
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA water-soluble copper-immobilized covalent organic framework functioning as an “OFF–ON” fluorescent sensor for amino acids†
Lamiaa Reda
Ahmed
a,
Ahmed F. M.
EL-Mahdy
 b,
Cheng-Tang
Pan
ac and
Shiao-Wei
Kuo
*bd
b,
Cheng-Tang
Pan
ac and
Shiao-Wei
Kuo
*bd
aInstitute of Medical Science and Technology, National Sun Yat-Sen University, Kaohsiung 80424, Taiwan
bDepartment of Materials and Optoelectronic Science, National Sun Yat-Sen University, Kaohsiung, 80424, Taiwan. E-mail: kuosw@faculty.nsysu.edu.tw
cDepartment of Mechanical and Electro-Mechanical Engineering, National Sun Yat-Sen University, Kaohsiung 80424, Taiwan
dDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 807, Taiwan
First published on 10th May 2021
Abstract
In this paper, we describe the construction of a new fluorescent hydroxyl- and hydrazone-based covalent organic framework (TFPB–DHTH COF) through the one-pot polycondensation of 1,3,5-tris(4-formylphenyl)benzene (TFPB) and 2,5-dihydroxyterephthalohydrazide (DHTH) under solvothermal conditions. The synthesized TFPB–DHTH COF possessed high crystallinity, a regular pore structure, excellent chemical stability, and sites featuring functional groups suitable for interactions with metal ions. In addition, the TFPB–DHTH COF was highly soluble in water and buffer and displayed a strong fluorescence emission under UV irradiation. The fluorescence of the TFPB–DHTH COF was quenched selectively by Cu2+ ions, forming a non-fluorescent copper-immobilized TFPB–DHTH COF (Cu@TFPB–DHTH COF); the off fluorescent state of the Cu@TFPB–DHTH COF was efficiently and selectively switched on upon the addition of cysteine (Cys) and L-histidine (L-His). Therefore, the Cu@TFPB–DHTH COF functioned as an “OFF–ON” fluorescent sensor for the detection of Cys and L-His. When incorporating N-ethylmaleimide and Ni2+ ions, we could separately and selectively detect Cys and L-His, respectively. Our new sensing assay could be completed within 30 min; it provided linear relationships with respect to concentrations of Cys and L-His of 2–100 and 2–200 μM, respectively, with limits of detection of 340 and 520 nM, respectively. Our strategy highlights the possibility of using water-soluble COFs as novel fluorescent sensors for amino acids, with promise for applications industrially or in environmental remediation.
Introduction
Cysteine (Cys) is an essential small thiol-functionalized amino acid for living cells, where it plays crucial roles in various pathological and physiological processes (e.g., redox-balance regulation, anti-aging, metabolism, biocatalysis, protein synthesis, and detoxification), while also being associated with many diseases involving cancers. Cys is a significant structural and functional component of many enzymes and proteins.1–5 Nevertheless, abnormal levels of Cys in the human body can place human health at risk. For example, a high level of Cys can be a vital factor for amyotrophic lateral sclerosis and Parkinson's, Alzheimer's, and cardiovascular diseases.6,7 In addition, a lack of Cys can lead to symptoms such as liver damage, hair discoloration, developmental delay, and hematopoietic dysfunction.8 Therefore, it is important to develop a simple selective, sensitive and reliable method for the detection of Cys. L-Histidine (L-His) is a natural amino acid featuring an aromatic imidazole unit; it is mainly present in the active-catalytic sites of various enzymes. L-His plays important roles in decreasing the risk of internal microtrauma bleeding and in monitoring the metal transport processes of essential biological bases.9,10 Chronic L-His deficiency is associated significantly with Friedreich's ataxia, epilepsy, Parkinson's disease, and the inability to develop normal erythropoiesis; the overexpression of L-His in biological fluids (serum and urine) can cause hereditary metabolic disorder.11–14 Therefore, reliable and highly sensitive detection and quantification of L-His is thus of considerable significance for therapeutic systems including clinical treatment and medicinal science.Several analytic techniques have been reported previously for the quantitative detection of Cys and L-His, including those based on UV-Vis absorption spectroscopy, electrochemical analysis, capillary electrophoresis, spectrofluorimetry, immunoassaying, and high-performance liquid chromatography (HPLC).15–21 These techniques, however, have various shortcomings: they can be time-consuming and/or operationally inconvenient, and they can require cumbersome and/or expensive instruments. Recently, photophysical analytical techniques, particularly fluorescence-based versions, have become widespread for their high sensitivity, low detection limits, low cost, convenience, and suitability for bioimaging.22–30 The design of novel fluorescent probes including fluorophoric molecules, metal nanoclusters, and carbon dots (CDs), has also gained recognition.31,32 The detection mechanisms of these fluorescent probes have depended mainly on interactions between the functional groups of the probes and the thiol/amino groups of Cys or the imidazole/amino groups of L-His. In addition, mechanisms involving the coordination displacement of ligand–metal probes by Cys and L-His have also been reported. For example, Park et al. described the label-free detection of Cys through the fluorescence turn-on of metallophilic Hg–Au nanoclusters upon coordination of its Hg2+ moieties with Cys through its thiol group.23 Moreover, Yan et al. demonstrated water-soluble CDs as a novel fluorescent probe for the detection of Cys through the turn-on fluorescence of a CD–Hg2+ complex after Hg2+–Cys coordination.24 Das et al. developed a ligand-displacement approach for the detection of L-His by using a fluorescent [Cu(LH2)Cl2]·2H2O probe (where LH2 is a pyridoxal-semicarbazide Schiff base) as a naked-eye fluorescent sensor.33 These few examples suggest that there remains much room for the development of rapidly indicating, easy-to-prepare, versatile and inexpensive fluorescent probes for the detection of Cys and L-His.
Covalent organic frameworks (COFs) are drawing interest as innovative porous crystalline polymers, formed from assembling organic building units into ordered structures through strong covalent bonds.34–37 Similar to conventional crystalline porous materials [e.g., zeolites and metal organic frameworks (MOFs)], COFs possess precise predesigned structures and tailorable functionality, thereby allowing structural and chemical control over their particular functions.37–39 In addition to overcoming the shortcomings of the weakly crystalline structures and irregular pore distributions of most inorganic porous materials, COFs can also operate without the potential for collapse that often occurs for MOFs in organic solvents and water.40 Moreover, COFs can display high thermal and chemical stabilities, low densities, extremely high surface areas, regular porosity, excellent crystalline forms, and tunable structures and pore sizes.41–44 Accordingly, COFs are promising candidate materials for various applications, including the removal of pollutants from wastewater, (photo)catalysis, energy storage, gas separation and uptake, sensing, solar cells, the detection and removal of toxic metal ions, and photodynamic therapy.45–55
Various COFs displaying fluorescence characteristics have been constructed previously and applied as chemosensors for pH, electron-rich arenes, mercury ions, 2,4,6-trinitrophenol, and anions.56–63 For biosensing applications, COF materials can have several attractive features, including high conductivity, high loading capacity, and biocompatibility. First, the uniform and crystalline structures of COFs can accelerate the migration of charged particles and, thereby, enhance their biosensor response rates.64 Second, the regular porosity of COFs can cause them to have large specific active surface areas and high surface free energies, leading to very high loading capacities when combined with organic molecules.65 Third, the biocompatibility of COF materials can provide appropriate microenvironments for biomolecules, thereby maintaining their activity. Together, these features appear to make COFs more designable than conventional porous materials based on organic polymers and inorganic materials.66,67
The distinctive photophysical and chemical properties of COFs encouraged us to develop one for the detection of L-His and L-Cys. Biosensors based on COFs remain rare and, to the best of our knowledge, the use of water-soluble COFs for the detection of amino acids has not been reported previously. In this paper, we describe the preparation of a water-soluble copper-immobilized COF (Cu@TFPB–DHTH COF) through the polycondensation of 1,3,5-tris(4-formylphenyl)benzene (TFPB–3CHO) and 2,5-dihydroxyterephthalohydrazide (DHTH) under solvothermal conditions (forming the TFPB–DHTH COF) and subsequent incorporation of Cu2+ ions (Scheme 1). The TFPB–DHTH COF displayed high chemical stability and a strong fluorescence emission, while the Cu@TFPB–DHTH COF exhibited a non-fluorescent emission. Interestingly, the addition of Cys or L-His to the fluorescence-off state of the Cu@TFPB–DHTH COF stimulated the snatching and seizure of the Cu2+ ions and, thereby, switched on the fluorescence emission of the TFPB–DHTH COF (Scheme 2). Therefore, we also studied the use of the Cu@TFPB–DHTH COF as a water-soluble “OFF–ON” fluorescent sensor for the detection of Cys and L-His.
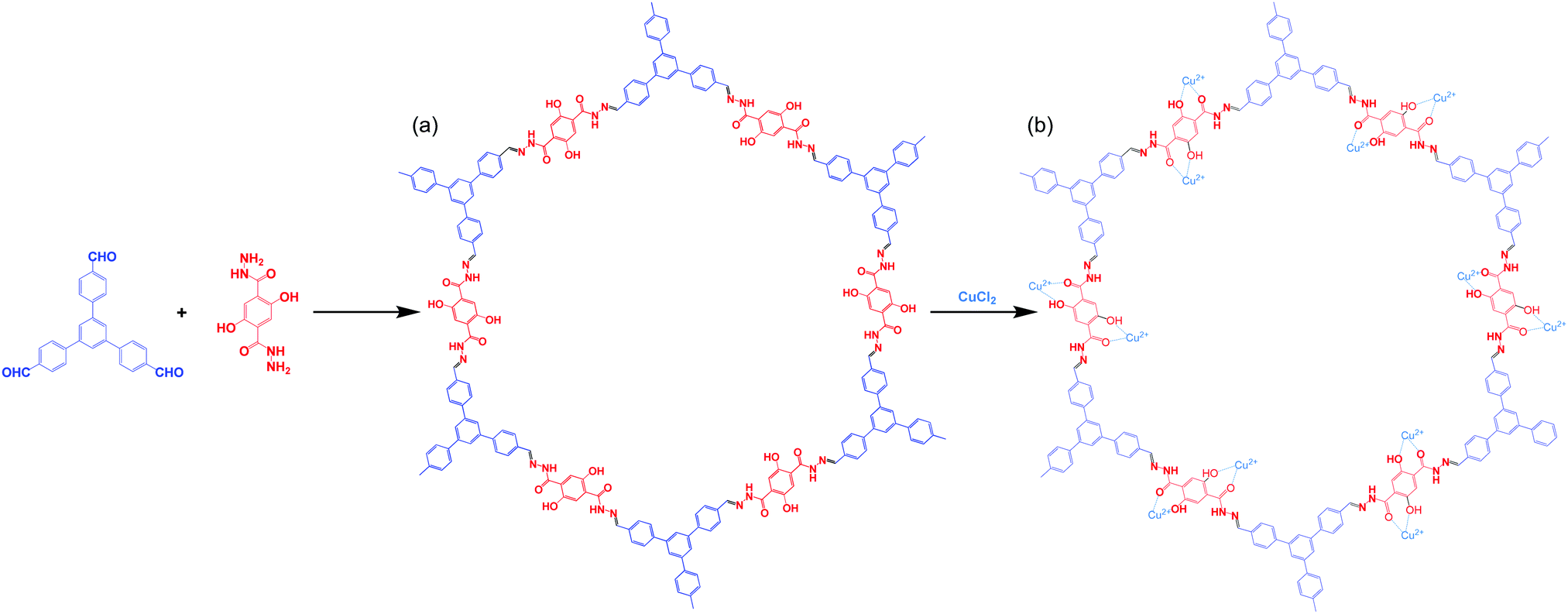 | ||
| Scheme 1 Synthesis of the TFPB–DHTH COF and the Cu@TFPB–DHTH COF. | ||
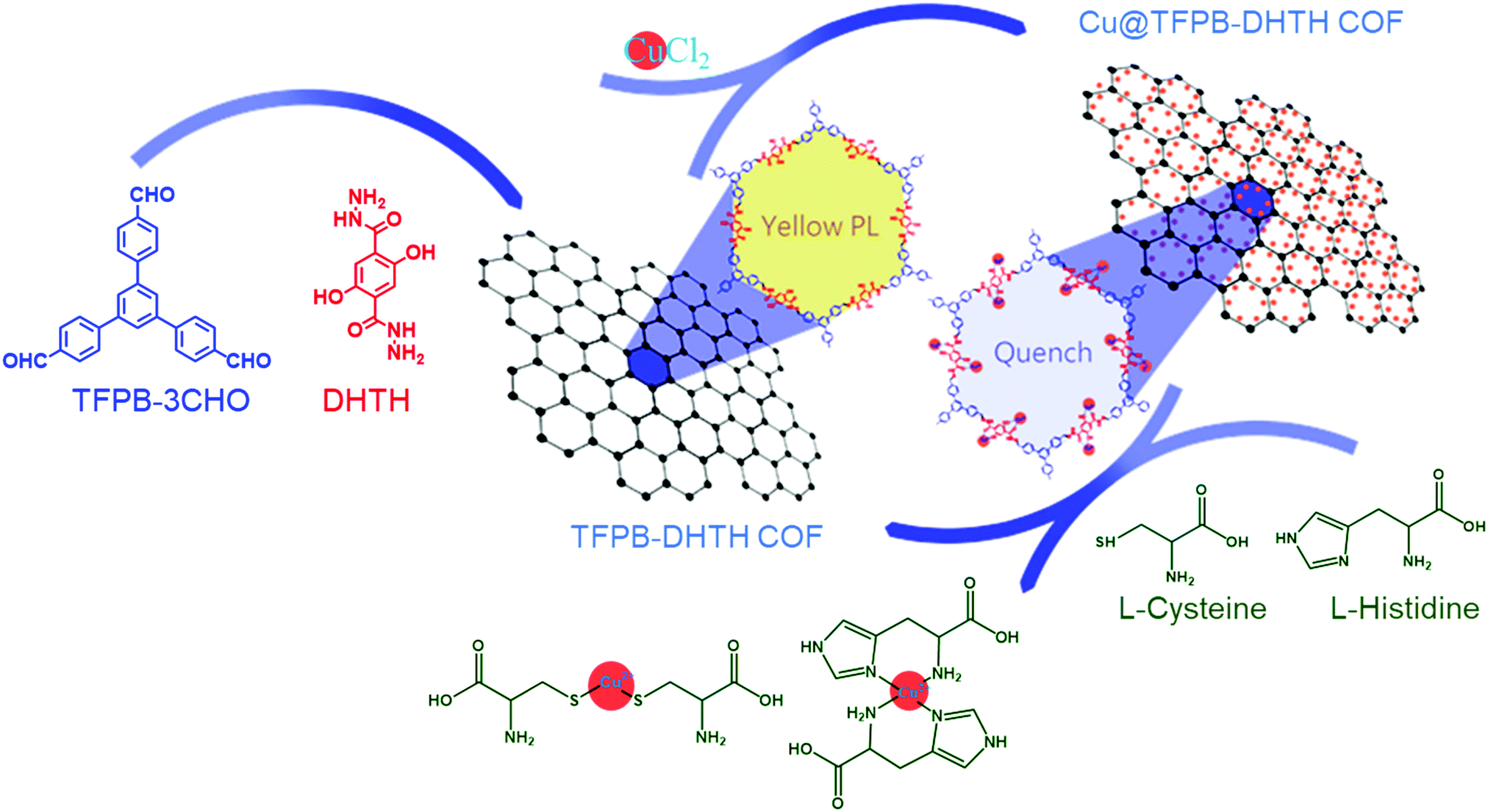 | ||
| Scheme 2 Schematic representation of the fluorescence sensing of Cys and L-His using the Cu@TFPB–DHTH COF. | ||
Experimental section
Materials
Tetrakis(triphenylphosphine)palladium(0), 2,5-dihydroxy-1,4-benzenedicarboxylic acid, and 1,3,5-tribromobenzene were purchased from Acros. 4-Formylphenylboronic acid, Cys, L-His, and 10× PBS buffer were obtained from Sigma-Aldrich. Potassium carbonate, mesitylene, and hydrazine hydrate (98%) were purchased from Alfa Aesar. Methanol (MeOH), ethanol (EtOH), tetrahydrofuran (THF), 1,4-dioxane, glacial acetic acid, acetone, and dichloromethane (DCM) were obtained in analytical grade and were used without purification.Measurements
Transmission electron microscopy (TEM) was performed using a JEOL-2100 scanning electron microscope, operated at an accelerating voltage of 200 kV. Field-emission scanning electron microscopy (FE-SEM) was conducted using a JEOL JSM-7610F scanning electron microscope; the COF sample was subjected to Pt sputtering for 100 s prior to observation. The TFPB–DHTH COF sample for UV-Vis adsorption spectroscopy was dissolved in water and transferred to a small quartz cell (0.2 × 1.0 × 4.5 cm3); the spectrum was recorded using a JASCO V-770 UV-visible-NIR spectrophotometer. Fluorescence emission spectra were recorded using a LabGuide X350 spectrometer; samples were placed in a quartz cuvette having a path length of 1 cm and a slit width of 10 × 10 nm. The photoluminescence quantum yield (PLQY) of the TFPB–DHTH COF was recorded using a HORIBA Fluorolog-3 photon counting spectrofluorometer system equipped with a Quanta-φ 6-inch integrating sphere. All pH measurements were performed using a SUNTEX SP-2100 pH meter.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) to yield TFPB–3CHO as a white solid (990 mg, 80%). FTIR: 3040, 2811, 2717, 1685, 1604, 1563, 1420, 1380, 1214, 1170 cm−1 (Fig. S4, ESI†). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.08 (s, 3CHO, 3H), 8.18 (d, J = 8.8 Hz, 6H), 8.16 (s, 3H), 8.05 (d, J = 8.8 Hz, 6H) (Fig. S5, ESI†). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 194.14, 146.07, 141.39, 136.08, 130.80, 128.53, 126.86 (Fig. S6, ESI†).
:3, 5 mL) and an aqueous solution of acetic acid (6 M, 0.4 mL). The tube was degassed through several freeze/pump/thaw cycles and then heated at 120 °C. After 72 h, the mixture was cooled to room temperature, producing a solid that was filtered off and washed several times sequentially with 1,4-dioxane, MeOH, acetone, and THF. The TFPB–DHTH COF (92% yield) was obtained as a yellow-brownish solid after drying under vacuum at 120 °C for 24 h (Scheme 2).
:9) to yield TFPB–3CHO as a white solid (990 mg, 80%). FTIR: 3040, 2811, 2717, 1685, 1604, 1563, 1420, 1380, 1214, 1170 cm−1 (Fig. S4, ESI†). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.08 (s, 3CHO, 3H), 8.18 (d, J = 8.8 Hz, 6H), 8.16 (s, 3H), 8.05 (d, J = 8.8 Hz, 6H) (Fig. S5, ESI†). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 194.14, 146.07, 141.39, 136.08, 130.80, 128.53, 126.86 (Fig. S6, ESI†).
:3, 5 mL) and an aqueous solution of acetic acid (6 M, 0.4 mL). The tube was degassed through several freeze/pump/thaw cycles and then heated at 120 °C. After 72 h, the mixture was cooled to room temperature, producing a solid that was filtered off and washed several times sequentially with 1,4-dioxane, MeOH, acetone, and THF. The TFPB–DHTH COF (92% yield) was obtained as a yellow-brownish solid after drying under vacuum at 120 °C for 24 h (Scheme 2).
Results and discussion
Scheme 1a displays the synthesis of our new TFPB–DHTH COF. The use of solvothermal conditions with TFPB–3CHO and DHTH in 1,4-dioxane and mesitylene (1:3, v/v), in the presence of a catalytic amount of acetic acid (6 M), produced TFPB–DHTH COF. Fourier transform infrared (FTIR) and solid state 13C nuclear magnetic resonance (NMR) spectroscopy verified the molecular structure of the TFPB–DHTH COF. The FTIR spectrum of DHTH featured a very broad signal in the range 3670–3307 cm−1 for the vibrations of the OH groups, overlapped with signals at 3397 and 3307 cm−1 for the vibrations of the hydrazide (NHNH2) group, a signal at 1630 cm−1 for the vibrations of the amide carbonyl (NHCO) group, and signals at 1613 and 1537 cm−1 for the vibrations of aromatic (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) bonds [Fig. 1A(a) and Fig. S1, ESI†]. The FTIR spectrum of the triformyl monomer TFPB–3CHO featured signals at 3040, 2811, 2717, 1685, and 1605 cm−1 for the vibrations of the aromatic C–H bonds, the Fermi double-resonance of the aldehydic C–H bonds, the aldehydic CO bonds, and the aromatic groups, respectively [Fig. 1A(b) and Fig. S4, ESI†]. The FTIR spectrum of the TFPB–DHTH COF did not feature the signals of the aldehydic C–H bonds (at 2811 and 2717 cm−1) nor the aldehydic CO groups (at 1685 cm−1) of TFPB–3CHO, nor the signals of the NHNH2 groups (at 3397 and 3307 cm−1) of DHTH, suggesting complete consumption of these monomers. Moreover, new signals appeared at 1644 and 1602 cm−1 for the vibrations of the CO and CN groups, respectively, consistent with the formation of the acylhydrazone Schiff base (CONH–NC) linkages and then the successful formation of the TFPB–DHTH COF [Fig. 1A(c)].
C) bonds [Fig. 1A(a) and Fig. S1, ESI†]. The FTIR spectrum of the triformyl monomer TFPB–3CHO featured signals at 3040, 2811, 2717, 1685, and 1605 cm−1 for the vibrations of the aromatic C–H bonds, the Fermi double-resonance of the aldehydic C–H bonds, the aldehydic CO bonds, and the aromatic groups, respectively [Fig. 1A(b) and Fig. S4, ESI†]. The FTIR spectrum of the TFPB–DHTH COF did not feature the signals of the aldehydic C–H bonds (at 2811 and 2717 cm−1) nor the aldehydic CO groups (at 1685 cm−1) of TFPB–3CHO, nor the signals of the NHNH2 groups (at 3397 and 3307 cm−1) of DHTH, suggesting complete consumption of these monomers. Moreover, new signals appeared at 1644 and 1602 cm−1 for the vibrations of the CO and CN groups, respectively, consistent with the formation of the acylhydrazone Schiff base (CONH–NC) linkages and then the successful formation of the TFPB–DHTH COF [Fig. 1A(c)].
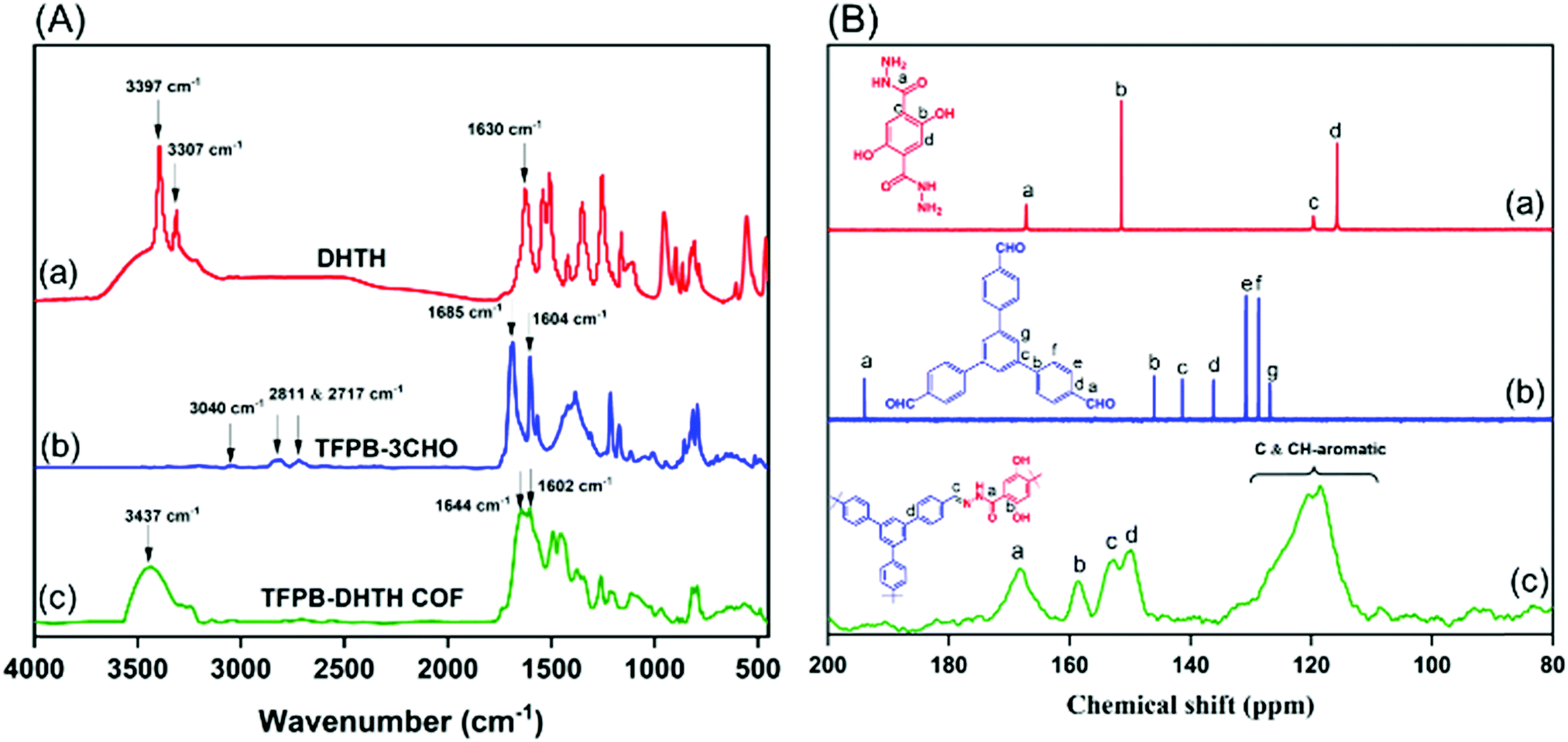 | ||
| Fig. 1 (A) FTIR spectra of (a) DHTH, (b) TFPB–3CHO, and (c) the TFPB–DHTH COF. (B) 13C NMR spectra of (a) DHTH and (b) TFPB–3CHO and (c) the solid state 13C NMR spectrum of the TFPB–DHTH COF. | ||
The 13C NMR spectrum of DHTH [Fig. 1B(a) and Fig. S3, ESI†] featured a signal at 167.37 ppm representing the carbon nuclei of its CO groups, as well as a high-intensity signal at 151.50 ppm for the aromatic carbon nuclei attached to the hydrazide group (C–CONHNH2). 7:25 PMhe signals of other aromatic carbon nuclei appeared at 119.73 and 115.59 ppm. The 13C NMR spectrum of TFPB–3CHO exhibited a peak at 194.14 ppm representing its aldehydic carbon nuclei [C(O)–H], as well as signals in the range from 146.07 to 128.86 ppm for the aromatic carbon nuclei [Fig. 1B(b) and Fig. S6, ESI†]. Solid state 13C NMR spectroscopy confirmed the successful condensation between DHTH and TFPB–3CHO to form the TFPB–DHTH COF, with the disappearance of the signal for the aldehydic carbon nuclei (at 194.14 ppm) of TFPB–3CHO and the appearance of new signals at 168.26 and 152.38 ppm for the CO and CN groups, respectively, of the formed acylhydrazone Schiff base (CONH–NC) [Fig. 1B(c)], in addition to signals of the other aromatic carbon nuclei at 158.38, 149.68, and 130.22–110.93 ppm.
To study the crystal structure of the TFPB–DHTH COF, we recorded its powder X-ray diffraction (PXRD) pattern with angles 2θ ranging from 1 to 30°. The as-synthesized TFPB–DHTH COF featured an experimental pattern (Fig. 2a, black curve) with an intense peak at a value of 2θ of 2.16°, corresponding to the [100] plane. In addition, the PXRD pattern of the COF featured a set of reflected peaks at low-angle range, at values of 2θ of 3.79, 4.40, and 5.83°, that we attribute to its [110], [200], and [210] planes, respectively. In the high-angle region, a broad reflected peak appeared at a value of 2θ of 25.74°, matching the [001] plane (Fig. 2a). This experimental PXRD pattern confirmed that our TFPB–DHTH COF possessed a microcrystalline framework and a long-range arrangement. We applied the Bragg equation to calculate the d-spacing between the [100] planes (d110) and the interlayer distance between the packed layers of the TFPB–DHTH COF. The as-synthesized COF exhibited a value of d100 and an interlayer distance of 4.01 nm and 3.45 Å, respectively (Table S1, ESI†). To gain insight into the two-dimensional (2D) packing of the TFPB–DHTH COF, we applied Material Studio software to simulate two possible crystalline packing models (completely eclipsed AA-stacking and staggered AB-stacking succession models) of our COF (Fig. 2b, c and Fig. S7, ESI†), and then estimated the theoretical PXRD patterns of these models. Notably, the locations of the experimental PXRD peaks of the TFPB–DHTH COF (Fig. 2a, black curve) matched well with those of the simulated PXRD pattern generated through Pawley refinement of the completely eclipsed AA-stacking model of our COF (Fig. 2a, red dots). This matching was confirmed by negligible differences (Fig. 2a, blue curve) between the experimental and simulated PXRD patterns. The optimized unit cell parameters of the TFPB–DHTH COF, estimated from this refinement of the eclipsed AA-stacking model, were a = b = 46.231 Å, c = 4.078 Å, α = β = 90°, and γ = 120° (Fig. 2b and Fig. S7a and S7b, Table S2, ESI†). Furthermore, comparisons of the experimental PXRD pattern of our TFPB–DHTH COF (Fig. 2a, black curve) with the calculated PXRD patterns of the completely eclipsed AA-stacking (Fig. 2a, green curve) and staggered AB-stacking (Fig. 2a, purple curve) succession models revealed consistency with the completely eclipsed AA-stacking model and high variance with the staggered AB-stacking model, suggesting completely eclipsed AA-stacking of the 2D layers in our TFPB–DHTH COF.
 | ||
| Fig. 2 (a) PXRD patterns of the TFPB–DHTH COF: experimental pattern (black); simulated Pawley refined pattern (red dots); their difference pattern (blue); and simulated patterns for the completely eclipsed AA-stacking (olive) and staggered AB-stacking (violet) succession models. (b and c) Top views of (b) the completely eclipsed AA-stacking and (c) staggered AB-stacking succession models. (d) Nitrogen adsorption–desorption isotherm of the TFPB–DHTH COF at 77 K. (e) Pore size distribution of the TFPB–DHTH COF. | ||
We measured N2 gas adsorption and desorption at 77 K to study the porous characteristics of our TFPB–DHTH COF. Fig. 2d reveals that the N2 adsorption curve of the TFPB–DHTH COF exhibited a maximum adsorption of 386 cm3 g−1. We estimated the surface area (360 m2 g−1) and total pore volume (0.58 cm3 g−1) of the TFPB–DHTH COF from its adsorption/desorption curve, by applying the Brunauer–Emmett–Teller (BET) model. In addition, we used nonlocal density functional theory (NLDFT) to investigate the pore size of the TFPB–DHTH COF, validating a narrow pore size distribution with a pore width of 3.77 nm (Fig. 2e and Table S1, ESI†). We visualized the morphology of the TFPB–DHTH COF by using FE-SEM and TEM. Fig. S8 (ESI†) reveals that the crystalline TFPB–DHTH COF underwent self-assembly during its formation to produce uniform micrometer-scale tubules. Almost all of these TFPB–DHTH COF tubules were assembled into a set of bundles, presumably arising because of strong hydrogen bonding among the surfaces of the COF. Nevertheless, after sonication in polar protic solvents (e.g., EtOH), separated COF tubules could be observed. TEM imaging of the TFPB–DHTH COF confirmed the micrometer-scale tubular morphology of our COF; it also revealed that the average length and diameter of the tubular TFPB–DHTH COF were 295 ± 32 and 750 ± 45 nm, respectively (Fig. S9, ESI†).
We used thermogravimetric analysis (TGA) to investigate the thermal and chemical stability of our synthesized TFPB–DHTH COF. Fig. S10 (ESI†) reveals that the TFPB–DHTH COF exhibited sufficient thermal stability, with decomposition temperatures (Td10) of 322 and 321 °C under N2 and air atmospheres, respectively. In addition, we tested the chemical stability of our TFPB–DHTH COF by dispersing 20 mg of the as-synthesized COF in various solvents [1,4-dioxane, THF, acetone, aqueous KOH (1 M), aqueous HCl (1 M), MeOH, DMF, water, and 1× PBS buffer] for 2 days, collecting the solid through filtration under vacuum, washing the sample several times with THF, and finally drying it at 100 °C for 20 h. The PXRD patterns and N2 gas adsorption and desorption isotherms of the collected COF samples revealed the high chemical stability of the TFPB–DHTH COF in these organic and aqueous solvents, as they revealed no changes in the intensities or positions of the signals (Fig. S11, ESI†) in addition to approximate similar surface area values (Table S3, ESI†). We attribute the high thermal stability of the TFPB–DHTH COF to the strong intramolecular hydrogen bonds between the CO groups of the hydrazone Schiff base (CONH–NC) units and the OH groups in the ortho positions of the aromatic rings; we suspected that these units would also act as bidentate sites for the selective bonding of metal ions.
We investigated the electronic excitation and luminescence of the TFPB–DHTH COF by measuring its fluorescence emission spectrum in various solvents. We prepared solutions of our COF crystallites in water at concentrations of 0.5, 0.25, 0.125, and 0.0625 mg mL−1, and then excited these solutions at 365 nm. Fig. 3a reveals that the fluorescence spectrum of each aqueous solution of the TFPB–DHTH COF exhibited an intense emission maximum at 520 nm. Interestingly, the fluorescence intensity of our COF increased from 16,430 to 31,161 to 41,453 a.u. upon decreasing the concentration of the COF from 0.5 to 0.25 to 0.125 mg mL−1 (Fig. 3b), respectively, indicative of aggregation-caused quenching (ACQ) of the TFPB–DHTH COF. We attribute the decrease in the fluorescence intensity to 29677 a.u. at a concentration of our COF of 0.0625 mg mL−1 to the very low amount of TFPB–DHTH COF in solution. Recently, we reported that fluorescent COFs having extended conjugated aromatic systems can display significant fluorescence emissions as dilute solutions; in contrast, in solution at high concentrations, their fluorescence emissions are normally quenched through the ACQ phenomenon.57 Furthermore, we recoded the time-dependent fluorescence intensity of an aqueous solution of TFPB–DHTH COF at a concentration of 0.125 mg mL−1 over a 60 min period; the maintained intensity suggested that our TFPB–DHTH COF did not display any photo-bleaching (Fig. S12, ESI†). We also studied the fluorescence behavior of the TFPB–DHTH COF in various solvents—THF, 1,4-dioxane, acetone, MeOH, DMF, water, and 1× PBS buffer (pH 7.4)—at a recording concentration of 0.0625 mg mL−1. The excitation of our COF solutions in THF, 1,4-dioxane, and acetone, using light from a common UV lamp at a wavelength of 365 nm, resulted in very weak (nearly dark) fluorescence colors that were clear to the naked eye (Fig. 4a). In contrast, the solutions of TFPB–DHTH COF in MeOH, DMF, water, and 1× PBS buffer displayed extremely bright yellow fluorescence. We also used a fluorescence spectrometer to record the fluorescence emission spectra of our COF in the same solvents under an excitation wavelength of 365 nm and open to the atmosphere. Fig. 4b reveals that the fluorescence spectra of the TFPB–DHTH COF in MeOH, DMF, water, and 1× PBS buffer show high-intensity emission maxima at 509, 512, 520, and 520 nm, respectively; in THF, 1,4-dioxane, and acetone they exhibited very weak emission maxima at 450, 449, and 502 nm, respectively. Thus, the emission wavelength of our COF was strongly influenced by the polarity of the solvent; upon increasing the polarity, the emission wavelength red-shifted. We attribute this solvatochromic effect of the emission wavelength of the TFPB–DHTH COF to strong noncovalent interactions (e.g., dipole effects, hydrogen bonding) between the COF and the solvent molecules of MeOH, DMF, water, and 1× PBS buffer, resulting in the stabilization of the excited state of the TFPB–DHTH COF and, therefore, facilitated charge-transfer. The absolute PLQY of our new COF in water, where it displayed very high fluorescence, was 22.6%. Furthermore, we estimated the band gap and the energy of the highest occupied molecular orbital (HOMO) of our new TFPB–DHTH COF by using UV-Vis absorption and photoelectron spectrometry. The UV-Vis spectrum of the TFPB–DHTH COF featured a characteristic band having its maximum adsorption at 354 nm (Fig. 4c). Converting the UV-Vis spectrum of our COF into a Tauc plot (Fig. S13, ESI†) revealed that the band gap of our TFPB–DHTH COF was 3.16 eV. The photoelectron spectrum of the TFPB–DHTH COF revealed a HOMO energy of 5.53 eV (Fig. 4d). Finally, we determined the energy of the lowest unoccupied molecular orbital (LUMO) of our TFPB–DHTH COF to be 2.37 eV, by subtracting the band gap from the HOMO energy.
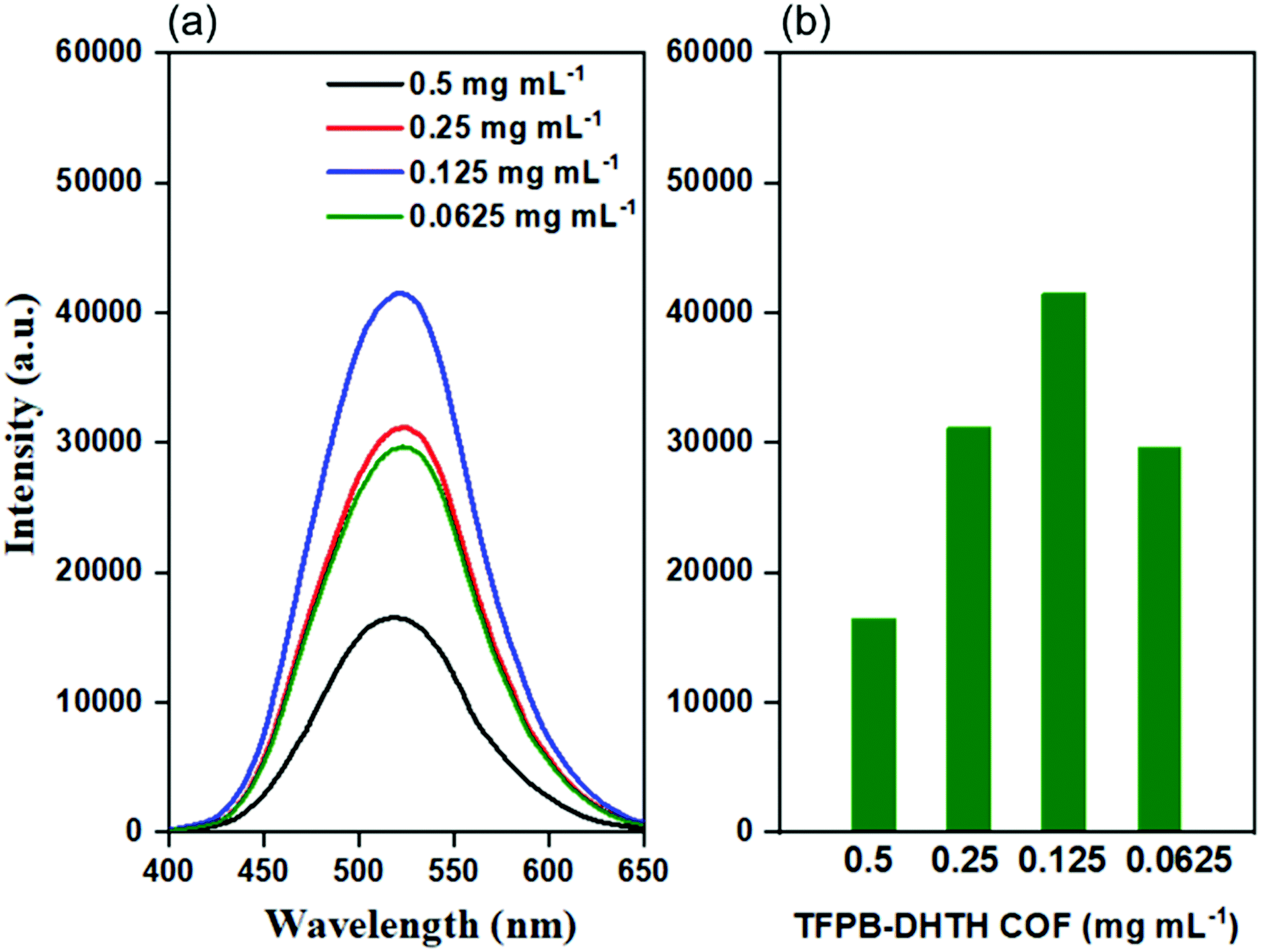 | ||
| Fig. 3 (a) Fluorescence emission spectra and (b) fluorescence emission intensities at a wavelength of 520 nm of the TFPB–DHTH COF dissolved in water at various concentrations (excitation at 365 nm). | ||
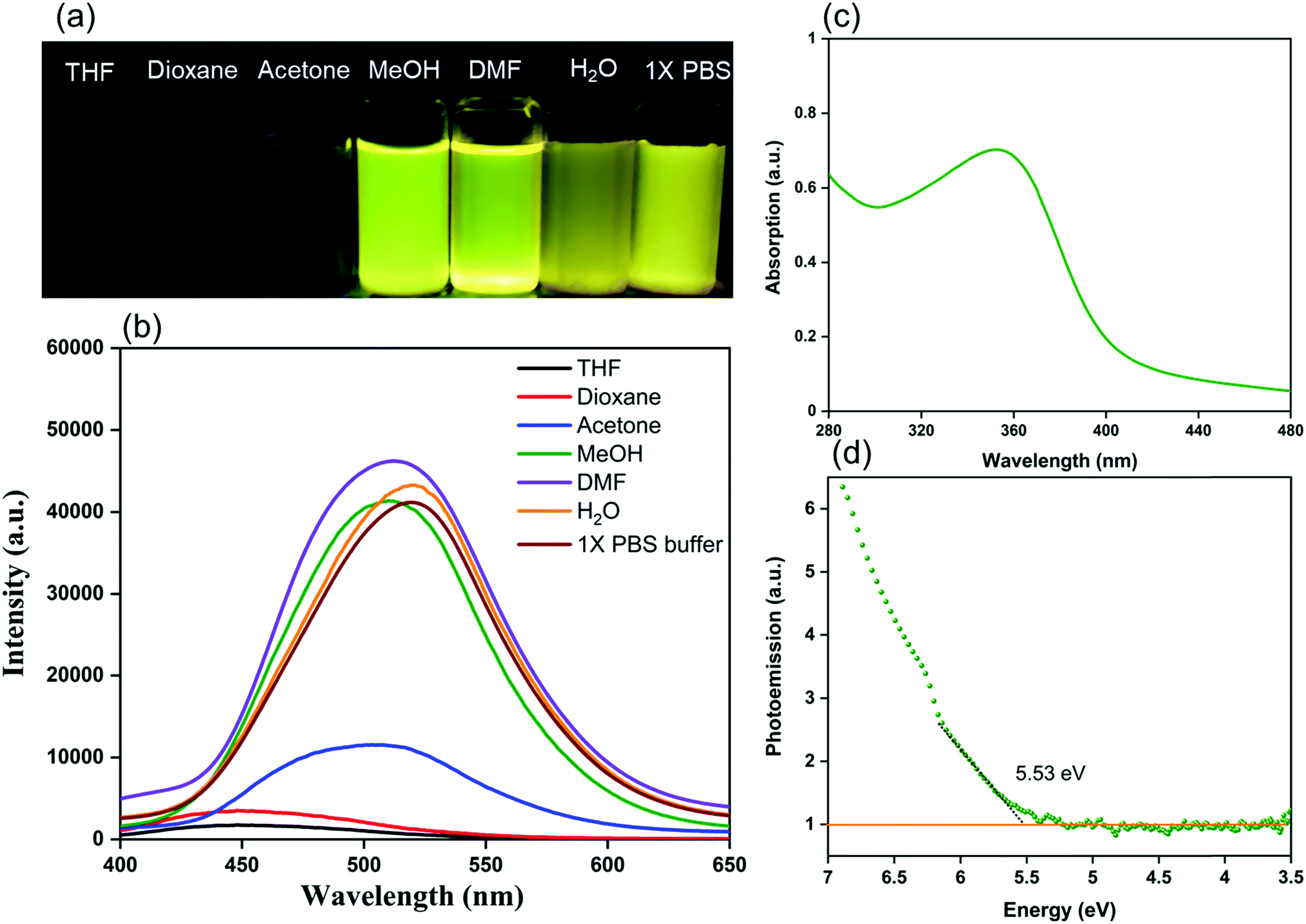 | ||
| Fig. 4 (a) Photographic images of the TFPB–DHTH COF dissolved/dispersed in various solvents upon irradiation with UV light at 360 nm. (b) Fluorescence emission spectra of the TFPB–DHTH COF dissolved/dispersed in various solvents (excitation at 365 nm). (c) UV-Vis absorption spectrum of the TFPB–DHTH COF dissolved in water (concentration: 0.125 mg mL−1). (d) Photoelectron spectroscopy of the TFPB–DHTH COF for calculation of the HOMO energy. | ||
Our new TFPB–DHTH COF displayed a high fluorescence emission, a uniform distribution of acylhydrazone and OH groups, excellent crystallinity, and good solubility in water and buffer; therefore, we were encouraged to investigate its applications for fluorescence sensing. First, we evaluated the quenching effect of the TFPB–DHTH COF dissolved in 1× PBS buffer (pH 7.4) after the addition of copper (Cu2+) ions. We added aqueous solutions of copper salts with different counter ions [Cu(OAc)2, Cu(NO3)2, CuCl2] to a solution of TFPB–DHTH COF in water. Fig. 5a reveals that the fluorescence emission intensity of the TFPB–DHTH COF was quenched dramatically after the addition of these copper salts. The approximately equal quenching efficiencies upon the addition of these copper salts suggested that the counter anions (OAc−, NO3−, Cl−) had only marginal effects on the recognition of the Cu2+ ions by the TFPB–DHTH COF.
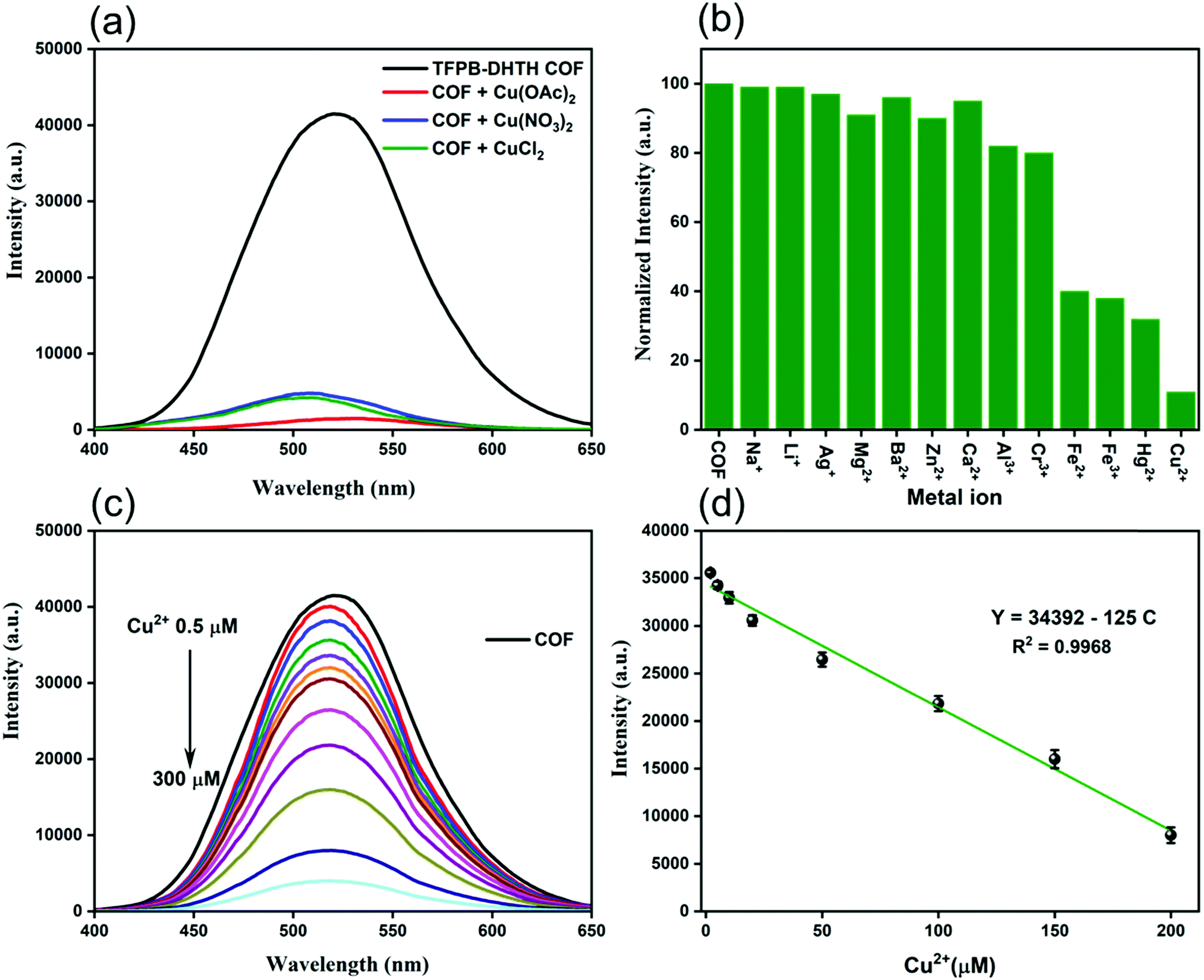 | ||
| Fig. 5 (a) Fluorescence emission spectra of the TFPB–DHTH COF dissolved in 1× PBS buffer (pH 7.4) in the presence of copper salts having various counter ions (Cu2+ concentration: 200 μM). (b) Fluorescence emission intensities at a wavelength of 520 nm of the TFPB–DHTH COF dissolved in 1× PBS buffer (pH 7.4) in the presence of Cu2+ and other metal ions (metal ion concentration: 200 μM). (c) Fluorescence emission spectra of the TFPB–DHTH COF dissolved in 1× PBS buffer (pH 7.4) in the presence of various concentrations of Cu2+ ions. (d) Calibration curve of the fluorescence emission intensity of the TFPB–DHTH COF with respect to the concentration of Cu2+ ions. Measurements were made at a TFPB–DHTH COF concentration of 0.125 mg mL−1 and under excitation at 365 nm. | ||
We investigated the selective quenching of the fluorescence of the TFPB–DHTH COF by Cu2+ ions by observing the change in the fluorescence emission intensity of our COF in the presence of 12 different metal ions (MClx; Mx+ = Na+, Li+, Ag+, Mg2+, Ba2+, Zn2+, Ca2+, Al3+, Cr3+, Fe2+, Fe3+, Hg2+, Cu2+) (Fig. 5b). Among these tested metal ions, only Cu2+ ions caused strong quenching of the fluorescence of the TFPB–DHTH COF; the other metal ions had only slight fluorescence quenching effects. Alkali metal ions (Na+, Li+, Ag+), alkaline earth ions (Mg2+, Ba2+, Ca2+), and the transition metal Zn2+ ion had negligible effects on the fluorescence emission intensity of our COF; Al3+ and Cr3+ ions quenched approximately 10% of the fluorescence intensity, while the Fe2+, Fe3+, and Hg2+ ions both quenched it by approximately 60%. These findings confirmed the outstanding selectivity and excellent quenching response of our TFPB–DHTH COF toward Cu2+ ions, with a quenching efficiency of approximately 90% (Fig. 5b). To optimize the fluorescence quenching with Cu2+ ions, we evaluated the fluorescence emission intensity of our COF in water in the presence of CuCl2 at various concentrations in 1× PBS buffer (pH 7.4). Fig. 5c reveals that the fluorescence emission intensity of an aqueous solution of TFPB–DHTH COF at a concentration of 0.125 mg mL−1 decreased gradually upon increasing the concentration of Cu2+ ions from 0.5 to 200 μM, but remained nearly unaffected thereafter when the concentration of Cu2+ ions increased to 300 μM. Approximately 90% of the fluorescence emission intensity of our COF was quenched in the presence of 200 μM of Cu2+ ions, representing a mole ratio of 1:6.8 (TFPB–DHTH COF/Cu2+). Thus, full quenching of the fluorescence of TFPB–DHTH COF required at least 6 mole equivalents of Cu2+ ions. The fluorescence emission of the COF and the concentration of Cu2+ ions had a linear relationship in the range from 2 to 200 μM, characterized by the equation Y = 34392 – 125C, where Y is the fluorescence intensity and C is the concentration of Cu2+ ions in solution; the correlation coefficient (R2) was 0.968 (Fig. 5d). We estimated the limit of detection (LOD) of Cu2+ ions to be 12 nM, based on 3σ/k (where σ is the standard deviation of six blanks and k is the slope of the calibration curve). The Cu2+ ion has significant coordination ability toward fluorescent materials and COFs having amino, CO, and OH groups in their structures; this coordinative binding induces electron transfer from the excited state of the fluorescent materials and COFs to the Cu2+ ion, leading to a dramatic quenching of fluorescence.69–73 Similarly, we attribute the fluorescence quenching of our TFPB–DHTH COF to strong binding between Cu2+ ions and the active sites (CO and OH groups) in the pore walls of our COF. This binding would induce electron transfer from the excited state of the COF to the Cu2+ ions. This electron transfer process and the coordination interaction between Cu2+ ions and the TFPB–DHTH COF were confirmed by X-ray photoelectron spectroscopy (XPS) measurements. Fig. S14 (ESI†) reveals that the TFPB–DHTH COF featured a set of XPS peaks of C1s, N1s, and O1s, while Cu@TFPB–DHTH COF featured a Cu2p peak in addition to other XPS peaks of C1s, N1s, and O1s. Fig. S15a and b (ESI†) also reveals that the binding energy of O1s orbital shifted from 532.37 eV in TFPB–DHTH COF to 533.26 eV in Cu@TFPB–DHTH COF. This positive shifting, therefore, strongly confirms the electron-transfer process from the excited state of the COF to the Cu2+ ions via the donor–acceptor coordination bonds formed between the active sites (CO and OH groups) in TFPB–DHTH COF and Cu2+ ions. Moreover, the Cu2p peak reveals Cu2p1/2 and Cu2p3/2 orbitals 954.24 and 934.71 eV, respectively (Fig. S15c, ESI†). Because we obtained 90% quenching of the fluorescence intensity of the aqueous COF solution (0.125 mg mL−1) upon the addition of 200 μM of Cu2+ ions, we prepared a corresponding stock solution of the Cu@TFPB–DHTH COF to test the turn-on fluorescence sensing of Cys and L-His.
The thiol functional groups of two Cys moieties can undergo coordination with a Cu2+ ion to form a stable 2:1 Cys/Cu2+ complex. The thiol–Cu2+ interaction is relatively stronger than the coordination between a Cu2+ ion and CO, OH and amino groups.71,74,75 In addition, the imidazole group of L-His can interact strongly with a Cu2+ ion to produce a stable 2:1 L-His/Cu2+ complex; again, the binding affinity is higher than that between a Cu2+ ion and CO, OH, and amino groups.71,74 Consequently, we suspected that addition of Cys or L-His to the Cu@TFPB–DHTH COF might induce snatching and seizure of its Cu2+ ions, thereby switching on the fluorescence emission of our TFPB–DHTH COF.
Therefore, we tested the Cu@TFPB–DHTH COF assembly as an “OFF–ON” fluorescence probe for the detection of Cys and L-His, operating through competitive coordination among the TFPB–DHTH COF, Cu2+ ions, and Cys/L-His influencing the fluorescence emission of the TFPB–DHTH COF. To determine the optimal conditions for the “OFF–ON” fluorescence detection of Cys and L-His, we varied the pH and the incubation time. Fig. S16a and S16c (ESI†) reveal that the fluorescence emission intensity of the aqueous solution of the Cu@TFPB–DHTH COF was recovered maximally, after the addition of aqueous solution of Cys (400 μM) or L-His (500 μM), for values of pH in the range from 4 to 7.4. Therefore, we selected pH 7.4, suitable for biological systems, as the optimal pH for the detection of Cys and L-His. Fig. S16b and S16d (ESI†) reveal the relationship between the fluorescence recovery intensity and the incubation time. The fluorescence intensity reached a maximum when the incubation time approached 15 min and then remained constant thereafter. Therefore, for subsequent detection of Cys and L-His, we employed an incubation time of 15 min.
Under these optimal conditions, we evaluated the “OFF–ON” fluorescence detection of Cys and L-His and examined the relationship between the fluorescence recovery intensity and the concentration of Cys and L-His. For Cys, the intensity of the fluorescence emission peak at 520 nm increased upon increasing of the concentration of Cys in the range from 2 to 400 μM in the presence of 300 μM of Ni2+ ions; it did not change any more upon increasing the concentration of Cys thereafter to 500, 1000, or 1500 μM (Fig. 6a and b). The inset of Fig. 6b reveals a linear relationship between the fluorescence intensity at 520 nm and the Cys concentration over the range from 2 to 100 μM. The regression equation for Cys detection was I = 8126.21 + 221.76[Cys], where I is the fluorescence intensity at 520 nm and [Cys] is the concentration of Cys (μM); the correlation coefficient (R2) was 0.9997. The LOD of Cys when using our new “OFF–ON” Cu@TFPB–DHTH COF fluorescence probe was equal to 340 nM, based on a signal-to-noise value (S/N) of 3. Likewise, upon incremental addition of L-His at concentrations in the range from 2 to 400 μM, the fluorescence intensity of our new Cu@TFPB–DHTH COF at 520 nm increased in the presence of 200 μM of NEM. The fluorescence intensity remained approximately unchanged when the concentration of L-His was greater than 500 mM (Fig. 6c and d). The inset of Fig. 6d reveals that the fluorescence intensity at 520 nm exhibited a linear relationship with the concentration of L-His within the range from 2 to 200 μM. The linear curve provided a regression equation for L-His detection of I = 7947.02 + 127.77 [L-His], where I is the fluorescence intensity at 520 nm and [L-His] is the concentration of L-His (μM); the correlation coefficient (R2) was 0.9993. We calculated the LOD of L-His to be 520 nM. Compared with other reported methods for the detection of Cys and L-His (Table S4, ESI†), our present method is among the most highly selective. In addition, our detection strategy has several advantages over those previous methods, including high thermal and chemical stability, no need for chemical modification, excellent crystallinity, high solubility in water, and low cost.
 | ||
| Fig. 6 (a) Fluorescence emission spectra of the Cu@TFPB–DHTH COF dissolved in 1× PBS buffer (pH 7.4) in the presence of various concentrations of Cys. (b) Calibration curve of the fluorescence emission intensity of the Cu@TFPB–DHTH COF with respect to the concentration of Cys. (c) Fluorescence emission spectra of the Cu@TFPB–DHTH COF dissolved in 1× PBS buffer (pH 7.4) in the presence of various concentrations of L-His. (d) Calibration curve of the fluorescence emission intensity of the Cu@TFPB–DHTH COF with respect to the concentration of L-His. Measurements were made at concentrations of the Cu@TFPB–DHTH COF and the Cu2+ ions of 0.125 mg mL−1 and 200 μM, respectively, under excitation at 365 nm. | ||
We examined the selectivity of our new Cu@TFPB–DHTH COF as an “OFF–ON” fluorescence probe by monitoring its fluorescence response after treatment with 13 different amino acids (Ala, Asp, Gln, Gly, Glu, Lys, Phe, Trp, Pro, Val, Tyr, Cys, L-His), each at a concentration of 200 μM. Fig. S17 (ESI†) reveals that, among the tested amino acids, only Cys and L-His could trigger a considerable recovery of the fluorescence intensity of the Cu@TFPB–DHTH COF probe; the other amino acids did not cause any major changes in the fluorescence intensity of our probe. To differentiate between Cys and L-His, we employed Ni2+ as a well-established L-His–binding metal ion and NEM as a Cys-masking reagent for the isolation of L-His and Cys, respectively, in our Cu@TFPB–DHTH COF–based detection assay. Because the Ni2+ ion binds selectively to L-His, we could detect Cys quantitatively when using our Cu@TFPB–DHTH COF probe; similarly, we could achieve the quantitative detection of L-His in the presence of NEM. Furthermore, Fig. S17 (ESI†) reveals that the presence of Ni2+ ions in the detection system did not affect the fluorescence recovery efficiency of Cys; neither did the presence of NEM affect the recovery efficiency of L-His. Therefore, our new detection strategy displayed good selectivity toward Cys and L-His, separately.
Conclusions
We have prepared a new fluorescent OH- and hydrazone-functionalized COF (TFPB–DHTH COF) displaying high crystallinity, excellent chemical stability, and suitable functional groups for interaction with metal ions; its solutions in water and buffer exhibited very high fluorescence emission under UV irradiation. The fluorescence emission of the TFPB–DHTH COF was switched off selectively upon the addition of Cu2+ ions, due to electron transfer from the excited state of the COF to the Cu2+ ions. The formed Cu@TFPB–DHTH COF functioned as an “OFF–ON” fluorescent sensor for the specific detection and differentiation of Cys and L-His in the presence of Ni2+ ions and NEM, respectively. Our new sensing assay could be completed within 30 min, with LODs of 340 and 520 nM for Cys and L-His, respectively. Thus, this facile “OFF–ON” detection assay for Cys and L-His would appear to have significant potential for use in a number of other domains.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported financially by the Ministry of Science and Technology, Taiwan, under contracts MOST 108-2638-E-002-003-MY2 and 108-2218-E-110-013-MY3.Notes and references
- Z. H. Fu, X. Han, Y. Shao, J. Fang, Z.-H. Zhang, Y.-W. Wang and Y. Peng, Anal. Chem., 2017, 89, 1937–1944 Search PubMed.
- H. Zhang, W. Li, J. Chen, G. Li, X. Yue, L. Zhang, X. Song and W. Chen, Anal. Chim. Acta, 2020, 1097, 238–244 Search PubMed.
- H. S. Jung, X. Chen, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2013, 42, 6019–6031 Search PubMed.
- X. Zhang, J. Lu, X. Ren, Y. Du, Y. Zheng, P. V. Ioannou and A. Holmgren, Biol. Med., 2015, 152, 192–200 Search PubMed.
- Q. Lin, K. Zhong, J. Zhu, L. Ding, J. Su, H. Yao, T. Wei and Y. Zhang, Macromolecules, 2017, 20, 7863–7871 Search PubMed.
- R. Janáky, V. Varga, A. Hermann, P. Saransaari and S. S. Oja, Neurochem. Res., 2000, 9, 1397–1405 Search PubMed.
- C. Yee, W. Yang and S. Hekimi, Cell, 2014, 4, 897–909 Search PubMed.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. D. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Nature, 2010, 7325, 790–795 Search PubMed.
- Y. Bessho, E. Iwakoshi-Ukena, T. Tachibana, S. Maejima, S. Taniuchi, K. Masuda, K. Shikano, K. Kondo, M. Furumitsu and K. Ukena, Neurosci. Lett., 2014, 578, 106–110 Search PubMed.
- L. D. Li, Z. B. Chen, H. T. Zhao and L. Guo, Biosens. Bioelectron., 2011, 26, 2781–2785 Search PubMed.
- M. Watanabe, M. E. Suliman, A. R. Qureshi, E. Garcia-Lopez, P. Bárány, O. Heimbürger, P. Stenvinkel and B. Lindholm, Am. J. Clin. Nutr., 2008, 87, 1860–1866 Search PubMed.
- H. Saito, L. T. Goodnough, J. M. Boyle and N. Heimburger, Am. J. Med., 1982, 73, 90175–90179 Search PubMed.
- K. V. R. Rao, P. V. B. Reddy, X. Tong and M. D. Norenberg, Am. J. Pathol., 2010, 176, 1400–1408 Search PubMed.
- H. Li, J. Liu, Y. Fang, Y. Qin, S. Xu, Y. Liu and E. Wang, Biosens. Bioelectron., 2013, 41, 563–568 Search PubMed.
- Q. Xiao, H. Gao, Q. Yuan, C. Lu and J. Lin, J. Chromatogr. A, 2013, 1274, 145–150 Search PubMed.
- X. Wang, C. Luo, L. Li and H. Duan, J. Electroanal. Chem., 2015, 757, 100–106 Search PubMed.
- K. B. A. Ahmed, M. Sengan, P. S. Kumar and A. Veerappan, Sens. Actuators, B, 2016, 233, 431–437 Search PubMed.
- C. Song, W. Zhao, H. Liu, W. Ding, L. Zhang, J. Wang, Y. Yao and C. Yao, J. Mater. Chem. B, 2020, 8, 494–7500 Search PubMed.
- Y. Che, H. Pang, H. Li, L. Yang, X. Fu, S. Liu, L. Ding and J. Hou, Talanta, 2019, 196, 442–448 Search PubMed.
- S. Ma, Q. Zhang, D. Wu, Y. Hu, D. Hu, Z. Guo, S. Wang, Q. Liu and J. Peng, J. Electroanal. Chem., 2019, 847, 113144 Search PubMed.
- R. Shen, L. Zou, S. Wu, T. Li, J. Wang, J. Liu and L. Ling, Spectrochim. Acta, Part A, 2019, 213, 42–47 Search PubMed.
- M. Tian, F. Guo, Y. Sun, W. Zhang, F. Miao, Y. Liu, G. Song, C.-L. Ho, X. Yu, J. Z. Sun and W.-Y. Wong, Org. Biomol. Chem., 2014, 12, 6128–6133 Search PubMed.
- K. S. Park, M. I. Kim, M. Woo and H. G. Park, Biosens. Bioelectron., 2013, 45, 65–69 Search PubMed.
- F. Yan, D. Shi, T. Zheng, K. Yun, X. Zhou and L. Chen, Sens. Actuators, B, 2016, 224, 926–935 Search PubMed.
- W. X. Ze, Z. Q. Zhang, R. Guo, Y. Y. Zhang, N. J. Zhu, K. Wang, P. P. Sun, X. Y. Mao, J. J. Liu, J. Z. Huo, X. R. Wang and B. Ding, Talanta, 2020, 217, 121010 Search PubMed.
- J. Wang, H. Jiang, H. Liu, L. Liang and J. Tao, Spectrochim. Acta, Part A, 2020, 228, 117725 Search PubMed.
- L. Zhao, X. He, Y. Huang, S. Zhang, H. Han, L. Xu, X. Wang, D. Song, P. Ma and Y. Sun, Anal. Bioanal. Chem., 2020, 412, 7211–7217 Search PubMed.
- J. Peng, P. Gong, S. Li, F. Kong, X. Ge, B. Wang, L. Guo, Z. Liu and J. You, Chem. Eng. J., 2020, 391, 123619 Search PubMed.
- P. Gong, L. Sun, F. Wang, X. Liu, Z. Yan, M. Wang, L. Zhang, Z. Tian, Z. Liu and J. You, Chem. Eng. J., 2019, 356, 994–1002 Search PubMed.
- P. Gong, S. Ji, J. Wang, D. Dai, F. Wang, M. Tian, L. Zhang, F. Guo and Z. Liu, Chem. Eng. J., 2018, 348, 438–446 Search PubMed.
- Y. Cai, J. Wang, C. Liu, S. Yang, Y. Zhang and A. Liu, ChemComm, 2020, 56, 11637–11640 Search PubMed.
- D. Sun, T. Liu, C. Wang, L. Yang, S. Yang and K. Zhuo, Spectrochim. Acta, Part A, 2020, 240, 118598 Search PubMed.
- C. Das, B. Pakhira, A. L. Rheingold and S. K. Chattopadhyay, Inorg. Chim. Acta, 2018, 482, 292–298 Search PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 Search PubMed.
- Y. Li, W. Chen, G. Xing, D. Jiang and L. Chen, Chem. Soc. Rev., 2020, 49, 2852–2868 Search PubMed.
- S. Yuan, X. Li, J. Zhu, G. Zhang, P. V. Puyvelde and B. V. Bruggen, Chem. Soc. Rev., 2019, 48, 2665–2681 Search PubMed.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 Search PubMed.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 Search PubMed.
- V. A. Kuehl, J. Yin, P. H. H. Duong, B. Mastorovich, B. Newell, K. D. Li-Oakey, B. A. Parkinson and J. O. Hoberg, J. Am. Chem. Soc., 2018, 140, 18200–18207 Search PubMed.
- Y. Meng, Y. Luo, J. Shi, H. Ding, X. Lang, W. Chen, A. Zheng, J. Sun and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 3624–3629 Search PubMed.
- A. F. M. EL-Mahdy, M. B. Zakaria, H. X. Wang, T. Chen, Y. Yamauchi and S. W. Kuo, J. Mater. Chem. A, 2020, 8, 25148–25155 Search PubMed.
- A. F. M. El-Mahdy, C. H. Kuo, A. Alshehri, C. Young, Y. Yamauchi, J. Kim and S. W. Kuo, J. Mater. Chem. A, 2018, 6, 19532–19541 Search PubMed.
- X. Chen, K. Geng, R. Liu, K. T. Tan, Y. Gong, Z. Li, S. Tao, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 5050–5091 Search PubMed.
- H. R. Abuzeid, A. F. M. EL-Mahdy and S. W. Kuo, Microporous Mesoporous Mater., 2020, 300, 110151 Search PubMed.
- A. F. M. El-Mahdy, Y. H. Hung, T. H. Mansoure, H. H. Yu, T. Chen and S. W. Kuo, Asian J. Chem., 2019, 14, 1429–1435 Search PubMed.
- A. F. M. El-Mahdy, C. Young, J. Kim, J. You, Y. Yamauchi and S. W. Kuo, ACS Appl. Mater. Interfaces, 2019, 11, 9343–9354 Search PubMed.
- L. Zhang, S. Wang, Y. Zhou, C. Wang, X. Z. Zhang and H. Deng, Angew. Chem., Int. Ed., 2019, 131, 14351–14356 Search PubMed.
- A. F. M. EL-Mahdy, Y. H. Hung, T. H. Mansoure, H. H. Yu, Y. S. Hsu, K. C. Wu and S. W. Kuo, J. Taiwan Inst. Chem. Eng., 2019, 103, 199–208 Search PubMed.
- H. R. Abuzeid, A. F. M. EL-Mahdy, M. M. M. Ahmed and S. W. Kuo, Polym. Chem., 2019, 10, 6010–6020 Search PubMed.
- W. R. Cui, C. R. Zhang, W. Jiang, F. F. Li, R. P. Liang, J. Liu and J. D. Qiu, Nat. Commun., 2020, 11, 436 Search PubMed.
- P. Das and S. K. Mandal, J. Mater. Chem. A, 2018, 6, 16246–16256 Search PubMed.
- A. F. M. EL-Mahdy, A. M. Elewa, S. W. Huang, H. H. Chou and S. W. Kuo, Adv. Opt. Mater., 2020, 8, 2000641 Search PubMed.
- X. Li, Q. Gao, J. Wang, Y. Chen, Z. H. Chen, H. S. Xu, W. Tang, K. Leng, G. H. Ning, J. Wu, Q. H. Xu, S. Y. Queck, Y. Lu and K. P. Loh, Nat. Commun., 2018, 9, 2335 Search PubMed.
- C. Wu, Y. Liu, H. Liu, C. Duan, Q. Pan, J. Zhu, F. Hu, X. Ma, T. Jiu, Z. Li and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 10016–10024 Search PubMed.
- B. Wang, X. Liu, P. Gong, X. Ge, Z. Liu and J. You, ChemComm, 2020, 56, 519–522 Search PubMed.
- M. G. Mohamed, C. C. Lee, A. F. M. EL-Mahdy, J. Lüder, M. H. Yu, Z. Li, Z. Zhu, C. C. Chueh and S. W. Kuo, J. Mater. Chem. A, 2020, 8, 11448–11459 Search PubMed.
- A. F. M. EL-Mahdy, M. Y. Lai and S. W. Kuo, J. Mater. Chem. C, 2020, 8, 9520–9528 Search PubMed.
- Y. F. Xie, S. Y. Ding, J. M. Liu, W. Wang and Q. Y. Zheng, J. Mater. Chem. C, 2015, 3, 10066–10069 Search PubMed.
- G. Chen, H. H. Lan, S. L. Cai, B. Sun, X. L. Li, Z. H. He, S. R. Zheng, J. Fan, Y. Liu and W. G. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 12830–12837 Search PubMed.
- S. L. Cai, Z. H. He, X. L. Li, K. Zhang, S. R. Zheng, J. Fan, Y. Liu and W. G. Zhang, Chem. Commun., 2019, 55, 13454–13457 Search PubMed.
- Z. Li, N. Huang, K. H. Lee, Y. Feng, S. Tao, Q. Jiang, Y. Nagao, S. Irle and D. Jiang, J. Am. Chem. Soc., 2018, 140, 12374–12377 Search PubMed.
- M. Li, Z. Cui, S. Pang, L. Meng, D. Ma, Y. Li, Z. Shi and S. Feng, J. Mater. Chem. C, 2019, 7, 11919–11925 Search PubMed.
- F. Z. Cui, J. J. Xie, S. Y. Jiang, S. X. Gan, D. L. Ma, R. R. Liang, G. F. Jiang and X. Zhao, Chem. Commun., 2019, 55, 4550–4553 Search PubMed.
- Y. H. Wang, K. J. Huang and X. Wu, Biosens. Bioelectron., 2017, 97, 305–316 Search PubMed.
- S. B. Wang, Z. X. Chen, F. Gao, C. Zhang, M. Z. Zou, J. J. Ye, X. Zeng and X. Z. Zhang, Biomaterials, 2020, 234, 119772 Search PubMed.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 Search PubMed.
- X. Liu, M. Hu, M. Wang, Y. Song, N. Zhou, L. He and Z. Zhang, Biosens. Bioelectron., 2019, 123, 59–68 Search PubMed.
- E. Cho, J. Choi, S. Jo, D. H. Park, Y. K. Hong, D. Kim and T. S. Lee, ChemPlusChem, 2019, 84, 1130–1134 Search PubMed.
- G. U. Reddy, H. Agarwalla, N. Taye, S. Ghorai, S. Chattopadhyay and A. Das, Chem. Commun., 2014, 50, 9899–9902 Search PubMed.
- Y. Chen, W. Y. Li, Y. Wang, X. D. Yang, J. Chen, Y. N. Jiang, C. Yu and Q. Lin, J. Mater. Chem. C, 2014, 2, 4080–4085 Search PubMed.
- J. Sun, F. Yang, D. Zhao, C. Chen and X. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 6860–6866 Search PubMed.
- C. Cui, Q. Wang, C. Xin, Q. Liu, X. Deng, T. Liu, X. Xu and X. Zhang, Microporous Mesoporous Mater., 2020, 299, 110122 Search PubMed.
- B. Guo, X. Pan, Y. Liu, L. Nie, H. Zhao, Y. Liu, J. Jing and X. Zhang, Sens. Actuators, B, 2018, 256, 632–638 Search PubMed.
- Q. H. You, A. W. M. Lee, W. H. Chan, X. M. Zhu and K. C. F. Leung, Chem. Commun., 2014, 50, 6207–6210 Search PubMed.
- J. F. Folmer-Andersen, V. M. Lynch and E. V. Anslyn, Chem. – Eur. J., 2005, 11, 5319–5326 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00234a |
| This journal is © The Royal Society of Chemistry 2021 |