
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tautomerization of single asymmetric oxahemiporphycene molecules on Cu(111)†
Simon
Jaekel‡
 a,
Emile
Durant
b,
Monika
Schied
a,
Mats
Persson
b,
Jakub
Ostapko
c,
Michał
Kijak
c,
Jacek
Waluk
cd and
Leonhard
Grill
*a
a,
Emile
Durant
b,
Monika
Schied
a,
Mats
Persson
b,
Jakub
Ostapko
c,
Michał
Kijak
c,
Jacek
Waluk
cd and
Leonhard
Grill
*a
aDepartment of Physical Chemistry, University of Graz, Heinrichstraße 28, Graz, Austria. E-mail: leonhard.grill@uni-graz.at
bSurface Science Research Centre, Department of Chemistry, University of Liverpool, Liverpool L69 3BX, UK
cInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
dFaculty of Mathematics and Science, Cardinal Stefan Wyszyński University, Dewajtis 5, 01-815 Warsaw, Poland
First published on 19th December 2022
Abstract
We have studied 22-oxahemiporphycene molecules by a combination of scanning tunneling microscopy at low temperatures and density functional theory calculations. In contrast to other molecular switches with typically two switching states, these molecules can in principle exist in three different tautomers, due to their asymmetry and three inequivalent binding positions of a hydrogen atom in their macrocycle. Different tautomers are identified from the typical appearance on the surface and tunneling electrons can be used to tautomerize single molecules in a controllable way with the highest rates if the STM tip is placed close to the hydrogen binding positions in the cavity. Characteristic switching processes are explained by the different energy pathways upon adsorption on the surface. Upon applying higher bias voltages, deprotonation occurs instead of tautomerization, which becomes evident in the molecular appearance.
Introduction
Molecular switches1 are molecules with at least two stable states, which can be realized reversibly by an external stimulus. They are of interest as model systems to study fundamental chemical processes, but also for applications in responsive molecular systems,2 for instance in molecular machines,3 as switchable catalyst,4 conductance switches5 or for switchable surfaces.6 The study of molecular switches at solid, well-defined single-crystal surfaces7–9 is advantageous, because it allows to use scanning tunneling microscopy (STM) for single-molecule imaging with complete access to the molecular switching state as well as its adsorption position and orientation with respect to the crystalline lattice underneath. In addition, the local surroundings of every single molecule can be imaged at the atomic scale. These are important as they can affect the molecular properties and consequently its switching behavior.10,11A prototypical class of molecular switches are based on intramolecular proton transfer, i.e. tautomerization, which plays an important role in the enzymatic catalysis in proteins and nucleic acids.12 Excited state tautomerization is responsible for the protecting effect in photo-stabilizers.13 Experimental studies of tautomerism allow detailed modeling of proton transfer, which includes the role of vibrational mode-selective tunneling.14 The family of porphines (free-base porphyrins)15 is a prominent example for tautomerization studies as the proton transfer occurs within the molecular cavity. Tautomerization in such molecules adsorbed on a surface has already been studied in porphyrins16 and (with various stimuli) in porphycenes,10,17–21 where the imaging of single molecules gives access to the proton positions within the chemical structure. Typically, these molecules are adsorbed as symmetric, bistable switches on the surface, exhibiting two equivalent states. However, realizing a molecular switch with multiple, i.e. more than two, states on a surface would be of great interest as such a system would offer multiple switching options beyond binary transitions. Such a case has been realized only once so far, but with symmetric and thus degenerate and equivalent states.16 Additionally, this was achieved in a deprotonated compound and thus not in the original chemical structure, which would exhibit only two switching states.16 Here, we have created three switching states by using porphyrin isomers that are asymmetric in their chemical structure of the molecular cavity, offering three inequivalent sites to bind the hydrogen atom.
As shown by Kumagai et al.,22 introducing a source of asymmetry to the switching system – either through the presence of the STM tip or molecules/adatoms nearby – can influence the switching process of porphycene molecules on Cu(110), which exhibits two-fold rotational symmetry. Due to the adsorption geometry with the molecular symmetry axis parallel to the close-packed rows of the surface, porphycene on Cu(110) has a two-fold rotational symmetry too. This can be circumvented through the adoption of an angle between the molecular symmetry axes and those of the substrate by choosing a surface on which a molecule adopts a chiral adsorption geometry – as phthalocyanine on Ag(100).23 Yet, this approach depends on the chosen surface and cannot be expected to be robust for different surfaces or adding substituents to the molecule. Hence, it is of particular interest to find a molecule that undergoes tautomerization and lacks rotational symmetry. For such a molecule, the intramolecular proton transfer rates to neighboring binding sites in either direction should be inequivalent for any of the possible binding sites. Of course, maintaining (meta-)stability of as many tautomers as possible is desirable.
One such candidate for a tautomerizing molecule without rotational symmetry is unsubstituted hemiporphycene (HPc; Fig. 1a).24 It is also a derivative of the basic porphyrin structure – like porphycene – and has the same number (20) of carbon atoms in the macrocycle as porphine and porphycene. Rotational asymmetry is introduced by three different links between the pyrrole units: two methine bridges (carbons 5 and 20, see Fig. 1a), one direct C–C bond (between carbons 9 and 10) and one ethylene bridge (carbons 14 and 15). The lack of rotational symmetry can be easily understood from the presence of bridging motifs which only appear once. This means that their position can only be reproduced by equivalents of the non-rotated structure. Under this consideration, corrole (three methine bridges, one direct link) would also be an interesting candidate for tautomerization. Synthesis of unsubstituted corroles has been reported recently,25 but the free-base form was found to be unstable.
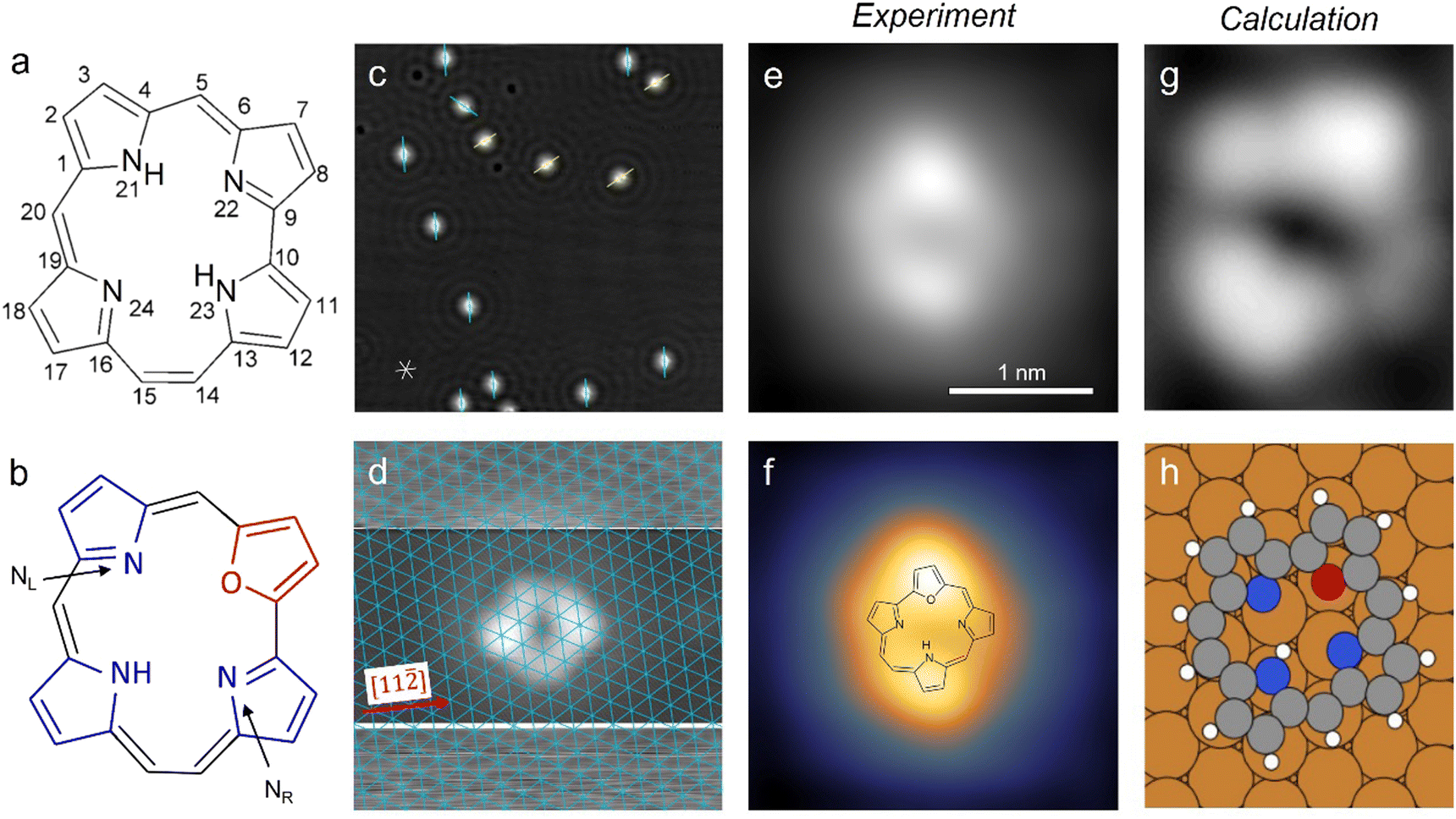 | ||
Fig. 1 Structure models of the unsubstituted hemiporphycene molecule (a) and the 22-oxahemiporphycene (O-HPc) molecule (b) with the pyrrole- and furan-groups highlighted in blue and red, respectively. (c) STM image showing O-HPc molecules on Cu(111) and the surrounding standing waves of the copper surface state, with marked preferred molecular orientations (40 × 40 nm2, 100 mV, 50 pA). The asterisk at the bottom left indicates the close-packed atomic rows (in [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0], [10] and [01] directions) of the Cu(111) surface. (d) STM image of a single O-HPc molecule, taken with increased contrast in the top and bottom sections to image the substrate with atomic resolution (see Section 3 in the ESI†). The thus obtained Cu(111) lattice is superimposed in blue. (e and f) STM image (both 2.55 × 2.55 nm2, 200 mV, 100 pA) of a single O-HPc molecule shown in a pure greyscale (e) and a two-color scale (f) for enhanced intramolecular contrast. The chemical structure of the O-HPc molecule is superimposed onto the STM image in (f). (g) Calculated STM image of a single O-HPc molecule with the corresponding adsorption geometry in (h). The molecule is oriented equally in (g and h). 0], [10] and [01] directions) of the Cu(111) surface. (d) STM image of a single O-HPc molecule, taken with increased contrast in the top and bottom sections to image the substrate with atomic resolution (see Section 3 in the ESI†). The thus obtained Cu(111) lattice is superimposed in blue. (e and f) STM image (both 2.55 × 2.55 nm2, 200 mV, 100 pA) of a single O-HPc molecule shown in a pure greyscale (e) and a two-color scale (f) for enhanced intramolecular contrast. The chemical structure of the O-HPc molecule is superimposed onto the STM image in (f). (g) Calculated STM image of a single O-HPc molecule with the corresponding adsorption geometry in (h). The molecule is oriented equally in (g and h). | ||
Tautomerization of hemiporphycene can be activated by pulses from an STM tip – the energy landscape of the different tautomers however appears to strongly favor one configuration.26 This is different for 22-oxahemiporphycene molecules (referred to as O-HPc from this point on) with a substitution of N with O at position 22 (Fig. 1b), which is reported here for the first time (details of its synthesis are provided in the ESI†). As a result of the substitution, the cavity of O-HPc binds only one hydrogen atom, which allows for three different tautomers of the molecule (see Fig. S8, ESI†), corresponding to the three nitrogen atoms.
Methods
Experimental
O-HPc molecules were deposited in situ through sublimation from a Knudsen cell onto a clean Cu(111) surface. The heating power of the cell was set to 468 mW, which resulted in crucible temperatures between 155 and 175 °C and rates of about 0.01 monolayers (ML) per minute, producing reproducible molecular coverages. Imaging was done by low temperature-scanning tunneling microscopy (LT-STM) in constant-current mode, with bias voltages applied to the sample (the STM tip is grounded), which was kept at around 5 K during imaging. All images have been sparsely filtered.Calculations
Calculations of equilibrium structures and minimum energy paths (MEPs) of O-HPc on the Cu(111) surface were obtained from periodic, plane-wave density functional theory (DFT) calculations using the Vienna ab initio simulation program (VASP).27 The Projector Augmented Wave (PAW) method28 was used to treat the electron-ion core interactions. The vdW-DF-cx29–31 version of the van der Waals density functional was used to treat the exchange–correlation effects. The Cu(111) surface was represented in a supercell by a four-layer slab with (5, 0; −3, 6) and (6 × 6) surface unit cells with a vacuum region of 30 Å. The (5, 0; −3, 6) cell was used to find the equilibrium structure configurations and calculate the MEPs between the different O-HPc tautomers using the climbing image nudged elastic band (CI-NEB) method,32–35 whereas the larger (6 × 6) cell was used in the simulation of STM images using the Tersoff-Hamann method at constant LDOS.36,37 For all calculations, the Brillouin zones of the (5, 0; −3, 6) and (6 × 6) cells were sampled with k-point meshes of (8 × 8 × 1) and (12 × 12 × 1), respectively. The plane-wave cut-off energy was 400 eV for all calculations. All geometrical optimizations were carried out until the ionic forces were less than 0.01 eV Å−1 while the bottom two layers of the Cu slab were constrained at the calculated lattice constant of 3.579 Å. Further computational details can be found in ref. 38.Results and discussion
The O-HPc molecules preferentially adsorb at the step edges of the Cu(111) surface, but at sufficient coverages they also appear as isolated molecules on the terraces (Fig. 1c and Fig. S1, S2 in the ESI†). The O-HPc molecule does not have any symmetry axes (see Fig. 1b), but its appearance in STM images shows two protrusions along the longer axis of the molecule (Fig. 1e and f), which can be used for assigning an orientation (as indicated in Fig. 1c). To determine the adsorption configuration, the substrate lattice can be atomically resolved (Fig. S3, ESI†) and – by modifying the tunneling parameters during scanning – it is also possible to simultaneously image the molecule and obtain the atomically resolved Cu(111) surface lattice in the very same image (Fig. 1d; see Section 3 in the ESI† for details). Consequently, the surface lattice can be superimposed over the molecular structure (blue grid in Fig. 1d), revealing the precise adsorption position and orientation with respect to the surface lattice. The result shows that the molecules orient approximately along the [11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ] direction of the substrate. Single molecules show a characteristic appearance in the STM image (Fig. 1e), which becomes even more evident using a multicolor scale (Fig. 1f) that also helps to separate the topography of the molecule and the surrounding standing wave of the copper surface state. It will be used from this point on to highlight changes to the appearance of the molecule.
] direction of the substrate. Single molecules show a characteristic appearance in the STM image (Fig. 1e), which becomes even more evident using a multicolor scale (Fig. 1f) that also helps to separate the topography of the molecule and the surrounding standing wave of the copper surface state. It will be used from this point on to highlight changes to the appearance of the molecule.
The characteristic presence of two bright lobes on the molecule can be used in combination with calculated images (Fig. 1g and h) to determine the orientation of the macrocycle as well as the binding site for the single hydrogen atom in the cavity. We find that one bright lobe corresponds to the pyrrole to which the hydrogen is bound, analogous to porphyrins,22,39 while the other lobe corresponds to the furan ring at the oxygen substitution. The sides between these two lobes correspond to the two remaining pyrroles and the four bridges (see superimposed structure in Fig. 1f). As one of the sides has a single rounded corner at equal distances to both bright lobes (the right side in Fig. 1f), this side can be interpreted as the pyrrole linked by the two methine-bridges (which have equal length). On the other side, there are two visible rounded corners at irregular distances. This contrast is assigned to the remaining pyrrole and the ethylene bridges of the molecule, which also fits to the results of the tautomerization experiments (see below). As the hydrogen is bound to the nitrogen position on the site opposite to the furan ring, it will be referred to as a trans state in this work. A detailed comparison of the experimental image in Fig. 1d with the simulated image of the trans tautomer (Fig. 1g and h) shows that the molecular orientation along a [11] direction and its relative position with respect to the underlying lattice is well-reproduced in the DFT calculations, thus confirming our assignment of Fig. 1d to the trans state.
According to this assignment of the trans configuration, the molecular appearance should change after tautomerization in the following way: the feature assigned to the furan ring should remain bright, the pyrrole ring to which the hydrogen moves should become brighter, and the pyrrole ring from which the hydrogen atom moved away should become darker. As shown in Fig. 2a, voltage pulses were first applied over the lobe identified with the initial binding position, because such a position had resulted in the highest switching rates for porphycene on Cu(111).40 Applying moderately increased bias voltages (e.g. −1.4 V) at the indicated position results in a bistability of the tunneling current (see Fig. 3a below) during the pulse and a noticeably changed molecule appearance (shown in Fig. 2b) if the pulse ends while the molecule is in the lower conductance state (the higher conductance state corresponds to the initial trans configuration). The appearance of the switched state exactly matches expectations: the lower lobe – which was interpreted as the pyrrole binding the hydrogen in the ground state – has become lower in apparent height, a lobe on the left side of the molecule has become higher in apparent height, and the lobe at the top – which was interpreted as the furan – has remained bright. Therefore, this switched state of O-HPc is assigned to the tautomer shown in Fig. 2f which will be referred to as the cis state in this work. The ground state of the molecule is identified as the trans state shown in Fig. 2e. It should be noted that in theory there are two distinct cis states with the hydrogen bound to position 21 and 23 respectively. However, only the one shown with the binding to position 23 was observed in our experiments.
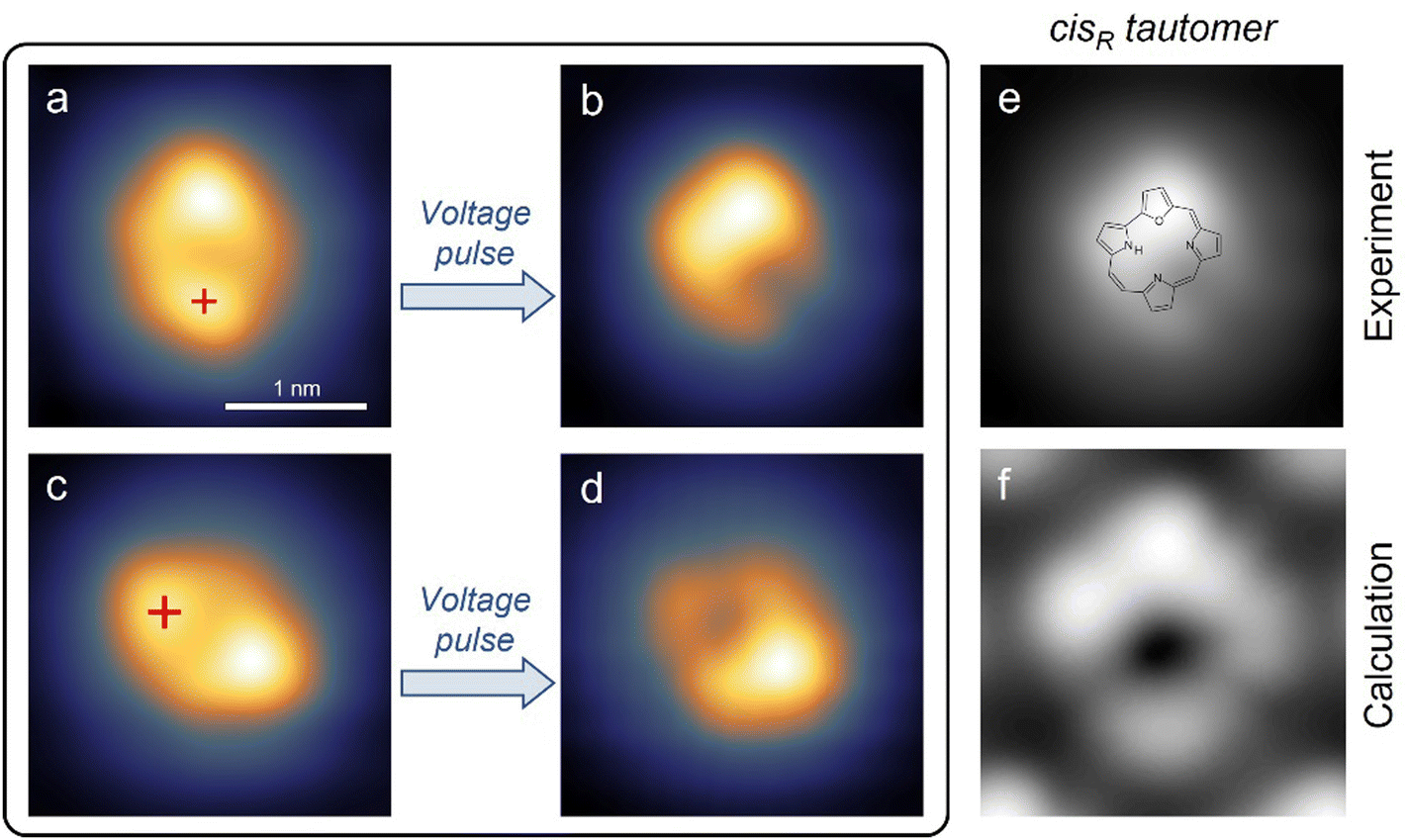 | ||
| Fig. 2 STM images (all 2.6 × 2.6 nm2) showing: (a) a single O-HPc molecule in the trans state with the tip position during voltage pulses marked with a red cross (200 mV, 100 pA). (b) The same molecule after a voltage pulse that ended in the low-conductance state, showing the appearance of the cis state (200 mV, 100 pA). (c) Single O-HPc molecule before the application of a voltage pulse with 2.0 V at the position indicated by the red cross (100 mV, 100 pA); (d) the deprotonated molecule after the 2.0 V pulse (100 mV, 100 pA). (e and f) Comparison of experimental and calculated STM images (both in the same orientation) of a single O-HPc molecule in the cisR state on the Cu(111) surface. | ||
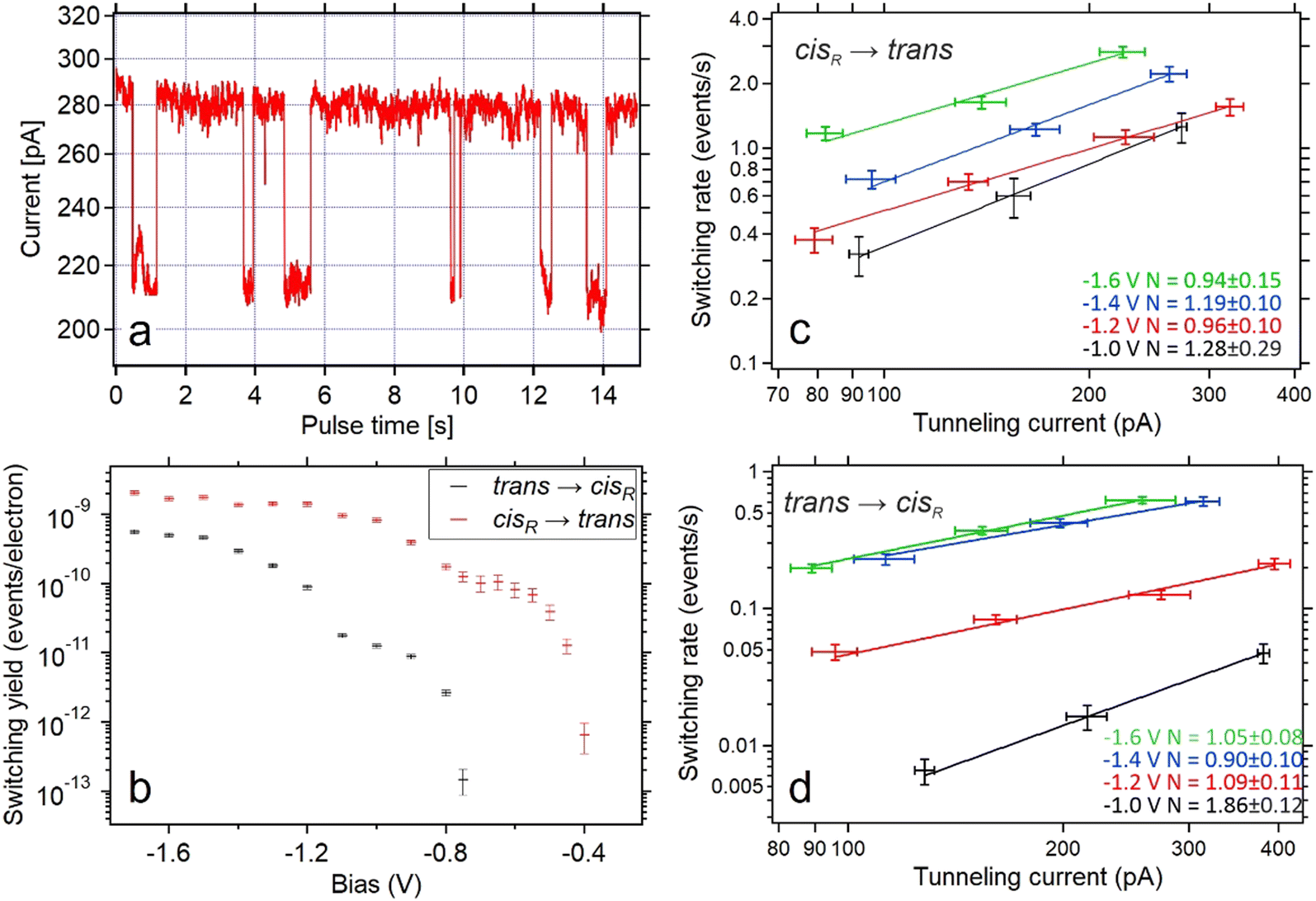 | ||
| Fig. 3 (a) Tunneling current as a function of time during application of increased bias voltage at the position indicated in Fig. 2a. The high-conductance state indicates that the molecule is in the trans state, while the low-conductance represents the cis tautomer. (b) The per-electron yield of both switching processes as a function of the bias voltages during the pulse. Typical currents during the switching events were ∼300 pA for trans-to-cis switching events and ∼220 pA for cis-to-trans switching events. Yields for bias voltages of at least −800 mV are based on 100 to 500 switching events. Yields at lower bias voltages are based on 5 to 20 switching events. (c and d) Switching rates of the cis-to-trans (c) and trans-to-cis (d) processes as a function of tunneling currents measured for different bias voltages (color coded with black (−1.0 V), red (−1.2 V), blue (−1.4 V) and green (−1.6 V)). Data points with the same bias voltage are fitted with a power law with a variable exponent N. | ||
In order to precisely determine which of the two cis tautomers is present, we have done DFT calculations of the geometric structures and the relative stabilities of the three tautomers of O-HPc both in the gas-phase and adsorbed on the Cu(111) surface. The interpretation of the topographical STM images of the adsorbed tautomers are corroborated by our simulated images. The optimized structures of these tautomers (shown in Fig. S8, ESI†) show that they are all planar in the gas phase. Henceforth, the two inequivalent cis tautomers are denoted by cisL and cisR, where the subscripts L(eft) and R(ight) reflect the positions of the H atom relative to the O atom (see Fig. S8a, ESI†). As shown in Table 1, the trans tautomer is the most stable one and cisR is more stable than cisL. The relatively large stability of the trans tautomer by 0.15 eV shows that the deposited O-HPc molecules should predominantly be trans tautomers, which confirms the experimental observations where all molecules are found in the trans state after deposition.
| Tautomer | ΔE (eV) | N–NL (Å) | N–NR (Å) | NL–NR (Å) |
|---|---|---|---|---|
| trans | 0.00 | 3.11 | 2.92 | 3.99 |
| cis R | 0.15 | 3.18 | 2.85 | 3.98 |
| cis L | 0.28 | 2.99 | 3.08 | 3.98 |
As shown in Table 1, the interatomic distances between the N atoms change substantially with the tautomer. A surprising finding is the result that the N–NL (N24–N21, see Fig. 1) distances are longer than the N–NR (N24–N23) interatomic distances for trans and cisR, despite the methine bridge between the pyrrole groups of the N and NL atoms containing only one atom while the ethylene bridge between the pyrrole groups of N and NR atoms contains two atoms (see Fig. S8a, ESI†). These interatomic distances are both within the range of hydrogen bonding (2.7–3.3 Å), whereas the NL–NR distance, being essentially the same for all three tautomers, is about 1 Å larger than the other N–N distances and is outside the range of hydrogen bonding.
The trend of the energetics over the tautomers in the gas phase can then be rationalized in terms of the differences in the hydrogen bonding in the cavity based on the N–N distances. The trans is now favoured over cisL and cisR, since hydrogen bonds can be formed with two N acceptor atoms for trans in contrast to the cis tautomers, where the H bond is formed by a single N acceptor atom. The cisR is now favoured over cisL by the N–NR interatomic distance being shorter than the N–NL interatomic distance. The relatively small energy differences between these tautomers reflects the energy scale of hydrogen bonding. Furthermore, the behaviour of the N–N interatomic distances suggests that the energy barrier for proton transfer between trans and cisR is lower than the corresponding barrier between trans and cisL and that the proton transfer between cisR and cisL is much less favourable than for the transfer between the trans and the cis tautomers. This suggestion is corroborated by our calculated energy barriers in Table 3.
The calculated geometric structure of the adsorbed tautomers are detailed in Fig. S10 (ESI†), whereas their energetics are summarized in Table 2. The relative ΔEads values follow the same trend as the relative energies of the gas phase tautomers in Table 1. However, the energy differences between the adsorbed tautomers are substantially reduced, but these differences are sufficiently large for trans being the thermodynamically stable tautomer on the surface.
| Tautomer | E ads (eV) | ΔEads (eV) |
|---|---|---|
| trans | −3.69 (−3.77) | 0.00 (0.00) |
| cis R | −3.65 (−3.73) | 0.04 (0.04) |
| cis L | −3.62 (−3.70) | 0.07 (0.07) |
A somewhat surprising finding is that the N–NR distances for the adsorbed tautomers are now longer for trans and cisR than for the N–NL distances of trans and cisL (Table S2, ESI†), in contrast to the tautomers in the gas phase (Table 1). This result would suggest, based on intramolecular H bonding, that cisL should be more stable than cisR and cannot rationalize that cisR is also more stable than cisL for the adsorbed tautomer. However, as shown for hemiporphycene, the distance is not the only parameter that controls the H bond strength as also the NHN angle must be considered.24 Additionally, in the case of the adsorbed tautomers there is also a competing contribution from the molecule–surface bonding by, for instance, the lone-pair bonding of the imine N atoms to the Cu surface (see Section 8 in the ESI†).
The reverse result for the N–NR and N–NL interatomic distances of the adsorbed tautomers compared to the ones in the gas phase might suggest that, in contrary to the gas phase, the energy barrier for the trans → cisR tautomerization should now be higher than the corresponding barrier for the trans → cisL tautomerization. However, the energy barriers E* in Table 3, as obtained from the calculated minimum energy paths (Section 11 in the ESI†), show that the trans → cisR tautomerization has the lowest energy barrier – both in the gas phase and on the surface. Thus, the N–N distance is not a good descriptor of the energy barriers for H transfer. In fact, the calculated straight path lengths ΔDH for the H transfers for the trans → cisR tautomerization is substantially shorter than for the trans → cisL tautomerization both in the gas phase and on the surface as shown in Table 3. This result suggests that ΔDH is a suitable descriptor for the potential energy barrier of the tautomerization. These path lengths are very similar for these tautomerizations in the gas phase and on the surface.
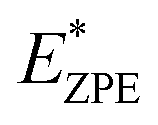 . The straight path length of the H atom between the two tautomers is given by ΔDH, whereas the value within the parenthesis represents the MEP length for the H atom. ΔDMol is the MEP path length calculated by including all the molecular atoms but not the substrate atoms
. The straight path length of the H atom between the two tautomers is given by ΔDH, whereas the value within the parenthesis represents the MEP length for the H atom. ΔDMol is the MEP path length calculated by including all the molecular atoms but not the substrate atoms
| Final tautomer | E* (eV) |
|
ΔDH (Å) | ΔDMol (Å) | |
|---|---|---|---|---|---|
| Gas phase | cis L | 0.87 | 1.7 (2.1) | 3.3 | |
| cis R | 0.36 | 1.0 (1.2) | 2.1 | ||
| Adsorbed | cis L | 0.86 | 0.71 | 1.6 (1.9) | 3.9 |
| cis R | 0.63 | 0.49 | 1.1 (1.5) | 4.1 |
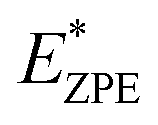
The values for the MEPs path lengths of the H atom (Table 3) show that these paths have some curvature. The path lengths for the MEPs that include all atoms of the molecule show that the tautomerizations involve distortions of the molecule. These two effects are most pronounced for the trans → cisR tautomerization on the surface and reflect the substantial increase of the energy barrier for this tautomerization on the surface compared to the gas phase. Finally, note that energy barriers for the reverse direction of the tautomerizations, cisR → trans and cisL → trans, will be reduced by the relative adsorption energies ΔEads of cisRL and cisL, respectively. This reduction is only 0.04 and 0.07 eV (Table 2) for the cisR → trans and the cisL → trans tautomerizations, respectively.
The calculated N–H stretch vibrational energy is about 400 meV (see Section 12 in the ESI†) and the loss of this mode at the transition structure suggests that the zero-point-energy (ZPE) corrections can have an important effect on the energy barrier. Consequently, the energy barriers are reduced by about 0.14 eV. The resulting ZPE corrected energy barriers 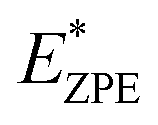 are shown in Table 3 (for details see Section 12 in the ESI†).
are shown in Table 3 (for details see Section 12 in the ESI†).
Our simulated topographical STM images (Fig. S10, ESI†) reveal that the molecular appearance of all three tautomers is dominated by two protrusions: One is caused by the furan ring, containing the oxygen atom, and the other one by the pyrrole ring that contains the amine N atom (NH), which is slightly brighter than the pyrrole groups with imine N atoms. These simulated images support the assignment of the experimental images, both for the trans tautomer in Fig. 1e–h and for the cis tautomer in Fig. 2e and f. In particular, the assignment of the cis tautomer (being either cisL or cisR, see Fig. S8b and c, ESI†) becomes clear from a comparison of our experimental STM data (in Fig. 2) with the calculated images (in Fig. S10, ESI†). We can therefore conclude that our switching experiments (Fig. 2a and b) lead to tautomerization from the trans to the cisR (and not the cisL) state. This agrees with the calculated energy barriers where the energy barrier for H transfer between trans and cisR is lower than the corresponding barrier for trans and cisL (Table 3). Accordingly, the trans molecule always chooses the lower barrier to cisR upon tautomerization.
By increasing the bias voltage beyond the threshold for tautomerization, it is also possible to dehydrogenate the adsorbed O-HPc molecule. At bias voltages above 2.0 V, a different behavior than from tautomerization appears: after a short time (seconds or less) of rapid switching between two conductances (trans ↔ cisR), a more pronounced drop in tunneling current and no further bistability is observed (Fig. S4, ESI†). This results in a topography different from the regular switching state, as shown in Fig. 2d. This third state is assigned to the deprotonated O-HPc molecule, as previous experiments on tautomerization switches showed that the bond of hydrogen atom(s) in the cavity is the first to be broken at increased bias voltages.39 The necessary bias voltages were 2.0 V for tetraphenylporphyrin,39,41 which precisely matches the threshold voltage of 2.0 V observed here, and 3.0 V respective 2.5 V for phthalocyanine,42 which is somewhat higher but overall comparable. The observation of similar dehydrogenation for O-HPc here confirms the initial assignments of the molecular appearance and supports the interpretation of the switching mechanism (Fig. 2).
Directly comparing the topography of the two states, as shown in Fig. 2e and f, confirms that for the chosen pulse position (see Fig. 2a) a high conductance identifies the trans state and a low conductance corresponds to the cisR state. This allows for easy collection of switching times for the trans and cisR states from the same I(t) traces (although at different currents) that show a characteristic bistability (Fig. 3a). To analyze large amounts of switching data, we employed the algorithm of Yuzhelevski et al.43 to fit the conductance data to a two-level model. The switching rates R(Ubias,I) for a given bias voltage Ubias and tunneling current in the trans-state I were evaluated according to
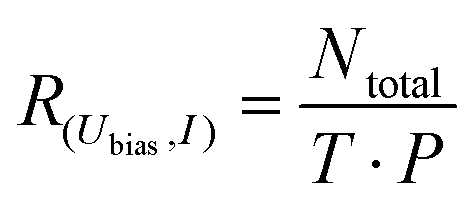
By testing various bias voltages during the pulses, we find that the threshold voltage for the trans → cisR tautomerization is lower than for the cisR → trans process. By using either the population of the trans state (PT) or the population of the cisR state (PC), it is possible to determine both the trans → cisR and the cisR → trans switching rates from the evaluation of the same voltage pulses, as long as the bias voltage is sufficiently large to activate both processes. This was found to be the case for voltages down to −750 mV. For smaller bias voltages (i.e. −750 mV < U < 0 V), no switching from the trans to the cisR state was observed. To collect data on the cisR → trans switching process for these bias voltage values, the molecule was first brought to the cisR state with a voltage pulse of −1.2 V, and then switched back into the trans state with the desired lower value. The switching rates were normalized by the tunneling current during the pulse to produce the switching yields shown in Fig. 3b.
It is clearly visible that both directions of tautomerization follow the same basic behavior: There is an initial threshold for the activation through tunneling electrons at −0.75 V for the trans → cisR and −0.4 V for the cisR → trans tautomerization. For lower bias voltages (i.e. larger moduli) the switching yields first reach a stable plateau before they increase by another order of magnitude after passing a second threshold at −1.2 V for the trans → cisR and −0.8 V for the cisR → trans direction. About the same threshold values are found for the opposite polarity (positive instead of negative voltages applied to the sample, STM tip always grounded).
A detailed study of the switching rates as a function of the tunneling current reveals a mostly linear dependence for both the cisR → trans and the trans → cisR switching processes. By fitting the cisR → trans switching data with rate ∝ currentN (with a variable exponent N) leads to a value of N ∼ 1 for various bias voltages (Fig. 3c and d), indicating a one-electron process as the origin for molecular excitation.44
Only for one case (trans → cisR) a non-linear dependence (N ≠ 1) is observed within the experimental accuracy. This is the case for the smallest applied bias voltage (−1.0 V), which is the only value that lies under the observed threshold (−1.2 V for this switching direction). This suggests that the tautomerization is activated by single inelastic tunneling electrons for all bias voltages larger than the threshold voltage (−1.2 V for trans → cisR and −0.8 V for cisR → trans) and multi-electron activated for smaller bias voltages,45 similar to previous works on porphycene tautomerization.22
These observations rule out resonant tunneling into (or out of) molecular electronic states, which caused switching of a bistable molecule on a Si(100) surface,46 as the origin for tautomerization since this would not result in symmetric behavior with the bias voltage polarity. Moreover, vibrational excitation, which was the basis for proton transfer in porphycene,17 is most likely not involved in the present case as the observed threshold voltages for one-electron processes are far above typical vibrational energies (e.g. 0.38 eV for the N–H stretch vibration in porphycene17). We therefore propose that the O-HPc tautomerization on Cu(111) here is induced by electronic excitation of the molecule. This also explains why the threshold voltage for the cisR → trans process is much smaller than for trans → cisR, despite the very similar total energies that differ only by 0.04 eV (Table 2), because the potential energy surface for tautomerization can be very different in the excited state.
Previous experiments on the tautomerization of porphyrins on surfaces39,40 showed that the location of the pulses influenced the switching rates. For this reason, pulses (at a bias voltage of −1.2 V) were applied at different positions over the trans molecule, as shown in Fig. 4a. The evaluation of the data proved difficult, because a clean two-level behavior (as shown in Fig. 3a) in the current/time traces is evident only for some tip positions. In many cases, the two conductance levels were either very close in the current signal and/or the current signal showed a strongly increased noise level. This noise could not be attributed to the tip or the detection system, because it was reliably found for certain tip positions, but not for others. Although a clear assignment was not possible, we believe that it is related to molecular motion around its adsorption position and orientation, but does not reflect proton transfer. These two factors rendered a clean timing of switching events very difficult. In combination with the large number of acquired data points (ultimately at least 4500 switching events) a simpler evaluation was therefore chosen. For each pixel the number of identifiable switching events was counted and divided by the sum of the durations of all pulses in this pixel. This rate was then normalized by the baseline tunneling current during the pulse. Fig. 4b shows the resulting map for this normalized rate, including both the trans → cisR and cisR → trans yields. It can be seen that the highest switching rate is centered on the pyrrole unit that binds the hydrogen atom in the trans configuration. Another peak in the switching rate is found approximately above the NR atom of the cavity (see Fig. S8a, ESI†). In general, our results show maxima near the two locations of the hydrogen in the two switching states, i.e. starting and end position of the proton during tautomerization, with the more pronounced maximum at the initial position. Such a preference for the initial location is in line with previous results of porphycene tautomerization.40 The relatively low switching rate near the pyrrole ring, where the proton would bind in the cisL tautomer, agrees with our observation that the hydrogen atom did not bind to this group.
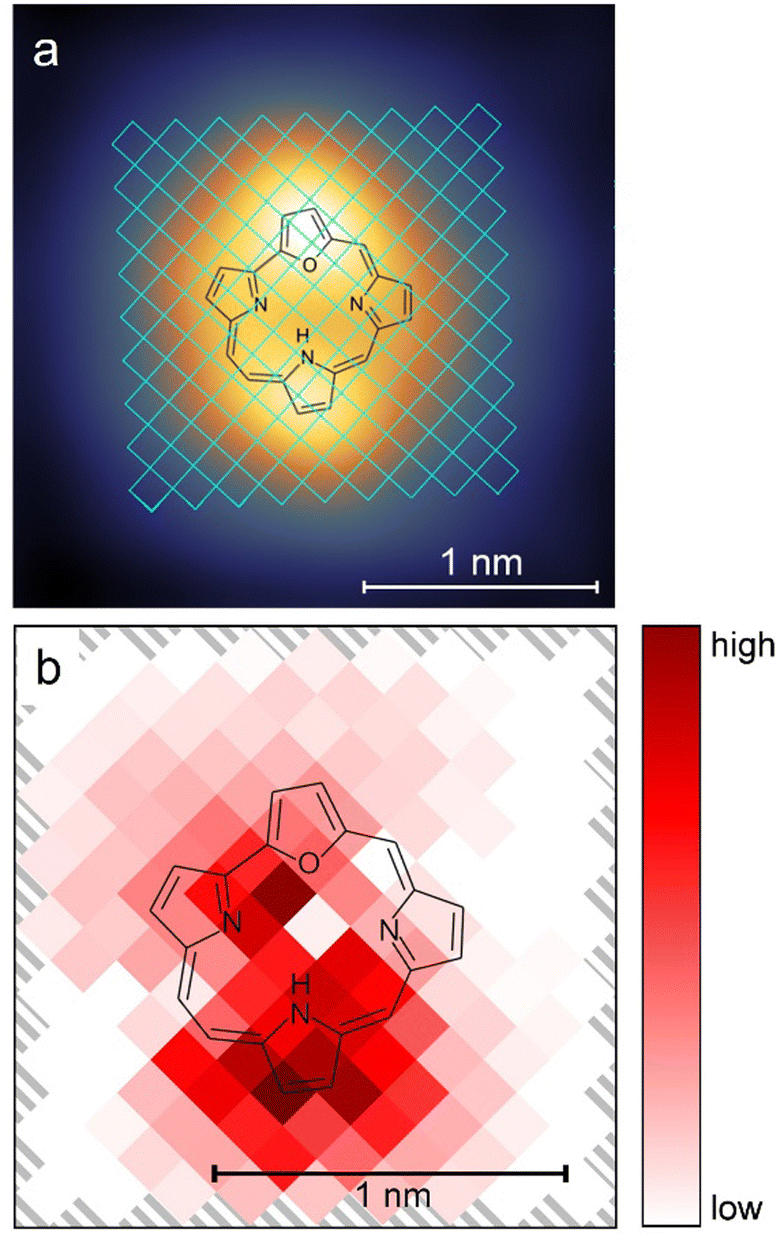 | ||
| Fig. 4 (a) The grid used for tip position dependent switching events is marked in blue over the STM image from Fig. 2a. Voltage pulses at −1.2 V with a duration of 28.83 s each were applied at the different positions of a 145 pixel grid over the trans molecule as shown. The pulses were applied at the center of the pixels with a size of 1.31 × 1.31 Å2. (b) Color-coded map of the switching activity dependent on the position of the tip over the molecule. | ||
In conclusion, two stable tautomers of O-HPc were characterized on the Cu(111) surface with the most stable being the trans state, both in the gas phase as well as upon adsorption. The calculations show that all adsorbed tautomers manifested bending away from the Cu(111) surface, which together with the adsorption energetics indicates chemisorption. By applying voltage pulses from the STM tip, single molecules can repetitively be switched between the trans and cis tautomers, the latter existing in the gas phase in two atomic arrangements (cisL and cisR). A detailed comparison of experimental and simulated STM images shows that the trans molecules are always switched into the cisR state, which is explained by the lower energy barrier for trans → cisR than trans → cisL, as revealed by calculations. Following the dependence on the experimental conditions, the O-HPc tautomerization on Cu(111) is likely induced by electronic excitation of the molecule. This occurs in one-electron processes for various bias voltages, while multi-electron processes seem to be involved if the energy of the tunneling electrons is below the characteristic threshold. The position dependence of the switching mechanism exhibits the highest rates if the STM tip is placed in vicinity to the two possible binding positions of the proton in the cavity and the rate is lowest at the position where no binding of the cavity proton occurs. Furthermore, and similar to other tautomerizing molecules, O-HPc can be deprotonated by applying a pulse with an elevated bias voltage of 2.0 V.
Author contributions
S. J., J. W. and L. G. conceived the experiments. S. J. did the measurements, supported by M. S. J. O. synthesized the molecules. E. D. and M. P. did the calculations. S. J., E. D., M. P. and L. G. analysed the data. L. G. wrote the manuscript with feedback from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Marie Curie Initial Training Network (ITN) through the ACRITAS project, the Polish National Science Centre (grant no. 2017/26/M/ST4/00872) as well as computer resources provided by Barkla at the University of Liverpool are gratefully acknowledged.References
- B. L. Feringa and W. R. Browne, Molecular switches, Wiley-VCH Weinheim, 2011 Search PubMed.
- Q. Zhang, D.-H. Qu, H. Tian and B. L. Feringa, Bottom-up: can supramolecular tools deliver responsiveness from molecular motors to macroscopic materials?, Matter, 2020, 3, 355–370 CrossRef.
- N. Koumura, R. W. J. Zijistra, R. A. van Delden, N. Harada and B. L. Feringa, Light-driven monodirectional molecular rotor, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- J. Wang and B. L. Feringa, Dynamic control of chiral space in a catalytic asymmetric reaction using a molecular motor, Science, 2011, 331, 1429–1432 CrossRef CAS PubMed.
- S. J. van der Molen and P. Liljeroth, Charge transport through molecular switches, J. Phys.: Condens. Matter, 2010, 22, 133001 CrossRef PubMed.
- Y. Liu, L. Mu, B. Liu and J. Kong, Controlled Switchable Surface, Chem. – Eur. J., 2005, 11, 2622–2631 CrossRef CAS PubMed.
- M. Alemani, M. V. Peters, S. Hecht, K.-H. Rieder, F. Moresco and L. Grill, Electric field-induced isomerization of azobenzene by STM, J. Am. Chem. Soc., 2006, 128, 14446–14447 CrossRef CAS PubMed.
- J. Henzl, M. Mehlhorn, H. Gawronski, K.-H. Rieder and K. Morgenstern, Reversible cis–trans isomerization of a single azobenzene molecule, Angew. Chem., Int. Ed., 2006, 45, 603–606 CrossRef CAS PubMed.
- B.-Y. Choi, S.-J. Kahng, S. Kim, H. Kim, H. W. Kim, Y. J. Song, J. Ihm and Y. Kuk, Conformational molecular switch of the azobenzene molecule: A scanning tunneling microscopy study, Phys. Rev. Lett., 2006, 96, 156106 CrossRef PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Controlling intramolecular hydrogen transfer by single atoms and molecules, Nat. Chem., 2014, 6, 41 CrossRef CAS PubMed.
- C. Dri, M. V. Peters, J. Schwarz, S. Hecht and L. Grill, Spatial periodicity in molecular switching, Nat. Nanotech., 2008, 3, 649–653 CrossRef CAS PubMed.
- G. J. Narlikar and D. Herschlag, Mechanistic aspects of enzymatic catalysis: lessons from comparison of RNA and protein enzymes, Ann. Rev. Biochem., 1997, 66, 19–59 CrossRef CAS PubMed.
- B. Marchetti, T. N. V. Karsili, M. N. R. Ashfold and W. Domcke, A ‘bottom up’, ab initio computational approach to understanding fundamental photophysical processes in nitrogen containing heterocycles, DNA bases and base pairs, Phys. Chem. Chem. Phys., 2016, 18, 20007–20027 RSC.
- J. Waluk, Spectroscopy and tautomerization studies of porphycenes, Chem. Rev., 2017, 117, 2447–2480 CrossRef CAS PubMed.
- L. E. Webb and E. B. Fleischer, Crystal structure of porphine, J. Chem. Phys., 1965, 43, 3100 CrossRef CAS.
- W. Auwärter, K. Seufert, F. Bischoff, D. Ecija, S. Vijayaraghavan, S. Joshi, F. Klappenberger, N. Samudrala and J. V. Barth, A surface-anchored molecular four-level conductance switch based on single proton transfer, Nat. Nanotech., 2012, 7, 41 CrossRef PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Thermally- and vibrationally induced tautomerization of single porphycene molecules on a Cu(110) surface, Phys. Rev. Lett., 2013, 111, 246101 CrossRef PubMed.
- J. N. Ladenthin, L. Grill, S. Gawinkowski, S. Liu, J. Waluk and T. Kumagai, Hot carrier-induced tautomerization within a single porphycene molecule on Cu(111), ACS Nano, 2015, 9, 7287–7295 CrossRef CAS PubMed.
- H. Böckmann, S. Liu, J. Mielke, S. Gawinkowski, J. Waluk, L. Grill, M. Wolf and T. Kumagai, Direct observation of photoinduced tautomerization in single molecules at a metal surface, Nano Lett., 2016, 16, 1034–1041 CrossRef PubMed.
- J. N. Ladenthin, T. Frederiksen, M. Persson, J. C. Sharp, S. Gawinkowski, J. Waluk and T. Kumagai, Force-induced tautomerization in a single molecule, Nat. Chem., 2016, 8, 935–940 CrossRef CAS PubMed.
- T. Kumagai, J. N. Ladenthin, Y. Litman, M. Rossi, L. Grill, S. Gawinkowski, J. Waluk and M. Persson, Quantum tunneling in real space: Tautomerization of single porphycene molecules on the (111) surface of Cu, Ag, and Au, J. Chem. Phys., 2018, 148, 102330 CrossRef PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Thermally and Vibrationally Induced Tautomerization of Single Porphycene Molecules on a Cu(110) Surface, Phys. Rev. Lett., 2013, 111, 246101 CrossRef PubMed.
- J. Kügel, M. Leisegang and M. Bode, Imprinting Directionality into Proton Transfer Reactions of an Achiral Molecule, ACS Nano, 2018, 12, 8733–8738 CrossRef PubMed.
- J. Ostapko, K. Nawara, M. Kijak, J. Buczynska, B. Lesniewska, M. Pietrzak, G. Orzanowska and J. Waluk, Parent, unsubstituted hemiporphycene: synthesis and properties, Chem. – Eur. J., 2016, 22, 17311–17320 CrossRef CAS PubMed.
- A. Kumar, P. Yadav, M. Majdoub, I. Saltsman, N. Fridman, S. Kumar, A. Kumar, A. Mahammed and Z. Gross, Corroles: The Hitherto Elusive Parent Macrocycle and its Metal Complexes, Angew. Chem., Int. Ed., 2021, 60, 25097–25103 CrossRef CAS PubMed.
- S. Jaekel, Azobenzene and Porphin Derivatives as Molecular Switches on Metallic and Insulating Surfaces, Dr. rer. nat. thesis, Institute for Chemistry, University of Graz, 2019 Search PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, van der Waals density functional for general geometries, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- G. Román-Pérez and J. M. Soler, Efficient implementation of a van der Waals density functional: application to double-wall carbon nanotubes, Phys. Rev. Lett., 2009, 103, 096102 CrossRef PubMed.
- K. Berland and P. Hyldgaard, Exchange functional that tests the robustness of the plasmon description of the van der Waals density functional, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 035412 CrossRef.
- G. Mills, H. Jónsson and G. K. Schenter, Reversible work transition state theory: application to dissociative adsorption of hydrogen, Surf. Sci., 1995, 324, 305–337 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- D. Sheppard, R. Terrell and G. Henkelman, Optimization methods for finding minimum energy paths, J. Chem. Phys., 2007, 128, 134106 CrossRef PubMed.
- S. Smidstrup, A. Pedersen, K. Stokbro and H. Jónsson, Improved initial guess for minimum energy path calculations, J. Chem. Phys., 2014, 140, 214106 CrossRef PubMed.
- J. Tersoff and D. R. Hamann, Theory and Application for the Scanning Tunneling Microscope, Phys. Rev. Lett., 1983, 50, 1998–2001 CrossRef CAS.
- N. Lorente and M. Persson, Theoretical aspects of tunneling-current-induced bond excitation and breaking at surfaces, Faraday Discuss., 2000, 117, 277–290 RSC.
- E. Durant, PhD Thesis, University of Liverpool, UK, 2021.
- W. Auwärter, K. Seufert, F. Bischoff, D. Ecija, S. Vijayaraghavan, S. Joshi, F. Klappenberger, N. Samudrala and J. V. Barth, A surface-anchored molecular four-level condcutance switch based on single proton transfer, Nat. Nanotechnol., 2011, 7, 41–46 CrossRef PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Controlling intramolecular hydrogen transfer in a porphycene molecule with single atoms or molecules located nearby, Nat. Chem., 2014, 6, 41–46 CrossRef CAS PubMed.
- L. Smykalla, P. Shukrynau, C. Mende, T. Rüffer, H. Lang and M. Hietschold, Manipulation of the electronic structure by reversible dehydrogenation of tetra(p-hydroxyphenyl)porphyrin molecules, Surf. Sci., 2014, 628, 92–97 CrossRef CAS.
- A. Sperl, J. Kröger and R. Berndt, Controlled Metalation of a Single Adsorbed Phthalocyanine, Angew. Chem., Int. Ed., 2011, 50, 5294–5297 CrossRef CAS PubMed.
- Y. Yuzhelevski, M. Yuzhelevski and G. Jung, Random telegraph noise analysis in time domain, Rev. Sci. Instr., 2000, 71, 1681–1688 CrossRef CAS.
- B. C. Stipe, M. A. Rezaei, W. Ho, S. Gao, M. Persson and B. I. Lundqvist, Single-molecule dissociation by tunnelling electrons, Phys. Rev. Lett., 1997, 78, 4410 CrossRef CAS.
- W. Ho, Single-molecule chemistry, J. Chem. Phys., 2002, 117, 11033 CrossRef CAS.
- M. Lastapis, M. Martin, D. Riedel, L. Hellner, G. Comtet and G. Dujardin, Picometer-scale electronic control of molecular dynamics inside a single molecule, Science, 2005, 308, 1000 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp04746b |
| ‡ Current address: Chair for Physical Chemistry II, Friedrich-Alexander-University Erlangen-Nürnberg, Egerlandstraße 3, Erlangen, Germany. |
| This journal is © the Owner Societies 2023 |