
Tuning the strength and swelling of an injectable polysaccharide hydrogel and the subsequent release of a broad spectrum bacteriocin, nisin A†
James
Flynn
a,
Edel
Durack
a,
Maurice N.
Collins
 b and
Sarah P.
Hudson
*a
b and
Sarah P.
Hudson
*a
aDepartment of Chemical Sciences, SSPC, SFI Research Centre for Pharmaceuticals, Bernal Institute, University of Limerick, Co., Limerick, Ireland. E-mail: sarah.hudson@ul.ie
bBernal Institute, School of Engineering, University of Limerick, Co., Limerick, Ireland
First published on 12th March 2020
Abstract
Bacteriocins, which are antimicrobial peptides, are a potential alternative to current ineffective antimicrobial therapies. They can inhibit the growth of clinically relevant pathogens but their proteinaceous nature renders them susceptible to degradation and deactivation in vivo. We have designed injectable polysaccharide hydrogels for the controlled release of an incorporated bacteriocin, nisin. Nisin was encapsulated into these hydrogels which were composed of varying percentages of oxidised dextran, alginate functionalised with hydrazine groups and glycol chitosan. The nisin gels exhibited antimicrobial activity against Staphylococcus aureus up to 10 days. The incorporation of a deacetylated chitosan and the reduction of alginate-hydrazine could be used to tune the gel's swelling behaviour, strength and the subsequent release profile of nisin. Glycol chitosan also shows synergistic inhibition of S. aureus with nisin.
1 Introduction
Antimicrobial resistant infections pose an urgent threat to public health, and although antimicrobial resistance is a natural phenomenon, cases have been fueled by the overuse or misuse of antimicrobial agents, thus exposing pathogens to sub-inhibitory levels of antibiotics, inducing resistance.1 A prevalence of resistant cases in strains of Streptococcus pneumoniae, methicillin resistant Staphylococcus aureus (MRSA), Mycobacterium tuberculosis (TB), Acinetobacter, Enterococci and multi drug resistant Gram-negative bacteria have resulted in the reduced efficacy of certain antimicrobial therapies – rendering patients, particularly those who are immunocompromised, susceptible to life-threatening infections.2 In order to replace these ineffective antimicrobial therapeutics, novel antimicrobial agents are required to combat the ever-increasing rates of antimicrobial resistance in pathogenic bacteria.2 According to the 2017 world surveillance report by the WHO, there are few new classes of antibiotics in the clinical pipeline against drug resistant clinical pathogens.3 One class of antimicrobial that have shown promising inhibitory activity against pathogenic bacteria, are bacteriocins.4–7Bacteriocins are peptides that exhibit an antimicrobial effect. They are produced by bacteria to inhibit competing bacteria. A number of bacteriocins have been identified to date, with inhibitory effects toward clinically relevant Gram positive and Gram negative bacteria, including strains that have shown resistance to other drug treatments, such as S. aureus, S. pneumonia, Clostridium difficile, vancomycin resistant enterococci and various mycobacteria.8–10 While bacteriocins show potential for therapeutic application, they exhibit a range of issues in vivo such as degradation by proteolytic enzymes, poor solubility and aggregation and unfolding during both formulation and storage.11 Nisin A, a 3.4 kDa peptide, is a bacteriocin produced by the Gram positive lactic acid bacterium (LAB) Lactococcus lactis subsp. lactis. The presence of lanthionine rings in its structure classifies nisin A as a lantibiotic. Nisin A has FDA approval for use as a food additive (E234) and is a certified GRAS excipient and shows potential for clinical application. Inhibitory activity has been demonstrated against pathogenic bacterial species including S. aureus, E. coli, S. pnemoniae, E. faecium and C. difficile.12–14 Nisin A, in addition to exhibiting antimicrobial properties also exhibits immunomodulatory and anticancer activity.15–17 However, the susceptibility of nisin A to degradation by the intestinal digestive enzymes trypsin and α-chymotrypsin has hindered the development of this bacteriocin as an antimicrobial therapeutic, thus no FDA approved clinical dosage form of this antimicrobial exists.18–24
Hydrogels prepared from natural biomaterials exhibit high biocompatibility, biodegradability and resemble the extracellular matrix (ECM).25,26 These natural biomaterials – namely dextran, chitosan, collagen, alginate, fibrin, elastin and hyaluronic acid have been extensively researched for their potential use in many different applications,27–29 from drug delivery to cell therapies.30,31In situ forming hydrogels offer a range of advantages such as tunable properties and local administration.32In situ crosslinking can be generated by either chemical or physicochemical association factors such as ion induced or thermally induced gelling.30 In this work, an injectable hydrogel, composed of dextran, alginate and glycol chitosan (GC), has been designed with tunable swelling and mechanical properties. The release of an incorporated bacteriocin, nisin, is subsequently controlled, with potential synergistic antimicrobial activity with the GC.
Dextrans are natural polysaccharides of glucose, with α-1,6 linkages and hydroxylated cyclohexyl units. They exhibit biocompatibility and biodegradability in the blood and in the gastrointestinal tract making them attractive materials for the delivery of drugs in vivo.33–35 Dextran can be cross-linked with divalent metal cations, and acid chlorides with UV induced gelation,36,37 functionalized with sodium periodate to dextran-dialdehyde, or by direct addition of glutaraldehyde with MgCl2.36,38–40 Alginate is typically utilized via crosslinking with multivalent cations such as Ca2+ or Ba2+, through interactions with carboxylic groups present in its structure, thus forming a gel network.41,42 These crosslinking methods however, possess drawbacks such as poor gel stability at neutral pH's, the need for induced polymerization with UV, and poor structural homogeneity caused by uncontrolled crosslinking with Ca2+, all of which limit their clinical application.43
Schiff base or hydrazone bond formation, has been utilized to produce polysaccharide based cross-linked hydrogels as this mode of crosslinking allows for variabilities in the microstructure of the gels by altering the hydrogel parameters.44,45 Hudson et al. developed an injectable dextran-aldehyde and carboxymethylcellulose-hydrazine hydrogel for the delivery of amphotericin B. They conjugated the amphotericin B to the dextran and injected the formulation. Conjugating the drug to the dextran allowed for antifungal drug release, killing Candida albicans for up to 3 weeks. The release profile was varied by changing the degree of functionalization of the dextran-aldehyde.33 Sharma and Taneja et al. investigated hydrazone link based xanthan-PEG gels for controlled drug delivery of doxorubicin and 3D cell culture. Xanthan was oxidized with sodium periodate, and the PEG was functionalized with hydrazine groups allowing for an injectable administration.46
Chitosan has been shown to exhibit antimicrobial activity both independently and synergistically, and is labelled as an antibacterial biopolymer.47 However, its poor solubility presents a hindrance in its application.48,49 In this manuscript, the addition of a deacetylated chitosan, glycol chitosan, was used as it is water soluble and capable of controlling the release of a cationic antimicrobial peptide, nisin A, from an injectable dextran-dialdehyde and alginate-hydrazine hydrogel. Controlling the swelling, mechanical properties and inhibitory activity of an antimicrobial peptide (AMP) loaded hydrogel has not been achieved previously through the use of glycol chitosan.
2 Experimental
2.1 Materials
Nisin A (95%, isolated from Lactococcus lactis in sauerkraut) was obtained from Handary, Belgium. Dextran (from Leuconostoc mesenteroides, 100–200 kDa), alginic acid (from brown algae, low viscosity), sodium (meta) periodate (>99%), ethylene glycol (99%), N-hydroxysulfosuccinimide sodium salt (S-NHS, > 98%), N-(3-dimethylaminopropyl)-N′- ethylcarbodiimide hydrochloride (EDC, crystalline), adipic acid dihydrazide (>98%), hydroxylamine hydrochloride, glycol chitosan (>60%), trifluroacetic acid (TFA, >99%), acetonitrile (ACN, >99.9%) and phosphate buffered saline (PBS) were all purchased from Sigma Aldrich, Ireland. Dimethyl sulfoxide (DMSO) was purchased from VWR International. Liquid nitrogen was supplied by BOC Gases (Ireland). Sodium taurocholate (>95%), L-α-lecithin (>99%), sodium chloride hexahydrate (98%), potassium chloride (99%), hydrochloric acid (37%, fuming) were all purchased from Fisher Scientific, Ireland. Pullulan/Dextran standards for Light Scattering/Tetra Detection (TDS3030) were provided by Particular Sciences, Dublin, Ireland. Staphylococcus aureus (DSM 20231) was purchased from the Leibniz Institute DSMZ – German collection of Microorganisms and Cell Cultures.Nisin hydrogels with no glycol chitosan. Following a similar method to Hudson et al.,33 hydrogels were prepared using a double barreled syringe set up. Dextran-dialdehyde was dissolved in KCl/HCl buffer (pH 2) at a concentration of 6% w/v. Alginate-hydrazine was dissolved in PBS (pH 7.4) at a concentration of 3% w/v. Both solutions were added to 1 ml syringes which were attached to a double barrel syringe apparatus. The solutions were extruded through a 21-gauge needle into a mold (6 mm × 3 mm, 100 μl) gelation occurring in situ within 5–10 seconds at ambient room temperature (∼23 °C). For hydrogels containing nisin, the dextran-dialdehyde was dissolved in a 20 mg ml−1 nisin solution (KCl/HCl, pH 2) such that each gel had a nisin concentration of approximately 1 mg.
Nisin hydrogels with glycol chitosan. Hydrogels with glycol chitosan were prepared as above, with the addition of glycol chitosan to the alginate solution at concentrations of 3% and 6% w/v, reducing the concentration of alginate from 3% to 1% and 0.5%, respectively. Gels containing glycol chitosan are denoted as Dex-Algα%-GCβ%, where ‘α%’ represents the w/v% of alginate-hydrazine and ‘β%’ represents the w/v% of glycol chitosan. ‘Dex-Alg-GC gels’ refers to the Dex-Alg3%-GC0%, Dex-Alg1%-GC3% and Dex-Alg0.5%-GC6% gels collectively, Table 1.
| Gel | Dextran-dialdehyde [mg ml−1] | Alginate-hydrazine [mg ml−1] | Glycol chitosan [mg ml−1] | Nisin (per gel) |
|---|---|---|---|---|
| Dex-Alg3%-GC0% | 60 | 30 | 0 | 1 mg |
| Dex-Alg1%-GC3% | 10 | 30 | ||
| Dex-Alg0.5%-GC6% | 5 | 60 | ||
| Dex-GC6% | 0 | 60 | ||
| Dex-Alg0.5% | 5 | 0 |
The swelling of the gels was studied by placing the pre-formed gels into a 24 well assay plate, 1 ml of m-FaSSGF (pH 1.6) was subsequently added. The plate was incubated at 37 °C and at specified time points the gels were removed with a spatula, blotted dry with filter paper and weighed. The diameter of the gels was also measured at each time point using a Vernier calipers. At each time point the buffer was totally replenished.
Mechanical characterization of the Dex-Alg-GC gels both with and without nisin was carried out using a compression test. Gels were formed, the surface area was determined and a pressure of 100 kPa was applied (relative to the area of each sample), at a speed of 0.25 μm min−1. The Young's modulus was determined based on the stress strain curve generated from the compression data.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 to 55:45 (A/B) over 52 minutes. The chromatogram for nisin A contained three peaks which accounted for the three components of nisin which were characterized in a previous study52 and the intact nisin peak was used to quantify nisin release.
:25 to 55:45 (A/B) over 52 minutes. The chromatogram for nisin A contained three peaks which accounted for the three components of nisin which were characterized in a previous study52 and the intact nisin peak was used to quantify nisin release.
IC50 of nisin A against S. aureus. The inhibitory concentration at 50% for nisin against S. aureus (DSM 20231) was determined as reported in a previous study.52 Briefly, a 1 mg ml−1 nisin solution (KCl/HCl) was diluted with PBS to concentrations of 10, 12, 15 and 20 μg ml−1 in the wells of a 96 well plate. 150 μl of an overnight S. aureus culture (OD595 0.1) was added to each test well. The plate was incubated for 24 hours at 37 °C in a Biotek Elx808 Ultra Microplate reader (Mason Technologies, Dublin, Ireland) with readings taken at 595 nm every 30 minutes.
Synergistic activity of glycol chitosan and nisin A. An overnight culture of S. aureus was cultured in BHI broth at 37 °C shaking at 140 rpm. Sterile BSA (10%, PBS pH 7.4) was added to the wells of a sterile 96 well plate (200 μl) and incubated at 37 °C for 30 minutes. The BSA was removed and the wells were rinsed with sterile PBS. The culture was diluted to an OD595 of 0.1. A 1 mg ml−1 nisin solution (KCl/HCl, pH 2) was filter sterilized through a 0.22 μm PES syringe filter. 3% and 6% GC solutions were prepared under sterile conditions, whereby GC was weighed out and UV irradiated for 30 minutes before being dissolved in sterile PBS. Dilutions were prepared at concentrations of 15 μg nisin combined with 0, 3 and 6% GC in the wells of the 96 well plate, whereby a volume of a 1 mg ml−1 nisin solution (KCl/HCl, pH 2) was added followed by the same volume of a 3%/6% GC (PBS, pH 7.4) solution to represent the ratio of nisin and dextran-CHO and GC in the gel formulation. 150 μl of the diluted culture was added to the wells. Buffer controls (KCl/HCl), blank controls (BHI) and positive controls (S. aureus) were run in conjunction. The plate was incubated for 24 hours at 37 °C in a Biotek Elx808 Ultra Microplate reader (Mason Technologies, Dublin, Ireland) with readings taken at 595 nm every 30 minutes. Blank readings were subtracted from test readings.
Antimicrobial activity of gels. Hydrogels were prepared as previously mentioned. The gels were frozen, thawed and UV irradiated for 30 minutes. The bioactivity study was carried out in a similar way to the release study (Section 2.2.6), whereby m-FaSSGF was filter sterilized through a 0.22 μm PES filter. Each sterile gel was administered into 1 ml of the sterile m-FaSSGF (n = 3) in a 24 well sterile culture plate. The plate was sealed and incubated at 37 °C. After 24 hours, the FaSSGF was removed from each gel sample under sterile conditions and frozen, the media was fully replenished and removed again at day 1, 3, 5 and day 10. Controls of m-FaSSGF and blank gels were also conducted. Optical density measurements were taken every 30 minutes for 24 hours, whereby 66.7 μl of each sample was added to 133 μl of the same S. aureus culture at an OD595 0.1, in the wells of a 96 well sterile culture plate. The plate was incubated at 37 °C in a Biotek ELx808 Ultra microplate reader (Mason Technologies, Dublin, Ireland) for 24 hours. Aliquots of the release media were kept under sterile conditions at all times. The test samples were not filtered before testing their antimicrobial activity.
000 cells were seeded per well of a 96 well assay plate, for 24 h to ensure adherence of HEK293 cells. Samples from day 14 of the stability study (Dex-Alg0.5%-GC6%) were UV treated for 30 minutes to ensure sterility. Media was changed in the wells prior to addition of the samples (10 μl). Live controls, dead controls (10% Triton X) and buffer controls (m-FaSSGF) were set up in conjunction. After an incubation period of 24 hours MTT reagent was added to each test well (10 μl). After 4 h, 100 μl propan-2-ol with 0.04 M HCl was added to each well to dissolve crystals as per manufacturers instruction. The plate was shaken and read at 570 nm. Test results were expressed as % cells that were viable after treatment when compared with the live controls.
3 Results
3.1 Characterization of hydrogels
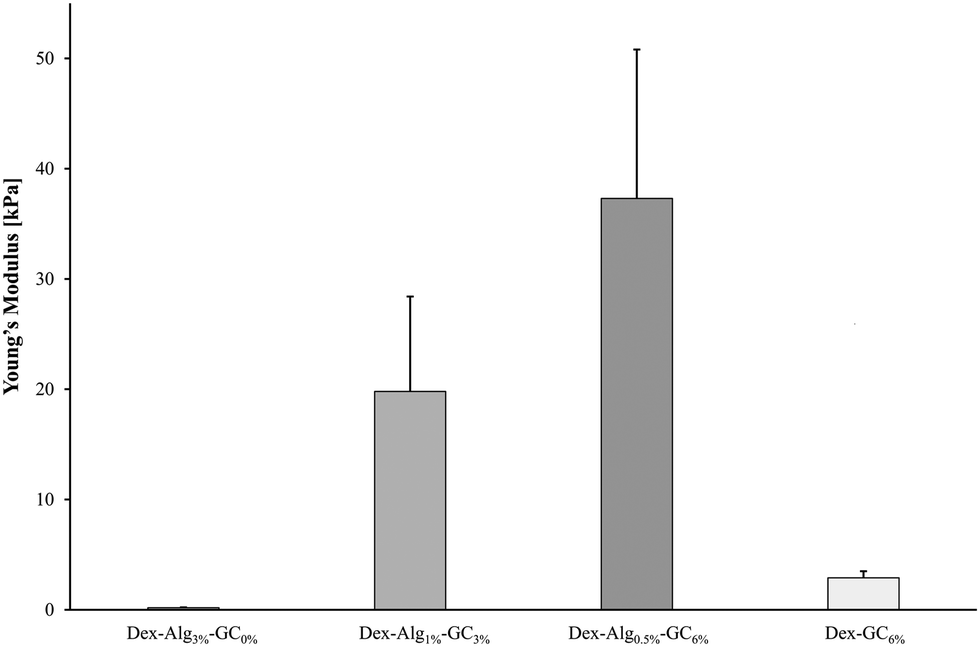 | ||
| Fig. 1 Young's moduli obtained for the Dex-Alg-GC and Dex-GC gels. | ||
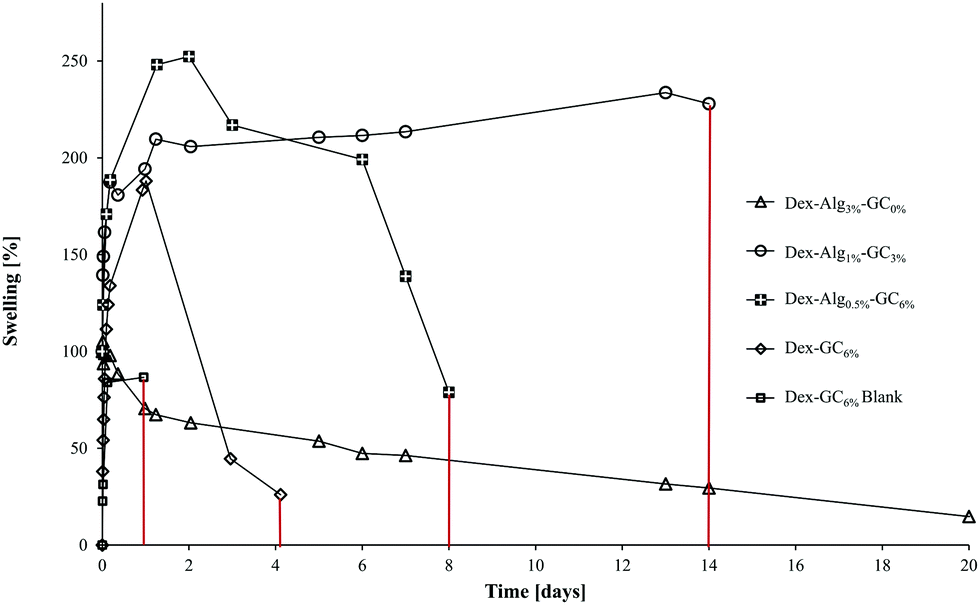 | ||
| Fig. 2 The swelling of the gels in m-FaSSGF (pH 1.6, 37 °C) was monitored until the gels disintegrated at which point the study was stopped (y axis error bars are included). | ||
| Hydrogel | Nisin [μg per gel] | Days to disintegration | Max. swell [%] | Max. diameter increase [%] |
|---|---|---|---|---|
| Dex-Alg3%-GC0% | 1000 | >22 days | 0 | ∼107 |
| Dex-Alg1%-GC3% | 1000 | ∼14 days | ∼200 | ∼133 |
| Dex-Alg0.5%-GC6% | 1000 | ∼8 days | ∼252 | ∼172 |
| Dex-GC6% | 1000 | ∼ 4–5 days | ∼180 | ∼56 |
| Dex-Alg0.5% | No gel formed | |||
| Dex-GC6%-blank | 0 | 1 day | ∼187 | ∼154 |
The swelling of the gels indicates that the substitution of alginate-hydrazine with GC increased the swelling behaviour of the gels, and reduced the length of time for the gels to disintegrate presumably via chain scission resulting from hydrolysis. When no GC was present (Dex-Alg3%-GC0%), no swelling was evident, however the gels did not appear to disintegrate even after 3 weeks of testing, as shown in Fig. 2. Upon introduction of GC, gels swelled to more than twice their original mass, and increased in diameter as GC concentrations increased, Fig. 3. The higher influx from the reduced degree of cross-linking however led to gels with higher GC concentrations disintegrating faster, as shown in Table 2 and Fig. 2. Gels with no alginate present (Dex-GC6%) disintegrated after just one week, and when nisin was not incorporated they disintegrated after 1 day, indicating a stabilisation effect through nisin interactions within the gel. Dex-Alg0.5% did not form gels.
 | ||
| Fig. 3 The change in diameter of the hydrogel for the duration of their swelling study in m-FaSSGF (pH 1.6, 37 °C). | ||
3.2 In vitro release of nisin
In vitro release studies show that increasing the concentration of GC slows the rate of nisin released into m-FaSSGF (pH 1.6, 37 °C), Fig. 4. m-FaSSGF was used as the release media as nisin exhibits a high degree of solubility in this media. The lower crosslink densities, and the replacement of stiffer alginate with more flexible GC chains, (see ESI† for characterisation of as received and functionalised polymers), allows nisin to interact with the gel more readily, while increasing the length of its diffusion path out of the gel. In gels with no GC, nisin was forced to the outer perimeters of the gel allowing for a faster release. Filtering for RP-HPLC analysis meant that any nisin bound to polymer fragments were filtered out before analysis. Thus the release results may not quantitatively represent the true nisin release from the gels, and 100% release was not observed.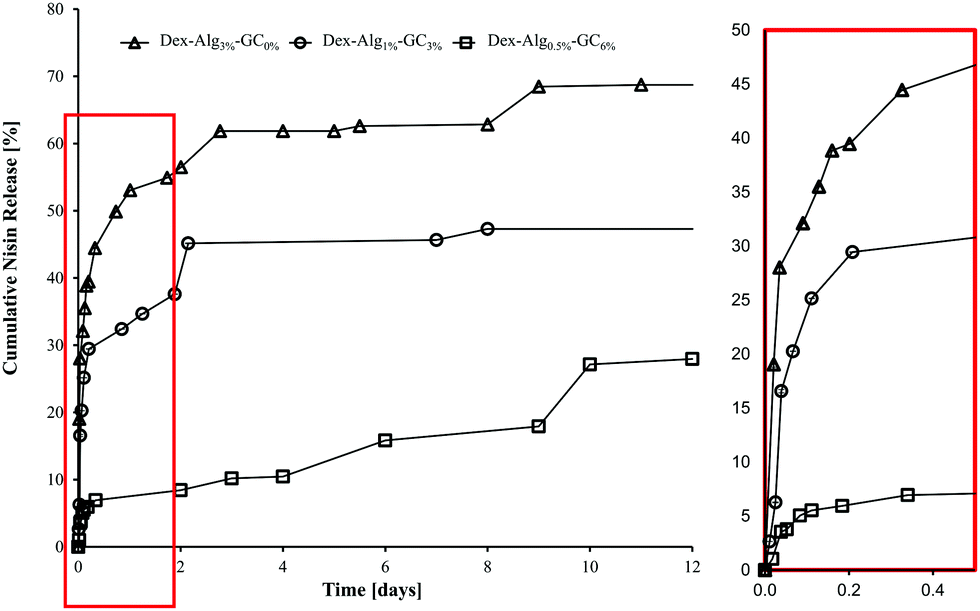 | ||
| Fig. 4 Cumulative release of nisin from the Dex-Alg-GC gels into m-FaSSGF (pH 1.6) over 12 days, before disintegration of the gels. | ||
3.3 In vitro antimicrobial activity
Blank Dex-Alg0.5%-GC6% gel controls, without nisin, indicated inhibition of a S. aureus culture, Fig. 5, whereas no such inhibition was observed with the Dex-Alg3%-GC0% gels. This indicated that the soluble GC polymer may have antimicrobial properties and therefore the activity of GC and nisin, both alone and in combination against S. aureus was tested. Growth curves demonstrated a synergistic inhibitory effect between glycol chitosan and nisin, Fig. 6. A nisin concentration of 15 μg ml−1 was tested with both 3% and 6% GC and independently (0% GC). It was found that a combination of 15 μg ml−1 nisin with 3% or 6% GC completely inhibited the growth of the culture. There was no difference in the inhibition of the culture between 3 or 6% GC indicating that 3% GC in combination with 15 μg ml−1 nisin is sufficient for the inhibition of S. aureus. Results indicated that a final O.D. of ∼0.4 for 15 μg ml−1 nisin alone, indicating ∼35% growth reduction, while in combination with 3% GC a final O.D. of ∼0.04 was obtained, indicating a reduction in growth of ∼93%, Fig. 6.
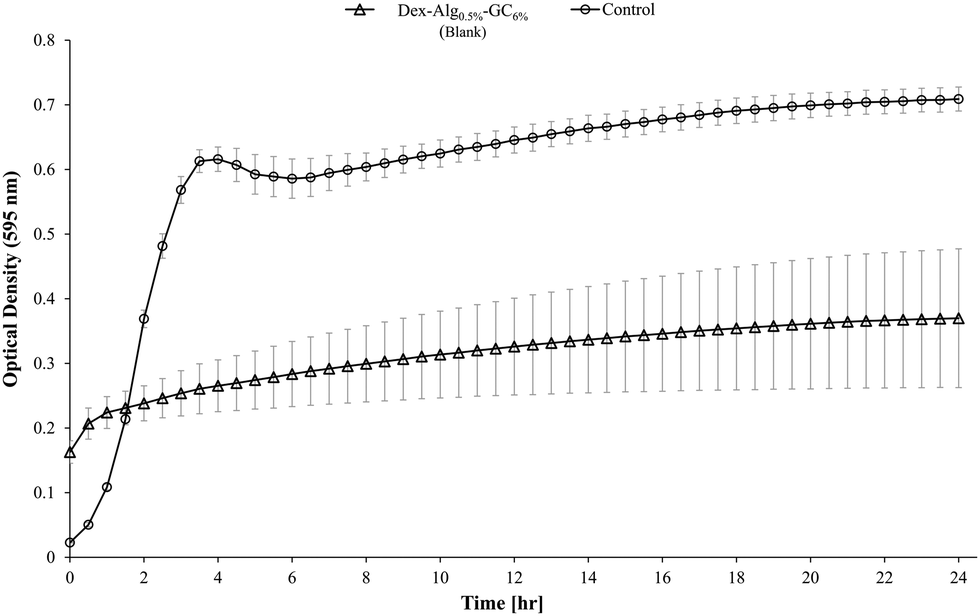 | ||
| Fig. 5 Inhibitory activity of the blank Dex-Alg0.5%-GC6% gel after 14 days, against S. aureus. | ||
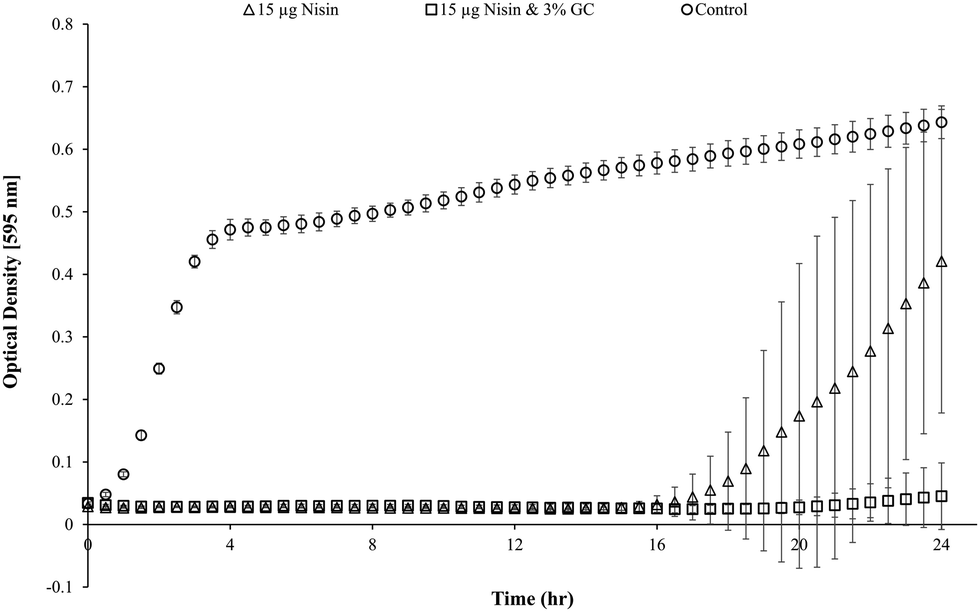 | ||
| Fig. 6 Synergistic inhibitory activity against the growth of S. aureus exhibited by nisin and GC, alone and combined. | ||
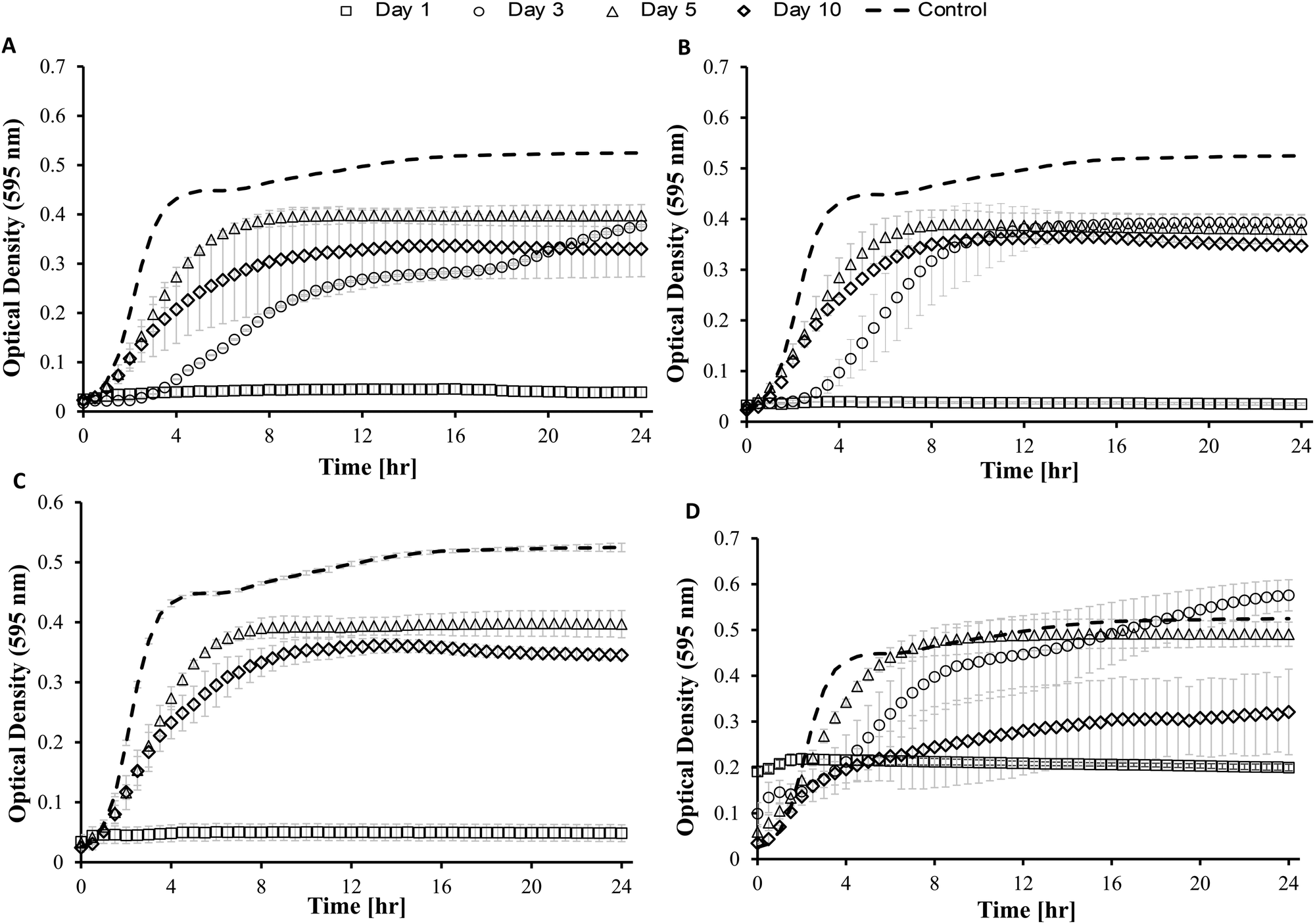 | ||
| Fig. 7 (a) Antimicrobial activity of Dex-Alg3%-GC0% gels on days 1, 3, 5 & 10, (b) Dex-Alg1%-GC3% on days 1,3,5 & 10, (c) Dex-Alg0.5%-GC6% gels on days 1, 5 & 10, (d) Dex-GC6% gels on days 1, 3, 5 &10, all against the growth of S. aureus. | ||
3.4 Biocompatibility of hydrogel degradation products
The biocompatibility of the degradation products of the Dex-Alg-GC6% gels at day 14 was assessed with HEK293 cells. MTT results demonstrated a lack of in vitro cytotoxicity after 24 hours. A variety of volumes of the swelling study media was used to ensure minimal pH change in the cell culture medium and buffer controls were run alongside these samples. The highest volume (15 μl) is presented in Fig. 8. All test samples show cell proliferation whereby the concentration of cells was ∼20% higher than that of the control, presumably due to the presence of the polysaccharides. By selecting the media (supernatant) of Dex-Alg0.5%-GC6% at day 14 during this assay, the HEK293 cells were exposed to the highest concentration of degraded polymers. No toxicity was observed and thus none would be expected at lower concentration of the degradation products at earlier timepoints. While the gels tested do not contain the highest concentration of alginate, they do contain the highest concentration of glycol chitosan used, which is the least studied of the polymers used in terms of toxicity.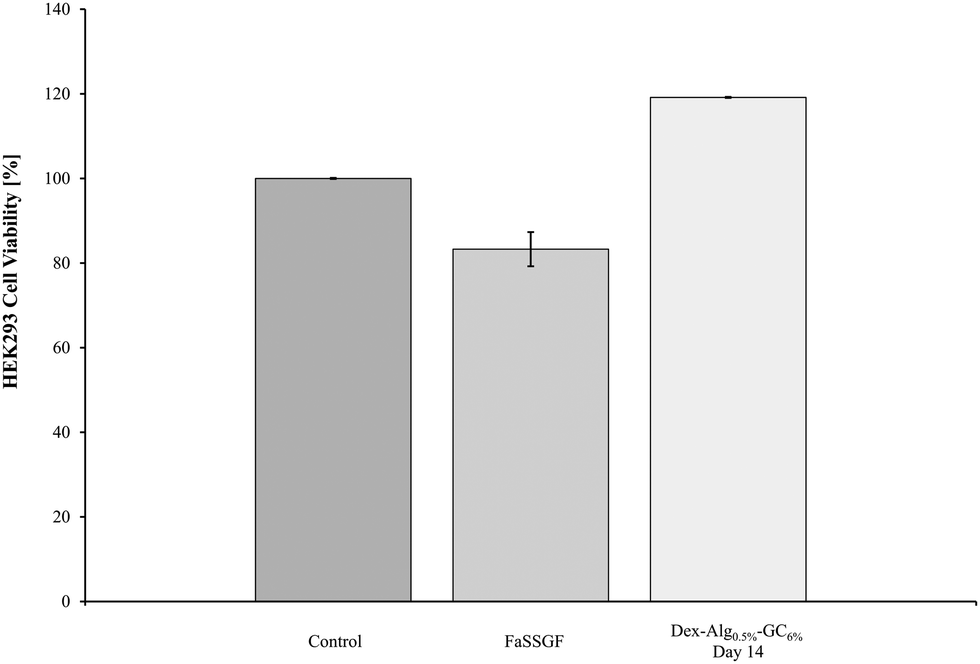 | ||
| Fig. 8 The biocompatibility of the Dex-Alg0.5%-GC6% gel was tested against HEK293 cells. The degradation products exhibited some proliferation of cells, but did not show any cytotoxicity. | ||
4 Discussion
The principle aim of this work was to develop an injectable polysaccharide based hydrogel system to encapsulate and control the release of a broad spectrum bacteriocin, nisin A. Dextran and alginate have been extensively employed in biomedical applications because of their biocompatibility, biodegradability and ease of production.54–56 Injectable hydrogels and other delivery platforms such as mesoporous materials have been explored for the encapsulation and delivery of sensitive proteins and peptides in previous studies.52,56 Whilst the mesoporous matrices facilitated protection of nisin against degradation by pepsin, and allowed nisin to be released into FaSSGF, retaining its antimicrobial activity, certain limitations such as difficulty in structural modification and in tuning the release/mechanical properties, alongside potential biocompatibility issues were present. Thus, innovative designs of delivery platforms that can be tailored to (1) control the release of a range of peptides and (2) tune their mechanical properties for specific applications, whilst satisfying the physicochemical conditions of the encapsulated peptide are needed.Here, it was hypothesized that the combination of (1) decreasing the degree of crosslink density by reducing the concentration of alginate-hydrazine and (2) the addition of a more flexible polymer (GC) in an injectable dextran alginate gel would tune the degree of swelling and mechanical properties while subsequently controlling the release of nisin. By allowing nisin to permeate deeper in the pores of the hydrogel, nisin is free to interact with the 3D gel structure thus reducing the rate of release. Zhu et al. have previously demonstrated the ability of chitosan to form electrostatic complexes with certain biomolecules such as lysozyme and nisin.49 The increase in porosity of the gels due to a reduction in covalent crosslinking (less alginate-ADH added) also allows for an increased influx of media and subsequent swelling degree. The incorporation of GC was hypothesized to increase the mechanical stiffness and the antimicrobial activity of the gels against a pathogenic strain of S. aureus and control the release of nisin.
Gels containing no GC with the lowest concentration of alginate-hydrazine (Dex-Alg0.5%) did not form a gel upon extrusion through a 21 G needle presumably due to the high reduction in hydrazine side groups required for cross linking. On the other hand, gels with GC incorporated, with 30 mg ml−1 alginate-hydrazine became too viscous for gel formation and as such the concentration of alginate-ADH had to be reduced with increasing GC content. Interestingly, as the concentration of GC is increased, the stiffness of the freshly made gel increases, perhaps due to a higher degree of polymer chain entanglement (more flexible chains), Fig. 1. Other studies have probed the tuning of the mechanical properties of hydrogels with particular focus on interpenetrating networks (IPN)57 and varying ionic concentration. Feng et al. demonstrated that by varying the ionic concentration in their peptide solutions from 0.1–2.1 M, they could modulate the stiffness of the gels, as measured by small angle X-ray scattering (SAXS).58 While this effect has been observed in other studies for both stiffness and swelling,59 there are certain limitations posed, particularly in terms of the suitability of the gel matrix for peptides/proteins. These limitations include reduced diffusion coefficients due to salt barrier formations, as well as the increased potential for ‘salting out’ or aggregation of biomolecules encapsulated in the gel.58 Through the alteration of the hydrogel network via reduction in the degree of covalent crosslinking and substitution with GC, the mechanical properties of the gels become tunable without increasing the risk of aggregation or precipitation of the encapsulated peptide. The presence of nisin in the gels did not appear to have any impact on the stiffness.
Swelling studies were conducted on the Dex-Alg-GC and Dex-GC6% gels. The reduction of covalent bonding and subsequent expansion of the gel network caused by the replacement of the hydrazine functionalized alginate by the glycol chitosan, allows for a higher solvent influx. Thus the higher % GC gels swell to a higher degree. The (1) reduction of alginate-hydrazine and (2) introduction of GC showed an increased degree of swelling potentially due to the reduction in covalent cross link densities which are replaced by physical chain entanglements.
It was hypothesized that alteration of the gel network through this reduction of covalent crosslinking would allow the nisin to diffuse further into the gels mesh, allowing nisin to interact more with the hydrogel matrix, thus slowing down its release rate. Indeed, nisin release appeared to be slowed by the introduction of increasing GC concentration and simultaneous reduction in alginate-hydrazine concentration, Fig. 4. The release was carried out over a period of two weeks for the 0% gels, 22 days for the 3% gels and 10 days for the 6% gels, after which the 3% and 6% GC gels had fully disintegrated. The respective final cumulative release detected by RP-HPLC was 70%, 62% and 27%. The release study was carried out for a more extended period than the swelling study as gels were not removed at each sampling point reducing additional stresses on the gel structures, thereby extending their stability. However, limitations due to the filtering of release samples mean that the release detected by RP-HPLC may not be indicative of the actual concentration of nisin released. As no nisin was detected in the release media after the gels had fully disintegrated, it was hypothesized that the nisin had bound to the soluble polymer fragments, and these were removed upon filtration. As such the release study was repeated under sterile conditions and the release media was tested for antimicrobial activity at days 1, 3, 5 and 10, without the filtration step, to prevent filtering out any polymer bound nisin.
GC, a cationic derivative of chitosan, was employed in these formulations for (1) its potential interactions with dextran-CHO and alginate-hydrazine to tune its swelling and mechanical properties, (2) potential electrostatic interactions between the GC and nisin to promote interactions with the gel structure and (3) it's hypothesized synergistic antimicrobial activity with nisin. GC was found to act synergistically with nisin at concentrations of 3% or 6% w/v, inhibiting a culture of S. aureus completely after 24 hours when combined with 15 μg nisin, Fig. 6. Blank gel (Dex-Alg-GC6% with no nisin) degradation products after 14 days were found to inhibit 50% of a S. aureus culture (OD 0.1) after 24 hours. He et al. demonstrated an in vitro anti-biofilm effect based on a combination of chitosan and nisin, whereby a concentration of 1% chitosan and 0.6% nisin was effective in the reduction of biofilm formation of Shewanella putrefaciens DHS01 and Shewanella algae DHS02.60 Nisin has also been shown to act synergistically with compounds other than chitosan such as monoglycerides and with the lacto peroxidase system.49 While the mechanism of action of chitosan as an antimicrobial still remains largely unknown, ionic interactions and subsequent membrane rupture have been hypothesized in other studies.61–63
While the antimicrobial activity of the gels did not differ greatly for the Dex-Alg-GC gels, it appeared that the Dex-GC6% gels exhibited antimicrobial activity on day 1 and after disintegration, rather than a sustained activity, Fig. 7. A common trend was observed whereby the activity decreased between days 3 and 5, and increased at day 10 where disintegration was evident in the Dex-Alg-GC gels. While a synergistic activity of inhibition was observed with solubilized nisin and GC outside of the gel system, this effect was not apparent in the gels antimicrobial activity. In vivo testing and a more sophisticated in vitro set up is required to account for this.
The Dex-Alg0.5%-GC6% gels were used to test the in vitro cytotoxicity of the hydrogel disintegration products in order to encompass the gel formulation with the highest concentration of GC. Alginate and dextran hydrogels have been previously shown to possess high biocompatibility.64–66 The MTT assay showed that no cell death was evident, in fact cell proliferation was present. This proliferation is presumably due to the presence of polysaccharides (dextran, alginate) in the disintegration media feeding the HEK293 cells and showing an increased cell density reading. While the in vitro cytotoxicity testing shows biocompatibility in the assay, in vivo testing is essential in rendering full cytotoxicity information including site of injection as well as hemocompatibility, however the polymers used in this study have been shown in other studies to exhibit a high degree of biocompatibility in vivo.67–70
5 Conclusions
The incorporation of glycol chitosan into an injectable polysaccharide gel allows for modulation of the gels swelling and mechanical properties without sacrifice of physiologically appropriate conditions for an encapsulated antimicrobial peptide. In previous investigations, the mechanical strength of hydrogels has been modulated through means of varying ionic concentrations, rendering the gels unsuitable for sensitive biologics. By introducing GC into these gels the mechanical strength of the gels was increased through varying GC concentrations. The incorporation of glycol chitosan also does not affect the biocompatibility, and it has been found to act synergistically with nisin in the inhibition of the growth of S. aureus. This study introduces a highly tuneable platform for the encapsulation and subsequent release of the AMP nisin for at least 10 days. Further development and in vivo testing will help define, develop, and utilise this system for a range of different clinical applications such as anti-biofilm coatings, coating for medical devices, controlled release delivery depots and for other AMP's.Funding
This research was funded through the help of Science Foundation Ireland grant 13/CDA/2122, SSPC, the SFI Research Centre for Pharmaceuticals (12/RC/2275_p2) and the Department of Chemical Sciences, University of Limerick, Ireland.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to acknowledge the Chemical Sciences Department and the Bernal Institute, in particular James Kelly. We also appreciate the help of Dr Eric Dalton and the staff in Stokes Research Institute for the facilities provided.References
- WHO, 2016, DOI: https://www.who.int/docs/default-source/searo/amr/global-antimicrobial-resistance-surveillance-system-(glass)-report-early-implementation-2016-2017.pdf?sfvrsn=ea19cc4a_2.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, A. O. Aboderin, S. S. Al-Abri, N. Awang Jalil, N. Benzonana, S. Bhattacharya, A. J. Brink, F. R. Burkert, O. Cars, G. Cornaglia, O. J. Dyar, A. W. Friedrich, A. C. Gales, S. Gandra, C. G. Giske, D. A. Goff, H. Goossens, T. Gottlieb, M. Guzman Blanco, W. Hryniewicz, D. Kattula, T. Jinks, S. S. Kanj, L. Kerr, M.-P. Kieny, Y. S. Kim, R. S. Kozlov, J. Labarca, R. Laxminarayan, K. Leder, L. Leibovici, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, V. Manchanda, L. Moja, B. Ndoye, A. Pan, D. L. Paterson, M. Paul, H. Qiu, P. Ramon-Pardo, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, W. Song, M. Steinbakk, J. Thomsen, G. E. Thwaites, J. W. M. van der Meer, N. Van Kinh, S. Vega, M. V. Villegas, A. Wechsler-Fördös, H. F. L. Wertheim, E. Wesangula, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef.
- WHO, 2017, http://entity/medicines/areas/rational_use/antibacterial_agents_clinical_development/en/index.html.
- H. D. Marston, D. M. Dixon, J. M. Knisely, T. N. Palmore and A. S. Fauci, JAMA, J. Am. Med. Assoc., 2016, 316, 1193–1204 CrossRef CAS PubMed.
- A. Fleming, in Les Prix Nobel en 1945, ed. A. Holmberg, Nobel Foundation, Stockholm, 1946 Search PubMed.
- E. Martens and A. L. Demain, J. Antibiot., 2017, 70, 520–526 CrossRef CAS PubMed.
- C. J. Michele, J. Langer and L. Slawomirski, Antimicrobial Resistance in G7 Counries and Beyond: Economic Issues, Policies and Options for Action, OECD, 2015 Search PubMed.
- CDC, Antibiotic Resistance Threats in the United States, 2013 | Antibiotic/Antimicrobial Resistance|CDC, 2013.
- P. N. Malani, JAMA, J. Am. Med. Assoc., 2014, 311, 1438–1439 CrossRef.
- B. B. Hui, D. M. Whiley, B. Donovan, M. G. Law and D. G. Regan, Sex. Transm. Infect., 2016, 93(3), 221–225, DOI:10.1136/sextrans-2016-052738.
- T. A. S. Aguirre, D. Teijeiro-Osorio, M. Rosa, I. S. Coulter, M. J. Alonso and D. J. Brayden, Adv. Drug Delivery Rev., 2016, 106, 223–241, DOI:10.1016/j.addr.2016.02.004.
- K. Kuwano, N. Tanaka, T. Shimizu, K. Nagatoshi, S. Nou and K. Sonomoto, Int. J. Antimicrob. Agents, 2005, 26, 396–402 CrossRef CAS PubMed.
- E. Severina, A. Severin and A. Tomasz, J. Antimicrob. Chemother., 1998, 41, 341–347 CrossRef CAS PubMed.
- A. Bartoloni, A. Mantella, B. P. Goldstein, R. Dei, M. Benedetti, S. Sbaragli and F. Paradisi, J. Chemother., 2004, 16, 119–121 CrossRef CAS PubMed.
- J. M. Shin, J. W. Gwak, P. Kamarajan, J. C. Fenno, A. H. Rickard and Y. L. Kapila, J. Appl. Microbiol., 2016, 120, 1449–1465 CrossRef CAS PubMed.
- C. Aranha, S. Gupta and K. V. R. Reddy, Contraception, 2004, 69, 333–338 CrossRef CAS PubMed.
- N. E. Joo, K. Ritchie, P. Kamarajan, D. Miao and Y. L. Kapila, Cancer Med., 2012, 1, 295–305 CrossRef CAS PubMed.
- C. Valenta, A. Bernkop-Schnürch and H. P. Rigler, J. Pharm. Pharmacol., 1996, 48, 988–991 CrossRef CAS PubMed.
- T. Ugurlu, M. Turkoglu, U. S. Gurer and B. G. Akarsu, Eur. J. Pharm. Biopharm., 2007, 67, 202–210 CrossRef CAS PubMed.
- A. D. van Staden, A. M. Brand and L. M. T. Dicks, J. Appl. Microbiol., 2012, 112, 831–840 CrossRef CAS PubMed.
- J. K. Dill, J. A. Auxier, K. F. Schilke and J. McGuire, J. Colloid Interface Sci., 2013, 395, 300–305 CrossRef CAS PubMed.
- K.-H. Jung, M.-W. Huh, W. Meng, J. Yuan, S. H. Hyun, J.-S. Bae, S. M. Hudson and I.-K. Kang, J. Appl. Polym. Sci., 2007, 105(5), 2816–2823, DOI:10.1002/app.25594.
- R. C. Correia, A. F. Jozala, K. F. Martins, T. C. V. Penna, E. A. D. R. Duek, C. D. O. Rangel-Yagui and A. M. Lopes, World J. Microbiol. Biotechnol., 2015, 31, 649–659 CrossRef CAS PubMed.
- K. Yamakami, H. Tsumori, Y. Sakurai, Y. Shimizu, K. Nagatoshi and K. Sonomoto, Pharm. Biol., 2013, 51, 267–270 CrossRef CAS PubMed.
- R. Barbucci, Hydrogels, Springer, Milan, Milano, 2009 Search PubMed.
- X. Bai, M. Gao, S. Syed, J. Zhuang, X. Xu and X.-Q. Zhang, Bioactive Mater., 2018, 3, 401–417 CrossRef PubMed.
- M. Liu, X. Zeng, C. Ma, H. Yi, Z. Ali, X. Mou, S. Li, Y. Deng and N. He, Bone Res., 2017, 5, 17014 CrossRef CAS PubMed.
- F. Zamboni, S. Vieira, R. L. Reis, J. Miguel Oliveira and M. N. Collins, Prog. Mater. Sci., 2018, 97, 97–122 CrossRef CAS.
- C. A. Murphy, J. B. Costa, J. Silva-Correia, J. M. Oliveira, R. L. Reis and M. N. Collins, Appl. Mater. Today, 2018, 12, 51–71 CrossRef.
- D. Y. Ko, U. P. Shinde, B. Yeon and B. Jeong, Prog. Polym. Sci., 2013, 38, 672–701 CrossRef CAS.
- F. Zamboni and M. N. Collins, Int. J. Pharm., 2017, 521, 346–356 CrossRef CAS PubMed.
- E. Bakaic, N. M. B. Smeets, T. Hoare, C. R. Nuttelman, C. N. Bowman, K. S. Anseth, M. Rheinstädter, T. Hoare, X. Li and H. Chen, RSC Adv., 2015, 5, 35469–35486 RSC.
- S. P. Hudson, R. Langer, G. R. Fink and D. S. Kohane, Biomaterials, 2010, 31, 1444–1452 CrossRef CAS PubMed.
- J. Varshosaz, Exp. Opin. Drug Delivery, 2012, 9, 509–523 CrossRef CAS PubMed.
- E. Markovsky, H. Baabur-Cohen, A. Eldar-Boock, L. Omer, G. Tiram, S. Ferber, P. Ofek, D. Polyak, A. Scomparin and R. Satchi-Fainaro, J. Controlled Release, 2012, 161, 446–460 CrossRef CAS PubMed.
- J. D. Kosmala, D. B. Henthorn and L. Brannon-Peppas, Biomaterials, 2000, 21, 2019–2023 CrossRef CAS PubMed.
- S.-H. Kim and C.-C. Chu, J. Biomed. Mater. Res., 2000, 49, 517–527 CrossRef CAS PubMed.
- A. Jeanes and C. A. Wilham, J. Am. Chem. Soc., 1950, 72, 2655–2657 CrossRef CAS.
- J. Maia, R. A. Carvalho, J. F. J. Coelho, P. N. Simões and M. H. Gil, Polymer, 2011, 52, 258–265 CrossRef CAS.
- X.-B. Yuan, H. Li, X.-X. Zhu and H.-G. Woo, J. Chem. Technol. Biotechnol., 2006, 81, 746–754 CrossRef CAS.
- A. D. Augst, H. J. Kong and D. J. Mooney, Macromol. Biosci., 2006, 6, 623–633 CrossRef CAS PubMed.
- S. K. Bajpai and R. Tankhiwale, Polym. Int., 2008, 57, 57–65 CrossRef CAS.
- C. K. Kuo and P. X. Ma, J. Biomed. Mater. Res., Part A, 2008, 84, 899–907 CrossRef PubMed.
- J. Karvinen, T. O. Ihalainen, M. T. Calejo, I. Jönkkäri and M. Kellomäki, Mater. Sci. Eng., C, 2019, 94, 1056–1066 CrossRef CAS PubMed.
- J. Karvinen, T. Joki, L. Ylä-Outinen, J. T. Koivisto, S. Narkilahti and M. Kellomäki, React. Funct. Polym., 2018, 124, 29–39 CrossRef CAS.
- P. K. Sharma, S. Taneja and Y. Singh, ACS Appl. Mater. Interfaces, 2018, 10, 30936–30945 CrossRef CAS PubMed.
- T. M. Tamer, M. A. Hassan, A. M. Omer, K. Valachová, M. S. M. Eldin, M. N. Collins and L. Šoltés, Carbohydr. Polym., 2017, 169, 441–450 CrossRef CAS PubMed.
- T. Jiang, R. James, S. G. Kumbar and C. T. Laurencin, in Natural and Synthetic Biomedical Polymers, ed. S. G. Kumbar, C. T. Laurencin and M. Deng, Elsevier, Oxford, 2014, pp. 91–113 DOI:10.1016/B978-0-12-396983-5.00005-3.
- X. Zhu, H. Wu, J. Yang, J. Tong, J. Yi, Z. Hu, J. Hu, T. Wang and L. Fan, React. Funct. Polym., 2015, 91–92, 71–76 CrossRef CAS.
- M. R. C. Marques, R. Loebenberg and M. Almukainzi, Dissolution Technol., 2011, 18, 15–28 CrossRef CAS.
- P. Bannigan, K. Stokes, A. Kumar, C. Madden and S. P. Hudson, Int. J. Pharm., 2018, 552, 180–192 CrossRef CAS PubMed.
- J. Flynn, S. Mallen, E. Durack, P. M. O'Connor and S. P. Hudson, J. Colloid Interface Sci., 2019, 537, 396–406 CrossRef CAS PubMed.
- M. N. Collins and C. Birkinshaw, J. Mater. Sci.: Mater. Med., 2008, 19, 3335–3343 CrossRef CAS PubMed.
- C. J. De Groot, M. J. A. Van Luyn, W. N. E. Van Dijk-Wolthuis, J. A. Cadée, J. A. Plantinga, W. D. Otter and W. E. Hennink, Biomaterials, 2001, 22, 1197–1203 CrossRef CAS PubMed.
- J. A. Cadée, M. J. A. van Luyn, L. A. Brouwer, J. A. Plantinga, P. B. van Wachem, C. J. de Groot, W. den Otter and W. E. Hennink, J. Biomed. Mater. Res., 2000, 50, 397–404 CrossRef.
- M. George and T. E. Abraham, J. Controlled Release, 2006, 114, 1–14 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- Y. Feng, M. Taraban and Y. B. Yu, Soft Matter, 2012, 8, 11723–11731 RSC.
- G. Nisato, J. P. Munch and S. J. Candau, Langmuir, 1999, 15, 4236–4244 CrossRef CAS.
- M. He, Q.-Y. Guo, W. Song, B.-G. Li and G.-W. Zhang, Food Control, 2017, 79, 349–355 CrossRef CAS.
- E. I. Rabea, M. E. T. Badawy, C. V. Stevens, G. Smagghe and W. Steurbaut, Biomacromolecules, 2003, 4, 1457–1465 CrossRef CAS PubMed.
- B. Stephen Inbaraj, T.-Y. Tsai and B.-H. Chen, Sci. Technol. Adv. Mater., 2012, 13, 015002 CrossRef PubMed.
- S. J. Jeon, M. Oh, W.-S. Yeo, K. N. Galvão and K. C. Jeong, PLoS One, 2014, 9, e92723 CrossRef PubMed.
- X. Yang, E. Bakaic, T. Hoare and E. D. Cranston, Biomacromolecules, 2013, 14, 4447–4455 CrossRef CAS PubMed.
- Z. Q. Liu, Z. Wei, X. L. Zhu, G. Y. Huang, F. Xu, J. H. Yang, Y. Osada, M. Zrínyi, J. H. Li and Y. M. Chen, Colloids Surf., B, 2015, 128, 140–148 CrossRef CAS PubMed.
- M. C. Straccia, G. G. Ayala, I. Romano, A. Oliva and P. Laurienzo, Mar. Drugs, 2015, 13, 2890–2908 CrossRef CAS PubMed.
- N. Artzi, N. Oliva, C. Puron, S. Shitreet, S. Artzi, A. bon Ramos, A. Groothuis, G. Sahagian and E. R. Edelman, Nat. Mater., 2011, 10, 890 CrossRef PubMed.
- S. R. Van Tomme and W. E. Hennink, Expert Rev. Med. Devices, 2007, 4, 147–164 CrossRef CAS PubMed.
- U. Ritz, M. Eberhardt, A. Klein, P. Frank, H. Götz, A. Hofmann, P. M. Rommens and U. Jonas, Gels, 2018, 4, 63 CrossRef PubMed.
- U. Rottensteiner, B. Sarker, D. Heusinger, D. Dafinova, S. N. Rath, J. P. Beier, U. Kneser, R. E. Horch, R. Detsch, A. R. Boccaccini and A. Arkudas, Materials, 2014, 7, 1957–1974 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The methods and results of the gel permeation chromatography analysis of the as-received and functionalized polymers, the degree of aldehyde substitution of the dextran and scanning electron micrographs of formed hydrogels. See DOI: 10.1039/d0tb00169d |
| This journal is © The Royal Society of Chemistry 2020 |