
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePairing cells of different sizes in a microfluidic device for immunological synapse monitoring†‡
Faruk Azam
Shaik
 ab,
Clara
Lewuillon
ac,
Aurélie
Guillemette
ac,
Bahram
Ahmadian
bd,
Carine
Brinster
ac,
Bruno
Quesnel
ac,
Dominique
Collard
be,
Yasmine
Touil
*ac,
Loïc
Lemonnier
*fg and
Mehmet Cagatay
Tarhan
*bde
ab,
Clara
Lewuillon
ac,
Aurélie
Guillemette
ac,
Bahram
Ahmadian
bd,
Carine
Brinster
ac,
Bruno
Quesnel
ac,
Dominique
Collard
be,
Yasmine
Touil
*ac,
Loïc
Lemonnier
*fg and
Mehmet Cagatay
Tarhan
*bde
aUniversity of Lille, Lille, France
bCNRS, IIS, COL, Univ. Lille SMMiL-E project, Lille, France
cUniv. Lille, CNRS, Inserm, CHU Lille, UMR9020-U1277 – Canther – Cancer Heterogeneity, Plasticity and Resistance to Therapies, F-59000 Lille, France. E-mail: yasmine.touil@inserm.fr
dUniv. Lille, CNRS, Centrale Lille, Junia, Univ. Polytechnique Hauts-de-France, UMR 8520 -IEMN -Institut d'Electronique de Microélectronique et de Nanotechnologie, F-59000 Lille, France. E-mail: cagatay.tarhan@junia.com
eLIMMS/CNRS-IIS IRL2820, The University of Tokyo, Tokyo, Japan
fUniv. Lille, Inserm, U1003 - PHYCEL - Physiologie Cellulaire, F-59000 Lille, France. E-mail: loic.lemonnier@inserm.fr
gLaboratory of Excellence, Ion Channels Science and Therapeutics, Villeneuve d'Ascq, France
First published on 26th January 2022
Abstract
Analyzing cell–cell interaction is essential to investigate how immune cells function. Elegant designs have been demonstrated to study lymphocytes and their interaction partners. However, these devices have been targeting cells of similar dimensions. T lymphocytes are smaller, more deformable, and more sensitive to pressure than many cells. This work aims to fill the gap of a method for pairing cells with different dimensions. The developed method uses hydrodynamic flow focusing in the z-direction for on-site modulation of effective channel height to capture smaller cells as single cells. Due to immune cells' sensitivity to pressure, the proposed method provides a stable system without any change in flow conditions at the analysis area throughout experiments. Paired live cells have their activities analyzed with calcium imaging at the immunological synapse formed under a controlled environment. The method is demonstrated with primary human T lymphocytes, acute myeloid leukemia (AML) cell lines, and primary AML blasts.
Introduction
Cell–cell interaction is crucial in the development and function of multicellular organisms. Beyond maintaining tissue homeostasis, understanding how these interactions work provides insightful information on different pathological contexts such as cancers,1 genetic diseases,2 immunological responses,3 and bacterial infections.4 They play an essential role in cell behavior and affect how cells migrate, proliferate, and differentiate.5Regarding immunological responses, these interactions during the formation of immunological synapses (IS) between immune cells and their partners or target cells could dictate cell fate diversity and, therefore, regulation of immune functions.3,6 In cancer, innate and adaptive immune responses strongly cooperate in fighting against malignant cells. Among the adaptive immune system, cytotoxic T cells expressing cell-surface CD8 (cluster of differentiation 8) are the most potent effectors in the anticancer immune response and form the backbone of current successful cancer immunotherapies.7 Nonetheless, dysfunctional immune responses of the T lymphocytes could lead to cancer progression.8 Investigating the bi-directional cell–cell interactions between T lymphocytes and cancer cells would reveal underlying mechanisms of the (i) resistance of cancer cells and (ii) the dysfunctions of T lymphocyte activities against the malignant cells. Heterogeneity of both cancer cells and immune effectors and their interactions is a hallmark of malignant diseases. In hematological malignancies, as in acute myeloid leukemia (AML), the immune biology is even more complex to study because leukemia cells share some immunological characteristics of their normal hematopoietic progenitor counterpart, and interaction may occur in various environments like the bone marrow niche or circulating blood.9 Thus, AML is a perfect model to highlight the importance of IS studies. The fine and dynamic monitoring of events (e.g., calcium mobilization) during IS formation highlights key mechanisms ruling cell fate. IS formation will result in events starting from the initial contact between T lymphocytes and leukemic cells and extending over several hours. While these events include calcium responses in both cell types, monitoring these responses remains challenging, especially when dealing with primary cells obtained from patients. Indeed, these cells are difficult to obtain in great quantity without adding an extra burden on the patients. Moreover, the heterogeneity of the T lymphocytes and leukemic cells with resistive properties, depending on the stage of the disease, demands IS analysis at a single pair level to characterize IS signatures. Also, analyzing specific conditions requires certain subpopulations of T lymphocytes and leukemic cells, which uses a limited number of cells from patients for analysis, thus limiting the use of conventional observation methods such as flow cytometry.
Conventional methods such as bulk coculture systems10 and poly-L-lysine-coated coverslips11 show significant limitations. These systems have no control over forming cell pairs.12 Low efficiency in pairing is inevitable, and single-cell pairing is only possible by chance. Such systems can provide information at the population level but will miss crucial single-cell responses to monitor the heterogeneity among cells.13 Precise information on the nature of the contact, e.g., duration and cells forming the contact, is essential to analyze the cellular response. In addition, observing the response under different experimental conditions, e.g., absence or presence of specific ions or molecules, provides valuable information to characterize the interaction at an IS. It is clear that conventional methods are inadequate for a detailed analysis of the immune response between two cells in contact, and alternative techniques providing high control on cell pairing are required.
Microtechnologies, e.g., micro-wells14–16 and droplet-based systems,17–19 attempted to overcome some of the limitations the conventional methods. Although these approaches improved performance over the conventional ones, they still have significant limitations on the spatial and temporal control over the contact, making sensitive analysis of early events unfeasible. The resulting low-throughput format leads to low spatiotemporal resolution.20 Another impractical point is the lack of control over the environmental conditions, which restricts multi-step assays, e.g., staining, washing, and drug testing. On the other hand, microfluidic systems provide practical solutions to major limitations. Due to the better handling capabilities, physical traps define precise positions of cell pairs,21 and controlling the flow in a microfluidic channel brings pairs in contact within a given time interval. Paired cells are monitored in a controlled environment by exchanging the solution in the channel. Elegant designs exhibit high efficiency in forming pairs of single cells by (i) applying a bi-directional flow to fill surrogate capture sites prior to the pairing step22,23 or (ii) inducing a lower flow rate for cells positioned at the narrow entrance of a trap before captured inside with a higher flow rate.24 A dense trap geometry allows a massive number of pairs to be formed in parallel without compromising single-pair monitoring. However, despite all demonstrated functionalities, these techniques still exhibit a critical limitation on the dimensions of the cells to pair. When physical traps are used, efficient pairing performance is available only for cells with similar dimensions. As a critical part of IS, T lymphocytes are significantly smaller than many other cells, e.g., one-third of the size of stem cells.23 Cells with significant size differences can cause pairs formed with multiple cells. Developing the channel and trap design for the larger cell can capture smaller cells, e.g., T lymphocytes, in doublets or triplets due to the height of the channel and traps. As a result, an alternative method, which is practical and efficient enough to work with patient samples, is needed to pair single cells of different dimensions.
Here, we develop a method to pair non-adherent cells having different dimensions without compromising the benefits of microfluidics. We specially designed physical traps to bring different types of cells in contact. Trap positions provide the exact locations of pairs formed in a given time. Continuous monitoring allows real-time measurements of the early and late events in a controlled environment. Cell pairing of different sizes is based on on-site channel height modulation and a 3D trap design capturing smaller cells only at the channel bottom so that the higher part of the channel is designed for capturing larger cells. The on-site channel height modulation ensures that small cells stay at the bottom of the channel through hydrodynamic flow focusing in the z-direction. The method is suitable for analyzing patient samples due to the simple unidirectional flow protocol reducing required steps after sample insertion and minimizing possible cross-contamination. To validate our method, we used primary human T lymphocytes from healthy donors and formed IS with two cell types, KG1 and primary human blasts obtained from acute myeloid leukemia (AML) patients, while monitoring Ca2+ responses in T lymphocytes.
Experimental
Fabrication
The microfluidic device is a PDMS slab with embedded channels bonded on a glass coverslip. The PDMS slab is fabricated in two steps: fabrication of the mold by SU8 photolithography and PDMS molding on the fabricated structures.The mold had three SU8 layers deposited and patterned on a silicon wafer. The first layer having 1.5–2.5 μm of thickness was created to pattern supporting pillars. SU8 2002 was spin-coated at 3500 rpm for 30 s on a 3 inch wafer and exposed with a maskless lithography system (100 mJ cm−2 with a 375 nm laser, Heidelberg MLA-150) after a soft bake of 2 minutes at 95 °C. The wafer was then post-baked for 2 minutes at 95 °C. The second layer, SU8 2005, was spun at 600 rpm for 30 s to have 4–6 μm of height for capturing small cells. After a soft bake of 1 minute at 65 °C followed by 2 minutes at 95 °C, the photoresist was patterned (120 mJ cm−2). The exposed wafer was baked for 1 minute at 65 °C followed by 2 minutes at 95 °C. The last layer was 12–14 μm of height. SU8 3010 photoresist was spun at 3000 rpm for 30 s, baked for 1 minute at 65 °C followed by 10 minutes at 95 °C, and exposed to UV light of 220 mJ cm−2. Following the third layer exposure, the wafer was post-baked at 1 minute at 65 °C followed by 5 minutes at 95 °C, developed in a SU8 developer solution and hard-baked at 150 °C for 15 minutes. As the last step, a thin layer of Teflon was deposited with an RIE machine (100 W, C4F8, 30 mTorr, 30 s, Oxford PlasmaPro80) for easy molding of the PDMS slab.
PDMS, having a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio for base elastomer:curing agent, was poured on the silicon wafer with SU8 structures after adequate mixing and degassing. The thickness of the PDMS slab was around 1 mm to keep the inlet reservoirs at a reasonable volume for rapid injection of the cell suspensions and other required solutions. The device was cured at 70 °C for 5 hours. The solidified PDMS was then peeled off, and individual devices were cut into proper dimensions. The inlets and the outlet were created using biopsy punchers (1.5 mm diameter for primary inlets and 0.5 mm diameter for the auxiliary inlet and the outlet to connect to pumps). The bottom surface of the PDMS slab was activated with a plasma cleaner (Harrick, Hi-level, 5 minutes), bonded to a glass coverslip (0.17 mm thickness), and baked at 90 °C for 30 minutes. The top surface of the device was covered with tape during the plasma exposure to protect the hydrophobic surface properties.
:1 ratio for base elastomer:curing agent, was poured on the silicon wafer with SU8 structures after adequate mixing and degassing. The thickness of the PDMS slab was around 1 mm to keep the inlet reservoirs at a reasonable volume for rapid injection of the cell suspensions and other required solutions. The device was cured at 70 °C for 5 hours. The solidified PDMS was then peeled off, and individual devices were cut into proper dimensions. The inlets and the outlet were created using biopsy punchers (1.5 mm diameter for primary inlets and 0.5 mm diameter for the auxiliary inlet and the outlet to connect to pumps). The bottom surface of the PDMS slab was activated with a plasma cleaner (Harrick, Hi-level, 5 minutes), bonded to a glass coverslip (0.17 mm thickness), and baked at 90 °C for 30 minutes. The top surface of the device was covered with tape during the plasma exposure to protect the hydrophobic surface properties.
Setup and protocol
Experiments were performed on an inverted microscope stage (Nikon Eclipse Ti2-E) in a controlled environment (i.e., 37 °C). A confocal microscope (ZEISS LSM 880) was used for monitoring side views of cell pairs. The microfluidic device was connected to a pressure pump (Fluigent, LineUp™ Push–Pull) with two control units. One of the controllers was connected to the outlet and the other to the auxiliary inlet. Each controller had a dedicated flow sensor (Fluigent, FLU-M-D), allowing precise control of the flow rates of the injected and withdrawn liquid.Prior to experiments, the microfluidic device surface was treated to avoid non-specific attachments by injecting 50 mg ml−1 pluronic (F-127) solution from the outlet using a syringe pump (Kd Scientific) with an initial flow rate of 25 μl min−1 for 3 min, followed by 5 μl min−1 for 7 min. Then, the device was washed with deionized (DI) water. Water was inserted via the primary inlets while the outlet pump was working in the withdraw mode at 25 μl min−1 for 5 min. Finally, the device channels were filled with culture medium injected via primary inlets while being withdrawn at 25 μl min−1 for 5 min. The washing step with water was essential to prevent aggregate/crystal formation when pluronic solution and media were mixed. While performing this surface treatment step, we used a surrogate PDMS layer (thin layer with 1.2 mm diameter openings) aligned with the primary inlets to keep the hydrophobic surface properties of the device's top surface. This step was crucial for efficient sample solution injection at the inlets during the experiments by preventing spread on the surface. After the surface treatment step, the device was placed on the microscope stage, proper microfluidic connections were made, and injection inlets were sealed with separate thin PDMS pieces.
The first step of the experiments was to capture cell type 1 (smaller cells), i.e., T lymphocytes. After removing the seal over primary inlet 1, 5 μl T lymphocyte suspension was dropped at the inlet 1 while the outlet flow was set to −2 μl min−1 and the auxiliary inlet flow to 1 μl min−1. Depending on the patient sample conditions, the flow rates were decreased to −1 μl min−1 and 0.5 μl min−1, respectively when cells were smaller or softer. The auxiliary inlet pushed T lymphocytes down to the channel bottom and provided single-cell capturing at the specifically designed trap layer, i.e., layer 1. After 5 minutes of flow, we increased the auxiliary inlet flow to 2.5 μl min−1 to prevent cell arrival at the trapping area and clean the channel without changing the flow conditions on the trapped cells. Then, the seal of primary inlet 2 was removed, and the primary inlet 1 was sealed. Similar to the first step, 5 μl of cell type 2 (KG1 cell lines) were dropped on inlet 2, and the auxiliary inlet was turned OFF due to the larger size of these cells. As the row and column spacing were designed according to cell type 2, these cells showed higher efficiency in capturing. Over 80% of the cells were paired after 3 minutes of flow. For long monitoring sessions, the auxiliary inlet can be turned ON (2.5 μl min−1) to seal the trapping area, or the flow can continue in the case of patient samples as such cases might not have the expected concentration levels. The trapping area was monitored with Ca2+ imaging protocol using the Fura 2 ratiometric calcium dye throughout the experiment. If post-pairing actions were required, the seal of primary inlet 3 was removed, and primary inlet 2 was sealed while the auxiliary flow was at 2.5 μl min−1. Paired cells could be exposed to any drugs or other solutions injected via primary inlet 3 after stopping the auxiliary flow. A demonstration was performed to stain the cytoskeleton after pairing.
Finite element fluidic modeling
The finite element method (FEM) was used to model and optimize the device. The microchannel geometry was drawn on COMSOL Multiphysics v.5.5 platform. The geometry used for the simulations was identical to the actual device. The user-defined proper meshing was optimized to reduce the simulation error and minimize the simulation time. Coarse or extremely coarse meshing introduces error in the results, while extremely fine meshing takes long simulation time and memory. The geometry-dependent selective meshing (position dependent) helps to improve the result in a time-efficient manner. The fluid velocity fields were simulated using the Navier–Stokes equation for an incompressible Newtonian fluid,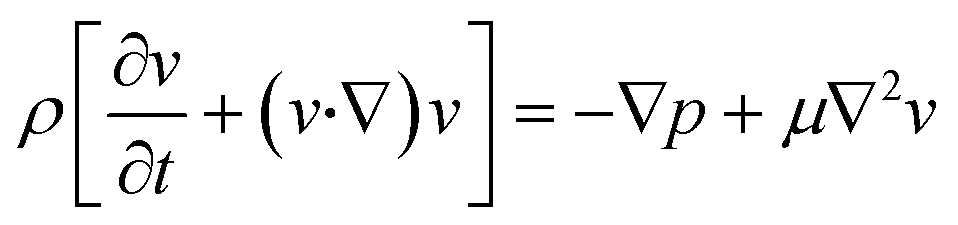 | (1) |
| ∇·v = 0 | (2) |
Channel height modulation was verified using the transport of the dilute species module and the Laminar flow module for the fluid velocity profile simulation. Primary inlet and auxiliary inlet solutions were simulated as fluids containing different concentrations. The parametric sweep study condition was chosen for auxiliary and outlet to alter the flow condition, thus different flow ratio (r) as in Table 1.
| Auxiliary flow | Outlet flow | Flow ratio (r) |
|---|---|---|
| (μl min−1) | (μl min−1) | (Auxiliary:outlet) |
| 0 | 2 | 0 |
| 0.5 | 2 | 0.25 |
| 1 | 2 | 0.5 |
| 1.5 | 2 | 0.75 |
| 2 | 2 | 1 |
| 2.5 | 2 | 1.25 |
The species concentration was monitored inside the microfluidic channel using convection–diffusion equation,
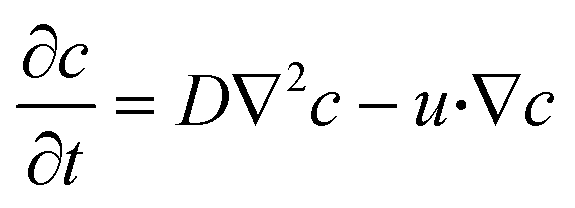 | (3) |
Cell culture and preparations
KG1 acute myeloid leukemia cell line was purchased from ATCC® (CCL-246™). This cell line was cultured at 37 °C in a humidified atmosphere with 5% CO2 in RPMI 1640 medium (Gibco) supplemented with 1% penicillin streptomycin antibiotic cocktail (Gibco) and 10% of fetal bovine serum (FBS) (Gibco). Healthy T lymphocytes and AML blasts were obtained from peripheral blood samples prepared by the French National Blood transfusion service (France) and Lille Hospital, respectively. PBMC were separated by Ficoll–Hypaque density gradient centrifugation, and CD8+ T cells were negatively selected using the EasySep Human CD8+ T Cell Isolation Kit and magnetic separation columns (Stem Cell Technology). AML blasts were isolated from peripheral blood after Ficoll separation and after lymphocyte depletion using immunomagnetic negative selection (CD3 for T-, and CD20 for B-lymphocytes). For microfluidic assays, samples were treated with DNase (50 U ml−1) to prevent long DNA segments from sticking in the device. Before injecting into the device, the cells were filtered through nylon mesh with 30 μm pore size (MACS® SmartStrainers) to eliminate any remaining aggregates. Cells were stained with membrane dyes DiI and DiO (Life Technologies) for pairing efficiency experiments following the manufacturer's instructions. The local ethics committee approved all studies, and all healthy and leukemia suffering patients signed an informed consent (EFS, CHRU de Lille, Tumorothèque du C2RC, approval numbers CSTMT079 and PLER/2021/005).Cell stimulation protocols
Ionomycin stimulation was performed by perfusing ionomycin (Invivogen) at 10 μM final concentration. Antibody stimulations were performed by pairing CD8+ T cells with anti-CD3/CD28-coated microbeads (Thermofischer Scientific). For SOC channels activity experiments, thapsigargin (focus biomolecules) was added at 2 μM final concentration, and then a 2 mM Ca2+ solution was subsequently injected 6 minutes later. Calcium channel inhibitors Synta66 (Sigma Aldrich) and BTP2 (Abcam) were added at a final concentration of 10 μM to assess the implication of SOC calcium channels in the thapsigargin-induced calcium entry.For the cell pairing experiments, first, allogenic healthy CD8+ T lymphocytes were activated by anti-CD3/CD28 beads at a 1:1 ratio for 3 days and cultured 7 days with 30 U ml−1 of human recombinant IL-2. Activated allogenic healthy CD8+ T lymphocytes were subsequently pulsed with or without 1 μg ml−1 of a cocktail of superantigens (SEA and Tsst-1; Sigma-Aldrich) for 30 min at 37 °C and AML blast cell was stained with membrane dye DiI (Thermo Fischer Scientific) before cell–cell pairing experiments.
Calcium imaging
Ratiometric dye Fura-2/AM (Interchim) was used as a Ca2+ indicator. Cells were loaded in serum-free RPMI 1640 (phenol red-free; Gibco) at 37 °C and 5% CO2 for 45 min. Cells were washed in RPMI 1640 by centrifugation and then resuspended in an extracellular solution containing (in mM): 140 NaCl, 5KCl, 1 MgCl2, 2 CaCl2, 10 glucose, 10 Hepes (pH 7.4 adjusted with NaOH). A similar solution devoid of calcium (0 Ca2+) was used during the imaging experiments, as indicated in the text and figures. All experiments were conducted at 37 °C in microfluidic devices installed on a microscope stage (Nikon Eclipse Ti2-E). Every 10 seconds, fluorescence was alternatively excited at 340 and 380 nm with an Optoscan monochromator (Roper Scientific) and captured at 510 nm by a CMOS camera (photometrics). Acquisition and analysis were performed with the MetaFluor software (Molecular Devices Corp.).On-chip actin staining and imaging
Actin filament staining was performed on the chip by adapting the manufacturer's protocols. After washing with PBS, cell pairs were fixed in 10% neutral buffered formalin for 10 min, rewashed with PBS, and then permeabilized with 0.1% Triton X-100 for an additional 10 min. Cells were rewashed with PBS before adding phalloidin-488 dye solution for actin filaments staining. Cells were imaged at corresponding wavelengths with an inverted fluorescence microscope.Results and discussion
Device design and optimization
Microfluidic systems provide practical solutions to investigate early signaling events of heterogeneous cell–cell interaction of single-cell pairs. These solutions allow analysis in a controlled environment with remarkable spatial and temporal controls. The critical requirement for such practical solutions is having complete control over the cell handling process. Cells must be positioned at predefined locations, and pairs must be formed within a limited time. These steps must be observed in real-time to measure cellular activity, e.g., monitoring intracellular Ca2+ concentrations using ratiometric dyes with fluorescence microscopy. Therefore, the proposed design uses specially designed physical traps on a glass coverslip to pair cells while monitoring fluorescence throughout an experiment.The microfluidic device is a fabricated PDMS slab placed on a coverslip. The device has three parts (Fig. 1a): an inlet area, an outlet area, and a trapping area. Inlet and outlet areas are used for cell handling by injecting a cell suspension in the channel and controlling the flow. The trapping area is crucial because both cell pairing and the activity measurements are performed in this area.
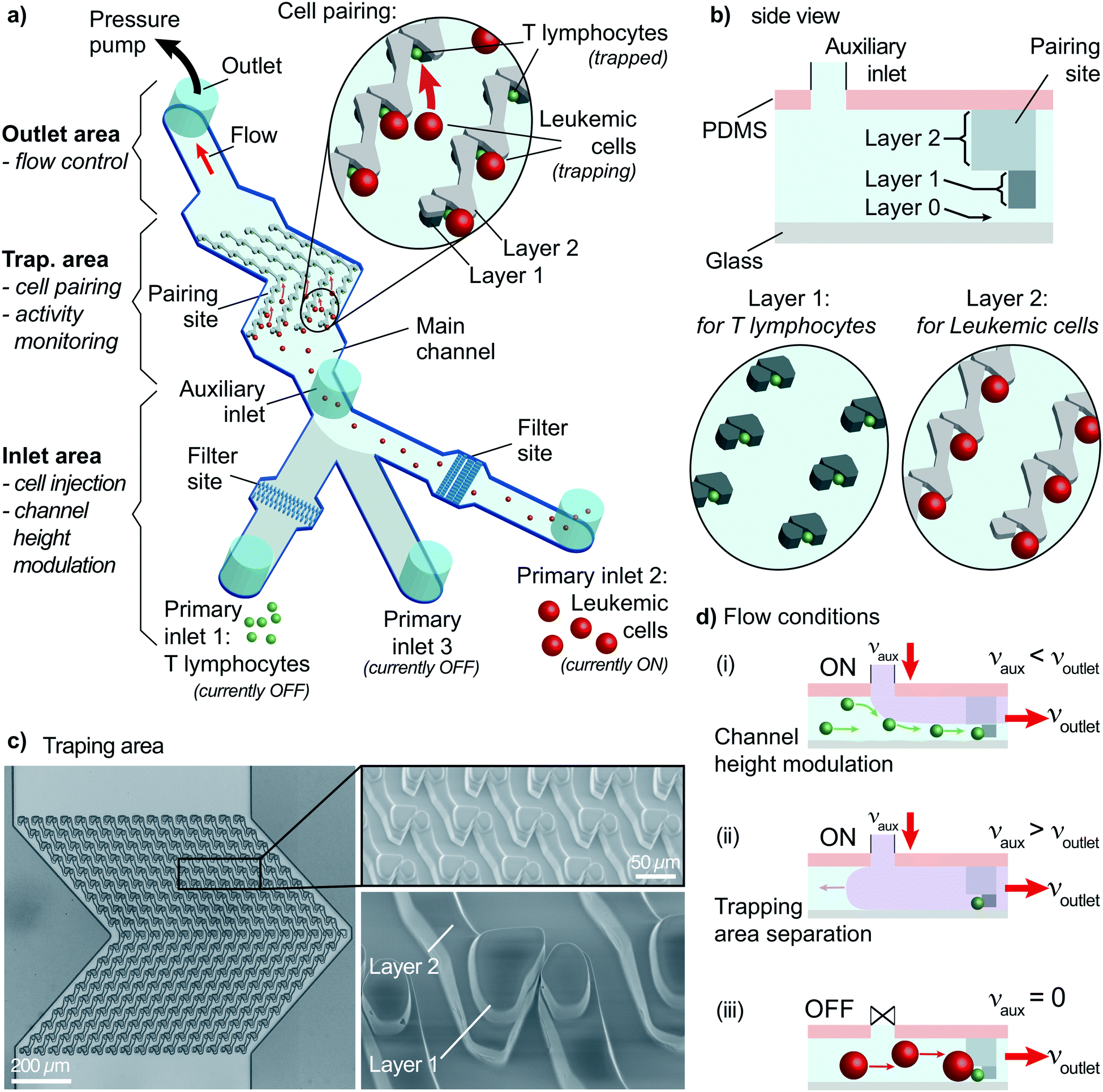 | ||
| Fig. 1 Overall view of the proposed method. a) The device has three areas: inlet area for cells/drugs injection and channel height modulation, trapping area for cell pairing and activity monitoring, and outlet area for flow control. A multi-layer PDMS device captures T lymphocytes (layer 1) and leukemic cells (layer 2) at the pairing sites. First, T lymphocytes are injected via primary inlet 1 and captured using auxiliary and outlet flows at the trap site. Then, primary inlet 2 is turned ON (others OFF), allowing layer 2 walls to guide leukemic cells to form pairs as demonstrated schematically. b) A side view of the device schematics demonstrates three layers: layer 0 for helping T lymphocytes capture at layer 1, and layer 2 walls directing leukemic cells to a pairing site. c) Brightfield image (left) and SEM images (right) show the trapping area and a close-up view of a trap with two layers on the PDMS device. d) The auxiliary inlet provides three flow conditions: (i) for channel height modulation, auxiliary flow is lower than the outlet flow allowing single-cell capturing of smaller cells. (ii) For separating the trapping area from the inlet area, auxiliary flow is set to a value higher than the outlet flow. This condition is used when changing the inlet in use. (iii) For allowing larger-cell capturing, the auxiliary flow is set to zero. The outlet flow, and thus, the flow condition in the trapping area, is set to a constant value throughout the experiments to minimize the mechanical stimulation of cells. | ||
The inlet area has three primary inlets for cells or drugs injection and an auxiliary inlet for on-site channel height modulation. Two primary inlets are assigned to the two types of cells to pair. The remaining primary inlet is required when post-pairing actions are taken, e.g., setting specific environmental conditions, staining, and drug insertion. These inlets are not connected to any pumps and allow direct injection of solutions into the channel to improve the practicality and provide rapid responses. The inlets are sealed to prevent cross-contamination and opened only during sample insertion (Fig. S1†). The auxiliary inlet is connected to a pressure pump with precise control of the induced flow. The sub-channels of primary inlets merge into the main channel before reaching the auxiliary inlet. An essential element of the inlet area is the filter site. Each cell-injection primary inlet has a cell-size specific filter area before the main channel (Fig. 1a and S2†). This filter prevents cell or debris aggregates from reaching the trapping area and clogging the channel.
The outlet area connects the main channel to a pressure pump to control the flow. The pump is first used to fill the channel prior to the experiments. After surface treatment, the channel is filled with a buffer solution (or media) until all air bubbles are removed from the channel using the “injection” mode (positive pressure). The second use of the pump is to control the flow in the “withdrawn” mode (negative pressure) to provide a unidirectional flow. Flow sensors connected to pumps provide precise monitoring and control of the constant unidirectional flow applied throughout the experiment.
The trapping area is the most critical part of the device, where cells are paired and monitored for analysis. An array of 3D traps is fabricated in the PDMS structure with dedicated layers for each cell type. There exist three layers (Fig. 1b). The first one (layer 0) at the bottom has a 2 μm height without any structures except for several columns maintaining structural integrity. The next layer (layer 1) captures cell type 1 (smaller cells) during the unidirectional flow from the primary inlet 1 to the outlet. Layer 1 structure starts from 2 μm until 8 μm above the bottom surface for capturing T lymphocytes. Traps are formed of two parts on each side of a narrow opening (Fig. 1b and c) to provide a flow path for cell capture. A second cell following the first one is forced to take another flow path as the narrow opening was blocked (demonstrated with velocity profile simulations; Fig. S3†). The channel height modulation is critical for cell type 1 (smaller cells) to be kept at the bottom surface for avoiding multiple cells captured on top of each other because the height of the trap can accommodate multiple cells (Fig. 1d). The last layer (layer 2) is designed as a wall from 8 μm height to 20 μm height to affect only cell type 2 (larger cells) (Fig. 1d). The wall is designed to have an angle (45°) with the initial flow direction to direct larger cells specifically to the pairing positions (Fig. 1a and S4†). The angle is reversed for the later part of the assay to improve the capture rate, which results in a >shaped array (Fig. S4†). Due to its small size, cell type 1 is not affected by layer 2 and can move under the wall structures, while the auxiliary flow is used for height modulation. The formed pair has a contact plane perpendicular to the visualization plane, allowing fluorescence imaging of each cell.
The trapping array is formed with specific spacing between each row and column. The array must be designed according to the size of larger cells, i.e., cell type 2, to prevent clogging. As a trade-off, the capturing efficiency of the smaller cells, i.e., cell type 1, decreases due to trap-site density being scarce for the size of cell type 1. Experimental evaluation showed that an optimum row spacing corresponds to ∼2 times and the column spacing to ∼1.4 times the cell diameter (cell type 2) (Fig. S5†). In the demonstrations, we used primary human CD8+ T lymphocytes (5–8 μm in diameter) and KG1 AML cell line (12–18 μm in diameter). Thus, the row and column spacings were chosen as 30 μm and 20 μm, respectively.
On-site channel height modulation
On-site channel height modulation is essential for providing pairs of single cells from each cell type. As the device must be designed with a height suitable for the larger cells (cell type 2) to avoid clogging, active on-site control is needed for adjusting the channel height for smaller cells (cell type 1) to sustain single-cell pairing without compromising practicality. We integrate the auxiliary-inlet 3D hydrodynamic focusing flow25 with the outlet cell handling flow when channel height is modulated. Positioned between primary inlets and the trapping area, the flow introduced at the auxiliary inlet forms a laminar flow pushing the injected type 1 cells (via inlet 1) to the bottom surface for trapping at the capture sites (Fig. 1d-i). This effective channel height can be modulated by collective control of the auxiliary inlet and outlet flow (Fig. 2a and S6†).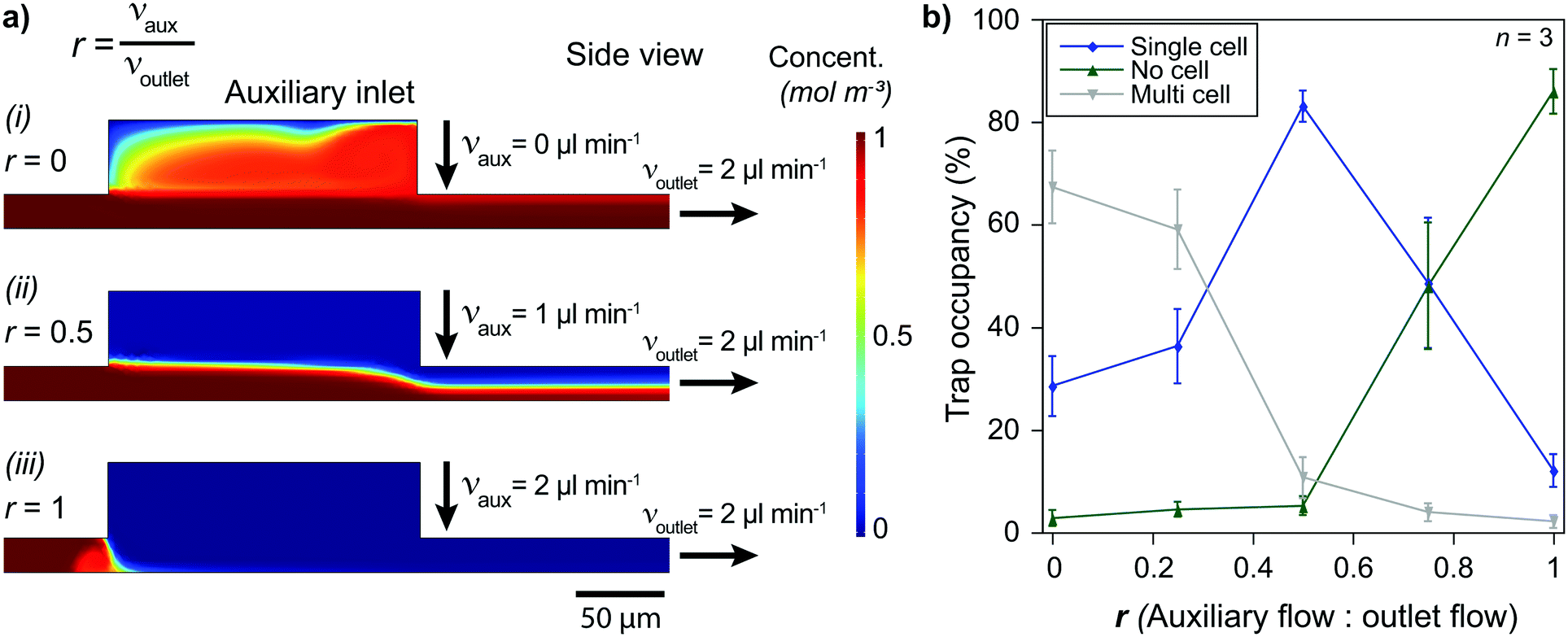 | ||
| Fig. 2 Single-cell capturing requires optimized flow conditions. a) Simulations show that solutions injected at the primary inlets can (i) cover all the trapping area (r = 0), (ii) flow only at a limited height above the surface (0 < r < 1), or (iii) be blocked at the auxiliary inlet (r = 1) by selecting auxiliary and outlet flows appropriately. b) Low auxiliary flow condition (r = ∼0) results in multiple T lymphocytes being captured in a single trap. On the other hand, high auxiliary flow (r = ∼1) prevents cells from moving to the trapping area and causes the vast majority of the trap sites to stay unoccupied. The auxiliary flow being around half of the outlet flow (r = ∼0.5) is optimum, resulting in >80% of the trap sites being filled with single T lymphocytes. | ||
The relation between the auxiliary inlet flow and the outlet flow plays a critical role in single-cell capturing for cell type 1 (smaller cells). We demonstrated the effect of on-site channel height modulation with primary human CD8+ T lymphocytes from healthy donors. When we did not use any auxiliary flow, only ∼30% of the trap sites were occupied with single T lymphocytes. The vast majority of the traps (∼70%) had multiple cells because the channel was too high for these small cells to capture as single cells. While keeping the outlet flow constant, increasing the auxiliary flow rate improved the single-cell capturing percentage. The auxiliary flow being half of the outlet flow (flow ratio, r = 0.5) provided the optimum condition with a single cell trapping rate being >80% and a multiple-cell trapping rate being ∼10%. Further increase in the auxiliary flow decreased the effective channel height preventing T lymphocytes from reaching the trapping area, resulting in fewer single-cells captured and increased empty traps (reaching ∼90% for r = 1). Therefore, for the given channel configuration, T lymphocyte experiments used half of the (withdrawn) outlet flow as the (injected) auxiliary flow (Fig. 2b).
Besides the single-cell capturing capability, the auxiliary flow could practically separate the trapping area from the inlet area. This separation helped stability when switching from using one primary inlet to another. Among three primary inlets, we used only one of them at once to provide practicality and prevent cross-contamination between inlet channels. Therefore, we sealed injection inlets with PDMS pieces when they were not used. Physically opening and sealing the primary inlets caused pressure fluctuations which could remove the captured cells from the traps. The auxiliary flow prevented any pressure fluctuation at the inlet area affecting the trapping area and separated them by making effective channel height zero (auxiliary inlet flow ≥ outlet flow, i.e., r ≥ 1, Fig. 1d-ii and S6†). As the outlet flow, and thus, the flow at the trapping area did not change, the captured cell condition stayed constant throughout the experiment.
Single-cell capturing
Cell pairing was performed between two cell types which required independent single-cell capturing of two different cell types at specific positions. Small type 1 cells were captured using the outlet and auxiliary flows, while type 2 cells were captured only with the outlet flow (Fig. 1d).Single-cell capturing of the smaller cells was demonstrated with primary human CD8+ T lymphocytes obtained from healthy donors. T lymphocytes with a concentration of 150000 cells ml−1 were injected into a fabricated device. The effect of the channel height modulation on single-cell capturing was investigated by comparing different pump configurations. An outlet flow rate of 2 μl min−1 was applied on the microfluidic device using a syringe pump (Kd Scientific) and a pressure pump (Fluigent, LineUp™ Push–Pull). These conditions were compared with a combined auxiliary and outlet flow case with 1 μl min−1 auxiliary and 2 μl min−1 outlet flow rates (flow ratio, r = 0.5 for auxiliary:outlet flows to stay in the optimum range). The results showed a clear improvement over the no auxiliary conditions after a 5 minute flow (Fig. 3a). Both syringe and pressure pumps showed similar results: only ∼25% of the traps were occupied with single cells, and double, triple, and quadruple occupations were all >20%. The auxiliary flow use improved the single-cell occupancy rate above 80%. Multiple-cell occupancy rates were <10%, where most of the multiple- cell occupancies were because of the arrival of cell aggregates rather than multiple single-cells being captured separately.
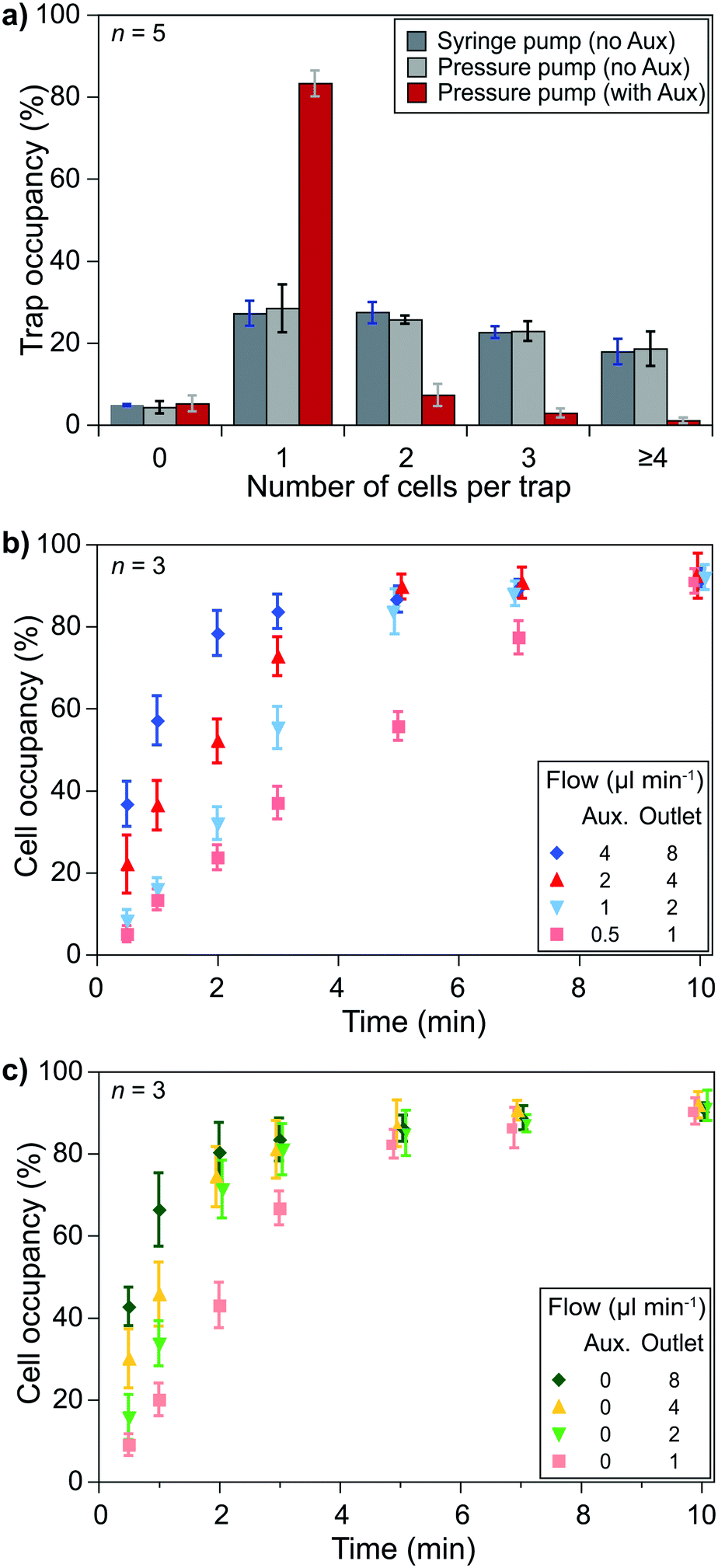 | ||
| Fig. 3 Single-cell capturing requires optimized flow conditions. a) Using auxiliary flow improves the capture of a single T lymphocyte drastically. b) Keeping the auxiliary to outlet flow ratio at 0.5, different flow rates show different trap occupancy characteristics. Obtaining 80% single-cell occupancy in 5 minutes is chosen as an acceptable condition (1 μl min−1 auxiliary and 2 μl min−1 outlet flow) to keep mechanical stimulation at low levels. c) KG1 cells are captured with only the outlet flow. Over 80% single-cell occupancy was achieved in 3 minutes with the same outlet flow (2 μl ml−1) due to the trap array design being better suited for the size of KG1 cells than T lymphocytes. | ||
Essential parameters to obtain optimum capturing performance were the auxiliary and outlet flow rates. We used the optimum flow rate ratio, i.e., 0.5, to test different flow conditions. Higher flow rates brought more cells and, thus, filled the trap sites in a shorter time. However, on the one hand, high flow rates provided higher drag forces on the cells that could cause cells to be compressed at the flow layer (the 2 μm layer at the bottom of the channel) and to slip away. We prefer minimizing mechanical stimulations as the pressure can activate signaling pathways and ion channels. On the other hand, lower flow rates did not exhibit notable mechanical effects but took longer to fill the trap sites. Injecting T lymphocytes (150000 cells ml−1) in the device with flow rates of 4 μl min−1 auxiliary and 8 μl min−1 outlet flows filled 80% of the traps with single cells in 2 minutes while flow rates of 0.5 μl min−1 auxiliary and 1 μl min−1 outlet flows took 10 minutes to reach >80% occupancy for single cells. We considered 5 minutes acceptable for >80% single-cell occupancy and chose 1 μl min−1 auxiliary and 2 μl min−1 outlet flows (Fig. 3b). However, when handling primary human CD8+ T lymphocytes from AML patients (not shown here) we observed a higher variation of the cells' size (and possibly stiffness). We had to use lower flow rates for some experiments to prevent cells from slipping away from the capture site.
Similar experiments were performed to capture larger cells, i.e., single KG1 cells, using only the outlet flow. Although an 80% single-cell capture rate was achieved in 2 minutes with 4 μl min−1 outlet flow, we decided to use 2 μl min−1 outlet flow to keep the same flow rate throughout the experiments while minimizing the potential mechanical stimulation of cells. According to the results, an 80% single-cell capture rate is achieved in 3 minutes of outlet flow (Fig. 3c).
The size difference between the two types of cells resulted in a decrease in the capturing efficiency of T lymphocytes. The row and column spacings were decided according to the KG1 cells to avoid clogging. Their diameters were 1.5–3 times larger than T lymphocytes. Consequently, there were large gaps between the T lymphocyte capture sites. We considered >80% of a single-cell trap occupancy as a sufficient level. The experimental results showed that around 30% of the T lymphocytes injected in the channel could be captured to reach this occupancy level in 5 minutes of combined auxiliary/outlet flow (Fig. S7a†). In contrast, >50% of the injected KG1 cells were captured in 3 minutes of outlet flow to reach >80% of a single-cell trap occupancy (Fig. S7b†).
Single-cell analysis: SOC monitoring
The previous sections demonstrated the single-cell capturing performance of the proposed method. A crucial step is to validate the functionality of the captured cells before applying for cell–cell interactions at an IS. We took this opportunity to perform a single-cell analysis of early signaling and functional responses of non-adherent cells. We performed experiments using cells loaded with the ratiometric Fura 2 calcium dye to validate cell viability and drug responsiveness. Cells were captured in our device while monitoring over time calcium concentrations [Ca2+]i in the cytoplasm with a dedicated calcium imaging rig. We used two representative cell types: primary human CD8+ T lymphocytes isolated from healthy donors and the KG1 AML cell line as proof of principle.The calcium response of CD8+ T-lymphocytes was monitored at 37 °C after being captured in the device (Fig. 4a). We first used the calcium ionophore ionomycin as a positive control to determine whether captured cell remained alive. The application of 2 μM ionomycin in the presence of 2 mM calcium in the extracellular solution elicited a rapid increase in [Ca2+]i expressed here as a ratio of fluorescence (F340/F380). This experiment showed that (i) cells remained viable in the device, and (ii) the extracellular solution around the cells could be exchanged within seconds.
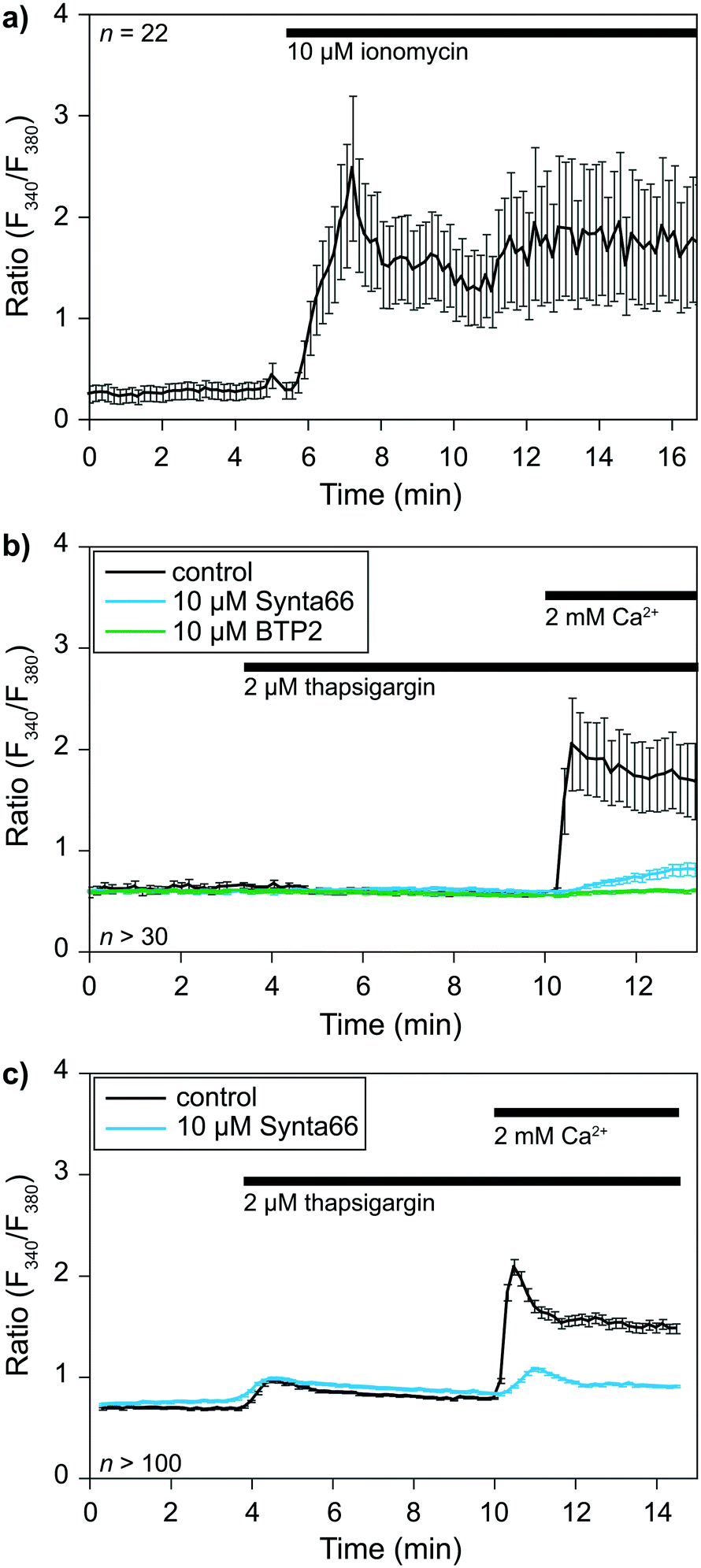 | ||
| Fig. 4 Calcium response of CD8+ T-lymphocytes and KG1 cells. a) Time course of T-lymphocytes calcium responses to the application of the calcium ionophore ionomycin. b) Time courses of T-lymphocytes responses to thapsigargin, either alone (control) or combined with Synta66 or BTP2, two inhibitors of SOC channels. c) The same experiment as (b), but with KG1 cells. n corresponds to the number of analyzed cells. | ||
We then evaluated cells' capacity to respond to a commonly used drug, namely thapsigargin, to further validate the potential application of our device to pharmacological studies. Thapsigargin is an inhibitor of SERCA (sarcoplasmic endoplasmic reticulum calcium ATPase) pumps. Application of this drug induces a calcium release from the endoplasmic reticulum (ER), leading to the activation of store-operated calcium (SOC) channels in the plasma membrane and the resulting capacitative calcium entry. SOCs represent the main calcium entry pathway in non-excitable cells, and their activation and roles are well documented in T cells and leukemia cells.26,27 We thus decided to use SOC channels as a readout for our device's practical applications to biological studies. Application of 2 μM thapsigargin at t = 3.5 min elicited a transient increase in [Ca2+]i corresponding to the ER calcium stores release (Fig. 4b and c). Subsequent application of 2 mM extracellular calcium at t = 10 min allowed us to determine SOC activity in both cell types. SOC activity resulted in a calcium entry into the cells, leading to an increase in [Ca2+]i visualized through an increase in the fluorescence ratio of the calcium dye Fura 2. Experiments were repeated in the presence of 10 μM BTP2 or 10 μM Synta66 (added with thapsigargin at t = 3.5 min), two well-known inhibitors of SOC channels, to further validate our ability to apply pharmacological modulators in our device. The perfusion of T lymphocytes and KG1 cells in separate devices with both compounds led to the expected inhibition of SOC activity. Indeed, in the presence of BTP2 and Synta66, extracellular calcium application only resulted in a slight increase in [Ca2+]i as shown by the reduced fluorescence variations compared to respective control conditions (Fig. 4b and c). Overall, these experiments show that this new device is compatible with biological systems studies.
Cell pairing and cellular activity monitoring
Investigating an immunological synapse requires a controlled cell pairing process between two cells, e.g., T lymphocytes and leukemic cells. We used primary human CD8+ T lymphocytes and KG1 AML cell lines for cell pairing demonstrations. As explained in the methods section, we first injected T lymphocytes via primary inlet 1 and captured them at the trapping area in the presence of an auxiliary flow. Then, the active primary inlet was changed from 1 to 2 while the auxiliary flow was separating the inlet area from the trapping area. KG1 cells were injected via inlet 2 and captured after stopping the auxiliary flow. The outlet flow was kept constant throughout the experiment to minimize mechanical stimulation of the cells. We considered >80% single-cell occupancy acceptable for both T lymphocytes and KG1 cells and performed cell pairing by operating the pump with the chosen flow parameters. The results showed that single T lymphocytes and single KG1 cells formed pairs at ∼70% of trap sites (Fig. 5a). An additional ∼10% of the sites were single-to-doublet cell pairs. Most of these multi-cell pairs (Fig. 5a and b) were formed by cells arriving in the trapping area as aggregates rather than being captured as two separate cells.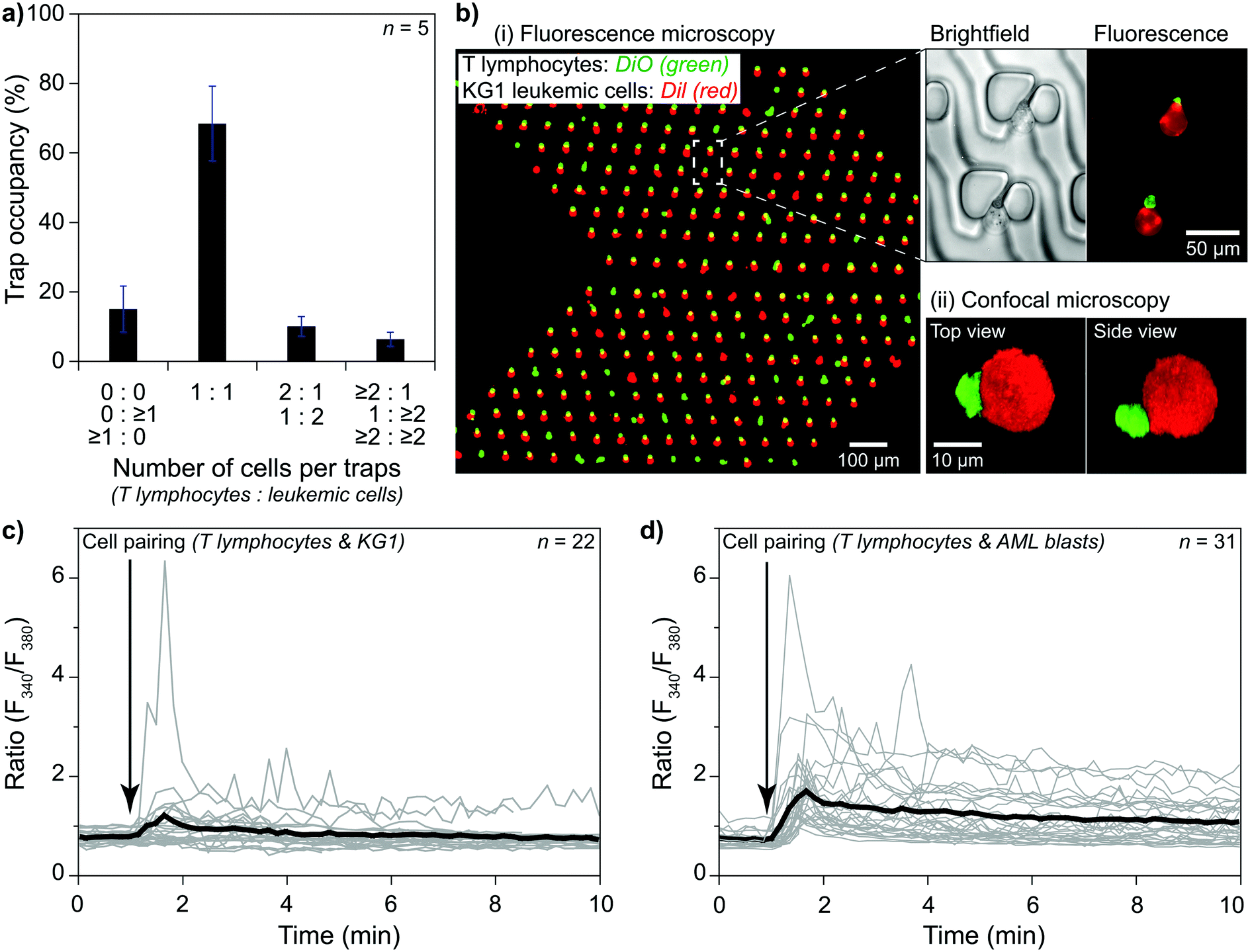 | ||
| Fig. 5 Cell pairing and monitoring their activities. a) The proposed method formed pairs of single primary human T lymphocytes and single KG1 cells captured at 70% of the trap sites. b) (i) A view of paired T lymphocytes (green) and KG1 cells (red). Most of the multi-cell pairs were captured as doublets. (ii) Imaging a pair with confocal microscopy. c) Ca2+ imaging experiments showing T cell activity following IS formation. Cell pairs were formed at t = 1 min. d) An allogenic condition was obtained by pairing primary human CD8+ T lymphocytes from healthy donors and primary AML blasts. | ||
Cell–cell interaction studies benefit from observing late events and the early ones. For monitoring late events, pairs must stay at each trap site they were captured until the end of the observation. We tested our protocol with two different designs for a 4 hour observation period under constant outlet flow at 37 °C. Slower flows, e.g., 0.02 μl min−1, resulted in losing pairs (<15%) mainly due to Brownian motion separating KG1 away, while faster flows resulted in losing pairs mainly due to T lymphocytes being slipped away through the narrow opening of the trap or the flow layer (layer 0) at the bottom. The narrow opening design affected the ratio of lost pairs. Continuous flow caused many pairs to slip away in a straight narrow opening design (80% at 4 μl min−1). Tilting the narrow opening at an angle of 45° increased the fluidic resistivity and decreased the adverse effects significantly. The lost pairs were only at ∼15% at 4 μl min−1 outlet flow in an angled narrow opening device. For long observation sessions, the optimum flow rate was obtained as 0.3 μl min−1, at which rate only <3% of the pairs were lost at the end of a 4 hour observation (Fig. S8†). The number of lost pairs did not change after 0.5 hours, which suggested that much longer observation was possible (Fig. S8†).
The ability to monitor cell–cell interactions from initial contact to an extended time opens a unique opportunity to perform single-cell analysis of the resulting early signaling and functional responses. We studied the early events that occurred during an IS formation between an effector immune cell with its target cell to validate the principle for our device's practical applications. We monitored early activation of the immune cell in real-time, reflected by the calcium mobilization at 37 °C. Here, the IS was formed by the interaction between one KG1 cell and one CD8+ T lymphocyte cell. Using our device coupled to a calcium imaging rig, we monitored in real-time basal [Ca2+]i, and [Ca2+]i variations, a rapid and sensitive readout of T cell receptor (TCR) engagement following IS formation between superantigen pulsed primary human CD8+ T lymphocytes and KG1. The results showed the induced calcium responses in T lymphocytes loaded with Fura2 calcium dye due to the pairs formed between CD8+ T lymphocytes and KG1 (Fig. 5c and S9a†). A similar unidirectional flow protocol was applied to pair CD8+ T lymphocytes and primary human leukemic blasts (Fig. 5d and S9b†). Positive control was performed by adding ionomycin at the end of all experiments to check T cells viability and responsiveness to stimuli (Fig. S9c†). Our results proved that the proposed method enables real-time analysis of cellular activities on pairs of single cells, even for size differences reaching three times.
The third primary inlet allows the proposed method to perform some post-pairing actions. After the formation of the IS and the assay period, the auxiliary inlet separates the inlet and the trapping areas without compromising the constant flow conditions at the trap sites. In this condition, the second primary inlet is sealed, and the third primary inlet is opened to inject solutions into the channel. We demonstrated the feasibility of our method to assay the early molecular events during lymphocyte activation. As actin filaments are reorganized during synapse formation, we stained them to monitor whether they were polarized at the cell contact point. We first fixed and then permeabilized the cells to perform staining. As the last step, we injected phalloidin-488 to finalize actin filaments fluorescent staining (Fig. S10†).
Discussion
The presented method provided pairs of non-adherent cells having different sizes to investigate early and late cellular activities at IS. Techniques such as droplet microfluidics could potentially bring cells of different sizes nearby but still cannot study early signaling dynamics and their correlation to subsequent functional behaviors.24 Demonstrated microfluidic channels show excellent performance in investigating early signaling and high efficiency in forming pairs of similar-sized cells.23 2D design changes on such systems could capture cells with different dimensions. However, when the smaller cell is as small as half of the larger cell, multiple cell capturing is inevitable in the z-direction. Therefore, we introduced a practical channel height modulation to provide pairs formed by single cells despite their size differences. Hydrodynamic flow focusing in the z-direction allowed us to flow the smaller cells by the bottom surface and capture them at the trap sites positioned at that level. As discussed below, this flow, named auxiliary flow, showed several other advantages.Pairing cells of different sizes required a device designed according to the larger cells. The channel height, trap density, and trap position were decided accordingly. Although we could overcome the channel height effect on smaller cells with an auxiliary flow, the trap density and positioning could not be designed according to the smaller cells. Consequently, such cells showed a lower capturing rate and efficiency. The overall capture efficiency can be improved by repeating the >shaped trap sites. This paper demonstrated the method with primary human samples after several sorting steps, which resulted in a limited number of cells. Moreover, to achieve sensitive Ca2+ imaging, we used relatively higher magnification (20×), limiting our observation area. As a result, we did not push higher efficiency rates but focused on pairs of single cells.
Pressure can activate the signaling pathways of a cell. It can also induce ATP release from the cells, activating SOC channels of the same and surrounding cells.27 T cells are not only sensitive to pressure28 but also smaller and more deformable than many other cells, e.g., cardiac myocytes28 and stem cells.23 As we monitor TCR with Ca2+ imaging to investigate cellular activities, stable and reliable protocols minimizing the pressure fluctuations applied on cells are crucial. A unidirectional flow format and single-step trapping minimize the disturbance to the captured cells, unlike other examples requiring bidirectional flows with consecutive two-trap-site capturing23 or multi-step trapping through physical constraints with different flow rates.24 Compressing cells (reaching 50%) during capturing24 may lead to unexpected cellular responses in addition to the ones due to immunological responses. The auxiliary inlet completely separates the inlet area from the trapping area without changing the flow conditions in the latter. Therefore, the auxiliary inlet flow and single-step cell capturing provide constant flow conditions at trap sites throughout the experiments minimizing potential external effects on cellular activity during actions taken at the front end of the device.
The proposed three primary inlet geometry may seem intricate; however, it minimizes washing steps and cross-channel contamination as only one inlet works at a time. When we increase the auxiliary flow rate above the outlet flow rate (flow ratio, r > 1) right before changing from inlet 1 to inlet 2, we induce a flow towards inlet 1 which cleans the main channel. The new cells injected in inlet 2 flow along the main channel without contamination from inlet 1. As a result, the auxiliary flow separating the inlet and trapping areas makes the system much more stable during the injection of new cells or solutions in the channel by preventing any flow fluctuation in the trapping area while cleaning the main channel simultaneously.
Conclusions
Studies on cell–cell interactions attracted many researchers, particularly cancer immunology, to develop cell pairing devices to investigate early events. Although some designs were demonstrated for analyzing pairs with similar-sized cells, these devices show difficulties maintaining pairs of single cells when the dimension of cells differs. We filled this gap by introducing 3D hydrodynamic flow focusing on changing the effective channel height while pairing cells with different dimensions. We used physical traps having different geometries to capture different cell types. Ca2+ imaging was performed in real-time to monitor early events at an immunological synapse formed by paired cells at specific trap sites. We demonstrated the method using primary human T lymphocytes, KG1 AML cell lines, and primary AML blasts. As T lymphocytes are sensitive to pressure, our method provided minimal disturbance to the cells and pairs by keeping the flow conditions at the trapping area constant throughout the experiments, including both cell capturing steps. The results showed that the proposed method could pair primary human cells having different dimensions forming an IS in a controlled environment.To sum up, the proposed method leads to deciphering mechanisms of IS formation between tumor cells and different types of immune cells (e.g., T and natural killer), which play a significant role in cancer immunology. In addition, despite the heterogeneity of immune-cancer cells interactions caused by their specific properties, our method allows detailed functional studies at the IS level reflecting the diversity of immune cells and malignant cells behaviors. The detailed tracking of immune–cancer cell interactions at the single pair level helps uncover key mechanisms to improve the efficiency of various immunotherapeutic strategies.
Author contributions
Conceptualization: D. C., B. Q., Y. T., L. L., M. C. T. Data curation: F. A. S, C. L, Y. T., L. L., M. C. T. Formal analysis: F. A. S, C. L, Y. T., L. L., M. C. T. Funding acquisition: D. C., B. Q., Y. T., L. L., M. C. T. Investigation: F. A. S, C. L, A. G., B. A., Y. T., L. L., M. C. T. Methodology: F. A. S, C. L, A. G., C. B., D. C., B. Q., Y. T., L. L., M. C. T. Project administration: Y. T., L. L., M. C. T. Resources: C. B., D. C., B. Q., Y. T., L. L., M. C. T. Supervision: C. B., D. C., B. Q., Y. T., L. L., M. C. T. Validation: F. A. S, C. L, A. G., B. A., Y. T., L. L., M. C. T. Visualization: F. A. S, B. A., L. L., M. C. T. Writing – original draft: F. A. S, C. L., Y. T., L. L., M. C. T. Writing – review & editing: F. A. S., C. L, D. C., B. Q., Y. T., L. L., M. C. T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is in the framework of SMMiL-E activities. F. A. Shaik is a recipient from a Contrat de Plan Etat-Région CPER Cancer 2015-2020 fellowship. This work is supported by grants from Contrat de Plan Etat-Région CPER Cancer 2015-2020, INSERM, CNRS, Ligue contre le cancer (Septentrion), Ligue nationale contre le cancer, ARC, and the Institut de Recherche sur le Cancer de Lille (IRCL). The authors acknowledge IRCL for hosting SMMiL-E. M. C. Tarhan acknowledges Fondation ARC. We would also like to thank Nathalie Jouy (Flow Cytometry Platform, Univ Lille, Lille, France) for her technical assistance, and Jean-Claude Gerbedoen (LIMMS, Lille, France) for his assistance on fabrication. Clara Lewuillon is financed by Lille Hospital and by Hauts de France Region.References
- Z. Wan, X. Gao, Y. Dong, Y. Zhao, X. Chen, G. Yang and L. Liu, Am. J. Cancer Res., 2018, 8, 1661–1673 CAS.
- M. Mittelbrunn and F. Sánchez-Madrid, Nat. Rev. Mol. Cell Biol., 2012, 13, 328–335 CrossRef CAS PubMed.
- H. Daneshpour and H. Youk, Curr. Opin. Syst. Biol., 2019, 18, 44–52 CrossRef PubMed.
- M. A. Kalliomäki and W. A. Walker, Clin. Gastroenterol., 2005, 34, 383–399 CrossRef PubMed.
- S. Cai, C. Wu, W. Yang, W. Liang, H. Yu and L. Liu, Nanotechnol. Rev., 2020, 9, 971–989 CrossRef CAS.
- E. C. Lai, Development, 2004, 131, 965–973 CrossRef CAS PubMed.
- H. Raskov, A. Orhan, J. P. Christensen and I. Gögenur, Br. J. Cancer, 2021, 124, 359–367 CrossRef CAS PubMed.
- D. E. Speiser, P. C. Ho and G. Verdeil, Nat. Rev. Immunol., 2016, 16, 599–611 CrossRef CAS PubMed.
- S. Khaldoyanidi, D. Nagorsen, A. Stein, G. Ossenkoppele and M. Subklewe, J. Clin. Oncol., 2021, 39, 419–432 CrossRef CAS PubMed.
- D. R. Bogdanowicz and H. H. Lu, Biotechnol. J., 2013, 8, 395–396 CrossRef CAS PubMed.
- H. Ge, L. Tan, P. Wu, Y. Yin, X. Liu, H. Meng, G. Cui, N. Wu, J. Lin, R. Hu and H. Feng, Sci. Rep., 2015, 5, 1–10 Search PubMed.
- Y. C. Chen, Y. H. Cheng, H. S. Kim, P. N. Ingram, J. E. Nor and E. Yoon, Lab Chip, 2014, 14, 2941–2947 RSC.
- H. Dolznig, C. Rupp, C. Puri, C. Haslinger, N. Schweifer, E. Wieser, D. Kerjaschki and P. Garin-Chesa, Am. J. Pathol., 2011, 179, 487–501 CrossRef PubMed.
- C. H. Kim, I. R. Suhito, N. Angeline, Y. Han, H. Son, Z. Luo and T. H. Kim, Adv. Healthcare Mater., 2020, 9, 1901751 CrossRef CAS PubMed.
- J. A. James, M. Sushmitha, R. Premkumar, V. Narayanamurthy and R. Kalpana, in 2017 International Conference on Microelectronic Devices, Circuits and Systems, ICMDCS 2017, 2017, vol. 2017, pp. 1–6 Search PubMed.
- Y. Zhou, N. Shao, R. Bessa de Castro, P. Zhang, Y. Ma, X. Liu, F. Huang, R. F. Wang and L. Qin, Cell Rep., 2020, 31, 107574 CrossRef CAS PubMed.
- T. Konry, S. Sarkar, P. Sabhachandani and N. Cohen, Annu. Rev. Biomed. Eng., 2016, 18, 259–284 CrossRef CAS PubMed.
- T. Lam, M. D. Brennan, D. A. Morrison and D. T. Eddington, Lab Chip, 2019, 19, 682–692 RSC.
- D. K. Kang, M. Monsur Ali, K. Zhang, E. J. Pone and W. Zhao, TrAC, Trends Anal. Chem., 2014, 58, 145–153 CrossRef CAS.
- S. Srikanth and Y. Gwack, Mol. Cells, 2013, 35, 182–194 CrossRef CAS PubMed.
- L. Li, H. Wang, L. Huang, S. Alan Michael, W. Huang and H. Wu, Anal. Chem., 2019, 91, 15908–15914 CrossRef CAS PubMed.
- A. M. Skelley, O. Kirak, H. Suh, R. Jaenisch and J. Voldman, Nat. Methods, 2009, 6, 147–152 CrossRef CAS PubMed.
- B. Dura, S. K. Dougan, M. Barisa, M. M. Hoehl, C. T. Lo, H. L. Ploegh and J. Voldman, Nat. Commun., 2015, 6, 1–13 Search PubMed.
- B. Dura, M. M. Servo, R. M. Barry, H. L. Ploeghf, S. K. Dougand and J. Voldman, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E3599–E3608 CrossRef CAS PubMed.
- H. Cabanas, T. Harnois, C. Magaud, L. Cousin, B. Constantin, N. Bourmeyster and N. Déliot, Oncotarget, 2018, 9, 26309–26327 CrossRef PubMed.
- N. Watkins, B. M. Venkatesan, M. Toner, W. Rodriguez and R. Bashir, Lab Chip, 2009, 9, 3177–3184 RSC.
- G. Dahl, Philos. Trans. R. Soc., B, 2015, 370, 1–11 CrossRef CAS PubMed.
- S. Faley, K. Seale, J. Hughey, D. K. Schaffer, S. Vancompernolle, B. McKinney, F. Baudenbacher, D. Unutmaz and J. P. Wikswo, Lab Chip, 2008, 8, 1700–1712 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc01156a |
| ‡ F. A. Shaik and C. Lewuillon contributed equally to this work. Y. Touil, L. Lemonnier, and M. C. Tarhan share the last authorship. |
| This journal is © The Royal Society of Chemistry 2022 |