DOI:
10.1039/D0PY00722F
(Review Article)
Polym. Chem., 2021,
12, 12-28
Reaction-induced phase transitions with block copolymers in solution and bulk
Received
16th May 2020
, Accepted 29th July 2020
First published on 6th August 2020
Abstract
Reaction-induced phase transitions (RIPTs) encompass a broad range of synthetic strategies that use chemical reactions to drive the organization of macromolecules into complex, nanoscale morphologies. A diverse set of chemical reactions are now available which can alter a polymer's composition and can provide access to a rich landscape of tailorable morphologies in both solution and bulk. In this review, we highlight advances in RIPTs that rely upon functionalization, deprotection, conjugation, chain-scission, grafting, and polymerization to induce disorder-to-order or order-to-order phase transitions in situ. To date, polymerization-induced self-assembly is the most well-established RIPT, but only represents one of a broader family of reactions for inducing macromolecular organization. This contribution explores the wider field of chemical reactions for inducing macromolecular self-assembly or reorganization and takes a comprehensive look at significant and recent developments in this flourishing area of research.
 Joshua Lequieu | Joshua Lequieu is an Assistant Professor of Chemical and Biological Engineering at Drexel University. He received a B.S. in Chemical Engineering from Cornell University, and a Ph.D. in Molecular Engineering from the University of Chicago under the guidance of Juan J. de Pablo. Following a postdoc in the Materials Research Lab at the University of California, Santa Barbara with Glenn H. Fredrickson, he joined Drexel University in 2020. His current research interests are on the theory and simulation of block polymers and on biophysical processes involving chromatin and DNA. |
 Andrew J. D. Magenau | Andrew J. D. Magenau is an Assistant Professor of Materials Science & Engineering at Drexel University. He received his bachelor's degree in Plastics Engineering from the Pennsylvania State University and PhD in Polymer Science & Engineering from the University of Southern Mississippi under the guidance of Professor Robson F. Storey. Afterward, he joined Professor Krzysztof Matyjaszewski at Carnegie Mellon University as a Visiting Assistant Professor. Following his postdoctoral studies, he joined the NIH's Molecular Biosensors & Imaging Center and then DuPont's central research & development. His current research interests are on developing new synthetic platforms for controlled/living polymerization, polymer functionalization, self-assembly, complex networks, and hybrid materials for additive manufacturing, advanced engineering materials, biomedical devices, and separation technologies. |
1. Introduction
Block copolymers are astonishing macromolecules that can organize into a myriad of equilibrium and metastable morphologies determined by a polymer's composition, microstructure, and processing.1–3 Owing to the versatile nature of block copolymers (BCPs), this class of material exhibits an immense scope of nanostructures, properties, and functions that facilitate prospective applications in drug delivery,4 water purification,5 microelectronics,6 and catalysis,7–9 amongst many others.1,2 This vast and venerable body of work has largely focused on the organization of BCPs whose fundamental macromolecular building blocks are chemically unmodified during the assembly process. However, in recent years, focus has begun to shift from static polymers to dynamic polymers whose composition can change during assembly. In dynamic polymers, chemical reactions are employed to alter a macromolecular building block's composition for triggering in situ self-assembly or a morphological transition. Dynamic processes have been shown to yield extensive structural and compositional versatility, well-defined nanostructures at exceptionally high concentrations (25–50 wt%),10,11 and morphologies that are challenging to achieve using traditional methods (e.g. bicontinuous morphologies).12 The ability to access such unique nanostructures originates from a thermodynamic and kinetic landscape that dynamically changes over the course of a reaction, which through judicious selection of conditions can be employed as a powerful tool to diverse and controllable morphologies. Interest in polymers with dynamic compositions also arises from the prospect of mimicking and understanding complex functions and structures achieved by nature. Within the cell for instance, biomacromolecules such as proteins and DNA are subjected to extensive covalent modifications that have been linked to the formation and organization of phase separated compartments in the cytoplasm and nucleus.13 Dynamic BCP systems thus provide a convenient and well-controlled platform for elucidating fundamental aspects of reaction-induced biological phase separation, the origins of biopolymer properties and performance, and a route to biologically-inspired soft materials.14–17
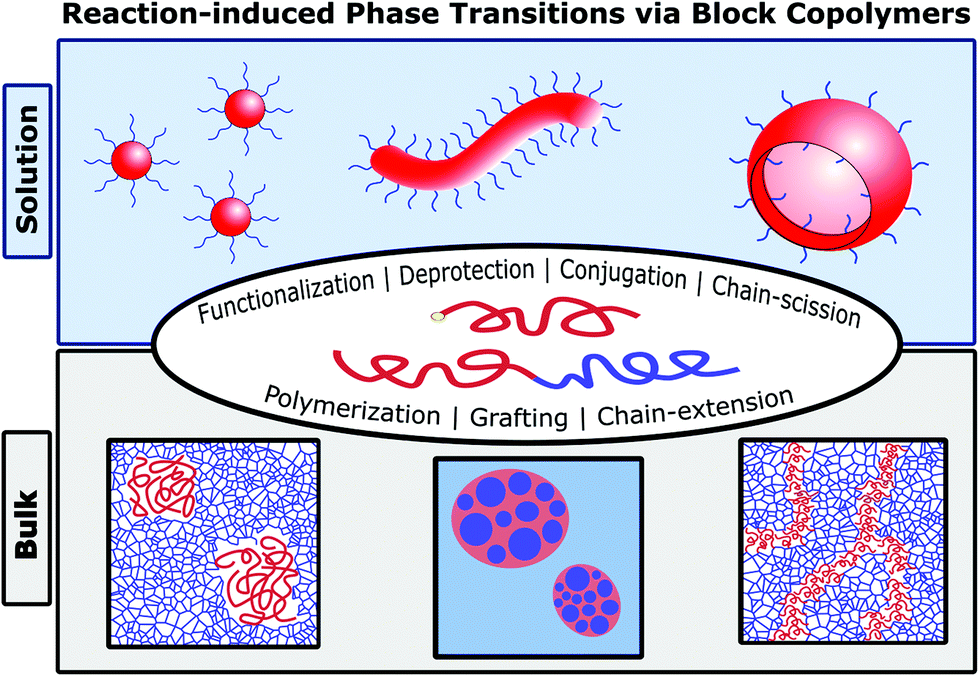
In this review, we highlight advances in the field of polymeric reaction-induced phase transitions (RIPTs),18 triggered by various mechanisms including functionalization, deprotection, conjugation, chain-scission, grafting, and polymerization (Fig. 1). This review covers RIPTs that impart order (disorder → order transition) or induce phase transformations (order → order transition) through primary bond formation or breakage. While the origins of this field can date back considerably (depending on the subtopic), a resurgence of new synthetic strategies for reaction-induced phase transitions has taken hold, facilitating the creation of new and exciting polymeric materials. Herein, we review two broad classifications of RIPTs by whether they occur in solution or in (near) bulk conditions, followed by further nuanced sorting into categories based upon the primary reaction mechanism driving each phase transition. In nearly all systems, the RIPTs originate from or create block-like (segregated) architectures, which we constitute as a BCP whether its structure is linear, graft, or crosslinked. Discussion of solution-based polymerization-induced self-assembly (PISA), though a flagship type of RIPT, has been excluded from this review as it is a standalone and burgeoning field summarized in many excellent reviews.11,19–23
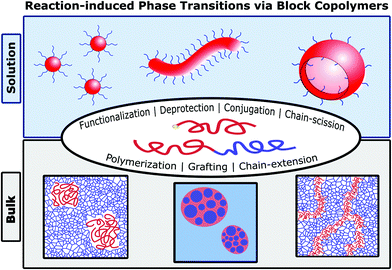 |
| | Fig. 1 Morphologies and reaction types for achieving RIPTs with BCPs in solution or bulk. | |
2. Reaction-induced phase transitions in solvent
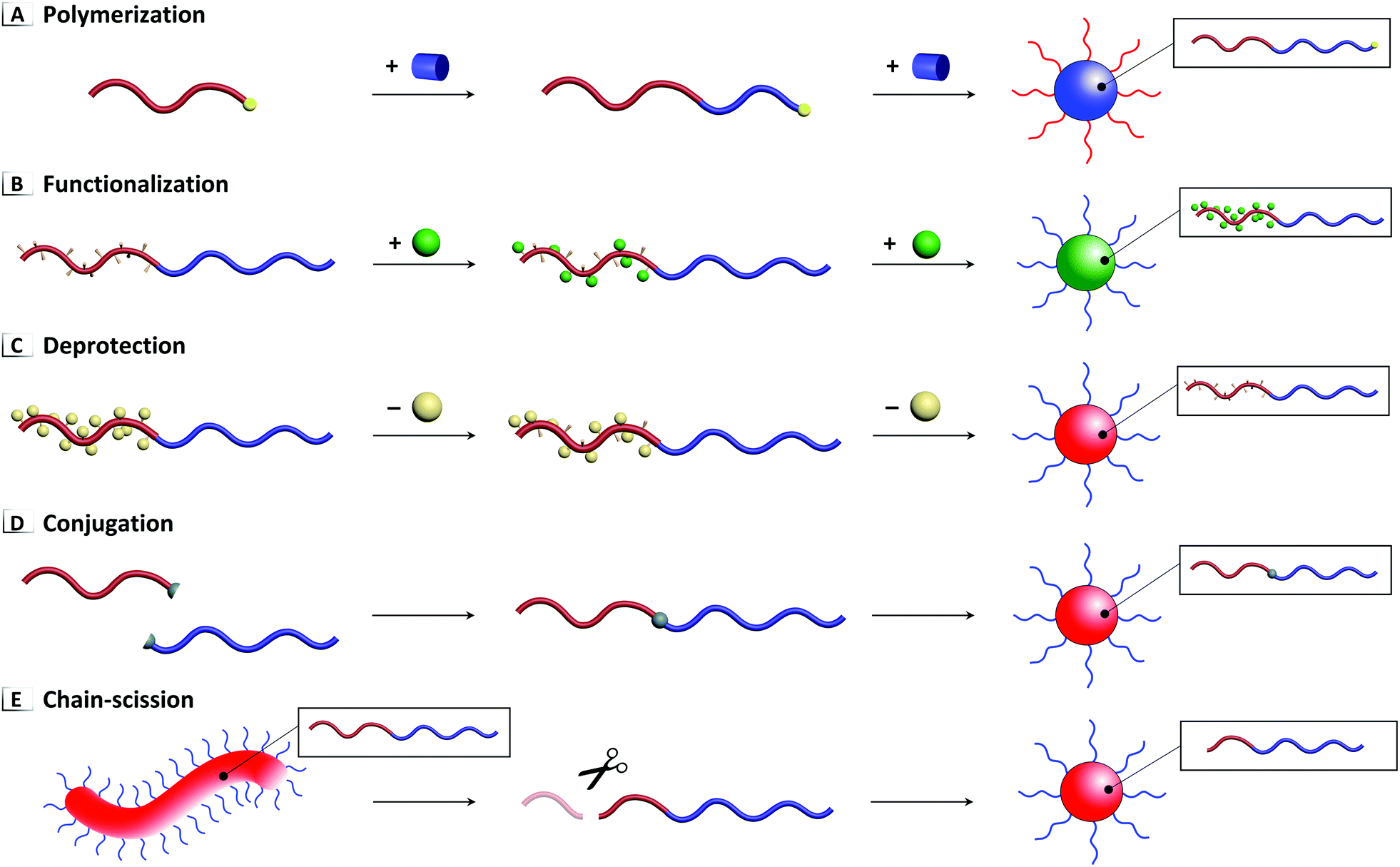
Chemical reactions performed on BCP unimers and pre-assembled nanostructures can serve as a powerful tool for inducing disorder-to-order or order-to-order transitions in solution. To date, five principal approaches to RIPTs have been demonstrated in solution using (1) polymerization, (2) functionalization, (3) deprotection, (4) conjugation, and (5) chain-scission as summarized in Fig. 2. As stated previously, polymerization driven RIPTs (Fig. 2A) or PISA have been excluded from this contribution because of their thorough treatment in various, detailed reviews.11,19–23 Each of these chemical reactions modifies the BCP's composition (with a concomitant change in molar mass) by forming or breaking covalent bonds within the polymer's pendent groups, backbone, or chain-ends. As these reactions proceed, interactions between the solvent and BCP are modified, which eventually trigger a phase transition where polymers assemble or reorganize into a variety of nanostructures ranging from spheres to worms to vesicles, among other complex morphologies. Additional processes for driving solution-based RIPTs have been excluded from this review due to scope, including those that (i) do not involve chemical modification, e.g. isomerization24 and external stimuli (pH, current, heat, etc.),25,26 or which (ii) involve crosslinking of unimers27,28 or (iii) more-frequent order-to-disorder transitions.24,29,30
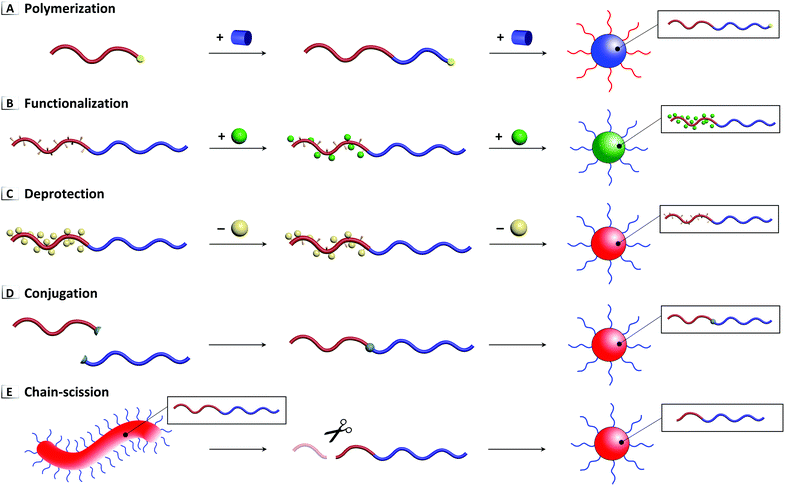 |
| | Fig. 2 Five primary approaches to RIPTs in solution through (A) polymerization, (B) functionalization, (C) deprotection, (D) conjugation, and (E) chain-scission. | |
2.1. RIPTs by functionalization (disorder → order)
The transition of disordered unimers to organized structures in solution can be accomplished through pendent group functionalization of BCPs (Fig. 2B), which grants access to an assortment of well-defined micellar structures. RIPTs by functionalization, or post-polymerization modification, have to the best of our knowledge only been reported using three chemical reactions via azo-coupling, Pd-catalyzed cross-coupling, and thiol–epoxide ring-opening. Feasibly, however, any post-polymerization modification could be employed to cause a RIPT, especially using a number of “click” chemistries or other highly-efficient transformations like activated esterification,31 Diels–Alder cycloaddition,32,33 thiol–ene/thiol–yne addition,34–36 and azide–alkyne cycloadditions.37–39 RIPTs by functionalization are thought to be driven by enthalpic changes, where favorable solvent–polymer interactions are exchanged for unfavorable interactions which induce micelle formation. Uniquely, functionalization-induced phase transitions, when using a universal precursor, can isolate side-chain chemistry as the defining parameter governing self-assembly since they maintain a fixed degree of polymerization, tacticity, and dispersity, from reactant to product BCP.
2.1.1. Azo-coupling.
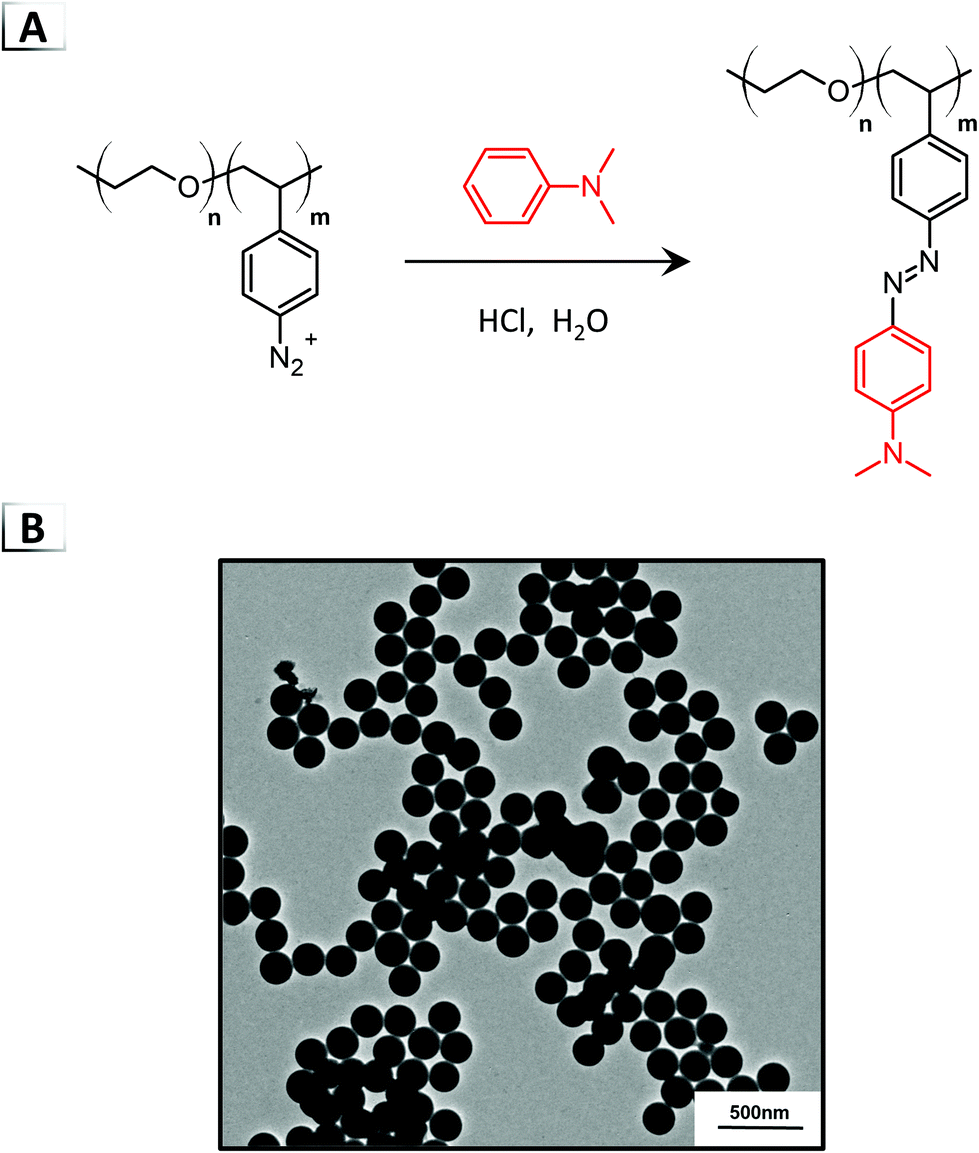
The first example of what we term functionalization-induced self-assembly (FISA) was reported in 2018 by He et al. using an azo-coupling reaction within aqueous media (Fig. 3A).40 In their report, a double hydrophilic BCP was employed that was composed of a stabilizing poly(ethylene oxide) block and a reactive poly(styrene diazonium) block, i.e. p(EO-b-SN2+). The p(EO-b-SN2+) building block was synthesized immediately prior to azo-coupling by diazotization of primary aromatic amines on a poly(vinylaniline) precursor. Subsequently, through reaction of the pendent aryldiazonium cations with N,N-dimethylaniline, approximately 80% of the pendent groups were converted to azobenzene moieties at sub-ambient temperatures. Amphiphilic character was generated in the BCP as the hydrophilic diazoniums salts were transformed into hydrophobic azobenzene groups, which consequently caused the self-assembly of spherical nanoparticles (Fig. 3B) visualized using transmission electron microscopy (TEM). The nanoparticle size could be tailored by changing the degree of polymerization (DP) of the reactive p(SN2+) block, yielding particle diameters from ca. 200 to 500 nm with a DP of 10 and 50, respectively. Furthermore, a dual functionalization was described using N,N-dimethylaniline and a fluorescent tetraphenylethene functional aniline capable of generating ∼200 nm nanoparticles for imaging applications in hypoxic tumors.
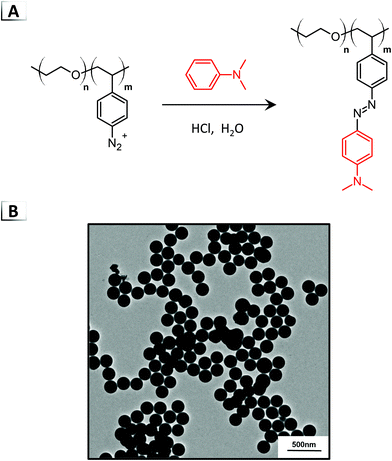 |
| | Fig. 3 (A) Azo-coupling of N,N-dimethylaniline on a BCP for functionalization-induced self-assembly. (B) TEM image of resultant nanoparticles. TEM data was adapted from ACS Macro Lett., 2018, 7, 437. | |
2.1.2. Pd-catalyzed cross-coupling.
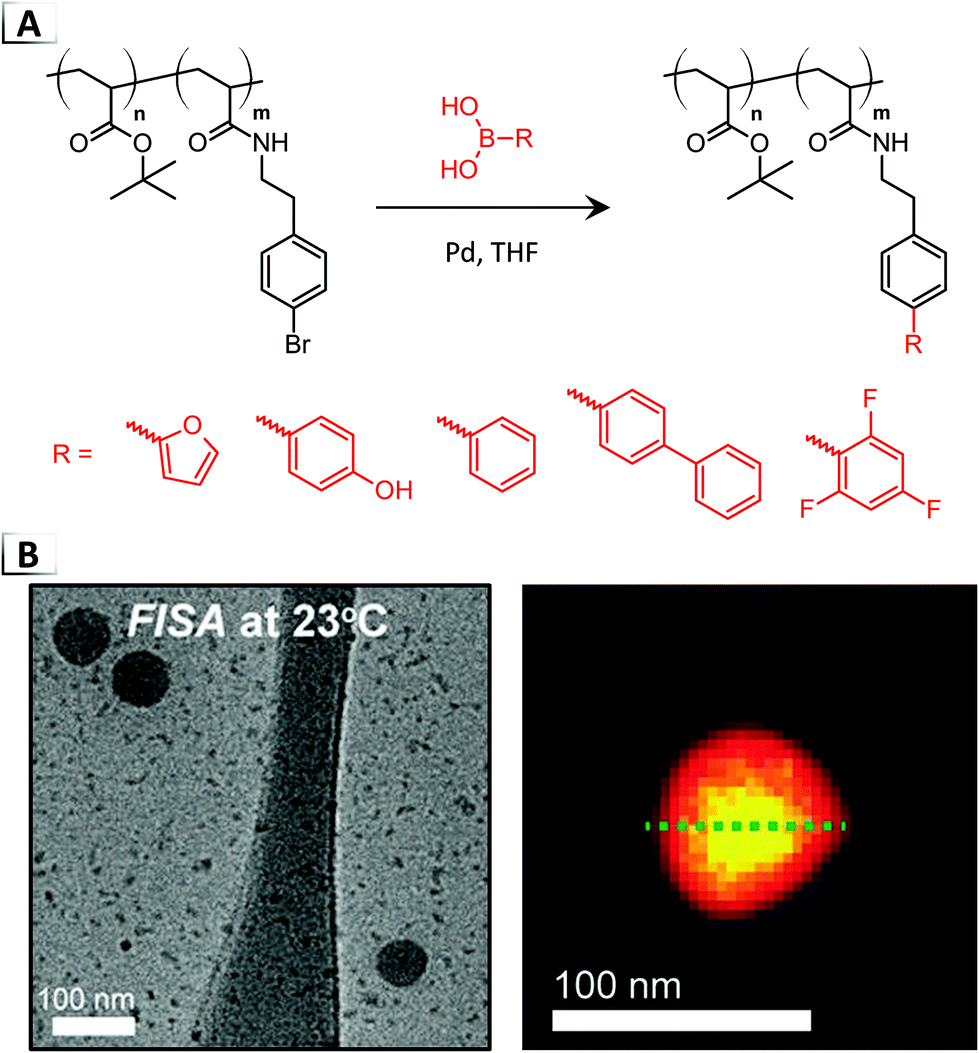
Later that year, Howe and Magenau reported the first instance of FISA in organic media through Pd-catalyzed cross-coupling.41,42 The researchers synthesized a custom BCP from tert-butyl acrylate and bromophenylethyl acrylamide, p(tBA-b-BPEA), utilizing the aryl bromide repeat unit for cross-coupling with a variety of functional boronic acids (Fig. 4A).41 Experimentation revealed that highly solvophobic functionalities were required to induce self-assembly, since the addition of pendent furan groups were incapable of producing a RIPT in THF whereas bi- and tri-aryl moieties could generate polymer micelles. After FISA, a core–shell morphology was visualized using fluorine mapping, revealing higher fluorine concentrations in the core of the micelles (Fig. 4B, yellow signal) via TEM electron energy-loss spectroscopy (EELS) in conjunction with FISA using trifluorophenyl boronic acid. Furthermore, self-assembly was found to proceed only after a critical degree of functionalization was reached and that the size of the micelle was dependent on the extent of functionalization, i.e. larger micelles were observed from increased degrees of functionalization. Moreover, the authors discovered that the nanoparticle size was impacted by the structure of the installed functional group, with progressively more solvophobic functionalities yielding larger micelles ranging from 40 to 100 nm in diameter according to dynamic light scattering (DLS).
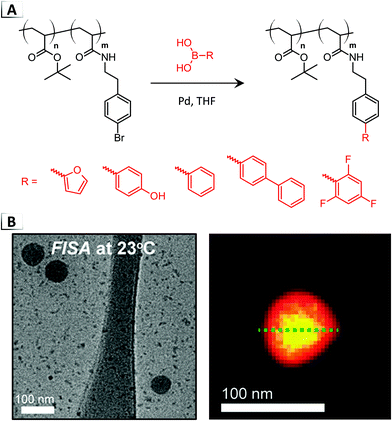 |
| | Fig. 4 (A) Pd-catalyzed cross-coupling of functional boronic acids to a BCP for functionalization-induced self-assembly. (B) TEM image of resultant nanoparticles and scanning TEM annual dark-field overlaid with colocalized EELS fluorine intensity on a resultant nanoparticle. TEM data was adapted from ACS Macro Lett., 2018, 7, 1503. | |
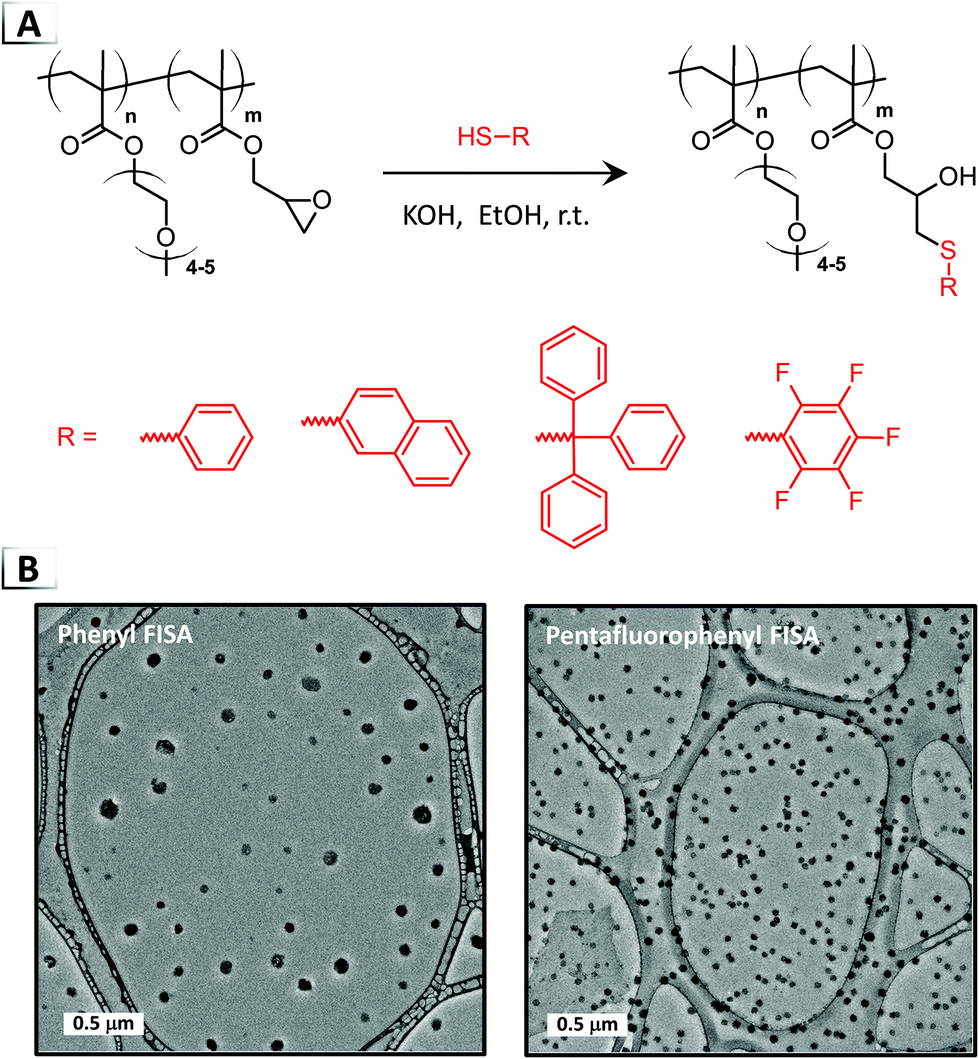
2.1.3. Thiol–epoxide ring-opening.
Recently, the application of “click” chemistry to FISA was reported by Magenau and coworkers using thiol–epoxide ring-opening (Fig. 5A).43 The utilization of thiol–epoxide ring-opening helped to circumvent drawbacks encountered with Pd-catalyzed cross-coupling (oxygen-sensitivity, heterogenous conditions, and a heavy-metal catalyst), while instead offering 100% atom efficiency, rapid rates, high conversions, and mild reaction conditions.44 The authors showed that FISA could be conducted under ambient conditions, in air and at room temperature, using an oligo(ethylene oxide)methoxy methacrylate and glycidyl methacrylate BCP, i.e. p(OEOMA-b-GMA). Under base catalyzed conditions, ring-opening of the pendent epoxides was demonstrated with a range of functional thiols in ethanol, achieving high to nearly quantitative functionalization (≥90%) of the epoxide groups within ca. 4 to 20 hours. FISA was found to be versatile over a variety of conditions (catalyst loadings and BCP concentrations) yielding nanoparticles ranging from ca. 40 to 160 nm (Fig. 5B). Notably, scaled-up conditions were effective up to 10 wt% BCP, yielding well-defined nanoparticles of ∼100 nm. Real-time DLS measurements during the phase transition also allowed for the particle size evolution to be correlated with percent functionalization which was used to extract the critical extent of reaction needed for self-assembly.
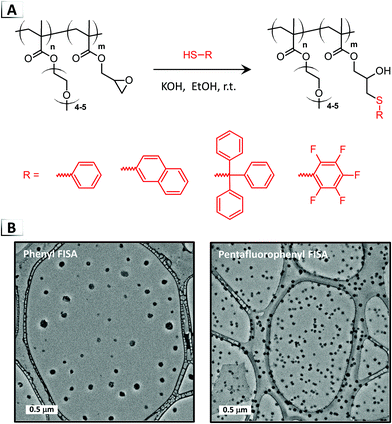 |
| | Fig. 5 (A) Thiol–epoxide ring-opening on a BCP for functionalization-induced self-assembly. (B) TEM image of resultant nanoparticles. TEM data was adapted from Polym. Chem., 2018, 9, 4605. | |
2.2. RIPTs by deprotection (disorder → order)
Reaction-induced phase transitions can also be triggered by the deprotection (or cleavage) of functional groups as means to assemble BCPs into higher-order structures (Fig. 2C). Through such functional group transformations, various research groups have shown that phase transitions can be driven by the resultant change in polarity of one block using chemical, photo-chemical, or thermal reactions. RIPTs in solution by deprotection, like functionalization, are thought to be enthalpic in nature, propelled by the replacement of favorable solvent–polymer interactions with unfavorable interactions that induce micellization.
2.2.1. Deacetylation.
The first instance of chemical deprotection for triggering a RIPT was reported by Jiang et al. with sugar-containing block copolymers (glyco-BCPs),45 a process later referred to as deprotection-induced self-assembly (DISA).46 In their seminal paper, acetyl groups on an α-mannopyranoside styrenic block were deprotected to hydroxy groups using an organic base in THF (Fig. 6A).45 Upon conversion of the less-polar acetyl groups to more-polar hydroxy groups, the glyco-BCPs spontaneously organized into micelles. Unlike other methods of self-assembly, DISA uniquely created self-assembled structures with the glyco-block residing within the micelle cores or the internal layer of vesicles (called “glycol-inside” structures), contrary to common “glyco-outside” nanoparticles with peripheral glyco-blocks.45 During DISA, the glyco-block DP was shown to impact the size and morphology of the self-assembled structures (i.e. spheres, vesicles, mixed phases). Larger glyco-blocks of DP ≈ 19 to 25 were found to produce vesicles, as opposed to spherical micelles (Fig. 6B), rationalized by a change in the geometric packing parameter.
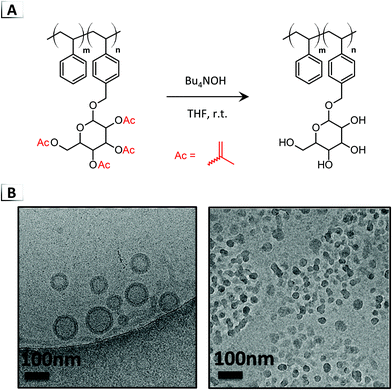 |
| | Fig. 6 (A) Deacetylation of a protected glyco-BCP for deprotection-induced self-assembly. (B) TEM images of resultant nanoparticles. TEM data was adapted from ACS Macro Lett., 2014, 3, 534. | |
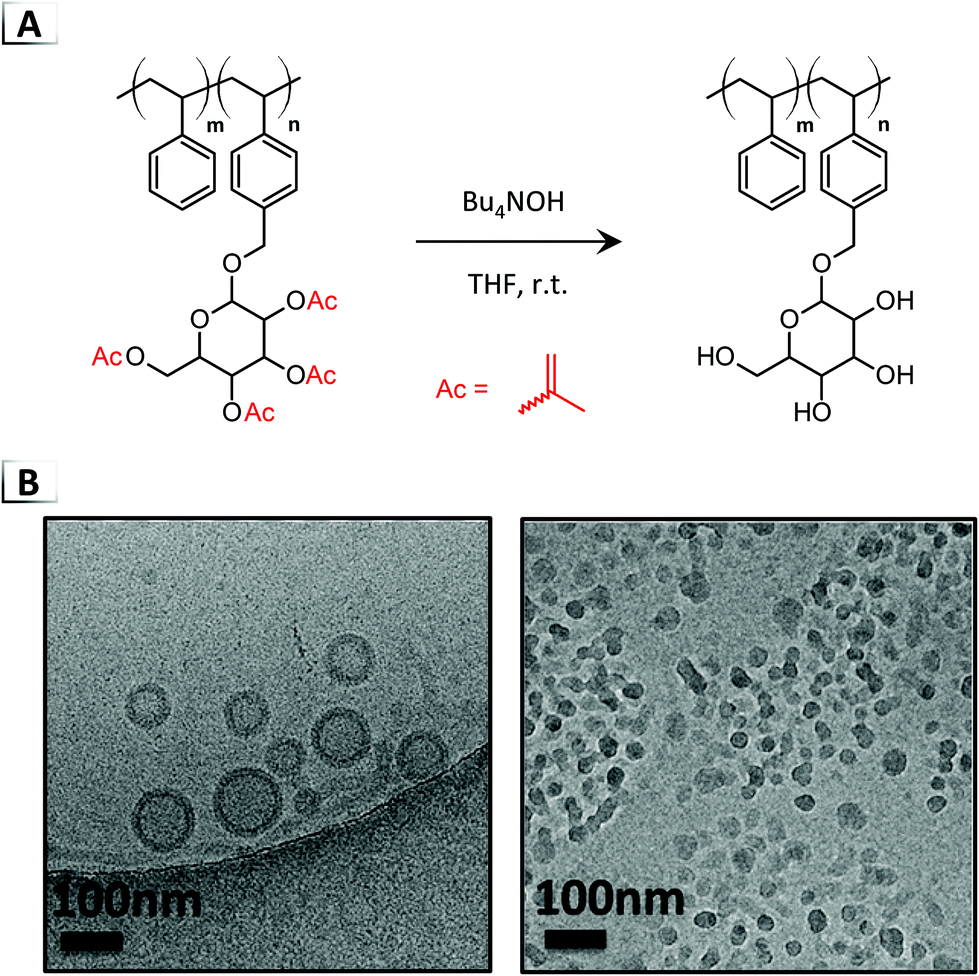
2.2.2. Debenzoylation.
Jiang and coworkers extended the DISA concept to benzoyl protecting groups using a similar deblocking protocol as in their initial report (Fig. 6A, Bu4NOH in THF).47 Through debenzoylation and liberation of the latent hydroxy groups, a considerable polarity change occurred in the glyco-block, trigging a disorder-to-order RIPT. Benzoyl groups were pursued because of their slower deprotection rate compared to acetyl groups, which enabled the authors to exert control over the reaction kinetics during self-assembly. Interestingly, not only did deblocking of the benzoyl groups give rise to different self-assembly kinetics, but it also provided access to different morphologies even when employing similar BCP compositions. With highly asymmetric BCPs and short glyco-blocks (DP ≈ 5–7), the acetyl groups yielded spherical micelles whereas the benzoyl groups produced vesicles. The authors reasoned that the slower deprotection rate of the benzoyl groups may produce structures that resemble equilibrium products, contrary to the rapid deprotection of the acetyl groups that led to kinetically trapped structures. When using a longer glyco-block of DP ≈ 15, vesicles were obtained with both deblocking groups, however, the diameter (46 vs. 116 nm) and aggregation numbers were significantly different. Mechanistic studies were also performed with in situ DLS and cryo-TEM, showing that the morphology evolved to vesicles during debenzoylation via formation and fission of a large intermediate aggregate.
2.2.3. Photolysis.
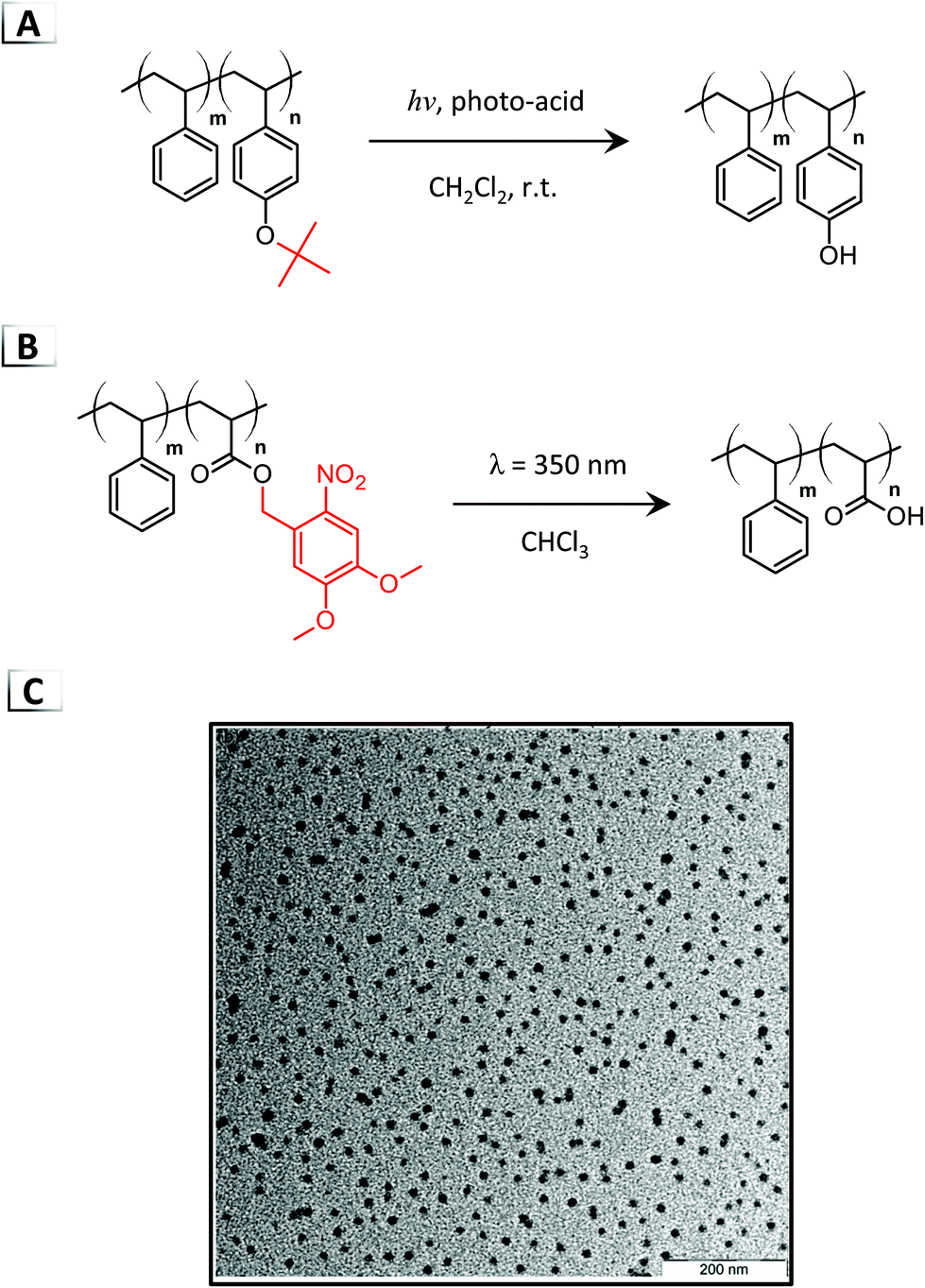
To date, there are only a handful of disorder-to-order RIPTs for the self-assembly of polymer micelles through photolysis of BCP pendent groups.48–51 Most photochemical RIPTs are order-to-disorder transitions (micelle disruption), order-to-order transitions, or transitions that occur through non-covalent processes, which have been described in an excellent review by Gohy and Zhao.24 To the best of our knowledge, in 2007, Kuwayama et al. made one of the first direct photolysis-based RIPTs of poly(tert-butoxystyrene)-block-poly(styrene) by photocleavage of the tert-butoxy groups using a photoacid generator at room temperature (Fig. 7A).48 Upon irradiation, photolysis converted the poly(tert-butoxystyrene) block into a poly(vinylphenol) block, generating micelles ∼63 nm in diameter. Experimentation revealed that a degree of photolysis of 50% was required to induce micellization. More recently, Gohy et al. demonstrated micellization through photocleavable ortho-nitrobenzyl esters under 350 nm light in organic media (Fig. 7B).51 This RIPT was performed with poly(dimethoxynitrobenzyl acrylate)-block-poly(styrene) eliminating the pendent dimethoxynitrobenzyl protecting groups to generate a hydrophilic poly(acrylic acid) block that was insoluble in chloroform.51 Micelles were observed with TEM after irradiation (Fig. 7C) and DLS measurements confirmed a range of nanoparticle sizes that were dependent on the BCP composition.
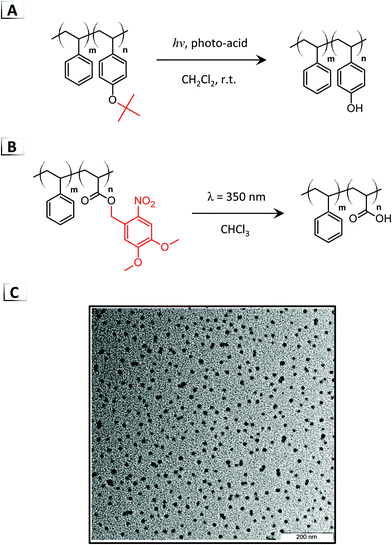 |
| | Fig. 7 Photolysis of (A) tert-butoxy groups and (B) ortho-nitrobenzyl esters on BCPs for photo-deprotection-induced self-assembly. (C) TEM image of the resultant nanoparticles from nitrobenzyl ester photolysis. TEM data was reproduced from Soft Matter, 2011, 7, 6891. | |
2.2.4. Thermolysis.
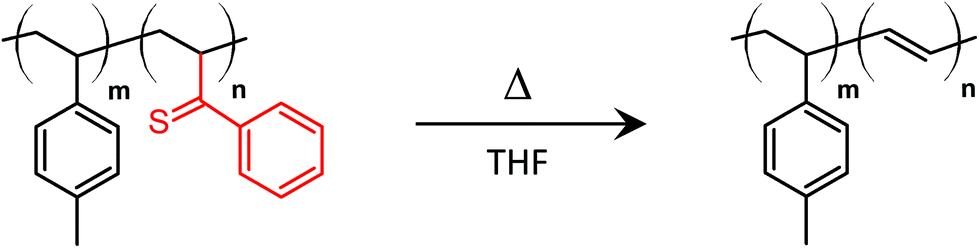
One of the only examples of a thermally driven RIPT was reported by Lam et al. through selective thermolysis of one block of a BCP.52 This thermolysis-induced self-assembly process occurred by an elimination reaction involving phenyl vinylsulfoxide repeat units which had been previously explored for the synthesis of conductive polymers.53 Specifically, the poly(phenyl vinylsulfoxide) block of a poly(para-methyl styrene) BCP was converted into poly(acetylene) through thermolysis at 55 °C in THF (Fig. 8). After 11–24 hours at elevated temperatures, the reactant BCP self-assembled into micelles with poly(acetylene) cores of 30 to 60 nm in radius. Outside of this example, thermally driven deprotection processes have been rarely reported for inducing the self-assembly of BCPs.
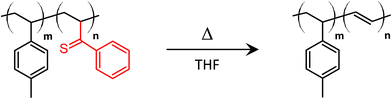 |
| | Fig. 8 Thermolysis of poly(para-methylstyrene)-block-poly(phenylvinylsulfoxide) for thermal deprotection-induced self-assembly. | |
2.3. RIPTs by conjugation (disorder → order)
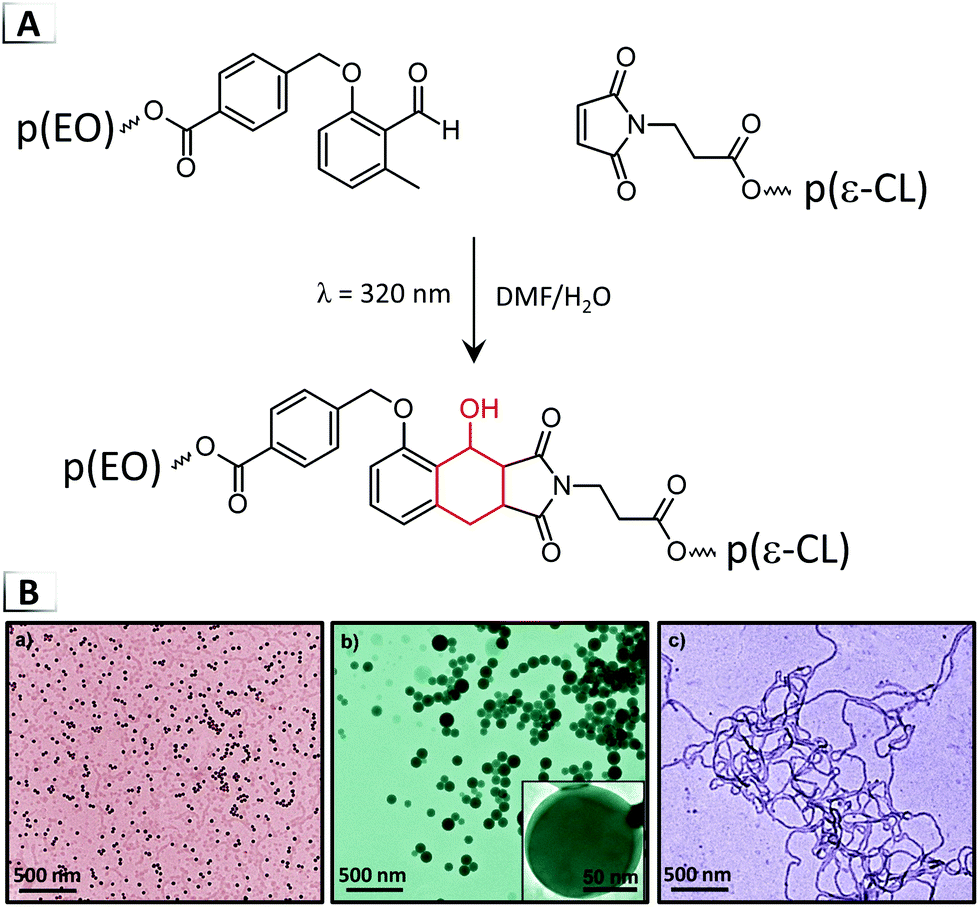
The transition of disordered unimers into organized nanostructures can also be caused by a third class of solution-based RIPT driven via chain-end coupling of homopolymers (Fig. 2D). As recently demonstrated by Yan et al., when solutions of homopolymers, each containing one of two complementary chain-ends are exposed to 310–365 nm light, a photoinitiated Diels–Alder coupling reaction can occur through isomerization of ortho-methylbenzaldehyde into ortho-quinodimethane chain-ends that subsequently undergo cycloaddition with maleimide chain-end functionalized homopolymers (Fig. 9A).54 The authors termed this type of conjugation-driven RIPT, light-initiated in situ self-assembly (LISA). In their report, binary and ternary homopolymer mixtures were coupled to form BCPs, which were observed to spontaneously generate spheres, worms, or vesicles (Fig. 9B) using maleimide-terminated poly(lactide) and poly(ε-caprolactone) with methylbenzaldehyde-terminated poly(ethylene oxide). Notably, the authors established reaction conditions that were relatively concentrated at 10 wt% and morphological phase diagrams as a function of different molar ratios of homopolymer and wavelengths of light. While literature has shown that macromolecular conjugation reactions can cause RIPTs, we envision that the general phenomena described in this report could encompass a number of unexplored coupling, ligation, and grafting-to reactions.
 |
| | Fig. 9 (A) Photoinitiated Diels–Alder coupling of binary mixtures of methylbenzaldehyde chain-end functionalized poly(ethylene oxide) with maleimide chain-end functionalized poly(ε-caprolactone) for conjugation-induced self-assembly. Ternary systems involved poly(lactic acid) as the third component. (B) TEM image of the resultant nanoparticles. TEM data was reproduced from Macromolecules, 2017, 50, 4276. | |
2.4. RIPTs with assembled nanostructures (order → order)
RIPTs via post-polymerization modification have also been successfully implemented as a means to reorganize nanostructures. Similar to the disorder-to-order transitions discussed in sections 1.1 and 1.2, the reorganization of nanostructures through order-to-order transitions using post-polymerization modification have been accomplished using functionalization and deprotection, but also uniquely with chain-scission reactions (Fig. 2E). While similar transitions on pre-assembled structures can be accomplished through other fascinating non-covalent processes and stimuli (thermal,55–58 guest–host,59–62 and others),25,26 this review focuses on chemical-reaction induced morphological transitions that involve the formation or breakage of covalent bonds.
2.4.1. Functionalization.
The functionalization of pre-assembled nanostructures is an effective means to decorate nanoparticles for various applications, however, initial reports performing such reactions failed to observe significant morphological changes when using thiol–ene and pentafluorophenyl–amine reactions.63,64 One early example of a functionalization driven RIPT on assembled nanostructures was reported in 2015 by Lowe et al. who utilized nucleophilic para-aromatic substitution reactions on the periphery of spheres, worms, and vesicles derived from PISA.65 Nanoparticle functionalizations were carried out in ethanol at 50 °C in the presence of an organobase by reacting exterior pentafluorophenyl groups with a range of functional thiols, the majority of which proceeded to quantitative levels of substitution. While functionalizations on spherical nanoparticles had minimal morphological impact, reactions carried out on worms caused a morphological transition into a mixed phase composed of spheres and worms when using a thiosugar, an outcome which likely spurred future investigations.
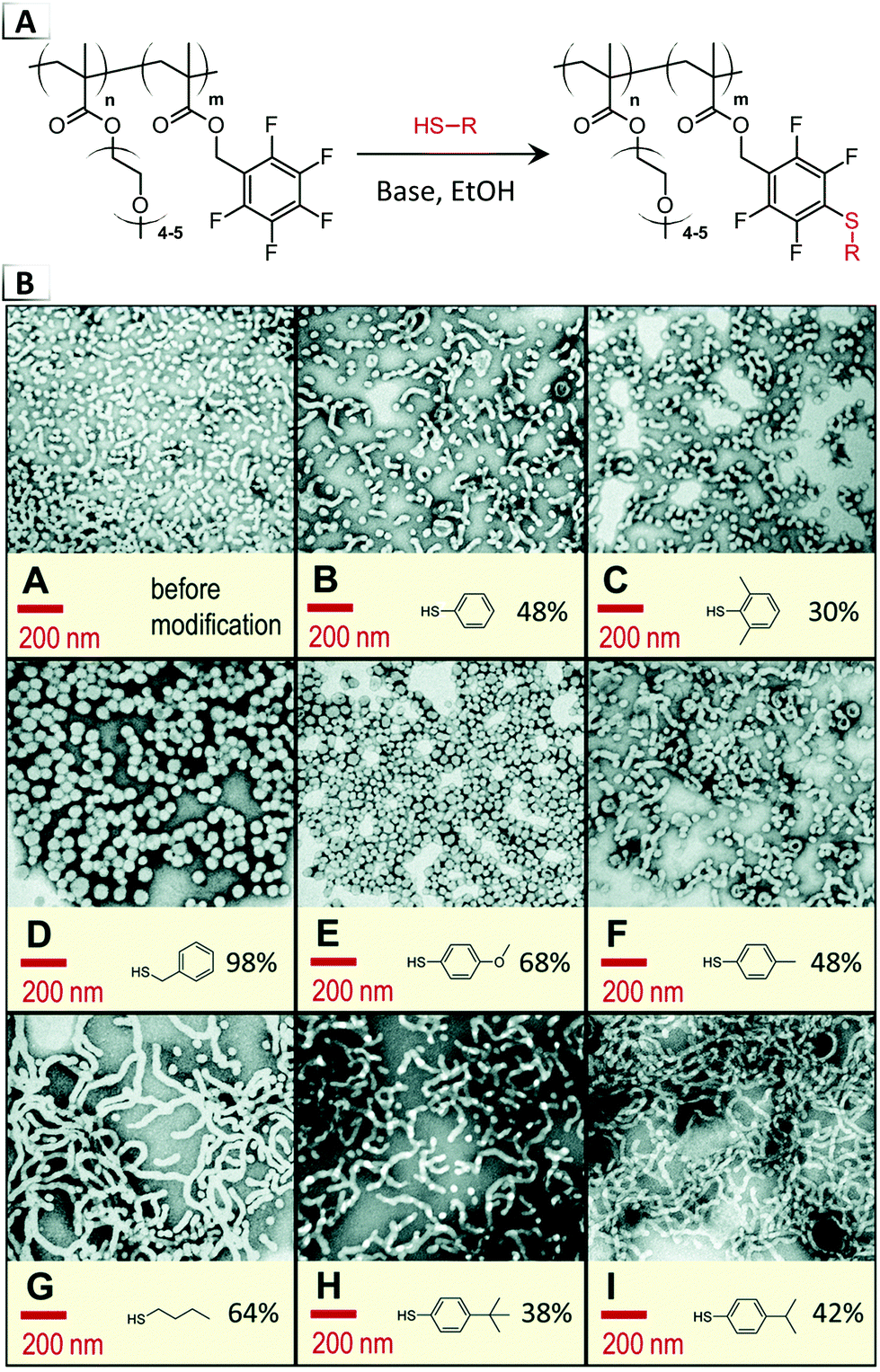
Later in 2020, Roth et al. attempted to induce RIPTs using the same thiol–pentafluorphenyl substitution reaction (Fig. 10A), but instead they targeted the core or internal block of assembled nanostructures.66 This work was motivated by the concept of a “universal precursor” that would allow one parent particle to be transformed into various sibling particles through alteration of the geometric packing parameter with irreversible chemical functionalizations.66 While many of the functionalization reactions caused order-to-disorder transitions, intriguingly, order-to-order transitions were observed with TEM from spheres to worms using a poly(oligo(ethylene oxide) methacrylate)-block-poly(pentafluorobenzyl acrylate) precursor and butanethiol, iso-propylbenzenethiol, or tert-butylbenzenthiol functionalities (Fig. 10B). In contrast, benzyl mercaptan and methoxybenzenthiol functionalities were found to result in larger diameter spherical morphologies, whereas the remaining thiols (benzenethiol, methylbenzene thiol, dimethylbenzene thiol) had a minimal impact on the morphology.
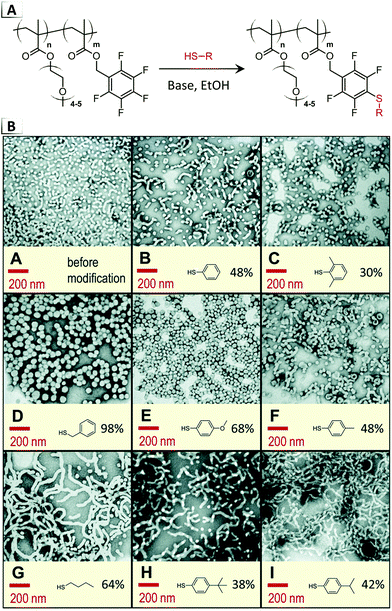 |
| | Fig. 10 (A) Reaction-induced phase transition of nanoparticles via functionalization using nucleophilic para-aromatic substitution of pentafluorophenyl groups on BCP pendant groups. (B) TEM images of resultant nanoparticles produced using various thiols. Figure B was reproduced from Polym. Chem., 2020, 11, 704. | |
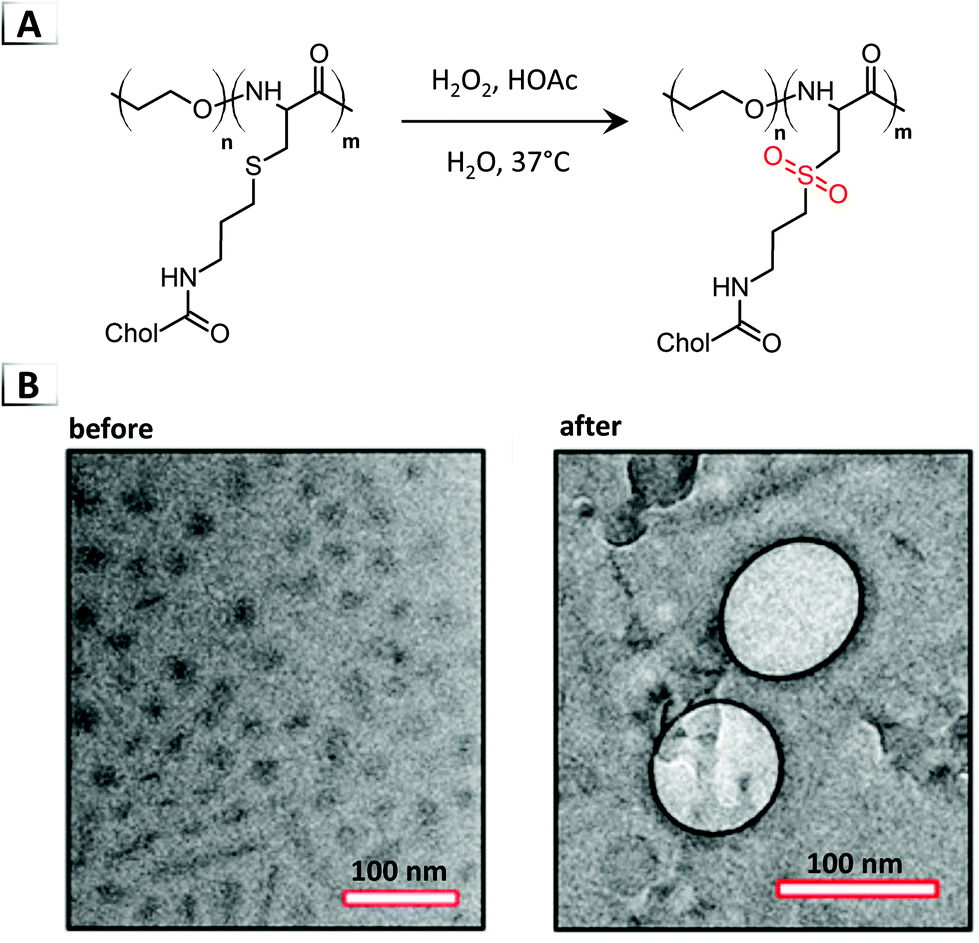
Another notable example of an order-to-order transition through post-polymerization modification was investigated by Fu et al. in 2018 using a polypeptide-based BCP.67 The BCP employed in this study consisted of a water soluble poly(ethylene oxide) block and a poly(L-cysteine) block that was decorated with Cholesterol (Chol). Oxidation of the BCP converted the thio–ether pendent groups to sulfones by using hydrogen peroxide and acetic acid for 16 hours in aqueous media (Fig. 11A). These reaction conditions yielded quantitative conversion to sulfone and a concomitant micelle-to-vesicle transition confirmed via TEM (Fig. 11B). Interestingly, not only did oxidation cause a micelle-to-vesicle transition, but the morphological transition was accompanied by a conformational change in secondary structure of the poly(L-cysteine) block from a β-sheet to an α-helix. The authors later investigated these assemblies in vivo/in vitro, and found their dynamic morphology enabled controlled payload release in addition to influencing cell interactions and nanoparticle fate.
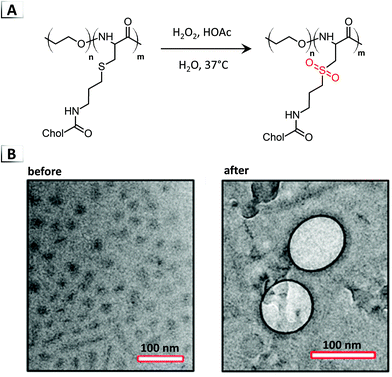 |
| | Fig. 11 (A) Reaction-induced phase transition of nanoparticles via functionalization using the oxidation of a thio–ether containing BCP. (B) TEM images of assembled structures before and after oxidation. TEM data was adapted from J. Am. Chem. Soc., 2018, 140, 6604. | |
2.4.2. Dynamic-covalent functionalization.
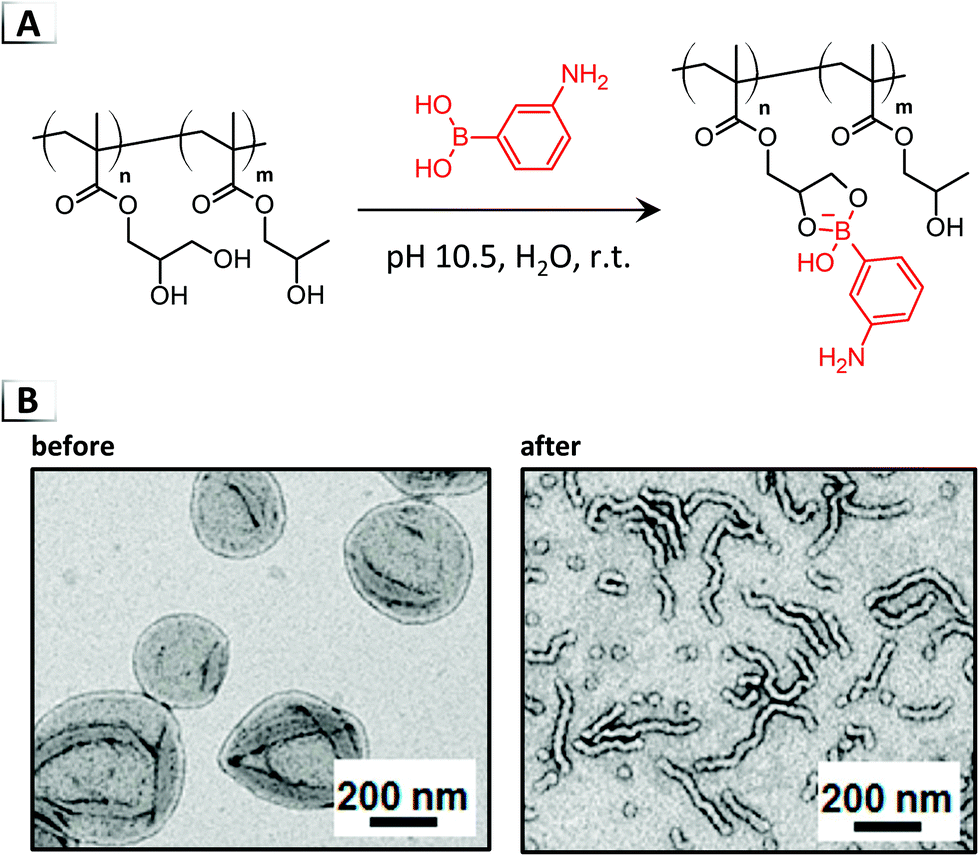
Not only have irreversible functionalization reactions been used to invoke RIPTs on assembled nanostructures, but in recent years, order-to-order morphological transitions have been demonstrated using dynamic-covalent chemistry.68,69 One of the first examples of dynamic-covalent functionalization of PISA-derived vesicles was demonstrated by Armes et al. through the reaction of aminophenylboronic acid with cis-diol pendent groups on a poly(glycerol monomethacrylate) BCP under alkaline conditions (Fig. 12A).68 Through the generation of phenylborate esters, the volume fraction of the stabilizer block increased, causing a corresponding reduction in the geometric packing parameter and a vesicles-to-sphere morphological transition (Fig. 12B). The same group was also able to show “reversible” morphological transitions between worms and spheres in a follow up article using the pH dependent binding of carboxyphenylboronic acid to diols.69
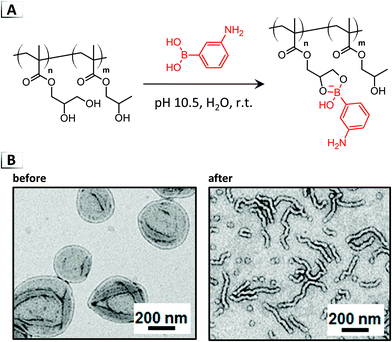 |
| | Fig. 12 (A) Reaction-induced phase transition of nanoparticles via dynamic-covalent functionalization using cis-diol and boronic acid binding. (B) TEM images of nanoparticles before and after functionalization. TEM data was adapted from J. Am. Chem. Soc., 2017, 139, 7616. | |
2.4.3. Deprotection.
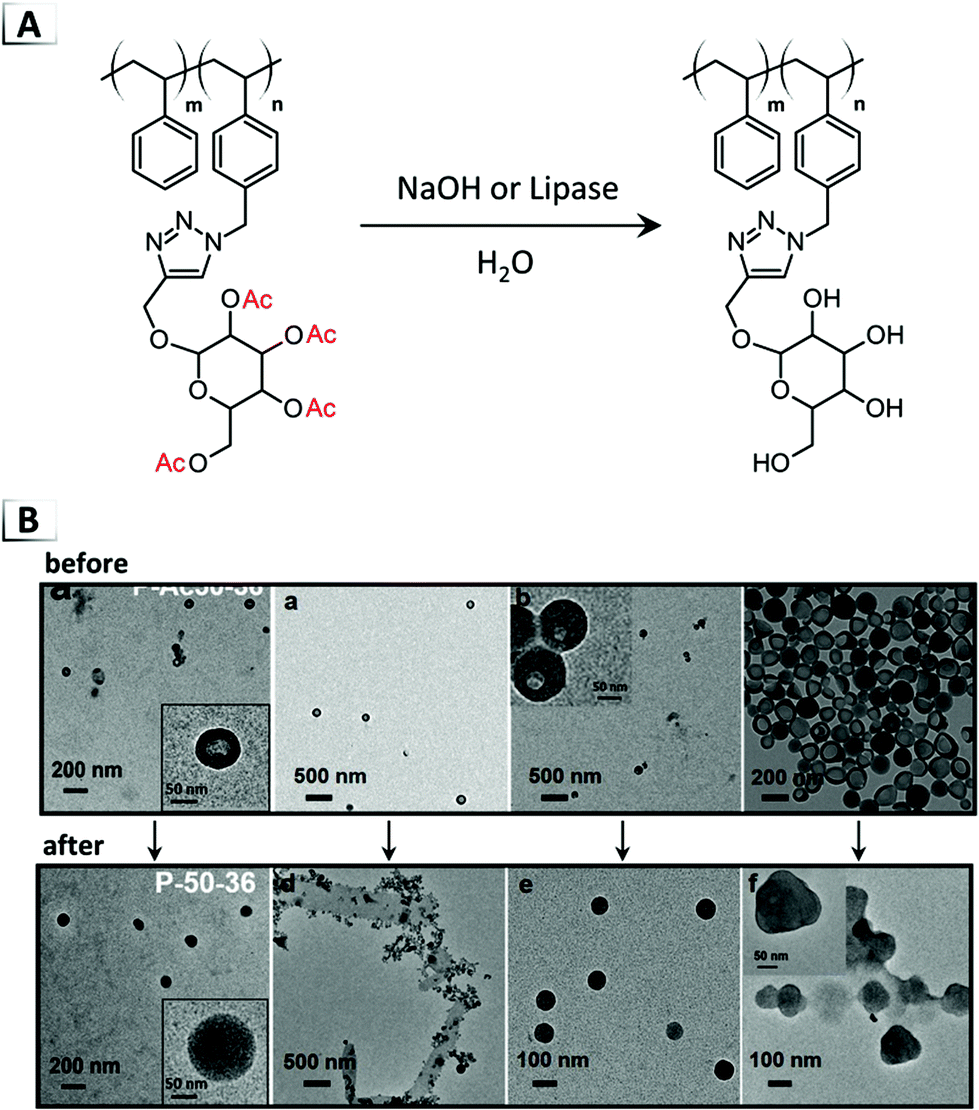
In addition to disorder-to-order transitions (see section 1.2), deprotection chemistry is a powerful handle for inducing order-to-order transitions. Jiang and coworkers demonstrated deacetylation of the internal glyco-block within vesicles as a useful tool for their conversion into numerous other morphologies.46 Deacteylation of vesicles was performed by two methods; the first was conducted under aqueous conditions with sodium hydroxide (unlike their prior examples in organic media)45,47 and the second was accomplished through enzymatic deprotection with a lipase (Fig. 13A). Depending on the composition of the glyco-BCP, vesicles were reported to transform into micelles, lamella sheets, and compound micelles (Fig. 13B). The authors investigated this morphological transition and presented evidence that the vesicle-to-micelle transition underwent a fusion mechanism followed by reorganization to form the final structure.
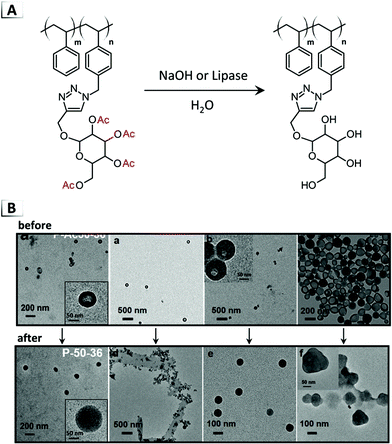 |
| | Fig. 13 (A) Reaction-induced phase transition of nanoparticles via deprotection using deacetylation. (B) TEM images of assembled structures before and after deprotection. TEM data was adapted from J. Am. Chem. Soc., 2018, 140, 8851. | |
2.4.4. Chain-scission.
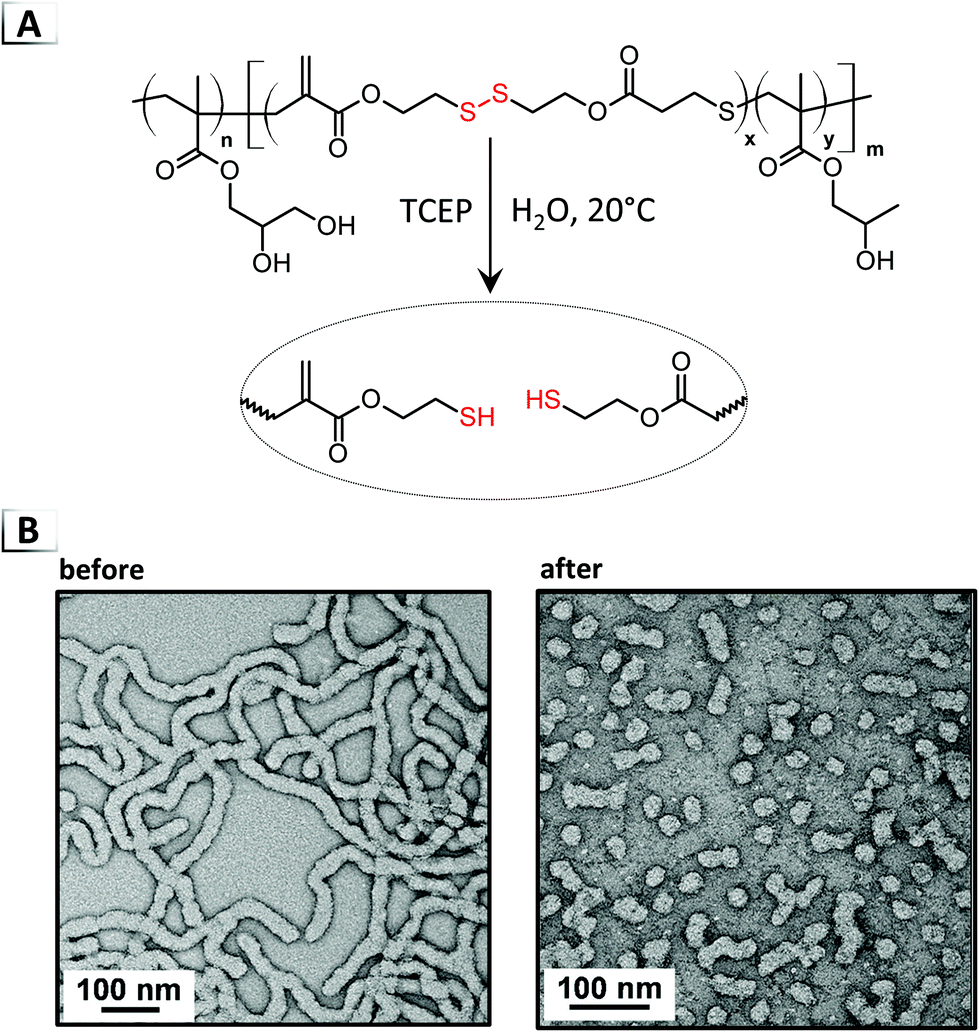
Uniquely, chain-scission reactions have been utilized with pre-assembled nanostructures to cause order-to-order RIPTs by altering the DP of one block within a BCP (Fig. 2E). From a geometric perspective, the morphological transition is driven by a change in the packing parameter, where chain-scission events effectively modify the volume fraction of the solvophobic component. To our knowledge, RIPTs via chain-scission of a solvophilic stabilizer block have yet to be reported. Two main strategies using either reductive-cleavage or metathesis have been employed to modify the chain-length of BCPs. In the first strategy, chain-scission was reported by Paulusse et al. in 2016 through the reduction of backbone disulfide bonds in PISA-derived particles (Fig. 14A).70 In their approach, a BCP was synthesized that contained backbone disulfide segments within the solvophobic, poly(hydroxypropyl methacrylate) block. Under reductive conditions using tris(carboxyethyl)phosphine (TCEP) at pH ≈ 8–9, the authors reported a transition from a worm-like morphology into a (mostly) spherical morphology in water at room temperature (Fig. 14B). Interestingly, the authors found that the reduction reaction occurred on a much shorter time scale than the morphological transition, i.e. the reduction to thiols occurred in 16 hours whereas the morphological transition required up to 8 days.
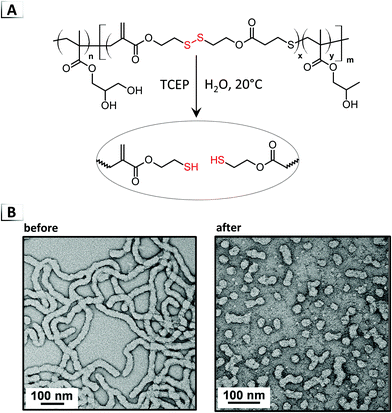 |
| | Fig. 14 (A) Reaction-induced phase transition of nanoparticles via chain-scission using reductive-disulfide cleavage. (B) TEM images of assembled structures before and after chain-scission. TEM data was adapted from Biomacromolecules, 2016, 17, 2277. | |
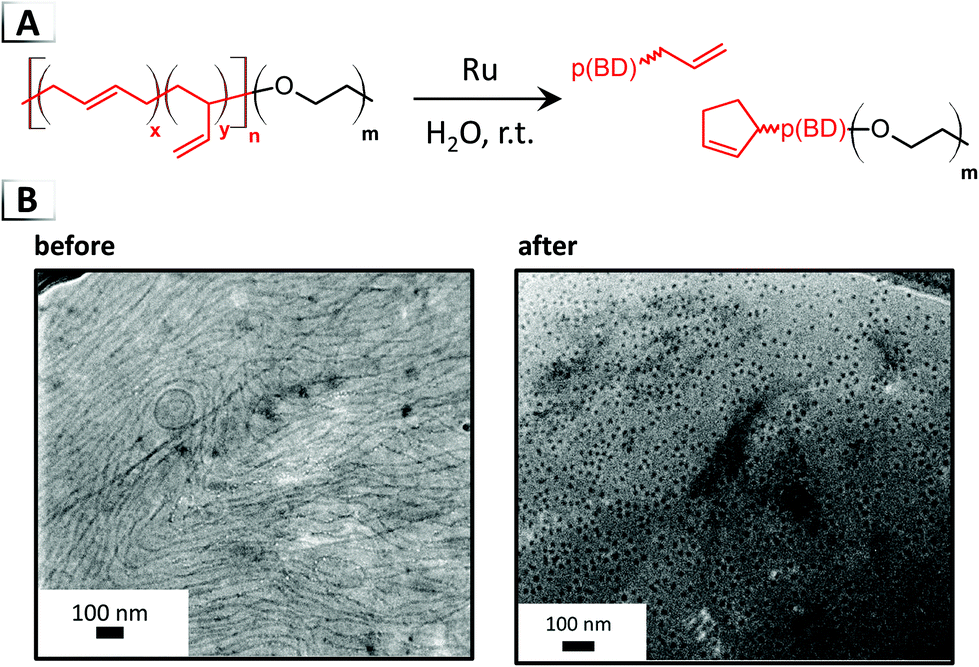
In the second strategy, Jones et al. utilized metathesis for chain-scission of poly(butadiene)-block-poly(ethylene oxide), p(BD-b-EO), to invoke order-to-order morphological transitions.71 Olefin metathesis reactions were performed on a p(BD) block with primarily 1,4-addition repeat units. Using the Hoveyda–Grubbs catalyst in aqueous dispersions, the hydrophobic butadiene block was degraded in pre-assembled nanostructures thereby altering the BCPs composition and hence its packing parameter. Chain-scission reactions within the p(BD) block are primary thought to occur through cross metathesis of backbone olefins or through ring-closing reactions involving side-chain olefins adjacent to backbone olefins (Fig. 15A), although other reaction pathways are possible.72 Morphological transitions took place within minutes, driving morphological transitions from vesicle to worm-like or worm-like to spherical micelles (Fig. 15B), depending on the weight fraction of poly(ethylene oxide) block.
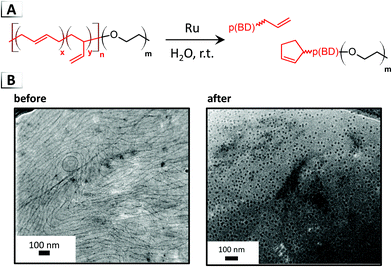 |
| | Fig. 15 (A) Reaction-induced phase transition of nanoparticles via chain-scission using olefin metathesis. (B) TEM images of assembled structures before and after chain-scission. TEM data was adapted from Macromolecules, 2018, 51, 6543. | |
3. Reaction-induced phase transitions in bulk
The most common reaction for bulk RIPTs is polymerization, which triggers in situ macro- or micro-phase separation, accomplished through grafting, crosslinking, and chain-extension (Fig. 1). Many widely used materials rely on bulk RIPTs to achieve their final properties including reaction–injection-molded polyurethanes,73–75 high-impact polystyrene,76 extended-wear contact lenses,77 separation media,78 and toughened epoxies.79,80 In the discussion that follows, we first describe the mechanism that underpins the formation of some of these historic materials, and then proceed to discuss more recent RIPTs that have been employed to obtain a range of exquisite nano-structured morphologies and materials.
One theme that will emerge in the following discussion is that nearly all structures formed by RIPTs in bulk result from kinetic arrest or gelation, and that it is precisely the in situ nature of the polymerization reaction that is responsible for the final structures that form. In many cases, this makes the bulk structures formed by RIPTs different from those that would form under non-reactive bulk assembly. Due to length limitations, our discussion here is limited to polymerization-based RIPTs and we will not discuss other fascinating examples of bulk RIPTs like photo-isomerization-induced phase transitions.81–84
3.1. Graft polymerization-induced phase separation
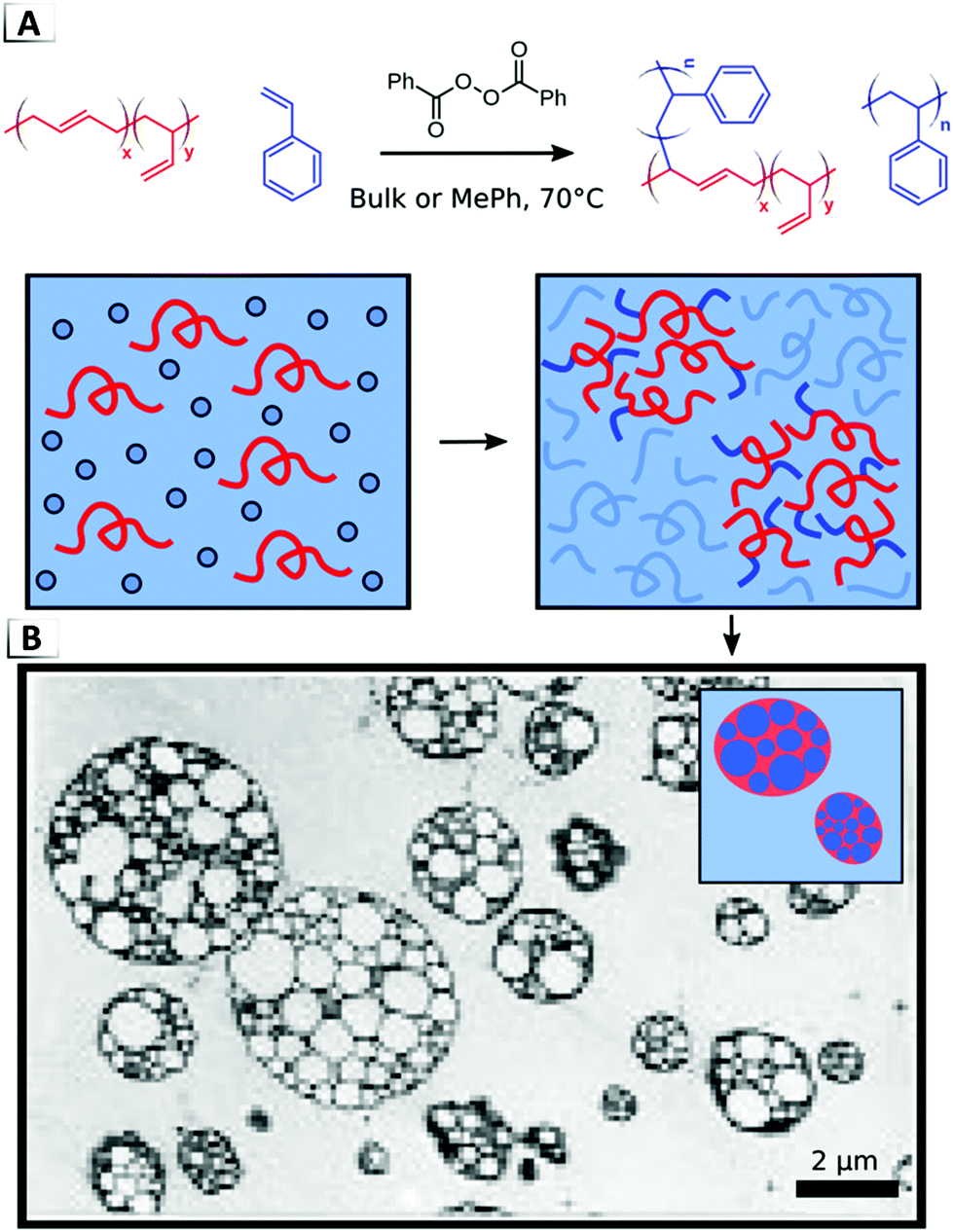
One of the most prominent examples of a material that results from a RIPT in the bulk is high-impact polystyrene (HIPS).76 HIPS is formed through the in situ polymerization of styrene in the presence of dissolved poly(butadiene), p(BD), as shown in Fig. 16. During polymerization, peroxide initiators attack the styrene monomers and p(BD) chains which results in the formation of both polystyrene homopolymer and polystyrene grafted polybutadiene (p(BD-g-S)), (Fig. 16A). While the grafting of poly(styrene) chains to poly(butadiene) may proceed through various pathways, evidence supports hydrogen abstraction from the allylic position and grafting-from the 1,4-addition repeat units of p(BD) when employing peroxide initiators.85–87 Homopolymerization and graft polymerization induce both phase separation and phase inversion, which results in the so-called “salami” morphology that is a signature of HIPS (Fig. 16B).76 It is important to note that this unique morphology arises from the in situ nature of the grafting reaction and it is not an equilibrium thermodynamic phase.76 The non-equilibrium origins of the salami morphology provide an important example of how RIPTs can gain access to morphologies inaccessible to static (non-reactive) systems.
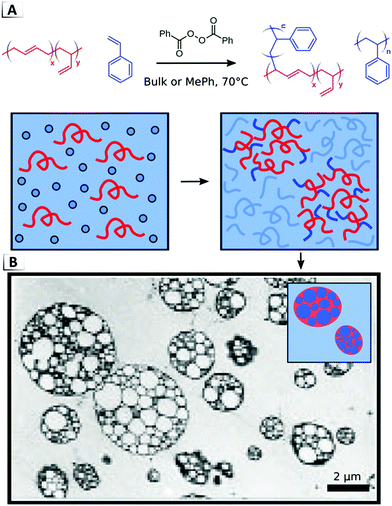 |
| | Fig. 16 (A) Formation of high-impact polystyrene via graft polymerization of styrene and poly(butadiene). (B) TEM image and illustration of salami morphology. TEM data was adapted from Macromolecules, 1996, 29, 2498. | |
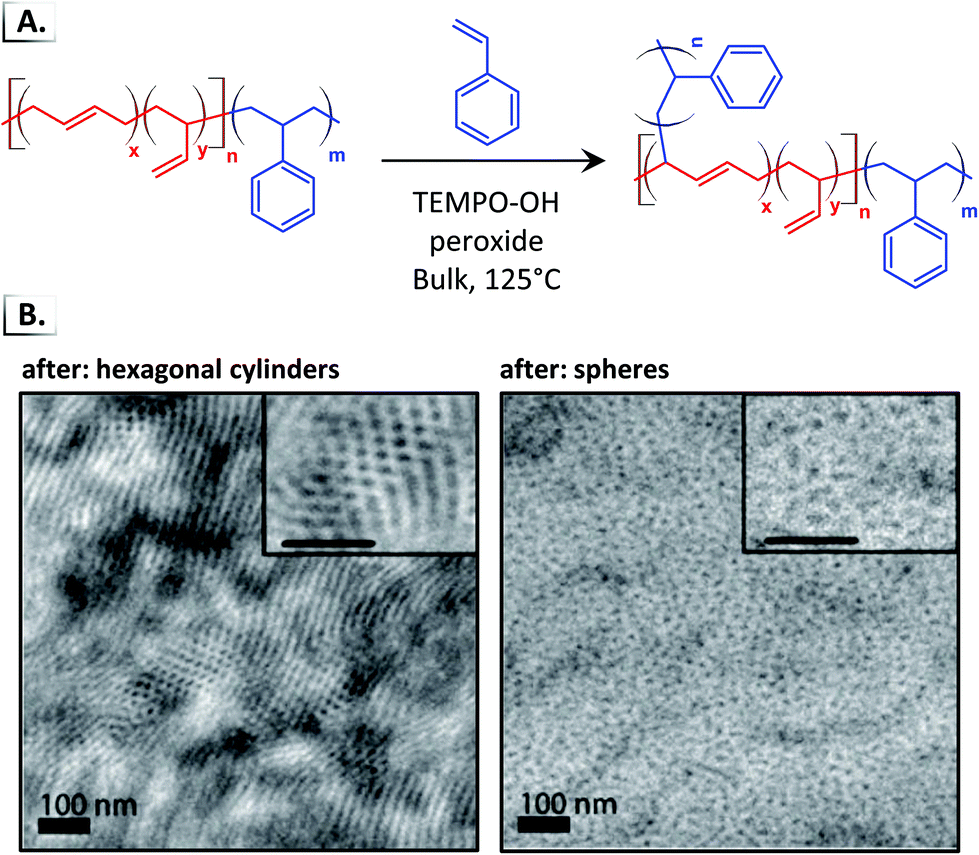
Despite the widespread use of HIPS, there are still many unanswered questions related to the phase transitions induced by graft polymerization. In recent work, Hickey et al. performed in situ polymerization of styrene from the p(BD) block of a lamella-forming p(S-b-BD) diblock copolymer (Fig. 17A).85 The growth of the p(S) grafts induced disorder-to-order or order-to-order phase transitions into different microphases (e.g. lamellar, hexagonal cylinders, disordered spheres) which could be controlled by changing the initial BCP-to-monomer ratio (Fig. 17B). Subsequent work showed that the order-to-order phase transition follows a complex phase path that passed through several metastable phases including the hexagonally perforated lamella phase.18 By trapping a system in one of these metastable phases, one can gain access to new phases that do not exist at equilibrium and that do not emerge directly from quenching a disordered sample, as shown recently with linear diblock copolymers.88
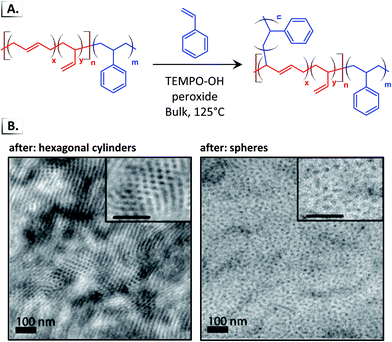 |
| | Fig. 17 (A) Graft polymerization of styrene from poly(butadiene)-block-poly(styrene). (B) TEM images of phase separated morphologies after graft polymerization. TEM data was adapted from ACS Macro Lett., 2018, 7, 822. | |
3.2. Polymerization-induced macrophase separation
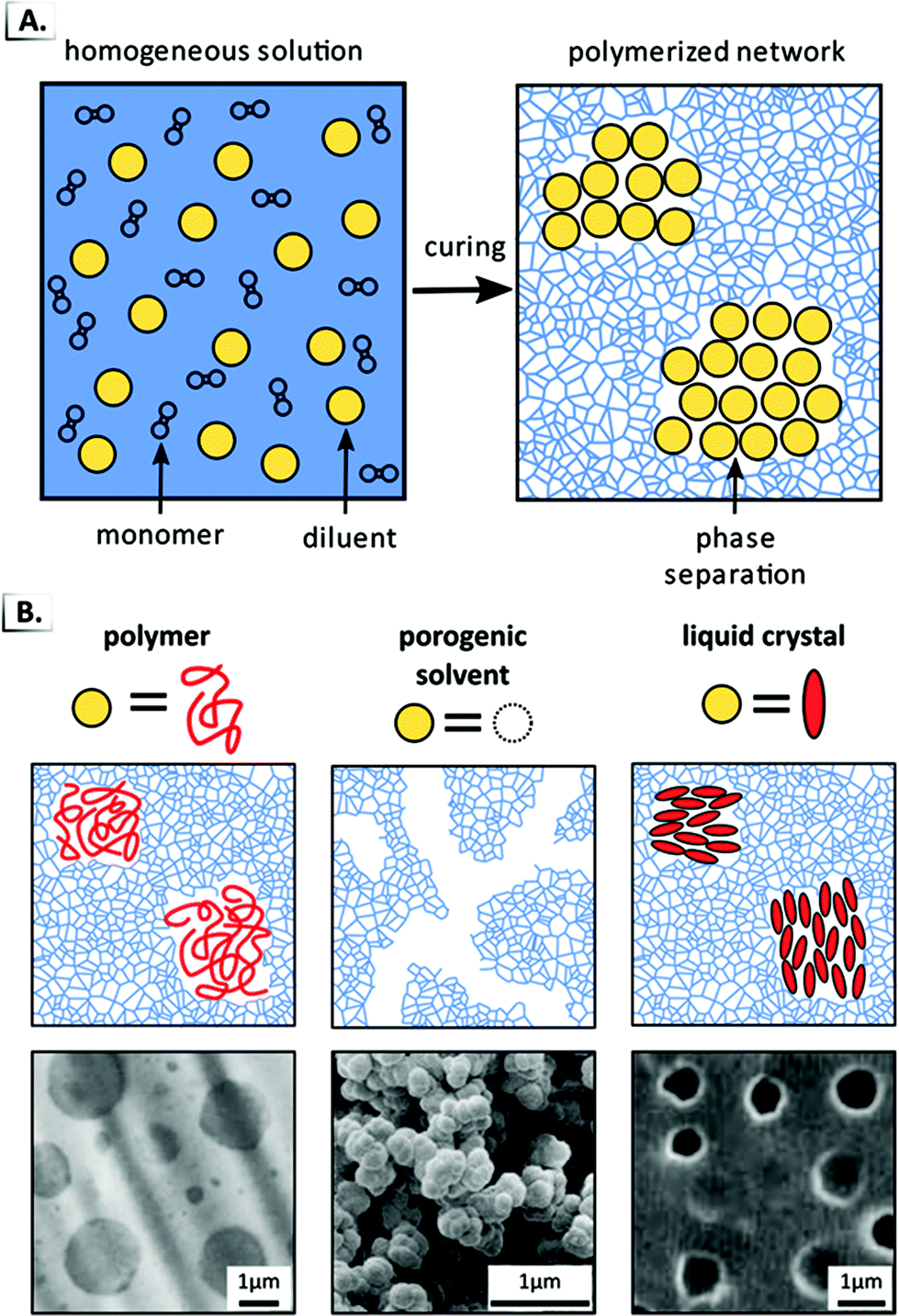
Another prominent and widely used example of a bulk RIPT involves the polymerization of a crosslinked polymer network in the presence of a non-reactive diluent (i.e. polymer, solvent, etc.). In these systems, an initially homogeneous mixture is polymerized that contains monomer, crosslinker, and non-reactive diluent (Fig. 18A). During polymerization, the favorable entropy of mixing between the network and diluent decreases, which triggers spinodal decomposition and the expulsion of the diluent from the network into its own phase. Though spinodal decomposition typically results in macrophase separation, the crosslinked network inhibits domain coarsening and results in a material with domains of diluent dispersed throughout the crosslinked network. As a consequence, there is a competition between the kinetics of macrophase separation and the kinetics of network formation, with the ratio of these two rates playing a critical role in the resultant structures. This technique has been referred to by many names in the literature including “reaction-induced”,79 “chemically-induced”,80 or “polymerization-induced” phase separation.89 Since the latter most clearly differentiates this approach from the others described in this review, we will refer to this process as polymerization-induced phase separation or PIPS.
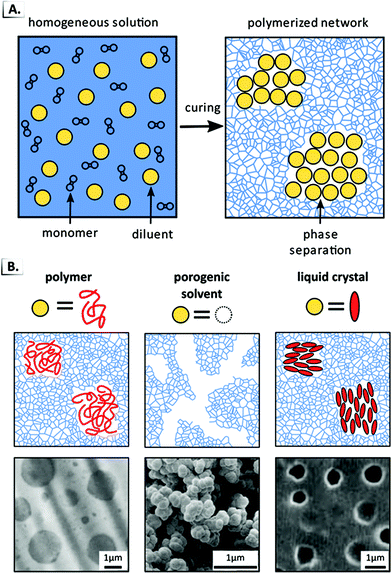 |
| | Fig. 18 (A) Schematic of polymerization-induced phase separation in the presence of a diluent. The non-reactive diluent is represented by yellow spheres and the polymerized network is represented by the blue scaffolding. (B) TEM data and illustrations of PIPS used to encapsulate polymers, porogenic solvent, or liquid crystals. Data was adapted from Polymer, 1989, 60, 1839; J. Sep. Sci., 2004, 27, 747; Mol. Cryst. Liq. Cryst., 1988, 165, 511. | |
The PIPS mechanism can be applied to a broad range of systems since it occurs due to the decreased entropy of mixing that results from polymerization. In principle, PIPS can occur in any system that combines polymerization with kinetic arrest and that does not depend on any chemical interactions between the polymerizing network and the diluent. Consequently, PIPS has been employed to create a wide range of phase-separated polymer alloys simply by varying the diluent molecule employed. Some common diluents include thermoplastics for toughened epoxy networks,79,80,90 porogenic solvents for separation media,78,91–100 or liquid crystals89,101 for light shutters,102 flexible displays,103 switchable photonic crystals,104 and polymer-dispersed blue-phases (Fig. 18B).105 More recently, PIPS has found applications in UV-cured dental materials,106–108 polymer–nanoparticle composites,109–113 nanostructured ceramics,114 supramolecular polymers,115 and shape memory composites.116,117 PIPS has been employed within microfluidic devices to produce microfibers118 and to create semi-permeable microcapsules for drug delivery.119 PIPS has also been used for emerging applications as magnetic sponges for oil spill reclamation,120 recyclable polyurethane blends,121 and polymer electrolytes in structural lithium ion batteries.122,123 As these examples illustrate, PIPS is an exceptionally versatile platform that is compatible with a wide range of diluents. In the following section, we highlight a recent extension of PIPS where the diluent is chosen to be a self-assembling BCP. As one might anticipate, the union of these complementary technologies has been particularly fruitful and has provided access to a wide range of nanostructured morphologies and functional materials.
3.3. Polymerization-induced microphase separation
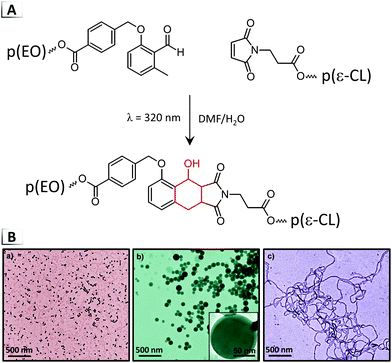
The main advantage that results from the combination of PIPS with BCP self-assembly is the ability to achieve highly-organized microphase separated domains. Domains in traditional PIPS result from a delicate balance between the rate of phase separation and the rate of kinetic arrest, which makes precise morphological control with PIPS challenging. In contrast, the domain size and morphology in self-assembling BCP systems are controlled through the molecular weight and volume fraction of each polymer block, which are synthetically easier to adjust. As a consequence, hybrid BCP/PIPS systems provide superior control over the size and morphology of the self-assembled domains, especially if nanoscale domains are desired. While processes that combine PIPS with BCP self-assembly are still appropriately referred to as PIPS throughout the literature, we will follow the nomenclature introduced by Seo and Hillmyer and refer to PIPS-based approaches that rely on BCPs as “polymerization-induced microphase separation” (PIMS).12 Owing to the enhanced level of morphological control offered by this RIPT, PIMS is being explored as technological platform for various application including the synthesis of polymer electrolyte membranes for use in fuel cells or lithium-ion batteries.124–127 PIMS systems are typically conducted using five main approaches (outlined in the following sections) involving BCPs that are (1) selective, (2) non-selective, or (3) reactive, in addition to, network forming chain-extensions that are conducted in the (4) absence or (5) presence of diluents.
3.3.1. Selective block copolymers.
Some of the earliest work that combined BCP self-assembly with PIPS involved blends of BCPs with epoxy resin.128,129 In one example, a series of poly(ethylene oxide)-block-poly(ethyl ethylene) p(EO-b-EE) copolymers were dispersed in an anhydride cured epoxy resin synthesized from a glycidyl end-capped poly(bisphenol-A-co-epichlorohydrin) prepolymer and phthalic anhydride hardener.128 The pre-cured epoxy resin was selective to the p(EO) block which resulted in the formation of highly-ordered BCP domains of ∼10 nm immersed in epoxy monomer (Fig. 19A). Upon curing, the structure remained largely intact and resulted in well-dispersed p(EO-b-EE) domains within the epoxy matrix. The only structural change observed upon curing was the expulsion of the p(EO) block from the epoxy network, resulting in p(EE) cores surrounded by a p(EO) corona (see Fig. 19A TEM). This indicates that the entropy-driven phase segregation associated with the epoxy matrix can still affect the final morphology, despite the initially favorable interactions between the p(EO) block and the pre-cured epoxy resin. Since domains were well below the wavelength of visible light, this procedure resulted in optically transparent materials that demonstrated an improved mechanical response relative to typical epoxy networks.130,131 Similar work employed poly(styrene)-block-poly(butadiene)-block-poly(methyl methacrylate) copolymers that resulted in many intriguing morphologies from an interplay between the nano-confinement induced by the epoxy network and the triblock copolymer's inherent molecular frustration.132,133
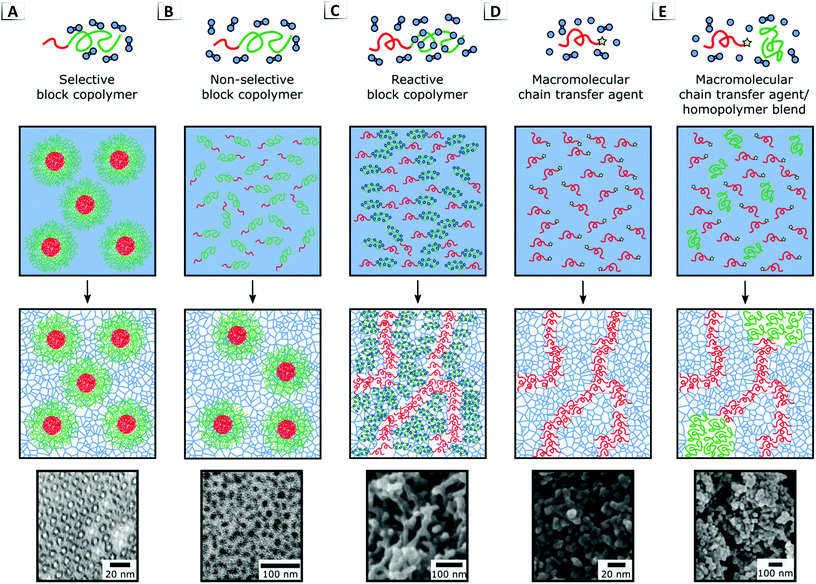 |
| | Fig. 19 Polymerization-induced microphase separation using BCPs and macro-chain transfers agents. (A) BCP with selective interactions with monomer solution.128 (B) BCP with non-selective interactions with monomer solution.134 (C) Reactive BCP with functional groups that are selectively polymerized into network.135 (D) Macro-chain transfer agent with mono- and di-functional monomers.12 (E) Hierarchical self-assembly achieved with macromolecular chain-transfer agent/homopolymer blends.136 TEM data was adapted from J. Am. Chem. Soc., 1997, 119, 2749; Macromolecules, 2006, 39, 711; J. Am. Chem. Soc., 2007, 129, 13786; Science, 2012, 336, 1422; J. Am. Chem. Soc., 2015, 137, 8896. | |
3.3.2. Non-selective block copolymers.
A common feature of these early studies was a two-step process whereby the BCPs were first self-assembled in selective media and then the epoxy resin was cured. Subsequent reports revealed that microphase separation and the epoxy cure could be combined into a single step by utilizing the principle of PIPS described above (Fig. 19B).134,137–140 One example of this approach involved blending a non-selective poly(ε-caprolactone)-block-poly(butadiene)-block-poly(ε-caprolactone) triblock copolymer with a bisphenol-A diglycidyl ether precursor and a 4,4′-methylenebis(2-chloroaniline) curing agent.134 The initially homogeneous blend was then cured at 150 °C, afterward forming a phase separated, nanostructured thermoset (see Fig. 19B TEM). While the approach did not form nanostructures that were as well-organized as earlier works,128,129 a wider range of polymer chemistries could be applied since the epoxy resin did not need to be selective for one block. This approach was subsequently employed with diblock, triblock, and star BCPs, which could be used to obtain a wide range of morphologies including dispersed and fused spherical particles, worm-like structures, bicontinuous structures, lamellae, and cylinders.134,137–140
3.3.3. Reactive block copolymers.
A potential disadvantage of the approaches described in sections 3.3.1 and 3.3.2 is that the BCP is not covalently incorporated into the network and is expelled upon network formation. Preventing the expulsion of at least one of the blocks could lead to improved mechanical properties130 and better control of the post-cure morphologies. One approach towards this goal relies upon a BCP that can be directly polymerized into the network to prevent BCP expulsion during cure.135 For instance, covalent incorporation was demonstrated using a diblock copolymer, composed of a polylactide block and a statistical block of styrene and para-norbornenylethyl styrene (i.e., p(LA-b-(S-s-N)), that was initially immersed in a dicyclopentadiene crosslinker and a small amount of solvent (Fig. 19C). By addition of Grubbs catalyst, copolymerization of the crosslinker and polymeric norbornenes occurred, which covalently incorporated the BCP into the network. In this system, polymerization triggered the microphase separation of the BCP and the network, but the phase separation was arrested due to the covalent bonds between the polymer and the network. This arrested phase separation yielded a disordered bicontinuous structure with ∼20 nm pores (Fig. 19C), which could be used to form an isoporous membrane with good mechanical properties upon p(LA) removal. Subsequent work showed that the pore size could be tailored by varying the p(LA) molecular weight and crosslinker weight percent, and that the formation of bicontinuous or discrete structures could be predicted from overlap concentrations in p(LA-b-(S-s-N)) crosslinker solutions.141
3.3.4. Network forming chain-extensions.
A particularly elegant RIPT relies upon simultaneous chain-extension and network formation (Fig. 19D).12,142–145 Seo and Hillmyer first demonstrated this approach by employing a poly(lactic acid) macro-chain transfer agent, p(LA)-CTA, in a statistical copolymerization of styrene (S) monomer and divinyl benzene (DVB) crosslinker under bulk conditions.12 As the crosslinked p(S-s-DVB) block grows, the driving force for segregation from the p(LA) block increases, which eventually triggers microphase separation. However, due to crosslinking, the polymer chains are polymerized into the matrix, which prevents their free diffusion and arrests the existing microphase separated morphology. This technique yields bicontinuous structures that can be converted into nanoporous matrixes upon p(LA) removal. The pore size achieved with this approach was ca. 4–8 nm, which could be controlled by varying the p(LA)-CTA's molecular weight. The authors described this system as “polymerization-induced microphase separation” (PIMS) since it resulted from a clear kinetic competition between the microphase separation of the growing BCP and the kinetic arrest caused by gelation.12 As a consequence of this competition, the structures generated by PIMS have been shown to depend on many parameters including reaction temperature, monomer-to-crosslinker ratio, p(LA)-CTA molar mass and mass fraction, and the reactivity ratio of the monomer and crosslinker.146 In more recent reports, the PIMS approach has been extended to the synthesis of hierarchical materials by incorporating dual-reactive monomers that can participate in hyper-crosslinking142–144 or rigid, sterically demanding monomers that can inhibit efficient chain packing.144,145
The ability of PIMS to form bicontinuous structures over a wide range of parameters is particularly notable, especially in contrast to the equilibrium self-assembly of linear diblock copolymers. At equilibrium, linear diblock copolymers only form bicontinuous phases in relatively small regions of phase space which correspond to the gyroid and O70 phases.1,128 By combining in situ polymerization with kinetic arrest due to crosslinking, PIMS can achieve bicontinuous structures over a much larger range of experimental parameters. Consequently, materials based on PIMS have received considerable attention in applications that benefit from bicontinuous morphologies such as polymer electrolyte membranes, catalytic supports, nanoscale templates, and separation media.
3.3.5. Network forming chain-extensions with diluent.
Another development has been to create hierarchically porous materials by combining microphase segregation associated with PIMS and macrophase segregation associated with traditional PIPS. One example of this approach has been to copolymerize S and DVB from a p(LA)-CTA in the presence of a nonreactive p(EO) homopolymer (Fig. 19E).136 Poly(ethylene oxide) is miscible with p(LA)147 and during the early stages of polymerization the p(EO) selectively partitions into the p(LA) domains. However, as the polymerization proceeds, the nanosized domains of p(LA) cannot accommodate the p(EO) chains above a certain molecular weight and macrophase separation begins to occur. In spite of this, full macrophase separation is inhibited due to the kinetically arrested p(S-s-DVB) matrix, and the final morphology contains a hierarchical pore structure that can be tuned by changing the p(EO) volume fraction (Fig. 19E).
4. Conclusions and future outlook
Reaction-induced phase transitions represent a broad and versatile platform for the organization of BCPs. By altering the chemical composition of a BCP in situ, RIPTs can trigger the formation of a myriad of sophisticated nanostructures in both solution and bulk. Herein, we have highlighted various types of reactions—functionalization, deprotection, conjugation, chain-scission, and polymerization—for inducing order in BCPs for access to an assortment of micellar, macrophase separated, and microphase separated materials. Even within this limited scope, the body of literature describing RIPTs is growing rapidly, a trend that will likely continue due to the expansive range of possibilities in how different chemical transformations and phase transitions might be combined.
Despite the vast potential of RIPTs, several underexplored areas remain, a few of which are outlined below. First, the field of RIPTs would be enriched through the application of new and powerful chemistries that are orthogonal, highly efficient, and simple to perform. For instance, orthogonal reaction handles would permit more complex phase transitions, multi-stage processes involving sequential/simultaneous RIPTs, or processes that could impart morphological change in addition to properties necessitated by a given application. Moreover, simple, yet highly efficient reactions would enable broader implementation and the reduction of residual contaminates (reagents, monomers, catalysts, crosslinkers, etc.). Second, the utilization of other advanced polymeric architectures (multi-block copolymers, bottlebrushes copolymers, miktoarm stars, etc.), beyond the typically employed diblock copolymer, are largely unexplored. Complex architectures in conjunction with the potential for non-equilibrium morphologies in RIPTs could unearth new, fascinating, and potentially useful nanostructures. Third, progress toward increased solids content, particularly in solution-based RIPTs, would enhance the processing of well-defined nanomaterials, enabling scalable quantities of materials, application development, and the minimization of VOCs or harmful reagents. PISA is an exemplary system that has achieved solids contents up to 50 wt%,11 whereas many other solution-based RIPTs are performed under significantly greater dilution. Last, the field requires a greater level of predictive capability, systematic experimentation, and a deeper understanding of the governing principles that underpin these complex and dynamic processes.
Conflicts of interest
There are no conflicts to declare.
References
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3–22 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS.
- W. A. Phillip, B. O'Neill, M. Rodwogin, M. A. Hillmyer and E. L. Cussler, ACS Appl. Mater. Interfaces, 2010, 2, 847–853 CrossRef CAS.
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS.
- I. P. Beletskaya, A. N. Kashin, A. E. Litvinov, V. S. Tyurin, P. M. Valetsky and G. van Koten, Organometallics, 2006, 25, 154–158 CrossRef CAS.
- S. Sugihara, M. Sudo and Y. Maeda, Langmuir, 2019, 35, 1346–1356 CrossRef CAS.
- J. Lü, Y. Yang, J. Gao, H. Duan and C. Lü, Langmuir, 2018, 34, 8205–8214 CrossRef.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS.
- M. Seo and M. A. Hillmyer, Science, 2012, 336, 1422–1425 CrossRef CAS.
- C. P. Brangwynne, P. Tompa and R. V. Pappu, Nat. Phys., 2015, 11, 899–904 Search PubMed.
- M. Buchanan, Nat. Phys., 2019, 15, 2–2 Search PubMed.
- C. F. Lee and J. D. Wurtz, J. Phys. D: Appl. Phys., 2018, 52, 023001 CrossRef.
- I. Santi, N. Dhar, D. Bousbaine, Y. Wakamoto and J. D. McKinney, Nat. Commun., 2013, 4, 2470 CrossRef.
- C. Ballestrem, B. Wehrle-Haller and B. A. Imhof, J. Cell Sci., 1998, 111, 1649–1658 CAS.
- E. S. Zofchak, J. A. LaNasa, V. M. Torres and R. J. Hickey, Macromolecules, 2020, 53, 835–843 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Soft Matter, 2012, 8, 7753–7767 RSC.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- J.-F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117–7129 RSC.
- C. Lu and M. W. Urban, Prog. Polym. Sci., 2018, 78, 24–46 CrossRef CAS.
- Y. Pei, A. B. Lowe and P. J. Roth, Macromol. Rapid Commun., 2017, 38, 1600528 CrossRef.
- L. Chen, R. Liu and Q. Yan, Angew. Chem., Int. Ed., 2018, 57, 9336–9340 CrossRef CAS.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS.
- H. Kim, Y. J. Kang, E. S. Jeong, S. Kang and K. T. Kim, ACS Macro Lett., 2012, 1, 1194–1198 CrossRef CAS.
- G. Liu, X. Wang, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2014, 136, 7492–7497 CrossRef CAS.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS.
- G. Delaittre, N. K. Guimard and C. Barner-Kowollik, Acc. Chem. Res., 2015, 48, 1296–1307 CrossRef CAS.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1355 RSC.
- A. J. D. Magenau, T. R. Hartlage and R. F. Storey, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5505–5513 CrossRef CAS.
- A. J. D. Magenau, J. W. Chan, C. E. Hoyle and R. F. Storey, Polym. Chem., 2010, 1, 831–833 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176–191 RSC.
- Y. Shi, X. Cao and H. Gao, Nanoscale, 2016, 8, 4864–4881 RSC.
- S. Li, J. Wang, J. Shen, B. Wu and Y. He, ACS Macro Lett., 2018, 7, 437–441 CrossRef CAS.
- D. H. Howe, R. M. McDaniel and A. J. D. Magenau, Macromolecules, 2017, 50, 8010–8018 CrossRef CAS.
- D. H. Howe, J. L. Hart, R. M. McDaniel, M. L. Taheri and A. J. D. Magenau, ACS Macro Lett., 2018, 7, 1503–1508 CrossRef CAS.
- D. H. Howe, K. J. Jenewein, J. L. Hart, M. L. Taheri and A. J. D. Magenau, Polym. Chem., 2019, 11, 298–303 RSC.
- E. Muzammil, A. Khan and M. C. Stuparu, RSC Adv., 2017, 7, 55874–55884 RSC.
- L. Su, C. Wang, F. Polzer, Y. Lu, G. Chen and M. Jiang, ACS Macro Lett., 2014, 3, 534–539 CrossRef CAS.
- W. Qi, Y. Zhang, J. Wang, G. Tao, L. Wu, Z. Kochovski, H. Gao, G. Chen and M. Jiang, J. Am. Chem. Soc., 2018, 140, 8851–8857 CrossRef CAS.
- X. Wu, L. Su, G. Chen and M. Jiang, Macromolecules, 2015, 48, 3705–3712 CrossRef CAS.
- E. Yoshida and S. Kuwayama, Colloid Polym. Sci., 2007, 285, 1287–1291 CrossRef CAS.
- E. Yoshida and S. Kuwayama, Colloid Polym. Sci., 2008, 286, 1621–1627 CrossRef CAS.
- J.-M. Schumers, O. Bertrand, C.-A. Fustin and J.-F. Gohy, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 599–608 CrossRef CAS.
- O. Bertrand, J.-M. Schumers, C. Kuppan, J. Marchand-Brynaert, C.-A. Fustin and J.-F. Gohy, Soft Matter, 2011, 7, 6891–6896 RSC.
- C. Wu, A. Niu, L. M. Leung and T. S. Lam, J. Am. Chem. Soc., 1999, 121, 1954–1955 CrossRef CAS.
- L. M. Leung and K. H. Tan, Polymer, 1994, 35, 1556–1560 CrossRef CAS.
- L. Chen, M. Xu, J. Hu and Q. Yan, Macromolecules, 2017, 50, 4276–4280 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS.
- M. J. Derry, O. O. Mykhaylyk and S. P. Armes, Angew. Chem., Int. Ed., 2017, 56, 1746–1750 CrossRef CAS.
- C. J. Mable, R. R. Gibson, S. Prevost, B. E. McKenzie, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 16098–16108 CrossRef CAS.
- Q. Chen, H. Schönherr and G. J. Vancso, Small, 2010, 6, 2762–2768 CrossRef CAS.
- H. Liu, Y. Zhao, C. A. Dreiss and Y. Feng, Soft Matter, 2014, 10, 6387–6391 RSC.
- H. Liu, S. Lin, Y. Feng and P. Theato, Polym. Chem., 2017, 8, 12–23 RSC.
- X. Ji, H. Wang, Y. Li, D. Xia, H. Li, G. Tang, J. L. Sessler and F. Huang, Chem. Sci., 2016, 7, 6006–6014 RSC.
- H. Yao, Y. Ning, C. P. Jesson, J. He, R. Deng, W. Tian and S. P. Armes, ACS Macro Lett., 2017, 6, 1379–1385 CrossRef CAS.
- B. Maiti, K. Bauri, M. Nandi and P. De, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 263–273 CrossRef CAS.
- Y. Pei, J.-M. Noy, P. J. Roth and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2326–2335 CrossRef CAS.
- Y. Pei, J.-M. Noy, P. J. Roth and A. B. Lowe, Polym. Chem., 2015, 6, 1928–1931 RSC.
- N. Busatto, J. L. Keddie and P. J. Roth, Polym. Chem., 2020, 11, 704–711 RSC.
- H. Liu, R. Wang, J. Wei, C. Cheng, Y. Zheng, Y. Pan, X. He, M. Ding, H. Tan and Q. Fu, J. Am. Chem. Soc., 2018, 140, 6604–6610 CrossRef CAS.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616–7623 CrossRef CAS.
- R. Deng, Y. Ning, E. R. Jones, V. J. Cunningham, N. J. W. Penfold and S. P. Armes, Polym. Chem., 2017, 8, 5374–5380 RSC.
- L. P. D. Ratcliffe, C. Couchon, S. P. Armes and J. M. J. Paulusse, Biomacromolecules, 2016, 17, 2277–2283 CrossRef CAS.
- B. H. Jones, G. D. Bachand, S. H. R. Shin, M. A. Firestone and W. F. Paxton, Macromolecules, 2018, 51, 6543–6551 CrossRef CAS.
- M. D. Watson and K. B. Wagener, Macromolecules, 2000, 33, 1494–1496 CrossRef CAS.
- A. J. Ryan, Polymer, 1990, 31, 707–712 CrossRef CAS.
- W. P. Yang and C. W. Macosko, Makromol. Chem., Macromol. Symp., 1989, 25, 23–44 CrossRef CAS.
- W. Li, A. J. Ryan and I. K. Meier, Macromolecules, 2002, 35, 5034–5042 CrossRef CAS.
- M. Fischer and G. P. Hellmann, Macromolecules, 1996, 29, 2498–2509 CrossRef CAS.
- P. C. Nicolson and J. Vogt, Biomaterials, 2001, 22, 3273–3283 CrossRef CAS.
- F. Svec, J. Sep. Sci., 2004, 27, 747–766 CrossRef CAS.
- T. Inoue, Prog. Polym. Sci., 1995, 20, 119–153 CrossRef CAS.
-
J. Kiefer, J. L. Hedrick and J. G. Hilborn, in Macromolecular Architectures, Springer Berlin Heidelberg, Berlin, Heidelberg, 1999, pp. 161–247 Search PubMed.
- O. Tsutsumi, T. Kitsunai, A. Kanazawa, T. Shiono and T. Ikeda, Macromolecules, 1998, 31, 355–359 CrossRef CAS.
- K. Okano, A. Shishido and T. Ikeda, Macromolecules, 2006, 39, 145–152 CrossRef CAS.
- X. Tong, L. Cui and Y. Zhao, Macromolecules, 2004, 37, 3101–3112 CrossRef CAS.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2016, 116, 15089–15166 CrossRef CAS.
- E. S. Zofchak, J. A. LaNasa, W. Mei and R. J. Hickey, ACS Macro Lett., 2018, 7, 822–827 CrossRef CAS.
- N.-J. Huang and D. C. Sundberg, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 2571–2586 CrossRef CAS.
- N.-J. Huang and D. C. Sundberg, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 2587–2603 CrossRef CAS.
- K. Kim, M. W. Schulze, A. Arora, R. M. Lewis, M. A. Hillmyer, K. D. Dorfman and F. S. Bates, Science, 2017, 356, 520–523 CrossRef CAS.
- J. W. Doane, A. Golemme, J. L. West, J. B. Whitehead and B. G. Wu, Mol. Cryst. Liq. Cryst. Incorporating Nonlinear Opt., 1988, 165, 511–532 CrossRef CAS.
- K. Yamanaka, Y. Takagi and T. Inoue, Polymer, 1989, 30, 1839–1844 CrossRef CAS.
- V. Pucci, M. A. Raggi, F. Svec and J. M. Frechet, J. Sep. Sci., 2004, 27, 779–788 CrossRef CAS.
- C. Viklund, F. Svec, J. M. J. Fréchet and K. Irgum, Chem. Mater., 1996, 8, 744–750 CrossRef CAS.
- N. Tsujioka, N. Ishizuka, N. Tanaka, T. Kubo and K. Hosoya, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3272–3281 CrossRef CAS.
- A. Palm and M. V. Novotny, Anal. Chem., 1997, 69, 4499–4507 CrossRef CAS.
- D. Moravcová, P. Jandera, J. Urban and J. Planeta, J. Sep. Sci., 2003, 26, 1005–1016 CrossRef.
- E. C. Peters, F. Svec, J. M. J. Fréchet, C. Viklund and K. Irgum, Macromolecules, 1999, 32, 6377–6379 CrossRef CAS.
- C. Viklund, A. Nordström, K. Irgum, F. Svec and J. M. J. Fréchet, Macromolecules, 2001, 34, 4361–4369 CrossRef CAS.
- F. Sinner and M. R. Buchmeiser, Macromolecules, 2000, 33, 5777–5786 CrossRef CAS.
- K. Kanamori, J. Hasegawa, K. Nakanishi and T. Hanada, Macromolecules, 2008, 41, 7186–7193 CrossRef CAS.
- K. J. Barlow, X. Hao, T. C. Hughes, O. E. Hutt, A. Polyzos, K. A. Turner and G. Moad, Polym. Chem., 2014, 5, 722–732 RSC.
- P. S. Drzaic, J. Appl. Phys., 1986, 60, 2142–2148 CrossRef CAS.
- D. K. Yang, L. C. Chien and J. W. Doane, Appl. Phys. Lett., 1992, 60, 3102–3104 CrossRef CAS.
- Z. Hong, Y. Jin, B. Y. Lee and S.-B. Kwon, Mol. Cryst. Liq. Cryst., 2015, 612, 193–200 CrossRef CAS.
- R. L. Sutherland, V. P. Tondiglia, L. V. Natarajan, S. Chandra, D. Tomlin and T. J. Bunning, Opt. Express, 2002, 10, 1074–1082 CrossRef.
- E. Kemiklioglu and L. C. Chien, Eur. Phys. J. E, 2017, 40, 37 CrossRef.
- C. R. Szczepanski, C. S. Pfeifer and J. W. Stansbury, Polymer, 2012, 53, 4694–4701 CrossRef CAS.
- C. R. Szczepanski and J. W. Stansbury, Polymer, 2015, 70, 8–18 CrossRef CAS.
- C. S. Pfeifer, Z. R. Shelton, C. R. Szczpanski, M. D. Barros, N. D. Wilson and J. W. Stansbury, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1796–1806 CrossRef CAS.
- J. Greener, T. H. van der Loop, C. Paquet, G. Scholes and E. Kumacheva, Langmuir, 2009, 25, 3173–3177 CrossRef CAS.
- S. Dubinsky, A. Petukhova, I. Gourevich and E. Kumacheva, Macromol. Rapid Commun., 2010, 31, 1635–1640 CrossRef CAS.
- H. E. Romeo, A. Vílchez, J. Esquena, C. E. Hoppe and R. J. J. Williams, Eur. Polym. J., 2012, 48, 1101–1109 CrossRef CAS.
- R. J. Williams, C. E. Hoppe, I. A. Zucchi, H. E. Romeo, I. E. dell'Erba, M. L. Gomez, J. Puig and A. B. Leonardi, J. Colloid Interface Sci., 2014, 431, 223–232 CrossRef CAS.
- C. Paquet and E. Kumacheva, Adv. Funct. Mater., 2007, 17, 3105–3110 CrossRef CAS.
- S. G. Rudisill, S. Shaker, D. Terzic, R. Le Maire, B. L. Su and A. Stein, Inorg. Chem., 2015, 54, 993–1002 CrossRef CAS.
- H. M. Keizer, R. P. Sijbesma, J. F. G. A. Jansen, G. Pasternack and E. W. Meijer, Macromolecules, 2003, 36, 5602–5606 CrossRef CAS.
- A. H. Torbati, H. B. Nejad, M. Ponce, J. P. Sutton and P. T. Mather, Soft Matter, 2014, 10, 3112–3121 RSC.
- Y. Zhang, X. Jiang, R. Wu and W. Wang, J. Appl. Polym. Sci., 2016, 133, 43534–43534 Search PubMed.
- W. Liu, Z. Xu, L. Sun, P. Guo, C. Zeng, C. Wang and L. Zhang, Chem. Eng. J., 2017, 315, 25–34 CrossRef CAS.
- B. Kim, T. Y. Jeon, Y. K. Oh and S. H. Kim, Langmuir, 2015, 31, 6027–6034 CrossRef CAS.
- Y. Wu, S. Xue, H. Yang, H. Zhang, T. Zhang and S. Gou, Chem. Eng. J., 2017, 328, 639–644 CrossRef CAS.
- A. Reghunadhan, J. Datta, N. Kalarikkal and S. Thomas, Polymer, 2017, 117, 96–106 CrossRef CAS.
- N. Ihrner, W. Johannisson, F. Sieland, D. Zenkert and M. Johansson, J. Mater. Chem. A, 2017, 5, 25652–25659 RSC.
- L. M. Schneider, N. Ihrner, D. Zenkert and M. Johansson, ACS Appl. Energy Mater., 2019, 2, 4362–4369 CrossRef CAS.
- M. W. Schulze, L. D. McIntosh, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2014, 14, 122–126 CrossRef CAS.
- L. D. McIntosh, M. W. Schulze, M. T. Irwin, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2015, 48, 1418–1428 CrossRef CAS.
- S. A. Chopade, S. So, M. A. Hillmyer and T. P. Lodge, ACS Appl. Mater. Interfaces, 2016, 8, 6200–6210 CrossRef CAS.
- S. A. Chopade, J. G. Au, Z. Li, P. W. Schmidt, M. A. Hillmyer and T. P. Lodge, ACS Appl. Mater. Interfaces, 2017, 9, 14561–14565 CrossRef CAS.
- M. A. Hillmyer, P. M. Lipic, D. A. Hajduk, K. Almdal and F. S. Bates, J. Am. Chem. Soc., 1997, 119, 2749–2750 CrossRef CAS.
- P. M. Lipic, F. S. Bates and M. A. Hillmyer, J. Am. Chem. Soc., 1998, 120, 8963–8970 CrossRef CAS.
- J. M. Dean, P. M. Lipic, R. B. Grubbs, R. F. Cook and F. S. Bates, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 2996–3010 CrossRef CAS.
- J. M. Dean, R. B. Grubbs, W. Saad, R. F. Cook and F. S. Bates, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2444–2456 CrossRef CAS.
- S. Ritzenthaler, F. Court, L. David, E. Girard-Reydet, L. Leibler and J. P. Pascault, Macromolecules, 2002, 35, 6245–6254 CrossRef CAS.
- S. Ritzenthaler, F. Court, E. Girard-Reydet, L. Leibler and J. P. Pascault, Macromolecules, 2003, 36, 118–126 CrossRef CAS.
- F. Meng, S. Zheng, W. Zhang, H. Li and Q. Liang, Macromolecules, 2006, 39, 711–719 CrossRef CAS.
- L. Chen, W. A. Phillip, E. L. Cussler and M. A. Hillmyer, J. Am. Chem. Soc., 2007, 129, 13786–13787 CrossRef CAS.
- S. A. Saba, M. P. Mousavi, P. Buhlmann and M. A. Hillmyer, J. Am. Chem. Soc., 2015, 137, 8896–8899 CrossRef CAS.
- F. Meng, S. Zheng, H. Li, Q. Liang and T. Liu, Macromolecules, 2006, 39, 5072–5080 CrossRef CAS.
- F. Meng, S. Zheng and T. Liu, Polymer, 2006, 47, 7590–7600 CrossRef CAS.
- Z. Xu and S. Zheng, Polymer, 2007, 48, 6134–6144 CrossRef CAS.
- S. Fan and T. Kyu, Macromolecules, 2004, 37, 1484–1491 CrossRef CAS.
- M. A. Amendt, L. Chen and M. A. Hillmyer, Macromolecules, 2010, 43, 3924–3934 CrossRef CAS.
- M. Seo, S. Kim, J. Oh, S.-J. Kim and M. A. Hillmyer, J. Am. Chem. Soc., 2015, 137, 600–603 CrossRef.
- S. Kim and M. Seo, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 900–913 CrossRef CAS.
- J. Lee and M. Seo, ACS Macro Lett., 2018, 7, 1448–1454 CrossRef CAS.
- M. B. Larsen, J. D. Van Horn, F. Wu and M. A. Hillmyer, Macromolecules, 2017, 50, 4363–4371 CrossRef CAS.
- M. W. Schulze and M. A. Hillmyer, Macromolecules, 2017, 50, 997–1007 CrossRef CAS.
- H. Mao and M. A. Hillmyer, Macromol. Chem. Phys., 2008, 209, 1647–1656 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
Andrew J. D.
Magenau
a and
Andrew J. D.
Magenau