
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–organic frameworks (MOFs) as host materials for the enhanced delivery of biomacromolecular therapeutics
Pei-Hong
Tong
 a,
Ling
Zhu
a,
Yi
Zang
b,
Jia
Li
*b,
Xiao-Peng
He
*a and
Tony D.
James
*cd
a,
Ling
Zhu
a,
Yi
Zang
b,
Jia
Li
*b,
Xiao-Peng
He
*a and
Tony D.
James
*cd
aKey Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Rd., Shanghai 200237, China. E-mail: xphe@ecust.edu.cn
bNational Center for Drug Screening, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, P. R. China. E-mail: jli@simm.ac.cn
cDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: t.d.james@bath.ac.uk
dSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China
First published on 27th September 2021
Abstract
Biomacromolecular drugs have become an important class of therapeutic agents for the treatment of human diseases. Considering their high propensity for being degraded in the human body, the choice of an appropriate delivery system is key to ensure the therapeutic efficacy of biomacromolecular drugs in vivo. As an emerging class of supramolecular “host” materials, metal–organic frameworks (MOFs) exhibit advantages in terms of the tunability of pore size, encapsulation efficiency, controllable drug release, simplicity in surface functionalization and good biocompatibility. As a result, MOF-based host–guest systems have been extensively developed as a new class of flexible and powerful platform for the delivery of therapeutic biomacromolecules. In this review, we summarize current research progress in the synthesis of MOFs as delivery materials for a variety of biomacromolecules. Firstly, we briefly introduce the advances made in the use of biomacromolecular drugs for disease therapy and the types of commonly used clinical delivery systems. We then describe the advantages of using MOFs as delivery materials. Secondly, the strategies for the construction of MOF-encapsulated biomacromolecules (Biomacromolecules@MOFs) and the release mechanisms of the therapeutics are categorized. Thirdly, the application of MOFs to deliver different types of biomacromolecules (e.g., antigens/antibodies, enzymes, therapeutic proteins, DNA/RNA, polypeptides, and polysaccharides) for the treatment of various human diseases based on immunotherapy, gene therapy, starvation therapy and oxidation therapy is summarized. Finally, the remaining challenges and available opportunities for MOFs as drug delivery systems are outlined, which we anticipate will encourage additional research efforts directed towards developing Biomacromolecules@MOFs systems for biomedical applications.
1. Introduction
With the rapid development of modern medical science and technology, the effective treatment of fatal human diseases such as cancer, neurodegenerative disorders and metabolic diseases and their secondary complications remains a global challenge. Over the past few decades, biomacromolecules with therapeutic functions including proteins, peptides, plasmid DNA, small-interfering RNA and polysaccharides have proven to be applicable in various clinical trials for disease therapy.1,2 Although biomacromolecular therapeutics have achieved remarkable success in the treatment of diseases, numerous challenges remain. For example, biomacromolecules are unstable during circulation in the blood, are easily degraded by enzymes, display short half-lives, exhibit immunogenicity, and can unselectively interact with healthy cells, and most are unable to cross biological barriers in vivo to reach target cells.3 As a result, researchers have developed a diverse range of drug delivery systems to protect biomacromolecules from being denatured and degraded in vivo. In addition, these systems facilitate the controlled release of biomacromolecular therapeutics in target tissues/cells.Currently used clinical delivery systems can be divided into viral and non-viral vectors.4 Compared with non-viral vectors, viral vectors exhibit low target-specificity, are of high cost and exhibit potential biosafety concerns. Non-viral vectors can be sub-divided into organic (e.g., liposomes and hydrogels) and inorganic (e.g., silica, carbon materials, inorganic nanoparticles, and organic polymers) carriers.5,6 However, problems still exist for non-viral vectors. For example, mesoporous silica displays low encapsulation efficiency, and the loaded guest molecules easily leak out. Inorganic nanoparticles can only adsorb biomacromolecules onto their surfaces through non-covalent bonding or covalent bioconjugation, which leads to minimal protection of biomacromolecules from in vivo degradation. In addition, liposomes as a clinically approved delivery system suffer from some drawbacks such as a low loading capacity for biomacromolecules, ease of oxidation and hydrolysis of phospholipids, and a short half-life.7 As a consequence, it is necessary to develop new vector systems for biomacromolecular therapeutics.
Metal–organic frameworks (MOFs) are porous materials formed by the coordination of metal cations/clusters with organic ligands.8,9 With the advancement of supramolecular host–guest chemistry, several structurally well-defined functional MOFs have been synthesized.10 In a mixed solution containing metal ions and organic ligands, biomacromolecules can be encapsulated into the interior space of a MOF material through self-assembly. Owing to the flexibility of coordination chemistry, such biomacromolecular (guests) can easily fit into the cavity of MOFs (hosts), producing structurally stable host–guest ensembles (Biomacromolecules@MOFs). Alternatively, the pores of mesoporous MOFs that have already been synthesized can be used to directly accommodate biomacromolecules. Using these approaches, the activity of biomacromolecules can be retained with enhanced stability under physiological conditions.11 To date, host–guest systems for biomacromolecules based on MOFs are much less explored compared to those based on cyclodextrins, crown ethers and calixarenes.12
The main advantages of MOFs as a carrier for biomacromolecules are as follows.13,14 (1) An extensive variety of MOF-based functional materials with different sizes, shapes and physical/chemical properties can be designed and prepared using available metal ions and organic ligands. (2) MOFs exhibit ultra-high porosity, large specific surface area and adjustable pore size, making them an excellent host matrix for encapsulating biomacromolecules. (3) The active sites that immobilize biomacromolecules are evenly distributed in the pores of MOFs, resulting in reduced drug leakage prior to reaching target tissues/cells. (4) The surface of MOFs is readily modifiable by a diverse range of targeting agents facilitating target-specific drug delivery. (5) MOFs exhibit good biocompatibility and low cytotoxicity and can effectively penetrate the cell membranes of different types of cells including liver cells, T cells, vascular cells, lung cells, germ cells, pancreatic cells, etc.15–21 This is because MOFs exhibit high porosity and large specific surface area, which facilitates enhanced contact with the cell membranes, thereby increasing their uptake by cells. In addition, positively charged MOF materials can bind to cell membranes through electrostatic interactions and thus can enter cells by endocytosis.
In this review, we summarize recent advances in the treatment of human diseases using Biomacromolecules@MOFs (Scheme 1). The host–guest strategies developed for the construction of Biomacromolecules@MOFs are summarized, and the application of MOFs for the controlled delivery of a diverse range of different biomacromolecules for disease therapy is introduced. Finally, we outline the future opportunities available and remaining challenges for Biomacromolecules@MOFs as delivery vectors.
 | ||
| Scheme 1 Biomacromolecules used for the host–guest interaction with metal–organic frameworks (MOFs) (Biomacromolecules@MOFs) and their application for the controlled release of biomacromolecular therapeutics achieving disease therapy via different therapeutic mechanisms. | ||
2. Host–guest strategies for the synthesis of Biomacromolecules@MOFs and their application in disease therapy
Biomacromolecular drugs exhibit high therapeutic efficacy for the treatment of a variety of human diseases including microbial infections, autoimmune diseases, metabolic disorders, neurodegenerative diseases and cancer.22–27 Compared with traditional small-molecule based drugs, they possess certain advantages over small-molecule based drugs in terms of better target specificity, unique therapeutic function, low susceptibility to multidrug resistance, and fewer side effects that can interfere with normal biological processes; in addition, the research and development cycle of biomacromolecular drugs is generally faster than that of small-molecule based therapeutics.28 The biomacromolecules used for disease therapy include DNA/RNA, antigens/antibodies and peptides/polysaccharides. MOFs represent an emerging class of delivery systems with the potential to substantially improve the efficacy of disease therapy.29 By using MOFs as the host molecule, biomacromolecules as the guest molecules can be more efficiently delivered to target tissues/cells and achieve enhanced therapeutic effect. The currently reported host–guest strategies to construct Biomacromolecules@MOFs can be divided into two different paths (Fig. 1).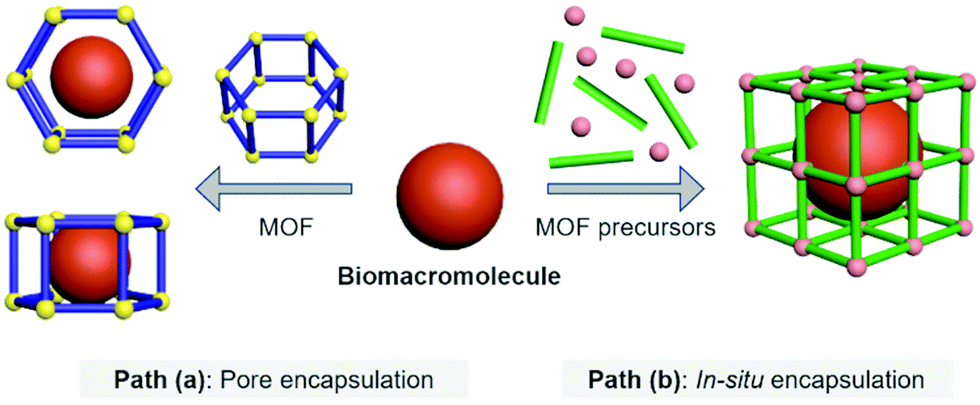 | ||
| Fig. 1 Host–guest strategies to construct Biomacromolecules@MOFs. | ||
Path (a)
Pore encapsulation is a host–guest strategy for encapsulating biomacromolecules using preformed MOFs.30 In recent years, many mesoporous MOFs have been successively used to accommodate biomacromolecular therapeutics based on pore encapsulation.31,32 Using this strategy, the pore shape and size of the MOF based material must match the three-dimensional structure of the biomacromolecules of interest to ensure the successful encapsulation. The driving forces by which biomacromolecules are immobilized into the pores of MOFs include van der Waals forces, hydrophobic/hydrophilic and electrostatic interactions, π–π stacking and hydrogen bonding. Moreover, to achieve pore encapsulation of biomacromolecules, the MOF material used should be soluble and water stable. Although pore encapsulation has been shown to be an effective method for loading biomacromolecules, this strategy has limitations in terms of the loading of large biomacromolecules.Path (b)
In situ encapsulation is an alternative strategy to pore encapsulation, which can accommodate a broader spectrum of biomacromolecules irrespective of the size of biomacromolecules.33,34 The principle of this strategy is that the MOF precursor undergoes a self-assembly process surrounding the biological macromolecules, and then the biomolecules are uniformly encapsulated inside the MOF material to obtain a host–guest supramolecular ensemble. However, only a limited number of MOF based materials have been used for in situ encapsulation of biomacromolecules (e.g., ZIF-8 and ZIF-90). This is because the conditions for synthesizing the MOF must be mild (e.g., low temperature, organic solvents) to ensure that the bioactivity of biomacromolecules is not compromised during the encapsulation process.35–38 The above-mentioned conditions hamper the use of a significant number of available MOFs. Furthermore, since in situ generation results in the complete encapsulation of the biomacromolecules within the MOFs, the loaded biomacromolecules can only be released when the MOFs are decomposed. As such, stimuli-responsive MOFs are the ideal choice for the controlled release of biomacromolecules.39,40 When stimulus-responsive MOFs are activated by specific intracellular triggers (e.g., acidic pH, reactive biospecies, ATP, etc.), their host–guest architecture will collapse, thereby releasing the biomacromolecules in a controlled manner.2.1 The host–guest materials of Proteins@MOFs
Therapeutic proteins have unique advantages in disease therapy.41 Protein-based drugs can directly induce cancer cell apoptosis through well-defined signaling pathways or indirectly inhibit tumor growth by regulating the tumor microenvironment or by stimulating an autoimmune response. In addition, the use of protein therapy does not modify the genetic information. Table 1 presents representative examples of currently established Proteins@MOFs for human disease therapy.42–53| Host | Guest | Size of composite particle | Release mechanism | Targeted cell lines | Type of disease | Ref. |
|---|---|---|---|---|---|---|
| a n. a. = not available. | ||||||
| ZIF-8 | GOx, HRP | ∼72 nm | pH | HeLa/U14 cells | Cervical cancer | 42 |
| NMIL-100 | GOx | ∼203 nm | GSH | 4T1 cells | Breast cancer | 43 |
| ZIF-90 | RNase A-NBC, CRISPR/Cas9 | 429 ± 27 nm, 583 ± 18 nm | ATP | HeLa cells | Cervical cancer | 44 |
| ZIF-8 | RNase A | ∼425 nm | pH | A549 cells | Lung cancer | 45 |
| Fe (SS) MOF | GOx | ∼190 nm | GSH | 4T1 cells | Breast cancer | 46 |
| ZIF-8 | GOx, CPO | ∼150 nm | pH | 4T1 cells | Breast cancer | 47 |
| ZIF-8 | Klenow (exo-)fragment polymerase | 162 ± 25 nm | pH | HeLa cells, MCF-7 cells | Cancer | 48 |
| UiO-AM | OVA | ∼437 nm | n. a. | L929 cells | n. a. | 49 |
| ZIF-8 | Cas9/sgRNA | ∼100 nm | pH | CHO cells | n. a. | 50 |
| NU-1000, PCN-222 | Insulin | ∼180 nm, ∼210 nm | Phosphate | SKOV-3 cells | n. a. | 51 |
| MIL-100 | Anti-EpCAM antibody | ∼110 nm | pH | MCF-7 cells | Cancer | 52 |
| ZIF-8 | Insulin/GOx | ∼500 nm | Glucose | n. a. | Diabetes | 53 |
Enzymes@MOFs materials are mainly prepared by the pore encapsulation strategy. For example, Ma et al. immobilized microperoxidase-11 (MP-11) into the pores of a mesoporous MOF.57 The selected Tb-TATB materials had pore sizes ranging from 3.0 nm to 4.1 nm, which provided sufficient space to accommodate MP-11 with a size of approximately 3.3 × 1.7 × 1.1 nm3. The host–guest MP-11@Tb-TATB material exhibited an excellent enzymatic activity. In addition, they reported that cytochrome c (Cyt c) could also enter the cavity of mesoporous MOFs, even though the size of the enzyme was larger than that of the MOF pore.58 This suggests that the conformation of the Cyt c enzyme molecule changed after being loaded into the MOFs, which might be caused by the surface contact of the enzyme with the MOF crystal. It was proposed that the tertiary structure of the protein undergoes partial unfolding to pass through the small pores of the nanocage.
Li et al. further developed a series of mesoporous Zr-based MOFs (NU-100x, x = 3, 4, 5, 6, 7) with pore sizes of 3.3 nm to 6.7 nm by precisely controlling the torsion angle associated with the organic ligand (Fig. 2).59 The extended NU-100x MOF structure was used to encapsulate lactate dehydrogenase (LDH), in which the larger channels were used to immobilize LDH, and the smaller channels and cavities facilitated the free diffusion of substrates and coenzymes. As such, the cascade-reaction activity of LDH@Nu-100x was demonstrated to be higher than that of free enzymes.
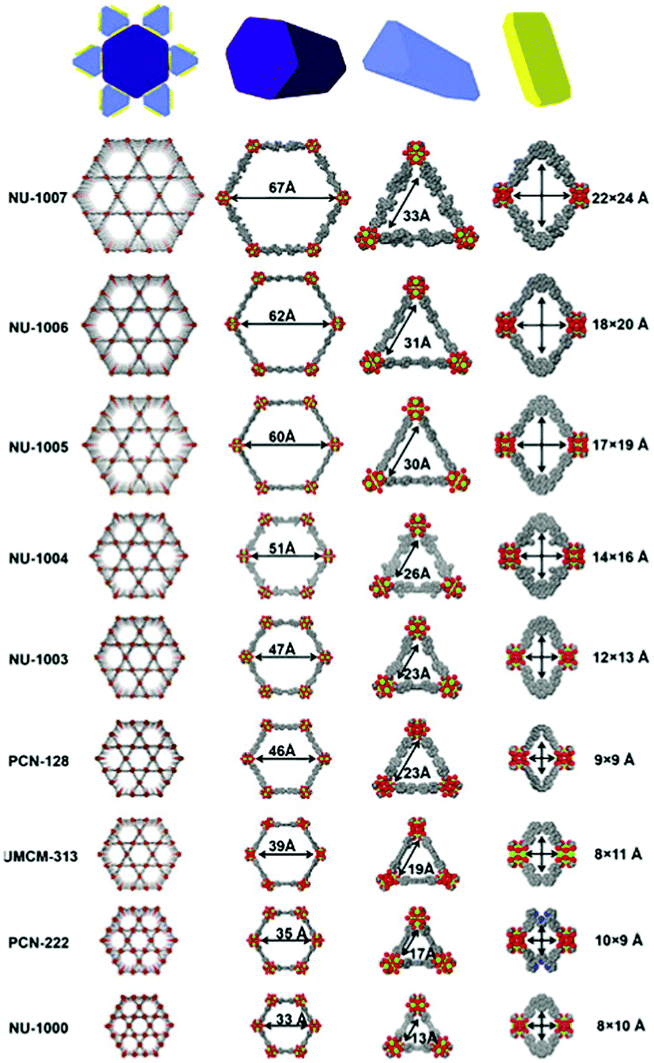 | ||
| Fig. 2 Comparison of the pore size in different zirconium-based MOFs. Reprinted with permission.59 Copyright 2021 Elsevier. | ||
Effective delivery and lysosomal release of enzymes in living cells remains a big challenge for protein therapeutics. As such, Chu et al. reported Caspase-3@MOFs as a universal strategy for the lysosomal delivery of enzymes (Fig. 3).60 Caspase-3, an important enzyme implicated in cell apoptosis,61 and 2-methylimidazole were pre-mixed, followed by slow addition of a Zn2+ solution. Then, polyvinylpyrrolidone (PVP) was added to modify the surface of the resulting caspase 3@ZIF-8, enhancing the stability of the carrier. Cellular experiments indicated that the ZIF-8 could release the encapsulated caspase-3 molecules under acidic conditions, inducing cell apoptosis. In order to further improve the targeted delivery of enzymes, Zhang et al. designed lysosomal-responsive nanoparticles (LYS-NPs) loaded with anti-cancer enzymes. With this research, perforin and granzyme B were co-loaded into the interior of ZIF-8 by in situ encapsulation, and then calcium ions were adsorbed onto the surface of ZIF-8 (Ca2+ is known to enhance the efficacy of perforin and granzyme B).62 Finally, a CD63 aptamer was coated onto the surface of the carrier as a targeting agent for the lysosomes of T cells. LYS-NPs were internalized by T cells, releasing the therapeutic agents in a pH-controlled manner within the lysosomes. Subsequently, when the major histocompatibility complex (MHC) of cancer cells activated the T cells, perforin, granzyme B, and Ca2+ were released from the lysosomes to kill the cancer cells. In vivo tests using breast cancer tumor-bearing mice indicated that treatment with T cells preloaded with LYS-NPs significantly suppressed the tumor growth with an inhibition of 70%. In contrast, the inhibition of T cells without the MOF pretreatment was only 27%.
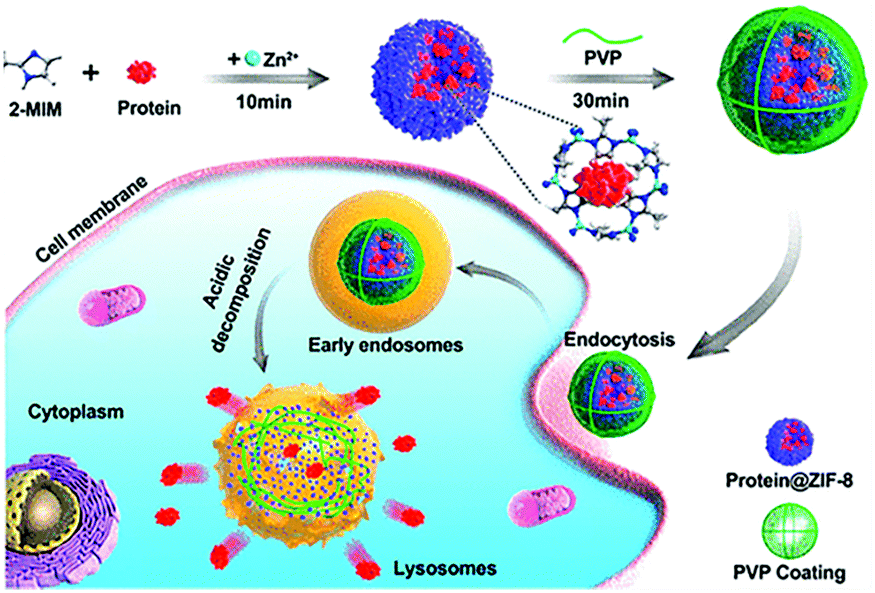 | ||
| Fig. 3 The construction of CRISPR/Cas9@ZIF-8 and its pH-controlled drug delivery in cells. Reprinted with permission.60 Copyright 2018 American Chemical Society. | ||
In recent years, biomimetic nanoparticles based on the surface coating of natural cell membranes have emerged as a promising strategy for disease therapeutics and vaccination.63 By introducing cell membranes to the surface of MOFs, the resulting delivery system can be used for homotypic targeting exhibiting low immunogenicity, which is a key requirement for personalized medicine.64 Zheng et al. designed a biomimetic host–guest nanosystem, in which gelonin (a plant toxin) was loaded into the interior of the ZIF-8 material using in situ encapsulation. Then, the membrane of MDA-MB-231 (human breast adenocarcinoma) cells was coated onto the surface of Gelonin@ZIF-8 to target breast cancer.65 The ZIF-8 exhibited an encapsulation efficiency of ∼94% and a loading efficiency of ∼41% for gelonin. In vitro and in vivo studies indicated that the Gelonin@ZIF-8/MDA-MB-231 cell membrane system protected the therapeutic proteins from protease digestion, evaded immune clearance, and promoted gelonin uptake by tumor cells. The internalized gelonin induced cell apoptosis by hydrolysis of rRNAs to disrupt protein synthesis. With the help of the biomimetic host–guest nanosystem, the therapeutic effect was increased 11-fold when compared to gelonin alone.
Similarly, Khashab et al. used the Michigan Cancer Foundation-7 (MCF-7) cell membrane of human breast cancer to functionalize a MOF delivery system (Fig. 4).66 A CRISPR/Cas9 genome editing system was loaded into the interior of ZIF-8 using in situ encapsulation to obtain the CRISPR/Cas9@ZIF-8 material (CC-ZIF). Then CC-ZIF was coated with the MCF-7 cell membrane resulting in a particle size of ∼150 nm. Amongst different cancer cells including MCF-7, the human cervical cancer Henrietta Lacks (HeLa) cell line, Human Dermal Fibroblast (HDFn) cells and activated Jurkat cells (ATC), the C3-ZIFMCF materials exhibited the highest uptake efficiency in MCF-7 cells. Subsequently, the authors demonstrated that the C3-ZIFMCF system significantly suppressed a model transfected gene (enhanced green fluorescent protein) exhibiting a 3-fold improved efficiency when compared with C3-ZIFHeLa. This biomimetic method enabled the specific targeting of cancer cells which should facilitate the clinical transformation of such gene-editing technology in the future.
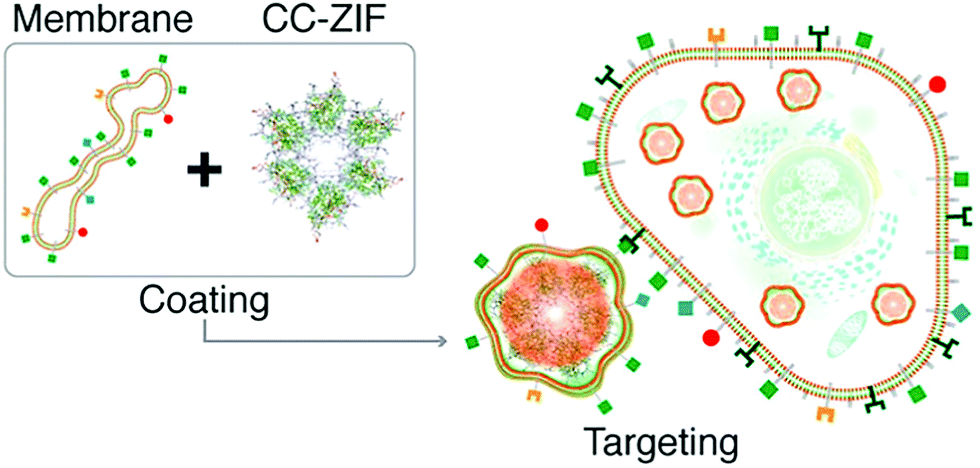 | ||
| Fig. 4 The preparation of C3-ZIF and its targeted entry into cells. Reprinted with permission.66 Copyright 2020 American Chemical Society. | ||
In addition to enzymes that directly kill cancer cells, those that play a complementary role with therapeutic agents have been used for MOF-based delivery systems.67,68 Tyrosinase (TYR) can activate the prodrug paracetamol in cancer cells. This process is accompanied by the production of reactive oxygen species (ROS) and the consumption of glutathione, while the enzymatically cleaved product (4-acetamido-o-benzoquinone) has a good therapeutic effect on drug-resistant cancer cells. Zhou et al. chose PCN-333(Al) as a carrier for TYR, which has the advantage of good enzyme loading capacity, simplicity of modification with fluorophores, and good chemical stability under a cellular environment.69 In addition, the MOF has two mesoporous cavities with diameters of 4.2 and 5.5 nm, respectively (Fig. 5). The host–guest materials TYR@PPCN-333 (Al) were obtained by encapsulating TYR within the larger mesopores (5.5 nm) of PPCN-333 (Al). The combined use of TYR@PCN-333 and paracetamol was more efficient at inducing cancer cell apoptosis and tumor ablation in vivo than using just paracetamol.
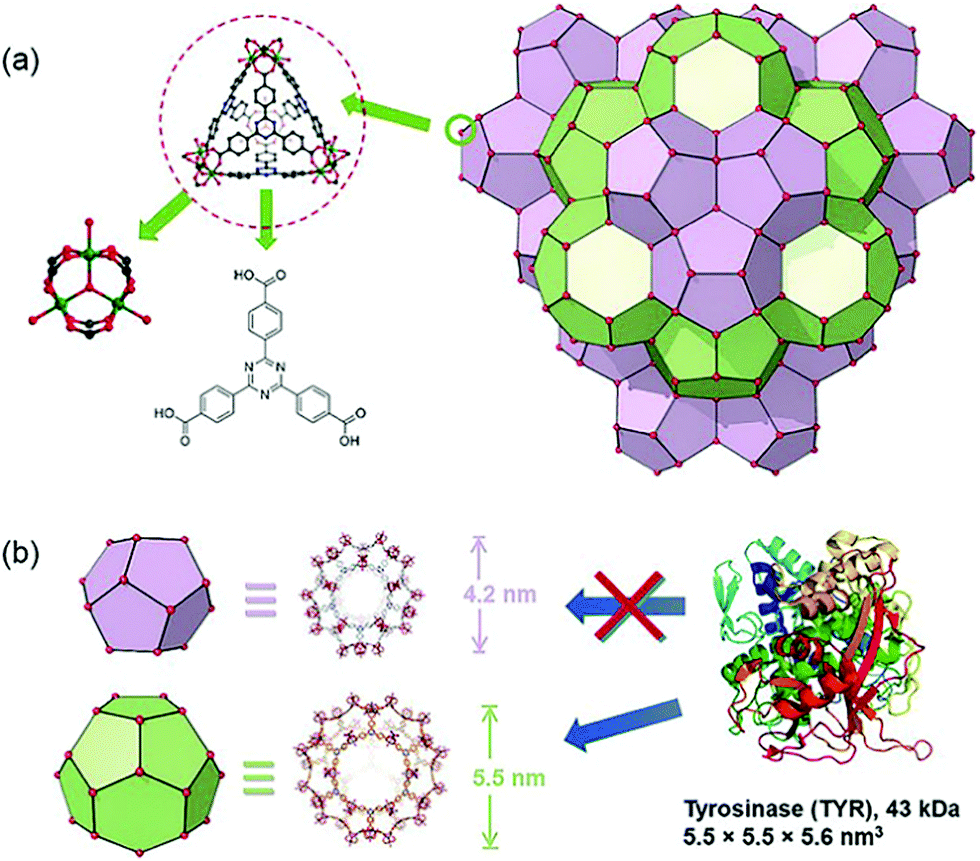 | ||
| Fig. 5 The structure of NPCN-333 with two kinds of mesoporous cavities. Reprinted with permission.69 Copyright 2018 John Wiley and sons. | ||
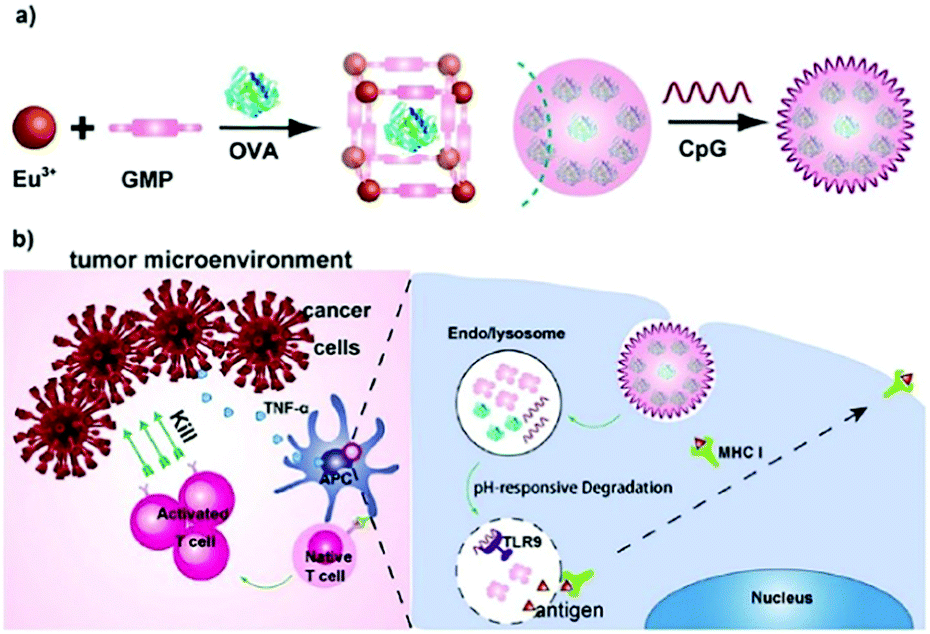 | ||
| Fig. 6 The construction of the OVA@Eu MOF/CpG material and its anti-tumor mechanism. Reprinted with permission.73 Copyright 2021 Elsevier. | ||
In immunotherapy, aluminum salts are the most common adjuvants because of good biocompatibility and the ability to stimulate immune responses to various antigens.74 Sung et al. developed an aluminum-based metal–organic framework (Al-MOF) as an antigen-delivery carrier as well as an adjuvant to effectively induce a durable immune response.75 The synthetic strategy was to use aluminum isopropoxide as the metal source, 2-aminoterephthalic acid as the organic ligand and the OVA antigen as the guest molecule, producing the OVA@Al-MOFs under mild ultrasonic conditions. The encapsulation efficiency of the OVA antigen was measured to be ∼95%, and the particle size of OVA@Al-MOFs was ∼70 nm. In addition, to overcome the mucosal barrier of oral antigens, OVA@Al-MOFs (positively charged) were loaded into yeast-derived capsules (negatively charged) via electrostatic interactions to obtain OVA@Al-MOFs/YCs composites. In vivo experiments indicated that the OVA@Al-MOFs/YCs delivery system targeted intestinal M cells and were subsequently endocytosed by macrophages to accumulate in mesenteric lymph nodes. In the presence of phosphate ions, Al-MOFs slowly released OVA antigens intracellularly, which induced the continuous production of high levels of mucosal S-IgA and serum IgG antibodies, realizing long-lasting immunotherapy in vivo.
Tumors can evade immune destruction and block antigen-activated immune responses through endogenous “immune checkpoints”,76 such as the immunosuppressive receptors expressed on T cells.77 As such, the immune response can be restored using immune checkpoint inhibitors (e.g., anti-PD-L1 and anti-PD-1 antibodies).78Antibodies@MOFs based on the host–guest strategy can protect antibodies from being degraded under various environmental conditions. Chen et al. used two zeolitic imidazolate frameworks (ZIF-8 and ZIF-90) as carriers for the immobilization of human immunoglobulin G (IgG) polyclonal antibodies (H-IgG) and adalimumab (Ada).79 They demonstrated that the encapsulated antibodies exhibited good thermal, chemical and mechanical stability. This study instigated the concept of protecting antibody-based drugs using host–guest chemistry.
Nivolumab (NV) is a monoclonal antibody-based checkpoint inhibitor approved by the U.S. Food and Drug Administration in 2014.80 Alsaiari et al. proposed the use of a biomimetic ZIF-8 material as a biocompatible carrier for delivery of NV (Fig. 7).81 NV was encapsulated within ZIF-8 by self-assembly to obtain NV@ZIF-8. Then, the cell membrane of MCF-7 cells was coated onto the surface of the NV@ZIF-8 to improve its tumor targeting ability. The average particle size of NV@ZIF-8/CC was determined to be ∼166 nm and the crystal shape was octahedral. Using in vivo experiments, compared with control materials, NV@ZIF-8/CC significantly improved the activation of CD8+ T cells in hematological malignancies, resulting in higher levels of interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) being generated. The lifespan of mice was also significantly prolonged. NV@ZIF-8/CC can specifically recognize tumors, reduce off-target delivery of therapeutic agents reducing side effects associated with immunotherapy, improve the sensitivity of the tumor microenvironment to NV, prolong the residence time of NV-ZIF in the tumor, and induce tumor-specific immunity.
 | ||
| Fig. 7 MOF targeted delivery of NV to activate T cells in a slightly acidic tumor microenvironment. Reprinted with permission.81 Copyright 2021 American Association for the Advancement of Science. | ||
The most used medicine for treating diabetes is insulin, and the effective delivery of insulin in vivo is key to controlling the patient's blood sugar levels.87 Farha et al. synthesized a zirconium-based porous MOF material denoted as NU-1000 for the encapsulation of insulin with a 40 wt% loading efficiency.88 The MOF “capsule” protected insulin from being degraded by gastric acid and digestive enzymes, and enabled the controlled release of insulin into the bloodstream (Fig. 8). The coordination bonds formed between the zirconium cluster and carboxylic acid-based ligands facilitated the stability of the MOF material at an environmental pH of 1.5–3.5. Since the size of folded insulin was about 13 Å × 34 Å, it could be encapsulated by the 34 Å pore of NU-1000. Once the insulin@NU-1000 nanocapsules entered blood circulation, phosphate ions triggered a slow decomposition of NU-1000, thereby enabling the buffered release of insulin. Zhao et al. constructed a glucose-responsive MOF composite as a nano-delivery system for insulin.89 The release rate of the system was dependent on the concentration of glucose. The host–guest material insulin-GOx@ZIF-8 was prepared by self-assembly. When the blood glucose concentration was high, glucose molecules could enter ZIF-8 and be oxidized by GOx, producing gluconic acid and H2O2. The ensuing decrease in microenvironmental pH resulted in decomposition of the MOFs and triggered the release of insulin.
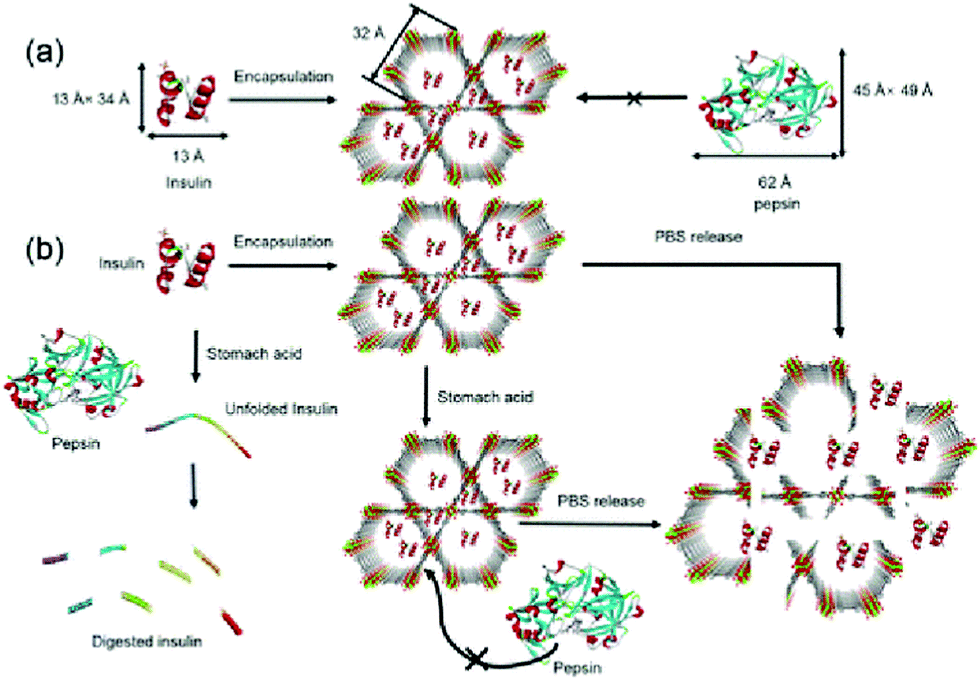 | ||
| Fig. 8 The pore encapsulation principle and release mechanism of Insulin@NU-1000. Reprinted with permission.88 Copyright 2018 American Chemical Society. | ||
Free hemoglobin (Hb), as a “carrier” of oxygen, exhibits poor stability and short blood circulation. For this reason, Yu et al. used ZIF-8 as a hard shell to protect Hb, which significantly enhanced its stability under alkaline, oxidative, high-temperature or enzymatic conditions.90 The Hb@ZIF-8 material with a particle size of 180 nm and a zeta potential of −2.1 mV prolonged the blood circulation time of Hb and reduced the non-specific distribution in organs. Hb@ZIF-8 was demonstrated to significantly increase the survival time of mice with hemorrhagic shock disease. This study provided an oxygen-carrying platform with good stability and extended circulation time.
2.2 Therapeutic Nucleic acid@MOFs
The development of DNAs and RNAs as therapeutic agents has attracted much interest in the field of disease therapy.91 Currently, small-interfering RNA (siRNA) and plasmid DNA (pDNA) are the most widely investigated therapeutic agents. However, it has been difficult to effectively deliver these genetic materials to target tissues. These negatively charged biomacromolecules also have difficulty in passing through the cell membrane, which is also negatively charged. In gene therapy, more than 70% of delivery systems are based on viral vectors, and only 11% are non-viral vectors.92,93 Due to their high loading capacity, ability for controlled drug release and good biodegradability, MOFs have been developed for enhanced delivery of DNAs and RNAs. Table 2 lists the representative and recently developed Nucleic acid@MOFs materials.94–103| Host | Guest | Size of composite particle | Release mechanism | Targeted cell lines | Type of disease | Ref. |
|---|---|---|---|---|---|---|
| a n. a. = not available. | ||||||
| MIL-101 | siRNA | 170 ± 10 nm | Phosphate | MCF-7/T cells | Breast cancer | 94 |
| Zn-NMOF | LNA-antisense miR-224 | ∼200 nm | pH | HCT116 cells | Colon cancer | 95 |
| MIL-100, MIL-101_NH2 | siRNA | ∼390 nm, ∼277 nm | Phosphate | Sw480 cell | Colorectal cancer | 96 |
| ZIF-8 | DNAzymes | ∼140 nm | pH | T cells, Huh-7 cells | Liver cancer | 97 |
| ZIF-8 | Fe-DNA | ∼132 nm | pH | 4T1 cells | Breast cancer | 98 |
| ZIF-8 | siRNA | ∼475 nm | pH | HUVEC cells | Lymphoma | 99 |
| MAF-7 | DNAzymes | ∼150 nm | ATP | K562/D cells | Leukemia | 100 |
| MIL-100 (Fe) | pDNA | 795 ± 26 nm | Phosphate | H1299 cells | Lung cancer | 101 |
| ZIF-C | siRNAs or CRISPR/Cas9 plasmid | ∼200 nm | pH | PC-3 cells | Prostate cancer | 102 |
| ZIF-8 | pDNA | ∼275 nm | pH | MCF-7 cells | n. a. | 103 |
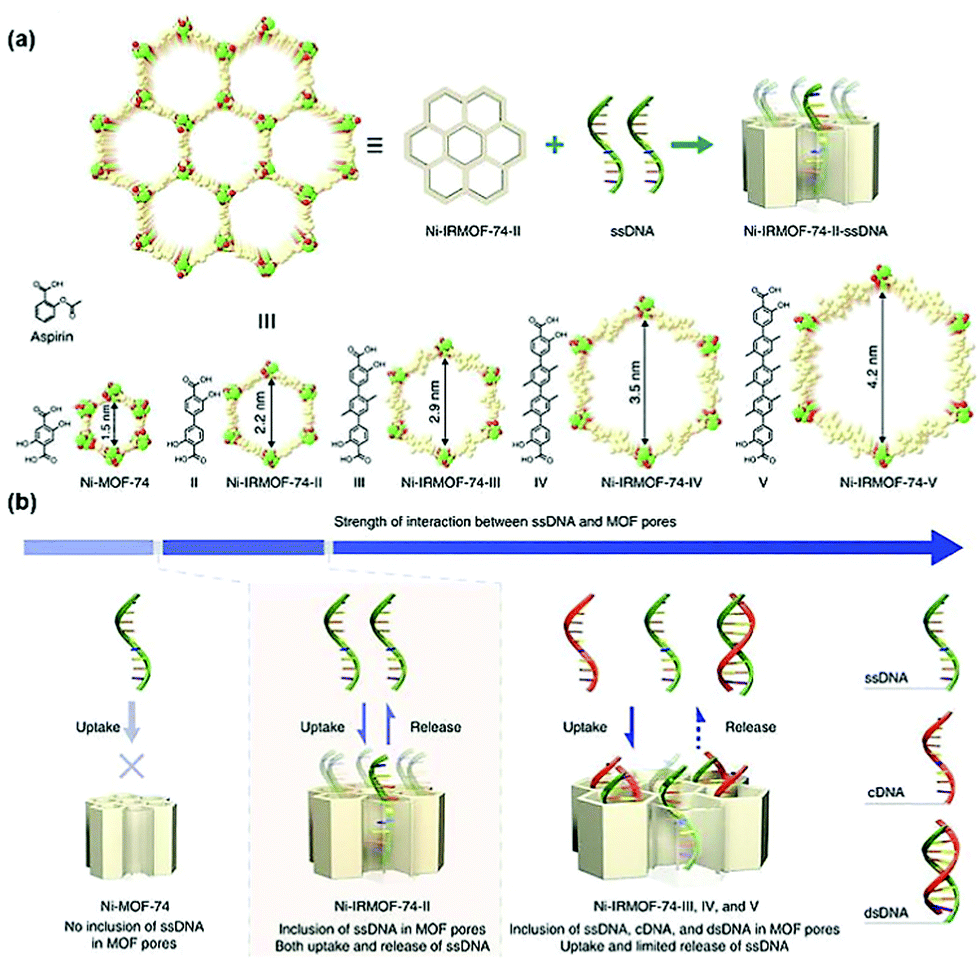 | ||
| Fig. 9 Precise control of the pore size of the MOF and the encapsulation and release mechanism of ssDNA. Reprinted with permission.104 Copyright 2018 Springer Nature. | ||
In addition to pore encapsulation, the in situ encapsulation strategy was also used to construct DNAs@MOFs. Single-stranded DNAzymes are effective for gene silencing. However, due to the lack of DNAzyme cofactors such as special ions, the activity of DNAzymes is insufficient in cells.105 To solve this problem, Wang et al. constructed a self-sufficient system by encapsulating a Ce6-modified DNAzyme into ZIF-8 nanoparticles.106 ZIF-8 acted as a pH-responsive nanocarrier as well as a supplier of an adequate amount of DNAzyme cofactors (the Zn2+ ions). The particle size of DNAzyme@ZIF-8 was determined to be 167 nm, and the loading capacity for DNAzyme was 10 wt%. The material spontaneously decomposed to release DNAzymes and Zn2+ ions under acidic conditions, and the released species acted as messenger RNA-targeting agents to activate DNAzyme cofactors required for gene therapy. In addition, the auxiliary photosensitizer Ce6 could be used both as a fluorescence imaging agent and in PDT. Similarly, Qu et al. designed a bimetallic MOF material loaded with DNAzymes to develop a host–guest platform for combined chemotherapy and gene therapy.107 MCF-7 tumor-bearing mice were intravenously injected with DNAzyme@Cu/ZIF-8. Then, the mice were treated with a prodrug combination (5-azidobenzene-1,3-diol and 4-ethynylphenol) and sodium ascorbate. The Cu2+ ions enriched in the tumor were reduced to Cu+ by sodium ascorbate to catalyze the copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction in situ between 5-azidobenzene-1,3-diol and 4-ethynylphenol, producing a resveratrol derivative toxic to cancer cells. Moreover, the released DNAzymes could be activated by Zn2+, which inhibited the proliferation and metastasis of cancer cells.
DNA plasmids are closed-loop DNA with the ability to self-replicate for DNA recombination. Shukla et al. used the in situ encapsulation method to encapsulate DNA plasmids into the interior of ZIF-8.108 The synthesis was achieved by the addition of the DNA plasmid to an aqueous solution of 2-methylimidazole, followed by the slow addition of an aqueous solution of zinc acetate at room temperature. A green fluorescent protein particle (plGFP) was used as a model genetic macromolecule to validate the transfection efficiency of ZIF-8 materials. Cell transfection experiments indicated that human prostate cancer epithelial cells (PC-3) were transfected with plGFP, and the transfection lasted for up to 4 days.
Ovarian cancer is one of the most malignant gynecological cancers with a high mortality rate. Gene therapy has proven to be an effective way to combat ovarian cancer. As such, Wang et al. constructed a new gene delivery system composed of MIL-88A nanoparticles and a microcircular DNA (MC) encoding the anti-CD3/anti-EpCAM bispecific T-cell engager (BiTE).109 The expressed products of MC induced T cell-mediated cytotoxicity to human ovarian cancer cells (SKOV3). The MC@MOF system was shown to have a high gene transfection efficiency in a mouse xenograft model for human peritoneal ovarian cancer. To further improve the transfection efficiency of DNA plasmids, cell-penetrating peptides (CPPs) are good capping systems and provide specific intracellular compartments without macrophage recognition and subsequent phagocytosis, cross the endothelial barrier and enter the cytoplasm of targeted cells.110
Langel et al. modified CPPs on the surface of ZIF-8 by electrostatic adsorption to improve the transfection efficiency of the plasmid (pGL3). HeLa cells were transfected with pGL3@ZIF-8/CPPs and pGL3@ZIF-8, respectively, and the results indicated that ZIF-8/CPPs increased the transfection efficiency of pGL3 by 5-fold.111 Conventional gene therapy relies on the introduction of therapeutically effective DNA fragments into target cells to correct disease-related genes. In contrast, Leong et al. developed a nanoparticle that can clear circulating free DNA (cfDNA) for the treatment of sepsis.112 The cationic metal–organic “nano-trap” (PEI-g-ZIF) was prepared in one pot to encapsulate cfDNA (Fig. 10). The particle size of PEI-g-ZIF was determined to be 100 nm. The PEI-g-ZIF captured cfDNA through electrostatic interactions, thereby suppressing cfDNA-induced activation of toll-like receptor (TLR) and the release of inflammatory cytokines. The biological evaluation indicated that PEI-g-ZIF NPs, as “nano traps”, exhibited good therapeutic efficiency for the clearance of cfDNA. The host–guest material effectively reduced inflammation and reversed the progress of sepsis.
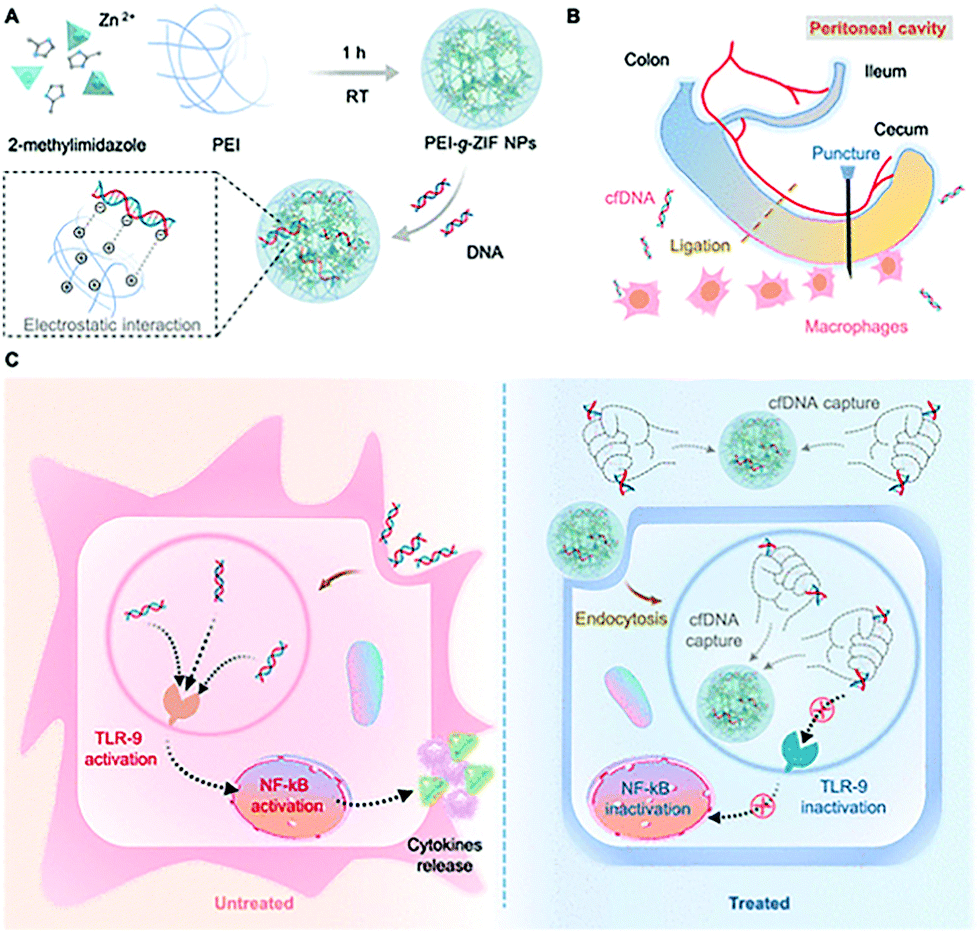 | ||
| Fig. 10 Schematic illustration of the preparation of PEI-g-ZIF and its capture of cfDNA. Reprinted with permission.112 Copyright 2021 American Chemical Society. | ||
siRNAs are short double-stranded RNA molecules that can be designed and synthesized for use in the RNAi pathway. siRNAs have evolved as an important tool for identifying the function of genes and are widely used in the treatment of human diseases.117 In 2014, Lin et al. developed a nanoscale MOF material (siRNA@University of Oslo/Cisplatin [UiO-Cis]) to deliver a combination of MDR gene-silencing siRNAs (Bcl-2, P-glycoprotein, and survivin) to cancer cells. The presence of the UiO-Cis host material facilitated the escape of the siRNAs from endosomes intracellularly to silence MDR genes in drug-resistant ovarian cancer cells. The host–guest composite reduced the expression of the three target proteins by 50%.118
Jimenez et al. used NU-1000 nanoparticles to enhance the delivery of siRNA. NU-1000 has a hexagonal mesoporous channel with a pore size of 3 nm, which is suitable for loading siRNAs.119 Computational simulations indicated that the binding energy of nNU-1000 with siRNA was −878 kJ mol−1. In addition, the siRNA@MOFs host–guest material was combined with various cofactors (proton sponge, KALA peptide and NH4Cl) to diminish endosomal retention and thus increase the gene-knockout efficiency.
Zhang et al. constructed a biomimetic nanoparticle for targeted delivery of siRNA in vivo (Fig. 11).120 First, the siRNA was loaded into the interior of the ZIF-8 through a one-pot method, and then the surface of the material was coated with a naturally derived platelet membrane. In vitro experiments indicated that the P-MOF-siRNA materials coated with platelet membranes had the capacity for tumor targeting, achieving efficient gene silencing of multiple target genes. The host–guest system also exhibited a good therapeutic effect in tumor-bearing breast cancer mice models. Similarly, Zhang et al. used HeLa cell membranes (CMHeLa) to camouflage ZIF-8 for targeted delivery of siRNA inhibiting the Polo-like kinase 1 (PLK1) gene of lung cancer cells.121 PLK1 belongs to a highly conserved protein family, PLKs, which regulate the cell cycle through different signaling pathways including triggering cell mitosis, facilitating spindle assembly and promoting centrosome maturation.122 siRNA@ZIF-8/CMHeLa resulted in a 72% reduction of PlK1 expression in tumor-bearing models. In contrast, siRNA alone exhibited a minimal effect on PLK1 suppression. This MOF biomimetic delivery system exhibited enhanced synergistic effects of homotopic targeting and gene silencing.
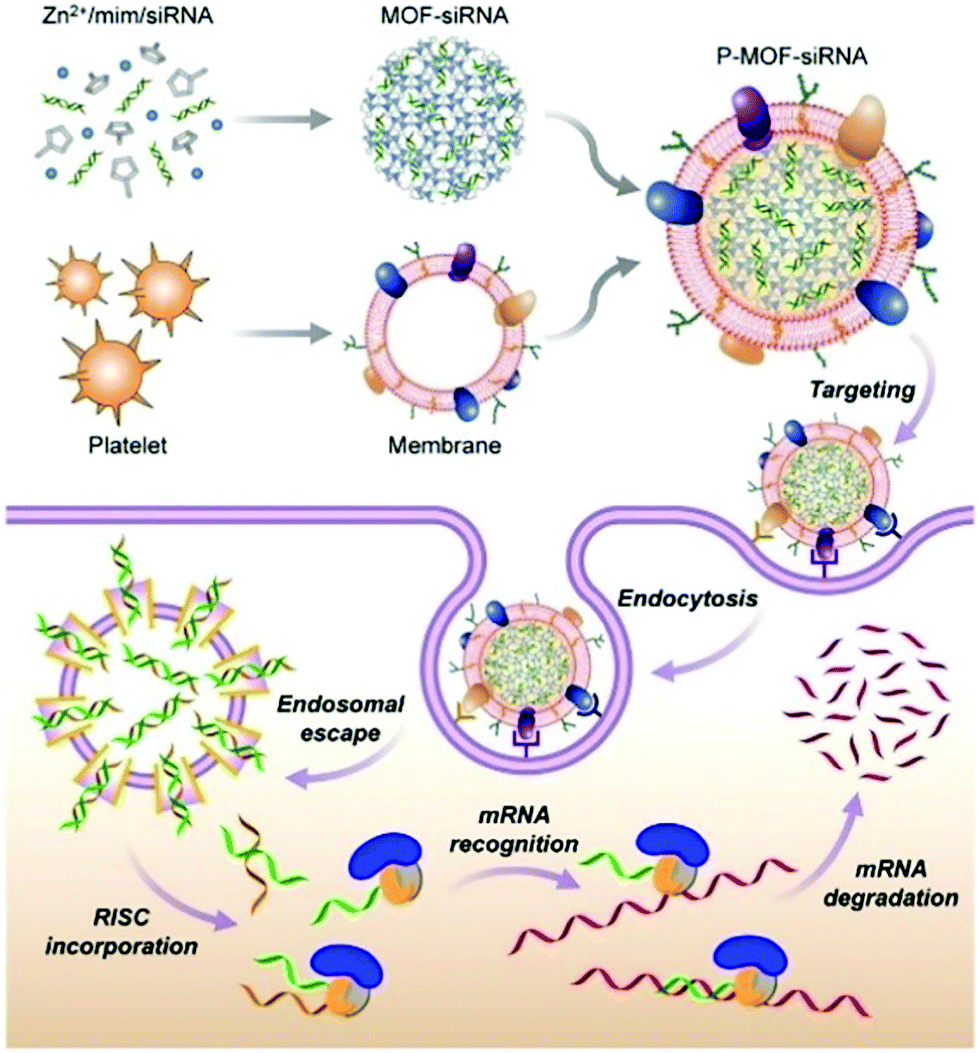 | ||
| Fig. 11 The construction of P-MOF-siRNA and its application in gene silencing. Reprinted with permission.120 Copyright 2020 American Association for the Advancement of Science. | ||
siSOX9 is a siRNA that down-regulates the expression of SOX9, which is an important transcription factor for glial cells. Qu et al. designed a cerium dioxide-doped MIL-100 material as a carrier to deliver siSOX9 and retinoic acid (RA) to neural stem cells (NSC).123 MIL-100 is a H2O2-responsive material that can release siSOX9 and RA in the lesion area, thereby modulating the differentiation of NSC into neurons. Furthermore, cerium dioxide acted as an antioxidant nanoenzyme that protected newborn neurons from oxidative damage in an inflammatory environment, thus enabling a longer survival rate of newborn neurons. The MIL-100 host–guest material systems exhibited excellent drug loading efficiency and promoted the directed differentiation of neural stem cells, improving the cognitive function in a triple-transgenic mouse model of Alzheimer's disease.
2.3 Host–guest materials of other Biomacromolecules@MOFs
In recent years, peptide-based therapeutics have played an important role in the treatment of diseases. Compared to therapeutic proteins, peptides can be more easily produced at a lower cost. However, peptides can also be easily degraded in vivo.124 Liang et al. synthesized a ZIF-8 material to encapsulate a therapeutic peptide (peptide 46), which can upregulate the expression of the tumor suppressor gene p53 in cancer cells.125 The hydrophobic cavities in ZIF-8 effectively eliminated polar solvents to prevent hydrolysis of peptides. The resulting Peptide 46@ZIF-8 significantly inhibited the proliferation of MCF-7 cells. Similarly, Liu et al. used ZIF-8 materials to encapsulate melittin in situ (Fig. 12).126 Melittin (MLT) is an amphiphilic cationic peptide, which can insert into the cell membrane producing transmembrane “holes” that lead to cell apoptosis. When melittin is delivered into cells, it interacts with the membrane of subcellular organelles to activate transcriptional regulation. In a series of transcriptome analyses, it was shown that MLT@ZIF-8 could regulate the expression of 3383 genes. For example, the phosphoinositide 3-kinase/serine-threonine kinase-regulated p53 pathway was activated in MLT@ZIF-8 treated A549 cells. This research represents the first example where the anti-cancer mechanism of MLT@ZIF-8 through transcriptomics analysis was elucidated.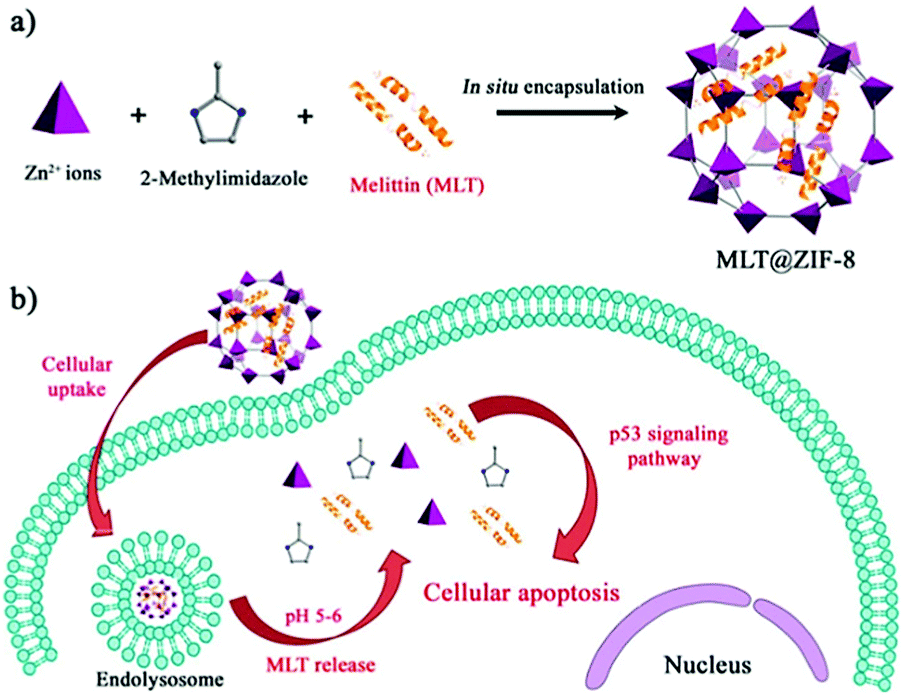 | ||
| Fig. 12 Preparation of MLT@ZIF-8 NPs and its application in anti-cancer therapy. Reprinted with permission.126 Copyright 2018 American Chemical Society. | ||
Osteogenic growth peptide (OGP) has been widely used to improve the proliferation and differentiation of osteoblast-related cells.127 Chen et al. prepared OGP@ZIF-67 nanoparticles using an in situ encapsulation approach and then deposited OGP@ZIF-67 onto the surface of titanium dioxide nanotubes (TNTs) using a cathodic electrophoretic deposition method to obtain TNT-ZIF-67@OGP.128 The composite material enhanced the proliferation and differentiation of mesenchymal stromal cells (MSCs) whilst inhibiting the inflammatory response of M1 macrophages. Using an infected femur animal model, TNT-ZIF-67@OGP materials exhibited excellent anti-inflammatory effects at the early stage of implantation and enhanced osseointegration of the bone-implant at a later stage. A comparison of the osseointegration capacities of TNT and TNT-ZIF-67@OGP in vivo indicated that the new-bone formation capacity around TNT-ZIF-67@OGP was 2-fold higher than that of TNT.
Positively charged peptides, such as the dodechistidine peptide (H12), can enhance the cellular uptake of MOF materials and/or facilitate their transport across cell membranes. Tang et al. prepared a 5-carboxyfluorescein (FAM)-labeled H12 (fH12)@ZIF-8 material as an efficient pH-responsive delivery system.129 Human hepatocellular carcinoma cells (HepG2) were incubated with 5-FAM/ZIF-8 (without the peptide) or fH12/ZIF-8 for 2 hours. The fluorescence of fH12/ZIF-8 was determined to be much stronger than that of 5-FAM/ZIF-8, suggesting that the cellular uptake of ZIF-8 was enhanced by doping with H12.
Polysaccharides play important functional roles in the biological system, and polysaccharide-based therapeutic agents have been increasingly developed.130 In order to achieve effective encapsulation and controllable release of polysaccharides, Falcaro et al. proposed the use of ZIF-8 encapsulated polysaccharides in situ.131 The authors demonstrated that an increase in the number of carboxyl groups in the polysaccharide structure led to a more effective coordination with metal cations, thus facilitating the self-assembly process of the host–guest materials. Hyaluronic acid (HA) is a therapeutic agent used for the treatment of osteoarthritis (OA). HA reduces inflammation in OA patients, protects chondrocytes from free-radical damage, and promotes cartilage regeneration.132 Kai et al. used MIL-100 (Fe) to simultaneously load HA and an anti-inflammatory protocatechuic acid (PCA) to obtain the MIL-100@HA@PCA composite for the treatment of OA.133 MIL-100@HA@PCA released HA and PCA slowly under acidic conditions, thereby reducing synovial inflammation in OA joints. Falcaro et al. used three types of pH-responsive MOF materials (ZIF-8, ZIF-90 and MAF-7) for the in situ encapsulation of a variety of glycosaminoglycans (GAG) for disease treatment (Fig. 13).134
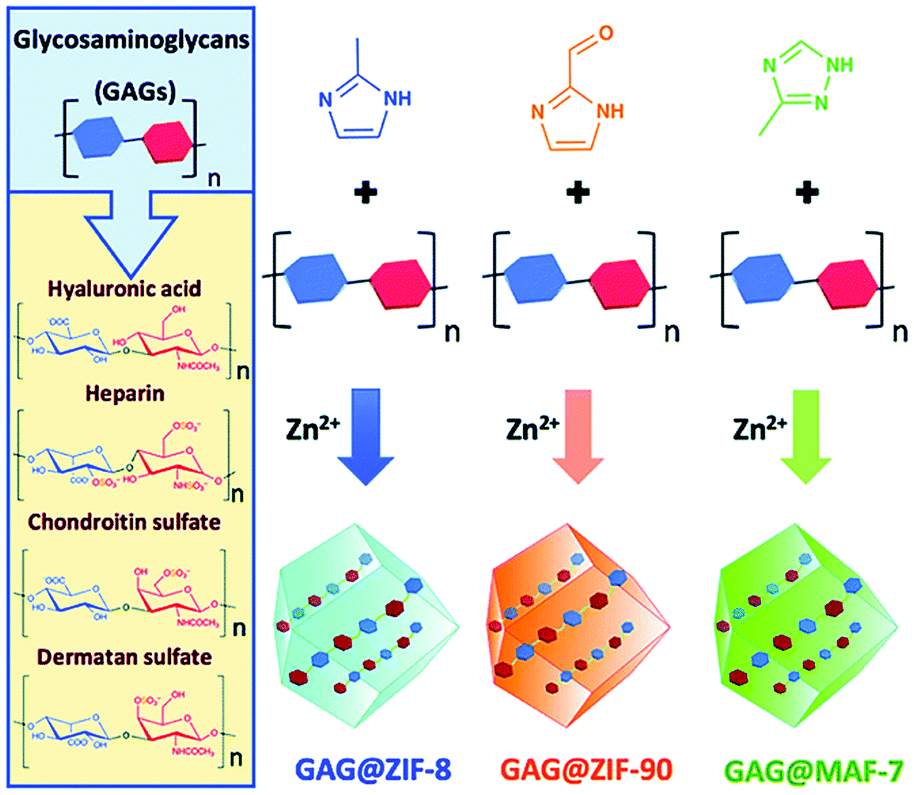 | ||
| Fig. 13 The construction of three types of pH-responsive GAG@MOF materials. Reprinted with permission.134 Copyright 2020 The Royal Society of Chemistry. | ||
The GAG family consists of hyaluronic acids, chondroitin sulfates, dermatan sulfates, keratin sulfates and heparan sulfates. GAGs play an important role in modulating cell development, repair, and replacement. For example, GAGs containing chondroitin sulfates are indispensable for the development of the brain, cartilage, and other tissues, and for maintaining the stability of neuronal synapses. Heparan sulfates are involved in the development of cells and angiogenesis, and in the regulation of blood clotting properties. GAGs are therefore important commercial therapeutics used for the treatment of human diseases including osteoarthritis, thrombosis, inflammation, and wound healing. The above strategy was successful in loading clinically approved GAG therapeutics (heparin, hyaluronic acid, chondroitin sulfate, dermatan sulfate, GM-1111, and HepSYL proteoglycan) into the MOF. Different GAG@MOF materials displayed varied loading efficiencies, guest space distribution and release dynamics. For example, GAG@ZIF-8 or GAG@MAF-7 host–guest materials were the best choices for treating osteomyelitis and infected wounds since they exhibited rapid release. However, GAG@ZIF-90 was the material of choice for controlling the drug release rate and reducing side effects. This research was the first to prepare GAGs@MOFs composites with tunable functions (e.g., encapsulation efficiency, protection mechanism, drug release rate).
The resistance of bacteria to antibiotics continues to increase, and to date it has been difficult to develop targeted vaccines for pathogenic bacteria. The difficulty mainly lies in the choice of suitable bacterial antigens for eliciting immune responses. Gassensmith et al. have used the in situ encapsulation method to load uropathogenic Escherichia coli (UPEC) to ZIF-8. The host–guest assembly was achieved through the weak interaction between Zn2+ and the peptide backbones on the membrane of the bacteria.135 The self-assembly did not change the conformation of proteins and saccharides on the membrane surface. However, the bacteria were deactivated. The protective layer of ZIF-8 was decomposed in vivo, and the re-exposed proteins and oligosaccharides were released to trigger immune responses. The UPEC@ZIF-8 host–guest material significantly increased the concentration of lgG in the serum, induced a strong humoral response, and prolonged the survival time of mice infected with fatal sepsis.
3. Conclusions and outlook
As an emerging class of delivery systems, MOFs have contributed substantially to the enhanced delivery of therapeutic biomacromolecules. Based on structurally well-defined pore networks and the mild host–guest chemistry used in their construction, different types of biomacromolecules (including antigens/antibodies, enzymes, therapeutic proteins, nucleic acids, polypeptides and polysaccharides) are efficiently encapsulated in a variety of nano-scale MOFs and exhibit sufficient retained bioactivity upon reaching the targeted cells. The two most popular host–guest strategies to construct Biomacromolecules@MOFs are the pore encapsulation and in situ encapsulation strategy. The internal/external stimuli in cells that activate the biomacromolecular release from MOFs include pH, ions, competitive binding agents, and redox-active species. In addition, by combining therapeutic biomolecules with other therapeutic agents and/or adjuvants, multimodal disease therapy has been achieved in vivo.Although significant progress has been made in the development of Biomacromolecules@MOFs host–guest delivery systems for disease treatment, several challenges remain that hamper the transformation of this emerging technique into clinical practice.
(1) Potential side effects and biosafety of the MOF delivery system should be systematically studied. These are important factors that determine whether MOFs can enter clinical trials. In the process of constructing MOFs, low-toxicity organic ligands and metal ions should be selected, and the tolerance of these materials by the human body should be considered. How each MOF is degraded and distributed in the human body should be carefully assessed by standard pharmacological and pharmacokinetic assays.
(2) Currently, only a limited number of MOF materials have been used for enhanced delivery of biomacromolecules. Structurally more diverse MOF systems for biomacromolecules with exquisitely tunable porous structure, chemical structure and morphology need to be developed. In particular, the size of Biomacromolecules@MOFs needs to be finely controlled to achieve good internal circulation in vivo. Another question is how to ensure the structural uniformity and repeatability of MOF materials when they are synthesized in bulk. In addition, to overcome the relatively harsh synthetic conditions of MOFs materials (e.g., high temperature, high pressure and the use of organic solvents), recently, a novel and generalizable approach to the encapsulation of small-molecule drugs in MOFs, known as the mechanochemical “SMART” strategy, has been developed.136–138 This method just requires a single mechanochemical process for drug loading, minimizing the harsh conditions required for the traditional solvothermal MOFs syntheses. We envision that the mechanochemical strategy can be used for Biomacromolecules@MOFs to help retain the structural and functional integrity of biomacromolecular therapeutics of interest.
(3) The mechanisms by which biomacromolecules bind MOFs remain largely unexplained. In-depth mechanistic studies can offer an important theoretical basis for the efficiency-enhanced loading and release of therapeutic biomacromolecules.
(4) The targeting specificity of MOF materials still needs to be improved using the surface functionalization of MOFs with appropriate targeting molecules, thereby improving the accumulation of the therapeutic agents at the targeted cells (reducing off-targeting).
(5) Due to the complexity and diversity of most human diseases, it is necessary to develop multifunctional Biomacromolecules@MOFs to achieve multi-target and multi-modal treatment of diseases. Significantly, the therapeutic results obtained in animal-based disease models may differ from those in humans. To enable clinical practice, several pre-clinical evaluations are required.
In conclusion, we anticipate that, once the problems outlined above have been addressed, Biomacromolecules@MOFs delivery systems will become one of the most promising materials for biomedical applications. Although big challenges remain, the rapid development of MOFs due to advancements in chemistry, materials science, and biotechnology will result in Biomacromolecules@MOFs suitable for the clinical translation of MOF material-based therapeutics in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 21788102, 91853201), the Shanghai Municipal Science and Technology Major Project (2018SHZDZX03) and the international cooperation program of Shanghai Science and Technology Committee (17520750100). T. D. J. wishes to thank the Royal Society for a Wolfson Research Merit Award and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University for support (2020ZD01).Notes and references
- H. J. Kong and D. J. Mooney, Nat. Rev. Drug Discovery, 2007, 6, 455–463 CrossRef CAS
.
- X. Liu, F. Wu, Y. Ji and L. Yin, Bioconjugate Chem., 2018, 30, 305–324 CrossRef
- S. Iqbal, M. Blenner, A. Alexander-Bryant and J. Larsen, Biomacromolecules, 2020, 21, 1327–1350 CrossRef CAS
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS
- A. Gigante, M. Li, S. Junghänel, C. Hirschhäuser, S. Knauer and C. Schmuck, MedChemComm, 2019, 10, 1692–1718 RSC
- A. Pachioni-Vasconcelos Jde, A. M. Lopes, A. C. Apolinário, J. K. Valenzuela-Oses, J. K. Valenzuela-Oses, J. S. Costa, O. Nascimento Lde, A. Pessoa, L. R. Barbosa and O. Rangel-Yagui Cde, Biomater. Sci., 2016, 4, 205–218 RSC
- S. Deodhar and A. K. Dash, Ther. Delivery, 2018, 9, 857–872 CrossRef CAS
- Q. L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC
- S. Wang, C. M. McGuirk, A. d'Aquino, J. A. Mason and C. A. Mirkin, Adv. Mater., 2018, 30, 1800202 CrossRef
- H. Cai, Y. L. Huang and D. Li, Coord. Chem. Rev., 2019, 378, 207–221 CrossRef CAS
- H. Sun, Y. Li, S. Yu and J. Liu, Nano Today, 2020, 35, 100985 CrossRef CAS
- J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC
- C. Y. Sun, C. Qin, X. L. Wang and Z. M. Su, Expert Opin. Drug Delivery, 2013, 10, 89–101 CrossRef CAS
- M. P. Auuçafy, B. L. da Silva, J. A. Oshiro-Junior, E. B. Manaia, B. G. Chiari-Andréo, R. A. M. Armando, R. C. G. Frem and L. A. Chiavacci, Curr. Pharm. Des., 2020, 26, 4174–4184 CrossRef
- M. X. Wu and Y. W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed
- D. Liu, B. Bai, Y. Sun and Y. Guo, Mater. Express, 2020, 10, 1197–1203 CrossRef CAS
- H. Zhang, J. Zhang, Q. Li, A. Song, H. Tian, J. Wang, Z. Li and Y. Luan, Biomaterials, 2020, 245, 119983 CrossRef CAS PubMed
- D. E. Al-Ansari, N. A. Mohamed, I. Marei, A. Zekri, Y. Kameno, R. P. Davies, P. D. Lickiss, M. M. Rahman and H. Abou-Saleh, Nanomaterials, 2020, 10, 1028 CrossRef CAS
- B. M. Jarai, Z. Stillman, L. Attia, G. E. Decker, E. D. Bloch and C. A. Fromen, ACS Appl. Mater. Interfaces, 2020, 12, 38989–39004 CrossRef CAS PubMed
- S. F. Li, H. Guo, Y. Huang, C. M. Li, Y. Liu and J. Han, J. Polym. Res., 2020, 27, 1–11 CrossRef
- Y. Sakamaki, J. Ozdemir, Z. Heidrick, A. Azzun, O. Watson, M. Tsuji, C. Salmon, A. Sinha, J. Batta-Mpouma, Z. McConnell, D. Fugitt, Y. Du, J. W. Kim and H. Beyzavi, ACS Appl. Bio Mater., 2021, 4, 1432–1440 CrossRef CAS PubMed
- B. Silver, K. Ramaiya, S. B. Andrew, O. Fredrick, S. Bajaj, S. Kalra, B. M. Charlotte, K. Claudine and A. Makhoba, Diabetes Ther., 2018, 9, 449–492 CrossRef CAS
- M. B. Parmar, R. B. Kc, R. Lobenberg and H. Uludağ, Biomacromolecules, 2018, 19, 4193–4206 CrossRef CAS PubMed
- X. Li, W. Yang, Y. Zou, F. Meng, C. Deng and Z. Zhong, J. Controlled Release, 2015, 220, 704–714 CrossRef CAS
- T. Wan and Y. Ping, Adv. Drug Delivery Rev., 2021, 168, 196–216 CrossRef CAS PubMed
- P. Yang, C. Lu, W. Qin, M. Chen, G. Quan, H. Liu, L. Wang, X. Bai, X. Pan and C. Wu, Acta Biomater., 2020, 104, 147–157 CrossRef CAS PubMed
- H. He, Q. Liang, M. C. Shin, K. Lee, J. Gong, J. Ye, Q. Liu, J. Wang and V. Yang, Front. Chem. Sci. Eng., 2013, 7, 496–507 CrossRef CAS
- B. Leader, Q. J. Baca, D. E. Golan and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed
- C. Chu, M. Su, J. Zhu, D. Li, H. Cheng, X. Chen and G. Liu, Theranostics, 2019, 9, 3134 CrossRef CAS PubMed
- X. Lian, A. Erazo-Oliveras, J. P. Pellois and H. C. Zhou, Nat. Commun., 2017, 8, 1–10 CrossRef CAS
- H. Y. Guan, R. J. LeBlanc, S. Y. Xie and Y. Yue, Coord. Chem. Rev., 2018, 369, 76–90 CrossRef CAS
- D. Liu, D. Zou, H. Zhu and J. Zhang, Small, 2018, 14, 1801454 CrossRef
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 1–8 Search PubMed
- G. Chen, S. Huang, X. Kou, F. Zhu and G. Ouyang, Angew. Chem., 2020, 132, 14051–14058 CrossRef
- F. Carraro, J. D. Williams, M. Linares-Moreau, C. Parise, W. Liang, H. Amenitsch, C. Doonan, C. O. Kappe and P. Falcaro, Angew. Chem., 2020, 132, 8200–8204 CrossRef
- Y. Pan, Y. Liu, G. Zeng, L. Zhao and Z. Lai, Chem. Commun., 2011, 47, 2071–2073 RSC
- F. K. Shieh, S. C. Wang, S. Y. Leo and K. W. Wu, Chem. – Eur. J., 2013, 19, 11139–11142 CrossRef CAS PubMed
- C. G. Jones, V. Stavila, M. A. Conroy, P. Feng, B. V. Slaughter, C. E. Ashley and M. D. Allendorf, ACS Appl. Mater. Interfaces, 2016, 8, 7623–7630 CrossRef CAS PubMed
- M. Wei, Y. Wan and X. Zhang, J. Compos. Sci., 2021, 5, 101 CrossRef CAS
- H. D. Lawson, S. P. Walton and C. Chan, ACS Appl. Mater. Interfaces, 2021, 13, 7004–7020 CrossRef CAS
- C. Wang, G. Sudlow, Z. Wang, S. Cao, Q. Jiang, A. Neiner, J. J. Morrissey, E. D. Kharasch, S. Achilefu and S. Singamaneni, Adv. Healthcare Mater., 2018, 7, 1800950 CrossRef PubMed
- J. Bai, C. Peng, L. Guo and M. Zhou, ACS Biomater. Sci. Eng., 2019, 5, 6207–6215 CrossRef CAS
- X. Wan, L. Song, W. Pan, H. Zhong, N. Li and B. Tang, ACS Nano, 2020, 14, 11017–11028 CrossRef CAS PubMed
- X. Yang, Q. Tang, Y. Jiang, M. Zhang, M. Wang and L. Mao, J. Am. Chem. Soc., 2019, 141, 3782–3786 CrossRef CAS
- J. Jia, S. Zhang, K. Wen and Q. Li, Int. J. Nanomed., 2019, 14, 9971 CrossRef CAS
- J. Yang, S. Ma, R. Xu, Y. Wei, J. Zhang, T. Zou, Z. Wang, H. Deng, N. Yang and Q. Shen, J. Controlled Release, 2021, 334, 21–33 CrossRef CAS PubMed
- C. Zhang, L. Zhang, W. Wu, F. Gao, R. Q. Li, W. Song, Z. N. Zhuang, C. J. Liu and X. Z. Zhang, Adv. Mater., 2019, 31, 1901179 CrossRef
- J. Zhang, M. He, C. Nie, M. He, Q. Pan, C. Liu, Y. Hu, T. Chen and X. Chu, Chem. Sci., 2020, 11, 7092–7101 RSC
- Y. Qi, L. Wang, H. Guo, Y. Pan, Z. Xie, N. Jin and Y. Huang, Biomater. Sci., 2019, 7, 4022–4026 RSC
- S. K. Alsaiari, S. Patil, M. Alyami, K. Alamoudi, F. Aleisa, J. Merzaban, M. Li and N. M. Khashab, J. Am. Chem. Soc., 2018, 140, 143–146 CrossRef CAS
- S. Wang, Y. Chen, S. Wang, P. Li, C. A. Mirkin and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 2215–2219 CrossRef CAS PubMed
- W. Xie, T. L. Yin, Y. L. Chen, D. M. Zhu, M. H. Zan, B. Chen, L. W. Ji, L. Chen, S. S. Guo, H. M. Huang, X. Z. Zhao, Y. Wang, Y. Wu and W. Liu, Nanoscale, 2019, 11, 8293–8303 RSC
- X. X. Yang, P. Feng, J. Cao, W. Liu and Y. Tang, ACS Appl. Mater. Interfaces, 2020, 12, 13613–13621 CrossRef CAS
- S. Datta, K. N. Rajnish, C. George Priya Doss, S. Melvin Samuel, E. Selvarajan and H. Zayed, Expert Opin. Biol. Ther., 2020, 20, 1151–1174 CrossRef CAS PubMed
- S. A. Farhadi, E. Bracho-Sanchez, S. L. Freeman, B. G. Keselowsky and G. A. Hudalla, Bioconjugate Chem., 2017, 29, 649–656 CrossRef PubMed
- H. A. D. Lagassé, A. Alexaki, V. L. Simhadri, N. H. Katagiri, W. Jankowski, Z. E. Sauna and C. Kimchi-Sarfaty, F1000Research, 2017, 6, 113 Search PubMed
- V. Lykourinou, Y. Chen, X. S. Wang, L. Meng, T. Hoang, L. J. Ming, R. L. Musselman and S. Ma, J. Am. Chem. Soc., 2011, 133, 10382–10385 CrossRef CAS PubMed
- Y. Chen, V. Lykourinou, C. Vetromile, T. Hoang, L. J. Ming, R. W. Larsen and S. Ma, J. Am. Chem. Soc., 2012, 134, 13188–13191 CrossRef CAS
- P. Li, Q. Chen, T. C. Wang, N. A. Vermeulen, B. L. Mehdi, A. Dohnalkova, N. D. Browning, D. Shen, R. Anderson, D. A. Gómez-Gualdrón, F. M. Cetin, J. Jagiello, A. M. Asiri, J. F. Stoddart and O. K. Farha, Chem, 2018, 4, 1022–1034 CAS
- T. T. Chen, J. T. Yi, Y. Y. Zhao and X. Chu, J. Am. Chem. Soc., 2018, 140, 9912–9920 CrossRef CAS
- J. A. Lopez, O. Susanto, M. R. Jenkins, N. Lukoyanova, V. R. Sutton, R. H. Law, A. Johnston, C. H. Bird, P. I. Bird, J. C. Whisstock, J. A. Trapani, H. R. Saibil and I. Voskoboinik, Blood, 2013, 121, 2659 CrossRef CAS PubMed
- Q. Zhao, Z. Gong, Z. Li, J. Wang, J. Zhang, Z. Zhao, P. Zhang, S. Zheng, R. J. Miron, Q. Yuan and Y. Zhang, Adv. Mater., 2021, 33, 2100616 CrossRef CAS PubMed
- M. Xuan, J. Shao and J. Li, Natl. Sci. Rev., 2019, 6, 551–561 CrossRef CAS
- W. Liu, Q. Yan, C. Xia, X. Wang, A. Kumar, Y. Wang, Y. Liu, Y. Pan and J. Liu, J. Mater. Chem. B, 2021, 9, 4459–4474 RSC
- G. Cheng, W. Li, L. Ha, X. Han, S. Hao, Y. Wan, Z. Wang, F. Dong, X. Zou, Y. Mao and S. Y. Zheng, J. Am. Chem. Soc., 2018, 140, 7282–7291 CrossRef CAS
- M. Z. Alyami, S. K. Alsaiari, Y. Li, S. S. Qutub, F. A. Aleisa, R. Sougrat, J. S. Merzaban and N. M. Khashab, J. Am. Chem. Soc., 2020, 142, 1715–1720 CrossRef CAS
- Y. Nivoix, D. Levêque, R. Herbrecht, J. C. Koffel, L. Beretz and G. Ubeaud-Sequier, Clin. Pharmacokinet., 2008, 47, 779–792 CrossRef CAS
- G. K. Meghwanshi, N. Kaur, S. Verma, N. K. Dabi, A. Vashishtha, P. D. Charan, P. Purohit, H. S. Bhandari, N. Bhojak and R. Kumar, Biotechnol. Appl. Biochem., 2020, 67, 586–601 CrossRef CAS PubMed
- X. Lian, Y. Huang, Y. Zhu, Y. Fang, R. Zhao, E. Joseph, J. Li, J. P. Pellois and H. C. Zhou, Angew. Chem., 2018, 130, 5827–5832 CrossRef
- R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Nat. Rev. Drug Discovery, 2019, 18, 175–196 CrossRef CAS PubMed
- Y. R. Murciano-Goroff, A. B. Warner and J. D. Wolchok, Cell Res., 2020, 30, 507–519 CrossRef
- N. Habibi, S. Christau, L. J. Ochyl, Z. Fan, A. H. Najafabadi, M. Kuehnhammer, M. Zhang, M. Helgeson, R. V. Klitzing, J. J. Moon, J. L. Moon and J. Lahann, Adv. Ther., 2020, 3, 2000100 CrossRef CAS
- F. Duan, X. Feng, X. Yang, W. Sun, Y. Jin, H. Liu, K. Ge, Z. Li and J. Zhang, Biomaterials, 2017, 122, 23–33 CrossRef CAS PubMed
- E. Jensen-Jarolim, World Allergy Organ., 2015, 8, 7 CrossRef
- Y. B. Miao, W. Y. Pan, K. H. Chen, H. J. Wei, F. L. Mi, M. Y. Lu, Y. Chang and H. W. Sung, Adv. Funct. Mater., 2019, 29, 1904828 CrossRef CAS
- S. M. Toor, V. S. Nair, J. Decock and E. Elkord, Semin. Cancer Biol., 2020, 65, 1–12 CrossRef CAS
- H. R. Kim, H. J. Park, J. Son, J. G. Lee, K. Y. Chung, N. H. Cho, H. S. Shim, S. Park, G. Kim, H. I. Yoon, H. G. Kim, Y. W. Jung, B. C. Cho, S. Y. Park, S. Y. Rha and S. J. Ha, J. Immunother. Cancer, 2019, 7, 1–13 CrossRef CAS PubMed
- L. S. Wood, N. P. Moldawer and M. S. N. Colleen Lewis, Clin. J. Oncol. Nurs., 2019, 23, 271–280 Search PubMed
- Y. Feng, H. Wang, S. Zhang, Y. Zhao, J. Gao, Y. Zheng, P. Zhao, Z. Zhang, M. J. Zaworotko, P. Cheng, S. Ma and Y. Chen, Adv. Mater., 2019, 31, 1805148 CrossRef PubMed
- F. Finkelmeier, O. Waidmann and J. Trojan, Expert Rev. Anticancer Ther., 2018, 18, 1169–1175 CrossRef CAS PubMed
- S. K. Alsaiari, S. S. Qutub, S. Sun, W. Baslyman, M. Aldehaiman, M. Alyami, A. Almalik, R. Halwani, J. Merzaban, Z. Mao and N. M. Khashab, Sci. Adv., 2021, 7, eabe7174 CrossRef CAS PubMed
- Y. Lu, W. Sun and Z. Gu, J. Controlled Release, 2014, 194, 1–19 CrossRef CAS
- K. Rehman, M. Sajid Hamid Akash, B. Akhtar, M. Tariq, A. Mahmood and M. Ibrahim, Curr. Drug Targets, 2016, 17, 1172–1188 CrossRef CAS PubMed
- J. Hall, S. Prabhakar, L. Balaj, C. P. Lai, R. A. Cerione and X. O. Breakefield, Cell. Mol. Neurobiol., 2016, 36, 417–427 CrossRef CAS PubMed
- C. A. Elena-Real, A. Díaz-Quintana, K. González-Arzola, A. Velázquez-Campoy, M. Orzáez, A. López-Rivas, S. Gil-Caballero, M. A. De la Rosa and I. Díaz-Moreno, Cell Death Dis., 2018, 9, 1–12 CrossRef CAS PubMed
- L. Ding, X. Lin, Z. Lin, Y. Wu, X. Liu, J. Liu, M. Wu, X. Zhang and Y. Zeng, ACS Appl. Mater. Interfaces, 2020, 12, 36906–36916 CrossRef CAS PubMed
- K. Kaul, M. Apostolopoulou and M. Roden, Metab., 2015, 64, 1629–1639 CrossRef CAS PubMed
- Y. Chen, P. Li, J. A. Modica, R. J. Drout and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 5678–5681 CrossRef CAS PubMed
- Y. Duan, F. Ye, Y. Huang, Y. Qin, C. He and S. Zhao, Chem. Commun., 2018, 54, 5377–5380 RSC
- S. Peng, J. Liu, Y. Qin, H. Wang, B. Cao, L. Lu and X. Yu, ACS Appl. Mater. Interfaces, 2019, 11, 35604–35612 CrossRef CAS PubMed
- S. Hager, F. J. Fittler, E. Wagner and M. Bros, Cells, 2020, 9, 2061 CrossRef CAS PubMed
- V. Gupta, S. P. Lourenço and I. J. Hidalgo, J. Pharm. Sci., 2021, 110, 1915–1920 CrossRef CAS
- S. Patil, Y. G. Gao, X. Lin, Y. Li, K. Dang, Y. Tian, W. J. Zhang, S. F. Jiang, A. Qadir and A. R. Qian, Int. J. Mol. Sci., 2019, 20, 5491 CrossRef CAS
- Q. Chen, M. Xu, W. Zheng, T. Xu, H. Deng and J. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 6712–6724 CrossRef CAS PubMed
- N. Mokri, Z. Sepehri, F. Faninam, S. Khaleghi, N. M. Kazemi and M. Hashemi, Gene Ther., 2021, 149, 1–11 Search PubMed
- T. Hidalgo, M. Alonso-Nocelo, B. L. Bouzo, S. Reimondez-Troitiño, C. Abuin-Redondo, M. de la Fuente and P. Horcajada, Nanoscale, 2020, 12, 4839–4845 RSC
- W. Wu, Y. Fan, B. Tan and H. Zhao, Microchi. Acta, 2020, 187, 1–9 CrossRef PubMed
- P. Wang, M. Xiao, H. Pei, H. Xing, S. H. Lou, C. K. Tsung and L. Li, Chem. Eng. J., 2021, 415, 129036 CrossRef CAS
- Y. L. Balachandran, X. Li and X. Jiang, Nano Lett., 2021, 21, 1335–1344 CrossRef CAS PubMed
- X. Li, Y. He, L. Yang, Z. He and J. J. Zhu, Chem. Commun., 2021, 57, 6776–6779 RSC
- A. Ringaci, A. V. Yaremenko, K. G. Shevchenko, S. D. Zvereva and M. P. Nikitin, Chem. Eng. J., 2021, 418, 129386 CrossRef CAS
- A. Poddar, S. Pyreddy, F. Carraro, S. Dhakal, A. Rassell, M. R. Field, T. S. Reddy, P. Falcaro, C. M. Doherty and R. Shukla, Chem. Commun., 2020, 56, 15406–15409 RSC
- Y. Li, K. Zhang, P. Liu, M. Chen, Y. Zhong, Q. Ye, M. Q. Wei, H. Zhao and Z. Tang, Adv. Mater., 2019, 31, 1901570 CrossRef PubMed
- S. Peng, B. Bie, Y. Sun, M. Liu, H. Cong, W. Zhou, Y. Xia, H. Tang, H. Deng and X. Zhou, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed
- F. Chen, M. Bai, K. Cao, Y. Zhao, X. Cao, J. Wei, N. Wu, J. Li, L. Wang, C. Fan and Y. Zhao, ACS Nano, 2017, 11, 11908–11914 CrossRef CAS PubMed
- H. Wang, Y. Chen, H. Wang, X. Liu, X. Zhou and F. Wang, Angew. Chem., 2019, 131, 7458–7462 CrossRef
- Z. Wang, J. Niu, C. Zhao, X. Wang, J. Ren and X. Qu, Angew. Chem., 2021, 133, 12539–12545 CrossRef
- A. Poddar, J. J. Conesa, K. Liang, S. Dhakal, P. Reineck, G. Bryant, E. Pereiro, R. Ricco, H. Amenitsch, C. Doonan, X. Mulet, C. M. Doherty, P. Falcaro and R. Shukla, Small, 2019, 15, 1902268 CrossRef PubMed
- J. Zhao, D. Lu, S. Moya, H. Yan, M. Qiu, J. Chen, X. Wang, Y. Li, H. Pan, G. Chen and G. Wang, Appl. Mater. Today, 2020, 20, 100701 CrossRef
- C. P. Cerrato, K. Künnapuu and Ü. Langel, Expet Opin. Drug Delivery, 2017, 14, 245–255 CrossRef CAS PubMed
- H. N. Abdelhamid, M. Dowaidar, M. Hällbrink and Ü. Langel, Microporous Mesoporous Mater., 2020, 300, 110173 CrossRef CAS
- F. Liu, S. Sheng, D. Shao, Y. Xiao, Y. Zhong, J. Zhou, C. H. Quek, Y. Wang, Z. Hu, H. Liu, Y. Li, H. Tian, K. W. Leong and X. Chen, Nano Lett., 2021, 21, 2461–2469 CrossRef CAS PubMed
- B. Mansoori, S. S. Shotorbani and B. Baradaran, Adv. Pharm. Bull., 2014, 4, 313 Search PubMed
- T. X. Lu and M. E. Rothenberg, J. Allergy Clin. Immunol., 2018, 141, 1202–1207 CrossRef CAS PubMed
- M. Acunzo, G. Romano, D. Wernicke and C. M. Croce, Adv. Biol. Regul., 2015, 57, 1–9 CrossRef CAS PubMed
- H. Zhao, T. Li, C. Yao, Z. Gu, C. Liu, J. Li and D. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 6034–6042 CrossRef CAS PubMed
- G. R. Devi, Cancer Gene Ther., 2006, 13, 819–829 CrossRef CAS PubMed
- C. He, K. Lu, D. Liu and W. Lin, J. Am. Chem. Soc., 2014, 136, 5181–5184 CrossRef CAS PubMed
- M. H. Teplensky, M. Fantham, C. Poudel, C. Hockings, M. Lu, A. Guna, M. Aragones-Anglada, P. Z. Moghadam, P. Li, O. K. Farha, S. B. D. Q. Fernández, F. M. Richards, D. I. Jodrell, G. K. Schierle, C. F. Kaminski and D. Fairen-Jimenez, Chem, 2019, 5, 2926–2941 CAS
- J. Zhuang, H. Gong, J. Zhou, Q. Zhang, W. Gao, R. H. Fang and L. Zhang, Sci. Adv., 2020, 6, eaaz6108 CrossRef CAS PubMed
- Y. Zhang, L. Yang, H. Wang, J. Huang, Y. Lin, S. Chen, X. Guan, M. Yi, S. Li and L. Zhang, Chem. Eng. J., 2021, 426, 131926 CrossRef CAS
- G. Combes, I. Alharbi, L. G. Braga and S. Elowe, Oncogene, 2017, 36, 4819–4827 CrossRef CAS PubMed
- D. Yu, M. Ma, Z. Liu, Z. Pi, X. Du, J. Ren and X. Qu, Biomaterials, 2020, 255, 120160 CrossRef CAS PubMed
- D. Wu, Y. Gao, Y. Qi, L. Chen, Y. Ma and Y. Li, Cancer Lett., 2014, 351, 13–22 CrossRef CAS PubMed
- J. Liu, Z. Guo, M. Kordanovski, J. Kaltbeitzel, H. Zhang, Z. Cao, Z. Gu, P. R. Wich, M. Lord and K. Liang, J. Colloid Interface Sci., 2020, 576, 356–363 CrossRef CAS PubMed
- Y. Li, N. Xu, W. Zhu, L. Wang, B. Liu, J. Zhang, Z. Xie and W. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 22974–22984 CrossRef CAS PubMed
- J. U. Liu, Y. U. Tang, W. Yang, B. Tao, Y. E. He, X. Shen, T. Shen, C. Lin and K. Cai, Biomater. Sci., 2019, 7, 1463–1476 RSC
- B. Tao, C. Lin, Y. He, Y. Z. Yuan, M. Chen, K. Xu, K. Li, A. Guo, K. Cai and L. Chen, Chem. Eng. J., 2021, 423, 130176 CrossRef CAS
- Y. Xu, Z. Li, D. Xiu, G. Sun, C. D. Snow, Y. Wang, Y. Wang, L. A. Belfiore and J. Tang, Mater. Des., 2021, 205, 109741 CrossRef CAS
- A. Rehman, S. M. Jafari, Q. Tong, T. Riaz, E. Assadpour, R. M. Aadil, S. Niazi, I. M. Khan, Q. Shehzad, A. Ali and S. Khan, Adv. Colloid Interface Sci., 2020, 284, 102251 CrossRef CAS PubMed
- E. Astria, M. Thonhofer, R. Ricco, W. Liang, A. Chemelli, A. Tarzia, K. Alt, C. E. Hagemeyer, J. Rattenberger, H. Schroettner, T. Wrodnigg, H. Amenitsch, D. M. Huang, C. J. Doonan and P. Falcaro, Mater. Horiz., 2019, 6, 969–977 RSC
- A. Migliore and S. Procopio, Clin. Cases Miner. Bone Metab., 2015, 12, 31 Search PubMed
- F. Xiong, Z. Qin, H. Chen, Q. Lan, Z. Wang, N. Lan, Y. Yang, L. Zheng, J. Zhao and D. Kai, J. Nanobiotechnol., 2020, 18, 1–14 CrossRef PubMed
- M. J. Velásquez-Hernández, E. Astria, S. Winkler, W. Liang, H. Wiltsche, A. Poddar, R. Shukla, G. Prestwich, J. Paderi, P. Salcedo-Abraira, H. Amenitsch, P. Horcajada, C. J. Doonan and P. Falcaro, Chem. Sci., 2020, 11, 10835–10843 RSC
- M. A. Luzuriaga, F. C. Herbert, O. R. Brohlin, A. Shahrivarkevishahi, Y. H. Wijesundara, K. Veera, C. E. Benjamin, S. Popal, M. D. Burton, M. A. Ingersoll, N. J. De Nisco and J. J. Gassensmith, BioRxiv, 2021, 14, 148452 Search PubMed
- J. Nawrocki, D. Prochowicz, A. Wiśniewski, I. Justyniak, P. Goś and J. Lewiński, Eur. J. Inorg. Chem., 2020, 796–800 CrossRef CAS
- D. Prochowicz, K. Sokołowski, I. Justyniak, A. Kornowicz, D. Fairen-Jimenez, T. Friščič and J. Lewiński, Chem. Commun., 2015, 51, 4032–4035 RSC
- D. Prochowicz, J. Nawrocki, M. Terlecki, W. Marynowski and J. Lewiński, Inorg. Chem., 2018, 57, 13437–13442 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2021 |