
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganoaluminum hydrides catalyzed hydroboration of carbonates, esters, carboxylic acids, and carbon dioxide†
Ben
Yan‡
a,
Sayan
Dutta‡
b,
Xiaoli
Ma
 *a,
Congjian
Ni
a,
Debasis
Koley
*b,
Zhi
Yang
*a and
Herbert W.
Roesky
*c
*a,
Congjian
Ni
a,
Debasis
Koley
*b,
Zhi
Yang
*a and
Herbert W.
Roesky
*c
aSchool of Chemstry and Chemical Engineering, Beijing Institute of Technology, Beijing, P. R. China. E-mail: maxiaoli@bit.edu.cn; zhiyang@bit.edu.cn
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur 741 246, India. E-mail: koley@iiserkol.ac.in
cDr. P. H. W. Roesky, Institut für Anorganische Chemie, Georg-August-Universität Göttin-gen, Tammannnstr. 4, 37077 Göttingen, Germany. E-mail: hroesky@gwdg.de
First published on 8th April 2022
Abstract
The reductive functionalization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O unit of carbonates, carboxylic acids, esters, and CO2, respectively has received great attention since its introduction. This method is often used industrially for the synthesis of high value-added energy products in chemistry. This opens up a new way forward to reduce greenhouse gases and the consumption of traditional energy sources. Herein, we report an earth-abundant, cheap, and readily available aluminum dihydride, which can catalyze the reduction of a range of carbonates, esters, carboxylic acids, and CO2, respectively in the presence of pinacolborane as a reducing agent. Moreover, we demonstrate that the reaction can proceed to obtain good yield products under mild conditions, with low catalyst loading and solvent-free reactions. The mechanism of the catalytic reduction of carbonates has been investigated.
O unit of carbonates, carboxylic acids, esters, and CO2, respectively has received great attention since its introduction. This method is often used industrially for the synthesis of high value-added energy products in chemistry. This opens up a new way forward to reduce greenhouse gases and the consumption of traditional energy sources. Herein, we report an earth-abundant, cheap, and readily available aluminum dihydride, which can catalyze the reduction of a range of carbonates, esters, carboxylic acids, and CO2, respectively in the presence of pinacolborane as a reducing agent. Moreover, we demonstrate that the reaction can proceed to obtain good yield products under mild conditions, with low catalyst loading and solvent-free reactions. The mechanism of the catalytic reduction of carbonates has been investigated.
Introduction
The reduction reaction plays a pivotal role in chemical transformation, synthesis, and recycling. Therefore, reduction reactions, especially hydrogenation reactions, have been widely used in traditional industrial, agricultural, and energy fields during the last decades. Hydroboration reaction has become a hot research topic due to its low toxicity, high reactivity, and high selectivity control in recent years as an alternative method to the traditional hydrogenation reduction process. However, it is significant to have a selective and efficient method to reduce the conversion of CO2 and derivatives into a wide range of C1 chemicals such as formaldehyde, methanol, etc.1 Therefore, the hydroboration of carbonates, carboxylic acids, esters, and carbon dioxide itself, containing CO units are great precursors.
The research on the synthesis and properties of compounds with low-valent elements and low coordination numbers of main group species are gradually developed over the past few years.2–6 In comparison with transition metals, most of the main group metals have the characteristics of earth-abundant, cheap, and environmentally friendly elements. Among them, organoaluminum compounds are attracting much attention due to the strong Lewis acidity of the central aluminum atom, which can be used for reactions with electron-rich compounds. This advantage has attracted chemists to struggle along with the development of new aluminum compounds for catalysts.7,8 In 2015 our group presented a method for the selective hydroboration of aldehydes and ketones using an organoaluminum hydride as an excellent catalyst.9 Afterward we expanded the substrates to alkynes,10 nitriles,11 and carbodiimides,12,13 while aluminum compounds for catalysis also made great progress.14–21 Given the growing interest in aluminum-catalyzed organic reactions and the fixation of carbon dioxide and its derivatives, we aimed our target at Al-catalyzed reductions. Surprisingly, we discovered that the hydroboration reactions of a broad range of cyclic and linear organic carbonates, esters, carboxylic acids, and even CO2 are efficiently catalyzed by aluminum hydrides. The reactions proceed under mild conditions. Herein, we report our results (Scheme 1).
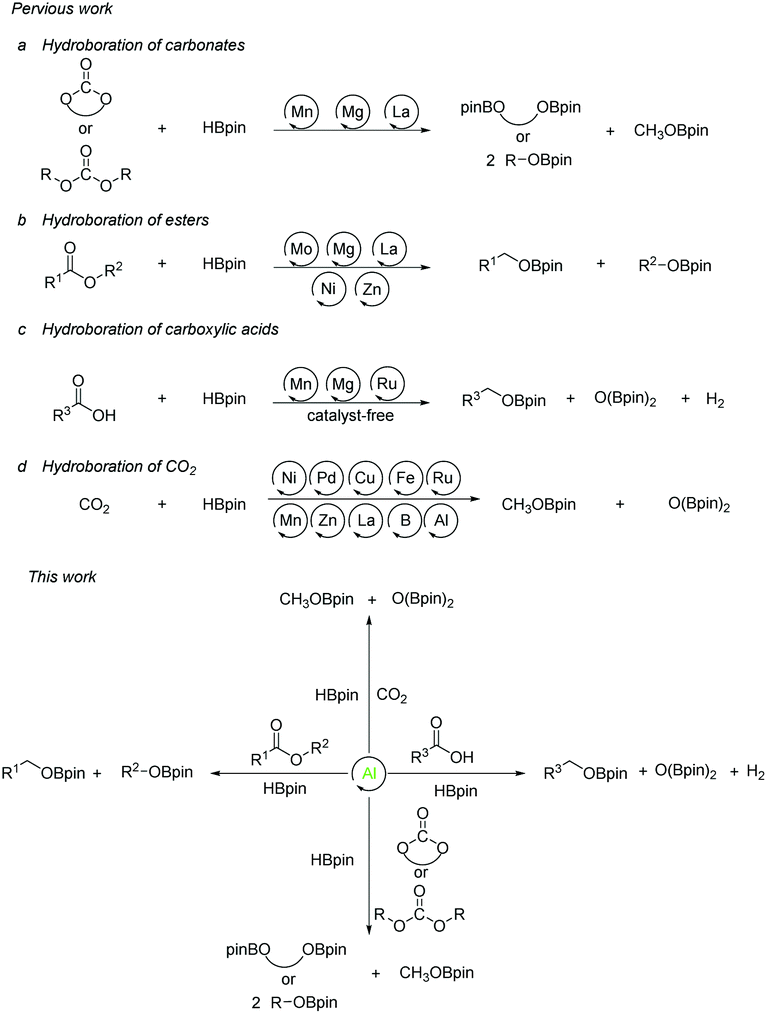 | ||
| Scheme 1 Previous approaches towards the hydroboration reactions of organic commodities and the progress of our strategy. | ||
Results and discussion
Reduction of carbonates
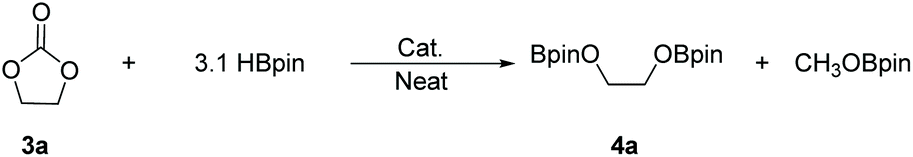
There is a common method employed in industry to reduce greenhouse gas emissions by reacting carbon dioxide with epoxides to form organic carbonates. Carbonates are widely used as solvents in various chemical reactions due to their excellent stability.22 Therefore, the reduction of carbonates becomes quite difficult. Carbonates are often directly catalyzed by hydrogenation enabling the conversion of CO2 into value-added chemicals such as methanol or diols. However, these reactions require either high temperature and pressure or transition metals as catalysts.23–30 Hence, the hydroboration reaction of carbonates can be used as an alternative method to avoid the use of flammable and explosive gases or transition metals as reductants. Nevertheless, there are a few reports on the hydroboration of carbonates (Scheme 1).29,31–34 Thus, it is crucial to develop hydroboration reactions of Al-catalyzed carbonates under mild conditions.We selected two reported aluminum hydrides LAlH2 (L = HC(CMeNAr)2 Ar = 2,6 – iPr2C6H3) (1, Fig. 1) and L′AlH2 (L′ = HC(CMeNAr)2, Ar = 2,6 – Et2C6H3) (2, Fig. 1) as catalysts and started our initial investigation by utilizing cyclic carbonate 3a as substrate and pinacolborane (HBpin) as reductant under the neat condition with catalyst loading of 5 mol%. In the first test, two aluminum hydrides were used as catalysts in the reactions which were not completed at 50 °C after 8 h (Table 1, entries 1 and 2). Extending the time to 10 h showed that large amounts of carbonate were still observed in the crude product even though the yield was improved (Table 1, entries 3 and 4). Based on the results, we noticed that aluminum dihydride 2 showed superior catalytic performance when compared with 1. The temperature of the reaction system was increased to 80 °C as a further step to achieve higher yields (Table 1, entry 5). To our delight, carbonate 3a was completely transformed at 100 °C after 10 h (Table 1, entry 6). Furthermore, when toluene and THF were selected as solvents, the yields even slightly decreased (Table 1, entries 7 and 8). With a lower loading of catalyst 2 to 3 mol%, only 91% yield was obtained (Table 1, entry 9). No expected product was formed when no catalyst was added in the control experiment (Table 1, entry 10).
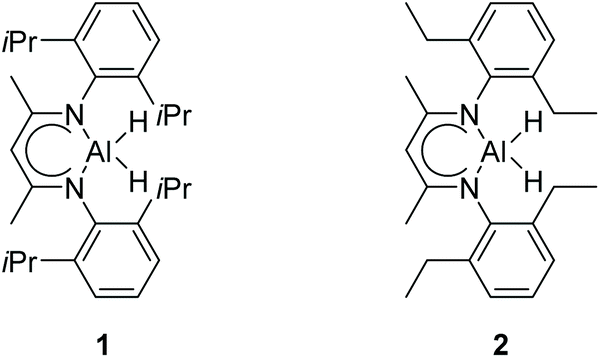 | ||
| Fig. 1 The structures of two aluminum hydrides. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | T (°C) | t (h) | Yieldb (%) |
| a Ethylene carbonate (3a) (1 mmol), HBpin (3.1 equiv.), catalysts (5 mol%). b The reaction was monitored by 1H NMR spectroscopy. c Toluene as a solvent. d THF as a solvent. e The loading of the catalyst was 3 mol%. | ||||
| 1 | 1 | 50 | 8 | 46 |
| 2 | 2 | 50 | 8 | 59 |
| 3 | 1 | 50 | 10 | 67 |
| 4 | 2 | 50 | 10 | 78 |
| 5 | 2 | 80 | 10 | 84 |
| 6 | 2 | 100 | 10 | 99 |
| 7c | 2 | 100 | 10 | 96 |
| 8d | 2 | 100 | 10 | 94 |
| 9e | 2 | 100 | 10 | 91 |
| 10 | — | 100 | 10 | 0 |
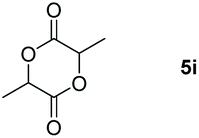
Following the optimal conditions, we investigated the substrate scope and limitation of the hydroboration of different cyclic and linear carbonates (Table 2). All reactions gave good yields using 5 mol% loadings of catalyst 2 at 100 °C under solvent-free conditions within 10 h. Five-membered ring carbonates bearing aliphatic groups such as Me and Et showed quantitative yield under the same condition of ethylene carbonate (Table 2, entries 2 and 3). When the branched-chain was replaced by the methoxy group the conversion of the hydroboration product is slightly reduced (Table 2, entry 4). Interestingly, we observed that the reduction of unsaturated carbonate occurred at the carbonate functional part rather than at the olefin group (Table 2, entry 5). 4-(Hydroxymethyl)-1,3-dioxolan-2-one bearing a hydroxyl group reacted with 4.1 equivalent of HBpin and resulted in a nearly quantitative yield of the corresponding boronate ester (Table 2, entry 6). Organic carbonates containing a six-membered ring were able to produce effective yields by reduction (Table 2, entries 7 and 9). However, 5,5-dimethyl-1,3-dioxane-2-one was observed only in 60% yield and this phenomenon may be related to the low solubility of the substrate in HBpin (Table 2, entry 8). The result is consistent with that reported in the article by Leitner et al. in 2018.29
|
|
|||
|---|---|---|---|
| Entry | Carbonate | Product | Yieldb (%) |
a Reaction conditions: 3 (1 mmol) and HBpin (3.1 mmol, 3.1 equiv.) and 2 (5 mol%), 100 °C for 10 h.
b The reaction was monitored by 1H NMR spectroscopy.
c 4.1 equiv. of HBpin was used.
d Average Mn of starting material: ∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. 000.
|
|||
| 1 |
|

|
99 |
| 2 |
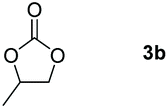
|
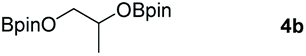
|
99 |
| 3 |
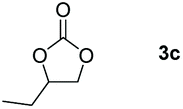
|
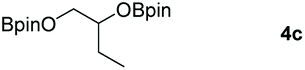
|
99 |
| 4 |
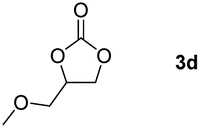
|
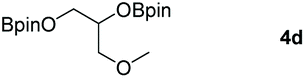
|
94 |
| 5 |
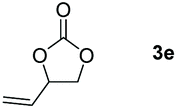
|
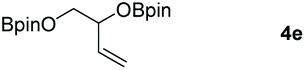
|
99 |
| 6c |
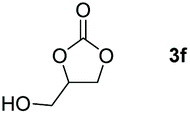
|
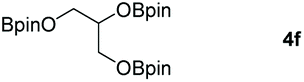
|
99 |
| 7 |
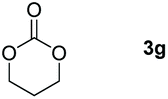
|
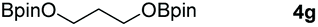
|
99 |
| 8 |
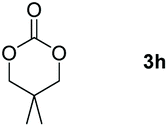
|
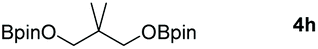
|
60 |
| 9 |
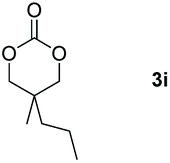
|
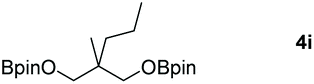
|
99 |
| 10 |
|
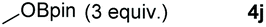
|
99 |
| 11 |
|

|
99 |
| 12 |
|

|
99 |
| 13 |
|

|
93 |
| 14 |
|

|
86 |
| 15 |
|

|
89 |
| 16d |
|
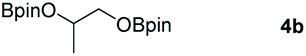
|
97 |
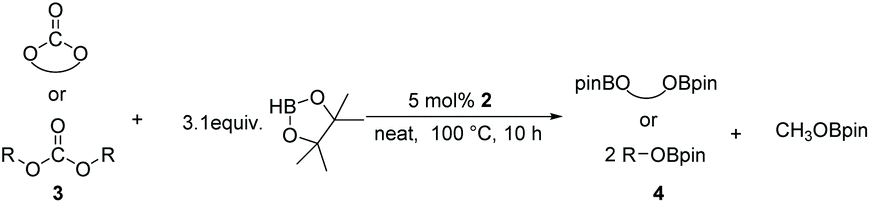
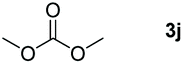
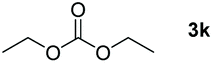
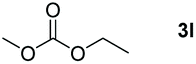
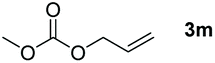
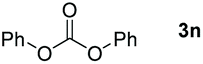
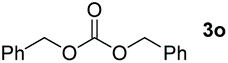
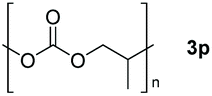
Typically, linear carbonates are more challenging to reduce than cyclic carbonates. Gratifyingly, dimethyl, diethyl, and ethyl methyl carbonates could achieve full conversion with excellent yields (Table 2, entries 10–12). The hydroboration of allyl methyl carbonate which contains the olefin group also accomplished 93% yield (Table 2, entry 13). Subsequently, we shifted our interest to the reduction of dibenzyl carbonate and diphenyl carbonate which underwent slightly decreased yields compared to the linear carbonates (Table 2, entries 14 and 15). To further investigate the applicability of aluminum hydride compound for catalyzing carbonates, we selected polypropylene carbonate as the reaction substrate which can be synthesized by propylene and CO2 industrially. Remarkably, the yield of the hydroboration product can achieve 97% conversion (Table 2, entry 16). The product can be further reacted with HCl and purified by flash chromatography to obtain the corresponding alcohol. The catalysts 1 and 2 can be easily synthesized by β-diimine and LiAlH4 or AlH3·NMe3 in yields greater than 90% at the gram scale. Even the Al-catalyzed protocol is not more effective than alkaline earth or transition metals systems (Al: 100 °C, 10 h, 99% yield vs. Mg/Mn: r.t. −90 °C, 3–8 h, 99% yield), it is also confirmed that our aluminum-based metal catalysts are based on low-cost and easily available when compared with existing protocols.29–32
Reduction of esters
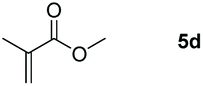
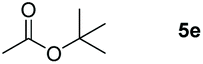
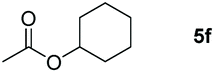
The ester linkages are both prevalent in cellulose and vegetable oils in nature and an essential transformation process in the synthesis of organic as well as of drug entities. Nevertheless, it is very challenging to reduce esters compared with the carbonyl groups of aldehydes and ketones on account of the thermodynamic inertness.35 Typically, the transformation is achieved by applying powerful reducing agents such as BH3 and LiAlH4 which require stringent reaction conditions and post-treatment processes.36–42 Therefore, the reduction of esters with boranes such as pinacolborane (HBpin) is comparatively stable and convenient to handle, thus avoiding unnecessary safety problems. As far as we know, only a few examples of hydroboration reactions of esters have been discussed in recent years and these catalysts are mainly made of rare earth and alkaline earth metals (Scheme 1).33,43–47 In some of the above descriptions, the ester is only served as a single example of substrates that have been published.43,45,48 Thus, it is necessary to develop a general protocol for the aluminum system to catalyze the hydroboration of esters.Initially, we explored whether the ester and pinacolborane would react spontaneously for lack of catalyst, unfortunately, no exciting results were detected even at temperatures up to 60 °C (see the ESI Table S1,† entries 1 and 2). Further, a detailed investigation by using aluminum dihydride 2 as a catalyst was investigated for the reduction of esters (Table S1,† entries 3–7). Noticeably, the yield increased with the rising temperature and the prolonged reaction time. Ultimately, the reaction yield achieved 99% within 12 h at a catalyst loading of 5 mol% with a temperature of 60 °C.
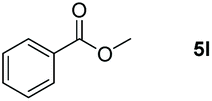
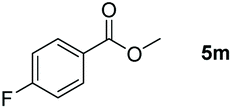
Inspired by the screening data, we evaluated the applicability of aluminum compound 2 to catalyze the hydroboration of varieties of ester substrates under optimized conditions. Table 3 shows the full scope of investigated esters. In the case of esters featuring different substituents, quantitative conversion was performed in 12 hours using 5 mol% of compound 2 at 60 °C no matter of the aliphatic or aromatic groups (Table 3, entries 1–7). It should be pointed out particularly that this catalytic system is more selective for ester groups than olefin groups (Table 3, entry 4). Next, we extended our substrate scope to cyclic esters including lactide which transformed the corresponding dialkoxy boronic esters in excellent yield (Table 3, entries 8–10). To obtain the information on the catalytic reaction of heterocyclic ester, we combined 2-coumaranone with HBpin to reach quantitative yield (Table 3, entry 11). Apart from the above substrates, a further range of electron-donating and electron-withdrawing group substrates proved to be suitable for this transition. In summary, we confirmed that this catalyst has good functional group tolerance (Table 3, entries 12–15). Similarly, esters can get the consistent conclusion with carbonates that the Al-based catalysts are not more efficient than alkaline earth metal systems (Al: 60 °C, 12 h, 99% yield vs. Mg r.t., 0.5–1 h, 99% yield).32,46
|
|
|||
|---|---|---|---|
| Entry | Ester | Product | Yieldb (%) |
| a Reaction conditions: 5 (1 mmol), HBpin (2.1 mmol, 2.1 equiv.) and 2 (5 mol%), 60 °C for 12 h. b The reaction was monitored by 1H NMR spectroscopy. c Product + EtOBpin. d Product + MeOBpin. e 4.2 equiv. of HBpin was used. | |||
| 1 |

|

|
99 |
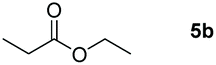
| 2c |
|

|
99 |
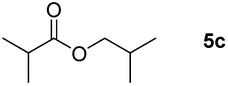
| 3 |
|
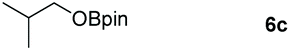
|
99 |
| 4d |
|

|
99 |
| 5c |
|

|
99 |
| 6c |
|

|
99 |
| 7 |
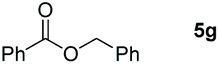
|
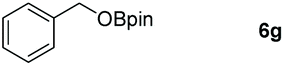
|
99 |
| 8 |
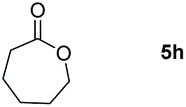
|
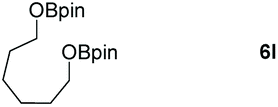
|
99 |
| 9e |
|
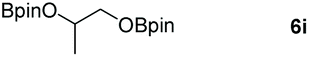
|
99 |
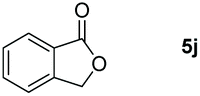
| 10 |
|

|
99 |
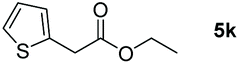
| 11c |
|
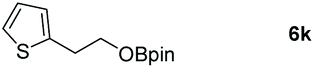
|
98 |
| 12d |
|

|
99 |
| 13d |
|
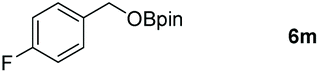
|
99 |
| 14d |
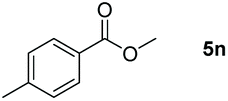
|
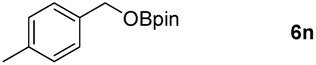
|
98 |
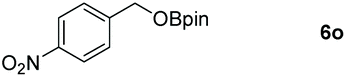
| 15d |
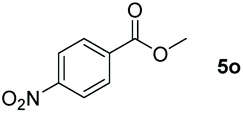
|
|
99 |
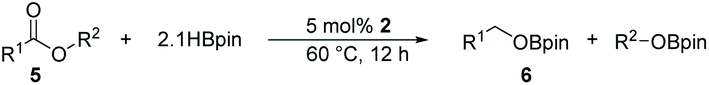
Reduction of carboxylic acids
As the broad basic unit in organic chemistry, alcohols have a wide range of applications in the fine chemical, agricultural, and pharmaceutical industries.49–52 It is important not only to generate novel functional groups in organic synthesis but also to enable biomass feedstocks for utilizing high-value chemicals. The conventional approach is to utilize metal hydrides such as LiAlH4 or Zn(BH4)2 as reducing agents, but the safety of the reaction and the subsequent disposal of the reactants pose problems.53,54 Moreover, hydrogen gas is considered to be one of the most efficient reagents for reduction. However, the extremely flammable nature of hydrogen and the demanding requirements for equipment to withstand high pressure and temperature conditions restrict its widespread application.30,55,56 Another methodology for the reduction of carboxylic acids to alcohols is through hydrosilylation, which suffers from the disadvantages of requiring transition metals (Ru, Rh, Ir, Fe, Mn, Cu, etc.) as catalysts that limit the range of substrates or require a light-mediated process.57–64Gunanathan and co-workers described the first example of a ruthenium-based catalyst acting on the reaction of the carboxylic acid with HBpin. The resulting alkyl boronate was treated hydrolytically to produce the primary alcohol. This procedure opened the way for the study of hydroboration of carboxylic acids.65 Previously, Leitner et al., as well as Maji et al., reported manganese-catalyzed borohydride reactions of carboxylic acids.29,66 However, to our knowledge, there is only one case of main group metal-catalyzed hydroboration reaction reported of carboxylic acids.67 With the increasing demand for transition metal substitution and green catalysts, there is a strong need to extend aluminum-catalyzed hydroboration reactions with unsaturated substrates to carboxylic acids, considering the interest in developing new catalysts for carboxylic acid reduction. During this protocol, there are several research groups, which have recently reported such reactions under catalyst-free and solvent-free conditions. Nevertheless, conditions such as excessive HBpin or heating are required to achieve a successful synthesis (Scheme 1).68–70
The reaction condition of the standard scheme which was obtained by screening the conditions using benzoic acid as the reaction substrate (see the ESI Table S2†). Firstly, the hydroboration reaction of benzoic acid with 3.1 equiv. of HBpin was carried out at room temperature under neat conditions. Only 57% yield was obtained after 1 hour (Table S2,† entry 1). Then, the HBpin was increased to 3.3 equiv. which resulted in a yield of 63% under the same condition (Table S2,† entry 2). We were gratified to find that the yield excellently increased after the reaction time was extended to 2 h (Table S2,† entries 3 and 4). In addition, a parallel experiment was conducted by decreasing compound 2 to 1 mol%, and the production was subsequently reduced (Table S2,† entry 5). When no catalyst was employed in the reaction, a sharp decline in the corresponding benzyl borate was detected (Table S2,† entries 6 and 7). Notably, the main group Al-catalyzed protocol is more effective when compared with transition metal systems (Al: r.t., 2 h, 99% yield vs. Ru/Mn: 60–115 °C, 20–24 h, 99% yield).29,65 Certainly, Al-catalyst has significant advantages over catalyst-free systems and lower reaction temperatures.
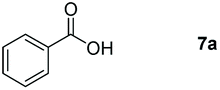
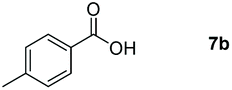
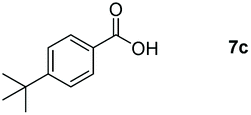
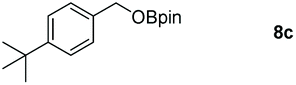
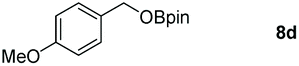
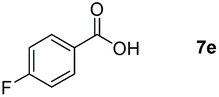
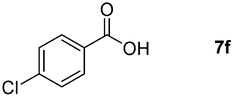
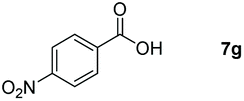
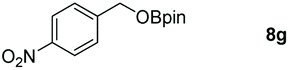
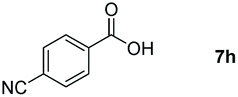
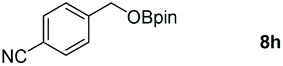
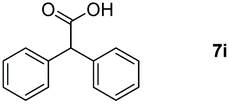
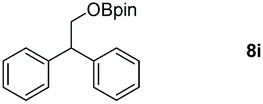
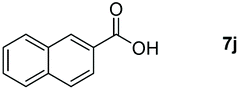
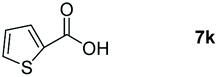
Taking the optimized reaction into account, we exploited the scope and limitations of the hydroboration of carboxylic acid. As shown in Table 4, we realized that either the aromatic acids with an electron-donating group (–Me, –tBu, –OMe) or electron-withdrawing substituents (–F, –Cl, –NO2, –CN), could afford quantitative yields of the corresponding boronate ester in 2 hours (Table 4, entries 2–8). Compounds with larger steric hindrances like diphenylacetic acid and 2-naphthoic acid also underwent a reduction reaction in excellent yield (Table 4, entries 9 and 10). Remarkably, 2-thiophenecarboxylic acid proceeded to hydroboration with HBpin, and a 99% yield was observed (Table 4, entry 11). Then, we expanded the substrate scope to the aliphatic carboxylic acids. Surprisingly, we observed that aliphatic carboxylic acids were obtained in quantitative yields under the same conditions which supported the suitability of the Al-catalyst for structurally diverse carboxylic acid substrates (Table 4, entries 12–18).
|
|
|||
|---|---|---|---|
| Entry | Carboxylic acid | Product | Yieldb (%) |
| a Reaction conditions: 7 (1 mmol), HBpin (3.3 mmol, 3.3 equiv.) and 2 (2 mol%), room temperature for 12 h. b The reaction was monitored by 1H NMR spectroscopy with mesitylene as an internal standard. | |||
| 1 |
|

|
99 |
| 2 |
|

|
99 |
| 3 |
|
|
99 |
| 4 |
|
|
96 |
| 5 |
|

|
98 |
| 6 |
|

|
99 |
| 7 |
|
|
97 |
| 8 |
|
|
99 |
| 9 |
|
|
98 |
| 10 |
|
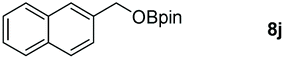
|
92 |
| 11 |
|
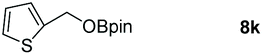
|
99 |
| 12 |

|

|
99 |
| 13 |

|

|
99 |
| 14 |
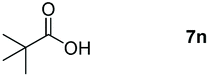
|
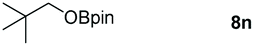
|
99 |
| 15 |
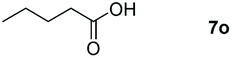
|
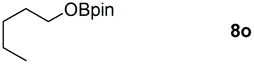
|
97 |
| 16 |
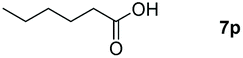
|

|
99 |
| 17 |
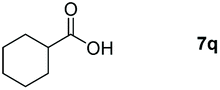
|

|
99 |
| 18 |
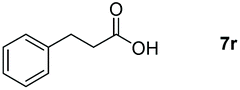
|
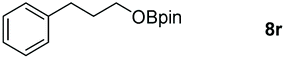
|
99 |
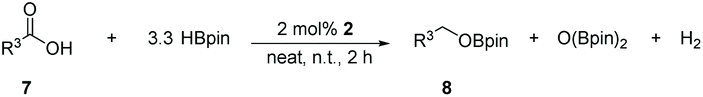
Reduction of carbon dioxide
The high-performance hydroboration catalysis of various substrates containing CO functional groups by aluminum compounds prompted us to explore directly the reduction of CO2. As a sustainable C1 feedstock, there is interest in converting from CO2 to products such as formaldehyde and methanol, which are currently synthesized from non-renewable feedstocks.71–80 Lately, the hydroboration of CO2 has received significant attention in organic synthesis. The majority of the reported catalytic systems focus on transition-metal complexes (including Ni,81,82 Pd,83–85 Cu,86 Fe,87 Ru,88,89 Co,90 Mn,29 and Zn91), alkali metal complexes,45,92 and other main-group catalysts.93–98 However, hydroboration of CO2 by using an aluminum catalyst is still rare (Scheme 1).99,100
In this case, we increased the reaction temperature and catalyst ratio to 10 mol% to ensure adequate consumption of HBpin. The experimental results showed that CO2 could be fully reduced with a yield of 95% after 48 h of stirring. With this exciting result, it offers a new approach for the direct reduction conversion of CO2 (Scheme 2).
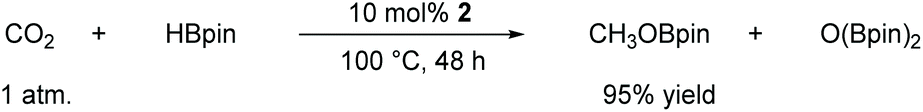 | ||
| Scheme 2 Aluminum-catalyzed hydroboration of CO2. | ||
It is interesting that the hydroboration of CO2 with HBpin using the similar organoaluminum hydrides catalyst was not observed in the previous report from Aldridge and co-workers.101 However, organoaluminum hydrides can catalyze CO2 this was observed by So and his colleagues, and they explained the reaction mechanism using DFT calculations.99 This phenomenon deserves to be further explored in our future work.
Proposed mechanism
To further understand the mechanism, we chose catalytic carbonate as the template and conducted several additional control experiments. An equimolar mixture of organoaluminum catalyst 2 with ethylene carbonate 3a and HBpin, respectively, was reacted at room temperature, and the results were analyzed by 1H NMR spectroscopy. We found that only the reaction of 2 with ethylene carbonate showed a new single resonance at 4.32 ppm in the 1H NMR with a 2H intensity by integration which is presumable to form an aluminum–oxygen compound (see ESI† for details). A detailed theoretical exploration of aluminum-catalyzed hydroboration of carbonates would be of great importance and necessary to unravel the complete mechanistic picture for this kind of fascinating system. Therefore, quantum chemical calculations were performed at the R-BP86-D3/def2-TZVP//R-BP86/def2-SVP level102 to explore the mechanistic avenues in aluminum dihydride 2-catalyzed hydroboration of cyclic carbonate 3a, leading to the formation of 4a and methoxyboronic acid pinacol ester (CH3OBpin). The reaction initiates with insertion of the carbonyl group of 3a into the Al–H bond in the catalyst to afford a significantly stable intermediate INT-1 (Fig. 2). This step needs to surmount an energy barrier of 18.8 kcal mol−1. INT-1 then rearranges to INT-2 to facilitate the approach of the oxygen (O2) center towards aluminum, which leads to the formation of an appreciably stable intermediate INT-3viaTS-2. The intramolecular hydride transfer from the aluminum center to the carbonyl carbon of INT-3 in the subsequent step promotes the liberation of formaldehyde (HCHO), giving rise to the generation of a remarkably stable intermediate INT-4. The incoming pinacolborane (HBpin) gets coordinated to the oxygen center (O3) in the resulting intermediate INT-4 to furnish a slightly less stable adduct INT-5viaTS-4 (Fig. 3).103 The subsequent hydride transfer from boron to aluminum with simultaneous Al–O3 bond rupture leads to the generation of INT-6. The step requires an intrinsic energy barrier of 5.9 kcal mol−1 and the single imaginary mode in the corresponding transition state TS-5 animates the breaking of Al–O3 (2.254 Å) and B–H (1.741 Å) bonds with concomitant Al–H (1.659 Å) bond formation. INT-6 then rearranges to INT-7 and the coordination of the second HBpin to the oxygen center (O2) in INT-7 results in INT-8viaTS-6. This step needs to overcome an energy barrier of 9.0 kcal mol−1, indicating a slightly higher energy barrier required for the coordination of the second pinacolborane compared to the first one. INT-8 then undergoes similar hydride transfer to afford the desired product 4a with the regeneration of the catalyst. This step corresponding to an overall activation barrier of 25.1 kcal mol−1 is the rate-determining step for the catalytic transformation. The calculated rate-limiting energy barrier is reasonable under the solvent-free, 100 °C reaction conditions.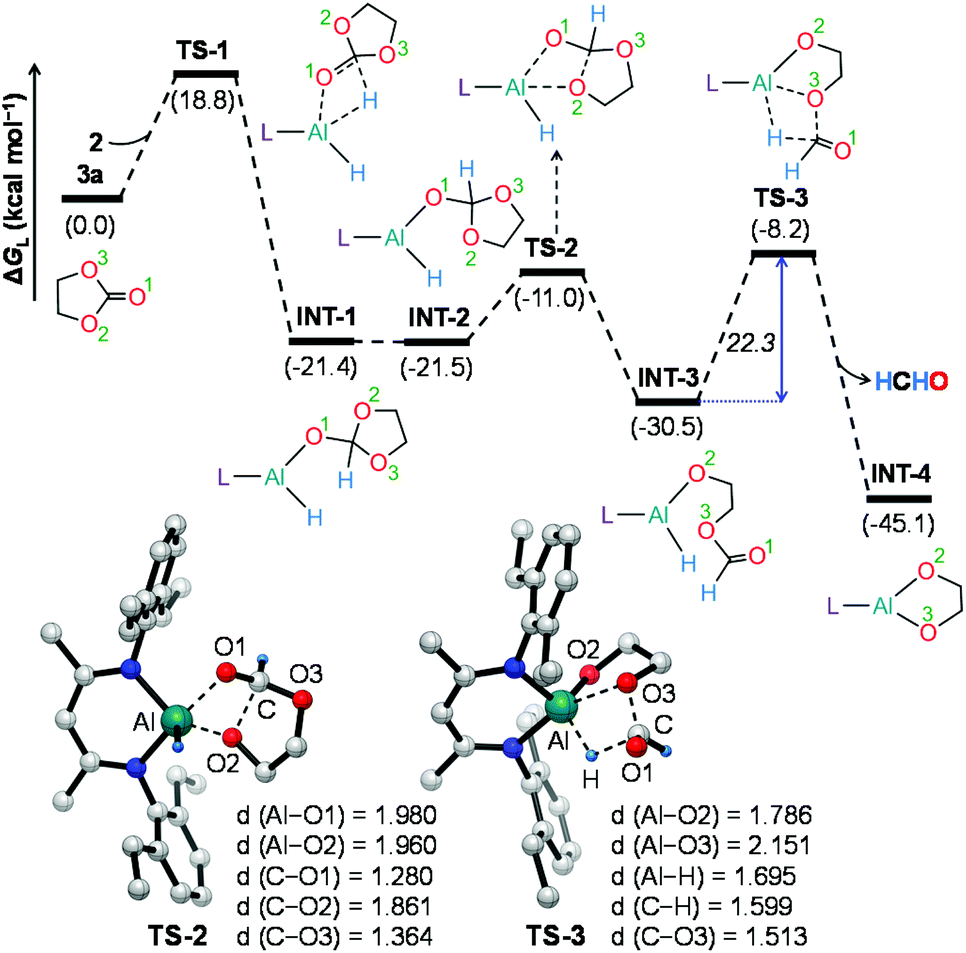 | ||
| Fig. 2 Energy profile for the formation of the intermediate INT-4 and formaldehyde. Optimized geometries of the transition states TS-2 and TS-3 with important geometrical parameters are also provided. Bond distances (d) are in angstroms (Å). Only key hydrogen atoms are shown for clarity. L = HC(CMeNAr)2, Ar = 2,6 – Et2C6H3. | ||
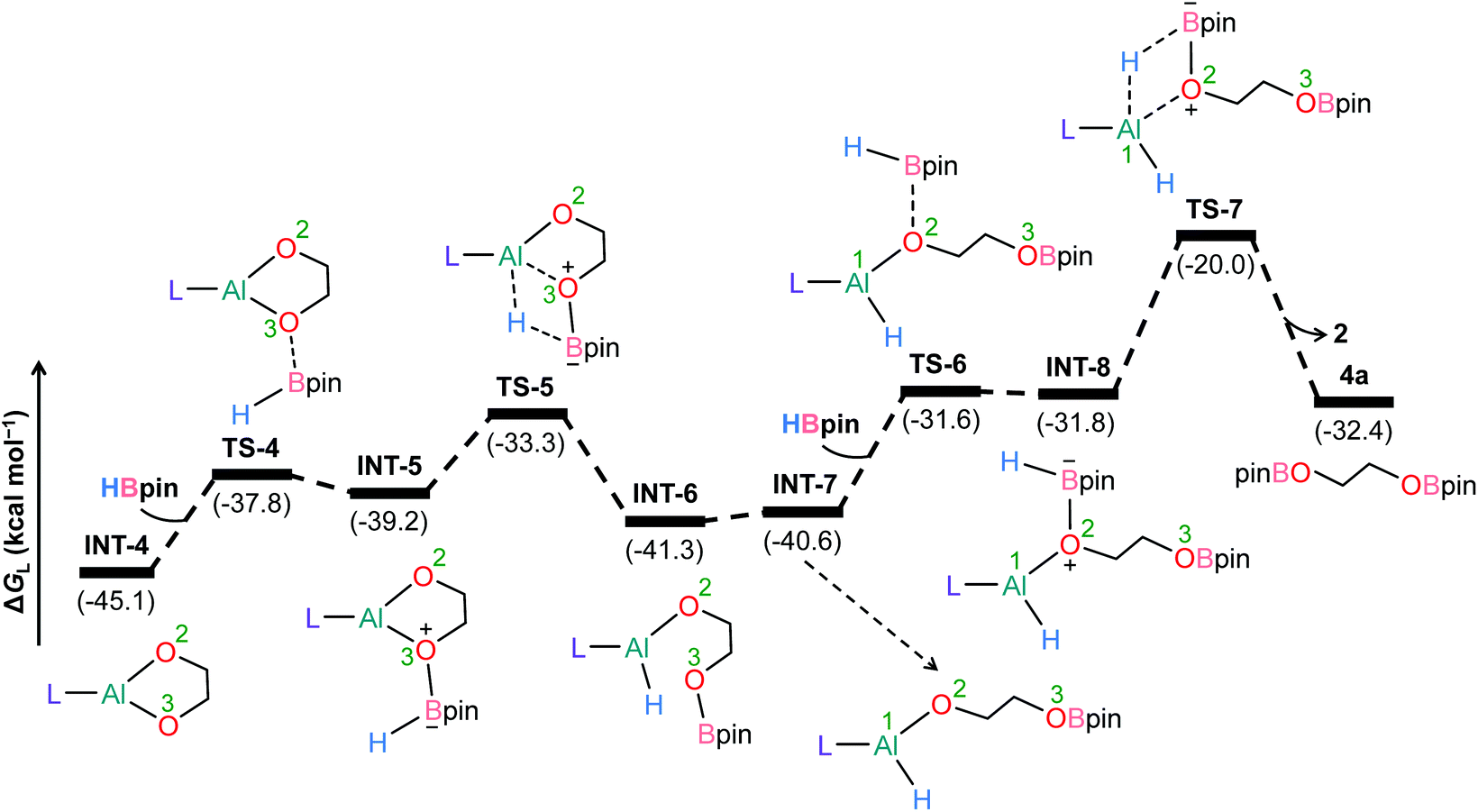 | ||
| Fig. 3 Energy profile for the 2-catalyzed formation of 4a. L = HC(CMeNAr)2, Ar = 2,6 Et2C6H3. | ||
We have also explored the alternative pathway leading to the generation of the intermediate INT-7 (Fig. 4). The intermolecular hydride transfer from the catalyst to the carbonyl carbon of INT-3 furnishes a remarkably stable intermediate INT-9 with the liberation of formaldehyde. This step needs to surmount an activation barrier of 25.9 kcal mol−1. Then similar HBpin coordination followed by hydride transfer to the aluminum center with the release of catalyst molecule affords INT-7. Though this reaction channel demands a slightly higher energy barrier compared to that of the intramolecular hydride transfer (22.3 kcal mol−1), participation of a second catalyst molecule in the reaction channel is less likely to operate under the experimental conditions. We have also checked another alternative route for the formation of INT-7 involving a substantially stable cyclic intermediate INT-13 (Fig. S5†). However, the second hydride transfer from aluminum to the carbonyl carbon in INT-2 with concurrent C–O2 bond breaking to afford INT-13 demands a drastically high energy barrier of 67.4 kcal mol−1. Additionally, participation of HBpin instead of 2 in the generation of formaldehyde from INT-3 also involves TS-18 which shows a reasonably high barrier height of 36.5 kcal mol−1 and therefore, can be safely discarded on the kinetic ground (Fig. S6†). Moreover, we have performed distortion–interaction analysis to cast light on the origin of activation barriers for TS-8 and TS-18 (Tables S3 and S4†).104–106 Though the distortion energy of the INT-3 fragment is appreciably lower in TS-18 than TS-8 [Δ‡Edist-INT-3: 27.7/22.7 kcal mol−1 in TS-8/TS-18], immensely larger distortion energy of the HBpin fragment in TS-18 compared to the catalyst fragment in TS-8 accounts for the substantially higher activation barrier in the former transition state [Δ‡Edist-2/Δ‡Edist-HBpin: 8.6/27.8 kcal mol−1 in TS-8/TS-18].
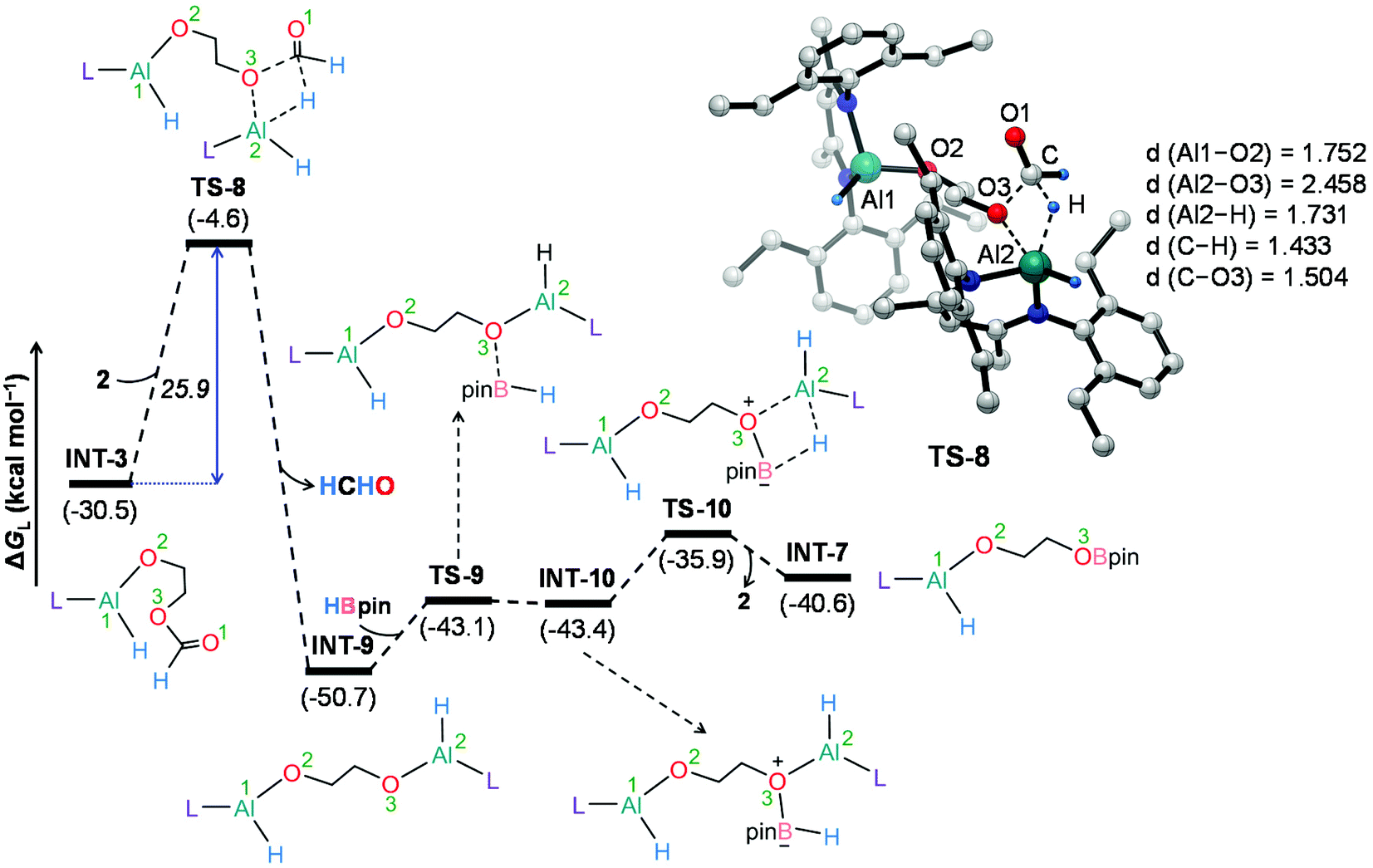 | ||
| Fig. 4 Energy profile for the alternative pathway leading to the formation of the intermediate INT-7. Optimized geometry of the transition state TS-8 with important geometrical parameters is also provided. Bond distances (d) are in angstroms (Å). Only key hydrogen atoms are shown for clarity. L = HC(CMeNAr)2, Ar = 2,6 Et2C6H3. | ||
On the other hand, 2-catalyzed hydroboration of formaldehyde furnishes CH3OBpin product (Fig. 5). The insertion of formaldehyde into the Al–H bond in the catalyst initiates this reaction channel to yield substantially stable intermediate INT-11, accompanying a moderate energy barrier of 13.6 kcal mol−1. The coordination of pinacolborane to the oxygen center in INT-11 generates a slightly less stable intermediate INT-12viaTS-12. Finally, the hydride transfer from boron to aluminum with concurrent Al–O bond rupture delivers CH3OBpin and the catalyst also gets regenerated. This step demands an intrinsic energy barrier of 6.9 kcal mol−1 and an overall energy barrier of 15.4 kcal mol−1 with respect to INT-11. Hence, DFT calculations reveal that the aluminum dihydride species not only catalyzes the formation of 4a but also plays a crucial role in generating formaldehyde in the catalytic system, which eventually furnishes CH3OBpin. The theoretical calculation results also verify our conjecture about the control experiment (Scheme S1†).
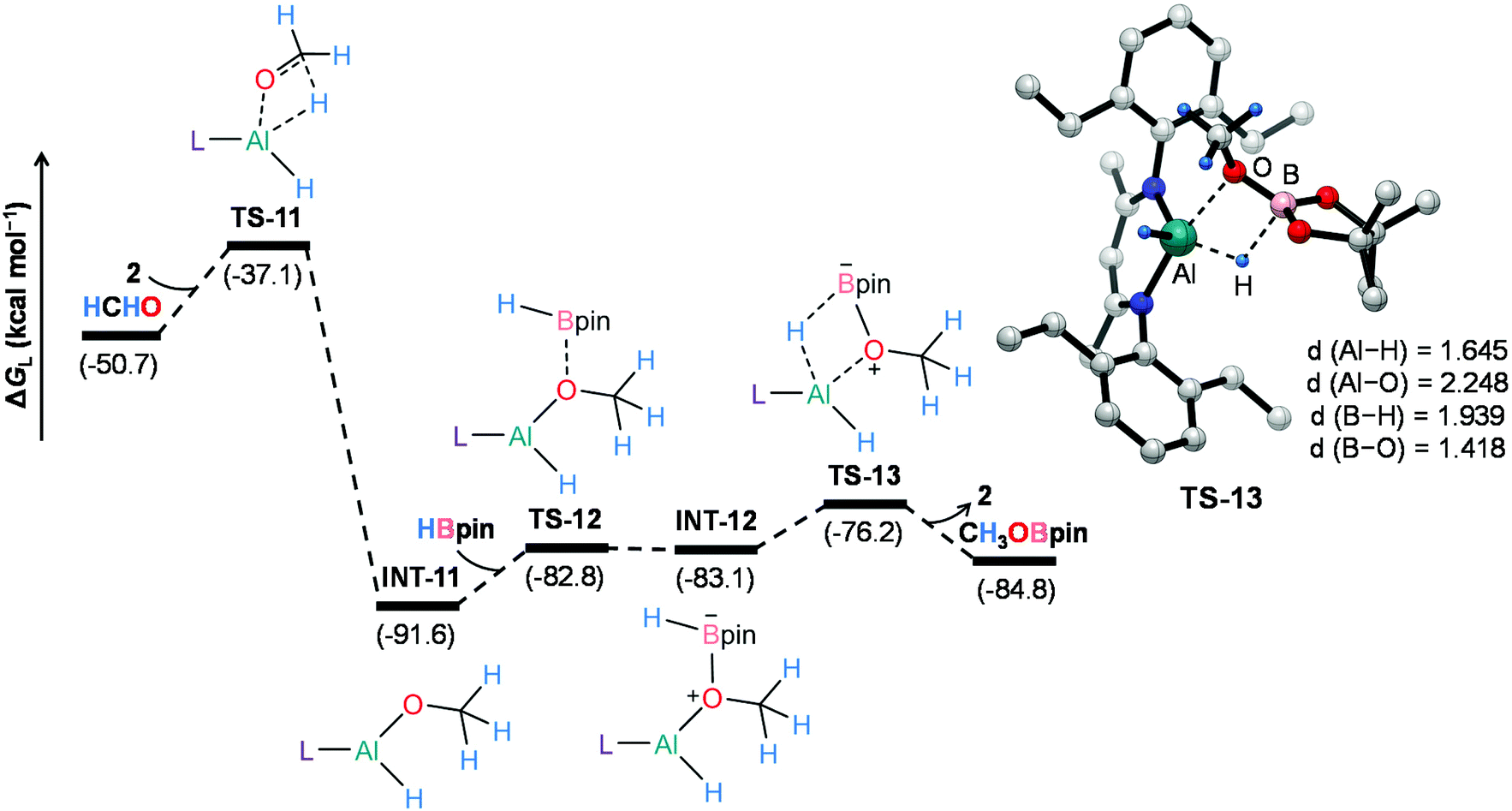 | ||
| Fig. 5 Energy profile for the 2-catalyzed formation of CH3OBpin. Optimized geometry of the transition state TS-13 with important geometrical parameters is also provided. Bond distances (d) are in angstroms (Å). Only key hydrogen atoms are shown for clarity. L = HC(CMeNAr)2, Ar = 2,6 Et2C6H3. | ||
Conclusions
In summary, we report the first excellent catalytic performance of reduction of a wide range of CO2-derived cyclic five- and six-membered carbonates, linear carbonates, polycarbonates, by using the cheap and environmentally friendly organic aluminum hydrides under neat condition. The new protocol shows excellent capabilities in hydroboration of esters, carboxylic acids, and carbon dioxide as well. Its simplicity and versatility make it a useful protocol compared to most the hydroboration strategies for challenging CO functionalities. In-depth mechanistic underpinning of aluminum dihydride 2-catalyzed hydroboration of carbonate 3a suggests a very unique reaction route with involvement of 2 in two parallel pathways, one giving rise to product 4a and the other to CH3OBpin via hydroboration of formaldehyde generated in the catalytic system. All transformations are carried out smoothly, under neat conditions with suitable catalyst loading, and showing they're sustainable applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (21872005, 21671018). H. W. R. thanks the Deutsche Forschungsgemeinschaft for financial support RO224/70-1. S. D. acknowledges the CSIR, India for the Senior Research Fellowship (SRF) and IISER Kolkata for the computational facility. D. K. acknowledges the generous funding from MoE-STARS (MoE-STARS/STARS-1/255) scheme. We are thankful to the reviewers for their valuable suggestions to improve the quality of the manuscript. Dedicated to Professor Ionel Haiduc on the occasion of his 85th birthday.Notes and references
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 RSC
.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed
- D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC
- A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2012, 51, 4526–4528 CrossRef CAS PubMed
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS
- P. Bag, C. Weetman and S. Inoue, Angew. Chem., Int. Ed., 2018, 57, 14394–14413 CrossRef CAS PubMed
- S. Dagorne and R. Wehmschulte, ChemCatChem, 2018, 10, 2509–2520 CrossRef CAS
- Z. Yang, M. Zhong, X. Ma, S. De, C. Anusha, P. Parameswaran and H. W. Roesky, Angew. Chem., Int. Ed., 2015, 54, 10225–10229 CrossRef CAS PubMed
- Z. Yang, M. Zhong, X. Ma, K. Nijesh, S. De, P. Parameswaran and H. W. Roesky, J. Am. Chem. Soc., 2016, 138, 2548–2551 CrossRef CAS PubMed
- W. Liu, Y. Ding, D. Jin, Q. Shen, B. Yan, X. Ma and Z. Yang, Green Chem., 2019, 21, 3812–3815 RSC
- Q. Shen, X. Ma, W. Li, W. Liu, Y. Ding, Z. Yang and H. W. Roesky, Chem. – Eur. J., 2019, 25, 11918–11923 CrossRef CAS PubMed
- Y. Ding, X. Ma, Y. Liu, W. Liu, Z. Yang and H. W. Roesky, Organometallics, 2019, 38, 3092–3097 CrossRef CAS
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed
- G. I. Nikonov, ACS Catal., 2017, 7, 7257–7266 CrossRef CAS
- V. A. Pollard, M. Á. Fuentes, A. R. Kennedy, R. McLellan and R. E. Mulvey, Angew. Chem., Int. Ed., 2018, 57, 10651–10655 CrossRef CAS PubMed
- Y. Liu, J. Li, X. Ma, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2018, 374, 387–415 CrossRef CAS
- W. Li, X. Ma, M. G. Walawalkar, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2017, 350, 14–29 CrossRef CAS
- L. C. Wilkins and R. L. Melen, Coord. Chem. Rev., 2016, 324, 123–139 CrossRef CAS
- A. Bismuto, M. J. Cowley and S. P. Thomas, ACS Catal., 2018, 8, 2001–2005 CrossRef CAS
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed
- E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609–614 CrossRef CAS PubMed
- Z. Han, L. Rong, J. Wu, L. Zhang, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 13041–13045 CrossRef CAS PubMed
- S. H. Kim and S. H. Hong, ACS Catal., 2014, 4, 3630–3636 CrossRef CAS
- V. Zubar, Y. Lebedev, L. M. Azofra, L. Cavallo, O. El-Sepelgy and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 13439–13443 CrossRef CAS PubMed
- A. Kumar, T. Janes, N. A. Espinosa-Jalapa and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 12076–12080 CrossRef CAS PubMed
- A. Kaithal, M. Hölscher and W. Leitner, Angew. Chem., Int. Ed., 2018, 57, 13449–13453 CrossRef CAS PubMed
- C. Erken, A. Kaithal, S. Sen, T. Weyhermüller, M. Hölscher, C. Werlé and W. Leitner, Nat. Commun., 2018, 9, 4521 CrossRef PubMed
- T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Hölscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed
- M. Szewczyk, M. Magre, V. Zubar and M. Rueping, ACS Catal., 2019, 9, 11634–11639 CrossRef CAS
- X. Cao, W. Wang, K. Lu, W. Yao, F. Xue and M. Ma, Dalton Trans., 2020, 49, 2776–2780 RSC
- X. Xu, Z. Kang, D. Yan and M. Xue, Chin. J. Chem., 2019, 37, 1142–1146 CrossRef CAS
- R. Thenarukandiyil, V. Satheesh, L. J. W. Shimon and G. Ruiter, Chem. – Asian J., 2021, 16, 999–1006 CrossRef CAS PubMed
- R. F. Nystrom and W. G. Brown, J. Am. Chem. Soc., 1948, 70, 3738–3740 CrossRef CAS PubMed
- M. Sutter, L. Pehlivan, R. Lafon, W. Dayoub, Y. Raoul, E. Métay and M. Lemaire, Green Chem., 2013, 15, 3020–3026 RSC
- J. Schörgenhumer, A. Zimmermann and M. Waser, Org. Process Res. Dev., 2018, 22, 862–870 CrossRef
- L. Le, J. Liu, T. He, D. Kim, E. J. Lindley, T. N. Cervarich, J. C. Malek, J. Pham, M. R. Buck and A. R. Chianese, Organometallics, 2018, 37, 3286–3297 CrossRef CAS
- D. Kim, L. Le, M. J. Drance, K. H. Jensen, K. Bogdanovski, T. N. Cervarich, M. G. Barnard, N. J. Pudalov, S. M. M. Knapp and A. R. Chianese, Organometallics, 2016, 35, 982–989 CrossRef CAS
- K. Junge, B. Wendt, A. Cingolani, A. Spannenberg, Z. Wei, H. Jiao and M. Beller, Chem. – Eur. J., 2018, 24, 1046–1052 CrossRef CAS PubMed
- T. J. Korstanje, J. I. v. d. Vlugt, C. J. Elsevier and B. D. Bruin, Science, 2015, 350, 298–302 CrossRef CAS PubMed
- J. Yuwen, S. Chakraborty, W. W. Brennessel and W. D. Jones, ACS Catal., 2017, 7, 3735–3740 CrossRef
- M. Arrowsmith, M. S. Hill, T. Hadlington, G. Kociok-Köhn and C. Weetman, Organometallics, 2011, 30, 5556–5559 CrossRef CAS
- D. Mukherjee, A. Ellern and A. D. Sadow, Chem. Sci., 2014, 5, 959–964 RSC
- D. Mukherjee, S. Shirase, T. P. Spaniol, K. Mashima and J. Okuda, Chem. Commun., 2016, 52, 13155–13158 RSC
- M. K. Barman, A. Baishya and S. Nembenna, Dalton Trans., 2017, 46, 4152–4156 RSC
- C. J. Barger, A. Motta, V. L. Weidner, T. L. Lohr and T. J. Marks, ACS Catal., 2019, 9, 9015–9024 CrossRef CAS
- A. Y. Khalimon, P. Farha, L. G. Kuzmina and G. I. Nikonov, Chem. Commun., 2012, 48, 455–457 RSC
- C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS
- B. T. Cho, Chem. Soc. Rev., 2009, 38, 443–452 RSC
- S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed
- J. V. B. Kanth and M. Periasamy, J. Org. Chem., 1991, 56, 5964–5965 CrossRef CAS
- Y. Suseela and M. Periasamy, Tetrahedron, 1992, 48, 371–376 CrossRef CAS
- X. Cui, Y. Li, C. Topf, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10596–10599 CrossRef CAS PubMed
- T. P. Brewster, A. J. M. Miller, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2013, 135, 16022–16025 CrossRef CAS PubMed
- M. Zhang, N. Li, X. Tao, R. Ruzi, S. Yu and C. Zhu, Chem. Commun., 2017, 53, 10228–10231 RSC
- Y. Corre, V. Rysak, X. Trivelli, F. Agbossou-Niedercorn and C. Michon, Eur. J. Org. Chem., 2017, 4820–4826 CrossRef CAS
- T. V. Q. Nguyen, W. J. Yoo and S. Kobayashi, Adv. Synth. Catal., 2016, 358, 452–458 CrossRef CAS
- J. A. Fernández-Salas, S. Manzini and S. P. Nolan, Adv. Synth. Catal., 2014, 356, 308–312 CrossRef
- J. Zheng, S. Chevance, C. Darcel and J.-B. Sortais, Chem. Commun., 2013, 49, 10010–10012 RSC
- L. C. Misal Castro, H. Li, J.-B. Sortais and C. Darcel, Chem. Commun., 2012, 48, 10514–10516 RSC
- K. Matsubara, T. Iura, T. Maki and H. Nagashima, J. Org. Chem., 2002, 67, 4985–4988 CrossRef CAS PubMed
- V. Gevorgyan, M. Rubin, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2001, 66, 1672–1675 CrossRef CAS PubMed
- S. Kisan, V. Krishnakumar and C. Gunanathan, ACS Catal., 2018, 8, 4772–4776 CrossRef CAS
- M. K. Barman, K. Das and B. Maji, J. Org. Chem., 2019, 84, 1570–1579 CrossRef CAS PubMed
- Y. Zheng, X. Cao, J. Li, H. Hua, W. Yao, B. Zhao and M. Ma, Chin. J. Org. Chem., 2020, 40, 2086–2093 CrossRef CAS
- A. Harinath, J. Bhattacharjee and T. K. Panda, Chem. Commun., 2019, 55, 1386–1389 RSC
- W. Wang, M. Luo, D. Zhu, W. Yao, L. Xu and M. Ma, Org. Biomol. Chem., 2019, 17, 3604–3608 RSC
- X. Xu, D. Yan, Z. Zhu, Z. Kang, Y. Yao, Q. Shen and M. Xue, ACS Omega, 2019, 4, 6775–6783 CrossRef CAS PubMed
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2017, 118, 434–504 CrossRef PubMed
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed
- W. H. Bernskoetter and N. Hazari, Acc. Chem. Res., 2017, 50, 1049–1058 CrossRef CAS PubMed
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2017, 118, 372–433 CrossRef PubMed
- S. Chakraborty, J. Zhang, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872–8873 CrossRef CAS PubMed
- L. J. Murphy, H. Hollenhorst, R. McDonald, M. Ferguson, M. D. Lumsden and L. Turculet, Organometallics, 2017, 36, 3709–3720 CrossRef CAS
- H.-W. Suh, L. M. Guard and N. Hazari, Chem. Sci., 2014, 5, 3859–3872 RSC
- S. Bontemps, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2014, 136, 4419–4425 CrossRef CAS PubMed
- M. R. Espinosa, D. J. Charboneau, A. Garcia de Oliveira and N. Hazari, ACS Catal., 2018, 9, 301–314 CrossRef
- R. Shintani and K. Nozaki, Organometallics, 2013, 32, 2459–2462 CrossRef CAS
- G. Jin, C. G. Werncke, Y. Escudié, S. Sabo-Etienne and S. Bontemps, J. Am. Chem. Soc., 2015, 137, 9563–9566 CrossRef CAS PubMed
- S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS PubMed
- T. Janes, K. M. Osten, A. Pantaleo, E. Yan, Y. Yang and D. Song, Chem. Commun., 2016, 52, 4148–4151 RSC
- S. R. Tamang and M. Findlater, Dalton Trans., 2018, 47, 8199–8203 RSC
- X. Wang, K. Chang and X. Xu, Dalton Trans., 2020, 49, 7324–7327 RSC
- D. Mukherjee, H. Osseili, T. P. Spaniol and J. Okuda, J. Am. Chem. Soc., 2016, 138, 10790–10793 CrossRef CAS PubMed
- M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2013, 135, 9326–9329 CrossRef CAS PubMed
- M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2014, 136, 10708–10717 CrossRef CAS PubMed
- T. Wang and D. W. Stephan, Chem. – Eur. J., 2014, 20, 3036–3039 CrossRef CAS PubMed
- N. von Wolff, G. Lefèvre, J. C. Berthet, P. Thuéry and T. Cantat, ACS Catal., 2016, 6, 4526–4535 CrossRef CAS
- A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS PubMed
- D. Franz, C. Jandl, C. Stark and S. Inoue, ChemCatChem, 2019, 11, 5275–5281 CrossRef CAS PubMed
- M.-A. Courtemanche, J. Larouche, M.-A. Légaré, W. Bi, L. Maron and F.-G. Fontaine, Organometallics, 2013, 32, 6804–6811 CrossRef CAS
- C.-C. Chia, Y.-C. Teo, N. Cham, S. Y.-F. Ho, Z.-H. Ng, H.-M. Toh, N. Mézailles and C.-W. So, Inorg. Chem., 2021, 60, 4569–4577 CrossRef CAS PubMed
- A. Caise, D. Jones, E. L. Kolychev, J. Hicks, J. M. Goicoechea and S. Aldridge, Chem. – Eur. J., 2018, 24, 13624–13635 CrossRef CAS PubMed
- D. Sarkar, C. Weetman, S. Dutta, E. Schubert, C. Jandl, D. Koley and S. Inoue, J. Am. Chem. Soc., 2020, 142, 15403–15411 CrossRef CAS PubMed
- D. Sarkar, S. Dutta, C. Weetman, E. Schubert, D. Koley and S. Inoue, Chem. – Eur. J., 2021, 27, 13072–13078 CrossRef CAS PubMed
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed
- S. Dutta, K. Singh and D. Koley, Chem. – Asian J., 2021, 16, 3492–3508 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt00785a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |