
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuneable CO2 binding enthalpies by redox modulation of an electroactive MOF-74 framework†
Patrick W.
Doheny
a,
Ravichandar
Babarao
 b,
Cameron J.
Kepert
*a and
Deanna M.
D’Alessandro
*a
b,
Cameron J.
Kepert
*a and
Deanna M.
D’Alessandro
*a
aSchool of Chemistry, The University of Sydney, New South Wales, 2006, Australia. E-mail: deanna.dalessandro@sydney.edu.au; cameron.kepert@sydney.edu.au; Tel: +61 2 93513777
bSchool of Science, Royal Melbourne Institute of Technology (RMIT University), Victoria, 3001, Australia
First published on 8th February 2021
Abstract
The [M2(dobdc)] series of frameworks (also known as MOF-74 and CPO-27, where M2+ = Zn, Mg, for example, and dobdc2− = 4,6-dioxido-1,3-benzenedicarboxylate) have been the subject of enormous interest in respect to their attractive gas adsorption properties that arise from the ultra-high concentration of open metal sites. An interesting prospect with respect to future applications of these materials is the possibility of generating a ‘switchable’ framework in which the guest adsorption can be modulated by virtue of the redox state of the framework. Herein, we report a closely related metal–organic framework (MOF), [Zn2(DSNDI)], where the redox state of the electroactive ligand DSNDI4− (N,N′-bis(4-carboxy-3-hydroxyphenyl)-1,4,5,8-naphthalenetetracarboxydiimide) can be modulated using both in situ and ex situ methods. The material is stable to both chemical and electrochemical reduction of its organic DSNDI units without structural collapse or degradation, and remains porous to N2, H2 and CO2. The chemically reduced frameworks demonstrate notable changes from the parent material, particularly with respect to CO2 binding. The CO2 isosteric heat of adsorption (Qst) increases from 27.9 kJ mol−1 in the neutral material to 43.9 kJ mol−1 after reduction due to enhanced host–guest interactions, the latter being comparable to the best performing member of the MOF-74 series, [Mg2(dobdc)]. The origin of the enhanced interaction was probed using Density Functional Theory (DFT) calculations which corroborated the presence of enhanced framework-CO2 interactions for the chemically-reduced MOF. The tuneable CO2 binding character provides an attractive mechanism to tune gas separations and capture.
Introduction
Metal–organic framework (MOF) materials have long been recognised as promising candidates for a wide range of applications including, but not limited to, CO2 storage,1–3 separations4–7 and electrocatalysis.8–11 In the field of gas sorption,12–14 storage and crude separations,7,15–17 the MOF-74/CPO-27 material,12 ([M2(dobdc)], where M2+ = Mg, Mn, Fe, Co, Ni, Cu and Zn and dobdc2− = 4,6-dioxido-1,3-benzenedicarboxylate), enjoys significant attention due to its porosity and ultra-high concentration of open metal sites that facilitate relatively strong guest binding compared with typical physisorption in framework materials.18 One attractive means by which multifunctional properties within a MOF may coexist or be tuned (such as gas sorption or selectivity) is by use of a redox ‘switch’, where certain characteristics emerge as a result of oxidation or reduction.19–22 Despite this, the study of electroactive MOF-74 materials is comparatively limited and mostly confined to redox-transformations of the metal ions such as Fe2+/3+ oxidation.23 Post-synthetic oxidation of the dobdc2− ligand has also been reported,24 in addition to the use of 2,5-disulfhydrylbenzene-1,4-dicarboxylate which led to an enhanced charge mobility relative to the [M2(dobdc)] material.25A recent report by Saha26 described the synthesis of a MOF-74 analogue incorporating the redox-active DSNDI ligand (where DSNDI4− = N,N′-bis(4-carboxy-3-hydroxyphenyl)-1,4,5,8-naphthalenetetracarboxydiimide) in which the band gap of the framework was modified by infiltrating tetrathiafulvalene (TTF) electron donors that interacted with the DSNDI acceptor units. An additional report by Dincă27 focused on an analogous framework utilising a regioisomer of the DSNDI ligand, NDISA (where NDISA4− = N,N′-bis(3-carboxy-4-hydroxyphenyl)-1,4,5,8-naphthalenetetradicarboximide) that was studied for electrochromic applications upon in situ reduction of the ligand.
Motivated by previous work suggesting that the gas sorption properties of MOFs can be switched by redox modulation,28–30 we now report the electroactive properties of the [Zn2(DSNDI)] framework, hereafter denoted 1, via a combination of in situ and ex situ techniques. Particular attention was focused on the modification of its gas sorption properties (N2, H2, CH4 and CO2), and the CO2 binding enthalpies by utilising post-synthetic chemical reduction. Density Functional Theory (DFT) calculations were employed to gain insights into the origins of the enhanced framework-CO2 interactions for the chemically-reduced materials. Such studies are important given the strong attention that the parent MOF-74 materials have received with respect to their guest sorption properties. Notably, the presence of 1D honeycomb pores and an ultra-high concentration of open metal binding sites upon activation of the materials makes them highly attractive for industrial gas capture and separations applications. The realisation of tuneable guest adsorption by virtue of a redox-active ligand enhances the multifunctionality of this family of frameworks.
Results and discussion
Electrochemical and spectroelectrochemical properties
The electrochemical properties of 1 were examined using cyclic voltammetry (CV) in 0.1 M [(n-C4H9)4N]PF6/DMF supporting electrolyte. The CV (Fig. 1) showed two processes in the cathodic region with a reversible, first process at −0.89 V vs. Fc/Fc+ and a second, quasi-reversible process at −1.41 V vs. Fc/Fc+ that became increasingly reversible at faster scan rates consistent with the electrochemistry of previously reported naphthalenediimide (NDI) containing frameworks.31 Given the redox-innocent nature of the Zn2+ nodes of the framework, the observed processes were assigned to the sequential electrochemical reduction of the NDI moiety of the DSNDI ligands within the MOF structure to the NDI radical anion and subsequent dianion species.27,28,32 This was supported by solution-state CV of the DSNDI ligand (Fig. S2, ESI†) which correlated well with that of the framework, showing that the redox properties of the ligand had been successfully transferred to the MOF.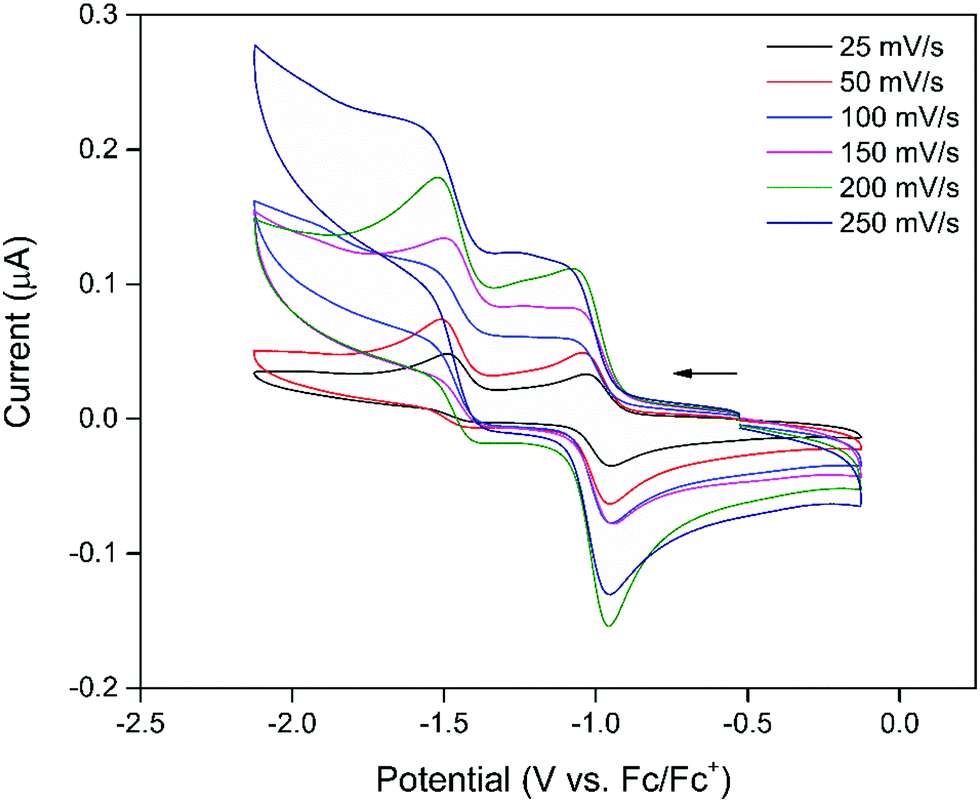 | ||
| Fig. 1 CV of 1 in 0.1 M [(n-C4H9)4N]PF6/DMF supporting electrolyte at scan rates of 25–250 mV s−1. The arrow indicates the direction of the forward scan. | ||
The optical properties of the neutral and reduced species of 1 were examined using in situ solid-state UV-Vis-NIR spectroelectrochemistry in 0.1 M [(n-C4H9)4N]PF6/DMF supporting electrolyte. The diffuse reflectance spectrum of the neutral material at 0 V (Fig. 2a) was characterised by a single band at 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 510 cm−1 that was attributed to the aromatic π → π* transition of the DSNDI ligand. Upon reduction at −1.2 V, bands in the visible region formed at 12810, 14260 and 16520 cm−1 in addition to a sharp band at 21050 cm−1; these were attributed to the formation of the DSNDI radical anion within the framework structure.33,34 The emergence of the new bands was concurrent with a colour change of the initially bright orange material to a deep brown and correlates well with the spectroelectrochemistry of the H4DSNDI ligand (Fig. S3 and S4, ESI†). Changing the potential further to −2.0 V (Fig. 2b) led to the sample turning black with a large increase in the spectral intensity alongside the appearance of bands at 17150, 18010, 19010 and 19940 cm−1 consistent with the formation of a dianionic NDI species.33,34
510 cm−1 that was attributed to the aromatic π → π* transition of the DSNDI ligand. Upon reduction at −1.2 V, bands in the visible region formed at 12810, 14260 and 16520 cm−1 in addition to a sharp band at 21050 cm−1; these were attributed to the formation of the DSNDI radical anion within the framework structure.33,34 The emergence of the new bands was concurrent with a colour change of the initially bright orange material to a deep brown and correlates well with the spectroelectrochemistry of the H4DSNDI ligand (Fig. S3 and S4, ESI†). Changing the potential further to −2.0 V (Fig. 2b) led to the sample turning black with a large increase in the spectral intensity alongside the appearance of bands at 17150, 18010, 19010 and 19940 cm−1 consistent with the formation of a dianionic NDI species.33,34
 | ||
| Fig. 2 Solid-state UV-Vis-NIR spectroelectrochemistry of 1 in 0.1 M [(n-C4H9)4N]PF6/DMF electrolyte showing potential changes from (a) 0 to −1.2 V and (b) −1.2 to −2.0 V where the arrows indicate the direction of the spectral progression. The step at 12500 cm−1 is due to a detector change. Insets: Photographs of the sample acquired in situ during the time-course of the experiment. | ||
Returning the potential to 0 V (Fig. S5, ESI†) led to the slow decay of the visible bands and restoration of the initial spectrum over the course of several hours. The slow decay of the radical bands after the removal of the applied potential implied the formation of a stable anionic radical. The restoration of the initial spectrum and sample colour upon returning the potential to 0 V was suggestive of a degree of reversibility and stability of 1 to electrochemical reduction.
The nature and delocalisation of the DSNDI radical within the anionic MOF was further examined using in situ X-band EPR spectroelectrochemistry. The initial spectrum at 0 V was characterised by a weak isotropic signal that was attributed to the formation of a photoradical during sample preparation that is characteristic of NDI containing materials.33,35,36 Upon application of an electrochemical potential the intensity of the signal was observed to increase to give a sharp, isotropic signal (Fig. S6a, ESI†) with a simulated (Fig. S6b, ESI†) g-value of 2.0014, consistent with the formation of an organic radical. The sharpness of the spectrum implied the presence of unresolved hyperfine splitting of the signal. The EPR spectrum of the electrochemical radical at 1.2 V was recollected (Fig. S7, ESI†) using a modulation amplitude of 0.1 G to yield a poorly resolved 13-line spectrum arising from hyperfine splitting of the signal. The latter is consistent with the EPR spectroelectrochemistry from previously reported studies of NDI radical anions that also exhibit a characteristic 13-line signal arising from hyperfine coupling of the NDI radical to the 14N and 1H nuclei of the NDI moiety.34
Porosity and gas sorption properties
The sorption properties of neutral 1 were examined using N2, H2, CO2 and CH4, extending upon the work of Saha who reported the N2 BET surface area of the material.26 The DMF solvated de novo material was first activated according to a similar procedure to that previously reported.26 The retention of structure and crystallinity after both solvent exchange and activation was confirmed using capillary PXRD (Fig. S8, ESI†) prior to gas sorption analysis.The N2 sorption properties at 77 K were characterised by Type IV isotherm behaviour (Fig. 3a) with a minor hysteresis observed in the high pressure region that was suggestive of a microporous structure with a high concentration of vacant adsorption sites. Applying BET theory yielded a surface area of 1748 ± 40 m2 g−1, a value comparable to those reported for structurally analogous MOF-74 materials,12,37 but slightly lower than that previously reported for 1 (cf. 2044 m2 g−1) by Saha and co-workers.26 Although not significantly different, the discrepancy may reflect differences in activation methods between this and the previous study.26 Further analysis of the N2 isotherm data enabled the characterisation of the pore size distribution (Fig. S9, ESI†) and pore volume, with a single pore size of 22.5 Å (in good agreement with the 23.8 Å pore width from the calculated model structure) and associated pore volume of 0.53 cm3 g−1 calculated.
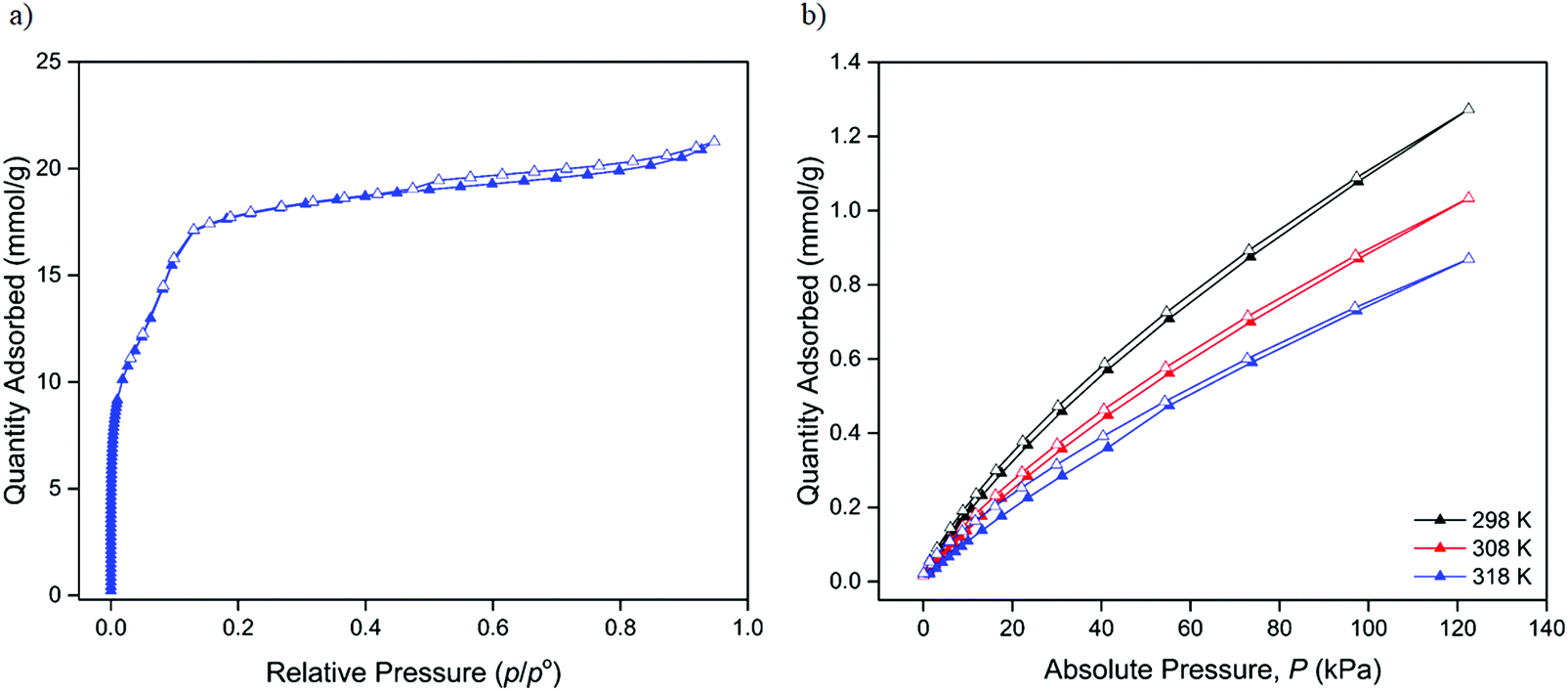 | ||
| Fig. 3 Gas sorption isotherms of 1 showing adsorption of (a) N2 at 77 K and (b) CO2 at 298, 308 and 318 K. Adsorption and desorption are indicated by closed and open triangles respectively. | ||
Given the strong interest in CO2 capture and storage with respect to MOF-74 materials, the CO2 adsorption properties of 1 were examined at three temperatures of 298, 308 and 318 K (Fig. S10, ESI†). The CO2 isotherms were indicative of Type I sorption behaviour and modest uptake with a maximum of 1.27 mmol g−1 of CO2 adsorbed at 1200 mbar and 298 K (Fig. 3b).
Type I behaviour was also observed with respect to the H2 (Fig. S11, ESI†) and CH4 (Fig. S12, ESI†) sorption properties (Table 1) of the MOF, with considerably lower uptake of CH4 observed at 298 K (0.24 mmol g−1 at 1200 mbar) relative to CO2. The variance in uptakes of CO2vs. CH4 were tentatively attributed to the differences in binding strength of the respective guests (Fig. S13–S15, ESI†). Previous literature reports have demonstrated a preference of MOF-74 materials to adsorb CO2 over CH4 due to its greater polarisability and steric influences that prevent effective binding of the CH4 molecule at the open metal sites.38 Conversely, the smaller size and linear geometry of CO2 facilitate stronger interactions with the open metal sites leading to enhanced uptake relative to CH4.
Upon completion of the gas sorption experiments the material was examined again using capillary PXRD (Fig. S16, ESI†) to assess the effects of multiple sorption and desorption cycles of guests on the framework stability and structure. PXRD confirmed that the structure had indeed been retained with minimal changes in crystallinity (Fig. S16, ESI†).
Chemical modulation of guest uptake
In order to examine the effects of the electroactivity of the framework on the sorption properties, chemical reduction of the activated material 1 was carried out using 0.1 M lithium naphthalenide (LiNP) in 0.5, 1.0, 2.0, 3.0, 4.0 and 5.0 stoichiometric equivalents to yield chemically reduced MOFs. All reduced samples were washed with dry THF to remove any residual LiNP or naphthalene from the framework post-reduction. The actual degree of framework reduction was determined using ICP-MS, with the measured ratio of Li to Zn used to quantify the extent of reduction (ESI,† Table S3). The reduced frameworks (hereafter denoted by their actual extent of reduction) were found to have retained both structure and crystallinity as confirmed by capillary PXRD (Fig. S17, ESI†). Solid state IR spectroscopy (Fig. S18, ESI†) and TGA (Fig. S34–S40, ESI†) of the reduced samples following the washing procedure also confirmed the removal of residual naphthalene from the pore space.The solid state UV-Vis-NIR spectra (Fig. 4) of the chemically reduced samples showed distinct changes relative to the neutral material. Highly intense visible bands were observed at 13070, 14460, 16210, 18480 and 20530 cm−1 and were attributed to the formation of the DSNDI radical anion.34 Additional higher energy transitions at 26840, 31560, 38070 and 43410 cm−1 were also found for these samples. The visible bands were assigned to the formation of the DSNDI radical anion and correlated well with those from the spectroelectrochemical experiment, while the higher energy bands were assigned to the aromatic transitions of the ligand.33,34 EPR spectroscopy of the reduced samples (Fig. S19–S24, ESI†) also confirmed the successful reduction of the framework with simulated signals (Fig. S25, ESI†) corresponding to the DSNDI radical anion (g = 2.0029) observed.
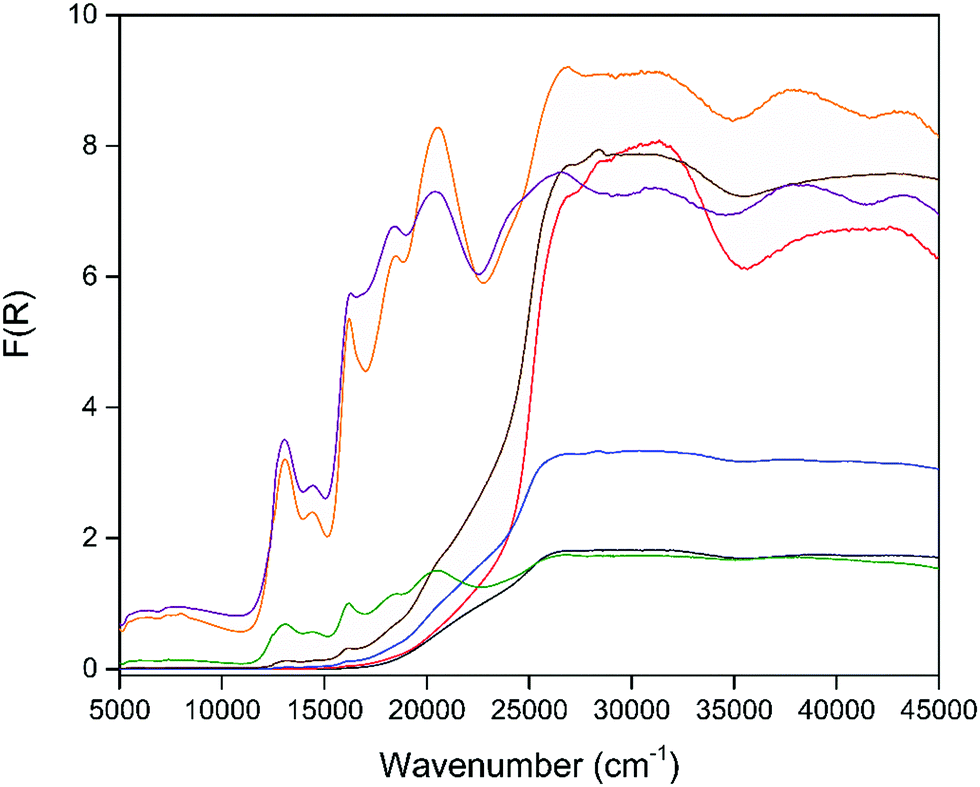 | ||
| Fig. 4 Solid-state UV/Vis/NIR spectra of [Zn2(DSNDI)], 1 (black) and its reduced analogues generated by chemical reduction with 0.39 (red), 0.47 (blue), 1.15 (brown), 1.82 (green), 2.27 (orange) and 2.83 equiv. (purple) of LiNP. | ||
The surface areas of the chemically reduced materials were calculated from the N2 isotherm data at 77 K (Fig. S26, ESI†). Although the N2 sorption isotherms for the reduced samples showed a qualitative similarity to that of the neutral material, the BET surface areas (Table 2) were found to be considerably lower, with increasing Li loading correlated with lower surface areas. The N2 uptake was also found to decrease dramatically, with the relatively low uptakes for the 2.27 and 2.82 equiv. samples attributed to the presence of Li+ counterions, required to charge balance the DSNDI radical anions, within the pore space. Interestingly, although a single pore was detected in the reduced samples, akin to the neutral material, the pore sizes were slightly larger than that of the neutral MOF. This may reflect a possible structural conditioning post-reduction to accommodate the newly introduced DSNDI radical ligands and corresponding Li+ counterions, as has been observed previously.31,39 Although not detected, some residual neutral naphthalene may be present in the reduced samples which could be influencing the BET surface areas; however, this contribution cannot be deconvoluted from the relatively larger effect of Li+ filling the pore space. Future work will focus on the effects of other reducing agents and the effects on CO2 binding.
| Material | BET surface area (m2 g−1) | Pore size (Å) |
|---|---|---|
| Neutral | 1748 ± 40 | 22.5 |
| 0.39 equiv. | 1462 ± 100 | 25 |
| 0.47 equiv. | 1298 ± 124 | 24.2 |
| 1.15 equiv. | 1129 ± 45 | 24.8 |
| 1.82 equiv. | 1084 ± 44 | 24.9 |
| 2.27 equiv. | 950 ± 79 | 24.7 |
| 2.83 equiv. | 889 ± 22 | 24.8 |
The adsorption properties with respect to CO2 at 298, 308 and 318 K were examined for the samples chemically reduced by 0.39, 0.47, 1.15 and 1.82 equiv. of LiNP. Although still maintaining modest porosity, the CO2 uptake of the 2.27 and 2.83 equiv. samples were characterised by very low uptakes and extremely long equilibration times. This was attributed to the occlusion of the pore space by Li+ counterions preventing diffusion and uptake of large, polarisable guests such as CO2. As such, only the sorption properties of the samples reduced by 0.39, 0.47, 1.15 and 1.82 equiv. are considered here, with all four materials demonstrating Type I sorption behaviour (Fig. 5a and Fig. S27–S29, ESI†). The uptakes of CO2 were found to be lower than those in the neutral material at all three temperatures, with a maximum uptake of 0.95 mmol g−1 recorded for the 0.47 equiv. sample at 298 K, a discrepancy attributed to the occupation of the pore space with Li+. Given previous reports demonstrating the tuning of the CO2 binding affinities of porous MOF materials using redox modification,24,28–30 the CO2 isosteric heats of adsorption (Qst) of the chemically reduced forms of 1 were of great interest. Applying the Clausius–Clapeyron equation (see ESI†) to the CO2 isotherms obtained at 298, 308 and 318 K for the 0.39, 0.47, 1.15 and 1.82 equiv. samples yielded Qst profiles (Fig. 5b and Fig. S30, S31, ESI†) for all four samples.
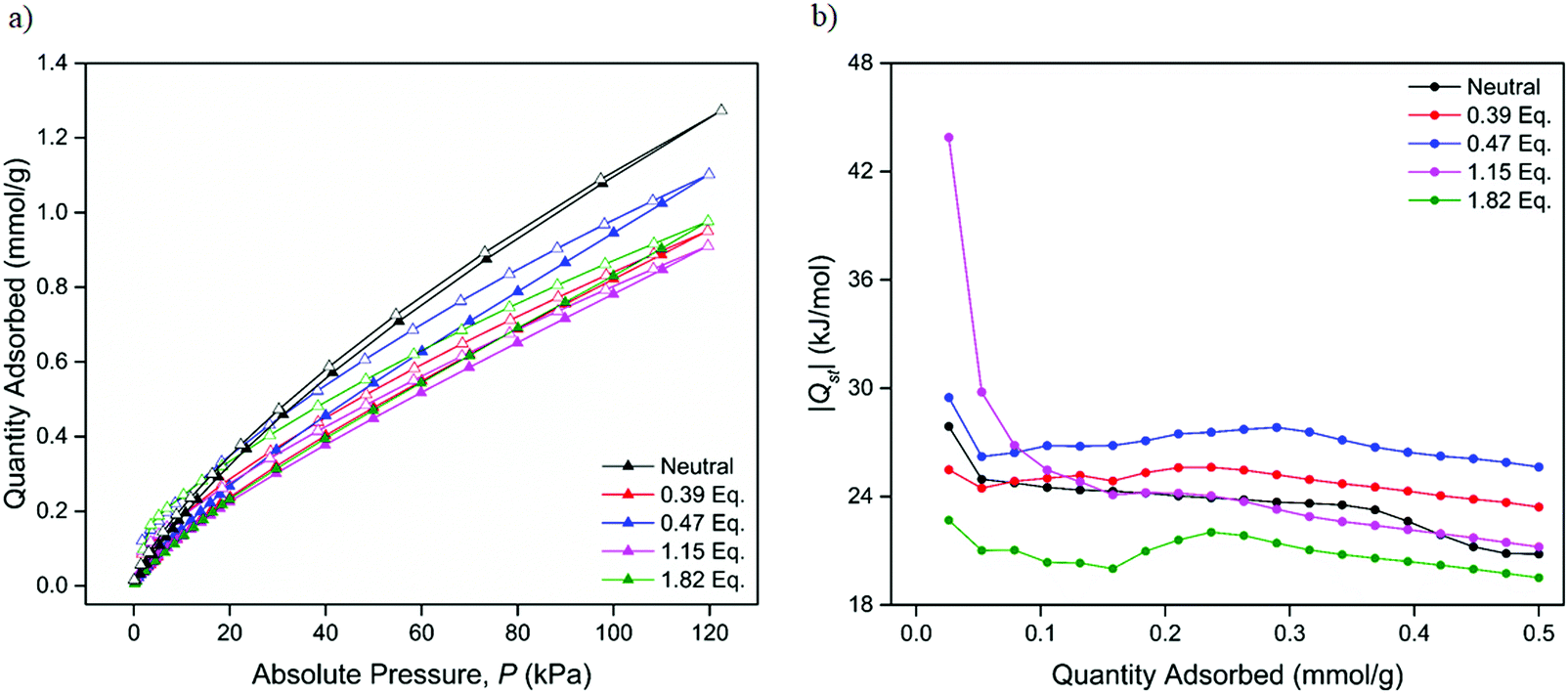 | ||
| Fig. 5 (a) CO2 adsorption (closed triangles) and desorption (open triangles) isotherms of the neutral and samples reduced by 0.39, 0.47, 1.15 and 1.82 equiv. of LiNP at 298 K and (b) CO2 isosteric heat of adsorption profiles for the neutral and chemically reduced samples. | ||
The CO2Qst value of the neutral form of 1 was calculated as 27.9 kJ mol−1, a value similar to that of the analogous Zn-MOF-74 analogue (30.6 kJ mol−1);40 this is unsurprising as the chemical environment of the parent system (Zn2+ open metal sites and an aromatic ligand system) resembles that of 1 with no major differences expected. The CO2Qst values (Table 3) of the 0.39 and 0.47 equiv. samples were similar to that of the neutral material, this being attributed to the relatively low degree of reduction that the framework had undergone. The sample reduced by 1.15 equiv. of LiNP demonstrated a larger increase in its CO2 binding affinity with a value of 43.9 kJ mol−1, while the sample reduced by 1.82 equiv. had a relatively lower Qst of 22.7 kJ mol−1. It should be noted that the Qst values for the reduced phases were calculated at the lowest pressure data point obtained. Thus, they do not strictly correspond to ‘zero-loadings’ (i.e., at 0 mbar), where the Qst may be higher.
The range of CO2 binding enthalpies recorded for the reduced materials range from weak (25–30 kJ mol−1) to moderately strong (30–50 kJ mol−1) physisorption interactions. Although this trend may suggest that higher Li loadings would lead to higher Qst values, the 1.82 equiv. sample was found to have an enthalpy of adsorption that was approximately half of the maximum value, and 5.2 kJ mol−1 lower than that of the neutral MOF. This suggests that a certain ‘optimal’ Li loading may exist, at which point the CO2Qst is maximised, and beyond which the CO2 binding interactions begin to decrease. While the binding affinities decrease at the higher loadings, the uptakes are still appreciable, suggesting that the high enthalpy sites become self-blocked at high loadings.
The observed modulation of CO2 binding enthalpies in the chemically reduced frameworks arises due to the changes in the chemical environment due to the formation of organic DSNDI radical anions and the presence of the associated Li+ counterions. To gain insight into the nature of the host–guest interactions between the framework and the CO2 and Li+ guests, DFT calculations (see ESI,† for simulation details) were employed (Fig. S32 and S33, ESI†). The static binding energy of CO2 in the neutral framework was 31.2 kJ mol−1. For the reduced framework, the initial location of one Li+ was initially identified from classical simulation, followed by a CO2 molecule. The latter yielded a binding energy of 55.5 kJ mol−1. The enhanced binding upon reduction is consistent with the experimental observations, and with previously reported examples of redox-modulated framework materials.28–30,41,42 These results demonstrate that the uptake and binding strengths of porous MOF materials can be modified by exploiting the counterion-guest interactions arising from ex situ chemical oxidation or reduction.
The successful modulation of the CO2Qst values demonstrated a successful proof-of-concept in using chemical reduction as a means to tune the guest affinities of electroactive MOF-74 analogues. Notably, the Qst value of 43.9 kJ mol−1 is significantly higher than that of the neutral framework and is comparable to the highest performing member of the MOF-74 series, [Mg2(dobdc)] in terms of its CO2 binding enthalpy.3 Given that the present study is concerned only with the Zn2+ material, this is both an encouraging result and provides motivation for further study of these electroactive MOF-74 analogues where their intrinsic redox capabilities and open metal binding sites.
Conclusions
A previously reported MOF-74 framework analogue incorporating the redox-active DSNDI ligand has been characterised with a particular focus on its CO2 sorption and electrochemical properties. The framework was found to successfully retain the electrochemical properties of the discrete DSNDI ligand, exhibiting two reduction processes without loss of electrochemical performance or structural collapse. The BET surface area and CO2 enthalpy of adsorption were calculated from N2 and CO2 adsorption isotherms as 1748 ± 40 m2 g−1 and 27.9 kJ mol−1 respectively, values that were comparable to the parent Zn-MOF-74 material. In order to assess the influence of the ligand's electroactive properties on the sorption functionalities of the framework, chemical reduction using LiNP was performed with the resulting materials containing radical DSNDI anionic species as confirmed by both UV-Vis-NIR and EPR spectroscopies. The reduced frameworks also demonstrated structural stability to guest desorption, allowing the gas sorption properties to be assessed. The porosity of the materials was successfully retained despite a decrease in BET surface area that was attributed to the occupation of the pore space by Li+ counterions. Notably, although the uptake of CO2 was slightly reduced, a substantial increase in the CO2 enthalpy of adsorption was observed, with a maximum Qst of 43.9 kJ mol−1 obtained for the sample reduced by 1.15 equiv. of LiNP. The enhanced CO2Qst value is comparable to the highest performing member of the MOF-74 series, Mg-MOF-74, in terms of its CO2 binding strength. This strengthening of CO2 binding from weak to strong physisorption interactions serves as a successful example of the ability to enhance and modulate host–guest affinities by the incorporation of electroactive components into porous framework materials.Experimental
[Zn2(DSNDI)] framework (1)
The [Zn2(DSNDI)] framework was synthesised according to a previously reported procedure with minor modifications.26 A screw-cap Pyrex jar was charged with H4DSNDI (510 mg, 0.95 mmol) and Zn2(NO3)2·6H2O (630 mg, 3.33 mmol) which was suspended and sonicated in a mixture of DMF (90 mL), EtOH (15 mL) and H2O (15 mL). The mixture was then heated at 90 °C for 24 h before cooling to room temperature, after which the supernatant was decanted and replaced with fresh DMF to remove residual precursors. After soaking for 24 h the target phase was obtained as an orange crystalline powder (0.41 g, 0.61 mmol, 64% based on DSNDI). Elemental Analysis: found: C 47.75%, H 3.81%, N 7.03%; calculated for C28H10N2O10·2.5DMF·2H2O: C 48.24%, H 3.59%, N 7.13%.Activation of the framework was achieved by exchanging the DMF solvent occupying the pore space with MeOH, with the solvent exchanged twice a day for three days. After heating under dynamic vacuum at 80 °C for 24 h, the activated framework was obtained as a bright yellow crystalline powder. At no point during the soaking or solvent exchanging steps was the sample allowed to go dry or be exposed to air, with the material kept under solvent at all times. The activated material was also protected from exposure to the atmosphere and was handled under an argon atmosphere in a glove box at all times prior to analysis.
Gas sorption analysis
Gas sorption and porosity analysis was performed using a Micromeritics 3-Flex over a 0–1.2 bar pressure range. Approximately 150–200 mg of sample was degassed at 80 °C under vacuum for 18 h prior to measurement. Nitrogen adsorption isotherms were collected at 77 K using an incremental dosing of N2 gas from 0–1 bar with the Brunauer, Emmett and Teller (BET) model applied to the data to extract the resulting surface area.H2 sorption studies were performed at 77 and 87 K while CO2 and CH4 sorption was carried out at 298, 308 and 318 K. Isosteric heats of adsorption for H2, CO2 and CH4 were calculated by applying the Clausius–Clapeyron equation to the sorption isotherms obtained at the corresponding temperatures.
Electrochemistry
Electrochemical measurements on solid materials were performed using a BASi Epsilon Electrochemical Analyser. Measurements were recorded using a three electrode set-up consisting of a glassy carbon working electrode, a platinum wire auxiliary electrode and an Ag/Ag+quasi-reference electrode in 0.1 M [(n-C4H9)4N]PF6/DMF supporting electrolyte that was degassed with Ar. The sample was mounted by dipping the glassy carbon working electrode into a paste of the powdered sample in DMF. Ferrocene was added at the completion of each experiment as an internal standard with all potentials quoted as V vs. Fc/Fc+.UV-Vis-NIR spectroelectrochemistry
UV-Vis-NIR spectroelectrochemistry of bulk materials was carried out using a CARY5000 spectrophotometer equipped with a Harrick Omni-Diff probe attachment. The Teflon spectroelectrochemical cell consisted of a platinum quasi-reference electrode, a silver wire auxiliary electrode and an Indium–Tin–Oxide (ITO) coated glass slide working electrode as reported previously.43 The sample was immobilised on the conductive side of the ITO slide using Teflon tape and the applied potential controlled using an eDAQ e-corder 410 potentiostat. 0.1 M [(n-C4H9)4N]PF6/DMF supporting electrolyte was used for all measurements with all spectral data reported as the Kubelka–Munk transform, where F(R) = (1 − R)2/2R (where R is the diffuse reflectance of the sample relative to the Teflon baseline).Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors gratefully acknowledge support from the Australian Research Council (FT170100283, DP190103130) for research funding. This research was supported by the use of the single crystal and powder X-ray diffraction services of the Sydney Analytical Core Facility at the University of Sydney. Access to EPR spectroscopy was also provided by the Sydney Analytical Core Facility. R. B. acknowledges the National Computing Infrastructure (NCI) facilities for the computational resources.References
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- A. Das and D. M. D’Alessandro, CrystEngComm, 2015, 17, 706–718 RSC.
- M. I. Gonzalez, M. T. Kapelewski, E. D. Bloch, P. J. Milner, D. A. Reed, M. R. Hudson, J. A. Mason, G. Barin, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2018, 140, 3412–3422 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS.
- T.-H. Bae and J. R. Long, Energy Environ. Sci., 2013, 6, 3565–3569 RSC.
- P.-Q. Liao, J.-Q. Shen and J.-P. Zhang, Coord. Chem. Rev., 2018, 373, 22–48 CrossRef CAS.
- M. B. Solomon, T. L. Church and D. M. D’Alessandro, CrystEngComm, 2017, 19, 4049–4065 RSC.
- P. M. Usov, B. Huffman, C. C. Epley, M. C. Kessinger, J. Zhu, W. A. Maza and A. J. Morris, ACS Appl. Mater. Interfaces, 2017, 9, 33539–33543 CrossRef CAS.
- F.-L. Li, Q. Shao, X. Huang and J.-P. Lang, Angew. Chem., Int. Ed., 2018, 57, 1888–1892 CrossRef CAS.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS.
- Y. Liao, L. Zhang, M. H. Weston, W. Morris, J. T. Hupp and O. K. Farha, Chem. Commun., 2017, 53, 9376–9379 RSC.
- D.-A. Yang, H.-Y. Cho, J. Kim, S.-T. Yang and W.-S. Ahn, Energy Environ. Sci., 2012, 5, 6465–6473 RSC.
- B. R. Barnett, S. T. Parker, M. V. Paley, M. I. Gonzalez, N. Biggins, J. Oktawiec and J. R. Long, J. Am. Chem. Soc., 2019, 141, 18325–18333 CrossRef CAS.
- E. D. Bloch, M. R. Hudson, J. A. Mason, S. Chavan, V. Crocellà, J. D. Howe, K. Lee, A. L. Dzubak, W. L. Queen, J. M. Zadrozny, S. J. Geier, L.-C. Lin, L. Gagliardi, B. Smit, J. B. Neaton, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2014, 136, 10752–10761 CrossRef CAS.
- J. E. Bachman, M. T. Kapelewski, D. A. Reed, M. I. Gonzalez and J. R. Long, J. Am. Chem. Soc., 2017, 139, 15363–15370 CrossRef CAS.
- W. L. Queen, M. R. Hudson, E. D. Bloch, J. A. Mason, M. I. Gonzalez, J. S. Lee, D. Gygi, J. D. Howe, K. Lee, T. A. Darwish, M. James, V. K. Peterson, S. J. Teat, B. Smit, J. B. Neaton, J. R. Long and C. M. Brown, Chem. Sci., 2014, 5, 4569–4581 RSC.
- D. M. D’Alessandro, Chem. Commun., 2016, 52, 8957–8971 RSC.
- I. Mjejri, C. M. Doherty, M. Rubio-Martinez, G. L. Drisko and A. Rougier, ACS Appl. Mater. Interfaces, 2017, 9, 39930–39934 CrossRef CAS.
- S. Wang, J. Liu, H. Zhao, Z. Guo, H. Xing and Y. Gao, Inorg. Chem., 2018, 57, 541–544 CrossRef CAS.
- M.-S. Yao, X.-J. Lv, Z.-H. Fu, W.-H. Li, W.-H. Deng, G.-D. Wu and G. Xu, Angew. Chem., Int. Ed., 2017, 56, 16510–16514 CrossRef CAS.
- E. D. Bloch, L. J. Murray, W. L. Queen, S. Chavan, S. N. Maximoff, J. P. Bigi, R. Krishna, V. K. Peterson, F. Grandjean, G. J. Long, B. Smit, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14814–14822 CrossRef CAS.
- A. F. Cozzolino, C. K. Brozek, R. D. Palmer, J. Yano, M. Li and M. Dincă, J. Am. Chem. Soc., 2014, 136, 3334–3337 CrossRef CAS.
- L. Sun, T. Miyakai, S. Seki and M. Dincă, J. Am. Chem. Soc., 2013, 135, 8185–8188 CrossRef CAS.
- Z. Guo, D. K. Panda, M. A. Gordillo, A. Khatun, H. Wu, W. Zhou and S. Saha, ACS Appl. Mater. Interfaces, 2017, 9, 32413–32417 CrossRef CAS.
- K. AlKaabi, C. R. Wade and M. Dincă, Chemistry, 2016, 1, 264–272 CrossRef CAS.
- C. F. Leong, T. B. Faust, P. Turner, P. M. Usov, C. J. Kepert, R. Babarao, A. W. Thornton and D. M. D’Alessandro, Dalton Trans., 2013, 42, 9831–9839 RSC.
- K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 9604–9605 CrossRef CAS.
- C. Hua, B. Chan, A. Rawal, F. Tuna, D. Collison, J. M. Hook and D. M. D’Alessandro, J. Mater. Chem. C, 2016, 4, 2535–2544 RSC.
- B. A. Johnson, A. Bhunia, H. Fei, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2018, 140, 2985–2994 CrossRef CAS.
- C. F. Leong, B. Chan, T. B. Faust and D. M. D’Alessandro, Chem. Sci., 2014, 5, 4724–4728 RSC.
- C. F. Leong, B. Chan, T. B. Faust, P. Turner and D. M. D’Alessandro, Inorg. Chem., 2013, 52, 14246–14252 CrossRef CAS.
- G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchison, M. A.-P. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup and C. P. Woodward, Aust. J. Chem., 2004, 57, 1011–1019 CrossRef CAS.
- H.-L. Zhang, J.-Z. Liao, W. Yang, X.-Y. Wu and C.-Z. Lu, Dalton Trans., 2017, 46, 4898–4901 RSC.
- A. Mallick, B. Garai, M. A. Addicoat, P. S. Petkov, T. Heine and R. Banerjee, Chem. Sci., 2015, 6, 1420–1425 RSC.
- D. Gygi, E. D. Bloch, J. A. Mason, M. R. Hudson, M. I. Gonzalez, R. L. Siegelman, T. A. Darwish, W. L. Queen, C. M. Brown and J. R. Long, Chem. Mater., 2016, 28, 1128–1138 CrossRef CAS.
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC.
- C. R. Wade, M. Li and M. Dincă, Angew. Chem., Int. Ed., 2013, 52, 13377–13381 CrossRef CAS.
- D. Yu, A. O. Yazaydin, J. R. Lane, P. D. C. Dietzel and R. Q. Snurr, Chem. Sci., 2013, 4, 3544–3556 RSC.
- Y. M. Litvinova, Y. M. Gayfulin, K. A. Kovalenko, D. G. Samsonenko, J. van Leusen, I. V. Korolkov, V. P. Fedin and Y. V. Mironov, Inorg. Chem., 2018, 57, 2072–2084 CrossRef CAS.
- H. J. Park and M. P. Suh, Chem. Sci., 2013, 4, 685–690 RSC.
- P. M. Usov, C. Fabian and D. M. D’Alessandro, Chem. Commun., 2012, 48, 3945–3947 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1868581 and 1868582. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ma00503g |
| This journal is © The Royal Society of Chemistry 2021 |