
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition metal(II) complexes of halogenated derivatives of (E)-4-(2-(pyridin-2-ylmethylene)hydrazinyl)quinazoline: structure, antioxidant activity, DNA-binding DNA photocleavage, interaction with albumin and in silico studies†
Chrisoula
Kakoulidou
a,
Christos T.
Chasapis
 b,
Antonios G.
Hatzidimitriou
a,
Konstantina C.
Fylaktakidou
*c and
George
Psomas
*a
b,
Antonios G.
Hatzidimitriou
a,
Konstantina C.
Fylaktakidou
*c and
George
Psomas
*a
aDepartment of General and Inorganic Chemistry, Faculty of Chemistry, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece. E-mail: gepsomas@chem.auth.gr
bNMR Facility, Instrumental Analysis Laboratory, School of Natural Sciences, University of Patras, Greece
cLaboratory of Organic Chemistry, Faculty of Chemistry, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece. E-mail: kfylakta@chem.auth.gr
First published on 18th October 2022
Abstract
Two novel halogenated (Br- and F-) quinazoline derivatives, namely [(E)-4-(2-((6-bromopyridin-2-yl)methylene)hydrazinyl)quinazoline] (L1) and [(E)-4-(2-((3-fluoropyridin-2-yl)methylene)hydrazinyl) quinazoline] (L2), were synthesized and characterized. Their interaction with a series of metal(II) ions (= Mn(II), Ni(II), Cu(II), Zn(II) and Cd(II)) resulted in the formation of six mononuclear complexes characterized by spectroscopic techniques and single-crystal X-ray crystallography. The complexes bear the formulae [Ni(L1)2](NO3)2 (1), [Zn(L2)2](NO3)(PF6) (2), [Cd(L2)(H2O)(CH3OH)(NO3)](NO3) (3), [Cu(L2)Cl2] (4), [Ni(L2)2](NO3)2 (5) and [Mn(L2)(CH3OH)(Cl)2] (6). The biological activity of the compounds was further evaluated in vitro regarding their interaction with calf-thymus DNA, their cleavage ability towards supercoiled circular pBR322 plasmid DNA in the absence or presence of irradiation at various wavelengths (UVA, UVB and visible light), their affinity to bovine serum albumin and their ability to scavenge 1,1-diphenyl-picrylhydrazyl and 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) radicals and to reduce H2O2. In silico molecular docking calculations were employed to study the behavior of the complexes towards calf-thymus DNA and bovine serum albumin.
1 Introduction
4-Hydrazonyl quinazolines are compounds that hybridize two pharmacophore cores: a quinazoline1,2 and a hydrazone.3,4 Quinazolines constitute a class of heterocycles that contain two fused aromatic benzene and pyrimidine rings and exhibit a plethora of biological activities.5–10 Hydrazones contain a characteristic R1R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NHR3 functional group which due to its isomerization and nucleophilic, electrophilic and acidic sites gives rise to versatile uses of their carrier compounds.10–13 In conjunction with well-situated electron donors, hydrazones are capable of forming metal complexes which were found to exhibit catalytic14–17 and biological activities.18–21 Although 4-quinazoline-hydrazone derivatives fulfill the structural criteria for metal coordination, very few derivatives are found in the literature. Specifically, 4-hydrazonylquinazolines in conjunction with the o-hydroxyazomethines of salicylaldehydes exhibited fluorescence selectively for Ga3+ and Zn2+ cations.22–24 Our group has designed and synthesized 2-pyridinyl-methylene-4-hydrazone quinazoline transition metal complexes which exhibit DNA binding and photocleavage as well as antioxidant activities.25,26 In addition, the above ligand was able to co-coordinate Zn2+ metal ions with the non-steroidal anti-inflammatory drug diclofenac and these complexes possessed enhanced antioxidant and radical scavenger activities compared to the commercial drug.27
N–NHR3 functional group which due to its isomerization and nucleophilic, electrophilic and acidic sites gives rise to versatile uses of their carrier compounds.10–13 In conjunction with well-situated electron donors, hydrazones are capable of forming metal complexes which were found to exhibit catalytic14–17 and biological activities.18–21 Although 4-quinazoline-hydrazone derivatives fulfill the structural criteria for metal coordination, very few derivatives are found in the literature. Specifically, 4-hydrazonylquinazolines in conjunction with the o-hydroxyazomethines of salicylaldehydes exhibited fluorescence selectively for Ga3+ and Zn2+ cations.22–24 Our group has designed and synthesized 2-pyridinyl-methylene-4-hydrazone quinazoline transition metal complexes which exhibit DNA binding and photocleavage as well as antioxidant activities.25,26 In addition, the above ligand was able to co-coordinate Zn2+ metal ions with the non-steroidal anti-inflammatory drug diclofenac and these complexes possessed enhanced antioxidant and radical scavenger activities compared to the commercial drug.27
Metal-based drugs (e.g. cisplatin) bind covalently to DNA bases via coordination sites resulting from the displacement of labile ligands.28–30 In addition, metal complexes that do not contain labile ligands and/or retain their stability may interact noncovalently with DNA via intercalation, groove-binding and/or electrostatic interactions.31–33 These interactions of metal complexes with DNA may cause its cleavage,33 whereas activation with light of appropriate wavelength may lead to photoapplications34 that include photocatalytic hydrogen evolution,35 creation of optical oxygen-sensitive probes,36 photocatalysis,37,38 DNA photocleavage25,27,39 and photodynamic therapies.40–42
A number of 4-aminoquinazolines bearing as a pendant a halogenated aromatic ring have been approved by the FDA as anticancer drugs (Fig. 1).6 Halogenated drugs, in general, gain more and more ground, with approximately 25% of all new drugs containing at least one halogen atom.43 These drugs benefit from the halogen bond that provides interactions with biological systems (protein systems and biological membranes).44,45 Halogen atoms have been introduced in known drugs46 as well as in various ligands that coordinate metal atoms in order to tune their activity.47–52 Based on a very recent publication, halogen bonds seem to exist between noncovalent ligands and natural nucleic acids, a fact that is expected to promote the use of halogens in drug design.43 Finally, halogen atoms in many cases increase photosensitization in photodynamic therapies due to the heavy atom effect.53,54
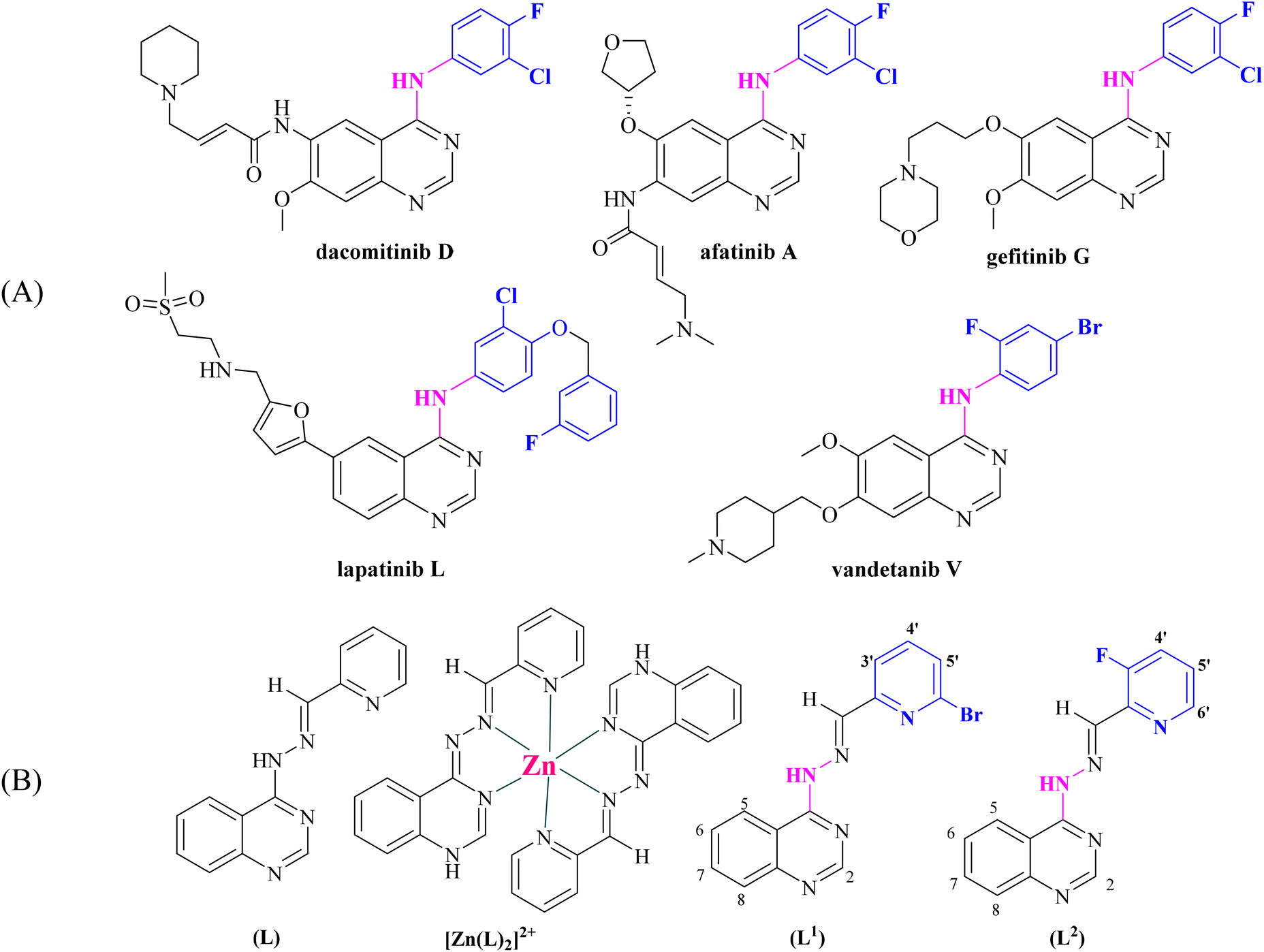 | ||
| Fig. 1 (A) Quinazoline derivatives approved by the FDA as anticancer agents. (B) Ligand (L) used in our previous studies25–27 and its Zn complex [Zn(L)2]2+.27 Evolution of the structure of [(E)-4-(2-(pyridin-2-ylmethylene)hydrazinyl)quinazoline] (L) into possible halogenated pharmacophores [(E)-4-(2-((6-bromopyridin-2-yl)methylene)hydrazinyl)quinazoline] (L1) and [(E)-4-(2-((3-fluoropyridin-2-yl)methylene)hydrazinyl)quinazoline] (L2) (aromatic H atoms numbering is also shown). | ||
Within this context and as a continuation of our previous project, we have designed, synthesized and characterized two novel halogenated derivatives of L (Fig. 1(B)), namely (E)-4-(2-((6-bromopyridin-2-yl)methylene)hydrazinyl)quinazoline (L1) and (E)-4-(2-((3-fluoropyridin-2-yl)methylene)hydrazinyl)quinazoline (L2) (Fig. 1(B)). These compounds were designed in order to satisfy certain structural features, such as the location of selected electron donors able to provide multiple coordination sites and rigidity that could enable DNA intercalation and halogen substitution on the linked heteroaromatic pyridine ring. We also report the synthesis and the characterization of six complexes of Mn(II), Ni(II), Cu(II), Zn(II) and Cd(II) ions with the novel compounds L1 and L2. The characterization of the complexes was achieved by spectroscopic techniques (IR, UV-vis, and 1H NMR) and single-crystal X-ray crystallography.
DNA is often a key target for the development of anticancer drugs and for photodynamic therapy.55–58 Therefore, the compounds were evaluated in vitro for (i) their affinity for calf-thymus (CT) DNA by UV-vis spectroscopy, viscosity measurements, and competitive studies with ethidium bromide (EB) by fluorescence emission spectroscopy and (ii) their ability to cleave supercoiled circular pBR322 plasmid DNA (pDNA) was monitored by agarose gel electrophoresis in the absence or presence of irradiation including UVA, UVB and visible light. Another significant activity that may be related to the potential anticancer activity is the potential antioxidant activity; within this context, the in vitro antioxidant activity of the compounds under study was assessed via their ability to scavenge the free radicals 1,1-diphenyl-picrylhydrazyl (DPPH) and 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) and to reduce H2O2. Albumins are proteins related to the potential transportation of bioactive compounds through the bloodstream towards their biological targets. Therefore, and as a supplement to the study of the biological activity of the compounds, their affinity for bovine serum albumin (BSA) was examined in vitro by fluorescence emission spectroscopy in order to calculate the corresponding binding constant and to determine the location of binding sites. In addition, in silico molecular docking studies on CT DNA and BSA were employed to evaluate the ability of the compounds to bind to these macromolecules and contribute to the understanding of the possible role they can play in the context of a plethora of diseases.
2 Experimental
2.1 Materials, instrumentation and physical measurements
All chemicals and solvents were reagent grade and used as purchased from commercial sources. 2-Aminobenzonitrile, 3-fluoro-2-pyridinecarboxaldehyde, and 6-bromo-2-formylpyridine were obtained from Fluorochem. Ammonium acetate, triethyl orthoformate, hydrazine hydrate, p-toluenesulfonic acid monohydrate, Ni(NO3)2·6H2O, CT DNA, EB, BSA, ABTS, K2S2O8, nordihydroguaiaretic acid (NDGA), butylated hydroxytoluene (BHT) were obtained from Sigma-Aldrich Co. 6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) was obtained from J&K. Sodium warfarin, ibuprofen, DPPH from TCI, MnCl2·4H2O, Zn(NO3)2·6H2O, CdCl2, Cd(NO)3·4H2O, CuCl2·2H2O, trisodium citrate dihydrate, NaCl, and NaH2PO4 were obtained from Merck. Supercoiled circular pBR322 plasmid DNA was obtained from New England Bioline. Tris base, boric acid, EDTA disodium salt dehydrate, loading buffer and H2O2 (30% w/v) were obtained from PanReac Applichem. L-Ascorbic acid, Na2HPO4 and all solvents were obtained from Chemlab. All reactions were monitored on commercially available pre-coated TLC plates (layer thickness 0.25 mm) of Kieselgel 60 F254. Yields were calculated after recrystallization.A CT DNA stock solution was prepared by the dilution of CT DNA with buffer (containing 150 mM NaCl and 15 mM trisodium citrate at pH 7.0) followed by exhaustive stirring at 4 °C for 2 days and kept at 4 °C for no longer than a week. The stock solution of CT DNA gave a ratio of UV absorbance at 260 and 280 nm (A260/A280) of ∼1.88, indicating that the DNA was sufficiently free of protein contamination.59 The DNA concentration per nucleotide was determined by the UV absorbance at 260 nm after 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 dilution using ε = 6600 M−1 cm−1.60
:20 dilution using ε = 6600 M−1 cm−1.60
The infrared (IR) spectra (400–4000 cm−1) were recorded using a Nicolet FT-IR 6700 spectrometer with the samples prepared as KBr pellets (abbreviation used: s = strong, vs = very strong, br = broad, m = medium, and w = weak) (Fig. S1†). The UV-visible (UV-vis) spectra were recorded in solution at concentrations in the range of 5 mM–50 μM using a Hitachi U-2001 dual beam spectrophotometer. The C, H and N elemental analyses were carried out using a PerkinElmer 240 B elemental analyzer. The 1H NMR spectra were recorded using an Agilent 500/54 (500 MHz) spectrometer using DMSO-d6 as the solvent. Chemical shifts (δ) are given in ppm and J values in Hz using solvent as an internal reference (H-numbering shown in Fig. 1; abbreviation used: s = singlet, d = doublet, t = triplet, br = broad, and m = multiplet). Molar conductivity measurements were carried out in 1 mM DMSO solution of the complexes using a Crison Basic 30 conductometer. The fluorescence spectra were recorded in solution using a Hitachi F-7000 fluorescence spectrophotometer. Viscosity experiments were carried out using an ALPHA L Fungilab rotational viscometer equipped with an 18 mL LCP spindle and the measurements were performed at 100 rpm.
2.2 Synthesis of (E)-4-(2-((6-bromopyridin-2-yl)methylene)hydrazinyl)quinazoline (L1) and (E)-4-(2-((3-fluoropyridin-2-yl)methylene)hydrazinyl)quinazoline (L2)
The synthesis of the compounds was performed in three steps, including the synthesis of 4-aminoquinazoline (I) and quinazolin-4-yl-hydrazine(II) as intermediates. The synthesis of intermediates I and II (Scheme 1) has been previously described in ref. 27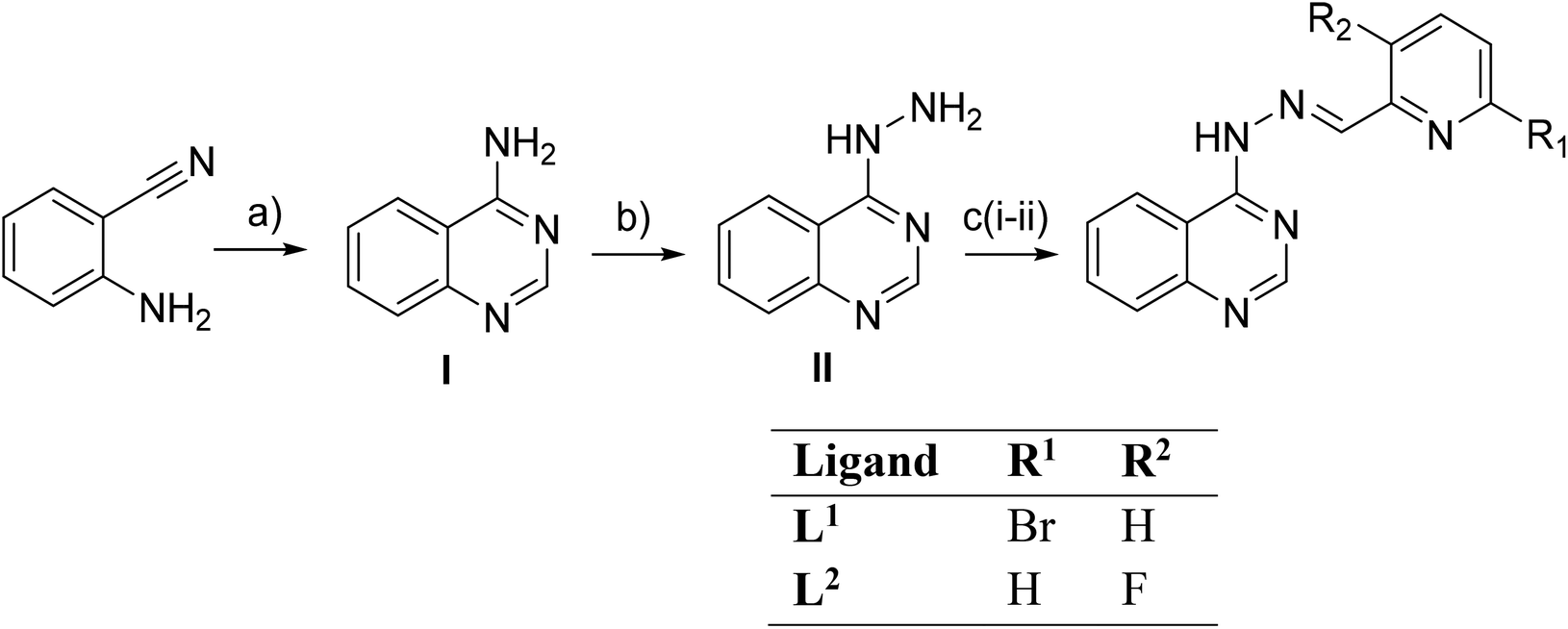 | ||
| Scheme 1 Synthesis of compounds L1 and L2. (a) NH4OAc, HC(OEt)3, EtOH reflux, 3.5 h, 78% yield; (b) NH2NH2, reflux, 24 h, 53% yield; (c) 6-bromo-2-formylpyridine or 3-fluoro-2-pyridinecarboxaldehyde, p-TsOH, EtOH reflux, 4 h and 2 h, respectively, 73% yield. | ||
N), 7.82 (t, J = 7.8 Hz, 1H, H4′), 7.90 (s, 1H, C(H)N), 7.67 (t, J = 7.2 Hz, 1H, H7), 7.62 (d, J = 8.0 Hz, 1H, H5′), 7.50 (d, J = 8 Hz, 1H, H8), 7.42 (t, J = 7 Hz, 1H, H6). IR (KBr disk), ν (cm−1): ν(N–H)secondary: 3348(m), ν(CN): 1631(vs), 1616(vs), w(N–H): 771(m). UV–vis in DMSO, λ (nm) (ε, M−1 cm−1): 413 (shoulder(sh)) (1600), 392 (2000), 372 (2100), 305 (10300).
N), 7.80 (t, J = 9.5 Hz, 1H, H4′), 7.66 (brt, 1H, H7), 7.51–7.47 (m, 2H, H8 and H5′), 7.41 (brt, 1H, H6). IR (KBr disk), ν (cm−1): ν(N–H)secondary: 3379 (br), ν(CN): 1630(vs), w(N–H): 775(s). UV–vis in DMSO, λ (nm) (ε, M−1 cm−1): 369 (9350), 303 (4300), 260 (6100).
2.3. Synthesis of the complexes
N): 1635 (s), 1615 (s); ν(C–C)aromatic: 1506 (vs), 1466 (s); ν3(NO3): 1383 (s); ν1(NO3): 800(w); w(N–H): 756 (s); ν(M–N): 561 (m). UV–vis in DMSO, λ (nm) (ε, M−1 cm−1): 780 (35), 590(sh) (45), 472 (30700), 434 (42900), 411 (sh) (37600), 390 (sh) (24800), 325(sh) (18000), 300 (25150). The complex is soluble in DMSO (ΛM = 80 S cm2 mol−1, 1 mM in DMSO).
N): 1637(s), 1617(s); ν(C–C)aromatic: 1541 (w), 1499 (s); ν3(NO3): 1384 (s); ν1(NO3): 843 (s); w(N–H): 763 (w); ν(M–N): 558 (m). UV–vis in DMSO, λ (nm) (ε, M−1 cm−1): 463 (23000), 436 (19700), 374 (20140), 293 (13560). The complex is soluble in DMSO (ΛM = 75 S cm2 mol−1, 1 mM in DMSO).
N): 1627 (s), 1610(s); ν(C–C)aromatic: 1533 (w); ν3(NO3): 1384 (vs); ν1(NO3): 828 (vs); w(N–H): 766 (w); ν(M–N): 532 (m). UV–vis in DMSO, λ (nm) (ε, M−1 cm−1): 468 (4820), 410 (sh) (9380), 370 (18960), 302 (9380). The complex is soluble in DMSO (ΛM = 55 S cm2 mol−1, 1 mM in DMSO).
N): 1637(s), 1611(s); ν(C–C)aromatic: 1566 (s), 1496 (s); w(N–H): 765 (s). UV–vis in DMSO, λ (nm) (ε, M−1 cm−1): 690 (85), 478 (45350), 446 (38250), 414(sh) (19650), 325(sh) (23700), 312 (27900), 272 (40550). The complex is soluble in DMSO and H2O (ΛM = 10 S cm2 mol−1, 1 mM in DMSO).
N): 1631 (vs), 1611 (vs); ν(C–C)aromatic: 1540 (w), 1497(vs); ν3(NO3): 1381(vs); ν1(NO3): 824 (w); w(N–H): 762 (s); ν(M–N): 536 (w). UV–vis in DMSO, λ (nm) (ε, M−1 cm−1): 783 (30), 633(sh) (40), 471 (42100), 438 (43750), 411 (sh) (32500), 347 (sh) (10600), 317 (sh) (19850), 293 (22750). The complex is soluble in DMSO (ΛM = 70 S cm2 mol−1, 1 mM in DMSO).
N): 1632 (s), 1608 (s); ν(C–C)aromatic: 1542(w), 1508 (s); w(N–H): 762 (s); ν(M–N): 527 (w). UV–vis in DMSO, λ (nm) (ε, M−1 cm−1): 468 (8770), 442 (sh) (6970), 408 (13233), 370 (27400), 296 (sh) (14500). The complex is soluble in DMSO (ΛM = 15 S cm2 mol−1, 1 mM in DMSO).
2.4 X-ray crystal structure determination
Suitable single-crystals of the complexes were mounted on thin glass fibers with the aid of epoxy resin. X-ray diffraction data were recorded using a Bruker Apex II CCD area-detector diffractometer, equipped with a Mo Ka (λ = 0.71073 Å) sealed tube source and a Triumph monochromator at 295 K, using the φ and ω scan techniques. The program Apex2 (Bruker AXS, 2006) was used for data collection and cell refinement. The collected data were integrated with the Bruker SAINT software package61 using a narrow-frame algorithm. Data were corrected for absorption using the numerical method SADABS,62 based on the crystal dimensions. The structures were solved using the SUPERFLIP package63 and refined using full-matrix least-squares on F2 using the Crystals program package version 14.61 build 6236.64 Anisotropic displacement parameters were applied to all non-hydrogen atoms of the complexes and the non-disordered solvent atoms, while hydrogen atoms were in general found and/or positioned geometrically and refined using a riding model. The details of crystal data and structural refinement parameters are shown in Table S1.†2.5 Study of the biological profiles of the compounds
All the procedures and relevant equations used in the in vitro study of the biological activity (antioxidant activity, interaction with CT DNA, plasmid DNA and BSA) of the compounds can be found in the ESI file (sections S1–S4).† A series of in silico studies were employed in order to study the interaction of the compounds with BSA and DNA. Details concerning the in silico computation procedures employed are given in the ESI file (section S5).†3 Results and discussion
3.1 Synthesis and characterization
The synthesis of the ligands was performed in three steps. The syntheses of 4-aminoquinazoline (step I) and quinazolin-4-yl-hydrazine (step II) have been previously reported.33,65,66 The quinazolin-4-yl-hydrazine reacted with 6-bromo-2-formylpyridine and 3-fluoro-2-pyridinecarboxaldehyde to give (E)-4-(2-((6-bromo-pyridin-2-yl)methylene)hydrazinyl)quinazoline (L1) and (E)-4-(2-((3-fluoro-pyridin-2-yl)methylene)hydrazinyl)quinazoline (L2), respectively. The yield in both reactions was 73%. Data obtained for the known compounds were in full accordance with the literature,27,65,66 while compounds L1 and L2 were fully characterized (Fig. S2 and S3†).All complexes were prepared via an aerobic reaction of the quinazoline solution with the corresponding metal salts of Ni(II), Zn(II), Cd(II), Cu(II) and Mn(II). The M:L ratio was 1:2 for complexes 1, 2 and 5 and 1:1 for complexes 3, 4 and 6. The coordination compounds were characterized by IR, 1H NMR and UV-vis spectroscopy. All crystal structures were characterized by single-crystal X-ray crystallography.
The complexes are stable in air, soluble in DMSO and insoluble in H2O. The molar conductivity values of the complexes were recorded in a 1 mM DMSO solution and are in accordance67 with the formulae found from single-crystal X-ray crystallography, i.e. complexes 4 and 6 have ΛM values of 10–15 S cm2 mol−1 (indicative of a non-electrolyte), complex 3 has a ΛM value of 55 S cm2 mol−1 indicative of a 1:1 electrolyte, while the ΛM values of complexes 1, 2 and 5 (= 70–80 S cm2 mol−1) may indicate a 1:2 electrolyte.67
The FT-IR spectra of the complexes are complicated due to the presence of similar N-heterocyclic groups of pyridine and quinazoline. The IR spectra of L1 and L2 show a weak band at 3348 cm−1 and 3379 cm−1, respectively, which is assigned to the ν(N–H) group, as well as an intense band at ∼1630 cm−1 which is assigned to the hydrazone ν(CN) group. In the IR spectrum of the complexes (Fig. S1†), similar bands are shown to be accompanied by slight shifts indicating the formation of the complexes. In the complexes, the ν(N–H) vibrations appeared at 3400–3442 cm−1 and the v(CN) vibrations at 1637–1608 cm−1. The formation of the complexes may be also verified by the appearance of medium-intensity bands in the area 527–561 cm−1 which is assigned to the ν(M–N) vibrations and are not observed on the IR spectra of ligand.68 In the IR spectra of complexes 1–3 and 5, two new bands are shown at 1381–1382 cm−1 and 800–843 cm−1 which are assigned to the ν3(NO3) and ν1(NO3) vibrations of the nitrate anions. In particular, for complex 3, the difference of Δν(NO3) (= ν3(NO3) − v1(NO3) = 147 cm−1 is in good agreement with a bidentate coordination mode of the nitrato ligand.68
The UV-vis spectra of the complexes and the ligands were recorded in a DMSO solution and as nujol mull. The UV-vis spectra of all compounds in the solid state (nujol) showed similar patterns to those recorded in the DMSO solution; therefore, the stability of the complexes in solution may be suggested. In these spectra, the bands observed in the UV or near-UV region of the spectrum located at 293–325 nm and 370–446 nm may be attributed to intra-ligand transitions, typical of CN and C bonds. In the spectra of the complexes, these bands are shifted indicating the formation of the complex. In particular, the bands of L1 and L2 at 372 nm and at 369 nm, respectively, which are associated with the hydrazone group, have suffered a bathochromic shift for complexes 1, 4 and 5 or a hypsochromic shift for complexes 2, 3 and 6. The bands at 463–478 nm may be attributed to charge-transfer transitions.
In the visible region of the spectra, characteristic bands are observed for complexes 1, 4 and 5. For Ni(II) complexes (1 and 5), three transition bands were observed which indicated an octahedral geometry. The three d–d bands located at 780–783 nm (ε = 30–35 M−1 cm−1), 590–633 nm (ε = 40–45 M−1 cm−1) and ∼470 nm (ε = 30700–42100 M−1 cm−1) may be attributed to the 3A2g → 3T2g, 3A2g → 3T1g and 3A2g → 3T1g(P) transitions, respectively. These bands are typical of octahedral high-spin Ni(II) complexes.65 For complex 4, a low-intense band appears at 690 nm (ε = 85 M−1 cm−1) which is attributed to a d–d transition which is typical of a square pyramidal geometry for copper(II) complexes.69
The 1H NMR spectra of complexes 2 and 3 were recorded in DMSO-d6. Complexes 2 and 3 seem to be stable as no significant changes were observed in the 1H NMR spectra when they were recorded at diverse time intervals up to 48 h (Fig. S4 and S5†). The full assignment was not accomplished as the peaks were very broad and overlapping.
3.2 Structure of the complexes
The crystal structures of complexes 1–6 were determined by single-crystal X-ray crystallography. The molecular structures are depicted in Fig. 2 and the selected bond distances and angles are cited in Table 1.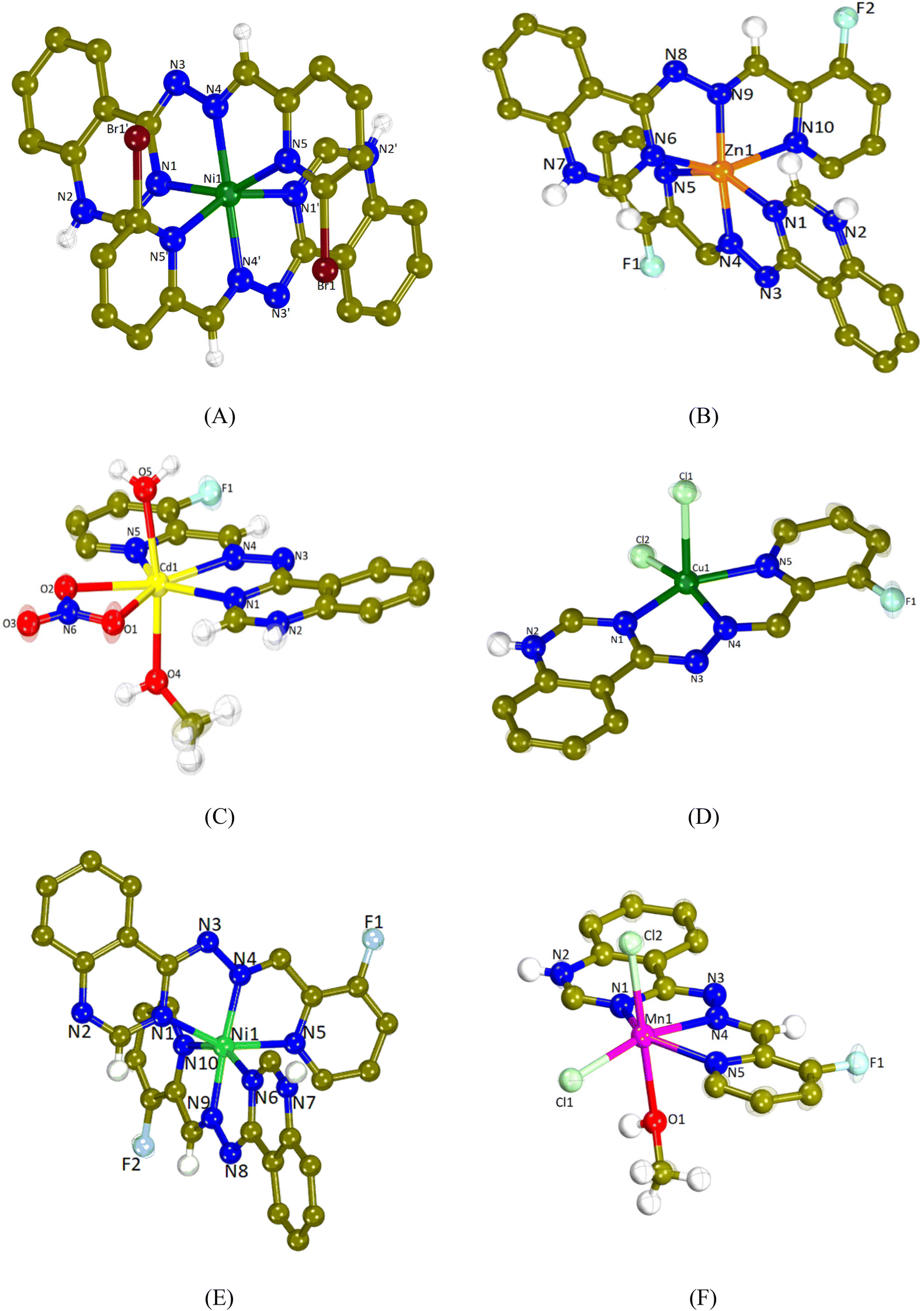 | ||
| Fig. 2 The molecular structure (the aromatic H atoms in all structures are omitted for clarity) of (A) the dication [Ni(L1)2]2+ in complex 1 (nitrate anions and solvate molecules are omitted for clarity), (B) the dication [Zn(L2)2]2+ in complex 2 (the anions NO3− and PF6− and solvate molecules are omitted for clarity), (C) the cation [Cd(L2)(H2O)(CH3OH)(NO3)]+ in complex 3 (nitrate anion is omitted for clarity), (D) [Cu(L2)Cl2] (complex 4), (E) the dication [Ni(L2)2]2+ in complex 5 (nitrate anions and solvate molecules are omitted for clarity), and (F) [Mn(L2)(CH3OH)Cl2] (complex 6) (solvate molecules are omitted for clarity). | ||
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| M = | Ni(II) | Zn(II) | Cd(II) | Cu(II) | Ni(II) | Mn(II) |
| Bond | Distance (Å) | Distance (Å) | Distance (Å) | Distance (Å) | Distance (Å) | Distance (Å) |
| M–Nqa | 2.088(3) | 2.139(2), 2.135(2) | 2.348(3) | 1.986(4) | 2.105(3), 2.097(2) | 2.254(3) |
| M–Nha | 2.007(3) | 2.070(2), 2.090(2) | 2.339(3) | 1.959(4) | 1.984(3), 1.988(2) | 2.234(2) |
| M–Npa | 2.189(3) | 2.271(2), 2.253(2) | 2.377(3) | 2.043(4) | 2.129(3), 2.141(3) | 2.299(3) |
| Bonds | Angle (°) | Angle (°) | Angle (°) | Angle (°) | Angle (°) | Angle (°) |
|---|---|---|---|---|---|---|
| a Nq = quinazoline N; Nh = hydrazone N; Np = pyridine N; Om = methanol O; Ow = water O; and On1/2 = nitrate O. | ||||||
| Nq–M–Nq | 95.64(18) | 99.64(9) | — | — | 94.24(9) | — |
| Nh–M–Nh | 166.1(2) | 162.19(9) | — | — | 174.37(11) | — |
| Np–M–Np | 92.53(17) | 91.83(9) | — | — | 94.00(10) | — |
| Nq–M–Nh | 76.02(12) | 74.35(9), 73.77(9) | 66.81(9) | 77.42(14) | 76.02(11), 76.31(10) | 69.37(9) |
| Nq–M–Np | 151.77(11) | 148.55(9), 148.25(9) | 136.71(9) | 156.04(15) | 154.60(10), 154.06(10) | 140.30(9) |
| Nh–M–Np | 76.47(12) | 74.33(9), 74.62(9) | 69.93(8) | 79.53(15) | 78.58(11), 78.01(11) | 71.44(9) |
| Om–M–Owa | — | — | 170.91(7) | — | — | |
| Nq–M–Oma | — | — | 90.74(9) | — | — | |
| On2–M–Oma | — | — | 90.21(9) | — | — | |
| On1–M–Owa | — | — | 89.83(11) | — | — | |
| Cl1–M–Np | — | — | — | 89.18(12) | 101.31(7) | |
| Cl2–M–Nh | — | — | — | 161.32(11) | 98.18(8) | |
| Cl2–M–Nq | — | — | — | 98.53(11) | 92.66(8) | |
| Cl2–M–Np | — | — | — | 101.22(12) | 98.94(8) | |
| Cl2–M–Om | — | — | — | — | 176.29(6) | |
| Cl1–M–Cl2 | — | — | — | 101.48(5) | 96.31(5) | |
All complexes are mononuclear and the L1 and L2 ligands are coordinated to each metal ion in a similar mode. They are tridentately bound via three different nitrogen atoms; the quinazoline N1, the hydrazine N4 and the pyridine N5 resulting in the formation of two fused five-membered chelate rings.
Compounds 1, 2 and 5 are dicationic complexes of the formula [M(L)2]2+, M = Ni(II), Zn(II) and Ni(II), respectively. Complexes 1 and 5 are neutralized by two nitrate anions and complex 2 by a NO3− anion and a PF6− anion. The metal-to-ligand ratio is 1:2 and the central metal is six-coordinated with a MN6 environment. The coordination geometry of the metal cation may be described as a distorted octahedron in which the hydrazine nitrogen atoms are present at the axial positions (the Nhydrazone–M–Nhydrazone angle is the largest angle around M in the range of 162.19(9)–174.37(11)°).
Compound 3 is a monocationic complex of the formula [Cd(L2)(H2O)(CH3OH)(NO3)]+ which is neutralized by one nitrate anion. The Cd(II) cation bears a coordination CdN3O4 sphere and it is hepta-coordinated and with a distorted pentagonal bipyramid geometry. The L2 ligand is tridentately bound to the metal center via N1, N4 and N5 atoms, the bidentate nitrate anion is bound via the O1 and O2 atoms, whereas the methanol and aqua ligands are bound via the O4 and O5 atoms, respectively. The axial positions of the pentagonal bipyramid are occupied by the O4 and O5 atoms (O4–Cd1–O5 = 170.91(7)°), whereas equatorial positions are occupied by the three nitrogen atoms of L2 and two oxygen atoms of the nitrato ligand.
Compounds 4 and 6 are neutral complexes with a metal-to-ligand ratio of 1:1. In complex 4, Cu1 is five-coordinated with a CuN3Cl2 coordination sphere and its geometry may be described as a distorted square pyramid as deduced by the trigonality index, τ5 (τ5 = (φ1 − φ2)/60°; φ1 and φ2 are the largest angles in the coordination sphere; τ = 0 is found for a perfect square pyramid and τ = 1 for a perfect trigonal bipyramid) which is 0.088 (Cl2–Cu1–N4 = 161.32(11)° and N1–Cu1–N5 = 156.04(15)°).70 The apical position of the pyramid is occupied by Cl1 (Cu1–Cl1 = 2.5595(16) Å) and the basal positions are occupied by the three N1, N4 and N5 atoms of FHL2 and the second chlorido ligand Cl2. In complex 6, the coordination sphere around the six-membered Mn1 is MnN3Cl2O leading to the distorted octahedral geometry. The axial positions are occupied by Cl2 and O1 (Cl2–Mn1–O1 = 176.29(6)°), while Cl1 lies in the plane formed by N1, N4 and N5 of L2. A series of hydrogen bonds offer further stabilization to the structures of complexes 1–3, 5 and 6 (Table S2†).
3.3 Interaction of the compounds with CT DNA
The interaction of all compounds with CT DNA was investigated by UV-vis spectroscopy and DNA viscosity measurements and indirectly via competitive studies with EB by fluorescence emission spectroscopy.UV-vis spectroscopy is a basic technique for the preliminary evaluation of the interaction mode of compounds with DNA. It may provide useful information to suggest the interaction mode (either covalent or noncovalent interactions or even cleavage) and to determine its strength by allowing us to calculate the DNA-binding constant (Kb). During the spectroscopic titrations, any change(s) in the charge-transfer or intraligand band(s) of the compounds may indicate the existence of interaction.71 These experiments may provide the first indication of interaction with DNA.
The UV-vis spectra of the compounds were recorded in the presence of increasing amounts of a CT DNA solution (Fig. S6†). In most cases, a slight hypochromism is observed with the simultaneous red-shift of most bands. In addition, the CT bands of the complexes located at 463–478 nm show a notable hyperchromism (Table 2). Therefore, a discrete conclusion of the DNA interaction mode of the compounds may not be safely suggested and more studies, such as DNA viscosity measurements and competitive studies with EB, were performed in order to define more firmly the interaction mode.
| Compounds | Band (ΔA/A0a (%), Δλb (nm)) | K b (M−1) |
|---|---|---|
| a “+” denotes hyperchromism and “—” denotes hypochromism. b “+” denotes red-shift and “—” denotes blue-shift. c “≫+” = high hyperchromism. d “elim” = eliminated. | ||
| L1 | 300 (−4, 0); 377 (−4, 0) | 5.32(±0.39) × 104 |
| L2 | 303 (−4, +2); 369 (−14, +2) | 3.64(±0.10) × 104 |
| [Ni(L1)2](NO3)2, 1 | 298 (+10, +3); 413 (−40, +25); 434 (−14, +13); 472 (≫+c, −2) | 4.70(±0.32) × 105 |
| [Zn(L2)2](NO3)(PF6), 2 | 293 (0, −3); 375 (−60, elimd); 463 (≫+, −2) | 5.91(±0.21) × 105 |
| [Cd(L2)(H2O)(CH3OH)(NO3)](NO3), 3 | 300 (−11, −6); 370 (−69, elim); 468 (≫+, −5) | 2.86(±0.22) × 106 |
| [Cu(L2)Cl2], 4 | 311(+10, +3); 414 (−28, elim); 446 (+7, +5); 478 (+17, −2) | 2.15(±0.25) × 106 |
| [Ni(L2)2](NO3)2, 5 | 293 (+16, −3); 411 (−43, elim); 438 (+1, +8); 471 (+40, −1) | 1.08(±0.45) × 106 |
| [Mn(L2)(CH3OH)Cl2], 6 | 296 (−2, −15); 370 (−28, +3); 468 (≫+, −2) | 4.94(±0.30) × 105 |
The DNA-binding constants (Kb) of the compounds (Table 2) have been calculated with the Wolfe–Shimer equation (eqn (S1)†)72 and the corresponding plots [DNA]/(εA − εf) versus [DNA] (Fig. S7†). In brief, the Kb values of the complexes are significantly higher (>10 times) than those of the corresponding ligands L1 and L2, with the Kb values of complexes 3 and 4 being the highest among the complexes suggesting the high affinity for CT DNA. Furthermore, the Kb constants of complexes 1–6 are higher than that of the classical intercalator EB (= 1.23 × 105 M−1) as calculated by Dimitrakopoulou et al.73 A comparison with the previously reported non-halogenated quinazoline L may reveal that the insertion of the halogens Br- or F- in the pyridine ring may result to lower Kb values for the organic compounds.27 In the case of the complexes, complexes 3–5 present higher Kb values than their analogues.25–27
Viscosity measurements of a CT DNA solution were carried out in order to define the type of interaction of the complexes with DNA. DNA-viscosity is sensitive to length variations being a very reliable technique to investigate the interaction mode between compounds and DNA. A significant increase in DNA-viscosity results from classic intercalation agents that penetrate DNA-bases and thus increase the overall DNA-length.74 Less-pronounced or no changes in DNA-viscosity are usually due to nonclassical intercalation (i.e. groove-binding or electrostatic interactions).75 The viscosity of a CT DNA solution (0.1 mM) was monitored upon the addition of increasing amounts of the compounds (up to the value of r = 0.36, Fig. 3(A)). The results demonstrate that all complexes induce a notable increase in relative DNA-viscosity, higher than the viscosity of free L1 and L2, with complex 5 inducing the higher increase. Therefore, intercalation may be suggested as the most prevailing interaction mode of the compounds with CT DNA.
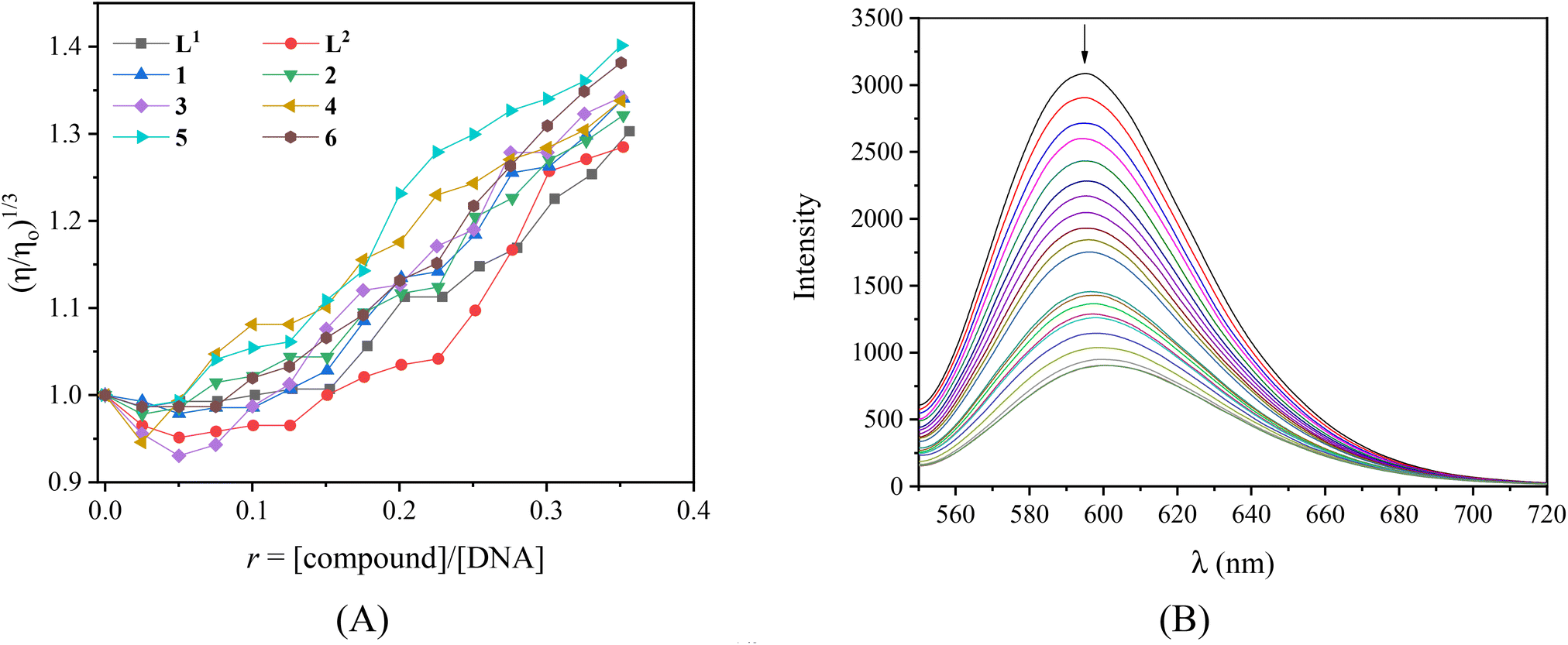 | ||
| Fig. 3 (A) Relative viscosity of CT DNA (η/η0)1/3 in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0) in the presence of the compounds at increasing amounts (r = [compound]/[DNA]). (B) Fluorescence emission spectra (λexc = 540 nm) of EB-DNA ([EB] = 20 μM, [DNA] = 26 μM) in buffer solution in the absence and presence of increasing amounts of [Zn(L2)2](NO3)(PF6) (complex 2) (r = [complex]/[DNA] = 0–0.2). The arrow shows the changes in intensity upon increasing the amounts of complex [Zn(L2)2](NO3)(PF6). | ||
As a typical DNA-intercalation marker, EB intercalates in-between neighboring DNA-base pairs. When a solution containing the EB-DNA adduct is excited at 540 nm, the intense emission band at 592–594 nm is typical of the intercalation of EB.76 Within this context, the competition of the compounds with EB for the DNA-intercalation sites may prove the intercalation of the compounds to DNA. The EB–DNA conjugate was formed after 1 h of pre-treatment of an EB solution ([EB] = 20 μM) with CT DNA ([DNA] = 26 μM) which exhibited an intense fluorescence emission band at 593 nm. The fluorescence emission spectra (λex = 540 nm) of the EB–DNA solution were recorded in the presence of increasing amounts of the compounds (Fig. S8,† and representatively shown for complex 2 in Fig. 3(B)) and showed significant quenching (up to 72.2% of the initial intensity for complex 6, Fig. S9,†Table 3).
| Compounds | ΔI/I0 (%) | K SV (M−1) | k q (M−1 s−1) |
|---|---|---|---|
| L1 | 44.5 | 6.22(±0.09) × 104 | 2.70(±0.04) × 1012 |
| L2 | 47.5 | 7.34(±0.08) × 104 | 3.19(±0.04) × 1012 |
| [Ni(L1)2](NO3)2, 1 | 35.4 | 9.36(±0.38) × 104 | 4.07(±0.17) × 1012 |
| [Zn(L2)2](NO3)(PF6), 2 | 71.3 | 1.97(±0.05) × 105 | 8.57(±0.19) × 1012 |
| [Cd(L2)(H2O)(CH3OH)(NO3)](NO3), 3 | 45.5 | 6.17(±0.19) × 104 | 2.68(±0.08) × 1012 |
| [Cu(L2)Cl2], 4 | 56.3 | 1.03(±0.02) × 105 | 4.49(±0.09) × 1012 |
| [Ni(L2)2](NO3)2, 5 | 56.1 | 2.45(±0.03) × 105 | 1.07(±0.01) × 1013 |
| [Mn(L2)(CH3OH)Cl2], 6 | 72.2 | 3.60(±0.13) × 105 | 1.57(±0.06) × 1013 |
The observed quenching of the EB-DNA fluorescence emission band is in agreement with the linear Stern–Volmer equation (eqn (S2)†)76 as shown in the corresponding Stern–Volmer plots (R ∼ 0.99, Fig. S10).† The EB-DNA quenching constants of the compounds (kq) have been calculated according to eqn (S3),† where the fluorescence lifetime of the EB-DNA system bears the value τ0 = 23 ns.77 The kq constants (Table 3) are significantly higher than 1010 M−1 s−1 suggesting that the quenching of the EB-DNA fluorescence induced from the complexes takes place via a static mechanism which leads to the formation of a new conjugate, obviously between DNA and the compound, verifying indirectly the EB-displacement and subsequently the intercalation of the compounds to CT DNA.
In conclusion, the complexes may interact with CT DNA via intercalation as suggested, while for complexes 1–3 and 5 an electrostatic interaction may also co-exist as a result of the cationic charge as found for their analogues with the non-halogenated quinazoline L as the ligand.25–27
3.4 Interaction of the compounds with plasmid DNA
The compounds L1, L2 and complexes 1–6 (DMSO solution of various concentrations) were mixed with pBR322DNA (tris buffer solution, 25 μM, pH = 6.8). The amount of DMSO within the final mixture never exceeded 10% v/v. Four experiments have been performed with plasmid DNA and the results were analyzed by gel electrophoresis on 1% agarose stained with EB: (a) in the dark upon incubation of the components for 150 min (Fig. 4(A) and Fig. S11†); (b) upon irradiation at 312 nm (UV-B) for 30 min (Fig. 4(B) and Fig. S12†); (c) upon irradiation at 365 nm (UV-A) for 120 min (Fig. 5(A) and Fig. S13†) and (d) upon irradiation under visible light for 120 min (Fig. 5(B) and Fig. S14†). All experiments were performed twice or more. In Fig. 4 and 5, the % average of total DNA cleavage or photocleavage is provided, whereas in Fig. S11–S14† the actual pictures of the plasmid DNA reactivity are depicted showing the results of one experiment of each compound at certain concentration(s).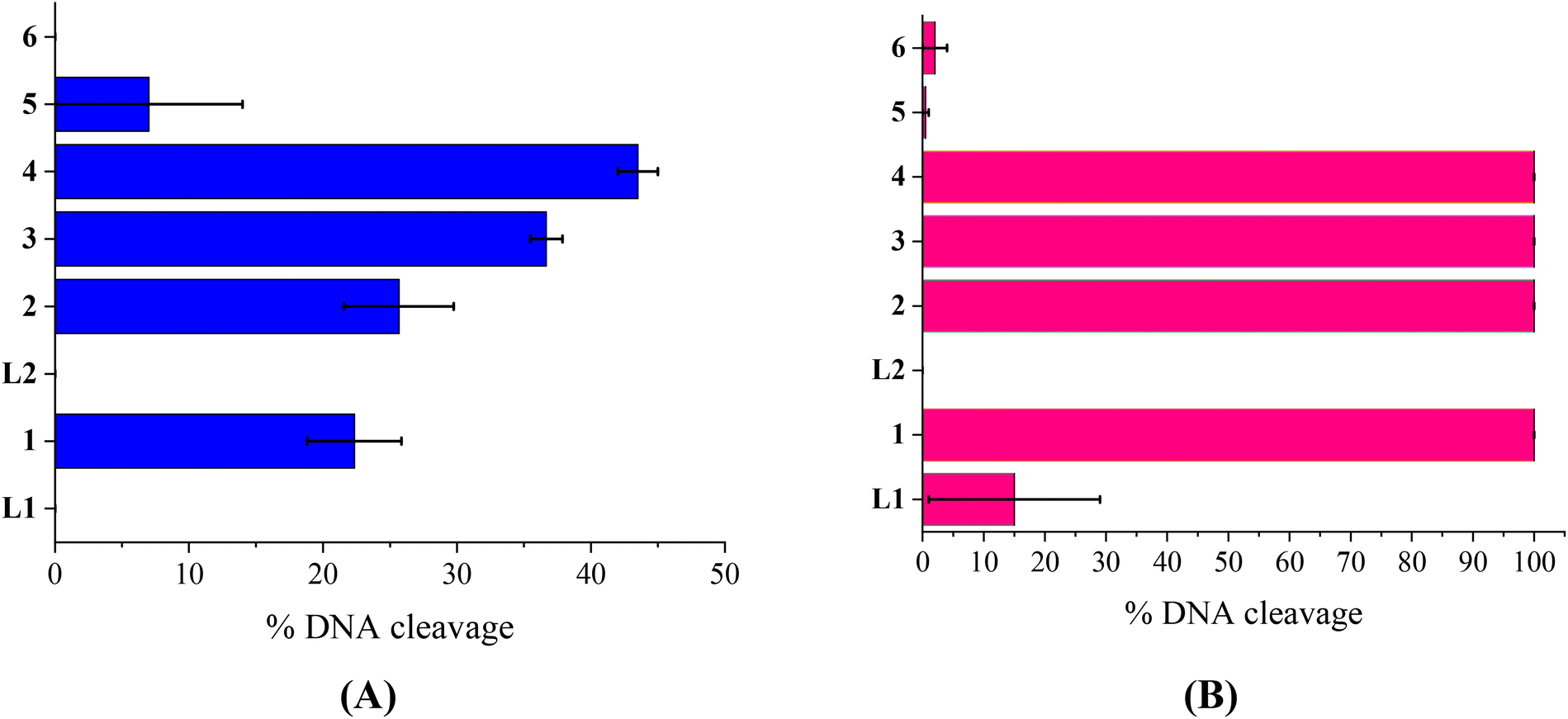 | ||
| Fig. 4 (A) Percentage (%) of total cleavage of plasmid DNA (pBR322 DNA) induced by the compounds at 500 μM. (B) Percentage (%) of total cleavage of plasmid DNA (pBR322 DNA) induced by the compounds at 500 μM upon irradiation at 312 nm. (Vertical axis shows the two ligands (L1 and L2) and complexes 1–6.) | ||
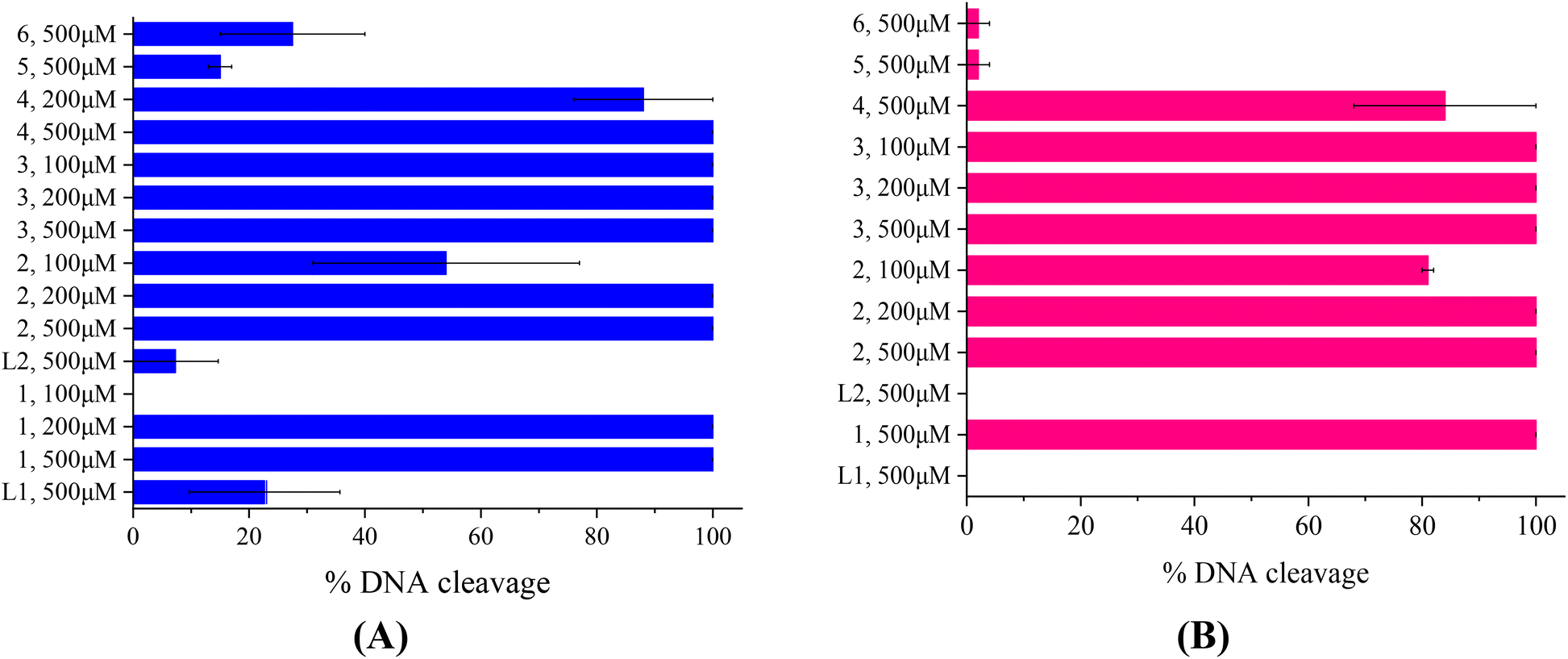 | ||
| Fig. 5 (A) Percentage (%) of total cleavage of plasmid DNA (pBR322 DNA) induced by the compounds at various concentrations (100–500 μM) upon irradiation at 365 nm after 60 min of electrophoresis. (B) Percentage (%) of total cleavage of plasmid DNA (pBR322 DNA) induced by the compounds at various concentrations (100–500 μM) upon irradiation with visible light, after 60 min of electrophoresis. (Vertical axis shows the two ligands (L1 and L2), complexes 1–6 and the corresponding concentrations.) | ||
The quinazoline compounds L1 and L2 and complex 6 have not shown any noteworthy effect on pDNA (Fig. 4(A)). On the other hand, complexes 3 and 4 have provoked single-stranded (ss) nicks up to ∼37(±1.2)% and 44(±1.5)%, respectively, reaching a close to 50% cleavage of the supercoiled plasmid at this concentration (Fig. 4(A)). Complexes 2 and 5 have shown lower cleavage activity inducing ss nicks, while complex 1 has provoked not only ss nicks but also dd nicks up to 26(±4.1)% overall (Fig. 4(A)).
Under UVB light, one may observe that L2 and complexes 5 and 6 did not cleave pDNA upon UVB irradiation, meaning that this irradiation did not cause any effect on them. On the other hand, L1 has shown some photocleavage whereas complexes 1–4 have shown remarkable photocleavage ability, exhibiting 100% photocleavage effect either with ss nicks or with ds nicks at 500 μM (Fig. 4(B) and Fig. S12†).
The photoirradiation results of the compounds with UVA light are shown in Fig. 5(A). One may observe that L2 does not show any significant cleavage effect on pDNA while L1 and complexes 5 and 6 show low damage. On the other hand, complexes 1–4 show high activity provoking total damage to pDNA at a high concentration of 500 μM (Fig. 5(A)), as the plasmid cannot be detected from EB in the case of complexes 1–3 (Fig. S13†). For these complexes, the concentration was lowered in order to reach approximately the 50% photocleavage activity of the plasmid. The best photocleavage effect was shown by complexes 1 and 2 at concentrations between 100 and 200 μM, by complex 3 much below 100 μM, and by complex 4 much below 200 μM (Fig. 5(A)).
Under visible light, compound L1 was inactive whereas L2 and complexes 5 and 6 continued to show no photocleavage activity towards pDNA. On the other hand, complexes 2 and 3 show high activity, completely destroying pDNA even at 100 μM concentration. Complexes 1 and 4 photocleave pDNA at higher concentrations (500 μM) up to 100% and 84(±16)% (Fig. 5(B)).
As a conclusion for the photocleavage of the ligands bearing the same metal but different halogen atoms (complexes 1 and 5), albeit at different positions, it seems that the brominated one (complex 1) shows extraordinary photocleavage at concentrations between 200 and 100 μM by UVA irradiation and much lower than 500 μM by visible light compared to the fluorinated complex 5 which is practically inactive. A similar nickel complex with the simple ligand L was inactive photochemically towards plasmid DNA.31 Comparing the literature data for the cleavage and photocleavage of the Zn complex ([Zn(L)2]2+) of the non-halogenated quinazolinone ligand L,27 which is a direct analogue to complex 2, it seems that fluorination of the ligand at position 3′ (L2) considerably enhanced the photocleavage activity using UVA and visible light ([Zn(L)2]2+ showed ∼ 90% pDNA photocleavage at 300 μM using both UVA and visible light27 and complex 2 100% photocleavage between 200 and 100 μM concentration using both UVA and visible light (Fig. 5)). As for compound 6 that is analogous to a similar complex of L26, the existence of Mn as a central metal seems to have diminished any photoreactivity in both complexes described herein and in the literature. Finally, a comparison of the photocleavage of complex 3 with the analogue of the non-halogenated ligand L25 favors the fluorinated one.
3.5 Radical scavenging activity of the compounds
Antioxidants protect living organisms from the damage that occurs from the uncontrolled presence of free radicals. Free radicals, such as reactive oxygen species and reactive nitrogen species, regulate the biological process conditions when they are imbalanced. If they are off-balance, oxidative stress occurs causing several human disorders, such as cancer, inflammation and aging processes. Most antioxidants exhibit free radical scavenging activity possessing interesting anticancer, anti-ageing and anti-inflammatory activities. In this study, we evaluated the antioxidant capacity of complexes 1–6 and L1 and L2via free radical scavenging studies, such as DPPH, ABTS and H2O2, and they were compared with selected reference compounds NDGA, BHT, Trolox and L-ascorbic acid (Table 4).| Compounds | DPPH%, 30 min | DPPH%, 60 min | ABTS% | H2O2% |
|---|---|---|---|---|
| L1 | 17.09 ± 0.27 | 18.17 ± 0.08 | 19.91 ± 1.39 | 69.54 ± 0.32 |
| L2 | 2.88 ± 0.25 | 3.51 ± 1.19 | 60.23 ± 0.83 | 70.97 ± 0.54 |
| [Ni(L1)2](NO3)2, 1 | 16.62 ± 0.82 | 16.84 ± 0.88 | 1.72 ± 0.93 | 98.05 ± 0.70 |
| [Zn(L2)2](NO3)(PF6), 2 | 51.32 ± 0.05 | 56.58 ± 0.65 | 83.71 ± 0.59 | 98.57 ± 0.19 |
| [Cd(L2)(H2O)(CH3OH)(NO3)](NO3), 3 | 8.80 ± 1.12 | 6.71 ± 0.20 | 35.98 ± 1.06 | 84.62 ± 0.47 |
| [Cu(L2)Cl2], 4 | 21.99 ± 2.35 | 23.31 ± 2.32 | 17.21 ± 0.65 | 84.72 ± 0.15 |
| [Ni(L2)2](NO3)2, 5 | 5.49 ± 0.33 | 3.12 ± 0.60 | 8.36 ± 0.41 | 87.46 ± 0.19 |
| [Mn(L2)(CH3OH)Cl2], 6 | 10.72 ± 0.20 | 12.27 ± 0.60 | 54.98 ± 0.41 | 96.45 ± 0.71 |
| NDGA | 87.08 ± 0.12 | 87.47 ± 0.12 | Not tested | Not tested |
| BHT | 61.30 ± 1.16 | 76.78 ± 1.12 | Not tested | Not tested |
| Trolox | Not tested | Not tested | 98.10 ± 0.48 | Not tested |
| L-Ascorbic acid | Not tested | Not tested | Not tested | 60.80 ± 0.20 |
The DPPH-scavenging activity is the most common method employed to evaluate the free radical scavenging activity of the compounds. DPPH has a characteristic absorption at 527 nm and the degree of the discoloration from the dark violet color demonstrates the scavenging potential of the compounds. The DPPH-scavenging activity of all complexes is low-to-moderate. Complex 2 shows the best activity among compounds (DPPH% = 56.58 ± 0.65%). All compounds show lower scavenging activity compared to the reference compounds NDGA and BHT.
ABTS radicals are used as a color reagent to determine the total oxidation capacity of a solution. ABTS˙ shows a strong absorption at 734 nm and its discoloration suggests the antioxidant properties of the compounds. As shown in Table 4, the scavenging activity of compounds L1, 1, 4 and 5 is rather low, while for L2, 3 and 6, it is moderate. Among the compounds, complex 2 shows the most remarkable ABTS-scavenging activity (ABTS% = 83.71 ± 0.59%).
Hydrogen peroxide is known to play an important role in killing several bacterial and fungal strains. There is increasing evidence that hydrogen peroxide either directly or indirectly via its reduction product OH˙ can act as a messenger molecule in the synthesis and activation of several inflammatory mediators. When a scavenger is incubated with H2O2 using a peroxidase assay system, the loss of H2O2 can be measured.78 All complexes show a high ability to reduce H2O2, higher than free ligands and reference compound L-ascorbic acid, and complexes 1 and 2 show the highest activity (= 98.05–98.57%).
In conclusion, all complexes seem to exhibit significant activity towards H2O2 showing a catalase-like activity. In total, complex 2 seems to have the best antioxidant activity among the present compounds.
3.6 Albumin-binding properties of the compounds
Serum albumins are among the most important proteins of the circulatory system due to their involvement in the transportation of drugs and other bioactive small molecules through the bloodstream.79,80 BSA is the most widely studied albumin and is a structural homologue to HSA. BSA has two tryptophan residues (Trp-134 and Trp-212 in the subdomains IB and IIA, respectively) that are responsible for the intense fluorescence emission band with λem,max = 343 nm for BSA, when its solutions are excited at 295 nm.76 When the compounds were added to the BSA solution (representatively shown for complex 1 in Fig. 6(A)), significant quenching was observed (up to 77.4% of the initial fluorescence for complex 2, Fig. S10,†Table 5). The inner-filter effect was evaluated using eqn (S6)81 and it was found to be negligible to affect the measurements.
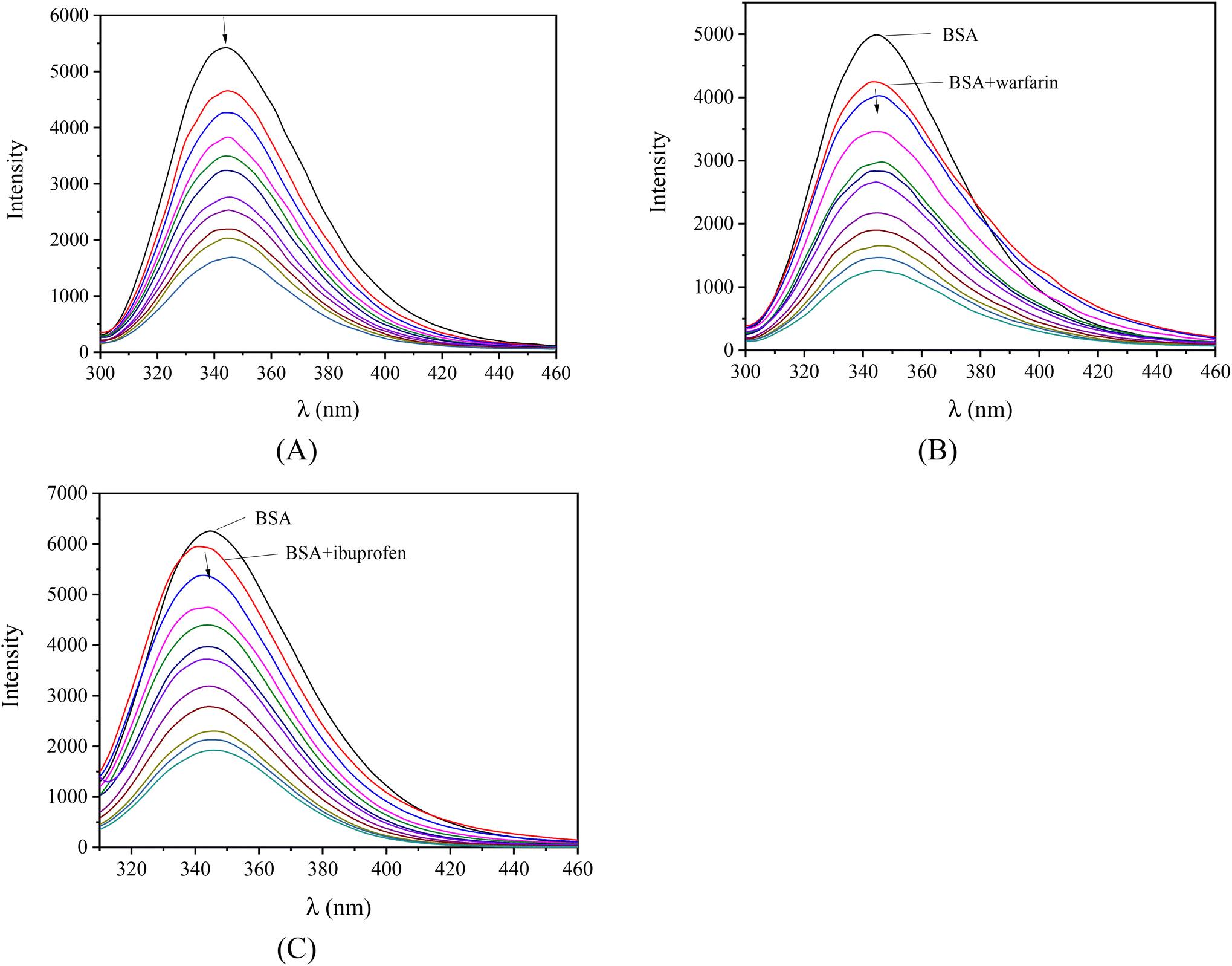 | ||
| Fig. 6 Fluorescence emission spectra (λexcitation = 295 nm) of BSA ([BSA] = 3 μM) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0) (A) in the presence of increasing amounts of [Ni(L1)2](NO3)2, (complex 1) (r = [complex]/[BSA] = 0–6), (B) in the presence of warfarin (3 μM) upon the addition of increasing amounts of [Ni(L1)2](NO3)2, (complex 1) (r = [complex]/[BSA] = 0–6), and (C) in the presence of ibuprofen (3 μM) upon the addition of increasing amounts of [Ni(L2)2](NO3)2, (complex 5) (r = [complex]/[BSA] = 0–6). The arrows show the changes in intensity upon increasing the amounts of the complexes. | ||
| Compound | ΔI/I0 (%) | k q (M−1 s−1) | K (M−1) |
|---|---|---|---|
| L1 | 61.5 | 8.36(±0.26) × 1012 | 2.07(±0.15) × 105 |
| L2 | 56.1 | 7.25(±0.15) × 1012 | 6.15(±0.39) × 104 |
| [Ni(L1)2](NO3)2, 1 | 69.0 | 1.02(±0.02) × 1013 | 8.18(±0.39) × 104 |
| [Zn(L2)2](NO3)(PF6), 2 | 77.4 | 1.83(±0.08) × 1013 | 3.46(±0.12) × 105 |
| [Cd(L2)(H2O)(CH3OH)(NO3)](NO3), 3 | 57.2 | 7.17(±0.21) × 1012 | 1.36(±0.08) × 105 |
| [Cu(L2)Cl2], 4 | 55.5 | 7.16(±0.25) × 1012 | 7.56(±0.40) × 104 |
| [Ni(L2)2](NO3)2, 5 | 66.7 | 1.12(±0.03) × 1013 | 6.54(±0.27) × 104 |
| [Mn(L2)(CH3OH)Cl2], 6 | 63.0 | 9.26(±0.27) × 1012 | 9.40(±0.82) × 104 |
The BSA-quenching constants (kq) concerning the interaction of the compounds with BSA were determined by the Stern–Volmer quenching equation (eqn (S2) and (S3)†), taking the fluorescence lifetime of tryptophan in BSA as τ0 = 10−8 s,76 and from the corresponding Stern–Volmer plots (Fig. S12†). The obtained values of kq (Table 5) are of the order 1012–1013 M−1 s−1 and are significantly higher than 1010 M−1 s−1 indicating the existence of a static quenching mechanism76 and, subsequently, may verify the interaction of the complexes with BSA leading to the formation of an adduct between the complexes and BSA.
The values of the BSA-binding constants (K) of the compounds were calculated by the Scatchard equation (eqn (S7)†) and plots (Fig. S13†). They were similar to the complexes containing the non-halogenated quinazoline L as the ligand.25–27 The BSA-binding constants of the compounds are relatively high (of the order 104–105 M−1) with complex 2 being the best BSA-binder among the compounds. Furthermore, the magnitude of the K values of the complexes (6.15 × 104–3.46 × 105 M−1) may indicate their tight and reversible binding to BSA, when compared with the limit value of K ≈ 1015 M−1 for the strongest known non-covalent interactions, i.e. among avidin and the diverse ligands and that they can get released upon arrival at the desired biotargets.82
Furthermore, competitive experimental studies with warfarin and ibuprofen were performed in order to determine the BSA subdomain in which the compounds may bind. Warfarin and ibuprofen were used as markers for sites I and II, respectively, which are the most important sites whereas drugs are bound at Sudlow's site I located in subdomain IIA and Sudlow's site II located in subdomain IIIA. Warfarin and ibuprofen are the most prevalent BSA binding site markers showing a high binding affinity for sites I and II, respectively. The addition of the compounds to the pre-treated solution containing BSA and the site-probe (warfarin or ibuprofen) resulted in a significant quenching of the initial fluorescence emission band (representatively shown in Fig. 6(B) and (C)).
The BSA-binding constants of the compounds in the presence of warfarin or ibuprofen (Table 6) were calculated using the Scatchard equation (eqn (S7)†) and the corresponding plots (Fig. S14 and S15†). These values are compared with those determined in the absence of any site marker. If the value of K decreases in the presence of the site marker, the binding of the compound to albumin is influenced by the presence of this marker resulting from the competition for the same binding site.83
| Compounds | No markers | Warfarin | Ibuprofen |
|---|---|---|---|
| L1 | 2.07(±0.15) × 105 | 3.83(±0.32) × 104 | 6.63(±0.30) × 104 |
| L2 | 6.15(±0.39) × 104 | 6.27(±0.34) × 104 | 8.82(±0.56) × 104 |
| [Ni(L1)2](NO3)2, 1 | 8.18(±0.39) × 104 | 5.08(±0.29) × 104 | 6.94(±0.56) × 104 |
| [Zn(L2)2](NO3)(PF6), 2 | 3.46(±0.12) × 105 | 6.87(±0.38) × 104 | 1.15(±0.08) × 105 |
| [Cd(L2)(H2O)(CH3OH)(NO3)](NO3), 3 | 1.36(±0.08) × 105 | 1.06(±0.07) × 105 | 9.07(±0.34) × 104 |
| [Cu(L2)Cl2], 4 | 7.56(±0.40) × 104 | 7.49(±0.59) × 104 | 7.58(±0.51) × 104 |
| [Ni(L2)2](NO3)2, 5 | 6.54(±0.27) × 104 | 4.76(±0.27) × 104 | 6.26(±0.42) × 104 |
| [Mn(L2)(CH3OH)Cl2], 6 | 9.40(±0.82) × 104 | 7.20(±0.33) × 104 | 7.83(±0.25) × 104 |
It may be noted that the BSA-binding constants of complexes 1, 2 and 5 are significantly decreased in the presence of warfarin, suggesting that these compounds may be bound to BSA at Sudlow's site I in subdomain IIA. The binding constant of complex 3 is decreased in the presence of ibuprofen, suggesting Sudlow's site II in subdomain IIIA as the most possible site of binding in BSA. In addition, L1 and complex 6 present lower K values in the presence of both markers which may indicate that they may bind to both sites without any obvious selectivity. On the other hand, the binding constants of L2 and 4 did not show notable changes suggesting that the compound does not show any selectivity upon binding at the sites Sudlow I and II.
3.7 Molecular docking calculations
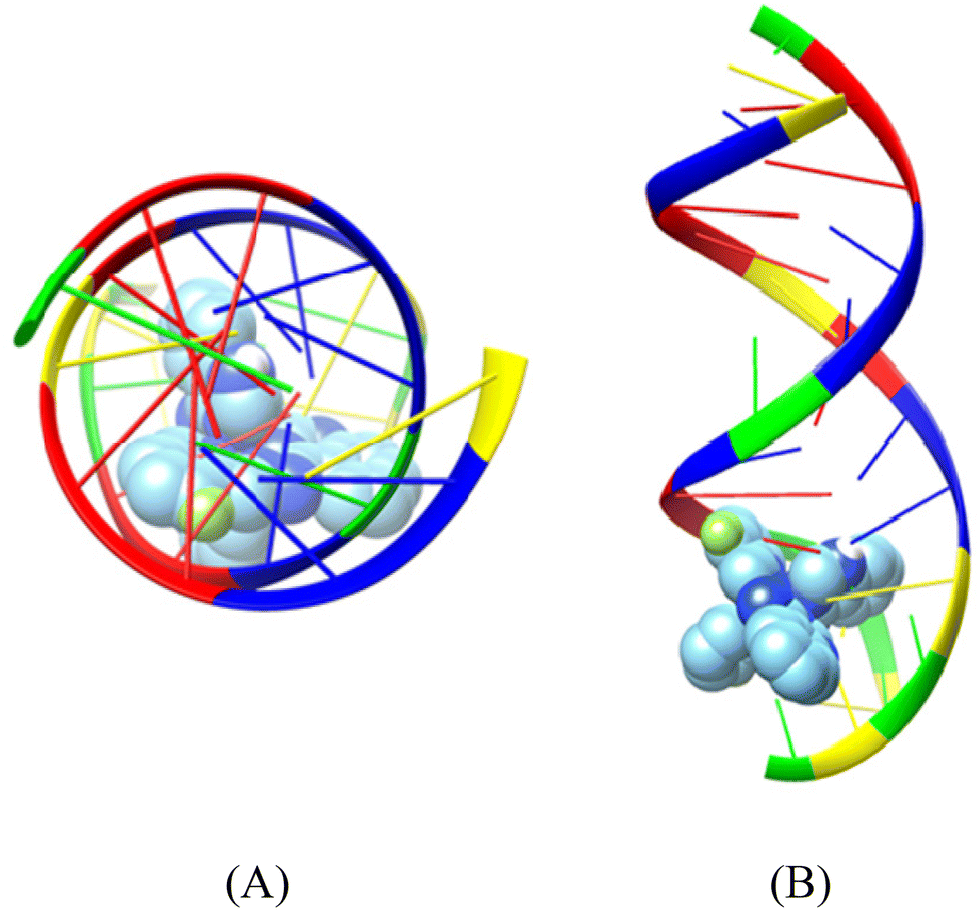 | ||
| Fig. 7 Binding pose of complex [Ni(L1)2](NO3)2 (complex 1) in the crystal structure of CT DNA (GpApApTpTpGpTpApApGpCpGpCp) (PDB ID: 2BJC) depicting its stabilization in the binding cavity of the minor grooves of DNA. (A) View above the axis of the helix, (B) Stereo view. DNA chains are illustrated as cartoon color codes (green G, red A, blue T and yellow C) and docked molecules are rendered in the spherical mode. | ||
| Complex | Minor groove |
|---|---|
| [Ni(L1)2](NO3)2, 1 | −5.8 |
| [Zn(L2)2](NO3)(PF6), 2 | −7.5 |
| [Cd(L2)(H2O)(CH3OH)(NO3)](NO3), 3 | −5.9 |
| [Cu(L2)Cl2], 4 | −6 |
| [Ni(L2)2](NO3)2, 5 | −8.1 |
| [Mn(L2)(CH3OH)Cl2], 6 | −6.3 |
The compounds are inserted between the hydrogen-bonded paired nucleotides, and the interrupted interstrand hydrogen-bond connections are listed in Table 8. All hydrogen bonds and hydrophobic contacts between the compounds and DNA are listed in Tables S3–S8.† Complex [Ni(L2)2](NO3)2 interrupts the interstrand hydrogen bond connections between the adenine–thymine pairs (the HN6 atoms of 108A and 109A and the O4 atoms of 15T and 14T, respectively) and the guanine-cytosine pairs (the HN2 atom of 110G and 12G and the O2 atom of 13C and 111C).
| Complex | Interrupted hydrogen-bonded paired nucleotides |
|---|---|
| [Ni(L1)2](NO3)2, 1 | GC |
| [Zn(L2)2](NO3)(PF6), 2 | AT, GC |
| [Cd(L2)(H2O)(CH3OH)(NO3)](NO3), 3 | AT, GC |
| [Cu(L2)Cl2], 4 | GC |
| [Ni(L2)2](NO3)2, 5 | AT, GC |
| [Mn(L2)(CH3OH)Cl2], 6 | GC |
| Complex | Site in subdomain IB | Site in subdomain IIB |
|---|---|---|
| [Ni(L1)2](NO3)2, 1 | — | −9 |
| [Zn(L2)2](NO3)(PF6), 2 | −10.2 | — |
| [Cd(L2)(H2O)(CH3OH)(NO3)](NO3), 3 | −8.8 | — |
| [Cu(L2)Cl2], 4 | −8.7 | — |
| [Ni(L2)2](NO3)2, 5 | −10.9 | — |
| [Mn(L2)(CH3OH)Cl2], 6 | — | −7.8 |
The hydrogen bonds and the polar and hydrophobic contacts that stabilize the complexes at the binding sites of BSA are listed in Tables S9–S14.† Complex [Ni(L1)2](NO3)2, 1 has the best docking score (lowest binding energy) at the binding site in subdomain IIB (Fig. 8). The binding site is located in the pocket between the helices of subdomain IIB and it is enclosed by the basic amino acid residues Arg208 and Lys350 (which stabilize the molecule via van der Waals interactions), the aromatic Phe205 (which makes strong π–π and polar interactions between the C atoms of complex and aromatic C atoms), and Ala209, Leu326 and Val481 (which are involved in hydrophobic interactions with the molecule). Complex 1 has the best docking score for the binding site in subdomain IB (Fig. 8). The binding site is located in the pocket between the helix h4 of domain IB, the long linker that precedes the helix h1 of domain IB and the helices h2 and h3 of the drug site 2 located in subdomain IIIA.84 The molecule is anchored inside this pocket due to strong interactions with a number of basic amino acid residues (such as Arg144, Arg185, Arg458, His145 and Lys114), hydrophobic residues (Ile141, Leu115, Leu189) and Ser428 (which makes polar interactions between the C atoms of the complex and the OH group).
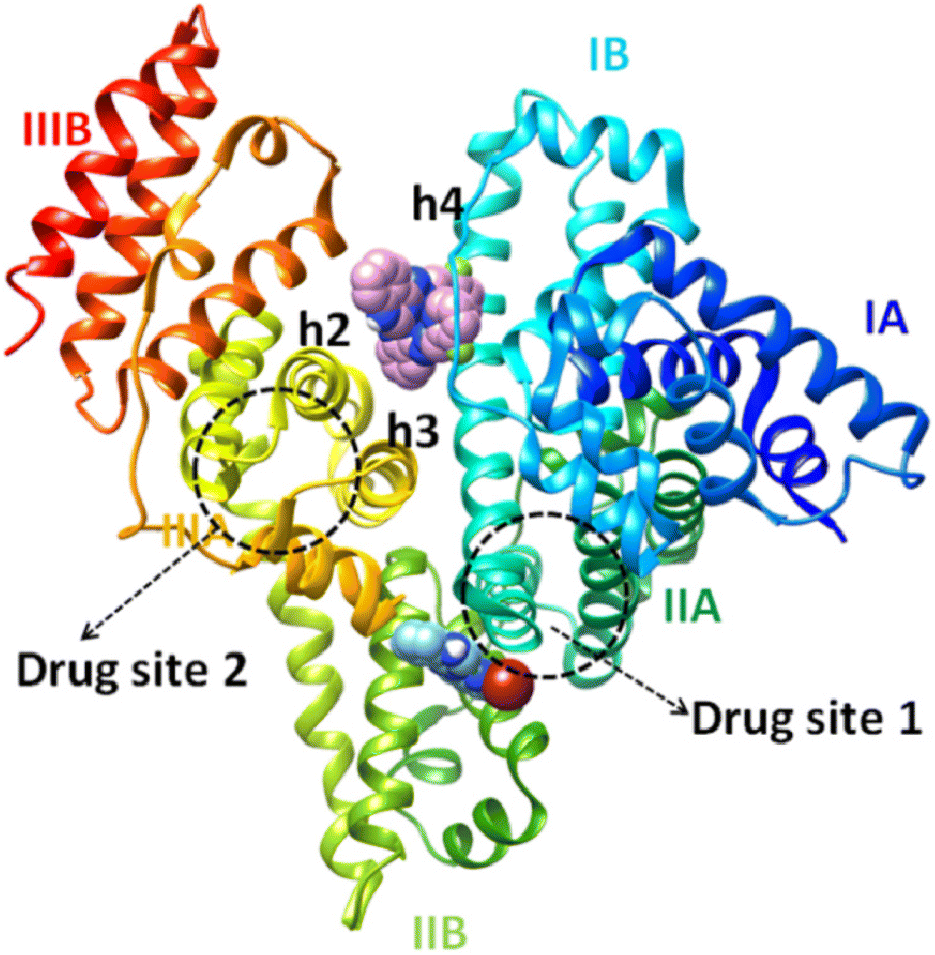 | ||
| Fig. 8 The binding sites of complexes [Zn(L2)2](NO3)(PF6) (complex 2) (upper complex) and [Ni(L1)2](NO3)2 |(complex 1) (lower complex), rendered in the spherical mode in subdomains IB and IIB of BSA protein, respectively. | ||
4 Conclusions
Two novel halogenated derivatives of (E)-4-(2-(pyridin-2-ylmethylene)hydrazinyl)quinazoline, namely (E)-4-(2-((6-bromopyridin-2-yl)methylene)hydrazinyl)quinazoline (L1) and (E)-4-(2-((3-fluoropyridin-2-yl)methylene)hydrazinyl)quinazoline (L2), were synthesized and characterized. These two compounds acted as ligands for a series of cationic or neutral transition metal(II) complexes, i.e. [Ni(L1)2](NO3)2, 1, [Zn(L2)2](NO3)(PF6), 2, [Cd(L2)(H2O)(CH3OH)(NO3)](NO3), 3, [Cu(L2)Cl2], 4, [Ni(L2)2](NO3)2, 5 and [Mn(L2)(CH3OH)Cl2], 6. The coordination of the tridentate ligands to the metal takes place via the quinazoline, the hydrazone and the pyridine nitrogen atoms.The DNA-cleavage induced by the compounds in the absence or presence of photoirradiation was studied by agarose gel electrophoresis experiments. In the absence of irradiation, the DNA cleavage is moderate and observed for concentrations above 500 μM. Upon irradiation, the DNA-cleavage is enhanced for all compounds and depends on the wavelength of the light used. The best photocleavage activities are observed for complexes 1–4 for UVA and visible light at a concentration of 100 μM with complex 3 being the most effective. Compared to the complexes of non-halogenated quinazoline L, the halogenation of the ligands either at position 6′ (Br in L1) or at position 3′ (F in L2) enhanced, in most cases, considerably DNA-photocleavage.
The interaction of the compounds with CT DNA was examined by diverse techniques, including UV–vis spectroscopy, viscosity measurements and EB-displacement studies. It has been concluded that all the compounds may interact with CT DNA via intercalation through the extended aromatic nature of the halogenated quinazoline ligands, while the cationic complexes may also interact electrostatically due to their cationic nature. Complexes 3 and 4 seem to be the tightest DNA-binders among the present compounds showing relevant high DNA-binding constants of a magnitude of 106 M−1.
The affinity studies of the compounds for BSA revealed their tight and reversible binding more selectively in Sudlow's site I in subdomain IIA.
In silico molecular modeling calculations were employed in order to provide useful information for understanding the mechanism of action of the complexes at the molecular level, indicating their ability to bind at the DNA minor groove. Further molecular docking calculations suggested the potential binding of the complexes on BSA which are mainly stabilized in subdomain IB or subdomain IIB.
Considering the antioxidant activity, the compounds have a low-to-moderate ability to scavenge DPPH and ABTS radicals with the exception of complex [Zn(L2)2](NO3)(PF6) which is a good radical scavenger. On the other hand, the results concerning the reduction of H2O2 revealed a promising catalase-like activity of the complexes.
In conclusion, the compounds presented significant radical scavenging and hydrogen peroxide reduction activity. They may bind tightly to CT DNA and may induce cleavage of plasmid DNA upon irradiation with UVA and visible light at significantly low concentrations (100 μM). These two types of biological activities constitute the components of potential anticancer activity. Thus, the present studies in combination with the ability of the compounds to bind tightly and reversibly to albumins may trigger a new group of potentially bioactive compounds which draw interest and deserve further and more elaborate biological studies.
Abbreviations
| ABTS | 2,2′-Azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) |
| BSA | Bovine serum albumin |
| BHT | Butylated hydroxytoluene |
| CT | Calf-thymus |
| DPPH | 1,1-Diphenyl-picrylhydrazyl |
| ds | Double-stranded |
| EB | Ethidium bromide, 3,8-diamino-5-ethyl-6-phenyl-phenanthridinium bromide |
| K | Albumin-binding constant |
| K b | DNA-binding constant |
| k q | Quenching constant |
| K SV | Stern–Volmer constant |
| L | (E)-4-(2-(Pyridin-2-ylmethylene)hydrazinyl)quinazoline |
| L1 | (E)-4-(2-((6-Bromopyridin-2-yl)methylene)hydrazinyl)quinazoline |
| L2 | (E)-4-(2-((3-Fluoropyridin-2-yl)methylene)hydrazinyl)quinazoline |
| NDGA | Nordihydroguaiaretic acid |
| pDNA | pBR322 plasmid DNA |
| r | [Compound]/[CT DNA] or [compound]/[BSA] |
| ss | Single-stranded |
| Trolox | 6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid |
| λ ex | Excitation wavelength |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CK acknowledges the financial support via a scholarship from the General Secretariat for Research and Technology (GSRT) and Hellenic Foundation for Research and Innovation (HFRI), Greek Ministry of Education, Research and Religion.References
- J. Dhuguru and O. A. Ghoneim, Molecules, 2022, 27, 2294 CrossRef CAS PubMed.
- R. Bansal and A. Malhotra, Eur. J. Med. Chem., 2021, 211, 113016 CrossRef CAS.
- J. V. Ragavendran, D. Sriram, S. K. Patel, I. V. Reddy, N. Bharathwajan, J. Stables and P. Yogeeswari, Eur. J. Med. Chem., 2007, 42, 146–151 CrossRef CAS.
- Banumathi, A. Sherapura, V. H. Malojirao, Zabiulla, B. S. Sharath, P. Thirusangu, R. Mahmood, N. S. Kumari, S. M. Baliga, S. A. Khanum and B. T. Prabhakar, Pharmacol. Rep., 2022, 74, 353–365 CrossRef CAS.
- N. A. Rehuman, A. G. Al-Sehemi, D. G. T. Parambi, T. M. Rangarajan, O. Nicolotti, H. Kim and B. Mathew, ChemistrySelect, 2021, 6, 7162–7182 CrossRef CAS.
- V. K. Sharma, A. Barde and S. Rattan, ARKIVOC, 2021, ix, 150–176 Search PubMed.
- W. Gan, C. Wang, Q. Pan, Y. Li, Y. Guo, D. Fan, Y. Peng, Z. Rao, S. Xu, P. Zheng and W. Zhu, Bioorg. Chem., 2022, 127, 105994 CrossRef CAS PubMed.
- T. Qin, J. Liu, J. Zhang, L. Tang, Y. Ma and R. Yang, Bioorg. Med. Chem. Lett., 2022, 72, 128877 CrossRef CAS PubMed.
- S. S. Darwish, P. Chen, M. M. Hamed, R. A. Wagdy, S. Chen, A. H. Abadi, M. Abdel-Halim, T. Hwang and M. Engel, Pharmaceuticals, 2022, 15, 778 CrossRef CAS.
- L. Liu, C. Guo, Q. Zhang, P. Xu, Y. Cui, W. Zhu, M. Fang and C. Li, J. Photochem. Photobiol., A, 2022, 423, 113593 CrossRef CAS.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- J. G. Betancourth, J. A. Castano, R. Visbal and M. N. Chaur, Eur. J. Org. Chem., 2022, e202200228 CAS.
- T. Gebretsadik, Q. Yang, J. Wu and J. Tang, Coord. Chem. Rev., 2021, 431, 213666 CrossRef CAS.
- Y. Wang, F. Zhang, Z. Liu and Z. Yao, Inorg. Chem., 2022, 61, 10310–10320 CrossRef CAS PubMed.
- S. Gayathri, P. Viswanathamurthi, K. Naveen and K. Murugan, Inorg. Chim. Acta, 2022, 537, 120957 CrossRef CAS.
- M. S. S. Adam, M. M. Makhlouf, M. A. Mohamed and A. D. M. Mohamad, Appl. Organomet. Chem., 2022, 36, e6688 CAS.
- T. T. Adejumo, N. V. Tzouras, L. P. Zorba, D. Radanovic, A. Pevec, S. Grubisic, D. Mitic, K. K. Andelkovic, G. C. Vougioukalakis, B. Cobeljic and I. Turel, Molecules, 2020, 25, 4043 CrossRef CAS PubMed.
- F. R. G. Bergamini, J. H. B. Nunes, C. M. Manzano, M. A. de Carvalho, M. A. Ribeiro, A. L. T. G. Ruiz, J. E. de Carvalho, W. R. Lustri, R. E. F. de Paiva, M. C. Portes, A. M. D. C. Ferreira and P. P. Corbi, J. Inorg. Biochem., 2022, 234, 111881 CrossRef CAS.
- C. Ghosh, D. Patra, N. Bala, I. Majumder, N. Sepay, P. Mukhopadhyay, S. Das, R. Kundu, M. G. B. Drew, A. R. Leσn, T. Ghosh and M. Pradhan, BioMetals, 2022, 35, 499–517 CrossRef CAS.
- E. A. Hassan, M. M. Ebrahium and A. M. Ebrahium, Appl. Organomet. Chem., 2022, 36, e6663 CAS.
- L. Dkhar, A. K. Verma, V. Banothu, W. Kaminsky and M. R. Kollipara, Appl. Organomet. Chem., 2022, 36, e6589 CrossRef CAS.
- J. Kimura, H. Yamada, H. Ogura, T. Yajima and T. Fukushima, Anal. Chim. Acta, 2009, 635, 207–213 CrossRef CAS PubMed.
- H. Yamada, A. Shirai, K. Kato, J. Kimura, H. Ichiba, T. Yajima and T. Fukushima, Chem. Pharm. Bull., 2010, 58, 875–878 CrossRef CAS PubMed.
- T. V. Trashakhova, E. V. Nosova, P. A. Slepukhin, M. S. Valova, G. N. Lipunova and V. N. Charushin, Russ. Chem. Bull., 2011, 60, 2347–2353 CrossRef CAS.
- C. Kakoulidou, V.-R. Kosmas, A. G. Hatzidimitriou, K. C. Fylaktakidou and G. Psomas, J. Inorg. Biochem., 2021, 219, 111448 CrossRef CAS.
- C. Kakoulidou, A. G. Hatzidimitriou, K. C. Fylaktakidou and G. Psomas, Polyhedron, 2021, 195, 114986 CrossRef.
- C. Kakoulidou, P. S. Gritzapis, A. G. Hatzidimitriou, K. C. Fylaktakidou and G. Psomas, J. Inorg. Biochem., 2020, 211, 111194 CrossRef.
- R. Ali, M. Aouida, A. A. Sulaiman, S. Madhusudan and D. Ramotar, Int. J. Mol. Sci., 2022, 23, 724 CrossRef PubMed.
- D. Tsvetkova and S. Ivanova, Molecules, 2022, 27, 2466 CrossRef PubMed.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565–4579 RSC.
- C. I. V. Ramos, A. R. Monteiro, N. M. M. Moura, M. A. F. Faustino, T. Trindade and M. G. P. M. S. Neves, Biomolecules, 2021, 11, 1404 CrossRef CAS PubMed.
- L. Andrezalova and Z. Orszaghova, J. Inorg. Biochem., 2021, 225, 111624 CrossRef CAS PubMed.
- I. M. M. de Carvalho, F. S. Gouveia, E. H. S. Sousa and L. G. F. Lopes, Metal Complexes as DNA Cleavage and Antimicrobial Agents, in Springer Handbook of Inorganic Photochemistry, ed. D. Bahnemann and A. O. T. Patrocinio, Springer Handbooks, Springer, 2022 Search PubMed.
- M. Wang and K. Ishii, Coord. Chem. Rev., 2022, 468, 214626 CrossRef CAS.
- D. N. Tritton, F. Tang, G. B. Bodedla, F. Lee, C. Kwan, K. C. Leung, X. Zhu and W. Wong, Coord. Chem. Rev., 2022, 459, 214390 CrossRef CAS.
- H. Yu, B. Yu and Y. Song, J. Coord. Chem., 2022, 75, 876–893 CrossRef CAS.
- E. M. Poland and C. C. Ho, Appl. Organomet. Chem., 2022, 36, e6746 Search PubMed.
- X. Zhao, Y. Hou, L. Liu and J. Zhao, Energy Fuels, 2021, 35, 18942–18956 CrossRef CAS.
- N. N. Rao, E. Kishan, K. Gopichand, R. Nagaraju, A. M. Ganai and P. V. Rao, Chem. Data Collect., 2020, 27, 100368 CrossRef CAS.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS.
- J. Karges, Angew. Chem., Int. Ed., 2022, 61, e202112236 CrossRef.
- S. R. Alves, I. R. Calori and A. C. Tedesco, Mater. Sci. Eng., C, 2021, 131, 112514 CrossRef.
- K. Mu, Z. Zhu, A. Abula, C. Peng, W. Zhu and Z. Xu, J. Med. Chem., 2022, 65, 4424–4435 CrossRef PubMed.
- R. S. Nunes, D. Vila-Viçosa and P. J. Costa, J. Am. Chem. Soc., 2021, 143, 4253–4267 CrossRef PubMed.
- Z. Xu, Z. Yang, Y. Liu, Y. Lu, K. Chen and W. Zhu, J. Chem. Inf. Model., 2014, 54, 69–78 CrossRef.
- J. Staron, W. Pietrus, R. Bugno, R. Kurczab, G. Satała, D. Warszycki, T. Lenda, A. Wantuch, A. S. Hogendorf, A. Hogendorf, B. Duszyńska and A. J. Bojarski, Eur. J. Med. Chem., 2021, 220, 113533 CrossRef.
- C. Wang, H. Yang, J. Du and S. Zhan, Appl. Organomet. Chem., 2021, 35, e6201 Search PubMed.
- H. A. Rudbari, A. Saadati, M. Aryaeifar, O. Blacque, J. V. Cuevas-Vicario, R. Cabral, L. R. Raposo and A. R. Fernandes, Bioorg. Chem., 2022, 119, 105556 CrossRef PubMed.
- N. Kordestani, H. A. Rudbari, A. R. Fernandes, L. R. Raposo, A. Luz, P. V. Baptista, G. Bruno, R. Scopelliti, Z. Fateminia, N. Micale, N. Tumanov, J. Wouters, A. A. Kajani and A. Bordbar, Dalton Trans., 2021, 50, 3990–4007 RSC.
- S. Keller, A. Prescimone, H. Bolink, M. Sessolo, G. Longo, L. Martínez-Sarti, J. M. Junquera-Hernández, E. C. Constable, E. Ortí and C. E. Housecroft, Dalton Trans., 2018, 47, 14263–14276 RSC.
- T. Nakanishi, A. Okazawa and O. Sato, Inorganics, 2017, 5, 53 CrossRef.
- S. Thakur, D. M. Gil, A. Frontera and S. Chattopadhyay, Polyhedron, 2020, 187, 114676 CrossRef CAS.
- P. Sarbadhikary, B. P. George and H. Abrahamse, Front. Pharmacol., 2022, 13, 921729 CrossRef CAS PubMed.
- Y. Li, G. Li, Q. Zhang, Y. Li, Q. Jia, W. Zhang, X. Feng, W. Xu and J. Liu, Chem. Phys. Lett., 2021, 784, 139091 CrossRef CAS.
- E. Ivens, M. M. D. Cominetti and M. Searcey, Bioorg. Med. Chem., 2022, 69, 116897 CrossRef CAS PubMed.
- E. Fontana and E. Ignatova, Curr. Opin. Oncol., 2022, 34, 395–402 CrossRef.
- S. L. Brown and S. Kendrick, Pharmaceuticals, 2021, 14, 96 CrossRef PubMed.
- A. Lauria, G. La Monica, A. Bono and A. Martorana, Eur. J. Med. Chem., 2021, 220, 113555 CrossRef PubMed.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–211 CrossRef.
- M. F. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- Bruker Analytical X-ray Systems, Inc., Apex2, Version 2 User Manual, M86- E01078, Madison, WI, 2006 Search PubMed.
- Siemens Industrial Automation, Inc., SADABS: Area-Detector Absorption Correction, Madison, WI, 1996 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- Y. Loidreau and T. Besson, Tetrahedron, 2011, 67, 4852–4857 CrossRef CAS.
- J. He, X. Wang, X. Zhao, Y. Liang, H. He and L. Fu, Eur. J. Med. Chem., 2012, 54, 925–930 CrossRef CAS.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, Wiley, New Jersey, 6th edn, 2009 Search PubMed.
- B. J. Hathaway, Comprehensive Coordination Chemistry, ed. G. Wilkinson, Pergamon Press, Oxford, 1987, vol. 5, pp. 533–773 Search PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verchoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051–3058 CrossRef CAS.
- A. Wolfe, G. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- A. Dimitrakopoulou, C. Dendrinou-Samara, A. A. Pantazaki, M. Alexiou, E. Nordlander and D. P. Kessissoglou, J. Inorg. Biochem., 2008, 102, 618–628 CrossRef CAS.
- F. Dimiza, F. Perdih, V. Tangoulis, I. Turel, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2011, 105, 476–489 CrossRef CAS PubMed.
- P. Zivec, F. Perdih, I. Turel, G. Giester and G. Psomas, J. Inorg. Biochem., 2012, 117, 35–47 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press, New York, third edn, 2006 Search PubMed.
- D. P. Heller and C. L. Greenstock, Biophys. Chem., 1994, 50, 305–312 CrossRef CAS.
- G. K. Jayaprakasha, L. Rao and K. Sakariah, Bioorg. Med. Chem., 2004, 12, 5141–5146 CrossRef CAS.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- R. E. Olson and D. D. Christ, Annu. Rep. Med. Chem., 1996, 31, 327–336 CAS.
- L. Stella, A. L. Capodilupo and M. Bietti, Chem. Commun., 2008, 4744–4746 RSC.
- O. H. Laitinen, V. P. Hytonen, H. R. Nordlund and M. S. Kulomaa, Cell. Mol. Life Sci., 2006, 63, 2992–3017 CrossRef CAS PubMed.
- M. Lazou, A. Tarushi, P. Gritzapis and G. Psomas, J. Inorg. Biochem., 2020, 206, 111019 CrossRef CAS PubMed.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2190744–2190749. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02622h |
| This journal is © The Royal Society of Chemistry 2022 |