K. Igawa, K. Tomooka, H. Fujii and K. Tanaka, Chem. – Eur. J., 2019, 25, 3369 CrossRef CAS PubMed.
- Y. Yamada, K. Morita, N. Mihara, K. Igawa, K. Tomooka and K. Tanaka, New J. Chem., 2019, 43, 11477 RSC.
- Y. Yamada, J. Kura, Y. Toyoda and K. Tanaka, New J. Chem., 2020, 44, 19179 RSC.
- Y. Yamada, J. Kura, Y. Toyoda and K. Tanaka, Dalton Trans., 2021, 50, 6718 RSC.
- Ü. İşci, A. S. Faponle, P. Afanasiev, F. Albrieux, V. Briois, V. Ahsen, F. Dumoulin, A. B. Sorokin and S. P. de Visser, Chem. Sci., 2015, 6, 5063 RSC.
- G. V. Nizova, G. Süss-Fink and G. B. Shul'pin, Chem. Commun., 1997, 397 RSC.
- G. B. Shul'pin, G. V. Nizova, Y. N. Kozlov, L. G. Cuervo and G. Süss-Fink, Adv. Synth. Catal., 2004, 346, 317 CrossRef.
- Q. Yuan, W. Deng, Q. Zhang and Y. Wang, Adv. Synth. Catal., 2007, 349, 1199 CrossRef CAS.
- Y. Naruta, F. Tani, N. Ishibara and K. Maruyama, J. Am. Chem. Soc., 1991, 113, 6865 CrossRef CAS.
- J. T. Groves and P. Viski, J. Org. Chem., 1990, 55, 3628 CrossRef CAS.
- S. A. Ikbal, C. Colomban, D. Zhang, M. Delecluse, T. Brotin, V. Dufaud, J.-P. Dutasta, A. B. Sorokin and A. Martinez, Inorg. Chem., 2019, 58, 7220 CrossRef CAS PubMed.
- D. Zhang, K. Jamieson, L. Guy, G. Gao, J.-P. Dutasta and A. Martinez, Chem. Sci., 2017, 8, 789 RSC.
- D. Zhang, J.-P. Dutasta, V. Dufaud, L. Guy and A. Martinez, ACS Catal., 2017, 7, 7340 CrossRef CAS.
- (a) H. Fujisaki, T. Ishizuka, Y. Shimoyama, H. Kotani, Y. Shiota, K. Yoshizawa and T. Kojima, Chem. Commun., 2020, 56, 9783 RSC; (b) H. Fujisaki, M. Okamura, S. Hikichi and T. Kojima, Chem. Commun., 2023, 59, 3265 RSC.
- H. Fujisaki, T. Ishizuka, H. Kotani, Y. Shiota, K. Yoshizawa and T. Kojima, Nature, 2023, 616, 476 CrossRef CAS PubMed.
- N. Branda, R. Wyler and J. Rebek Jr., Science, 1994, 263, 1267 CrossRef CAS PubMed.
- L. Garel, J. P. Dutasta and A. Collet, Angew. Chem., Int. Ed. Engl., 1993, 32, 1169 CrossRef.
- J. Nakazawa, J. Hagiwara, M. Mizuki, Y. Shimazaki, F. Tani and Y. Naruta, Angew. Chem., Int. Ed., 2005, 44, 3744 CrossRef CAS PubMed.
- A. V. Leontiev, A. W. Saleh and D. M. Rudkevich, Org. Lett., 2007, 9, 1753 CrossRef CAS PubMed.
- Y. Ruan, P. W. Peterson, C. M. Hadad and J. D. Badjić, Chem. Commun., 2014, 50, 9086 RSC.
- R. A. Fernandes, A. K. Jha and P. Kumar, Catal. Sci. Technol., 2020, 10, 7448 RSC.
- Y. Jiang, Y. Fan, S. Li and Z. Tang, CCS Chem., 2023, 5, 30 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |