
Bismuth phosphate nanoparticle catalyst for direct oxidation of methane into formaldehyde†
Aoi
Matsuda
a,
Kazuhiko
Obara
a,
Atsushi
Ishikawa
 b,
Meng-Hsuan
Tsai
c,
Chia-Hsin
Wang
c,
Yu-Chuan
Lin
d,
Michikazu
Hara
a and
Keigo
Kamata
*a
b,
Meng-Hsuan
Tsai
c,
Chia-Hsin
Wang
c,
Yu-Chuan
Lin
d,
Michikazu
Hara
a and
Keigo
Kamata
*a
aLaboratory for Materials and Structures, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8503, Japan. E-mail: kamata.k.ac@m.titech.ac.jp
bDepartment of Transdisciplinary Science and Engineering, School of Environment and Society, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan
cNational Synchrotron Radiation Research Center, 101 Hsin-Ann Rd., 30076, Hsinchu, Taiwan
dDepartment of Chemical Engineering, National Cheng Kung University, 1 University Rd., Tainan, 70101, Taiwan
First published on 25th July 2023
Abstract
The direct oxidation of methane (CH4) to formaldehyde (HCHO) with molecular oxygen (O2) as the sole oxidant was studied over various bismuth-based catalysts (BiPO4, α-Bi2O3, β-Bi2O3) using a fixed-bed flow reactor. The catalytic activity of monoclinic BiPO4 nanoparticles (BiPO4-DEG) synthesized in a mixed solvent of diethylene glycol (DEG) and water for the direct oxidation of CH4 was the highest among the catalysts tested. In high temperature region, BiPO4-DEG was more selective for HCHO formation than FePO4 nanoparticles. Based on mechanistic studies including the catalyst effect, kinetics, pulse-reaction experiments, infrared (IR) spectroscopy, operando near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS), and density functional theory (DFT) calculations, surface active oxygen species generated on BiPO4 possibly react with CH4 to give HCHO as the primary product. In contrast, the phosphate units of FePO4 nanoparticles react with CH4 followed by rapid reoxidation to FePO4.
1. Introduction
Methane (CH4) has attracted attention as an important raw material due to its abundance around the world as a main component of natural gas and a low emitter of greenhouse gases such as carbon dioxide (CO2) compared to coal and oil.1 However, CH4 is mainly used as an energy source for heating, cooking, and power generation. The use of CH4 as a chemical feedstock is very small due to the difficulty in activating the strong C–H bond (440 kJ mol−1).2 Current CH4 conversion into commodity chemicals and fuels is limited to indirect pathways via synthesis gas (CO + H2) produced by energy- and capital-intensive reforming processes.3 Therefore, the development of catalytic processes that can directly convert CH4 into useful chemicals and fuels is an important and challenging subjects, and various effective thermocatalytic,2,4–8 electrocatalytic,9 and plasma- or electric-field assisted systems10 have been reported for oxidative CH4 upgrading and non-oxidative dehydrogenative aromatization.11,12Various effective solid materials based on metal-exchanged zeolites,14–18 metal oxides,19–22 supported catalysts,23–25 metal phosphates,26–36 and metal organic frameworks37,38 can function as efficient catalysts for oxidative upgrading of CH4 such as oxidative coupling to ethylene (C2H4), oxygenation to methanol (CH3OH) and/or formaldehyde (HCHO), and oxidative halogenation using O2, H2O2, and N2O as oxidants. In particular, V- and Mo-based catalysts and B2O3/Al2O3 are efficient catalysts for the oxidation of CH4 into HCHO which is an important raw material as resins and polyfunctional alcohols.39–42 The performance of V- and Mo-based catalysts is improved by the metal loading amounts and space velocity, and Cu–MoOx catalysts selectively give HCHO in 1.0% yield (62% select.) for the CH4 oxidation at 700 °C in the presence of water vapor.41 Non-metallic B2O3-based catalysts exhibit high HCHO yield and selectivity, and molecular O2 bonded to tri-coordinated BO3 centers on B2O3 surfaces has been proposed as an active oxidant for CH4 activation.42
Among the solid catalyst materials, we have also focused on metal phosphates with unique surface redox and acid–base properties for the direct oxidation of CH4 with O2 as the sole oxidant.28,43,44 Monoclinic cerium orthophosphate (CePO4) nanorods efficiently catalyze the oxidative coupling of CH4 in an electric field without the need for external heating.28,43 Trigonal iron phosphate (FePO4) nanoparticles exhibits high activity for the selective oxidation of CH4 into HCHO. The weakly-basic phosphate units likely contribute to the suppression of complete oxidation to CO2.44 While the redox-active Lewis acidic metal sites of metal phosphates play an important role in the activation of CH4 and O2, the effect of metals and the reaction mechanism on the direct oxidation of CH4 is still unclear. Herein, we focus on bismuth, which is used as the main catalyst component in industrial propylene oxidation processes.45 Although bismuth phosphates (Fig. 1) have been mostly investigated for photocatalytic reactions,46–51 their application to other reactions (e.g., oxidative dehydrogenation,52–54 ammoxidation,55 decomposition,56 isomerization,57 aldol condensation58) including the direct oxidation of CH4 has not been sufficiently explored.23,26
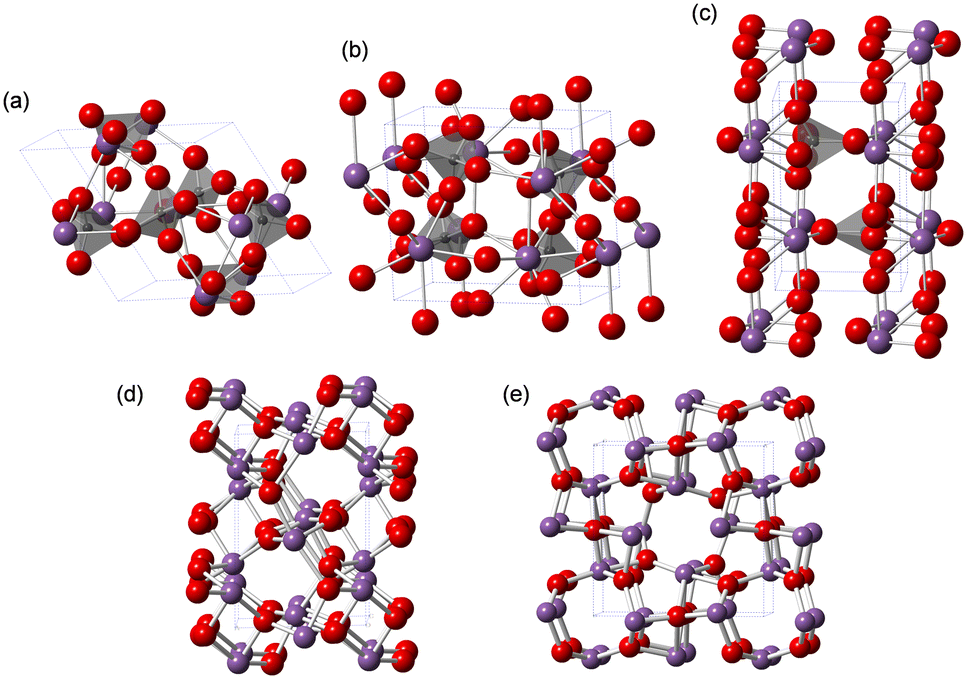 | ||
| Fig. 1 Structures of (a) trigonal BiPO4·(H2O)0.67, (b) monoclinic BiPO4 (P21/n), and (c) monoclinic BiPO4 (P21/m), (d) monoclinic α-Bi2O3, and (e) tetragonal β-Bi2O3. Purple, gray, and red spheres represent Bi, P, and O atoms, respectively. | ||
In this paper, we report the selective oxidation of CH4 to HCHO with molecular oxygen (O2) as the sole oxidant over a BiPO4 catalyst using a fixed-bed flow reactor. BiPO4 nanoparticles (BiPO4-DEG) synthesized in a mixed solvent in DEG/water exhibit higher catalytic activity and selectivity for HCHO than other bismuth-containing catalysts (Fig. 1) and FePO4 nanoparticles.44 The mechanistic studies including the catalyst effect, pulse experiments, kinetics, operando analysis and density functional theory (DFT) calculations indicate that surface oxygen species generated on BiPO4 possibly react with CH4 to give HCHO as a primary product.
2. Experimental section
2.1. Instruments
X-ray diffraction (XRD), energy dispersive X-ray fluorescence spectroscopy (ED-XRF), Fourier transform infrared spectroscopy (FT-IR), thermogravimetry-differential thermal analysis (TG-DTA), H2 temperature-programmed reduction (H2-TPR), nitrogen adsorption–desorption, scanning electron microscopy (SEM), transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS) were performed using previously described instruments.44,59–61 The details are described in the ESI.†2.2. Synthesis of BiPO4-DEG
NH4H2PO4 (1.035 g, 9 mmol) was dissolved in a mixed solution of diethylene glycol (DEG)/water (75 mL, 1/1 v/v). A DEG solution (30 mL) containing Bi(NO3)3·5H2O (4.37 g, 9 mmol) was added dropwise into this phosphate solution followed by stirring for 60 min at room temperature. The resulting precipitates were collected by centrifugation, washed with ethanol (40 mL × 3), and dried at 110 °C overnight to obtain BiPO4·(H2O)0.67 as a precursor. This precursor was calcined at 600 °C for 5 h to give BiPO4-DEG (2.54 g, 93% yield).2.3. Synthesis of BiPO4-HT
BiPO4-HT was synthesized by a hydrothermal (HT) reaction. Bi(NO3)3·5H2O (6.00 mmol, 2.91 g) and (NH4)2HPO4 (6.00 mmol, 0.792 g) were added to water (70 mL) and stirred for 30 min at room temperature. The resulting solution was transferred into a stainless steel autoclave with a Teflon vessel liner (TAF-SR type, Taiatsu Techno Corporation). After the solution was heated at 180 °C for 18 h, the precipitates were collected by filtration and washed with water (200 mL). The resulting precipitates were calcined at 600 °C for 5 h and the BiPO4-HT catalyst was obtained (1.75 g, 96% yield).2.4. Catalytic oxidation of CH4 with O2
The oxidation of CH4 with O2 over various bismuth-based catalysts was conducted in a fixed-bed continuous-flow reactor operated at atmospheric pressure. All of the catalysts were pressed into pellets, crushed, and sieved to 32–42 meshes before the reaction. The catalyst (100 mg) was loaded into a quartz reactor (2 mm inner diameter at the catalyst bed portion) over a plug of quartz wool. When using a quartz reactor with larger inner diameter (4 mm) under the conditions in Fig. 3(d), the HCHO yield was not changed but the CO yield increased, which likely indicates quenching effect. A reaction gas containing CH4/O2/N2 in a molar ratio of 20/20/60 was used and the total gas flow rate was 10 sccm (i.e., CH4/O2/N2 = 2/2/6 sccm). After introduction of the reaction gas flow to the reactor, the reaction temperature was increased from room temperature to 600 °C at a heating rate of ca. 40 °C min−1 and then held for 60 min. After the first sampling of the reaction gas, the temperature was decreased by 10 °C to 470 °C (420 °C in the case of FePO4) every 60 min with periodic analysis of the reaction gas. The products (CO, CO2, CH3OH, HCHO, CH3OCH3, C2H6, C2H4) were analyzed using on-line gas chromatography (Shimadzu GC-8A) with a thermal conductivity detector (TCD) and two packed columns (a mixed column of Porapak-QS/-N and molecular sieves 5A). All of the lines and valves between the reactor exit and the gas chromatograph were heated to 120–130 °C to prevent condensation of the products. Details of the calculations of CH4 conversion, yield, and selectivity are as follows: CH4 conversion (%) = carbon of (CO, CO2, HCHO)/carbon of input CH4 × 100. Yield (%) = carbon of product/carbon of input CH4 × 100. Selectivity (%) = yield/CH4 conversion × 100. Carbon balance (%) = (carbon of (CO, CO2, HCHO) + carbon of output CH4)/carbon of input CH4 × 100. In each case, the carbon balance was in the range of 98.7 ± 1.4%.2.5. Pulse-reaction experiments for CH4 oxidation
The pulse-reaction experiments were performed using a quartz reactor (4 mm inner diameter at the catalyst bed portion) over a plug of quartz wool. The exit gas was directly connected to a gas chromatograph with a TCD (Shimadzu, GC-8A), and HCHO/CO2 and CO were analyzed using Porapak-N and molecular sieves 5A columns, respectively. The details of the pulse-reactions of He-treated BiPO4-DEG with (i) CH4 or (ii) CH4/O2 pulse are as follows. Prior to the pulse-reaction experiments, BiPO4-DEG (100 mg) was pretreated in a He flow (20 sccm) at 600 °C for 30 min followed by (i) CH4 pulse (1 mL) or (ii) CH4/O2 pulse (1 mL, 1/1 v/v) through the catalyst bed at 600 °C.2.6. Computational details
The energy evaluation and geometry optimization of the atomic positions were carried out according to the following procedure. The initial atomic positions for the oxides were obtained from the experimental structure of monazite-type monoclinic BiPO4 (P21/n) taken from the inorganic crystal structure database (ICSD) with ID = 426![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 231. The unit cell consists of a 2 × 2 × 3 supercell, and the lower 2/3 of the unit cell was fixed during the geometry optimization. The core electrons were represented by the projector augmented-wave (PAW) method,62 and the valence electrons were expanded by the plane wave basis set up to a cutoff energy of 400 eV. The meta-GGA SCAN functional was used as the exchange–correlation functional in the DFT calculations.63 The Gaussian smearing method with σ = 0.1 was used throughout for the smearing of the electron occupation near the Fermi level. The convergence thresholds for the electronic state calculation and geometry optimization were set to 1.0 × 10−5 eV and 0.03 eV Å−1 in energy and force, respectively. Integration in the reciprocal lattice space was performed by numerical integration using k-points, which were placed such that the spacing between them was 0.3 Å−1. The gamma point was always included. For isolated molecule calculations (H2, O2, CH3, and CH4), a single k-point was placed on the gamma point. A vacuum layer with a thickness of 12 Å was placed between the slabs, and a dipole correction in the z-direction was introduced to remove the artificial interaction between slabs. All the calculations were performed with the Vienna ab initio simulation package (VASP) version 5.4.64,65
231. The unit cell consists of a 2 × 2 × 3 supercell, and the lower 2/3 of the unit cell was fixed during the geometry optimization. The core electrons were represented by the projector augmented-wave (PAW) method,62 and the valence electrons were expanded by the plane wave basis set up to a cutoff energy of 400 eV. The meta-GGA SCAN functional was used as the exchange–correlation functional in the DFT calculations.63 The Gaussian smearing method with σ = 0.1 was used throughout for the smearing of the electron occupation near the Fermi level. The convergence thresholds for the electronic state calculation and geometry optimization were set to 1.0 × 10−5 eV and 0.03 eV Å−1 in energy and force, respectively. Integration in the reciprocal lattice space was performed by numerical integration using k-points, which were placed such that the spacing between them was 0.3 Å−1. The gamma point was always included. For isolated molecule calculations (H2, O2, CH3, and CH4), a single k-point was placed on the gamma point. A vacuum layer with a thickness of 12 Å was placed between the slabs, and a dipole correction in the z-direction was introduced to remove the artificial interaction between slabs. All the calculations were performed with the Vienna ab initio simulation package (VASP) version 5.4.64,65
2.7. Operando near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) experiment
NAP-XPS analysis was performed at the Taiwan Light Source (TLS) beamline 24A of the National Synchrotron Radiation Research Center (NSRRC), Taiwan. The tested sample was pelleted and the spectra were recorded at 550 °C under evacuation and in O2 atmosphere (0.1 mbar) in the NAP-XPS chamber. P 2p signal at 133.2 eV was used to correct the energy shift.3. Results and discussion
3.1. Synthesis and characterization of bismuth phosphate nanoparticles
The low solubility of Bi species (e.g., Bi(NO3)3) in aqueous media typically requires severe reaction conditions such as hydrothermal treatment to obtain bismuth phosphate (BiPO4) catalyst materials, which results in low specific surface areas. Thus, solvothermal and related synthesis methods are more effective for the synthesis of BiPO4 nanoparticles. We also synthesized BiPO4 nanoparticles (BiPO4-DEG) by the calcination of a precursor, which was prepared by the reaction of Bi(NO3)3·5H2O and NH4H2PO4 in a mixed solution of diethylene glycol (DEG)/water, in air at 600 °C.The XRD patterns for the precursor and BiPO4-DEG showed the formation of hydrated bismuth phosphate in the trigonal system (BiPO4·(H2O)0.67) and a mixture of monoclinic BiPO4 (P21/n and P21/m), respectively (Fig. 2(a)). Braque and co-workers reported that the trigonal phase of BiPO4·(H2O)0.67 (Fig. 1(a)) is metastable and irreversibly transforms to the monazite-type structure (P21/n, Fig. 1(b)) and that this monazite-type BiPO4 slowly transforms into the high-temperature monoclinic form (P21/m, Fig. 1(c)) when heated above 600 °C.66 No impurity phases such as bismuth phosphates, bismuth oxides, or phosphorous oxides were observed. Elemental analysis of BiPO4-DEG using energy dispersive X-ray fluorescence spectroscopy (ED-XRF) showed that the molar ratio of Bi/P was 1/1, which also supports the high purity. On the other hand, only the monazite-type structure was observed for BiPO4-HT synthesized by the hydrothermal reaction of Bi(NO3)3 and (NH4)2HPO4 at 180 °C followed by calcination at 600 °C in a similar manner to that of previous reports (Fig. S1†).47,49
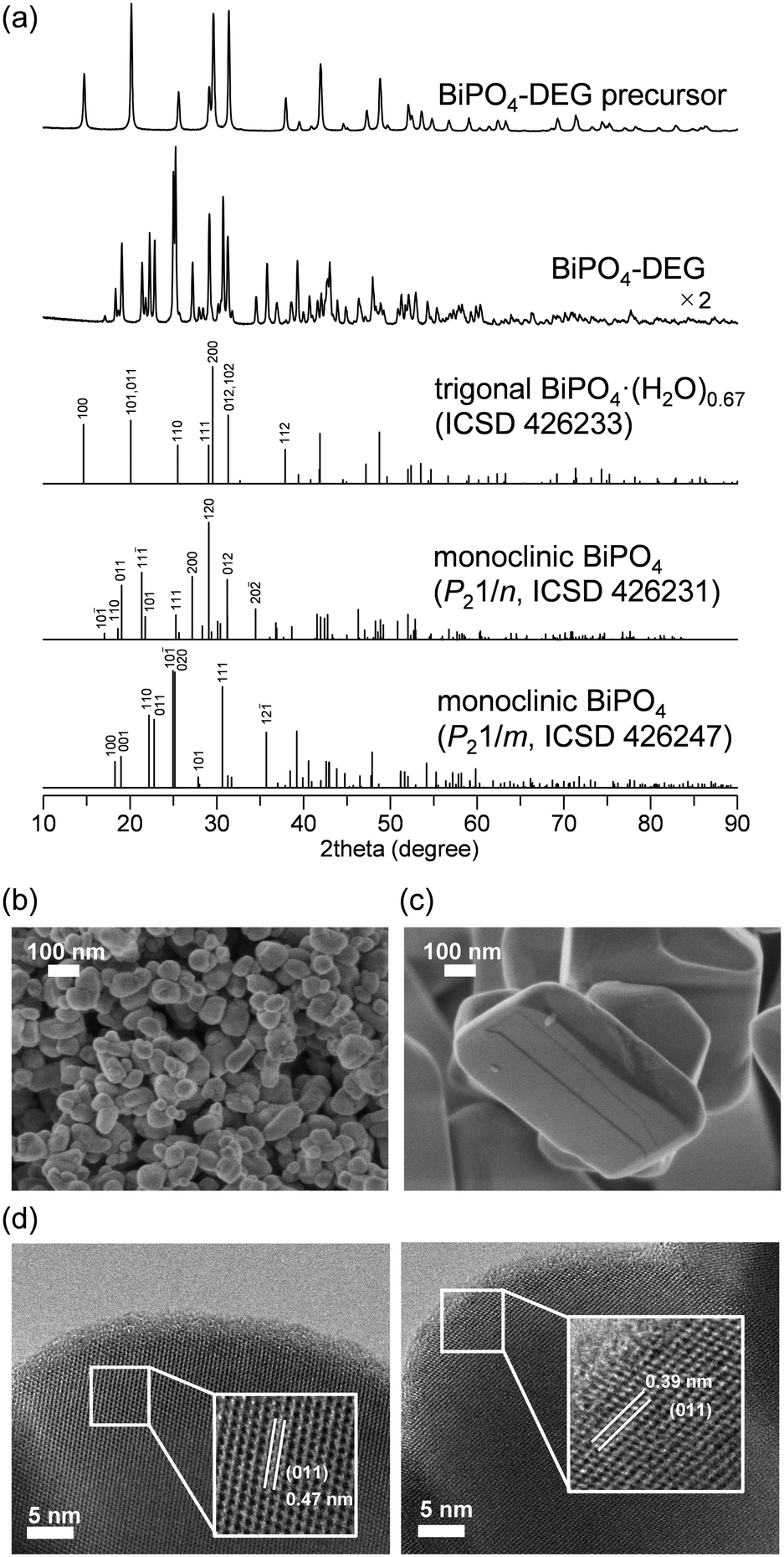 | ||
| Fig. 2 (a) XRD patterns for BiPO4-DEG precursor, BiPO4-DEG, trigonal BiPO4·(H2O)0.67 (ICSD 426233), monoclinic BiPO4 (P21/n, ICSD 426231), and monoclinic BiPO4 (P21/m, ICSD 426247). SEM images for (b) BiPO4-DEG and (c) BiPO4-HT. (d) TEM images for BiPO4-DEG. | ||
The specific surface area of BiPO4-DEG was 10 m2 g−1, which is much larger than that of BiPO4-HT (<1 m2 g−1) (Table 1). SEM observations of BiPO4-DEG showed the formation of spherical nanoparticles with estimated particle sizes of ca. 50–100 nm (Fig. 2(b)), while large particles with sizes of hundreds of nanometers were observed for BiPO4-HT (Fig. 2(c)). These nanoparticle sizes were in reasonable agreement with the grain sizes (d = 46 and 51 nm for BiPO4 (P21/n and P21/m), respectively) calculated from the (10−1) and (12−1) diffraction lines, respectively, using Scherrer's equation. A similar morphology and size distribution of the nanoparticles were observed by TEM, with clear lattice fringes throughout the particles indicating their crystallinity (Fig. 2(d)). The distances between fringes of different particles were 0.47 and 0.39 nm, assigned to the d-spacing for the (011) planes of monazite-type (P21/n) and high-temperature monoclinic BiPO4 (P21/m), respectively.47,50
| Entry | Catalyst | S BET (m2 g−1) | Crystallite sizea (nm) |
|---|---|---|---|
| a Calculated by the XRD peaks using Rigaku PDXL2 software. b Data from ref. 44. | |||
| 1 | BiPO4-DEG precursor | 14 | 38 (110) |
| 2 | BiPO4-DEG | 10 | P 21/n: 46 (10−1) |
| P 21/m: 51 (12−1) | |||
| 3 | BiPO4-HT | <1 | 93 (10−1) |
| 4 | α-Bi2O3 | 1 | 97 (002) |
| 5 | β-Bi2O3 | 10 | 70 (201) |
| 6b | FePO4 | 22 | 33 (100) |
| 22 (200) | |||
3.2. Catalytic oxidation of CH4 into HCHO with O2
The catalyst effect of bismuth-based materials (BiPO4-DEG, BiPO4-HT, α-Bi2O3 (Fig. 1(d)), and β-Bi2O3 (Fig. 1(e))) was investigated for the oxidation of CH4 with O2 as the sole oxidant. There were three main products, i.e., such as formaldehyde (HCHO), carbon monoxide (CO), and carbon dioxide (CO2). Other products such as CH3OH were not observed (Fig. S2†). Fig. 3(a) shows the HCHO yield as a function of reaction temperature under a CH4/O2/N2 (2/2/6 sccm) flow in the temperature range of 450–600 °C. BiPO4-DEG selectively gave HCHO with little formation of CO2. The formation of CO2 was mainly observed using other bismuth-based catalysts such as BiPO4-HT, α-Bi2O3, and β-Bi2O3 (Fig. 3(a) and S2†). There was no significant change in the XRD patterns for any of the Bi-based catalysts recovered after CH4 oxidation (Fig. S3†), although some β-Bi2O3 was changed to α-Bi2O3, which suggests that the catalytic performance is derived from their structures. Although the water vapor plays an important role in the oxidative conversion of CH4 in some cases,67 the presence of water vapor did not enhance the present CH4 oxidation and HCHO yield/selectivity. The space time yield of BiPO4-DEG at 600 °C was 0.45 mmolHCHO g−1 h−1 (HCHO selectivity: 40%), and the value and selectivity for HCHO were higher than those for previously-reported Bi-based catalysts such as Bi2O3–B2O3/SiO2 (4.3 × 10−2 mmolHCHO g−1 h−1, HCHO selectivity: 30%, 550 °C)23 and Bi–P–O (7.7 × 10−2 mmolHCHO g−1 h−1, HCHO selectivity: 4%, 700 °C).26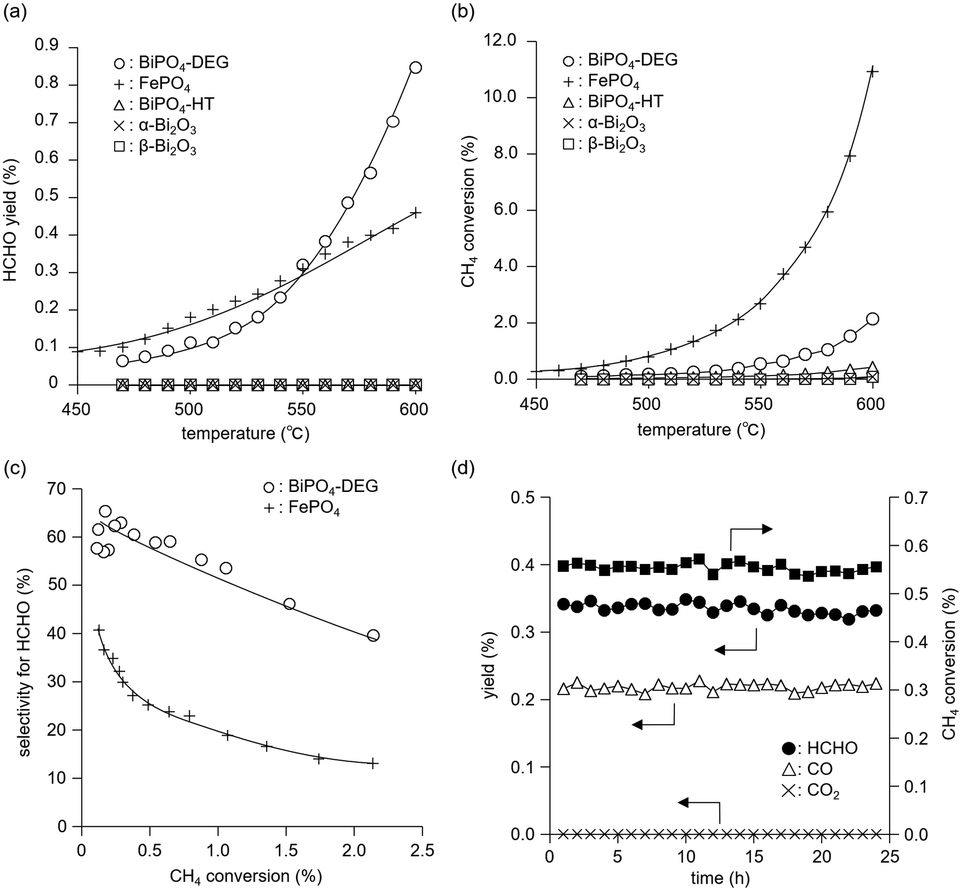 | ||
| Fig. 3 (a) HCHO yield and (b) CH4 conversion as a function of reaction temperature and (c) selectivity for HCHO as a function of CH4 conversion over BiPO4-DEG, FePO4, BiPO4-HT, α-Bi2O3, and β-Bi2O3 catalysts. Reaction conditions: catalyst (100 mg), CH4/O2/N2 (2/2/6 sccm), 420–600 °C. (d) Time-on-stream performance of BiPO4-DEG for oxidation of CH4. Reaction conditions: BiPO4-DEG (100 mg), CH4/O2/N2 (2/2/6 sccm), 550 °C. | ||
Next, the catalytic performance of BiPO4-DEG was compared with that of FePO4 nanoparticles, which are effective heterogeneous catalysts for the direct oxidation of CH4 into HCHO.44 BiPO4-DEG functioned as a solid catalyst to efficiently give HCHO at high reaction temperatures compared to FePO4 (Fig. 3(a)). The HCHO yield of BiPO4-DEG exceeded that of FePO4 above 550 °C, while FePO4 showed higher HCHO yield than BiPO4-DEG below 550 °C. Notably, a significant difference in the selectivity for HCHO was observed between these two catalysts. Fig. 3(b) and (c) show the dependence of CH4 conversion on the reaction temperature and the relationship between selectivity to HCHO and CH4 conversion, respectively. In the case of FePO4, the CH4 conversion increased up to ca. 10% at 600 °C and the selectivity for HCHO was ca. 15–40% in the range of ∼2% CH4 conversion. On the other hand, BiPO4-DEG showed ca. 40–70% selectivity for HCHO in the same CH4 conversion region, which suggests that BiPO4-DEG more selectively promotes the direct oxidation of CH4 into HCHO than FePO4. BiPO4-DEG also exhibited durability without significant change in the selectivity for HCHO (60.6 ± 1.6%) and CO (39.4 ± 1.6%) and CH4 conversion (0.55 ± 0.02%) at 550 °C for 24 h time-on-stream as shown in Fig. 3(d). On the other hand, the CO yield gradually decreased from 1.29% (1 h) to 0.52% (12 h) for the CH4 oxidation at 600 °C (Fig. S4(a)†), whereas the HCHO yield was not significantly changed for 12 h and CO2 was hardly formed in a similar way to the oxidation at 550 °C. Despite such a decrease in the CO yield, the HCHO yield and selectivity after 12 h were 0.74% and 59%, respectively, and higher than those of FePO4 (0.42% yield and 4% selectivity) at 600 °C. From the XRD pattern for the recovered BiPO4-DEG catalyst after the reaction at 600 °C (Fig. S4(b)†), there was a structural change in the grain sizes for BiPO4-DEG calculated from the diffraction lines using Scherrer's equation (from 46 to 82 nm for P21/n; from 51 to 56 nm for P21/m) and the ratio of P21/n to P21/m (from 50/50 to 20/80); thus, the bulk structures of BiPO4 gradually changed during the CH4 oxidation at 600 °C in sharp contrast to the reaction at 550 °C.
The temperature dependence of CH4 conversion and product selectivity in CH4 oxidation with BiPO4-DEG is shown in Fig. 4(a). HCHO was selectively formed in the low-temperature region (∼550 °C), and the CH4 conversion and selectivity for CO gradually increased with increasing reaction temperature. Fig. 4(b) shows the dependence of the CH4 conversion and selectivity for each product on the contact time (W/FCH4). HCHO selectivity decreased with increasing W/FCH4 but the CH4 conversion and CO selectivity increased, which suggests that HCHO is the primary product and CO is formed by sequential oxidation of HCHO. Such over-oxidation of HCHO into CO and CO2 is also observed in the direct oxidation of CH4 with FePO4.44
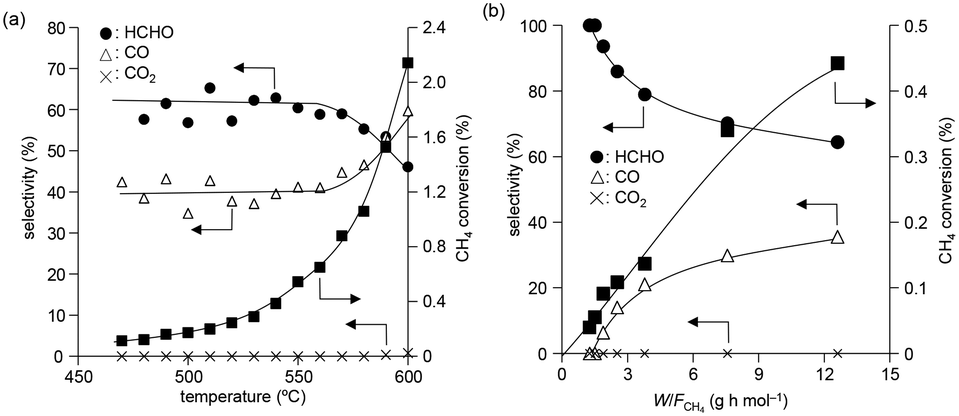 | ||
| Fig. 4 (a) Product selectivity and CH4 conversion as function of reaction temperature with O2 over BiPO4-DEG. Reaction conditions: BiPO4-DEG (100 mg), CH4/O2/N2 (2/2/6 sccm). (b) Effect of W/FCH4 on product selectivity and CH4 conversion. Reaction conditions: BiPO4-DEG (100 mg), CH4/O2/N2 (38:8:54 molar ratio; total 7.8–78 sccm), reaction temperature (550 °C). | ||
3.3. Mechanistic studies on direct oxidation of CH4 into HCHO over BiPO4
We previously reported that not only the oxidizing ability of metal phosphates but also the surface acid–base properties are key factors in the direct and selective oxidation of CH4.28,43,44 To investigate the oxidizing ability of Bi-based catalysts, H2-temperature-programmed reduction (H2-TPR) profiles were measured from 50 to 650 °C (Fig. 5). The reduction of β-Bi2O3 started around 250 °C, but the reduction of bismuth phosphates occurred at higher temperatures (from ∼350 °C). The H2 consumption estimated from the H2-TPR profiles below 550 °C decreased in the order of β-Bi2O3 (6.01 mmol g−1) > BiPO4-DEG (3.53 mmol g−1) > α-Bi2O3 (3.13 mmol g−1) > BiPO4-HT (1.03 mmol g−1) > FePO4 (0.93 mmol g−1),44 which is significantly different from the order of CH4 conversion at 550 °C (FePO4 (2.69%) > BiPO4-DEG (0.54%) > BiPO4-HT (0.12%) > α-Bi2O3 (<0.01%) > β-Bi2O3 (0%)) (Fig. S5†). In addition, the onset reduction temperature increased in the order of β-Bi2O3 (270 °C) < BiPO4-DEG (320 °C) < α-Bi2O3 (350 °C) < FePO4 (450 °C) < BiPO4-HT (490 °C), and the order is also inconsistent with the order of reactivity for CH4 oxidation. A good correlation between H2 consumption and CH4 conversion has been reported in the case of iron-based phosphates and oxides.44 Thus, the present discrepancy cannot be explained by the reaction mechanism in which lattice oxygen atoms of metal phosphates and oxides are involved in CH4 oxidation.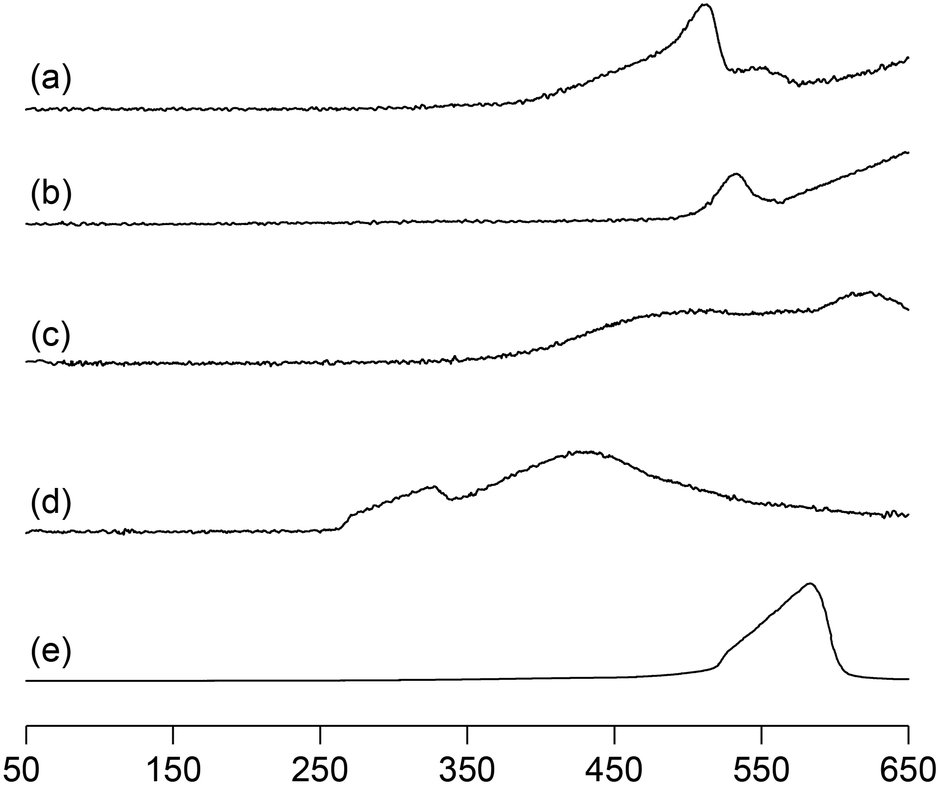 | ||
| Fig. 5 H2-TPR profiles for (a) BiPO4-DEG, (b) BiPO4-HT, (c) α-Bi2O3, (d) β-Bi2O3, and (e) FePO4. | ||
Pulse-reaction experiments for BiPO4-DEG pretreated in He at 600 °C were conducted to determine the origin of the oxygen atoms incorporated in the oxygenated products (i.e., from the oxygen atoms in BiPO4 solid or the surface oxygen species generated from gaseous O2) (Table 2). No formation of oxygenated products was observed in the reaction of only the CH4 pulse with BiPO4-DEG in sharp contrast to the FePO4 nanoparticles that reacted with CH4 to give C1 products (HCHO and COx) and partially reduced FePO4−δ.44 On the other hand, HCHO, CO, and CO2 were formed with selectivities of 44%, 54%, and 2%, respectively, at 0.9% CH4 conversion when the CH4 + O2 pulse (1/1, v/v) was passed through the catalyst bed at 600 °C. The formation of C2-coupling products such as ethane and ethylene was not observed for both the pulse reaction and catalytic oxidation of CH4 over BiPO4-DEG. Oxidative coupling of CH4 (OCM) typically proceeds under the harsh reaction conditions (>650 °C) to cleave the strong C–H bonds of CH4, and it is well accepted that the generation of methyl radicals by the solid-catalyst surface with subsequent gas-phase propagation and termination is involved in the OCM mechanism.2,4,13 These results suggest that the surface oxygen species generated from O2 and BiPO4 likely reacted with CH4 to yield C1 products (HCHO and COx) without the formation of methyl radicals in the gas-phase. The kinetics of CH4 oxidation over BiPO4-DEG at 550 °C were investigated. Fig. 6(a)–(c) show the dependence of the reaction rate (CH4 conversion) on the partial pressures of CH4 (PCH4) and O2 (PO2), and the catalyst loading of BiPO4-DEG. A first-order dependence of the reaction rates on PCH4 and catalyst loading was observed (Fig. 6(a) and (c)). The dependence of the reaction rate on PO2 exhibited saturation kinetics (Fig. 6(b)), which is consistent with the Langmuir–Hinshelwood mechanism through adsorption (i.e., activation) of O2.68–71
| Entry | Pulse | CH4 conversion (%) | Yield (%) (selectivity (%)) | ||
|---|---|---|---|---|---|
| HCHO | CO | CO2 | |||
| a Reaction conditions: BiPO4-DEG (100 mg), He (20 sccm), pulse volume (1 mL), reaction temperature (600 °C). | |||||
| 1 | CH4 | <0.01 | <0.01 (—) | <0.01 (—) | <0.01 (—) |
| 2 | CH4 + O2 (1/1, v/v) | 0.93 | 0.41 (44) | 0.50 (54) | 0.02 (2) |
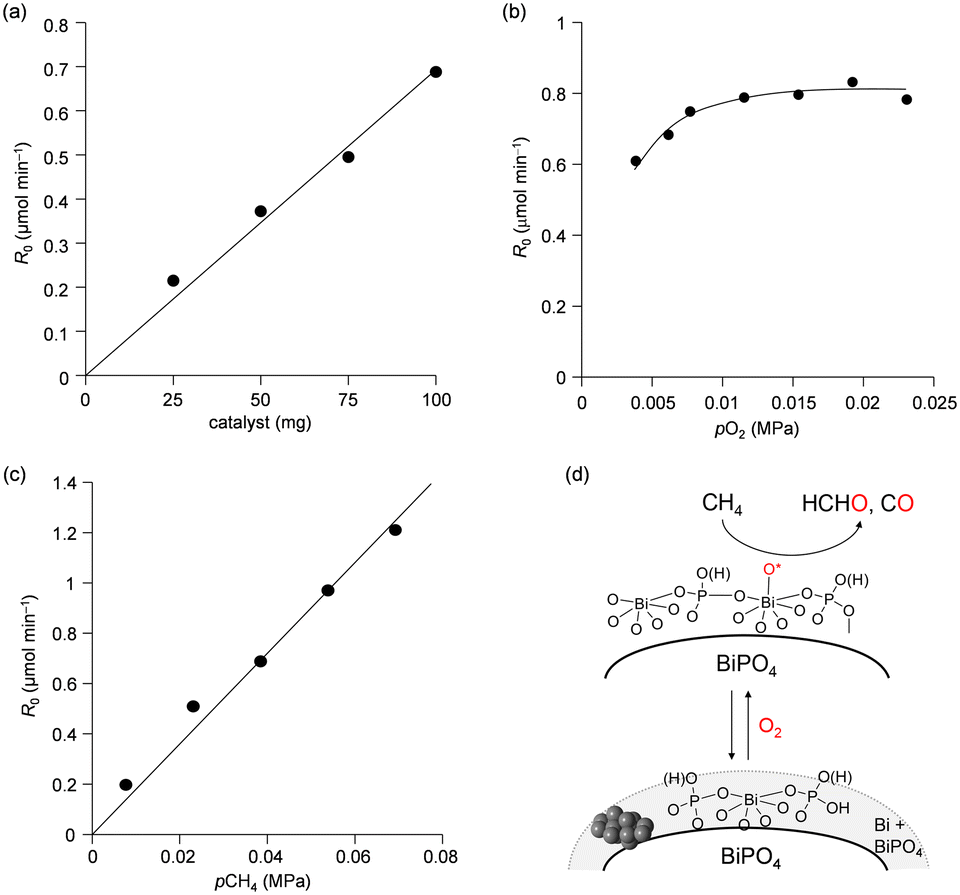 | ||
| Fig. 6 Effect of (a) catalyst amount and partial pressure of (b) O2 and (c) CH4 on reaction rate. Reaction conditions for (a): BiPO4-DEG (25–100 mg), CH4/O2/N2 (5/1/7 sccm; total 13 sccm), reaction temperature (550 °C). Reaction conditions for (b): BiPO4-DEG (100 mg), CH4/O2/N2 (5/0.5–3/7.5–5 sccm; total 13 sccm), reaction temperature (550 °C). Reaction conditions for (c): BiPO4-DEG (100 mg), CH4/O2/N2 (1–9/1/11–3 sccm; total 13 sccm), reaction temperature (550 °C). (d) Proposed reaction mechanism for oxidation of CH4 with O2 over BiPO4-DEG. | ||
XPS measurements were performed to further investigate the surface structures of BiPO4-DEG. For the XPS Bi 4f spectrum of BiPO4-DEG at room temperature, a main peak around 159.8 eV (Bi 4f7/2) assignable to Bi3+ species in BiPO4 and a slight peak around 155.9 eV of metallic Bi species were observed (Fig. 7(a)).72,73 Such formation of metallic Bi species has also been reported for BiPO4 synthesized in ethylene glycol solvent probably due to the weak reducing ability of ethylene glycol.48 It has also been reported that the formation of surface oxygen vacancies of BiPO4 leads to the disordered edge of BiPO4 particles observed by TEM analyses.51 In the case of BiPO4-DEG, the edge of the nanoparticles become disordered (thickness ∼1 nm, Fig. 2(d)), which supports the formation of metallic Bi species. The XPS O 1s spectrum of BiPO4-DEG also showed a peak around 533 eV which corresponds to adsorbed water in addition to the main peak around 531 eV assignable to lattice oxygen of BiPO4.
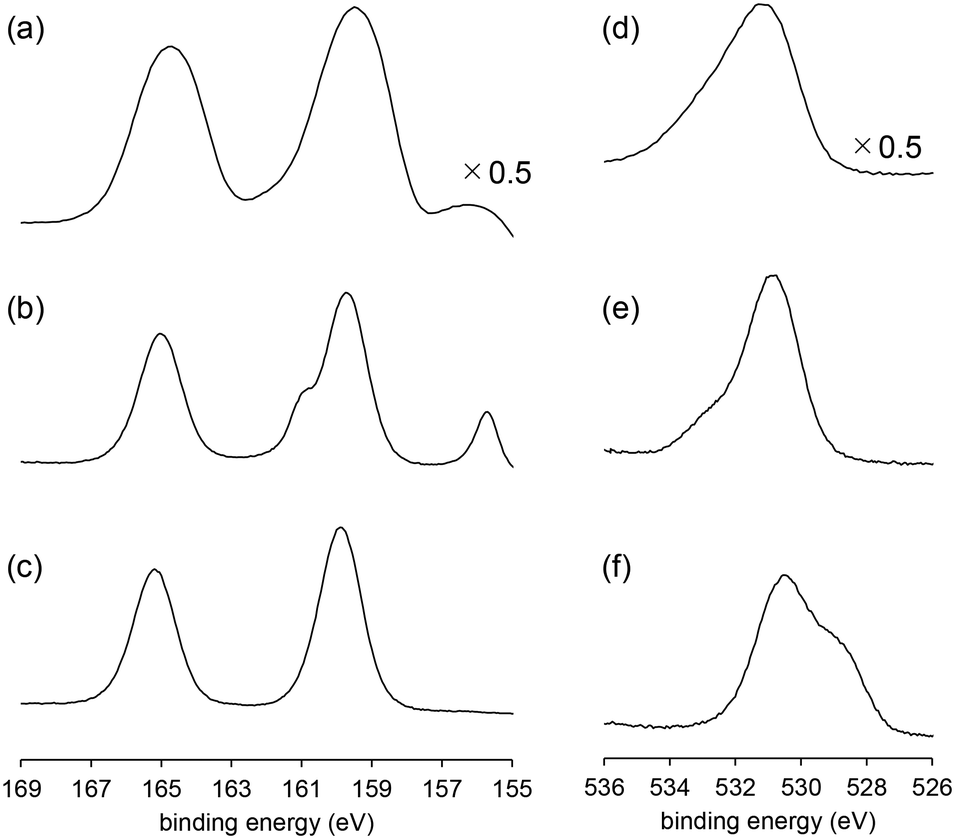 | ||
| Fig. 7 XPS (a–c) Bi 4f and (d–f) O 1s spectra of BiPO4-DEG. (a and d) BiPO4-DEG at room temperature under evacuation, (b and e) BiPO4-DEG at 550 °C under evacuation, and (c and f) BiPO4-DEG at 550 °C under O2 (0.1 mbar). | ||
Surface oxygen species are formed on a CePO4 catalyst through the activation of the O2 molecule assisted by the electric field. Such active oxygen species possibly facilitate the oxidative coupling of the CH4 reaction even at low temperature. In this case, the surface Lewis acidic Ce sites would play an important role in the activation of O2.28,43 Therefore, the surface acid properties of the present BiPO4-DEG catalyst were investigated by IR measurements of samples adsorbed with pyridine and acetone. The IR spectrum for pyridine-adsorbed BiPO4-DEG showed a band at 1446 cm−1 assignable to the pyridine species coordinated to the Lewis acid sites, and no band at 1540 cm−1 due to pyridinium ions bonded to the Brønsted acid sites (Fig. 8(a)). The amount of Lewis acid sites was estimated to be 26 μmol g−1 from the intensity of the band at 1446 cm−1. The density of surface Bi cations on BiPO4-DEG was calculated to be 1.6 nm−2 from the BET surface area of BiPO4-DEG and the amounts of Lewis acid sites measured using pyridine-adsorbed IR. This value was comparable to that for monoclinic CePO4 nanorods (1.6 nm−2) with monazite-type structure and the calculated value estimated from the main surface structure obtained from HAADF-STEM observation.28,29 The IR spectrum of acetone adsorbed on BiPO4-DEG (Fig. 8(b)) showed one strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band due to acetone molecules coordinated to Lewis acid sites, with the band position at a lower wavenumber (1683 cm−1) than those for acetone in the gas phase (1731 cm−1), CePO4 (1699 cm−1), and FePO4 (1685 cm−1).28,43,44 All these results support the presence of uniform surface Lewis acidic Bi species on the BiPO4-DEG nanoparticles. Such coordinatively unsaturated sites would be involved in the O2 activation. In addition, the basicity was also evaluated by IR spectroscopy with adsorbed CHCl3 and CH3OH (Fig. 8(c) and (d)). The presence of weakly-basic phosphate units on BiPO4 was confirmed,‡ which likely contributes to the suppression of complete oxidation to CO2 in a similar way to the CH4 oxidation over FePO4.44
O stretching band due to acetone molecules coordinated to Lewis acid sites, with the band position at a lower wavenumber (1683 cm−1) than those for acetone in the gas phase (1731 cm−1), CePO4 (1699 cm−1), and FePO4 (1685 cm−1).28,43,44 All these results support the presence of uniform surface Lewis acidic Bi species on the BiPO4-DEG nanoparticles. Such coordinatively unsaturated sites would be involved in the O2 activation. In addition, the basicity was also evaluated by IR spectroscopy with adsorbed CHCl3 and CH3OH (Fig. 8(c) and (d)). The presence of weakly-basic phosphate units on BiPO4 was confirmed,‡ which likely contributes to the suppression of complete oxidation to CO2 in a similar way to the CH4 oxidation over FePO4.44
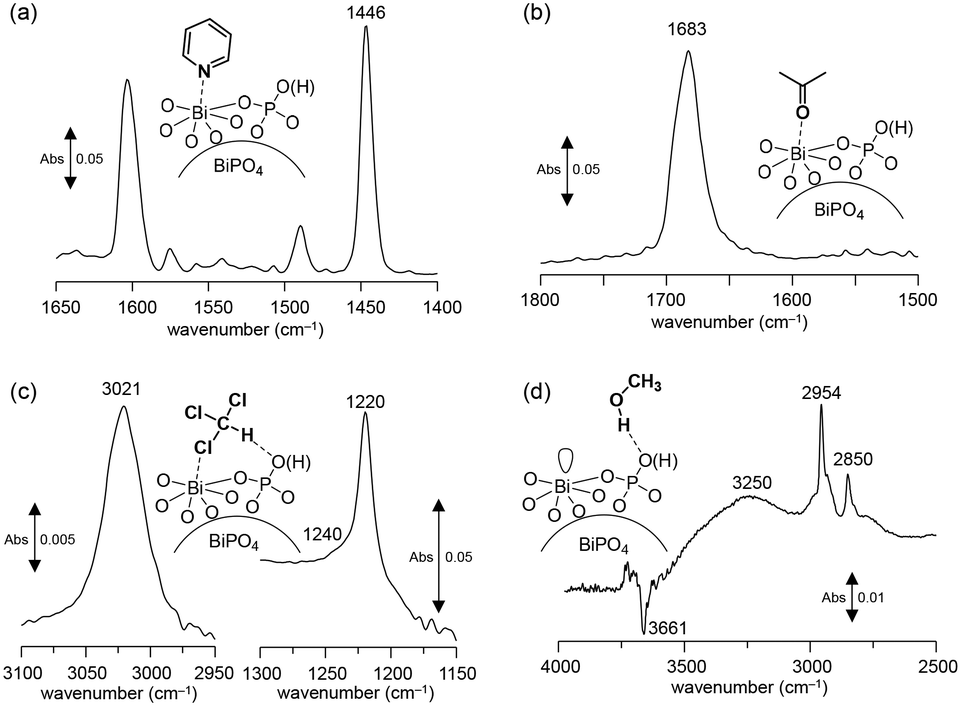 | ||
| Fig. 8 Difference IR spectra for (a) pyridine-, (b) acetone-, (c) chloroform-, and (d) CH3OH-adsorbed BiPO4-DEG at 25 °C. | ||
To obtain information on possible active oxygen species formed on surface Bi sites, an operando near-ambient-pressure X-ray photoelectron spectroscopy (NAP-XPS) experiment was conducted under O2 atmosphere. When BiPO4-DEG was exposed to O2 (0.1 mbar) at 550 °C, only the peak corresponding to BiPO4 was observed in the XPS Bi 4f spectra with the disappearance of the peak of metallic Bi, which suggests reconstruction of the surface structure. The XPS O 1s spectra also changed during the reaction of BiPO4-DEG with O2. While the main peak around 531 eV assignable to lattice oxygens of BiPO4 was unchanged, the peak around 533 eV corresponding to adsorbed oxygen atoms disappeared and a new peak around 529 eV appeared upon exposure of O2. It has been reported that the peaks due to bismuth oxides are in the range of 528.8–529.9 eV and that the negative shift of O 1s peaks is likely caused by the presence of non-bridging Bi–O species.74–78 Thus, the new peak may be caused by surface oxygen species such as Bi–O species generated on BiPO4, which possibly react with CH4 to give HCHO as a primary product. There was no significant difference in the XPS Bi 4f spectra between the fresh and recovered BiPO4-DEG catalysts after the CH4 oxidation under the conditions in Fig. S6,† which suggests that the in situ generated oxygen species observed in the reaction temperature range play an important role in the present CH4 oxidation.
To further investigate possible involvement of surface active oxygen species on BiPO4, density functional theory (DFT) calculations were carried out. Since the surface Bi density of BiPO4-DEG was comparable to that of monoclinic CePO4 nanorods as described above, the monazite-type structure (P21/n) was used as a model catalyst for the CH4 oxidation. Surface energy calculations for the (100), (110), and (111) surfaces of BiPO4 identified that the (100) surface is the most stable one. Furthermore, the DFT-based thermodynamic analysis have shown that the partially oxidized BiPO4 (100) surface (0.25 monolayer) in which O atom adsorbed on Bi is the stable surface under the reaction environment (see the details in ESI,† Fig. S7 and S8). The following two pathways the CH4 activation were compared: (i) H-abstraction by surface O atom and CH3 adsorption on PO4 units, and (ii) H-abstraction by lattice O atom and CH3 adsorption on PO4 units. Fig. 9 shows the optimized geometry and the reaction energies for CH4 activation by these two pathways. The reaction energy (ΔE) is defined as the dissociative adsorption energy of CH4. H-abstraction using a surface O atom is an exothermic reaction, since the calculated ΔE is −1.65 eV. On the other hand, H-abstraction by a lattice O atom is an endothermic reaction (ΔE = 0.94 eV). Thus, our calculations indicate that the CH4 activation takes place using surface O atoms, and not lattice O atoms. The surface O atoms are formed from gaseous O2 in the inlet gas, and therefore CH4 activation requires the presence of O2. This is consistent with the observed reaction rate dependence on PO2 as seen in Fig. 6. In addition to the possible involvement of surface oxygen species generated on BiPO4, the weak basicity on BiPO4 (Fig. 8(c) and (d)),‡ similar to those on FePO4 and CePO4, likely leads to high HCHO selectivity of BiPO4-DEG.79
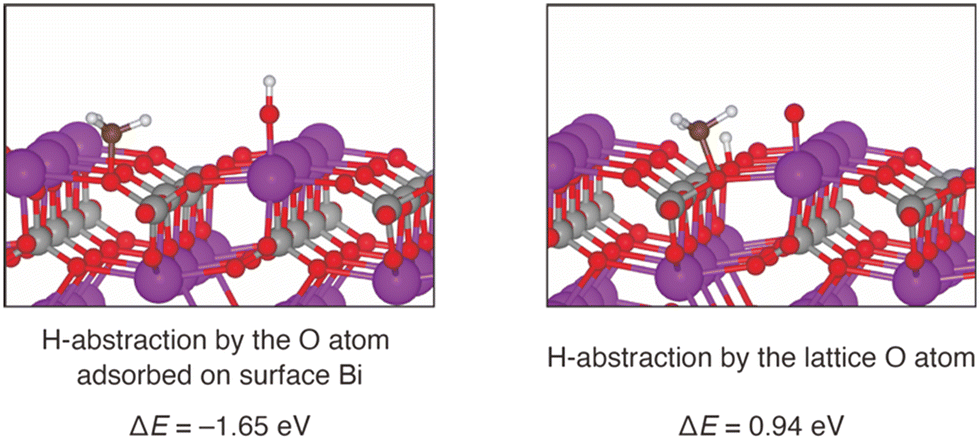 | ||
| Fig. 9 Optimized structure of the product state of the CH4 activation on 0.25 ML Bi-oxidized BiPO4 (100) surface. Two reaction pathways are considered, and their reaction energies (ΔEs) calculated with the DFT are shown. | ||
Conclusions
In summary, bismuth-based phosphate and oxide catalysts were investigated for the oxidation of CH4 with O2 as the sole oxidant. Monoclinic BiPO4 nanoparticles (BiPO4-DEG) exhibited higher catalytic performance for the direct oxidation of CH4 into HCHO than BiPO4 synthesized by the hydrothermal method (BiPO4-HT), α-Bi2O3, and β-Bi2O3. The lack of correlation between the catalytic activity and oxidizing ability estimated by H2-TPR and pulse reaction experiments suggested that CH4 oxidation did not proceed with lattice oxygen supplied from the BiPO4-DEG solid. On the basis of mechanistic studies including the catalyst effect, kinetics, NAP-XPS, and DFT calculations, the oxidation of CH4 may proceed at surface oxygen species generated on surface Bi atoms of BiPO4. IR spectroscopy measurements of BiPO4-DEG with adsorbed probe molecules indicate the presence of uniform Lewis acid sites and weak basic sites, which possibly activate O2 and suppress subsequent oxidation into CO2, respectively. Such a possible O2 activation mode for BiPO4-DEG would result in high selective formation of HCHO at high temperatures in sharp contrast to FePO4 nanoparticles with the redox mechanism.Author contributions
A. M. and K. O. designed the experiments, performed the experimental investigation, and conducted data analyses with the help of K. K. A. I. performed the DFT calculations. M. T., C. W., Y. L. performed the NAP-XPS analyses. K. K., A. I., and Y. L. wrote the paper. The draft was reviewed by A. M., A. I., Y. L., M. H. and K. K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded in part by a Grant-in-Aid (21H01713) for Scientific Research from the Japan Society for the Promotion of Science (JSPS), and the PRESTO (JPMJPR15S3 and JPMJPR17S1), CREST (JPMJCR16P3), and A-STEP (JPMJTR20TG) programs of the Japan Science and Technology Agency (JST), and the “Design and Engineering by Joint Inverse Innovation for Materials Architecture” program of the Japan Ministry of Education, Culture, Sports, Science and Technology (MEXT).Notes and references
- D. Saha, H. A. Grappe, A. Chakraborty and G. Orkoulas, Chem. Rev., 2016, 116, 11436–11499 CrossRef CAS PubMed.
- R. Horn and R. Schlögl, Catal. Lett., 2015, 145, 23–39 CrossRef CAS.
- A. Abdulrasheed, A. A. Jalil, Y. Gambo, M. Ibrahim, H. U. Hambali and M. Y. Shahul Hamid, Renewable Sustainable Energy Rev., 2019, 108, 175–193 CrossRef CAS.
- C. Hammond, S. Conrad and I. Hermans, ChemSusChem, 2012, 5, 1668–1686 CrossRef CAS PubMed.
- K. Otsuka and Y. Wang, Appl. Catal., A, 2001, 222, 145–161 CrossRef CAS.
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- A. V. Vekki and S. T. Marakaev, Russ. J. Appl. Chem., 2009, 82, 521–536 CrossRef CAS.
- A. H. Bagherzadeh Mostaghimi, T. A. Al-Attas, M. G. Kibria and S. Siahrostami, J. Mater. Chem. A, 2020, 8, 15575–15590 RSC.
- S. Ogo and Y. Sekine, Chem. Rec., 2017, 17, 726–738 CrossRef CAS PubMed.
- I. Vollmer, I. Yarulina, F. Kapteijn and J. Gascon, ChemCatChem, 2018, 11, 39–52 CrossRef.
- K. Sun, D. M. Ginosar, T. He, Y. Zhang, M. Fan and R. Chen, Ind. Eng. Chem. Res., 2018, 57, 1768–1789 CrossRef CAS.
- H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 10096–10115 CrossRef CAS PubMed.
- M. H. Mahyuddin, Y. Shiota and K. Yoshizawa, Catal. Sci. Technol., 2019, 9, 1744–1768 RSC.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F.-S. Xiao, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- P. Xiao, Y. Wang, T. Nishitoba, J. N. Kondo and T. Yokoi, Chem. Commun., 2019, 55, 2896–2899 RSC.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- B. E. R. Snyder, M. L. Bols, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Chem. Rev., 2018, 118, 2718–2768 CrossRef CAS PubMed.
- P. Schwach, M. G. Willinger, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 11381–11384 CrossRef CAS PubMed.
- J. H. Lunsford, Angew. Chem., Int. Ed. Engl., 1995, 34, 970–980 CrossRef CAS.
- B. P. C. Hereijgers and B. M. Weckhuysen, ChemSusChem, 2009, 2, 743–748 CrossRef CAS PubMed.
- M. Bugnola, R. Carmieli and R. Neumann, ACS Catal., 2018, 8, 3232–3236 CrossRef CAS.
- K. Otsuka and M. Hatano, J. Catal., 1987, 108, 252–255 CrossRef CAS.
- K. Harrath, X. Yu, H. Xiao and J. Li, ACS Catal., 2019, 9, 8903–8909 CrossRef CAS.
- C. Williams, J. H. Carter, N. F. Dummer, Y. K. Chow, D. J. Morgan, S. Yacob, P. Serna, D. J. Willock, R. J. Meyer, S. H. Taylor and G. J. Hutchings, ACS Catal., 2018, 8, 2567–2576 CrossRef CAS.
- T. Ohno and J. B. Moffat, Catal. Lett., 1992, 16, 181–190 CrossRef CAS.
- A. V. Annapragada and E. Gulari, J. Catal., 1990, 123, 130–146 CrossRef CAS.
- A. Sato, S. Ogo, K. Kamata, Y. Takeno, T. Yabe, T. Yamamoto, S. Matsumura, M. Hara and Y. Sekine, Chem. Commun., 2019, 55, 4019–4022 RSC.
- V. Paunović, G. Zichittella, M. Moser, A. P. Amrute and J. Pérez-Ramirez, Nat. Chem., 2016, 8, 803–809 CrossRef PubMed.
- G. Zichittella, V. Paunović, A. P. Amrute and J. Pérez-Ramírez, ACS Catal., 2017, 7, 1805–1817 CrossRef CAS.
- Y. Wang and K. Otsuka, J. Catal., 1995, 155, 256–267 CrossRef CAS.
- Y. Wang, X. Wang, Z. Su, Q. Guo, Q. Tang, Q. Zhang and H. Wan, Catal. Today, 2004, 93–95, 155–161 CrossRef CAS.
- G. O. Alptekin, A. M. Herring, D. L. Williamson, T. R. Ohno and R. L. McCormick, J. Catal., 1999, 181, 104–112 CrossRef CAS.
- Y. Wang and K. Otsuka, J. Mol. Catal. A: Chem., 1996, 111, 341–356 CrossRef CAS.
- Y. Wang and K. Otsuka, J. Chem. Soc., Faraday Trans., 1995, 91, 3953–3961 RSC.
- V. Gomonaj and H. Toulhoat, ACS Catal., 2018, 8, 8263–8272 CrossRef CAS.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y.-S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS PubMed.
- T. Ikuno, J. Zheng, A. Vjunov, M. Sanchez-Sanchez, M. A. Ortuno, D. R. Pahls, J. L. Fulton, D. M. Camaioni, Z. Li, D. Ray, B. L. Mehdi, N. D. Browning, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and J. A. Lercher, J. Am. Chem. Soc., 2017, 139, 10294–10301 CrossRef CAS PubMed.
- V. Fornés, C. López, H. H. López and A. Martínez, Appl. Catal., A, 2003, 249, 345–354 CrossRef.
- H. Launay, S. Loridant, A. Pigamo, J. L. Dubois and J. M. M. Millet, J. Catal., 2007, 246, 390–398 CrossRef CAS.
- T. Akiyama, R. Sei and S. Takenaka, Catal. Sci. Technol., 2021, 11, 5273–5281 RSC.
- J. Tian, J. Tan, Z. Zhang, P. Han, M. Yin, S. Wan, J. Lin, S. Wang and Y. Wang, Nat. Commun., 2020, 11, 5693 CrossRef CAS PubMed.
- S. Kanai, I. Nagahara, Y. Kita, K. Kamata and M. Hara, Chem. Sci., 2017, 8, 3146–3153 RSC.
- A. Matsuda, H. Tateno, K. Kamata and M. Hara, Catal. Sci. Technol., 2021, 11, 6987–6998 RSC.
- P. Sprenger, W. Kleist and J.-D. Grunwaldt, ACS Catal., 2017, 7, 5628–5642 CrossRef CAS.
- W. K. Darkwah, B. B. Adormaa, M. K. C. Sandrine and Y. Ao, Catal. Sci. Technol., 2019, 9, 546–566 RSC.
- C. Pan, J. Xu, Y. Chen and Y. Zhu, Appl. Catal., B, 2012, 115–116, 314–319 CrossRef CAS.
- F. Tian, H. Zhao, G. Li, Z. Dai, Y. Liu and R. Chen, ChemSusChem, 2016, 9, 1579–1585 CrossRef CAS PubMed.
- J. Xu, L. Li, C. Guo, Y. Zhang and W. Meng, Appl. Catal., B, 2013, 130–131, 285–292 CrossRef CAS.
- Y. Guo, P. Wang, J. Qian, Y. Ao, C. Wang and J. Hou, Appl. Catal., B, 2018, 234, 90–99 CrossRef CAS.
- Y. Lv, Y. Zhu and Y. Zhu, J. Phys. Chem. C, 2013, 117, 18520–18528 CrossRef CAS.
- F. Qiu, L. T. Weng, P. Ruiz and B. Delmon, Appl. Catal., 1989, 47, 115–123 CrossRef CAS.
- N. Abadzhjieva, P. Tzokov, I. Uzunov, V. Minkov, D. Klissurski and V. Rives, React. Kinet. Catal. Lett., 1994, 53, 413–418 CrossRef CAS.
- M. Ruwet, S. Ceckiewicz and B. Delmon, Ind. Eng. Chem. Res., 1987, 26, 1981–1983 CrossRef CAS.
- T.-S. Chang, L. Guijia, C.-H. Shin, Y. K. Lee and S.-S. Yun, Catal. Lett., 2000, 68, 229–234 CrossRef CAS.
- Y. Takita, M. Ninomiya, R. Matsuzaki, H. Wakamatsu, H. Nishiguchi and T. Ishihara, Phys. Chem. Chem. Phys., 1999, 1, 2367–2372 RSC.
- B. Gallace and J. B. Moffat, J. Catal., 1982, 76, 182 CrossRef CAS.
- F. Pazoki, S. Bagheri, M. Shamsayei, M. J. Nejad and A. Heydari, Mater. Chem. Phys., 2020, 253, 123327 CrossRef CAS.
- T. Aihara, W. Aoki, S. Kiyohara, Y. Kumagai, K. Kamata and M. Hara, ACS Appl. Mater. Interfaces, 2023, 15, 17957–17968 CrossRef CAS PubMed.
- M. Koutani, E. Hayashi, K. Kamata and M. Hara, J. Am. Chem. Soc., 2022, 144, 14090–14100 CrossRef CAS PubMed.
- Y. Yamaguchi, R. Aono, E. Hayashi, K. Kamata and M. Hara, ACS Appl. Mater. Interfaces, 2020, 12, 36004–36013 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. Sun, A. Ruzsinszky and J. P. Perdew, Phys. Rev. Lett., 2015, 115, 036402 CrossRef PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- B. Romero, S. Bruque, M. A. G. Aranda and J. E. Iglesias, Inorg. Chem., 2002, 33, 1869–1874 CrossRef.
- K. Takanabe and E. Iglesia, Angew. Chem., Int. Ed., 2008, 47, 7689–7693 CrossRef CAS PubMed.
- E. Hayashi, T. Tamura, T. Aihara, K. Kamata and M. Hara, ACS Appl. Mater. Interfaces, 2022, 14, 6528–6537 CrossRef CAS PubMed.
- K. Sugahara, K. Kamata, S. Muratsugu and M. Hara, ACS Omega, 2017, 2, 1608–1616 CrossRef CAS PubMed.
- S. Kawasaki, K. Kamata and M. Hara, ChemCatChem, 2016, 8, 3247–3253 CrossRef CAS.
- V. D. Makwana, Y. C. Son, A. R. Howell and S. L. Suib, J. Catal., 2002, 210, 46–52 CrossRef CAS.
- P. Deng, H. Wang, R. Qi, J. Zhu, S. Chen, F. Yang, L. Zhou, K. Qi, H. Liu and B. Y. Xia, ACS Catal., 2020, 10, 743–750 CrossRef CAS.
- Q. Jing, L. Huang, Q. Li, Y. Song and L. Chen, J. Mater. Sci.: Mater. Electron., 2020, 31, 20954–20963 CrossRef CAS.
- J. Gong, C. S. Lee, E. J. Kim, J. H. Kim, W. Lee and Y. S. Chang, ACS Appl. Mater. Interfaces, 2017, 9, 28426–28432 CrossRef CAS PubMed.
- J. Liu, S. Guo, H. Wu, X. Zhang, J. Li and K. Zhou, J. Mater. Sci. Technol., 2021, 85, 1–10 CrossRef CAS.
- J. Sun, J. Wen, G. Wu, Z. Zhang, X. Chen, G. Wang and M. Liu, Adv. Funct. Mater., 2020, 30, 2004108 CrossRef CAS.
- Q. Chen, Y. Wang and H. Wang, J. Non-Cryst. Solids, 2018, 481, 85–93 CrossRef CAS.
- L. N. Elliott, D. Austin, R. A. Bourne, A. Hassanpour, J. Robb, J. L. Edwards, S. Sutcliffe and T. N. Hunter, Langmuir, 2023, 39, 5697–5709 CrossRef CAS PubMed.
- T. Komanoya, K. Nakajima, M. Kitano and M. Hara, J. Phys. Chem. C, 2015, 119, 26540–26546 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00590a |
| ‡ The basicity was confirmed by IR spectroscopy with adsorbed CHCl3 (Fig. 8(c) and (d)). The red-shift of the original C–H stretching mode of the CHCl3 molecule from 3034 cm−1 to 3021 cm−1 indicates the presence of basic sites on the surface.74 The band shift (13 cm−1) was larger than that for FePO4 (∼1 cm−1) but lower than that for CePO4 (26 cm−1); thus, the basicity of BiPO4 was stronger than that of FePO4 but weaker than that of CePO4.39,40 In the IR spectrum of BiPO4-DEG with adsorbed CH3OH, broad bands between 3000 and 3500 cm−1 appeared with negative v(O–H) bands, and bands at 2954 and 2850 cm−1 assignable to the asymmetric and symmetric CH3 stretching modes of molecularly adsorbed CH3OH, respectively, were observed. All these results are consistent with previous reports for the interaction of probe molecules on metal phosphates with uniform Lewis acid sites and weak base sites. |
| This journal is © The Royal Society of Chemistry 2023 |