
A nonconjugated radical polymer with stable red luminescence in the solid state†
Zhaoyu
Wang‡
 a,
Xinhui
Zou‡
a,
Yi
Xie‡
b,
Haoke
Zhang
a,
Lianrui
Hu
a,
Christopher C. S.
Chan
a,
Ruoyao
Zhang
a,
Jing
Guo
c,
Ryan T. K.
Kwok
a,
Jacky W. Y.
Lam
a,
Ian D.
Williams
a,
Zebing
Zeng
c,
Kam Sing
Wong
a,
C. David
Sherrill
b,
Ruquan
Ye
*d and
Ben Zhong
Tang
*e
a,
Xinhui
Zou‡
a,
Yi
Xie‡
b,
Haoke
Zhang
a,
Lianrui
Hu
a,
Christopher C. S.
Chan
a,
Ruoyao
Zhang
a,
Jing
Guo
c,
Ryan T. K.
Kwok
a,
Jacky W. Y.
Lam
a,
Ian D.
Williams
a,
Zebing
Zeng
c,
Kam Sing
Wong
a,
C. David
Sherrill
b,
Ruquan
Ye
*d and
Ben Zhong
Tang
*e
aDepartment of Chemistry, Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, Institute for Advanced Study, Guangdong-Hong Kong-Macao Joint Laboratory of Optoelectronic and Magnetic Functional Materials, Department of Chemical and Biological Engineering, and Department of Physics, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
bCenter for Computational Molecular Science and Technology, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, Georgia 30332-0400, USA
cState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China
dDepartment of Chemistry, State Key Laboratory of Marine Pollution, City University of Hong Kong, Hong Kong, China. E-mail: ruquanye@cityu.edu.hk
eSchool of Science and Engineering, Shenzhen Institute of Aggregate Science and Technology, The Chinese University of Hong Kong, Shenzhen, Guangdong, 518172, China. E-mail: tangbenz@cuhk.edu.cn
First published on 12th July 2022
Abstract
Organic radicals are unstable and stable radicals usually display non-luminescent properties. Luminescent radicals possess the all-in-one properties of optoelectronics, electronics, and magnetics. To date, the reported structures of luminescent radicals are limited to triphenylmethyl radical derivatives and their analogues, which are stabilized with extended π-conjugation. Here, we demonstrate the first example of a nonconjugated luminescent radical. In spite of the lack of delocalized π-stabilization, the radical polymer readily emits red luminescence in the solid state. A traditional luminescent quencher, 2,2,6,6-tetramethylpiperidin-1-yl turned into a red chromophore when grafted onto a polymer backbone. Experimental data confirm that the emission is associated with the nitroxide radicals and is also affected by the packing of the polymer. This work discloses a novel class of luminescent radicals and a distinctive pathway for luminescence from open-shell materials.
New conceptsIt has been often observed that stable organic radicals with open-shell structures are non-luminescent, due to the favourable non-radiative decay after excitation. These radicals can function as luminescence quenchers even when they are attached to chromophores. Hence, the phenomenon of luminescence in radicals is rare and unusual. As yet, all reported luminescent radicals are categorized as π-radicals, featured with the presence of aromatic rings and extended conjugation. In this work, the concept of nonconjugated and nonaromatic luminescent radicals was demonstrated for the first time. The structure is distinctive from those of the common triphenylmethyl radical derivatives. Moreover, the emission mechanism is also different from that of any previous luminescent radical, most of which show the characteristic of aggregation-caused quenching (ACQ). Our work shows that the feature of aggregation-induced emission (AIE) and the luminescence comes from aggregation and molecular interactions, instead of single molecules. In a nutshell, the work will bring instructive insights into the structural design of luminescent radicals and inspire future research in mechanisms and applications. |
Introduction
Most of the organic radicals with open-shell structures are highly reactive and diligent efforts have thus been made to stabilize the unpaired electron by delicate structure design.1,2 Stable radicals have been widely applied in many fields, such as electrochemical catalysis,3 batteries,4 and organic synthetic catalysis.5 However, due to the enhanced intersystem crossing (ISC) effect of radicals, they typically go through a non-radiative relaxation pathway upon excitation, thereby showing non-luminescence properties.6 Even though a highly luminescent chromophore was covalently attached to a stable radical, the luminescence from the chromophore would be easily quenched, since radicals usually function as a luminescence quencher, rather than as an emitter.7,8 It was not until 2006 that a highly luminescent radical was first reported,9 which garnered wide attention and aroused great interest among researchers. Intriguingly, the unpaired electron in radicals generates a singly occupied molecular orbital (SOMO) in the ground state, resulting in the overall molecule spin as a doublet.10 The fascinating open-shell structure enables luminescent radicals to possess optoelectronic, electronic, and magnetic properties simultaneously.11 Stable radicals have been widely investigated from excited-state mechanisms12–16 to applications.17–21 In particular, taking advantage of the emission from a spin doublet, instead of a singlet, the luminescent radical readily breaks the efficiency limitations in organic light-emitting diodes (OLEDs), which shows the potential to raise the internal quantum efficiency to 100%.22 Li and coworkers demonstrated a series of highly efficient red and infrared LEDs through the use of luminescent radicals as emitters, among which the maximum external quantum efficiency reached 27% at a wavelength of 710 nm.17In spite of this, luminescence from stable radicals remains a sporadic phenomenon.11 To date, all reported luminescent radicals are π-radicals, which are featured with extended π systems and the presence of phenyl rings.6 Furthermore, the structures are mainly restricted to triphenylmethyl radical derivatives and their analogues (Fig. S1, ESI†).6,9,17 Some other structures with radicals attached to aromatic hydrocarbons might also emit light, but the examples are rare.23 The radicals illustrated in Fig. S1 (ESI†) are only emissive in an isolated state, such as a diluted solution, but their emission would be weakened once in the aggregated and solid states.24 Therefore, discovering new systems is highly desirable.
In nature, non-covalent interaction and self-assembly play a critical role in the photophysical properties.25,26 It was reported that a series of biomolecules that are nonconjugated and nonaromatic could also emit light at short wavelength upon irradiation, such as cellulose, starch, serum albumin, and amino acids and so on, which was accounted for by intermolecular interactions and through-space conjugation.27 The series of nonconjugated luminescent materials were termed as clusteroluminogens.28 Moreover, modern photophysics suggests that in addition to the intrinsic energy states of chromophores, their luminescence properties are affected or even could be reversed by the surrounding environment.29,30 For example, green fluorescent proteins are fluorescent when the chromophore, 4-(p-hydroxybenzylidene)imidazolidin-5-one, is buried at the centre of protein. If the protein is denatured, the fluorescence from the exposed chromophore would vanish promptly.31
Herein, we report the first example of a nonconjugated luminescent radical, poly(4-glycidyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl) (PGTEMPO), which was synthesized based on 2,2,6,6-tetramethylpiperidin-1-yl (TEMPO). First, TEMPO is a classical stable radical that has been widely applied in various fields, such as catalysis in polymerization32 and electrochemical materials.33 However, it was generally used as a luminescence quencher of chromophores34 by the effect of enhanced intersystem crossing.35 Serendipitously, we find that TEMPOs can be turned into emitters by a simple polymerization. Second, although the TEMPO-containing polymer is nonconjugated and possesses no aromatic rings, the narrow band gap between the highest occupied molecular orbital (HOMO) and SOMO of the nitroxide radical enables the polymer to emit at the long wavelength. In comparison with this polymer, conventional red-emissive chromophores have to depend on complicated π-system to achieve narrow band gap and long-wavelength emission.36 Third, experimental and theoretical data underscore the significance of intermolecular non-covalent interactions among the TEMPO units, which is completely different from previous working mechanisms of luminescent radicals. Fourth, living organisms are filled with aqueous medium, which impede the biological applications of previous luminescent radicals.24 In contrast, we achieved the luminescence of PGTEMPO in the aggregated and solid state and successfully demonstrated the biomedical application of PGTEMPO on intracellular detection of the antioxidant. Last but also important, unlike the recently reported radicals (Fig. S1, ESI†), this is the first time that the luminescent radicals without delocalized π-stabilization were achieved. Our results disclose an unusual luminescence phenomenon and advance the development of luminescent radicals.
Results and discussion
Synthesis, characterization, and photophysical properties
The nonconjugated radical polymer, poly(4-glycidyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl) (PGTEMPO), was synthesized via the ring-opening polymerization of stable radical monomers initiated by potassium tert-butoxide (Fig. 1a). The TEMPO derivative, 4-glycidyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl (GTEMPO), was used as the monomer as it is stable at a wide range of temperatures and crystallizes easily. PGTEMPO is orange and it has a number-average molecular weight of 5.4 kg mol−1 and a narrow molecular weight distribution (Đ = 1.63). It is readily soluble in common organic solvents, such as tetrahydrofuran (THF), chloroform, dichloromethane, and dimethylsulfoxide. Electron paramagnetic resonance (EPR) spectroscopy and attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy confirms the existence of stable radicals and the nitroxide functional group respectively (Fig. S4 and S5, ESI†). In comparison with the EPR intensity of GTEMPO, the intensity of PGTEMPO in THF solution with the same concentration shows little change, which indicates that most of the stable radicals are still active after the polymerization (Fig. S4, ESI†).37 Furthermore, the thermal properties of PGTEMPO was characterized by thermogravimetric analysis (TGA), presenting a degradation temperature (Td) of ∼246 °C (Fig. S6, ESI†).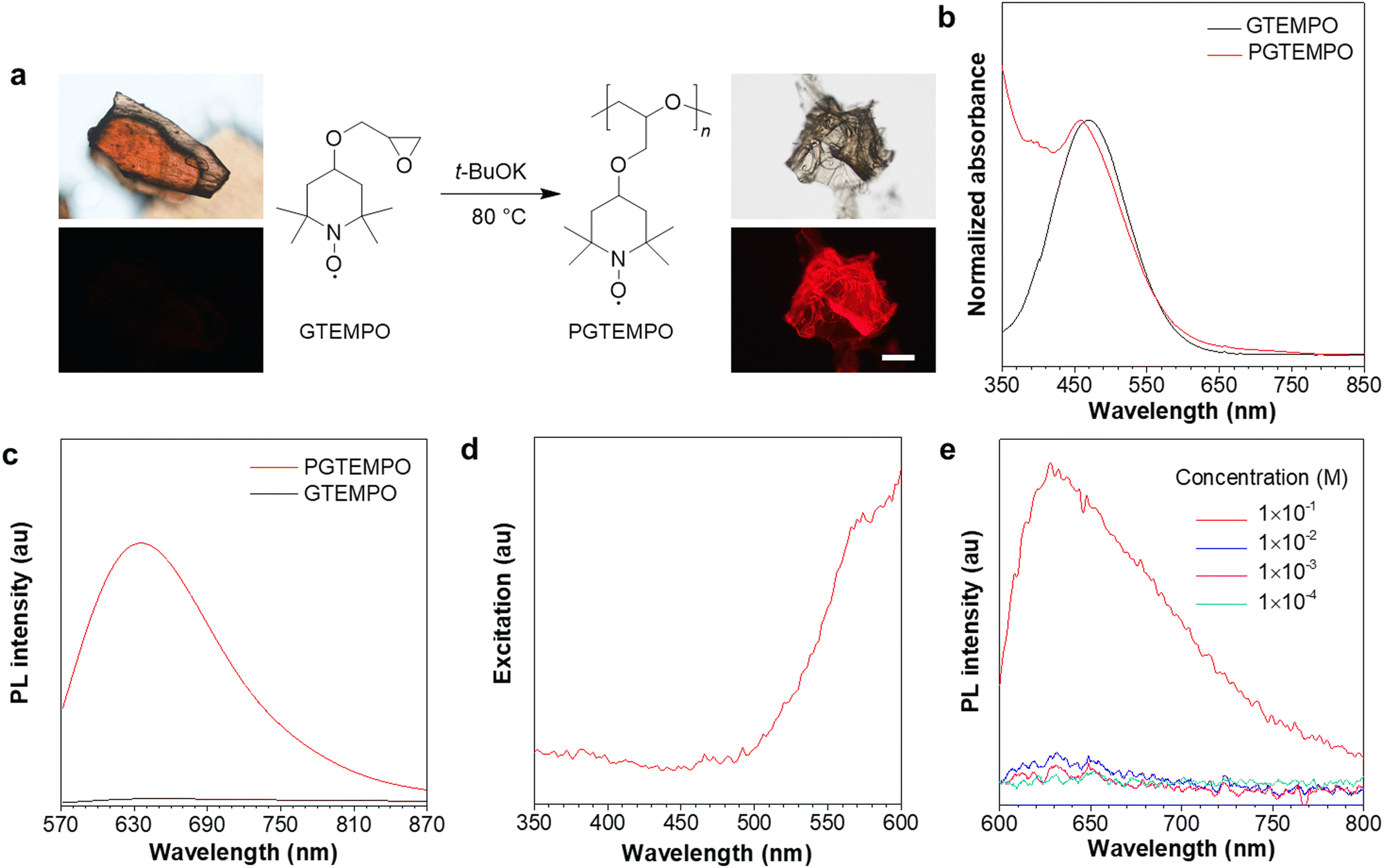 | ||
| Fig. 1 Synthesis and photophysical properties. (a) Synthetic route of the PGTEMPO radical polymer and its photos taken under (up) ambient light and (down) 510–550 nm excitation. Scale bar: 200 μm. (b) Normalized absorption spectra of PGTEMPO and GTEMPO in THF solution. (c) PL spectra of PGTEMPO and GTEMPO in the solid state excited at 532 nm. (d) Excitation spectrum of PGTEMPO in the solid state at the emission peak of 635 nm. (e) PL spectra of PGTEMPO in THF solution at various concentrations. Excitation: 532 nm. | ||
The absorption spectra of PGTEMPO and GTEMPO in THF solution showed similar absorption maxima (λabs), which are located at about 459 nm and 468 nm, respectively (Fig. 1b). No PL emission signal was detected from the GTEMPO monomer. Yet for PGTEMPO, a red emission band with peak intensity at ∼635 nm, a quantum yield of 1.3% and a lifetime of 0.198 ns emerged in the solid state under 532 nm excitation (Fig. 1c). The excitation spectrum of PGTEMPO was obtained at the fixed emission peak of 635 nm as shown in Fig. 1d, which does not align with the absorption spectra. The photographs were taken under various excitation channels (Fig. S7, ESI†), which further confirm that the monomer is non-luminescent under a broad range of irradiation. Then, the PL spectra of PGTEMPO were measured in THF at various concentrations from 0.1 mM to 100 mM (Fig. 1e). At low concentration, PGTEMPO displayed negligible emission. However, when the concentration increased to a threshold of 100 mM, the emission intensity suddenly showed a 10-fold increase. This phenomenon is analogous to aggregation-induced emission (AIE), which describes a photophysical effect in which materials show luminescence under irradiation in the aggregated or the solid state, but become weakly emissive or non-emissive in the diluted or the isolated state.38,39 This phenomenon indicates that the restriction of motion will be beneficial to luminescence.40 However, unlike the conventional AIE chromophore, PGTEMPO does not contain classical extended conjugated structures; from the PL data, it was hypothesized that the emission of PGTEMPO comes from intermolecular interactions, which is similar to those of clusteroluminescence. At low concentration, the population of intermolecular interactions is low, which accounts for the faint emission. When the concentration reaches the threshold, the intermolecular interactions are enhanced, which turns on the luminescence.
Mechanism study
To confirm that the luminescence is associated with the radical on TEMPO, we first designed an experiment to chemically quench the radical site on PGTEMPO with ascorbic acid (vitamin C, VC)41 and observed the subsequent luminescence change. After reacting with VC, the nitroxide group (N–O) was reduced into a hydroxylamine group (N–OH), generating poly(4-glycidyloxy-2,2,6,6-tetramethylpiperidine-1-hydroxyl) (PGTEMPOH) (Fig. 2a). The suppression of the EPR signal (Fig. 2b) and the emergence of the NMR spectrum of PGTEMPOH (Fig. S10, ESI†) suggested the successful quenching of the radical. The response to VC is also very sensitive and rapid since the orange color of the PGTEMPO solution quickly fades to colorless after reduction into PGTEMPOH (Fig. 2c and d). As we expected, the red-emissive peak of PGTEMPO could no longer be observed along with the quenching of radicals (Fig. 2e). On the other hand, the quenching of radicals generates PGTEMPOH, which is a classic clusteroluminogen.28 Previous study suggests that the clustering effect from the inter/intramolecular hydrogen bond interactions could trigger the emission.42 As expected, we observed a blue emission from PGTEMPOH under an excitation of 360 nm, which is absent in PGTEMPO (Fig. 2f). The luminescence quenching experiment proves that radicals play a crucial role in the unusual red-luminescence property of PGTEMPO.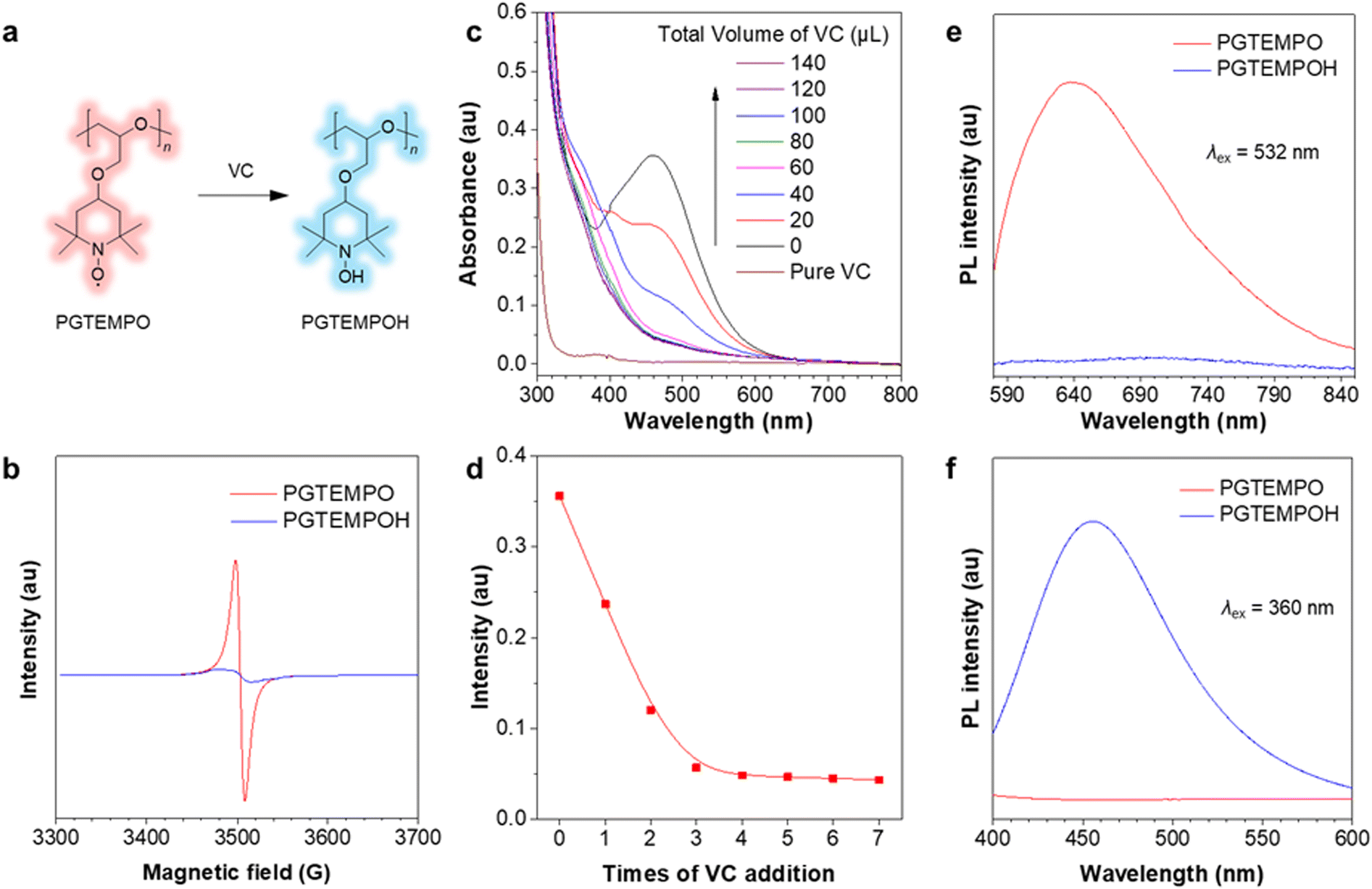 | ||
| Fig. 2 Mechanism study of the role of nitroxide radical in the photophysical property of PGTEMPO. (a) Reaction scheme of PGTEMPO with VC to generate PGTEMPOH. (b) The EPR signal of PGTEMPO and PGTEMPOH in the solid state. (c) UV-Vis spectra of PGTEMPO in DMSO (0.02 M, 1.5 mL) before and after addition of VC DMSO solution (0.6 M, each time 20 μL). (d) Plot of UV intensity versus times of VC addition. (e and f) PL spectra of PGTEMPO and PGTEMPOH in the solid state under an excitation wavelength of (up) 532 nm and (down) 360 nm respectively. | ||
To understand the origin of luminescence from nitroxide radicals, we studied the dependence of luminescence on the polymer packing. A cycle of temperature-dependent PL was performed between −20 and 50 °C, and compared to the differential scanning calorimetry (DSC) results (Fig. 3a and b). The PGTEMPO presents a glass transition temperature (Tg) of 17.40 °C. In general, the luminescence intensity decreases as the temperature increases, which is because of the favourable non-radiative decay pathways at higher temperatures.43 Surprisingly, there is a significant drop in the PL intensity between 7 and 17 °C, where the polymer undergoes a glassy transition. It is hypothesized that the glassy transition breaks the rigidity of the polymer, which decreases the intensity.44 In addition, the real-time monitoring of the luminescence of PGTEMPO at 80 °C under N2 was performed to probe the dynamic structural evolution of PGTEMPO (Fig. 3c). As high temperatures will boost non-radiative decay, the maximum PL intensity decreased rapidly within the first 20 min. Serendipitously, the PL intensity rebounded afterwards. Previous Monte Carlo simulations suggested that the annealing will form a continuous percolation network between TEMPO units for charge transport.45 Therefore, it is plausible that the increasing population of through-space interaction among the TEMPO units stimulated by the annealing process accounts for the escalating PL intensity. This is also supported by the observation of a red shift from 635 to 647 nm during the real-time annealing.
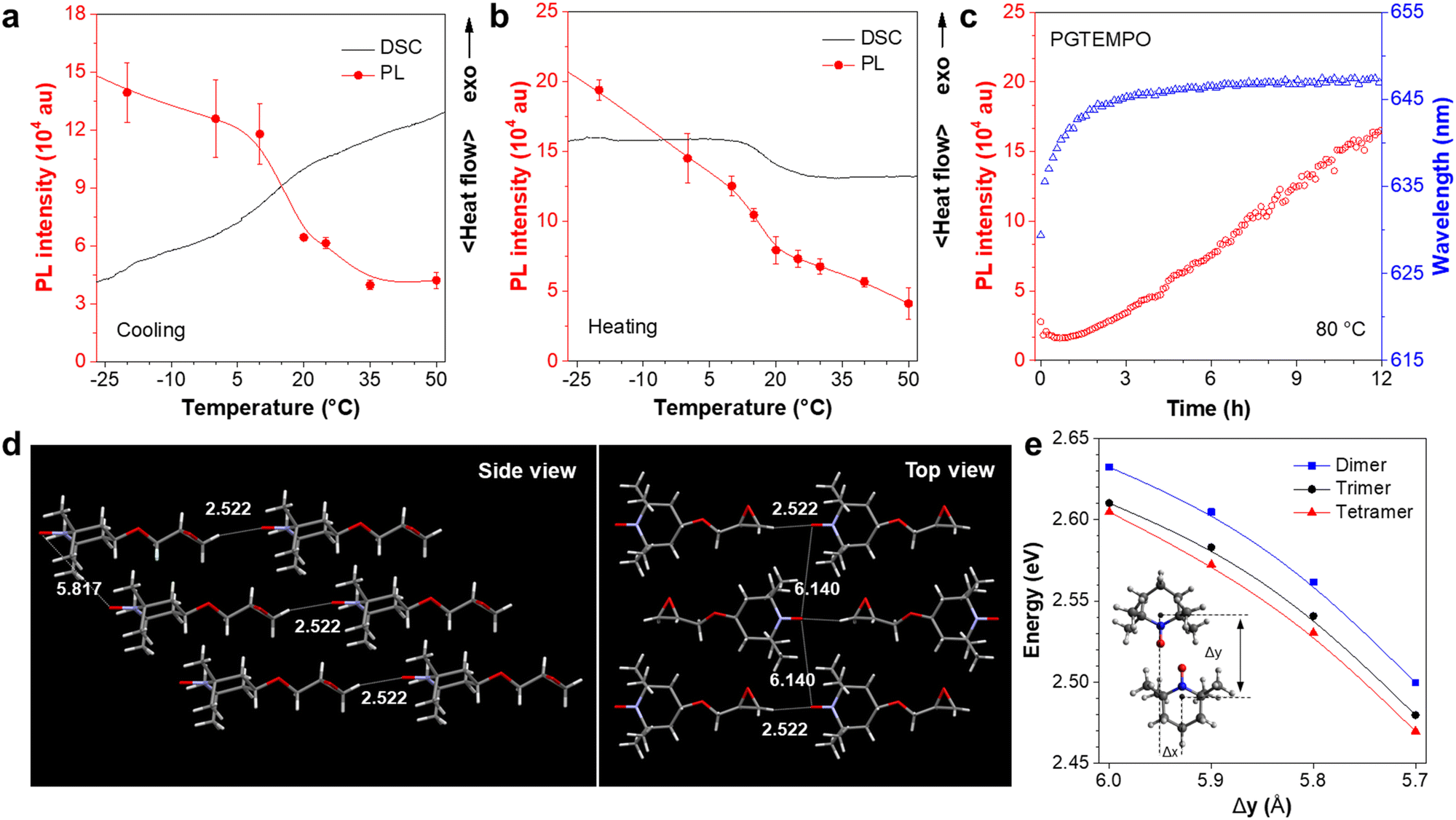 | ||
| Fig. 3 Structure-dependent photophysical properties of PGTEMPO. (a and b) The PL intensity of unannealed PGTEMPO at various temperatures in combination with differential scanning calorimetry thermograms recorded under nitrogen at a rate of 10 °C min−1. (c) The real-time annealing of PGTEMPO at the temperature of 80 °C under nitrogen. Excitation: 532 nm. (d) Single crystal X-ray structure of GTEMPO from the side view and top view. Distances were expressed in the unit of Å. (e) First excitation energy of TEMPO cluster (dimer, trimer, and tetramer) at various separation distances. | ||
To further understand the luminescence mechanism from radical interactions, we analyzed both the structural information and calculations for the monomer and the polymer. We first investigated the properties of the monomer, GTEMPO. The X-ray diffraction analysis revealed that the GTEMPO powder is orderly packed (Fig. S13, ESI†). The single crystal structure of GTEMPO was obtained as shown in Fig. 3d. In the side view, the nitroxide groups are sterically hindered by the surrounding methyl groups. The nearest distance between two nitroxide groups is 5.817 Å. If we term the nitroxide site as the head of the molecule, from the top view, one can observe that GTEMPO adopts a head-to-tail model, and the distance between adjacent nitroxide groups is 6.140 Å. Then we use time-dependent density functional theory (TDDFT) calculations using the B3LYP functional and the def2-TZVP basis set via Q-Chem, to reveal the electronic structure. The ground state of TEMPO is a doublet (D0) due to the existence of an unpaired electron. As depicted in Fig. S14 (ESI†), 44α refers to the SOMO. The calculated D1 energy of TEMPO is 2.754 eV (459 nm), which agrees with the experimental absorption data (Fig. 1b, 468 nm) and is attributed to the HOMO–SOMO transition.
In comparison, for PGTEMPO, the backbone of the polymer readily breaks the orderly packed conformation, as shown by the powder X-ray diffraction (XRD) results in which the fresh PGTEMPO sample lost the fine peaks and became completely amorphous after annealing at 80 °C (Fig. S15, ESI†). To understand the electronic states of PGTEMPO, we first simulated the structure by aligning the TEMPO dimer to form a parallelogram.46 We further added extra TEMPO units near the parallelogram to form trimer and tetramer clusters to model the effect of multi-unit clusters. For convenience, we defined the y-axis of the geometry as the line running through the atoms N1 and C4, and the x-axis as the line running through atoms C3 and C5 (Fig. S16a, ESI†). We used the displacement between the TEMPO unit on the x-axis (Δx) and the y-axis (Δy) to specify the configuration of TEMPO dimers (Fig. S16b, ESI†). Trimers and tetramers are constructed by placing the extra TEMPO unit above and below the nitroxide radical parallelogram plane of the dimer (Fig. S16c, ESI†). For all dimers in this section, we chose the value of Δx to be 1.5 Å and Δy to be between 5.5 Å and 6.0 Å, so that the two TEMPO units can approach each other to form the parallelogram between nitroxide radicals, while avoiding direct collision between the radicals and methyl groups on C2 and C6. The ground state frontier orbitals of the TEMPO dimer, trimer, and tetramer are shown in Fig. S17–S19 (ESI†), respectively. The excitation energy of the cluster at various distances was calculated and is plotted as shown in Fig. 3d. The results show that the energy gap decreases as the value of Δy decreases and the clustering size increases. This suggests that the existence of through-space interaction will form a new through-space cluster in the polymer with a narrower HOMO–SOMO gap. It agrees with the excitation spectrum that the luminescence is induced by excitation of long wavelength (Fig. 1d). In addition, it also explains the red-shift emission of annealed PGTEMPO (Fig. 3c), as according to the Monte Carlo simulation45 that annealing will favor the TEMPO clustering and result in a narrower energy gap.
To investigate if TEMPO-doped and PGTEMPO-doped commercial polymer could also be emissive, TEMPO and PGTEMPO were doped in polystyrene (PS) respectively. PGTEMPO-doped PS remains emissive under the excitation of 510–550 nm, while TEMPO-doped PS does not, as shown in Fig. S20 (ESI†). It highlights the intrinsic photophysical property of PGTEMPO. Besides, it also demonstrates that the polymerization of TEMPO, rather than simply doping in commercial polymers, is crucial for turning on the luminescence.
Application of antioxidant detection
Free radicals in the form of reactive oxygen/nitrogen species (ROS/RNS) widely exist in cells of living bodies and some of them could attack and modify biologically essential molecules such as proteins, DNA, and carbohydrates.47 Radical scavenging is one of the most significant functions of antioxidants and, to date, several small molecules have been reported and recognized as major radical scavenging antioxidants in vivo, such as vitamin C (VC),48 glutathione (GSH),49 and vitamin E. The imbalance between the antioxidant defense system and the production of ROS, would give rise to oxidative stress, which is related to many neurodegenerative disorders, including Parkinson's disease (PD) and Alzheimer's disease (AD).50 Thus, extracellular and intracellular detection and visualization of antioxidants is of great significance in fundamental research as well as practical application.As a proof-of-concept demonstration, we used PGTEMPO as a potential fluorescent sensor for antioxidant detection and chose VC as the representative for antioxidants. To render the hydrophobic PGTEMPO with good intracellular biocompatibility and solubility in water, we encapsulated PGTEMPO with the assistance of a surfactant, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy-(polyethylene glycol)-2000] (DSPE-PEG2000) via nanoprecipitation,51 as schematically illustrated in Fig. 4a. Briefly, the PGTEMPO and DSPE-PEG were dissolved in THF and added dropwise into an aqueous solution. After sonication, PGTEMPO self-assembled into nanoparticles (NPs). The hydrodynamic diameters of nanoparticles center around 95 nm as revealed by dynamic light scattering (DLS) (Fig. S21, ESI†).
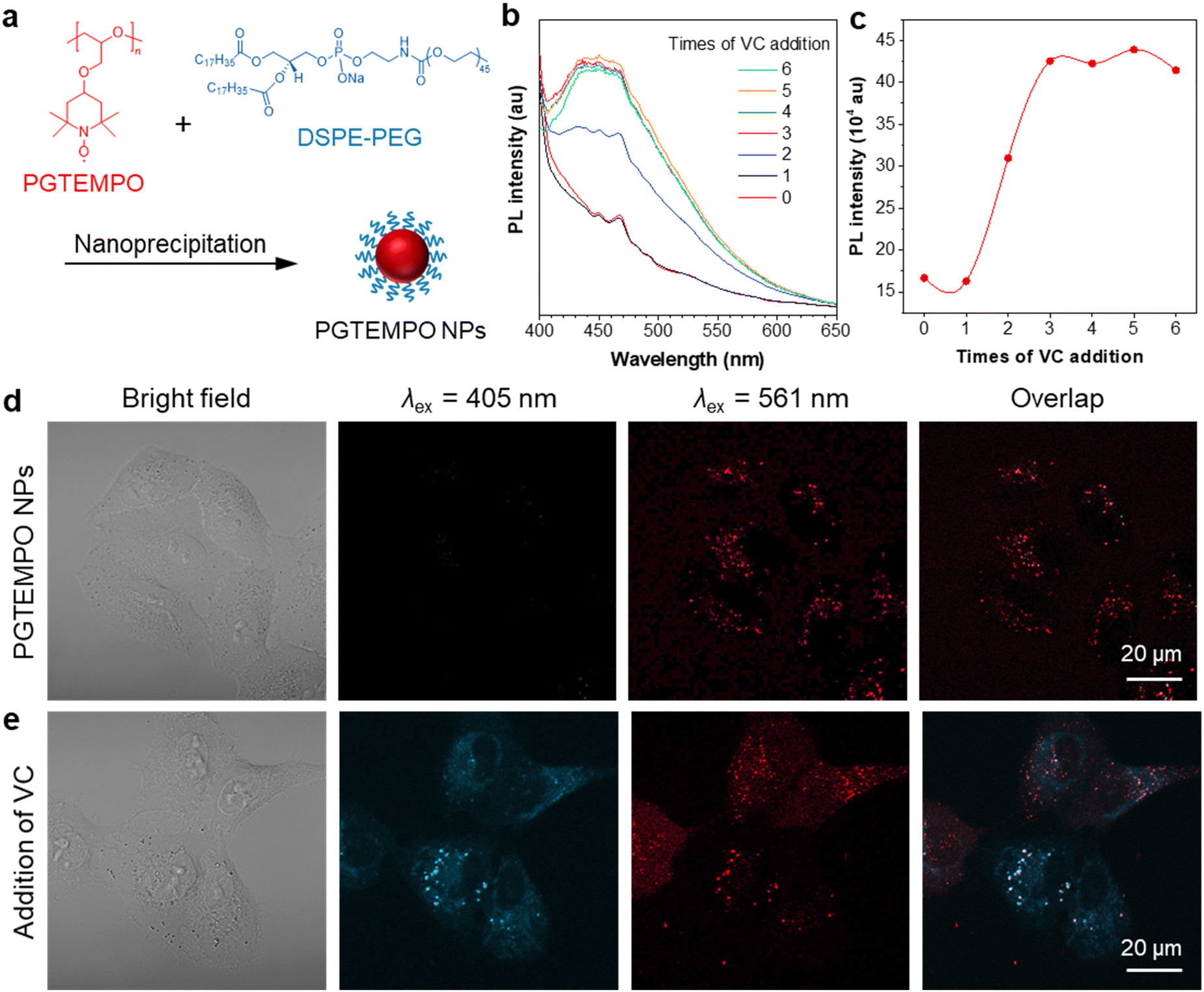 | ||
| Fig. 4 Demonstration of antioxidant detection. (a) Schematic preparation of PGTEMPO NPs via a nanoprecipitation method by using an amphiphilic block copolymer DSPE-PEG as the encapsulation materials. (b) PL spectra of PGTEMPO NPs solution (1 mL, 2 mg mL−1) before and after addition of VC aqueous solution (0.2 M, each time 10 μL). Excitation: 360 nm. (c) Plot of intensity versus times of VC addition. (d and e) Confocal laser scanning microscope images of A549 cells after incubation with (d) PGTEMPO NPs (10 μg mL−1) for 4 h and (e) after addition of VC medium solution (1 mg mL−1) and 15 min incubation. Excitation: 405 nm for the second column and 561 nm for the third column. | ||
To demonstrate the viability of VC sensing in cells, we first tested the fluorescence response of PGTEMPO NPs to VC in water. The reaction of PGTEMPO NPs with VC was very fast at room temperature and in aqueous solution. We obtained a peak at around 450 nm after the addition of VC solution to PGTEMPO NPs (Fig. 4b and c). Afterwards, we explored the in vitro cellular uptake and VC mapping performance of PGTEMPO NPs using A549 lung cancer cells as an example. After incubation for 4 h, we observed a substantial accumulation of PGTEMPO NPs inside the A549 cells via confocal laser scanning microscope. In the Fig. 4d image under excitation of 561 nm, red luminescence was observed. Subsequently, we added VC to the medium and incubated the cells for 15 min. The fluorescent signal in cells under irradiation of 405 nm emerged in Fig. 4e, confirming that PGTEMPO NPs could serve as a turn-on probe for intracellular mapping of VC distribution. To further confirm that PGTEMPO NPs targeted at lysosome, a colocalization experiment was carried out with commercial LysoTracker Green (LTG), a lysosome marker with short excitation/emission of 488/511 nm as the control. As shown in Fig. S22 (ESI†), the fine lysosome structures from PGTEMPO NPs greatly overlap with those from LTG, confirming the specific lysosome targeting of PGTEMPO NPs. The fluorescent signal in cells indicated that VC diffused in cells swiftly (<15 min) and can enter lysosomes. In comparison with reported VC fluorescent probe from one-channel imaging,52 the dual fluorescent signals (under 561 nm and 405 nm channel) from PGTEMPO NPs improve the imaging reliability.
Conclusions
We have synthesized a nonconjugated radical polymer showing luminescence in the solid state. To our knowledge, this is the first report of a stable luminescent radical without any π-delocalized stabilization. Luminescence quenching experiments confirm the key role of nitroxide radicals. Combining the structural information and calculations, we propose that the intermolecular interactions of TEMPO clusters account for the long-wavelength emission, which could also bring inspiring ideas to the study of mysterious luminescence from living organisms. Taking advantage of the AIE property, a biological application for intracellular antioxidant detection was demonstrated. There are still opportunities for future improvement, in areas such as the low quantum yield. Nonetheless, this work provides a novel strategy to turn on the luminescence from organic radicals and expands the family of luminescent radicals to a large extent. We expect that future work will improve this disadvantage via better molecular structure design. We also envision that further experimental and theoretical research on this unconventional luminescence phenomenon will provide insights into principles governing radical luminescence and find applications in broad fields.Author contributions
Z. W., R. Y., and B. Z. T. conceived the idea. Z. W. synthesized the materials and completed the characterization. X. Z., Z. W., C. C. S. C., and K. S. W. performed the photophysical experiments. Y. X., L. H., and D. S. carried out the theoretical calculations and result analyses. R. Z. and Z. W. carried out the biological application experiments. J. G. and Z. Z. conducted the EPR measurements. I. W. carried out the single crystal X-ray diffraction. H. Z., R. Y. and B. Z. T. initiated and supervised the work. Z. W., R. Y., and B. Z. T. wrote the manuscript. H. Z., R. T. K. K., J. W. Y. L., and K. S. W. revised the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (21788102), the Research Grants Council of Hong Kong (16305618, 16304819, C6009-17G, and N-HKUST 609/19), the University Grants Committee of Hong Kong (AoE/P-02/12), the Innovation and Technology Commission (ITC-CNERC14SC01), and the Natural Science Foundation of Guangdong Province (2019B121205002). R. Y. acknowledges the funding support from Hong Kong Research Grant Council under Early Career Scheme (Project No. 21300620). We would also like to thank Dr Shunjie Liu, Dr Qingqing Gao, and Zaiyu Wang for their kind assistance. Besides, we are grateful for Dr Herman H. Y. Sung who conducted single crystal X-ray diffraction in this work.Notes and references
- O. Armet, J. Veciana, C. Rovira, J. Riera, J. Castañer, E. Molins, J. Rius, C. Miravitlles, S. Olivella and J. Brichfeus, J. Phys. Chem., 1987, 91, 5608–5616 CrossRef CAS.
- M. Ballester, J. Riera, J. Castafier, C. Badía and J. M. Monsó, J. Am. Chem. Soc., 1971, 93, 2215–2225 CrossRef CAS.
- Y. Wu, H. Yi and A. Lei, ACS Catal., 2018, 8, 1192–1196 CrossRef CAS.
- H. Nishide, S. Iwasa, Y. J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
- R. A. Miller and R. S. Hoerrner, Org. Lett., 2003, 5, 285–287 CrossRef CAS PubMed.
- Y. Teki, Chem. – Eur. J., 2020, 26, 980–996 CrossRef CAS PubMed.
- R. E. Schwerzel and R. A. Caldwell, J. Am. Chem. Soc., 1973, 95, 1382–1389 CrossRef CAS.
- S. K. Chattopadhyay, P. K. Das and G. L. Hug, J. Am. Chem. Soc., 1983, 105, 6205–6210 CrossRef CAS.
- V. Gamero, D. Velasco, S. Latorre, F. López-Calahorra, E. Brillas and L. Juliá, Tetrahedron Lett., 2006, 47, 2305–2309 CrossRef CAS.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS PubMed.
- X. Ai, Y. Chen, Y. Feng and F. Li, Angew. Chem., Int. Ed., 2018, 57, 2869–2873 CrossRef CAS PubMed.
- S. Kimura, T. Kusamoto, S. Kimura, K. Kato, Y. Teki and H. Nishihara, Angew. Chem., 2018, 57, 12711–12715 CrossRef CAS PubMed.
- H. Guo, Q. Peng, X. K. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Credgington, V. Coropceanu, R. H. Friend, J. L. Brédas and F. Li, Nat. Mater., 2019, 18, 977–984 CrossRef CAS PubMed.
- K. Kato, S. Kimura, T. Kusamoto, H. Nishihara and Y. Teki, Angew. Chem., Int. Ed., 2019, 58, 2606–2611 CrossRef CAS PubMed.
- A. Ito, A. Shimizu, N. Kishida, Y. Kawanaka, D. Kosumi, H. Hashimoto and Y. Teki, Angew. Chem., 2014, 126, 6833–6837 CrossRef.
- S. Kimura, S. Kimura, K. Kato, Y. Teki, H. Nishihara and T. Kusamoto, Chem. Sci., 2021, 12, 2025–2029 RSC.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef CAS PubMed.
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS PubMed.
- A. Shimizu, A. Ito and Y. Teki, Chem. Commun., 2016, 52, 2889–2892 RSC.
- A. Rajca, Y. Wang, M. Boska, J. T. Paletta, A. Olankitwanit, M. A. Swanson, D. G. Mitchell, S. S. Eaton, G. R. Eaton and S. Rajca, J. Am. Chem. Soc., 2012, 134, 15724–15727 CrossRef CAS PubMed.
- L. Ji, J. Shi, J. Wei, T. Yu and W. Huang, Adv. Mater., 2020, 32, 1908015 CrossRef CAS PubMed.
- A. Abdurahman, T. J. H. Hele, Q. Gu, J. Zhang, Q. Peng, M. Zhang, R. H. Friend, F. Li and E. W. Evans, Nat. Mater., 2020, 19, 1224–1229 CrossRef CAS PubMed.
- Y. Beldjoudi, M. A. Nascimento, Y. J. Cho, H. Yu, H. Aziz, D. Tonouchi, K. Eguchi, M. M. Matsushita, K. Awaga, I. Osorio-Roman, C. P. Constantinides and J. M. Rawson, J. Am. Chem. Soc., 2018, 140, 6260–6270 CrossRef CAS PubMed.
- A. Abdurahman, Q. Peng, O. Ablikim, X. Ai and F. Li, Mater. Horiz., 2019, 6, 1265–1270 RSC.
- O. Shimomura and F. H. Johnson, Nature, 1970, 227, 1356–1357 CrossRef CAS PubMed.
- O. Shimomura, F. H. Johnson and Y. Saiga, J. Cell. Comp. Physiol., 1962, 59, 223–239 CrossRef CAS PubMed.
- Y. Wang, Z. Zhao and W. Z. Yuan, ChemPlusChem, 2020, 85, 1065–1080 CrossRef CAS PubMed.
- H. Zhang, Z. Zhao, P. R. McGonigal, R. Ye, S. Liu, J. W. Y. Lam, R. T. K. Kwok, W. Z. Yuan, J. Xie, A. L. Rogach and B. Z. Tang, Mater. Today, 2020, 32, 275–292 CrossRef CAS.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- P. Sun, Q. Wu, X. Sun, H. Miao, W. Deng, W. Zhang, Q. Fan and W. Huang, Chem. Commun., 2018, 54, 13395–13398 RSC.
- R. Y. Tsien, Angew. Chem., Int. Ed., 2009, 48, 5612–5626 CrossRef CAS PubMed.
- Y. Miwa, K. Yamamoto, M. Sakaguchi and S. Shimada, Macromolecules, 2001, 34, 2089–2094 CrossRef CAS.
- T. Hagemann, M. Strumpf, E. Schröter, C. Stolze, M. Grube, I. Nischang, M. D. Hager and U. S. Schubert, Chem. Mater., 2019, 31, 7987–7999 CrossRef CAS.
- J. Nie, D. Yang, X. Tang, L. Zhang, Z. Ni and X. Gui, Dyes Pigm., 2019, 170, 107644 CrossRef CAS.
- S. A. Rivera and B. S. Hudson, J. Am. Chem. Soc., 2006, 128, 18–19 CrossRef CAS PubMed.
- W. Xu, D. Wang and B. Z. Tang, Angew. Chem., 2020, 133, 7552–7563 CrossRef.
- Y. Joo, V. Agarkar, S. H. Sung, B. M. Savoie, B. W. Boudouris, H. Zhang, X. Zheng, R. T. K. Kwok, J. Wang, N. L. C. Leung, L. Shi, J. Z. Sun, Z. Tang, J. W. Y. Lam, A. Qin and B. Z. Tang, Science, 2018, 359, 1391–1395 CrossRef CAS PubMed.
- Q. Wang, X. Dou, X. Chen, Z. Zhao, S. Wang, Y. Wang, K. Sui, Y. Tan, Y. Gong, Y. Zhang and W. Z. Yuan, Angew. Chem., 2019, 58, 12667–12673 CrossRef CAS PubMed.
- H. Zhang, Z. Zhao, A. T. Turley, L. Wang, P. R. McGonigal, Y. Tu, Y. Li, Z. Wang, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2020, 32, 2001457 CrossRef CAS PubMed.
- X. Wang, X. Qiao, X. Yin, Z. Cui, P. Fu, M. Liu, G. Wang, X. Pan and X. Pang, Chem. – Asian J., 2020, 15, 1014–1017 CrossRef CAS PubMed.
- Y. Tang, F. He, M. Yu, S. Wang, Y. Li and D. Zhu, Chem. Mater., 2006, 18, 3605–3610 CrossRef CAS.
- R. Ye, Y. Liu, H. Zhang, H. Su, Y. Zhang, L. Xu, R. Hu, R. T. K. Kwok, K. S. Wong, J. W. Y. Lam, W. A. Goddard and B. Z. Tang, Polym. Chem., 2017, 8, 1722–1727 RSC.
- Y. Cui, F. Zhu, B. Chen and G. Qian, Chem. Commun., 2015, 51, 7420–7431 RSC.
- N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 47, 15349–15353 CrossRef PubMed.
- Y. Joo, V. Agarkar, S. H. Sung, B. M. Savoie and B. W. Boudouris, Science, 2018, 359, 1391–1395 CrossRef CAS PubMed.
- H. Zhang, X. Zheng, R. T. K. Kwok, J. Wang, N. L. C. Leung, L. Shi, J. Z. Sun, Z. Tang, J. W. Y. Lam, A. Qin and B. Z. Tang, Nat. Commun., 2018, 9, 4961 CrossRef PubMed.
- E. Niki, Free Radical Biol. Med., 2014, 66, 3–12 CrossRef CAS PubMed.
- S. J. Padayatty, A. Katz, Y. Wang, P. Eck, O. Kwon, J. H. Lee, S. Chen, C. Corpe, M. Levine, A. Dutta and S. K. Dutta, J. Am. Coll. Nutr., 2003, 22, 18–35 CrossRef CAS PubMed.
- M. M. Nemat Alla and N. M. Hassan, Acta Physiol. Plant., 2007, 29, 247–258 CrossRef.
- P. K. Chatterjee, S. Cuzzocrea, P. A. J. Brown, K. Zacharowski, K. N. Stewart, H. Mota-Filipe and C. Thiemermann, Kidney Int., 2000, 58, 658–673 CrossRef CAS PubMed.
- X. Cai, D. Mao, C. Wang, D. Kong, X. Cheng and B. Liu, Angew. Chem., 2018, 130, 16634–16638 CrossRef.
- K. Ishii, K. Kubo, T. Sakurada, K. Komori and Y. Sakai, Chem. Commun., 2011, 47, 4932–4934 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2045204. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2mh00808d |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |