
A self-healing, recyclable and conductive gelatin/nanofibrillated cellulose/Fe3+ hydrogel based on multi-dynamic interactions for a multifunctional strain sensor†
Haocheng
Fu
,
Bin
Wang
 *,
Jinpeng
Li
*,
Jun
Xu
*,
Jun
Li
,
Jinsong
Zeng
,
Wenhua
Gao
and
Kefu
Chen
*,
Jinpeng
Li
*,
Jun
Xu
*,
Jun
Li
,
Jinsong
Zeng
,
Wenhua
Gao
and
Kefu
Chen
Plant Fiber Material Science Research Center, State Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: febwang@scut.edu.cn; ljp@scut.edu.cn; xujun@scut.edu.cn
First published on 10th March 2022
Abstract
Conductive hydrogels have emerged as promising material candidates for multifunctional strain sensors, attributed to their similarity to biological tissues, good wearability, and high accuracy of information acquisition. However, it is difficult to simultaneously manufacture conductive hydrogel-based multifunctional strain sensors with the synergistic properties of reliable healability for long-term usage and environmental degradability/recyclability for decreasing the electronic waste. This work reports a facile strategy to engineer a self-healing, recyclable and conductive strain sensor by virtue of molecular-level multi-dynamic interactions (MMDIs) including Schiff base complexes, hydrogen bonds, and coordination bonds, which were fabricated using a dialdehyde TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl)-oxidized nanofibrillated cellulose (DATNFC) pre-reinforced gelatin nanocomposite hydrogel (gelatin/DATNFC hydrogel, GDH) followed by dipping in an Fe3+ aqueous solution. The MMDI strategy allows synchronous regulation of both bulk and interfacial interactions to obtain exciting properties that outperform those of conventional hydrogels, including extraordinary compressive stress (1310 kPa), intriguing self-healing abilities, and remarkable electrical conductivity. With these outstanding merits, the as-prepared gelatin/DATNFC/Fe3+ hydrogel (GDIH) is developed to be a multifunctional strain sensor with appealing strain sensitivity (GF = 2.24 under 6% strain) and compressive sensitivity (S = 1.14 kPa−1 under 15 kPa), which can be utilized to manufacture electronic skin and accurately discern subtle bodily motions, handwriting and personal signatures. Notably, this GDIH-based sensor also exhibited reliable self-healing properties for long-term usage, environmental degradability and complete recyclability for decreasing the electronic waste. In consideration of the extremely facile preparation process, biocompatibility, satisfactory functionalities, remarkable self-healing properties and recyclability, the emergence of the GDIH-based sensor is believed to propose a new strategy for the development of sustainable-multifunctional strain sensors and healthcare monitoring.
New conceptsWe proposed a facile strategy to produce a stretchable, compressible, self-healable, degradable and recyclable hydrogel-based strain sensor by virtue of molecular-level multi-dynamic interactions(MMDIs) including Schiff base reactions, hydrogen bonds, and coordination bonds between dialdehyde TEMPO-oxidized nanofibrillated cellulose (DATNFC), gelatin and Fe3+. By introducing MMDIs to double network structures, the preparation of multifunctional strain sensors with synergistic properties was achieved. Based on this MMDI strategy, the gelatin/DATNFC/Fe3+ hydrogel (GDIH) showed significant increases in tensile (990.7%) and compressive strength (3822.7%) compared with pure gelatin hydrogel. The GDIH also exhibited intriguing self-healing abilities, reliable recyclability and remarkable electrical conductivity. With these outstanding merits, the as-prepared GDIH was developed to be a multifunctional strain sensor with appealing strain sensitivity (GF = 2.24 under 6% strain) and compressive sensitivity (S = 1.14 kPa−1 under 15 kPa), which can accurately discern subtle bodily motions and handwriting, and be utilized to produce electronic skin. |
Introduction
With the continuous development of intelligent electronic equipment, hydrogel-based sensors have drawn enormous attention in various fields such as bioelectricity interfaces, artificial tissues, wearable electronics and personal healthcare monitoring due to their outstanding flexible, lightweight and sensor characteristics.1–9 However, the preparation of hydrogel-based sensors with high sensitivity, remarkable mechanical properties, excellent sensing stability, rapid response and low detection limit has been challenging for a long time due to the deficiency of the association mechanism between crosslinking structures and properties.5 Simultaneously, the existing conductive hydrogels have poor mechanical properties and are inevitably damaged during the deformation process, which has a negative effect on their practical applications in flexible strain sensors. Moreover, there is gradually increasing electronic waste of strain sensors owing to their non-degradability under natural conditions and incomplete recyclability. Hence, it is urgent to develop conductive hydrogel-based strain sensors with outstanding mechanical strength, remarkable self-healing capability, environmental degradation ability and recyclability to detect subtle deformations.To address these issues, much effort has been made to fabricate such hydrogel-based sensing devices. Recently, the introduction of sacrificial bonds (e.g., hydrogen bonds, metal coordination bonds, and Schiff base reactions) in synthetic hydrogels provided a novel strategy with a wide range of attractive properties. The existence of sacrificial bonds could consume the mechanical energy during mechanical processes, and further enhance the strength of the hydrogels. Various strategies have been reported based on this theory, such as double network (DN) structures,10–15 and topological cross-linking by slide-ring and dynamic cross-linking systems.16,17 In recent years, DN hydrogels, which consist of two interpenetrating crosslinking networks including a first network composed of weak bonds and a second network composed of strong bonds (covalent bonds, non-covalent bonds), have emerged as sensor materials with tunable mechanical properties and sensing performance. For instance, Gao et al. introduced sodium caseinate and carboxymethyl chitosan into a polyacrylamide hydrogel and achieved satisfactory performance.18 The results showed that the elastic modulus and compressive strength of the composite were nearly 16 kPa and 8 kPa, respectively. Gong et al. designed a series of classical covalent DN hydrogels utilizing fish collagen and hydroxyapatite (HAp) nanoparticles.19 The classical covalent DN hydrogels exhibited remarkable mechanical strength. Nevertheless, synchronously achieving prospective mechanical behavior and a tunable structure still remains challenging due to the fact that it is impeded by some inherent bottlenecks. On the one hand, most reported hydrogels consist of conventional covalent structures and hence have a relatively rigid, irreversible and non-adaptive nature,10 resulting in poor energy dissipation within the bulk polymer networks when subjected to large deformation and impeding the interfacial interplay of the hydrogel with the substrate. On the other hand, the as-obtained hydrogels suffer from insufficient dynamic interaction points within both the bulk and the interface, thus posing difficulties in improving stretchability and elasticity. Therefore, there is an urgent need to open up a new approach by virtue of multiple dynamic interactions to fabricate DN hydrogels with remarkable mechanical properties, excellent sensing performance, and intriguing degradability.
Natural polymer hydrogels, a three-dimensional network structure containing mostly water,20 have a considerable application prospect in the field of strain sensors attributed to their excellent biocompatibility.10 Most natural polymer hydrogels have poor mechanical strength and it is difficult for them to meet the requirements of multifunctional strain sensors. Gelatin, a natural protein produced from partially denatured collagen by several processes, has great potential for degradable hydrogels with appealing cost-effectiveness because of its outstanding biodegradability, biocompatibility, non-toxicity and cheapness.21–26 However, pure gelatin hydrogels generally have poor mechanical strength, which greatly limits their extensive use for multifunctional strain sensors. Fortunately, the mechanical performance of gelatin can be tailored because of outstanding compatibility with the reinforcement phase. Nanofibrillated cellulose (NFC), derived from cellulose with prominent biocompatibility, environmental degradation ability and excellent mechanical properties,27–31 has been utilized to enhance the strength of polymer hydrogels due to the existence of hydrogen bonds. For example, Han et al. fabricated a series of DN natural polymer hydrogels with a prospective Young's modulus (257 kPa) utilizing TEMPO-oxidated nanofibrillar cellulose and covalent polyacrylamide.32 Jiang et al. used NFC, polyvinyl alcohol and DMSO to fabricate a hydrogel with two asymmetric network structures, which can be used at −70 °C.20 Tang et al. used dialdehyde NFC to construct a Schiff base structure between –CHO and –NH2 of gelatin, proving the remarkable dialdehyde NFC-induced enhancement of the strength.24 Generally, two main details should be considered in the preparation of NFC-reinforced polymer hydrogels: the uniformity of NFC and the interaction between NFC and the matrix. The reasonable utilization of surface groups is significant to ensure the stable cross-linking between NFC and other polymers. Although the enhancement mechanism of NFC on the properties of hydrogels has been reported by researchers, the synergistic effect of hydrogen bonds, aldehyde groups and carboxyl groups of the oxidized NFC on the double network structure of the natural polymer hydrogel is still unclear.
Herein, we propose a new and facile fabrication method for degradable and recyclable multifunctional strain hydrogel-based sensors with prominent mechanical strength and self-healing, which are assembled using gelatin, DATNFC, and Fe3+, inspired by the virtue of molecular-level multi-dynamic interactions (MMDIs). The GDIH was obtained by introducing Fe3+ into the first network formed by gelatin and DATNFC, which was achieved via an easy synthetic procedure without complex molecular design or multistep functionalization. The as-obtained GDIH had robust degradability because of the existence of gelatin and DATNFC. Benefiting from the MMDI among the amino-groups of gelatin, different kinds of surface-terminated groups (–OH, –COOH, –CHO, etc.) of DATNFC, and Fe3+, the GDIH possessed an outstanding elasticity and excellent self-healing capability. In addition, compared with the previously reported results, the encapsulated GDIH with a slight water loss (Fig. S1 and S2, ESI†) can be assembled as a stretchable, compressible, healable, degradable and recyclable strain sensor to detect tiny deformation-induced electrophysiological signals with sensing stability and fast compression-response time (200 ms). The present research sheds light on the fabrication of healable, recycled, degradable, and electrophysiological signal-sensitive multifunctional strain sensors with potential applications in human−machine interactions, disease diagnosis, smart robot prostheses and writing anti-counterfeiting devices.
Results and discussion
MMDI hydrogels were prepared via an effective sol–gel reaction followed by an ion-immersing approach, as shown in Fig. 1a, and were fabricated using a facile synthetic procedure without complex molecular design and complicated chemical reactions. The basic recipe mainly consists of gelatin, DATNFC, and Fe3+. DATNFC was added into the pre-dissolved gelatin suspension, and then cooled at −4 °C for 24 h to obtain the GDH with different contents of DATNFC (GDHx, where x represents the mass percentage of DATNFC). Subsequently, the GDH was immersed in an FeCl3 solution to obtain GDIH. The GDIHs containing different Fe3+ contents were designated as GDIH10-a, where a represents the concentration of FeCl3 (see ESI,† for more details). Here, DATNFC acts as a key bridge to design multiple dynamic interactions with the other components. Briefly, DATNFC can leverage its abundant aldehyde/carboxyl groups to interplay with –NH2, –OH and Fe3+ to form Schiff base reactions, hydrogen bonds, and coordination bonds, respectively, all of which are dynamic and capable of reversibly breaking and reconstructing (Fig. 1b).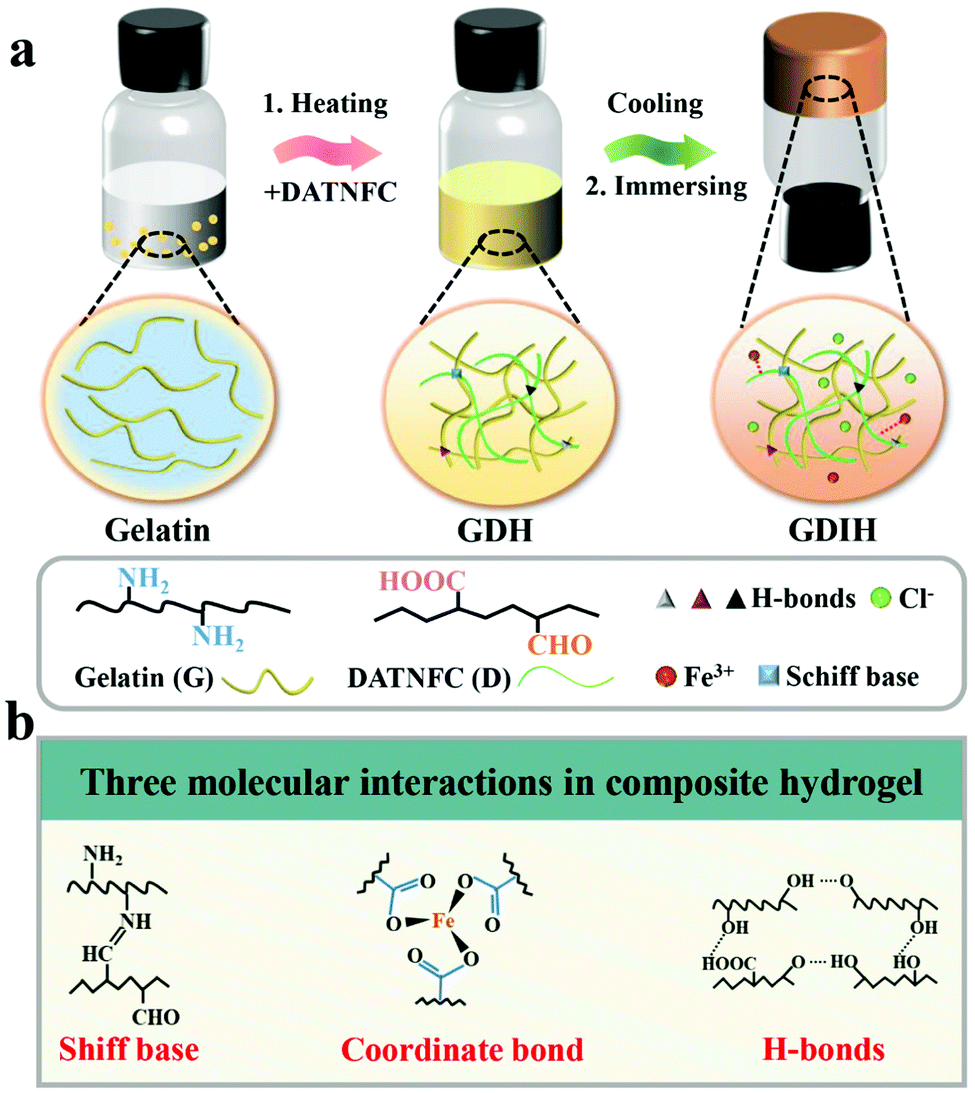 | ||
| Fig. 1 (a) Schematic illustration of the preparation and internal structure of multifunctional hydrogel-based sensors. (b) Schematic description of the three molecular interactions in the sensors. | ||
By weakening the hydrogen bonds and strengthening the electrostatic repulsion between the cellulose chains, DATNFC was synthesized by selective carboxylation accompanying mechanical trituration, followed by dialdehyde treatment. As shown in Table S1 (ESI†), the aldehyde content of DATNFC was 2.55 mmol g−1. Fig. S3 (ESI†) shows the AFM image of cellulosic nanofibers in DATNFC with an average diameter of 37.4 nm. Abundant polar functional groups involving –CHO, –COOH, and –OH on DATNFC provided excellent compatibility and cohesiveness with other matrixes and good dispersion in aqueous systems, providing sufficient crosslinking sites during the gelation procedure without conventional chemical crosslinking. Moreover, the stress could be effectively transferred to the strong DANFC frameworks under external deformation, where the DATNFC could serve as an integrated platform for fine regulation of the bulk/interfacial properties of the hydrogels, to provide superior mechanical properties and excellent self-healing abilities.
The FTIR spectrum in Fig. S4 (ESI†) proves the successful crosslinking of gelatin and DATNFC. In order to estimate the appropriate addition amount of DATNFC in the nanocomposite hydrogel, the effect of DATNFC on the mechanical properties of the nanocomposite hydrogel was explored. The illustration of the self-assembly process in Fig. 2a indicated that gelatin and DATNFC were crosslinked together by hydrogel bonds and the Schiff base after adding DATNFC. As shown in Fig. 2b, it was apparent that the color of the mixture got dark after adding DATNFC. In addition, the crosslinking index could be utilized to reflect the formation of the Schiff base. The crosslinking index of GDH had an upward trend with increasing DATNFC addition (Fig. 2c), indicating the successful crosslinking between –NH2 of gelatin and –CHO of DATNFC. As expected, the tensile-stress and elastic modulus of the pure gelatin hydrogel were 15.1 kPa and 56.1 kPa, respectively, while the gelatin/DATNFC composite hydrogel with 15% DANFC (GDH15) exhibited approximately four-fold tensile strength (53.7 kPa) and pentaploid elastic modulus (253.4 kPa) (Fig. 2d and Fig. S5, ESI†). This was ascribed to sufficient hydrogen bonds and Schiff base reactions in GDH. After immersing in a FeCl3 solution, the structural properties of GHID were analyzed by XRD, EDS, XPS, transmittance and thermal stability. The result verified that the hydroxyl, carboxyl and aldehyde groups in DATNFC could form dynamic interfacial interactions with hydroxyl groups, amino groups (–NH2) in gelatin and Fe3+, including Schiff base reactions, hydrogen bonds, and coordination bonds (Fig. S6–S9, ESI†).
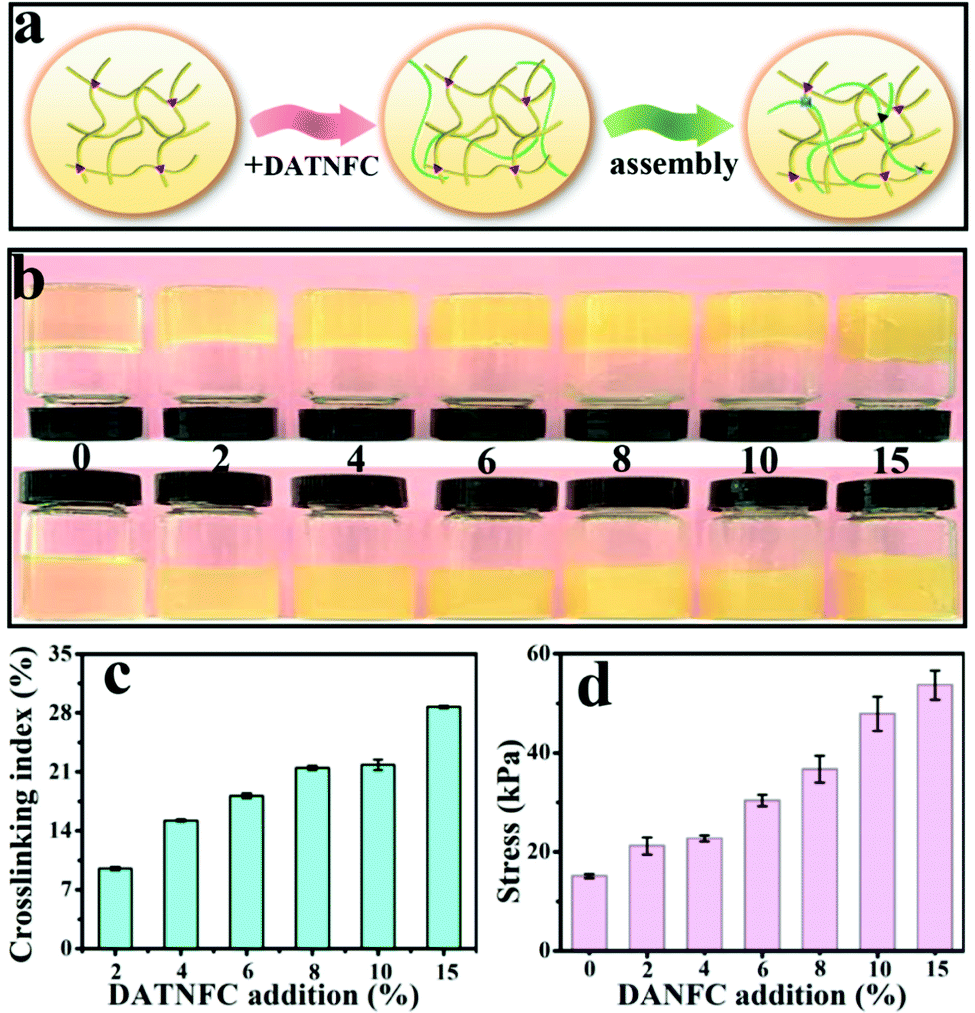 | ||
| Fig. 2 (a) The illustration of the self-assembly process, (b) optical photos, (c) crosslinking indexes and (d) tensile stress of GDH. | ||
The outstanding mechanical performance is critical for soft wearable sensors and electronic skins. The synthesized GDIH showed an intriguing elasticity that can be extensively stretched and pressed. Fig. 3a and Fig. S10 (ESI†) show that the formed dynamic chemical bonds (Schiff base, hydrogen bonds and coordination bonds) could be broken and recombined upon continuous strain through an efficient energy dissipation mechanism. As illustrated in Fig. 3b and Fig. S11 (ESI†), the GDIH10-0.06 displayed prospective tensile strength and elastic modulus of 164.7 and 501.1 kPa, which were higher than those of GDIH10 (Fig. 2d and Fig. S5, ESI†). It was apparent that the significant enhancement effect of Fe3+ on mechanical strength was attributed to the sufficient Fe3+-carboxyl coordinative bonds and hydrogen bonds. In addition, the fatigue resistance test during the stretching process indicated that the tensile strength of the GDIH10-0.06 after the pre-cycle was lower than that with no pre-stretching (Fig. 3c), while the tensile strength after the pre-cycle could be nearly constant after 10 successive loading–unloading cycles at the strain of 35%, showing an outstanding fatigue resistance against cyclic loads. The durability in tensile properties mainly originated from simple breaking of the multiple cross-linking-mediated dynamic interactions under deformation to achieve highly-efficient energy dissipation (Fig. 3a). Moreover, the as-obtained GDIH also showed prominent compressive strength. As shown in Fig. 3d, the compressive strength of GDIH had an upward trend with the increase of Fe3+ due to the existence of the stable double network (DN) in the nanocomposite hydrogel. The cycle loading–unloading compressive tests were conducted to evaluate the fatigue resistance of the nanocomposite hydrogel in the compression process (Fig. 3e). The optical photos (insets) indicated that the nanocomposite hydrogel could restore to the original height after being pressed by a finger. As shown in Fig. 3e, the compressive strength of GDIH10-0.06 after the pre-cycle was lower than that of the original GDIH10-0.06, while the compressive stress of GDIH10-0.06 could be nearly constant with the increasing of the cycle time. Fig. 3f summarizes the compressive stress of different kinds of hydrogels. It suggested that the compressive strength of GDIH10-0.10 (1310 kPa) in the present work surpassed that of many hydrogels published previously.
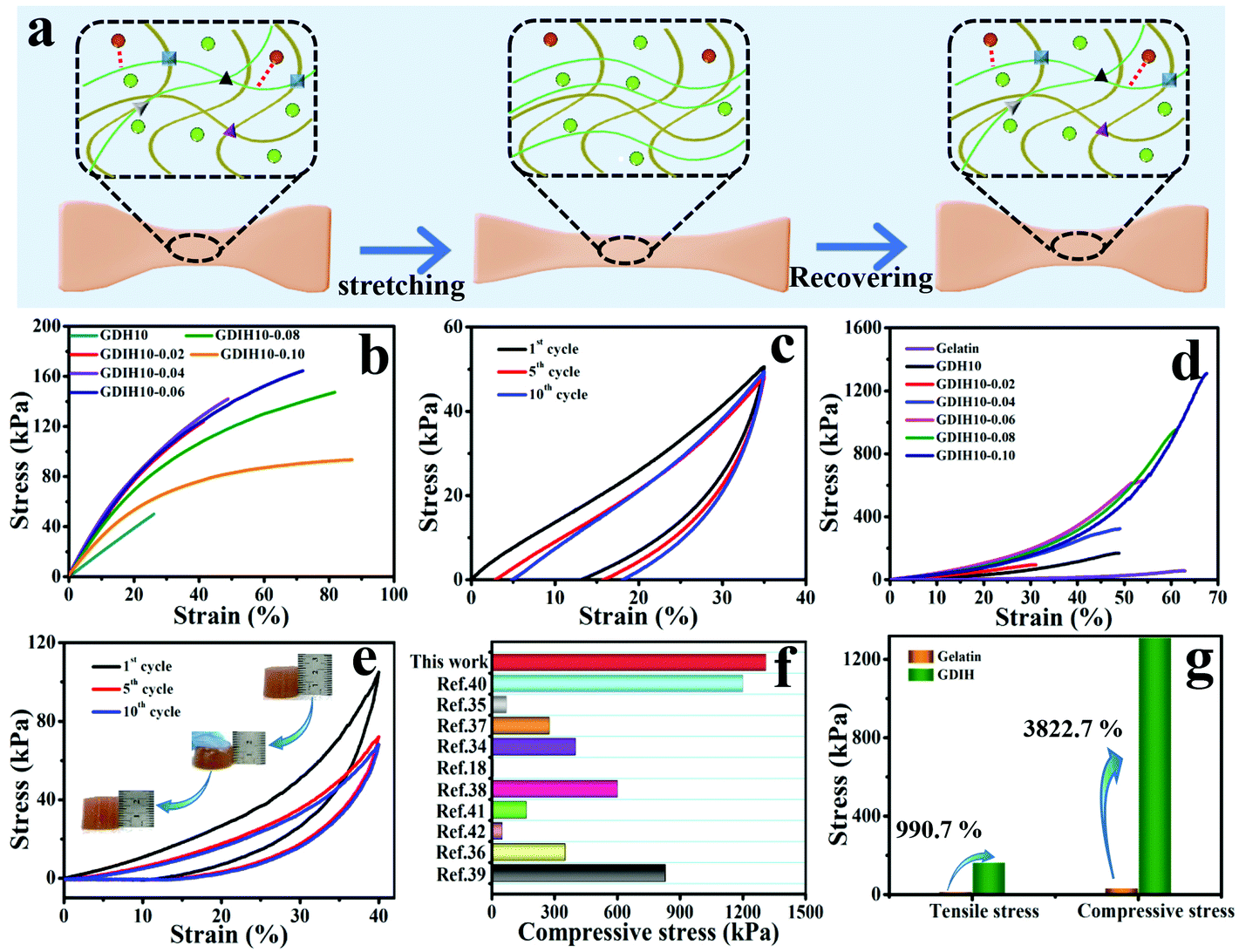 | ||
| Fig. 3 (a) Energy dissipation mechanism of the double network during the tensile test. Mechanical properties of GDIH: (b) tensile curves with no pre-cycle of the nanocomposite hydrogel and (c) cyclic tensile curves of GDIH10-0.06 after three pre-cycles. (d) Compressive curve with no pre-cycle of GDIH. (e) Cyclic compressive curves of GDIH10-0.06 after three pre-cycles. The insets are the optical photos of GDIH10-0.06 during compressing and recovering process. (f) Comparison of the compressive stress for GDIH10-0.10 with those of the reported hydrogels.18,34–42 (g) The enhancement of GDIH in mechanical strength compared with pure gelatin. | ||
In general, energy dissipation is crucial to improve the mechanical properties of the nanocomposite hydrogel. The energy during mechanical deformation could be consumed by the Schiff base and sacrificial bonds including Fe3+-carboxyl coordinative bonds and hydrogen bonds. The resultant GDIH showed an excellent mechanical performance, and the tensile strength and compressive strength increased by 990.7 and 3822.7% compared with pure gelatin, respectively (Fig. 3g) due to the existence of the multiple crosslinked first-network ascribed to the combined advantages of the Schiff base, coordination bonds and hydrogen bonds. Furthermore, the cyclic compression tests indicated that a hysteresis loop was shown in the compression tests, demonstrating the fast reformation of MMDI-mediated interactions upon unloading for the effective dissipation of energy and recovery of mechanical deformation. As a result, the GDIH showed remarkable extensibility, elasticity and good mechanical stability because of the MMDI strategy of the Schiff base reactions, hydrogen bonds, and coordination bonds, enabling various interesting applications of GDIH. The internal structure of GDIH10-0.06 after external force action was analyzed by FE-SEM. As shown in Fig. S12a and b (ESI†), the structure of GDIH10-0.06 after one cyclic stretching-recovering was similar to that of the original sample due to the outstanding recombination of the Schiff base, hydrogel bonds and coordination bonds of GDIH10-0.06. After breaking, obvious pores appeared in the internal structure of GDIH10-0.06 (Fig. S12c, ESI†). The GDIH10-0.06 showed the same phenomenon after the compression treatment (Fig. S12d–f, ESI†). It is worth noting that large membrane regions and large pores appeared in GDIH10-0.06 due to the high water absorption of the system.33
The impedance of GDIH was measured by using EIS, as sketched in Fig. 4a. Because of the existence of bulk resistance, the positive/negative charges of FeCl3 can move freely in the double network structure under the action of an external electric field.43 The ionic conductivity of the nanocomposite hydrogel increased from 0.31 × 10−2 to 2.27 × 10−2 S m−1 along with the content of Fe3+ increasing from 0.04 to 0.10 mol L−1. For instance, the GDIH10-0.06 hydrogel was utilized as a conductor to build a conductive circuit, and then the LED bulb in the circuit is lit (Fig. 4b). As shown in Video V1 (ESI†), the LED bulb showed different brightness under distinct pressures. Cyclic loading-unloading tensile tests and compressive tests of GDIH10-0.06 under 15–40% strain and 3–5% strain indicated the excellent sensitivity and stability to different strains (Fig. 4c and d). Because of the strain-induced resistance changes of GDIH10-0.06, it could be potentially applied as a resistivity-type sensor, and then the sensitivity and reliability of the GDIH10-0.06-based sensor were further explored. Fig. 4e shows that the increased relative resistance changes were proportional to the tensile strain, showing a linear increment with the strain sensitivity (GF) of 2.242 (R2 = 0.996) and 1.819 (R2 = 1.000) under 6% and 40%, respectively. The relative resistance changes were attributed to the ionic transport. When the hydrogel was stretched, the length of the conductive paths grew longer and the conductive network became loose. The ionic transport was limited resulting in the increase of the bulk resistance.8,44 The result showed that the calibration process could be simplified due to this good linear relationship, resulting in better signal accuracy. Simultaneously, this good linear relationship also enabled the GDIH-based sensor to avoid the interference of external factors, ensuring the reliability of detecting human motions and electrophysiological signals. Apart from the strain sensitivity, the GDIH-based sensor could also respond to pressure, and the pressure sensitivity (S) was defined as the slope of the compressive curves. The compressive curve in Fig. 4f was composed of four regions, and the S of GDIH10-0.06 in the range of 0–15 kPa is 1.14 kPa−1 (R2 = 0.982), which was far superior to those of other reported studies (Fig. 4g). In contrast to the tensile sensing behavior, the decrease of the resistance was attributed to the shortening of the ionic transport pathway during the compression process. The response time is also an important property to explore the sensing performance. As presented in Fig. 4h, the response and recovery times of GDIH10-0.06 were 400 and 200 ms, showing a rapid response to the recovery of subtle deformations. In addition, the stability of the nanocomposite hydrogel under a cyclic loading-unloading compressive test of up to 3% strain indicated that the strain sensing property was stable for over 150 cycles with almost no attenuation in the relative resistance changes (Fig. 4i). It was apparent that our nanocomposite hydrogel acting as a sensing material has a good repeatability. The above results showed that our GDIH with considerable conductivity, remarkable sensitivity and reliable stability was suitable for preparing strain sensors. On the other hand, our nanocomposite hydrogel also exhibited an outstanding adhesion property after adding Fe3+ (Video V2 and V3, ESI†), which created conditions for the application of the nanocomposite hydrogel in the field of multifunctional strain sensors.
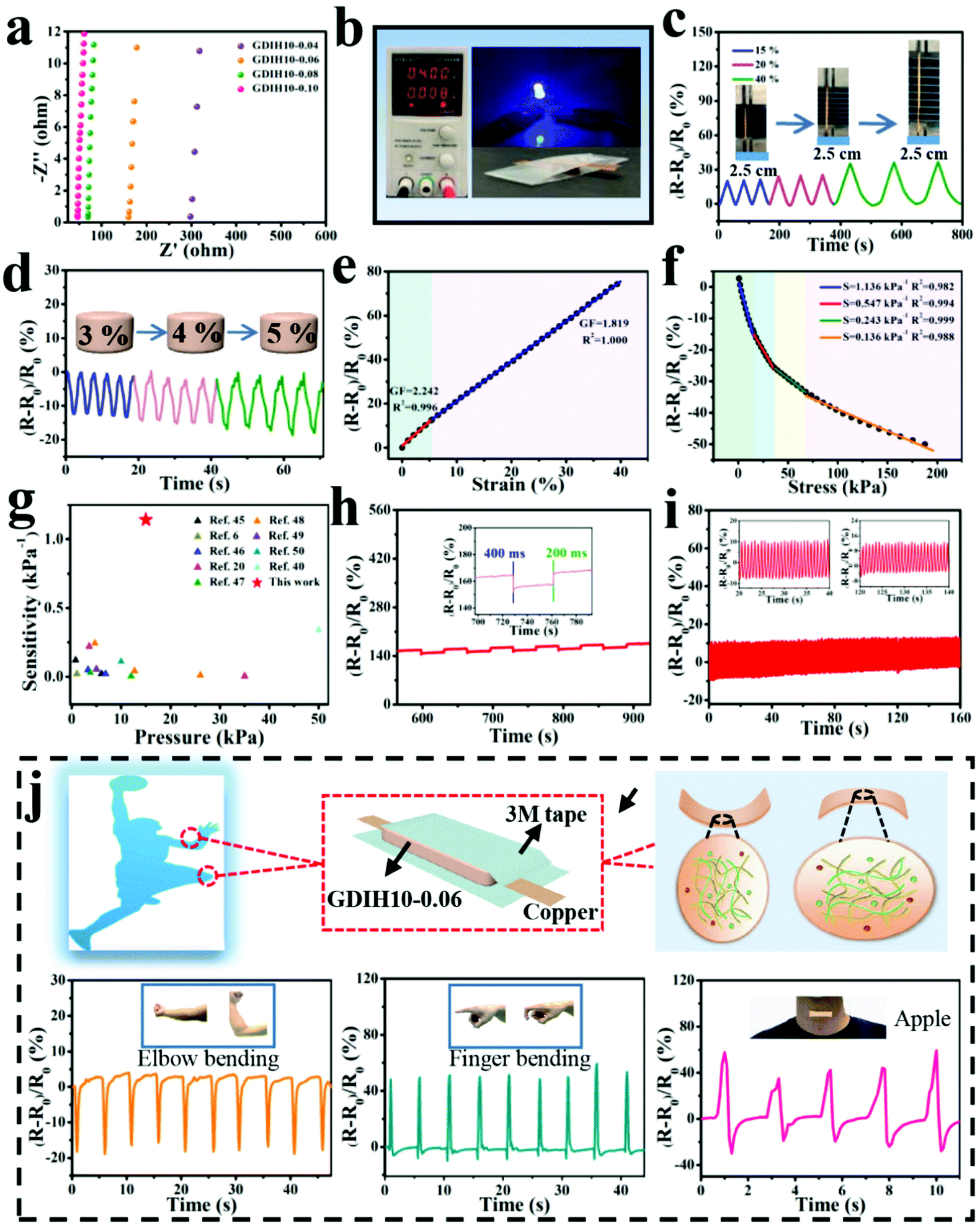 | ||
| Fig. 4 (a) EIS plots of GDIH. Sensitivity of GDIH10-0.06-based electronic devices: (b) Electric circuit constructed via the GDIH10-0.06. (c) Time-dependent relative resistance changes with different tensile strains. (d) Real-time relative resistance changes for different compressive stresses. (e) Relative resistance changes and GF with the increase of tensile strain. (f) Relative resistance change and pressure sensitivity at various compressive pressures. (g) Comparison of sensitivity for GDIH10-0.06 with reported hydrogel sensors.6,20,40,45–50 (h) Response time and recovery time of the GDIH10-0.06 at 3% compressive strain. The inset is particle enlarged drawing. (i) The stability of the resistance changes for GDIH10-0.06 under cyclic pressing-releasing tests over 150 cycles at 3% compressive strain. The insets are particle enlarged drawings. (j) Illustration of the assembly of GDIH10-0.06-based wearable sensors and the recorded relative resistance changes of the strain for elbow bending, forefinger bending and throat vibration, respectively. | ||
Inspired by the aforementioned advantages, GDIH-based sensors exhibited great prospects in green wearable sensors. As illustrated in Fig. 4j and Fig. S13 (ESI†), GDIH10-0.06 was assembled into a wearable strain sensor to detect complex body movements (elbow joint, forefinger, knee and throat) in real time. As expected, the GDIH10-0.06-based sensors displayed distinct characteristic signal peaks when the elbow joint was completely bent to 90°, and the relative resistance changes immediately restored to the original state when the elbow joint was straightened. It was obvious that the GDIH10-0.06-based sensor exhibited outstanding stability in the cyclic straightening-bending test. In addition, the changes in internal structure during the shrinkage and stretching led to a difference in the peak direction. The GDIH10-0.06-based sensor also showed an excellent sensitivity and stability during the cycling test of 90° knee-bending and natural walking (Fig. S13a and b, ESI†). Moreover, further to the discussed sensing performance of the GDIH-based sensor to detect subtle deformations, the detection of throat motion during speaking was carried out. Phonation recognition of the GDIH10-0.06-based sensor was accurately presented by the similar valleys and peaks in the process of speaking the word “Nihao” or “Apple” five times, respectively (Fig. 4j and Fig. S13, ESI†). Therefore, the remarkable sensing ability of monitoring micro and large deformations showed that the GDIH-based sensor could be regarded as a promising wearable strain sensor to detect human motion in the full range.
In addition, the assembled GDIH10-0.06-based sensor could be applied to detect handwriting by utilizing PET films as the encapsulation layers (Fig. 5a). As shown in Fig. 5b–e, separate letters (A, B, C, and D) showed obvious differences in the number of peaks. The signal of the combined letters (ABCD) could also be recognized, and the respective signals of A, B, C, and D in the combined letters were consistent with the signals of separate letters (Fig. 5f). The GDIH10-0.06-based sensor exhibited remarkable repeatable recognition of writing, indicating its outstanding ability to record handwriting as an electronic device. As listed in Fig. 5g–i, the words “ok”, “yes” and “paper” were handwritten by two different researchers. It was apparent that the relative resistance changes and peak shapes of the same word were different from each other. Due to the excellent ability of recording handwriting and distinguishing handwriting, our GDIH10-0.06-based sensor showed great potential in the field of writing anti-counterfeiting. Finally, a 4 × 4 hydrogel array was assembled to detect the 2D distribution of force and strain, which is widely used by electronic skin. Each hydrogel (5 × 5 × 4 mm3) acted as a sensor unit (Fig. 5j). As shown in Fig. 5k, remarkable relative resistance changes of this array were recorded using a computer when the B2, B3, C2 and C3 units were pressed by a weight of 50 g. The result showed that this strategy can be extended to fabricate highly integrated arrays of small gel pixels for the application of electronic skin, as reported by Fu et al. and Bao et al. who introduced advanced array sensors.51,52
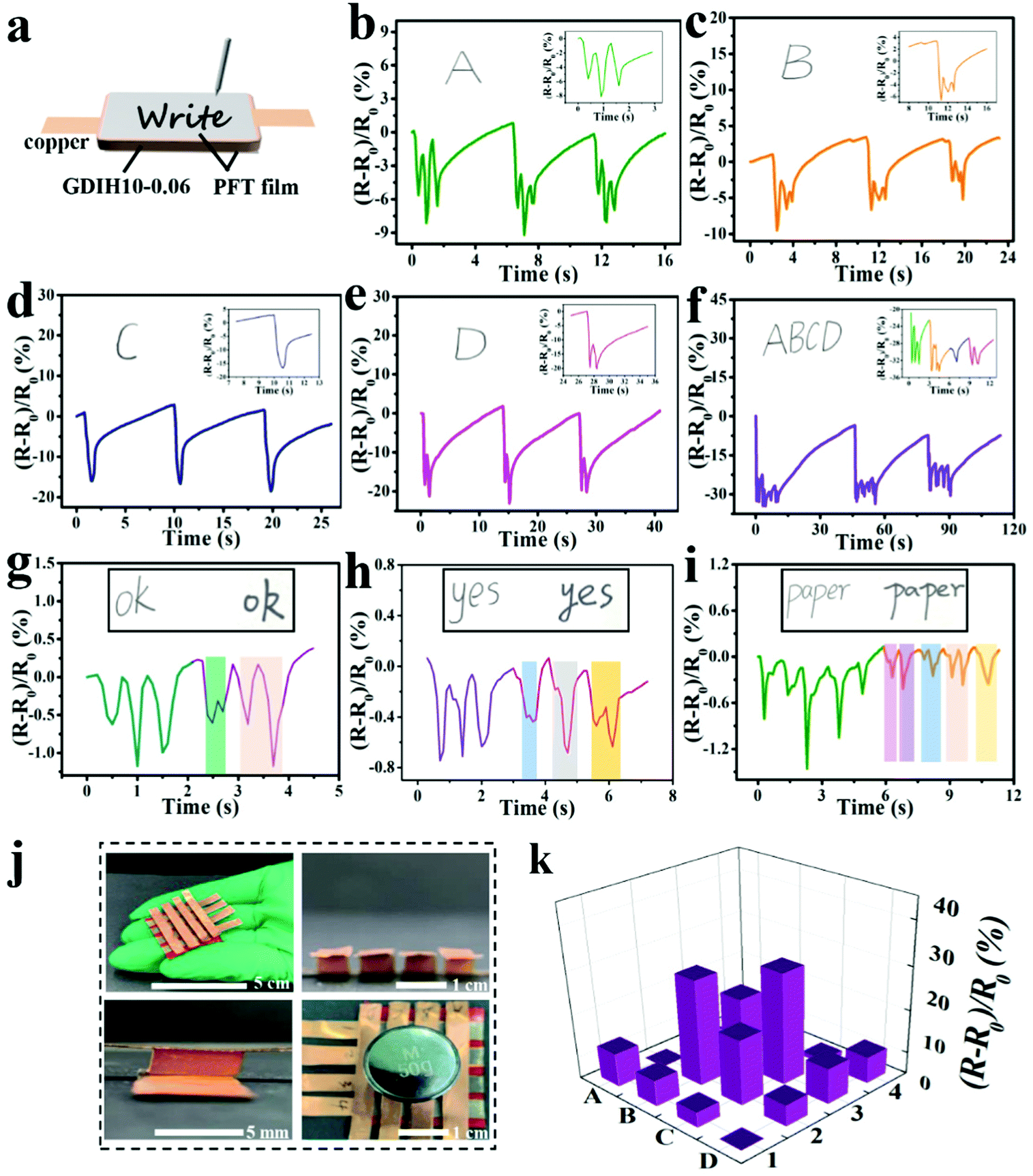 | ||
| Fig. 5 (a) Schematic diagram of signature sensing during writing by the GDIH10-0.06-based sensor. (b–f) Relative resistance changes of “A”, “B”, “C”, “D”, and “ABCD” written in the same handwriting. (g–i) Relative resistance changes of “ok”, “yes” and “paper” written in different handwritings. The insets are particle enlarged drawings. (j) A flexible 4 × 4 array of GDIH10-0.06 connected with copper tapes. During the test, the mass of the weight is 50 g. (k) Prism map of the array pressed on B2, B3, C2 and C3. | ||
Due to the dynamic interactions between the different kinds of surface-terminated groups (–OH, –COOH, and –CHO) of DATNFC, –NH2 of gelatin and Fe3+, GDIH had reliable self-healing capability, enabling the hydrogel sensor to resist external damage and extend its service life (Fig. 6a).53–56 As listed in Fig. 6b–g, the LED could light-up quickly when the broken sample contacted without any external assistance showing that the ionic transport was ensured by the self-healed dynamic network. Specifically, the response time of self-healing for the GDIH10-0.06-based sensor was 100 ms (Fig. 6h), which indicated that the sensor had a fast self-healing response.57 Additionally, the healing process was also carried out under 40 °C for 60 s followed by a standing process at room temperature for 10 min to obtain the self-healed sample, and then its mechanical and electrical properties were explored. The tensile stress and elastic modulus of the self-healed GDIH10-0.06-based sensor were nearly 60.5 and 227.7 kPa, respectively. This illustrated that nearly 36.7 and 45.3% of the original tensile strength and elastic modulus were restored by the self-healed GDIH10-0.06, respectively (Fig. 3b, 6i and Fig. S11, ESI†). The GF values of the self-healed hydrogel (2.455 at 5.49% and 2.477 at 30.00%) were higher than those of the original hydrogel (2.242 at 5.49% and 1.819 at 30.00%) (Fig. 4e and Fig. S14, ESI†). In addition, the real-time relative resistance changes of the GDIH10-0.06-based sensors demonstrated that the self-healed sensor displayed an outstanding response to different strains (5, 10 and 15%) (Fig. 6j), showing the remarkable sensitivity and stability of the self-healed sensor. Based on the above results, the self-healed sensor was applied in the field of wearable strain sensors and writing anti-counterfeiting materials, respectively (Fig. S15 and S16, ESI†).58 The result showed that our self-healed hydrogel could be re-utilized to prepare strain sensors.
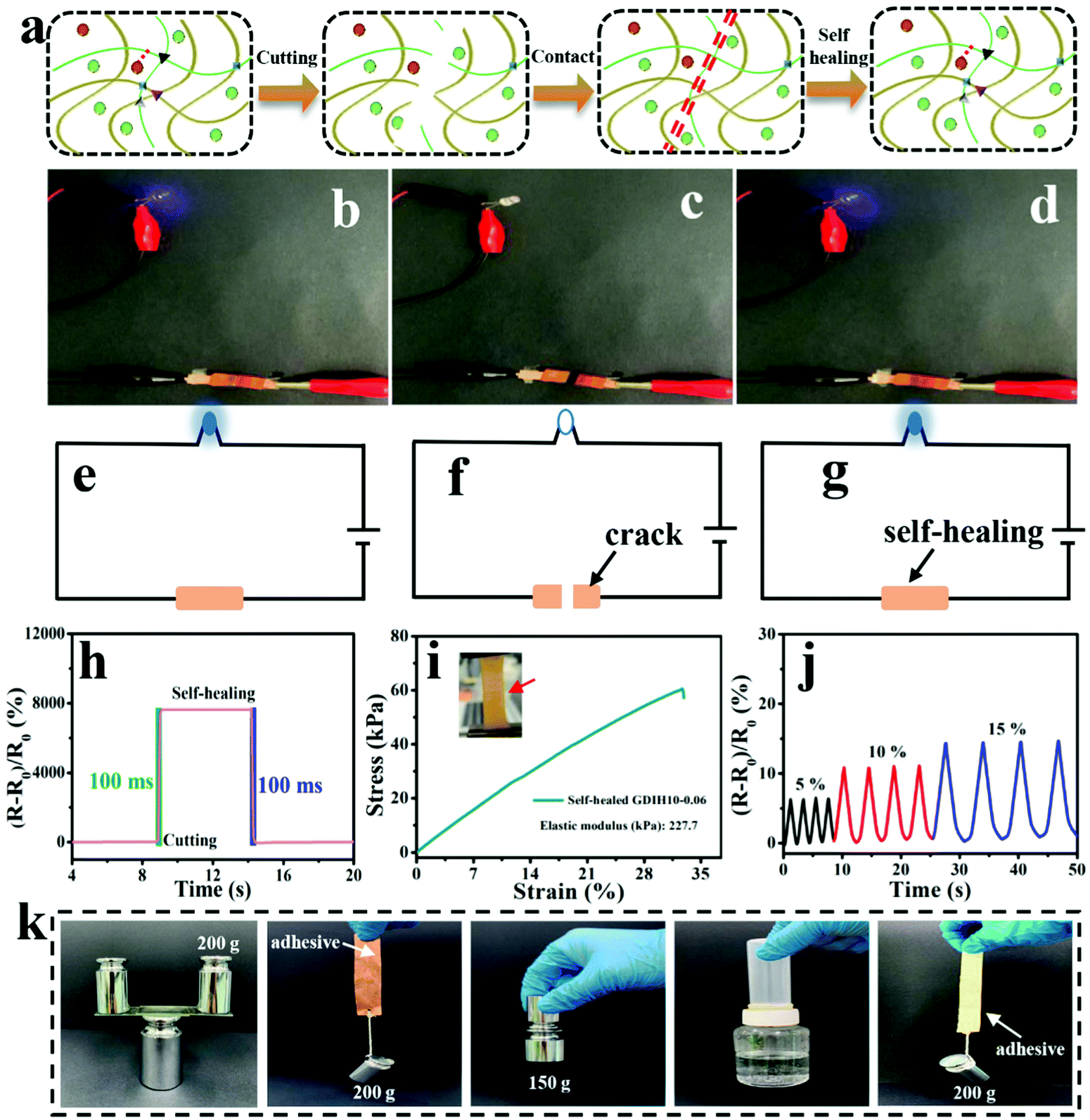 | ||
| Fig. 6 (a) Illustration of the cutting, contact and self-healing of the GDIH10-0.06-based sensor. (b–d) Comparisons of the luminance of LEDs (working voltage of 4 V) by using the GDIH10-0.06-based sensor. (e–g) The closed circuits of the LEDs. (h) The responses of cutting and self-healing of the GDIH10-0.06-based sensor. (i) Tensile strength of the self-healed GDIH10-0.06-based sensor. The inset is the optical photo of self-healed GDIH10-0.06 during tensile test. (j) Real-time relative resistance changes of the self-healed GDIH10-0.06-based sensor under different strains. (k) Digital images of the adhesion behavior of the recycled GDIH10-0.06 adhesives. | ||
Nowadays, a large amount of electronic waste with low degradability aggravates the environmental burden, resulting in increasing demand for degradable electronic devices. The development of recyclable electronic materials has attracted great attention for various electronic devices. Significantly, our GDIH10-0.06-based sensors could be recycled and reused. The abandoned hydrogel sensors could be easily dispersed in water to enhance their fluidity for 1 h at 45 °C under 800 rpm, and could be reused for preparing recycled G6DIH10-0.06, where “G6” represented the concentration of recycled gelatin (6.0 wt%). The “G” in GDIH10-0.06 represented the concentration of gelatin (10.0 wt%). The recycled sample could be obtained within 1 h, indicating that it is extremely simple and eco-friendly. As shown in Fig. S17 (ESI†), the tensile strength of the recycled G6DIH10-0.06 (24.8 kPa) was lower than that of the original G6DIH10-0.06. This might be caused by the decreasing interactions of the Schiff base, hydrogel bonds and coordinate bonds.8 Although the tensile strength showed a downward trend, it was still similar to the strength of some hydrogel sensors reported in previous studies.15,36,59,60 The ionic conductivity of the recycled G6DIH10-0.06 (0.11 × 10−3 S m−1) was similar to that of the original G6DIH10-0.06 (0.14 × 10−3 S m−1), showing that the ionic transport was ensured by the recycled network structure (Fig. S18a, ESI†). The GF of recycled G6DIH10-0.06 was decreased in the recycling process (Fig. S18b and c, ESI†), while its GF was higher compared with that of the original GDIH10-0.06. In addition, as listed in Fig. S18d (ESI†), the recycled G6DIH10-0.06 had an outstanding sensitivity and stability to different tensile strains. The recycled G6DIH10-0.06 was assembled for preparing strain sensors, and the sensing performance of the recycled G6DIH10-0.06-based sensors was further tested. As shown in Fig. S19 (ESI†), the relative resistance change of the recycled G6DIH10-0.06-based sensor was similar to that of the original G6DIH10-0.06-based sensor, and the recycled sensor also exhibited a sensitive and stable sensing signal. The results showed that our recycled G6DIH10-0.06 was still suitable for preparing strain sensors. The abandoned hydrogels could also be reused for preparing adhesives (Fig. S20, ESI†). The adhesive had an outstanding adhesion on both hydrophilic and hydrophobic surfaces including glass, copper, iron, plastic and paper due to the recombination of the polymer networks, exhibiting strong adhesion in both directions parallel to and perpendicular to the bonding surface (Fig. 6k). It was obvious that our adhesive exhibited an excellent adhesion performance compared with glue stick and glue (Fig. S21, ESI†). Therefore, the abandoned GDIH-based sensors have great prospects in the field of attractive material candidates for decreasing electronic waste.
Conclusions
In summary, a novel GDIH-based sensor was fabricated through a facile two-step process involving the sol–gel method followed by an ion penetration approach. Owing to the existence of a supramolecular-dynamic crosslinked DN structure formed by a Schiff base, hydrogen bonds and Fe3+-carboxyl coordinative bonds, the tensile and compressive strengths of the nanocomposite hydrogel were nearly 990.7 and 3822.7% higher than those of the pure gelatin hydrogel. Based on the MMDI strategy, the as-fabricated hydrogel-based sensor exhibited remarkable mechanical properties (compressive stress up to 1310 kPa), excellent sensitivity (strain sensitivity up to 2.24 under 6% strain, compressive sensitivity up to 1.14 kPa−1 under 15 kPa), outstanding sensing stability, considerable self-healing and full recyclability. This hydrogel-based sensor design verified the synergistic effects of DANFC and Fe3+ in improving the structural stability, providing a feasible scheme to solve the long-standing dilemma between mechanical strength and sensing. In addition, the composite hydrogel exhibited excellent performance of anti-aging and biocompatibility, indicating that there were infinite possibilities for the GDIH-based sensor in the field of detecting full-range human body movements, writing anti-counterfeiting and electronic skin with high sensing stability, sensitivity and durability, which were attributed to the multiple cross-linking structures. All these characteristics showed that the as-developed strain sensor is a good electronic device, for potential commercialization with mass production for broad medical applications in healthcare monitoring and the development of soft robotics, artificial throats and artificial intelligence devices.Author contributions
Haocheng Fu: conceptualization, investigation, and writing – original draft; Bin Wang, Jinpeng Li, and Jun Xu: conceptualization, writing – review and editing; Jun Li, Jinsong Zeng, and Wenhua Gao: formal analysis, drawing – original figures; Kefu Chen: writing – review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the support of the National Natural Science Foundation of China (22078113), the Natural Science Foundation of Guangdong Province (2019A1515010996, 2021A1515010899), China National Postdoctoral Program for Innovation Talents (BX20200134), China Postdoctoral Science Foundation (2021M701251), and Science and technology project of Guangzhou (202102080416, 202102020713).References
- M. Amjadi, A. Pichitpajongkit, S. Lee, S. Ryu and L. Park, ACS Nano, 2014, 8, 5154–5163 Search PubMed.
- M. Amjadi, K.-U. Kyung, I. Park and M. Sitti, Adv. Funct. Mater., 2016, 26, 1678–1698 Search PubMed.
- M. Amjadi, Y. J. Yoon and I. Park, Nanotechnology, 2015, 26, 375501 Search PubMed.
- D. Buenger, F. Topuz and J. Groll, Prog. Polym. Sci., 2012, 37, 1678–1719 Search PubMed.
- R. Cheng, J. S. Zeng, B. Wang, J. P. Li, Z. Cheng, J. Xu, W. H. Gao and K. F. Chen, Chem. Eng. J., 2021, 424, 130565 Search PubMed.
- X. H. Zhang, N. N. Sheng, L. N. Wang, Y. Q. Tan, C. Z. Liu, Y. Z. Xia, Z. H. Nie and K. Y. Sui, Mater. Horiz., 2019, 6, 326–333 Search PubMed.
- Z. W. Wang, Y. Cong and J. Fu, J. Mater. Chem. B, 2020, 8, 3437–3459 Search PubMed.
- C. Dang, F. Peng, H. C. Liu, X. Feng, Y. Liu, S. N. Hu and H. S. Qi, J. Mater. Chem. A, 2021, 9, 13115–13124 Search PubMed.
- H. Yi, S. H. Lee, H. Ko, D. Lee, W. G. Bae, T. I. Kim, D. S. Hwang and H. E. Jeong, Adv. Funct. Mater., 2019, 29, 1902720 Search PubMed.
- X. W. Xu, V. V. Jerca and R. Hoogenboom, Mater. Horiz., 2021, 8, 1173–1188 Search PubMed.
- A. Nakayama, A. Kakugo, J. P. Gong, Y. Osada, M. Takai, T. Erata and S. Kawano, Adv. Funct. Mater., 2004, 14, 1124–1128 Search PubMed.
- Z. Q. Cao, H. L. Liu and L. Jiang, Mater. Horiz., 2020, 7, 912–918 Search PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 Search PubMed.
- J. Y. Sun, X. H. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. G. Suo, Nature, 2012, 489, 133–136 Search PubMed.
- X. Liu, Z. J. Ren, F. F. Liu, L. Zhao, Q. J. Ling and H. B. Gu, ACS Appl. Mater. Interfaces, 2021, 13, 14612–14622 Search PubMed.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 Search PubMed.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 Search PubMed.
- G. Y. Wang, Q. Zhang, Q. Wang, L. B. Zhou and G. H. Gao, ACS Appl. Mater. Interfaces, 2021, 13, 24173–24182 Search PubMed.
- M. T. I. Mredha, N. Kitamura, T. Nonoyama, S. Wada, K. Goto, X. Zhang, T. Nakajima, T. Kurokawa, Y. Takagi, K. Yasuda and J. P. Gong, Biomaterials, 2017, 132, 85–95 Search PubMed.
- Y. H. Ye, Y. F. Zhang, Y. Chen, X. S. Han and F. Jiang, Adv. Funct. Mater., 2020, 30, 2003430 Search PubMed.
- S. Poveda-Reyes, L. R. Mellera-Oglialoro, R. Martínez-Haya, T. C. Gamboa-Martínez, J. L. G. Ribelles and G. G. Ferrer, Macromol. Mater. Eng., 2015, 300, 977–988 Search PubMed.
- L. R. Zhang, J. Liu, X. J. Zheng, L. Zhang, X. L. Zhang and K. Y. Tang, Carbohydr. Polym., 2019, 216, 45–53 Search PubMed.
- C. L. Liu, H. J. Zhang, X. Y. You, K. P. Cui and X. C. Wang, Adv. Electron. Mater., 2020, 6, 2000040 Search PubMed.
- Q. X. Liu, J. Liu, S. F. Qin, Y. Pei, X. J. Zheng and K. Y. Tang, Int. J. Biol. Macromol., 2020, 164, 1776–1784 Search PubMed.
- J. Babaei, M. Mohammadian and A. Madadlou, Food Chem., 2019, 270, 189–195 Search PubMed.
- N. Li, W. Chen, G. X. Chen and J. F. Tian, Carbohydr. Polym., 2017, 171, 77–84 Search PubMed.
- T. Z. Yuan, J. S. Zeng, B. Wang, Z. Cheng and K. F. Chen, Carbohydr. Polym., 2021, 254, 117441 Search PubMed.
- S. C. Patankar and S. Renneckar, Green Chem., 2017, 19, 4792–4797 Search PubMed.
- M. Cheng, Z. Y. Qin, Y. Y. Chen, J. M. Liu and Z. C. Ren, Cellulose, 2017, 24, 3243–3254 Search PubMed.
- P. F. Li, J. S. Zeng, B. Wang, Z. Cheng, J. Xu, W. H. Gao and K. F. Chen, Carbohydr. Polym., 2020, 247, 116721 Search PubMed.
- J. P. Li, L. Kang, B. Wang, K. F. Chen, X. J. Tian, Z. Ge, J. S. Zeng, J. Xu and W. H. Gao, ACS Sustainable Chem. Eng., 2018, 7, 1146–1158 Search PubMed.
- J. Yang, F. Xu and C. R. Han, Biomacromolecules, 2017, 18, 1019–1028 Search PubMed.
- S. Spoljaric, A. Salminen, N. D. Luong and J. Seppälä, Eur. Polym. J., 2014, 56, 105–117 Search PubMed.
- L. Guan, S. Yan, X. Liu, X. Y. Li and G. H. Gao, J. Mater. Chem. C, 2019, 7, 5230–5236 Search PubMed.
- J. Q. Han, Q. Q. Ding, C. T. Mei, Q. L. Wu, Y. Y. Yue and X. W. Xu, Electrochim. Acta., 2019, 318, 660–672 Search PubMed.
- X. Jing, H. Y. Mi, Y. J. Lin, E. Enriquez, X. F. Peng and L. S. Turng, ACS Appl. Mater. Interfaces, 2018, 10, 20897–20909 Search PubMed.
- J. Lv, C. C. Kong, C. Yang, L. Yin, I. Jeerapan, F. Z. Pu, X. J. Zhang, S. Yang and Z. M. Yang, Beilstein J. Nanotechnol., 2019, 10, 475–480 Search PubMed.
- R. P. Tong, G. X. Chen, D. H. Pan, J. F. Tian, H. S. Qi, R. A. Li, F. C. Lu and M. H. He, ACS Sustainable Chem. Eng., 2019, 7, 14256–14265 Search PubMed.
- H. Lee, A. Erwin, M. L. Buxton, M. Kim, A. V. Stryutsky, V. V. Shevchenko, A. P. Sokolov and V. V. Tsukruk, Adv. Funct. Mater., 2021, 31, 2103083 Search PubMed.
- X. L. Yang, L. Q. Cao, J. D. Wang and L. P. Chen, ACS Sustainable Chem. Eng., 2020, 8, 10726–10739 Search PubMed.
- R. Zhao, X. Y. Xu and L. Hu, ACS Appl. Polym. Mater., 2021, 3, 5155–5161 Search PubMed.
- C. J. Zhou and Q. L. Wu, Colloids Surf., B, 2011, 84, 155–162 Search PubMed.
- C. Dang, M. Wang, J. Yu, Y. Chen, S. H. Zhou, X. Feng, D. T. Liu and H. S. Qi, Adv. Funct. Mater., 2019, 29, 1902467 Search PubMed.
- M. Wang, R. A. Li, X. Feng, C. Dang, F. L. Dai, X. Q. Yin, M. H. He, D. T. Liu and H. S. Qi, ACS Appl. Mater. Interfaces, 2020, 12, 27545–27554 Search PubMed.
- X. M. Fu, J. X. Li, C. Q. Tang, S. L. Xie, X. M. Sun, B. J. Wang and H. S. Peng, Adv. Funct. Mater., 2020, 31, 2008355 Search PubMed.
- G. Ge, Y. Z. Zhang, J. J. Shao, W. J. Wang, W. L. Si, W. Huang and X. C. Dong, Adv. Funct. Mater., 2018, 28, 1802576 Search PubMed.
- Q. Chang, Y. F. He, Y. Q. Liu, W. Zhong, Q. Wang, F. Lu and M. Xing, Adv. Funct. Mater., 2020, 30, 1910080 Search PubMed.
- G. Ge, W. Yuan, W. Zhao, Y. Lu, Y. Z. Zhang, W. J. Wang, P. Chen, W. Huang, W. L. Si and X. C. Dong, J. Mater. Chem. A, 2019, 7, 5949–5956 Search PubMed.
- M. Wu, J. S. Chen, Y. H. Ma, B. Yan, M. F. Pan, Q. Y. Peng, W. D. Wang, L. B. Han, J. F. Liu and H. B. Zeng, J. Mater. Chem. A, 2020, 8, 24718–24733 Search PubMed.
- X. Hu, X. X. Xia, S. C. Huang and Z. G. Qian, Biomacromolecules, 2019, 20, 3283–3293 Search PubMed.
- Z. W. Wang, J. Chen, Y. Cong, H. Zhang, T. Xu, L. Nie and J. Fu, Chem. Mater., 2018, 30, 8062–8069 Search PubMed.
- S. H. Wang, J. Xu, W. C. Wang, G.-J. N. Wang, R. Rastak, F. Molina-Lopez, J. W. Chung, S. M. Niu, V. R. Feig, J. Lopez, T. Lei, S.-K. Kwon, Y. Kim, A. M. Foudeh, A. Ehrlich, A. Gasperini, Y. J. Yun, B. Murmann, J. B. H. Tok and Z. N. Bao, Nature, 2018, 555, 83–88 Search PubMed.
- R. Tutika, A. B. M. T. Haque and M. D. Bartlett, Commun. Mater., 2021, 2, 1–8 Search PubMed.
- Y. Cao, T. G. Morrissey, E. Acome, S. I. Allec, B. M. Wong, C. Keplinger and C. Wang, Adv. Mater., 2017, 29, 1605099 Search PubMed.
- L. Yin, H. Y. Cheng, S. M. Mao, R. Haasch, Y. H. Liu, X. Xie, S. W. Hwang, H. Jain, S. K. Kang, Y. W. Su, R. Li, Y. G. Huang and J. A. Rogers, Adv. Funct. Mater., 2014, 24, 645–658 Search PubMed.
- S. T. Lin, H. Yuk, T. Zhang, G. A. Parada, H. Koo, C. J. Yu and X. H. Zhao, Adv. Mater., 2016, 28, 4497–4505 Search PubMed.
- S. M. Zhang and F. Cicoira, Adv. Mater., 2017, 29, 1703098 Search PubMed.
- M. D. Bartlett, M. D. Dickey and C. Majidi, NPG Asia Mater., 2019, 11, 1–4 Search PubMed.
- Y. G. Yan, L. X. Wei, X. Y. Qiu, J. L. Shao, H. L. Liu, X. Cui, J. Huang, L. Xie, Z. F. Hu and C. Z. Huang, ACS Appl. Mater. Interfaces, 2021, 3, 1479–1487 Search PubMed.
- L. Zhao, Z. J. Ren, X. Liu, Q. J. Ling, Z. J. Li and H. B. Gu, ACS Appl. Mater. Interfaces, 2021, 13, 11344–11355 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2mh00028h |
| This journal is © The Royal Society of Chemistry 2022 |