
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Materials promoting viral gene delivery
Kübra
Kaygisiz
 and
Christopher V.
Synatschke
*
and
Christopher V.
Synatschke
*
Max Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: synatschke@mpip-mainz.mpg.de
First published on 29th September 2020
Abstract
Therapeutic viral gene delivery is an emerging technology which aims to correct genetic mutations by introducing new genetic information to cells either to correct a faulty gene or to initiate cell death in oncolytic treatments. In recent years, significant scientific progress has led to several clinical trials resulting in the approval of gene therapies for human treatment. However, successful therapies remain limited due to a number of challenges such as inefficient cell uptake, low transduction efficiency (TE), limited tropism, liver toxicity and immune response. To adress these issues and increase the number of available therapies, additives from a broad range of materials like polymers, peptides, lipids, nanoparticles, and small molecules have been applied so far. The scope of this review is to highlight these selected delivery systems from a materials perspective.
 Kübra Kaygisiz | Kübra Kaygisiz studied Chemistry at the Johannes Gutenberg University of Mainz, where she received her Bachelor's (2017) and Master's (2019) degree. After completing her Master's thesis on photo-patterned peptide gradients, she continued her work in the group of Prof. Tanja Weil at the Max Planck Institute for Polymer Research for her PhD studies. Her research interests focus on peptides as viral transduction enhancers and biofunctional self-assembled nanostructures. |
 Christopher V. Synatschke | Christopher Synatschke studied chemistry at the University of Bayreuth, Germany, and in 2013 received his PhD with distinction (summa cum laude) working on therapeutic applications of polyelectrolyte nanostructures in the group of Prof. Axel Müller. Then, as a Feodor Lynen Postdoctoral Fellow at Northwestern University, USA, he explored peptide–polymer hybrid biomaterials in the group of Prof. Samuel Stupp. After returning to Germany in 2017, he joined the group of Prof. Tanja Weil at the Max Planck Institute for Polymer Research where he now heads the “Biomaterials” group. His research interests include bioinspired, nanostructured materials to control cellular behavior. |
The promise of viral gene delivery
Due to their inherent infectivity, non-pathogenic viral particles (VPs) are highly interesting vectors for gene delivery. In therapeutic applications they replace or deactivate disease-causing genes or introduce new genes to treat a disease.1 Since the first commercial production in 20032 the amount of gene therapies entering clinical trials and eventually earning approval is growing each year3,4 with promising future prospects.5 Since 2017, the European Medicine Agency (EMA) and the U.S. Food and Drug Administration (FDA) approved eight new therapeutics,6,7 amongst them for example Luxturna®, the first adeno-associated virus-based gene therapy8 and recently Zolgensma® a one-time injection gene therapy to treat spinal muscular atrophy.9Novel tailor-made gene therapeutic strategies pave the way towards personalized medicine, in which the patient's individual genetic circumstances are considered. Viral gene delivery promises to enable various new treatments and current research is directed at areas such as oncolytic virotherapy,10 tissue regeneration and formation (bones,11 spinal cord,12 eye,13 cardiac muscles14), treatment of hemophilia,15 neurodegenerative diseases16 and cystic fibrosis.17
Unfortunately, therapies in clinical studies have faced serious setbacks through side effects such as serious liver injuries.9 A lethal case due to high dose adenovirus administration in 199918 raised attention to safety issues like immunogenicity, range of infectable host cells (tropism) as well to biological limitations, such as loading capacity of genetic information and batch-to-batch variation of viral vectors (VV).
Consequently, alternatives to VVs have become popular in the last 20 years. Non-viral gene delivery tries to mimic VVs by packaging genetic information in synthetic carriers like cationic polymers or lipids or by using physical methods for direct delivery to host cells as reviewed elsewhere.19 The advantages of these approaches are a lower immunogenicity due to the lack of preformed antibodies and an easier targeted delivery owing to the modular design of synthetic carriers.20 However, non-viral gene delivery methods often require more laborious preparation and suffer from low efficacy compared to viral methods.21 Viruses, optimized through millions of years of evolution, still continue to be the most efficient gene carriers also for therapeutic approaches and are hence used in more than 70% of clinical trials22 and in the majority of approved gene therapy drugs.7
Viral vectors
Viruses are highly ordered nanoassemblies of nucleic acids and proteins and can infect a broad range of organisms from bacteria to humans, both in pathogenic and non-pathogenic contexts.23,24 Therapeutic VVs are obtained by replacing original gene sequences with beneficial ones.25,26 These nucleic acids are packaged in a virus capsid, which is composed of proteins displaying functional groups like amines on the surface and can interact with specific receptors on cell membranes. Some virus capsids are additionally enveloped by a lipid bilayer.27 The virus mediated delivery of nucleic acids (transduction) consists of several steps involving diffusion and translocation through the cell membrane, followed by disruption of the endosome (endosomal escape), release and integration of viral genomes to the nucleus.26,28In clinical trials, adenoviruses (Ad) are the most frequently used VVs for gene transfer. Ad provides high transduction efficiency (TE) to a broad range of dividing and non-dividing cells.29 Ad tropism is mainly determined by capsid proteins, hexones and fibers,30,31 which contain fiber knob domains for coxsackievirus and adenovirus receptor (CAR) interaction.32 The Ad serotype 5 (Ad5) is one of the most common vectors in clinical trials, however, they accumulate in the liver and show strong immunogenicity due to high prevalence of pre-existing immunity to Ad.33
Adeno-associated viruses (AAV) have emerged as promising vectors with low immunogenicity and long-term stability, resulting in the recently approved therapies, marketed as Luxturna® and Zolgensma®.34,35
As part of the retrovirus (RV) family, lentiviruses (LV) have been increasingly applied in recent years due to their inherent ability to infect both dividing and non-dividing cells, efficient integration into host genetic information and possibility for large scale production.36–39
Finally, oncolytic viruses are attracting growing interest as versatile cancer therapeutics.40–45 They can selectively replicate in cancer cells, eventually inducing cell-lysis and further activating antitumor immune response.46,47 Promising oncolytic vectors are genetically modified from adenovirus, herpes virus, reovirus and measles virus.48–51
The properties of the most common VVs discussed in this review are summarized in Table 1.
| Name | Avg. Rh | Zeta potential | Enveloped | Genetic payload | Ref. |
|---|---|---|---|---|---|
| Ad | 100 nm | −30 mV | No | 37 kb | 47, 52 and 53 |
| AAV | 29 nm | −9 mV | No | 4.7 kb | 54–56 |
| LV | 166 nm | −18 mV | Yes | 8 kb | 57,59 |
Limitations
Despite intense research efforts, VVs still face several limitations. An immune response by the host can lead to rapid inactivation of viral particles and clearance from the blood stream. Subsequent inflammatory processes can lead to liver injury or even multiorgan failure, in severe cases.60,61 The production of neutralizing antibodies is a lasting humoral immune response, which hinders any further viral gene delivery.62 To minimize an immune response, low vector doses are used which in turn results in reduced efficiency.Moreover, the entry to cells can be limited by the natural tropism of VVs. For example, Ad cannot transduce to CAR-negative CD34+ stem cells.63 Further, transduction can also be hindered by low passive diffusion rates to cell membrane,64 poor translocation to the cellular endosome, slow endosomal escape and inefficient nuclear genome integration.65,66
Strategies to promote viral gene delivery
Over several decades various techniques to promote the delivery of VVs have been developed. These can be categorized into (i) physical methods, (ii) genetic bioengineering of viruses, and (iii) chemical methods, namely material additives. The main aims of these delivery strategies include enhancing transduction efficiency (TE), long-term release of VPs, reduction of immune response, broadening of tropism and targeted delivery.6,67Physical methods
Physical methods such as microinjection,68,69 microfluidic,70,71 sonication,72,73 centrifugation,74 cellular deformation,75 laser irradiation76,77 or electroporation78,79 induce or facilitate cell entry of vectors by mechanical force, mainly through disrupting the plasma membrane. They are commonly applied in non-viral gene delivery because they transport the genetic information easily without the need for carriers and the otherwise low infectivity. These methods are also viable for viral gene delivery with electroporation being a noteworthy exception.80 A downside of using VVs with physical methods is the lack of protection from immune response as well as the invasive and cell-damaging procedure. Very comprehensive overviews of membrane disruptive methods for cargo delivery have recently been published elsewhere.81,82Genetic engineering
Genetic engineering of virus capsids is laborious but gives access to vectors with higher safety profile, modified tropism, and no batch-to-batch variability. Especially in recent years, highly efficient vectors could be obtained using genetic manipulation.Strategies include amino acids mutations, peptide domain insertions and incorporation of chemical functional groups83–85 as recently reviewed for AAV.86–88 For example, introduction of unnatural amino acid residues such as azides on the capsid surface can give access to selective click chemistry.89,90 Further, genetically modified viruses can enhance TE,91,92 broaden the tropism,63,93 enable fluorescent imaging,85 escape neutralizing antibodies,94,95 generate stimuli responsive vectors,96e.g. by light97,98 or enzymes,99 and target cells.100,101 Comprehensive recent reviews on bioengineering AAV can be found elsewhere.102,103
Chemical methods
Viral gene delivery can be enhanced by synthetic additives mostly by promoting attractive virus–cell interactions. To this end, VPs are either chemically modified on the outer sphere or non-covalently bound to additives.Covalent attachment of additives to virus capsid yields stable and irreversible modifications.104 However, the covalent attachment might interfere with transduction and deactivate viruses by shielding epitopes on the capsid surface necessary for adhesion.105
Chemical modification of the virus surface requires mild conditions to maintain the integrity of VPs.106 Due to the abundance of lysine in the capsid protein,107 most bioconjugation methods are mild amine functionalizations at physiological conditions, e.g. using N-hydroxysuccinimide (NHS).108 Other approaches apply azide–alkyne click chemistry109,110 and utilize thiol-displaying bioengineered capsids for Michael addition.111
Non-covalent attachment is simpler and generally obtained via attractive electrostatic interactions or incorporation in various scaffolds. This can result in different architectures such as coatings,112–114 complexes,115–120 capsules,121–125 and matrices126–128 as illustrated in Fig. 1. Table 2 provides a summary of these architectures and highlights their respective benefits and drawbacks.
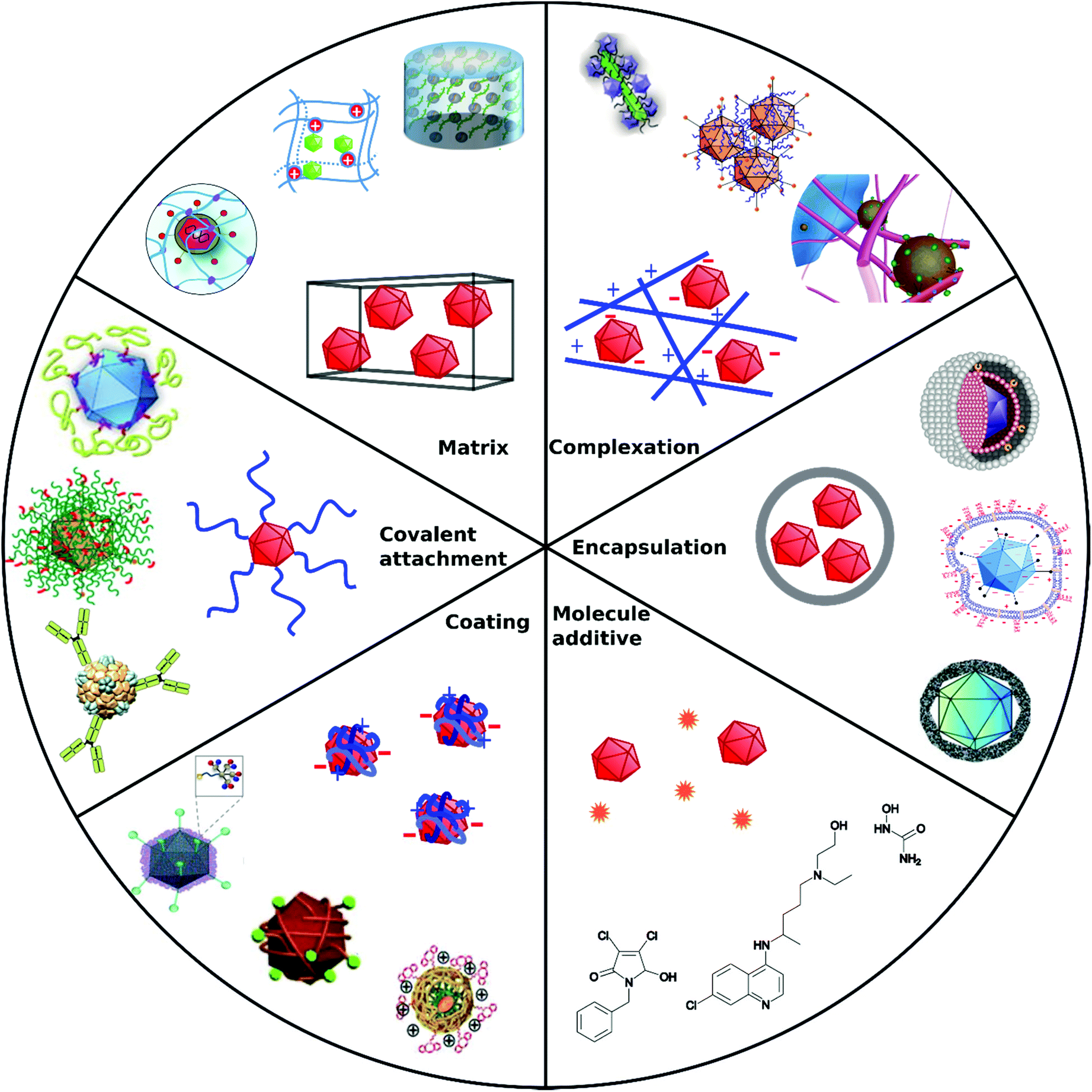 | ||
| Fig. 1 Overview of different architectural formations for chemical delivery methods of VVs. Figure insets in clockwise order starting from top adapted with permissions from: ref. 274. Copyright 2014 The Royal Society of Chemistry, ref. 307. Copyright 2013 Elsevier, ref. 348. Copyright 2020 Springer Nature, ref. 421. Copyright 2019 American Chemical Society, ref. 422. Copyright 2014 Elsevier, ref. 112. Copyright 2012 John Wiley & Sons, ref. 208. Copyright 2019 John Wiley & Sons, ref. 209. Copyright 2012 John Wiley & Sons, ref. 254. Creative Commons BY 4.0. 2020 John Wiley & Sons, ref. 378. Copyright 2020 American Chemical Society, ref. 311. Copyright 2015 Elsevier, ref. 138. Copyright 2016 Elsevier, ref. 263. Copyright 2019 The Royal Society of Chemistry, ref. 291. Creative Commons BY 4.0. 2020 MDPI, ref. 282. Copyright 2012 Elsevier. | ||
| Approach | Main formation pathway | Main function | Benefits | Limitations | Examples | Ref. |
|---|---|---|---|---|---|---|
| Covalent attachment | Bioconjugation chemistry on capsid proteins | Target cell receptors by specific capsid modifications, shield from immune response | Permanent, stable | Interference with capsid functions | Polymers, peptides, | 104–106 and 108–110 |
| Coating | Electrostatic interactions, precipitation | Overcome charge repulsion | Maintain virus capsid functions | Non-permanent, elimination due to cationic charge in vivo | Calcium phosphate, silica, flexible polymers | 112–114 |
| Complexation | Electrostatic interactions | Overcome charge repulsion | Maintain virus capsid functions | Non-permanent, elimination due to cationic charge in vivo | Peptide fibrils, iron nanoparticles | 115–120 |
| Encapsulation | Incorporation in capsules | Shield from immune response, overcome charge repulsion | Stealth VPs | Insulation of capsid functional domains | Liposomes, polymers | 121–125 |
| Matrix | Coincubation/coincorporation/cogelation | Local administration Implantable or injectable matrix | Spatiotemporal release | Degradability, unwanted accumulation | Hydrogels | 126–128 |
| Molecule additive | No formation with VPs | Manipulate cellular processes | Synergistic mode of action, easy to apply | Cytotoxicity, no directed VP diffusion to cell | Cytostatic drugs | 135 |
Enhanced interactions between cells, VPs and additives can be obtained via various pathways.
For cationic delivery agents, binding is mainly driven by attractive electrostatic interactions.129 This results in directed diffusion and colocalization of viral particles and cell membranes, thereby facilitating cell entry. Some materials can further enhance the fusion of viral and cellular membranes.130
Cationic as well as anionic additives with high molecular weight can sediment with VPs onto cells in culture, which increases TE due to increased contact between VPs and cells.131,132
VVs can also be immobilized on surfaces prior to cellular adhesion.133,134
Chemically facilitated cell entry can also be achieved by manipulating the host cell itself. Especially small cytostatic molecules can affect viral infectivity and TE by intervening with cell cycle processes.135
Applying delivery systems able to bypass receptor mediated cell entry can broaden tropism. For example, CAR is downregulated in cancer cells, which makes oncolytic Ad delivery challenging.136 Delivery systems can improve addressing VPs to CAR negative cells and circumvent CAR-mediated endocytosis.116,137–140
A drawback of chemical delivery systems are cytotoxic reactions due to high additive concentrations.114,141 Furthermore, non-degradable additives, e.g. high molecular weight polymers can result in unwanted accumulation and health risks in vivo.142
Delivery systems
Viral gene delivery systems including polymers, peptides, lipids, nanoparticles, and small molecules display diverse modes of action on a wide range of size scales (Fig. 2). The main focus of this review is to highlight materials for promoting viral gene delivery.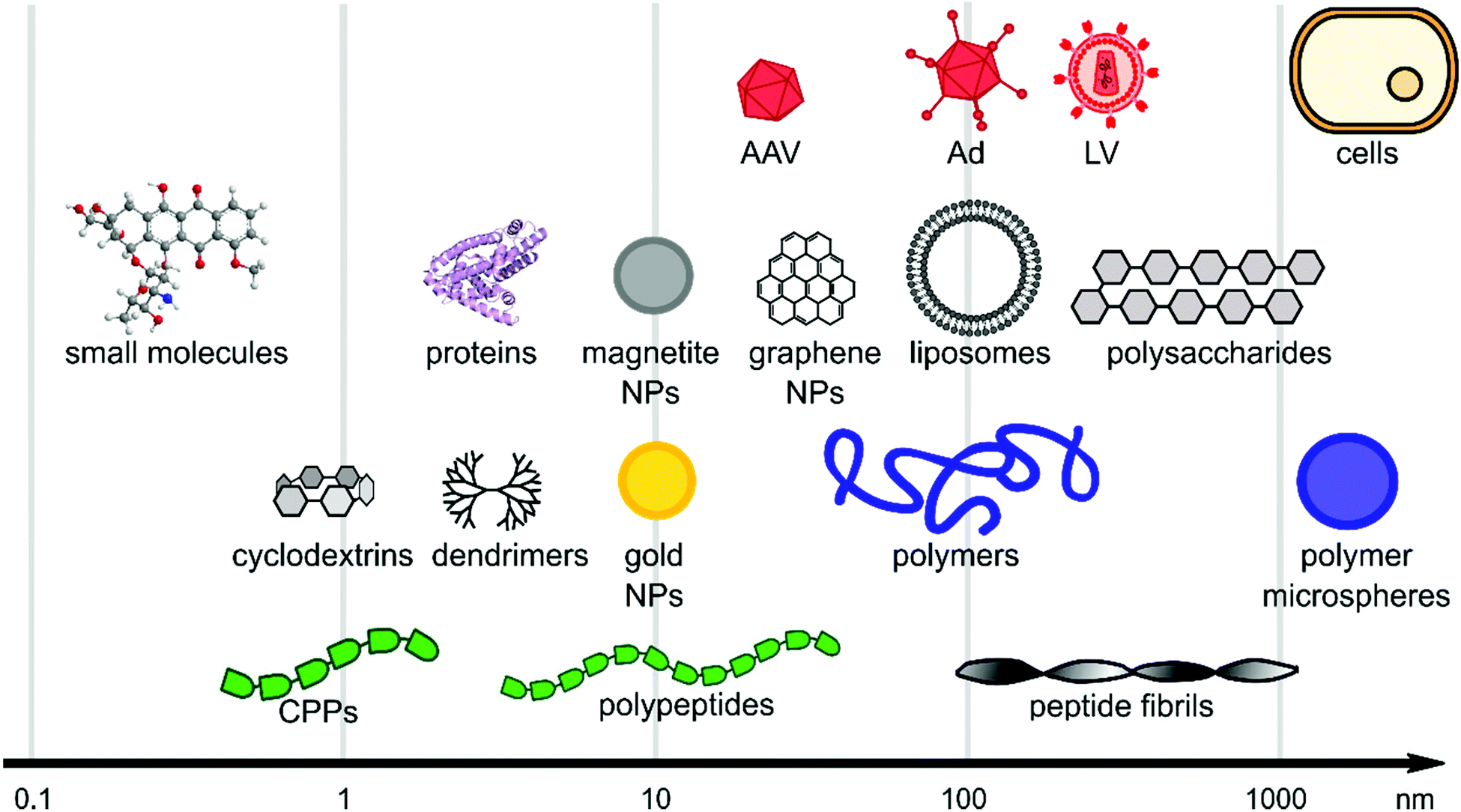 | ||
| Fig. 2 Size scales of selected materials of interest for viral delivery. The x-axis is in logarithmic scale and schematic figures point out size range of material classes. | ||
Polymers
Polymers are the most extensively studied delivery systems for VVs. They typically consist of covalently connected repeating monomeric units and reach a molecular mass of several kDa. This section provides an overview on common and emerging polymeric materials of natural and synthetic origin.Research on polymeric delivery agents began with DEAE-D as one of the first reagents used for VV delivery to mammalian cells in 1965.143 Due to its advantageous ease of use and high transduction efficiency, it has long been a popular delivery agent for various virus types.144,145 Nevertheless, one of the major issues with DEAE-D is cellular toxicity at high concentrations needed for efficient transduction,143 which resulted in a declining interest for its use as a delivery agent for VVs.
Beside DEAE-D, the cationic polymer polybrene was heavily used as an enhancer of viral delivery in vitro in early days.146 Due to its cost efficiency as well as its simple and safe handling it remains a popular additive,147e.g. for enhancing delivery of Ad to human mesenchymal stem cells (hMSCs)148 and as an additive in ultrasound-enhanced RV delivery to the retina.72
In general, it can be summarized that polycationic compounds like DEAE-D, polybrene, poly-L-lysine and protamine sulfate enhance TE, whereas polyanionic compounds like heparin, pyran or cyclodextrin modified with carboxylic groups inhibit TE and reverse the effect of polycationic additives, when combined.146,149–151 However, anionic dextran sulfate in certain concentrations could promote focus formation of Rous sarcoma virus and thereby slightly enhance TE.149 In contrast to these findings, dextran sulfate inhibited TE of HIV-1152 and neutralized enhancing effects of cationic additives.149 The anionic polysaccharide chondroitin sulfate C also showed unexpected increase of TE in combination with polybrene in vitro. This observation was explained by sedimentation of larger polymer–virus complexes on cells in contrast to free diffusion of unmodified viruses in the supernatant solution.131,153
In order to yield VVs with improved safety profile, e.g. protect them from neutralizing antibodies and evade immune response, polymers have been coated154,155 or covalently bound156–158 to viruses. The safety of polymer-coated VVs has been increased by changing tropism and retargeting cells.156 Further, natural and synthetic hydrogels from polymers have been investigated for spatially controlled VV delivery.159,160 Amongst them macroporous hydrogels were utilized for long-term release of VVs,161–164 and thermoresponsive polymer gels were applied as injectable systems.127,163 A selected overview of general structural motifs of the polymers and a summary of all polymers discussed in the following subsections are provided in Chart 1 and Table 3, respectively.
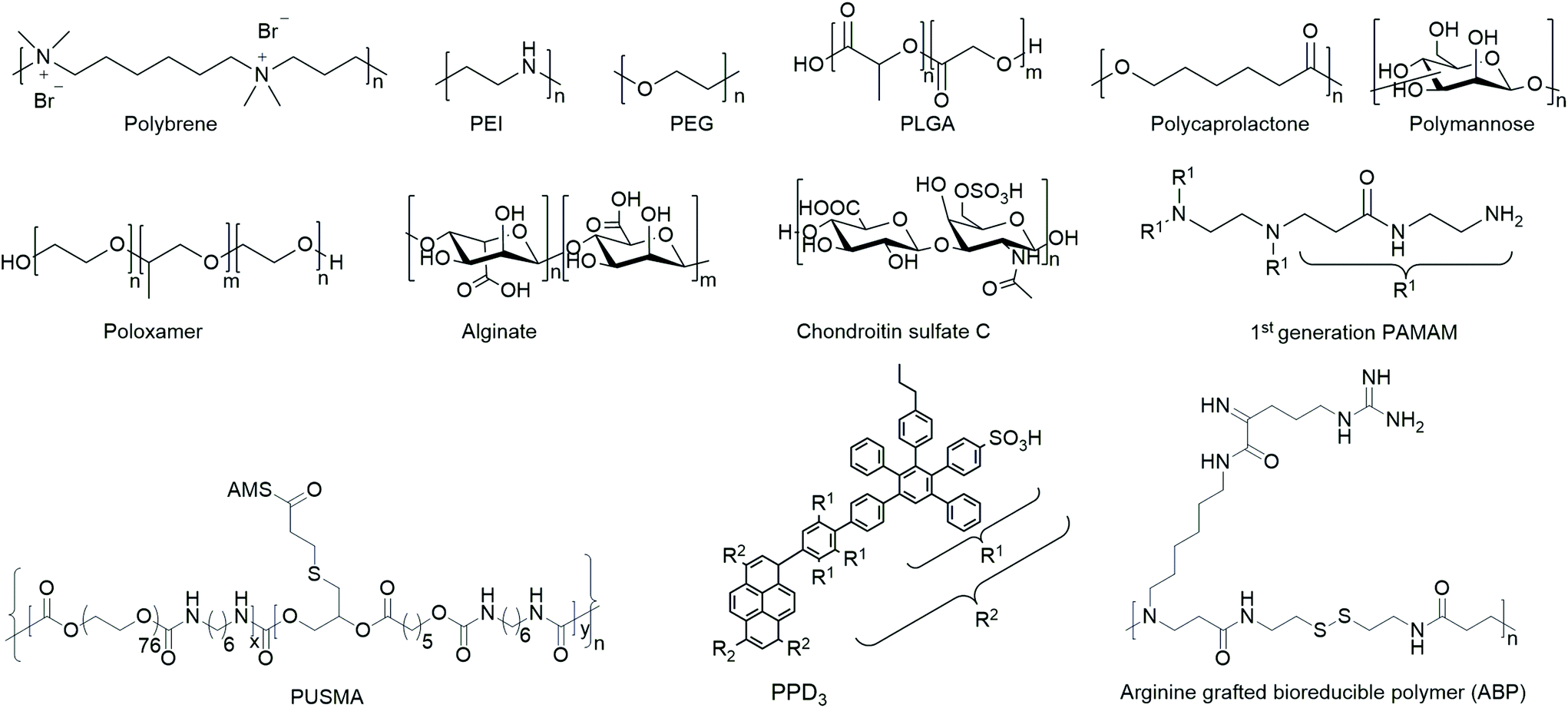 | ||
| Chart 1 Overview of general structural motifs of selected polymers. | ||
| Abbreviation | Compound | Properties | Fabrication of VP-peptide | Ref. |
|---|---|---|---|---|
| Abbreviations: cationic (c.), hydrophilic (h.), hydrophobic (p.), amphiphilic (amph.), anionic (a.), incubation (inc.), encapsulation (enc.) covalent conjugation (cov. conj.), adenovirus (Ad), adeno-associated virus (AAV), lentivirus (LV), retrovirus (RV), tobacco mosaic virus (TMV), murine leukemia viruses (MLV). | ||||
| DEAE-D | Diethylaminoethyl-dextran | c., h. | Inc. (poliovirus/avian sarcoma virus/simian virus 40) | 143, 149, 144, 149 and 145 |
| Polybrene | 1,5-Dimethyl-1,5-diazaundecamethylene polymethobromide | c. | Inc. (sarcoma virus/RV/Ad) | 146, 147 and 148 |
| PEG | Polyethylene glycol | h. | Cov. conj. (Ad/AAV) | 157, 173, 177, 178, 83, 174 and 176 |
| PELA | Poly-DL-lactide-poly(ethylene glycol) | Amph. | Inc. in microspheres (Ad) | 121 |
| PEGbPHF | Poly(ethylene glycol)-b-poly(L-histidine-co-L-phenylalanine) | pH sensitive, h. | Inc., nanoplex formation (Ad) | 185 |
| Poly-histidine/PEG | h., pH sensitive | Inc. (AAV) | 282 | |
| HA–PEG | Hyaluronic acid cross-linked with PEG | Porous scaffold | Inc. (LV) | 162 |
| CDPCP | β-Cyclodextrin–PEI-MMP-cleavable-PEG (MMP-cleavable = GPLGIAGQC) | c. | Inc. (Ad) | 138 |
| APP | PEGylated and taxol-conjugated polymeric arginine grafted poly(disulfide amine) | c., h. | Inc. (Ad) | 187 |
| PEGDA/PLL | Poly(ethylene glycol) diacrylate blended with PLL | h. matrix | Inc. (LV) | 283 |
| PEI | 25 kDa poly(ethylene imine) | c. | Inc. (Ad) | 114 |
| PEI–DEG-bis-NPC | 2 kDa PEI cross-linked with diethylene glycol | h., c. | Inc. (Ad) | 200 |
| PEI–CyD–FA | 600 Da PEI cross-linked with cyclodextrin and folic acid | c., h. | Inc. (Ad) | 199 |
| PEI–DA | PEI conjugated with deoxycholic acid | c., h. | Inc. (Ad) | 207 |
| rPEI | 1.8 kDa PEI cross-linked with cystamine | Branched polymers | Inc. (Ad) | 196 |
| PCDP | PEI cross-linked with cystamine derivative | Bioreducible, c. | Inc. (Ad) | 203 |
| PEI–pGMA | PEI-b-poly(glycidyl methacrylate) | c. | Inc. (LV) | 210 |
| PEI–DPA | PEI functionalized with 3-(3,4-dihydroxy-phenyl) propionic acid (catechol groups) | c., h. | Inc. (AAV) | 209 |
| PEI–DBCO | 1.8 kDa PEI–dibenzocyclooctyl | c., h. | Cov. conj. (LV) | 208 |
| PEI/hyaluronic acid | c., a. | Layer by layer deposition (Ad) | 201 | |
| PEI/chondroitin sulfate | c., a. | Layer by layer deposition (measles virus) | 202 | |
| PEG-g-PEI | 2 kDa PEG grafted on 25 kDa PEI | c. | Inc. (Ad) | 139 |
| Poloxamer 407 | PEO101–PPO56–PEO101 | Viscous oil/gel | Inc. (Ad/LV/AAV)/coinjection (Ad/LV) | 230, 232, 233, 238, 229, 231 and 160 |
| Poloxamer 407/polycarbophil | Viscous oil/gel | Inc. (Ad) | 239 | |
| Poloxamer PF68 and T908 | Viscous oil/gel | Inc. (AAV) | 236 and 237 | |
| LentiBOOST | Poloxamer 338 (PEO141–PPO44–PEO141) | Viscous oil/gel | Inc. (LV) | 240–245 |
| PAMAM, EGFR targeting peptide, PEG | Polyamidoamine G3, G4, G5, peptide: CYHWYGYTPQNVI, 2 kDa PEG | c., dendrimer | Inc. (Ad) | 140 |
| PAMAM, antibody, PEG | PEGylated polyamidoamine G4, 63 kDa, Erbitux® | c., dendrimer | Inc. (Ad) | 251 |
| PPD3 | Polyphenylene 3 | Amph., dendrimer | Inc. (Ad) | 116 |
| PPD3-dendron | One quarter of amphiphilic polyphenylene 3 | Amph. dendron | Inc. (Ad) | 254 |
| PCL | Poly(ε-caprolactone) | Nanofibers | Electrospinning (Ad) | 164 |
| PCL/ELP | PCL blended with elastin like pentapeptide (VPGVG)128 | Nanofibers | Electrospinning (AAV) | 223 |
| PCL–AAV protein tagged | 80 kDa PCL tagged with AAV protein binding to AAV | Microspheres or electrospun fibers | PCL–AAV protein binding | 224 |
| PLGA | Poly(lactide-co-glycolic acid) | p. (LA)/h. (GA) | Enc. (Ad)/lyophilization (LV, Ad) | 155, 133 and 215 |
| PLL/PLGA | Poly (lactic-co-glycolic) acid and poly-L-lysine | Inc. in PLL and enc. with PLGA (Ad) | 213 | |
| PLGA/PLL | 75/25 DL-PLGA 9.4 kDa/PLL 56 kDa | p. (LA)/h. (GA), cat., h. (PLL) | Inc. (Ad) | 213 |
| PLGA/PEG | 50/50 | p. (LA)/h. (GA), h. (PEG) | Cov. conj. of PEG–SPA and enc. in PLGA (Ad) | 214 |
| Alginate | Linear copolymer of [D-mannuronate (β1→4) L-guluronate (α1→4)]n | h. gel | Emulsion (Ad)/cogelation (Ad)/cogelation (AAV, LV)/cogelation (LV) | 258, 259, 294 and 260–262 |
| Alginate/poloxamer 407 | Enc. (rAAV) | 122 | ||
| Chitosan | Poly β-(1→4)-linked D-glucosamine | c., linear polymer | Inc. (MLV) | 269 |
| Chitosan/β-glycerol phosphate | Crosslinked hydrogel | p., c. | Cogelation (LV) | 127 |
| Polymannose | Poly (1→6)-linked α-D-mannose | Linear and branched polymer network | Cov. conj. oxidative via NaIO4 (Ad)/cov. conj. reductive amination (Ad) | 266 and 265 |
| Cellulose-g-P(QDMAEMA) | Cellulose-grafted poly(N,N-dimethylaminoethyl methacrylate) | c., brush polymer | Inc. (cowpea chlorotic mottle virus and norovirus-like particles) | 274 |
| Polyaminoglycoside | Poly hydroxyethyl disulfide diglycidyl ether and tobramycin | Branched c. polymer | Inc. (LV) | 276 |
| β-Cyclodextrin | h. and p. | Cov. conj. (TMV) | 272 | |
| α-Cyclodextrin | α-Cyclodextrin with pluronic PF68 and chondroitin sulfate or hyaluric acid | c., gel mixtures | Inc. (rAAV) | 271 |
| EGDE 3,3′ | Ethylene glycol diglycidyl ether (EGDE) and 3,3′-diamino-N-methyl dipropylamine (3,3′) | Branched c. copolymer | Inc. (Ad) | 295 |
| Polydopamine | Inc. (AAV) on upside down polydopamine coated surface | 290 | ||
| Catecholamines | Polynorepinephrine or polydopamine | Seeding onto surface (AAV) | 289 | |
| pHPMA | Poly(N-(2-hydroxypropyl)methacrylamide) | Polyvalent, h. | Inc. (Ad)/cov. conj. (Ad) | 280, 156, 158 and 279 |
| PUSMA | PEG cross-linked with 1,6-hexamethylene diisocyanate and epsilon caprolactone sulfamethazine | Biodegradable, thermoresponsive sol–gel polymer | Inc. (Ad) | 163 |
| Poly-arginine-g-polydisulfide amine | c. | Inc. (Ad) | 114 | |
| Polyester urethane urea | Copolymer of polycaprolactone diol, butyl diisocyanate, and putrescine blended PEG | Nanofibers | Electrospinning (AAV) | 222 |
| Polystyrene | Polystyrene coated with methyl methacrylate and divinylbenzene | Nanocups | Inc. (VV) | 293 |
| pNaSS | Poly(ε-caprolactone) grafted poly(sodium styrene sulfonate) | c. film | Inc. and immobilization (Ad) | 225 |
| HEMA/APMA | Hydroxyethyl methacrylate (HEMA) with aminopropyl methacrylamide (APMA) | c. hydrogel | Inc. (rAAV) | 291 |
| PVA–VEA | Vinyl ether acrylate-functionalized poly(vinyl alcohol) | pH degradable hydrogel network | Inc. (Ad) | 292 |
| Poly(2-ethyl-2-oxazoline) | Thermoresponsive, h. | Cov. conj. via EDC/NHS (hepatitis B VLP) | 278 | |
| Polyketal | Cross-linked amino ketal methacrylamide and ketal bis methacrylamide mixed with siRNA | Acid degradable | Polymerized on Eosin-5-isothiocynate conjugated AAV | 284 and 285 |
| DNA-aptamer | sgc8-aptamer | Polynucleotide | Cov. conj. (AAV) | 288 |
| 5′-ATCTAACTGCTGCGCCGCCGGG AAAATACTGTACGGTTAGA-3′ | Polynucleotide | Cov. conj. (MS2 bacteriophage) | 286 | |
| Example: TGTGCCAAAGAGAGTGGTGGGGGGGTGGGCGGAACTCGCG | Polynucleotide | Inc. (vesicular stomatitis virus) | 287 | |
Covalent attachment of functional PEG derivatives to Ad capsid proteins, first introduced by O'Riordan in 1999,106 protected the virus from neutralizing antibodies106,157 and displayed reduced clearance rates by Kupffer cells.171 However, PEG-coated stealthy VPs are hindered in cell membrane interaction, which can be tackled by bifunctional multipurpose PEG linkers. Cell specific bioactive peptides attached to these PEG linkers introduced targeted high affinity interaction while showing low immunogenicity even in systemic administration.172,173 Protection from neutralizing antibodies has been shown for PEGylated AAV at certain cross-linking ratios.174 Interestingly, TE of VPs varied depending on the grafting efficiency of the activation agents for covalent attachment of PEG and their susceptibility to induce aggregation of VPs.175 Genetic modifications of AAV resulting in expression of thiol groups on the capsid surface extended bioconjugation methods with thioether or disulfide bonds, e.g. for selective conjugation of NHS-functionalized PEG.83 Moreover orthogonal conjugation of PEG derivatives can also be achieved by click chemistry, when genetically modified AAV capsids presenting azide moieties are used.176
PEGylated Ad with low molecular weight of 5 kDa were successfully applied for accumulation in tumor tissue and showed enhanced permeability and retention (EPR) effect when injected intraveneously.177 Noteworthy, in other studies, PEGylation of Ad with 20 kDa PEG instead of 5 kDa resulted in reduced liver transduction and hepatotoxicity.178,179 PEGylation also shields Ad from pre-existing Ad antibodies. This is relevant for applications in vaccines or gene therapy due to possible pre-existing immunity against Ad in humans.180In vitro TE of PEGylated Ad was reduced compared to in vivo. This observations was traced back to higher cellular uptake with integrin and proteoglycan interaction, which is more strongly induced by pharmacodynamic force in vivo.181 Beside neutralization of Ad by the serum, blood cells can also inhibit Ad activity after systemic administration.182 Addressing this need, PEGylated Ad has been used to circumvent endothelial cell activation from blood cells and thereby improve TE and safety.183
However, a crucial issue is the so-called PEG-dilemma. Concurrent to EPR effect, PEGylated viruses show lower cellular uptake and lower endosomal escape eventually leading to reduced TE. In order to overcome this problem, PEG was combined with other polymers as reviewed elsewhere for non-viral gene delivery.184 Copolymers from PEG include spherical poly-DL-lactide particles,121 pH-sensitive poly(L-histidine-co-L-phenylalanine),185 micellar poly(disulfide amine),186,187 degradable gelatin161 to name just a few examples. Fan et al. reported an elaborate design of a β-cyclodextrin–PEI-MMP-cleavable-peptide–PEG polymer (CDPCP, Fig. 3). This polymer design aimed to overcome liver accumulation by using PEG 5 kDa, and further to electrostatically coat Ad by grafting PEG onto a high-molecular weight, branched PEI-cyclodextrin copolymer via matrix metalloproteinase (MMP) sensitive peptide linkers (GPLGIAGQC). These peptides are cleaved in the environment of the tumor and uncover positive charge of PEI thus enhancing cellular uptake and Ad gene delivery. Thereby CDPCP avoids liver accumulation, inhibits Ad blood cell interaction and prolongs circulation time.138
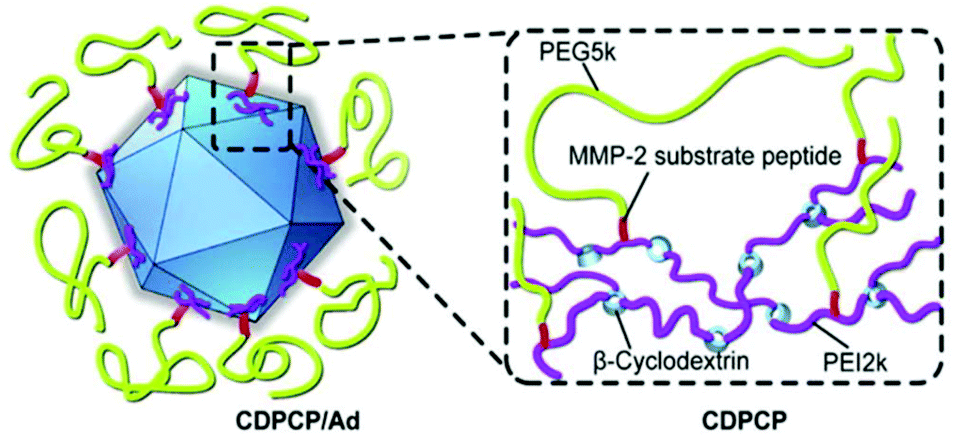 | ||
| Fig. 3 Schematic diagram of Ad conjugated to enzymatically cleavable PEG–PEI–β-cyclodextrin (CDPCP). Adapted with permission from ref. 138. Copyright 2016 Elsevier. | ||
These advances achieved in stimuli-responsive targeted delivery have the potential to be a valuable tool for systemic administration of viruses, e.g. of oncolytic viruses to tumor tissue, in future clinic applications. However, despite extensive use of PEG as a delivery system, in the 2010s, unexpected immune response and accelerated blood clearance of PEGylated therapeutics were reported.188,189 Consequently there is a need for alternative polymeric delivery systems.190
Moreover cholesterol conjugated to PEI can facilitate cell entry via an energy- and endocytosis-independent membrane translocation pathway, originally known from cell penetrating peptides.204,205 The amphiphilic combination of bile acids like deoxycholic acid and the cationic PEI efficiently transport cargo206 and further enhance TE of Ad in CAR negative cells.207
An elaborate approach of PEI assisted gene delivery was applied by Pan et al. for genetic manipulation of primary T-cell to target malignant tissue as new potent immunotherapeutics. In a stepwise process, they first labelled T-cells with azide groups by metabolic incorporation of azide-glucose onto the membrane, then adding PEI-dibenzocyclooctyl-complexed LV. Conjugation of azide-dibenzocyclooctyl via click chemistry resulted in colocalization of complexed LV with T-cells, which facilitate interaction and robustly enhance TE in T-cells (Fig. 4A). In vivo experiments with tumor-bearing mice displayed significantly longer survival times than the control group or polybrene-assisted delivery.208
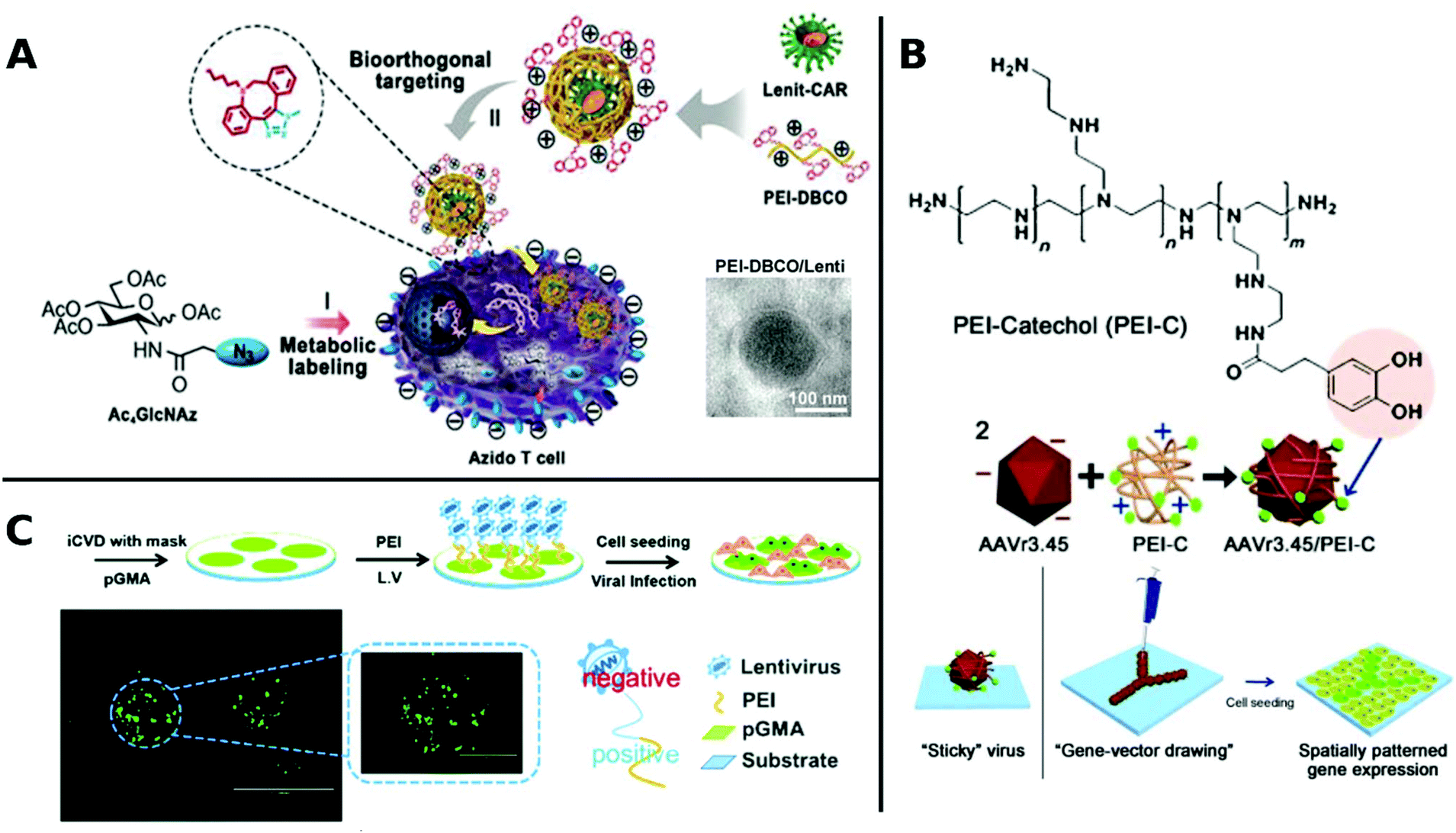 | ||
| Fig. 4 A. Schematic illustration of PEI-DBCO/azide-glucose system in T-cells. LV are coated with PEI-DBCO, which is conjugated to azide-glucose pretreated T-cells via click-chemistry. Inlet TEM-image shows morphology of coated LV (scale bar: 100 nm). Adapted with permission from ref. 208. Copyright 2019 John Wiley & Sons. B. Formation of AAV/PEI-C hybrid particles and schematic illustration of immobilization and patterning of sticky vectors. Adapted with permission from ref. 209. Copyright 2012 John Wiley & Sons. C. Schematic fabrication process of viral delivery platform by subsequently coating of pGMA, PEI and LV attached by electrostatic interactions. Representative image of spatially patterned GFP-expression of transduced HUVECs (scale bar: 200 μm, inlet image: 400 μm). Adapted with permission from ref. 210. Copyright 2019 The Royal Society of Chemistry. | ||
Delivery systems with spatially resolved gene expression have the potential to mimic the complexity of biological systems. An interesting example is branched PEI, which was functionalized with catechol groups and coated onto AAV. In this way, viral particles were obtained which could be placed and immobilized on tissue culture plates by a micropipette (Fig. 4B). Increased amounts of polymer coatings led to more stable immobilization resisting several washing steps and allowing to pattern viral particles. Thus, spatially resolved gene expression and cell manipulation could be achieved.209 Recently, spatially defined transduction has been further simplified by coating poly(glycidyl methacrylate) copolymerized PEI as viral affine substrate. LVs were immobilized simply via electrostatic interactions and colocalized with HUVECs by incubation on this platform, resulting in enhanced TE and maintained cell functions (Fig. 4C). Further, patterned transduction and GFP expression could be generated by covering the substrate with a mask during fabrication.210
In recent years, PEI has evolved from a simple polymeric additive to a popular element in copolymeric formulations for enhancing gene delivery, as copolymers can mediate inherent drawbacks of PEI such as high toxicity.
While Ad encapsulated in PLGA displayed greater cell viability and lower immunogenicity in vivo, it did not enhance TE.155 The TE could be enhanced by PEGylation214 before the encapsulation step and by surface modification of PLGA spheres with polysaccharides.12
In a tissue engineering approach, LV or Ad was immobilized in a PLGA scaffold and ensured long-term transgene expression over a period of four weeks in vivo. LV or Ad were incorporated in the PLGA scaffold by consecutive lyophilization of both components together with sucrose. Additional fibronectin and collagen could further enhance binding but not TE for LV delivery.133 Zhu et al. incorporated Ad in PLGA scaffolds for bone regeneration in vivo. The electrospun PLGA yielded a nanofibrous scaffold, which enabled spatially defined, long-term gene expression of Ad encoding hBMP2 to promote osteogenic differentiation. This technique was able to cover more than 80% of the bone defects that could not be repaired in the control groups within eight weeks.215 Another electrospinning approach coated oncolytic vaccinia virus onto PLGA nanofibers to maintain their antitumor activity at the tumor tissue site.216 Furthermore, melt-processing was reported as a solvent- and additive-free approach for fabrication of PLGA–VP scaffolds. This homogenous dispersed composite materials for prolonged VP release are created by heating up PLGA and VP.217
Another example of polyesters is polycaprolactone (PCL) which is widely used as long-term implants in biomedical applications due to their good mechanical properties, slow degradability and biocompatibility.218 In recent years, electrospinning techniques emerged as a promising process to create PCL fibrous structures for sustained and localized gene delivery.219,220 In order to incorporate cargo to the fibers, co-axial spinning techniques can be utilized, in which an inner jet with cargo molecules and outer jet with polymer solution overcome the limitations of single stream electrospinning.221
In one such approach, Ad was encapsulated in PCL in a core–shell fashion for subsequent porogen-mediated release. Pores of approx. 3 μm size were created by leaching out incorporated low molecular weight PEG particles. The fibrous scaffolds enabled efficient local TE and reduced macrophage activation to attached cells.164 Similar results were obtained with PCL containing copolymers of polyester urethane urea and polyester ether urethane urea electrospun together with low and high molecular weight PEG and AAV to yield various fibrous nanostructures for sustained delivery to cardiac tissue over a period of 2 months in vitro (Fig. 5A).222
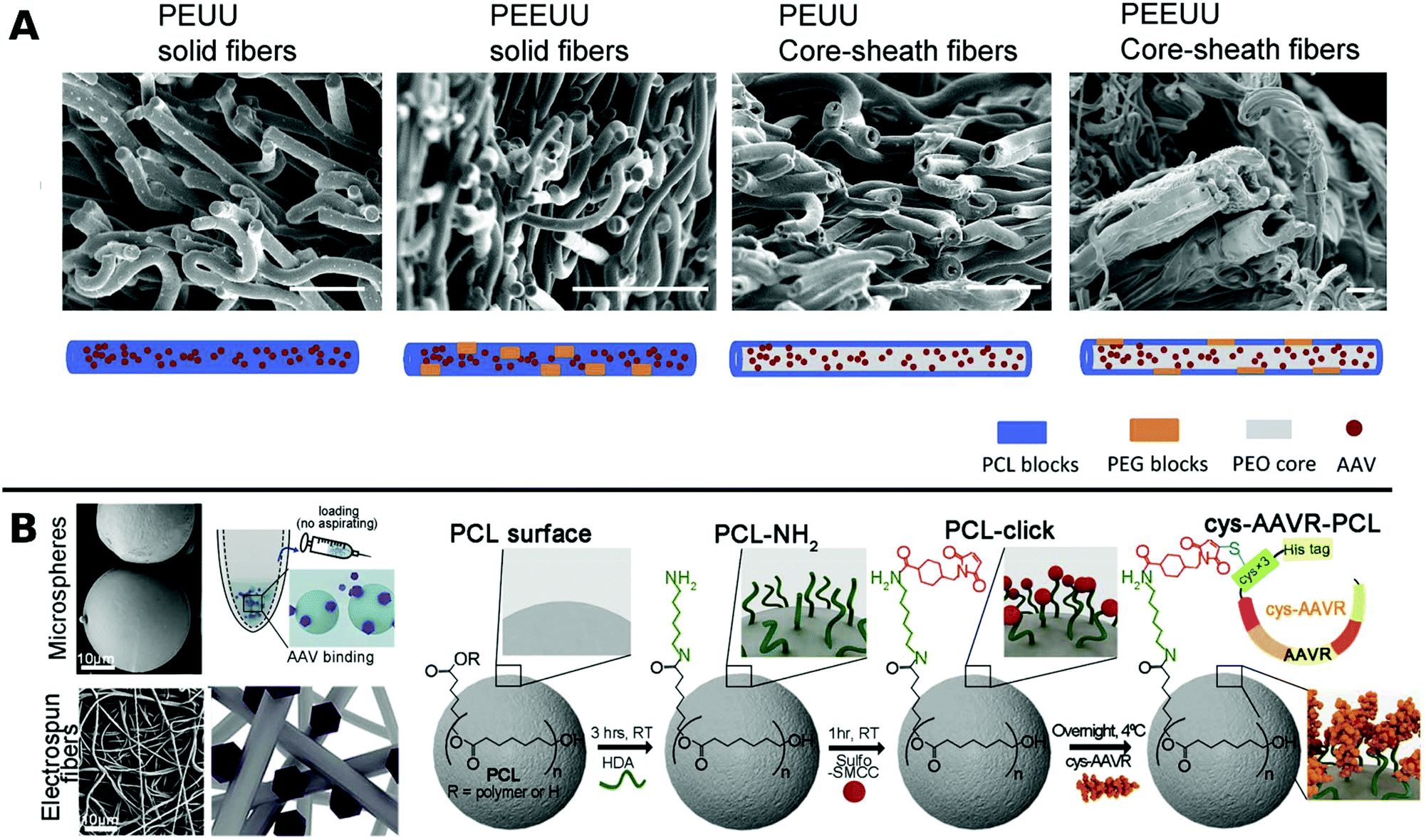 | ||
| Fig. 5 A. SEM images at the cross sections of freeze dried polyester urethane urea (PEUU) and polyester ether urethane urea (PEEUU) scaffolds and schematic illustration of rAAV encorporation strategy (scale bar: 10 μm). Adapted with permission from ref. 222. Copyright 2017 Elsevier. B. SEM images representing PCL microspheres (top) and electrospun fibers (bottom) (scale-bar: 10 μm) with schematic diagram of AAV–PCL complexation. Reaction scheme shows steps to fabricate AAV tagged PCL. Adapted with permission from ref. 224. Copyright 2019 Elsevier. | ||
Blending PCL with elastine-like pentapeptide (ELP) together with AAV resulted in nanofibrous scaffolds with adjustable degradability dependent on the polymer ratio for controlled AAV release.223 Moreover, cysteine tagged proteins binding selectively to AAV, were attached to maleimide-displaying PCL microspheres or electrospun fibers and gave access to a therapeutic platform for intramuscular injection or subcutaneous implantation with reduced off-target (Fig. 5B).224
Recently, poly(ε-caprolactone)-grafted poly(sodium styrene sulfonate) films immobilized rAAV and enhanced gene delivery to hard-to-transduce hMSCs in vivo, thus enabling less invasive, effective treatment of focal cartilage lesions.225
PLGA and PCL were successfully applied as highly versatile platforms mainly as electrospun fibers and microspheres with controllable spatiotemporal gene delivery, especially considering applications in tissue engineering. Due to its biocompatibility and slow degradation rate both materials, PLGA and PCL has proven to be a reliable scaffold and a promising material especially for local, long-term gene delivery in vivo.
Poloxamers are sold in various block lengths and commonly used in pharmaceutical and cosmetic applications as surfactants with no safety concerns.227 More recently, poloxamers have also been applied as gels for gene delivery, as they exhibit thermal gelation behavior at concentrations higher than 20%.228 For example, suspensions of VPs cooled at 4 °C can be gelated at physiological temperature around 37 °C, which gives access to homogeneous, injectable, spatially precise viral delivery in vivo.229 However, a potential adverse effect of poloxamer injection concerning the temporal accumulation of microclots in vessels and organs has been reported.142
Several studies have shown enhanced TE of localized viral delivery by poloxamer 407 (PEO101–PPO56–PEO101) amongst others to vascular smooth muscle cells in vitro,230 to arteries in vivo,231 to the central nervous system,232 to solid tumors in vivo,229 and for treatment of spinal cord injury.160
Rey-Rico et al. applied a series of poloxamers for enhanced AAV delivery to hMSCs even in the presence of host cell receptor binding inhibitory heparin or VP neutralizing antibodies. Prior to gelation, the VPs were encapsulated in poloxamer micelles to shield from adverse conditions, and cell viability was maintained to 100% over a period of 21 days during gene transfer in culture.233 Long-term expression of AAVs to hMSCs have previously been reported for fibrin or RAD16-1 encapsulated viruses for shorter periods compared to poloxamer encapsulation.234,235 Linear poloxamer PF68 and four branched poloxamer T908 encapsulated rAAV in a micellar architecture and enhanced spatiotemporal gene delivery efficiency to osteoarthritis chondrocytes in vitro.236,237
Recently, treatment of cartilage damage was applied in a clinically relevant in vivo minipig model with rAAV delivered by poloxamer 407 (Fig. 6). Controlled release of the therapeutic SOX9 from the poloxamer gel improved repair of full-thickness chondral defects.238
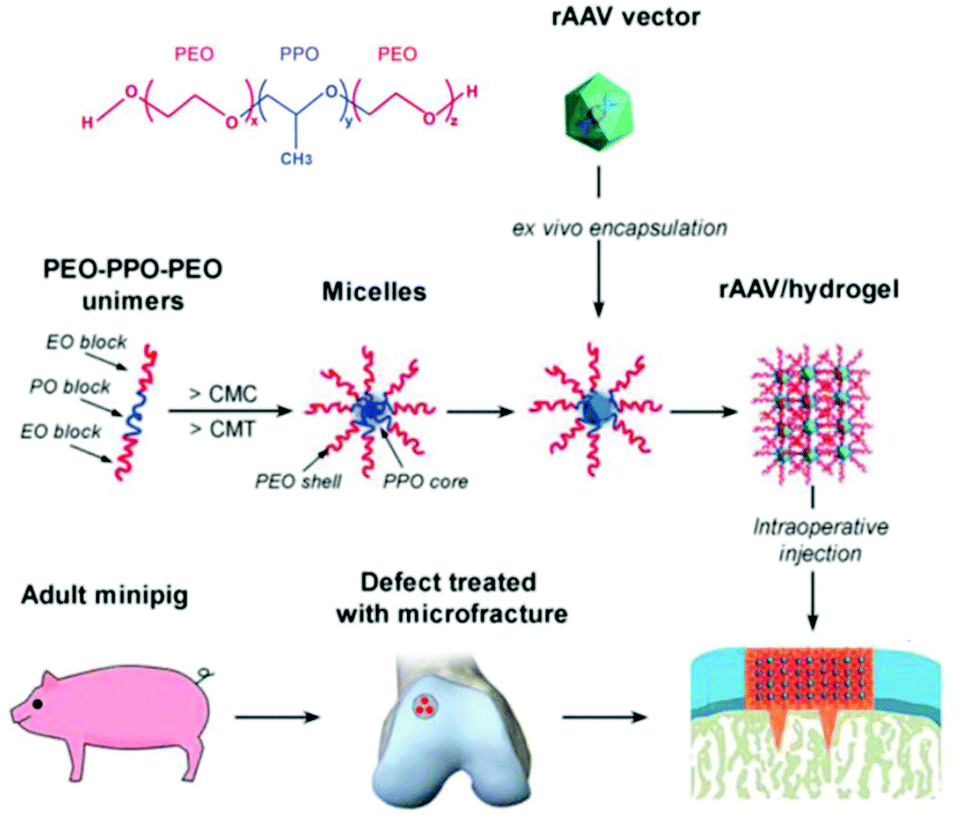 | ||
| Fig. 6 Structure of poloxamer 407 and flowchart to generate rAAV poloxamer hydrogel systems for controlled release in knee implantations for minipigs. Adapted with permission from ref. 238. Creative Commons BY 4.0. 2019 John Wiley & Sons. | ||
To further enhance adhesiveness of poloxamer 407 for local viral delivery to organ surfaces, it was blended with >1% polycarbophil, a polyacrylic acid cross-linked with divinyl glycol. Addition of polycarbophil changed the sol–gel transition temperature and led to higher adhesiveness due to numerous carboxylic groups, which can easily form bonds with surrounding molecules. Spatially resolved and stable Ad delivery to heart tissue was shown in vitro and in vivo without having adverse impact to TE or cell viability.239 Poloxamer 338 (PEO141–PPO44–PEO141), also traded as synperonic F108, enhanced LV delivery in difficult-to-transduce T-cells more efficiently than polybrene. In combination with polybrene the TE was further elevated, which was explained by the distinct modes of each adjuvant, polybrene compensating electrostatic repulsion and poloxamer 338 fluidization the membrane.240
More recently, poloxamer 338 was commercialized as LentiBOOST™, and applied for LV delivery to CD34+ stem cells,241–243 CD4+ and CD8+ stem cells,244 and T-cells.245 Enhanced TE, non-toxicity and clinical relevance were emphasized in all of these studies. The evolving progress in poloxamers as adjuvants for viral delivery in clinical application holds great promise for improved gene-based treatments.
Early works focused on inhibiting virus infections248 or were used to control assembly249 of viruses without any relation to gene delivery.
Polyamidoamine (PAMAM) is a common cationic dendrimer frequently used in non-viral gene delivery250 and was applied for the first time to enhance TE of Ad by Vetter et al.140 An epidermal growth factor receptor (EGFR) targeting peptide sequence was coupled to the dendrimer via a PEG-linker and coated on Ad, which resulted in increased specific infection to EGFR overexpressing tumor cells. The PAMAM complexed Ad showed lower cellular toxicity and higher TE than branched PEI.140 EGFR targeting antibodies attached to PEGylated PAMAM also showed enhanced TE of Ad and tumor suppression in vivo.251
Further, polyphenylene dendrimers (PPD) were applied for viral delivery. PPD have amphiphilic properties forming a rigid globular architecture in solution with functional end groups organized like a shell in the periphery.252 These features make PPD a promising protein-mimicking drug delivery agent.253 Recently, Wu et al. reported PPD3 with engineered amphiphilic surface patches mimicking a protein corona, which enabled non-electrostatic interactions with Ad. The capsid proteins were complexed by PPD due to amphiphilic interactions, which resulted in enhanced TE to CAR negative cells and protection from neutralizing antibodies and the coagulation factor X.116 Further remodelling of PPD3 led to one amphiphilic dendron branch, which was one quarter of the original PPD3 size (Fig. 7). This minimized dendron branch allowed Ad binding and non-covalent post-modification of viral capsids while maintaining advantageous properties of PPD3.254 One disadvantage in using dendrimers is their laborious synthesis. Branched polymers carry a large number of functional groups, similar to dendrimers, but are often easier to synthesize. One example is the cationic copolymer EGDE 3,3′ containing the monomer ethylene glycol diglycidyl ether (EGDE) and 3,3′-diamino-N-methyl dipropylamine (3,3′), which induced higher TE of Ad to bladder cancer cells compared to the transfection reagent PEI.255
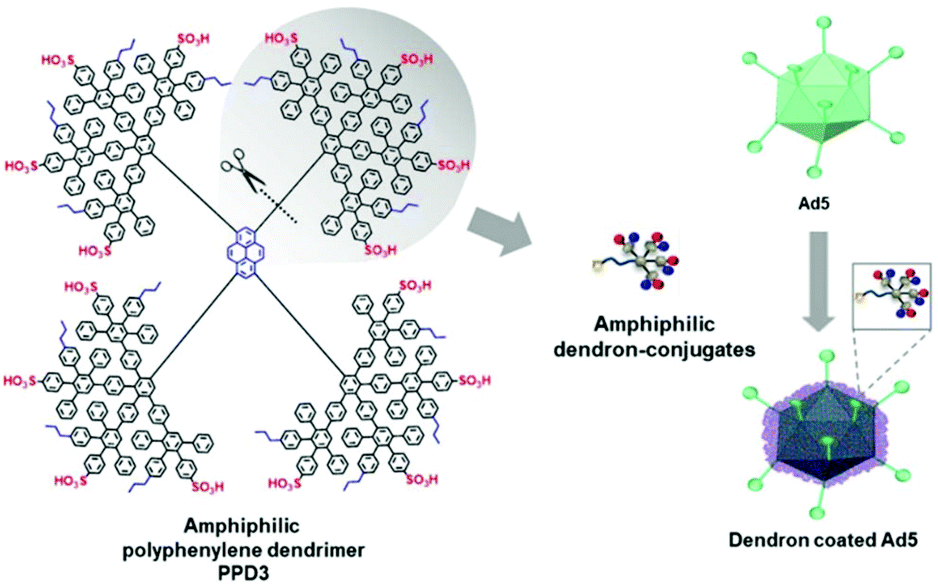 | ||
| Fig. 7 Structure of amphiphilic polyphenylene dendron derived by desymmetrization of PPD3 dendrimer. Schematic chart shows dendron coated Ad5. Adapted with permission from ref. 254. Creative Commons BY 4.0. 2020 John Wiley & Sons. | ||
Alginate is a linear copolymer consisting of β-D-mannuronate and α-L-guluronate and is commonly used as a hydrogel in food additives or pharmaceutical applications. Even though unmodified alginate hydrogels cannot interact specifically with mammalian cells,257 a lot of progress has been made using such gels in controlled VV release in recent years. Ad encapsulated in alginate circumvent immune response in vivo258 and augment long-term oncolytic Ad infection to tumor cells.259 Distinct hydrogel-rAAV capsules were formed by tuning the composition and cross-linking temperature for alginate and poloxamer 407 containing systems. All of these rAAV capsules showed enhanced targeted delivery to hMSCs.122 The spatiotemporal release kinetics of VVs can be stunted or enhanced by hydrogel physiochemical properties.260,261 For example fabrication methods determine hydrogel mesh size, matrix affinity interactions and degradability and thus result in varying release kinetics for different sizes of VVs. LV delivery from CaCO3 or CaCl2 gelated alginate microcapsules generated by a microfluidic technique correlated with mechanical properties like hydrogel network mesh size and degradation rate and was controllable by the gelation method.261 Moreover, low molecular weight alginate gels show faster degradation and release of LV, compared to more sustained release from high molecular weight alginate.260 On-chip fabrication of alginate microgels gave access to blends of adjustable alginate formulations in a bottom up approach (Fig. 8A). Depending on the composition of different alginate molecular weights the degradation and release time of LV could be controlled in vivo.262 Building on these results, Madrigal et al. reported that physical properties of various formulated alginates impact AAV and LV delivery to a different extent (Fig. 8B). LV release was tuned by initial strength and degradation rate of alginate gel, whereas AAV delivery remained unchanged independent of formulation. This result was interpreted as release of AAV by diffusive transport, whereas LV release is mainly controlled by degradation rate of alginate, which may be due to mesh/VP size relation. Consequently fast degradable alginate gels lead to higher TE for both AAV and LV.263
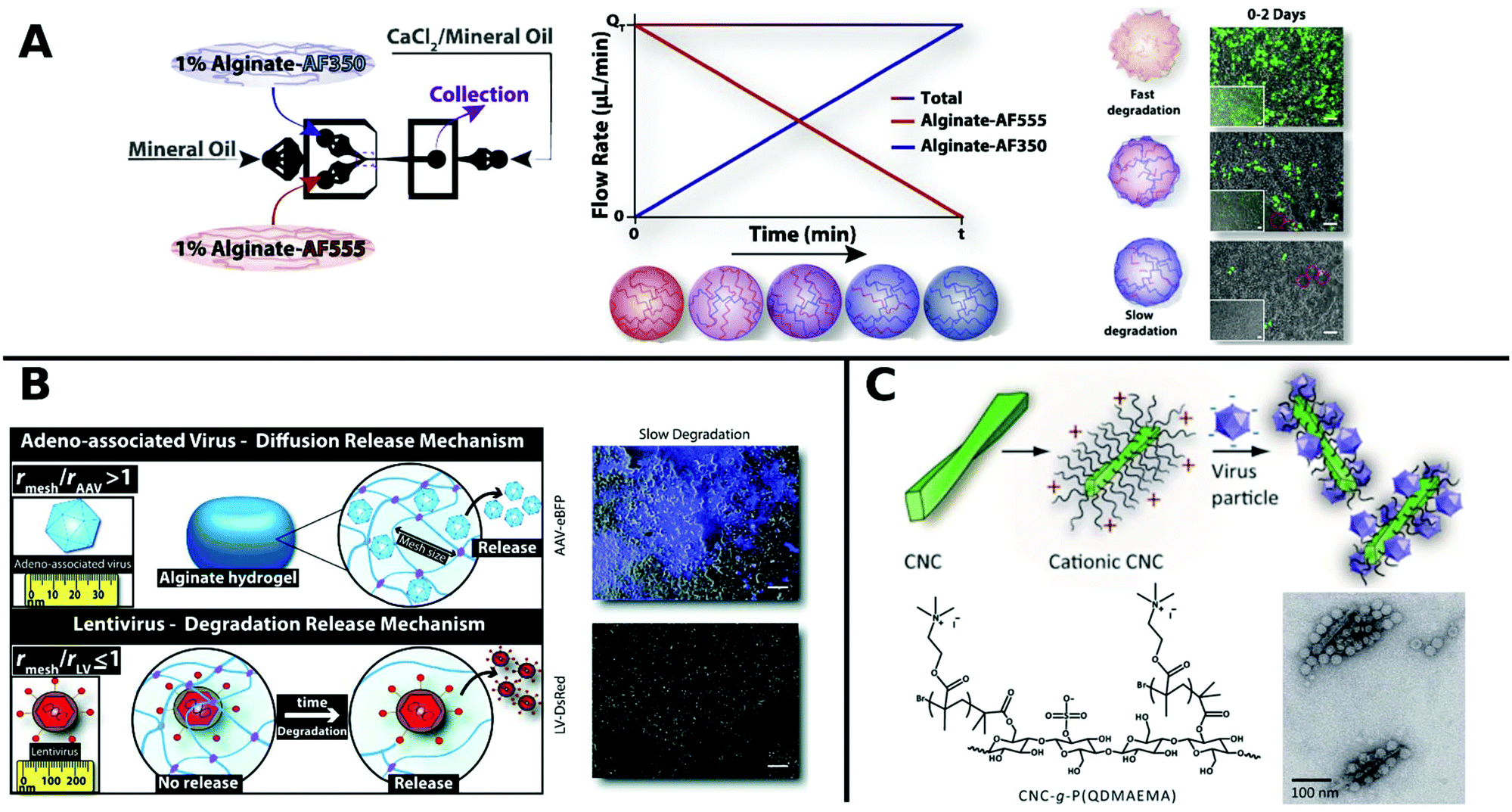 | ||
| Fig. 8 A. Generation of gradient alginate microgel suspension via microfluidic blending. Gradual replacement of degradable alginate-AF555 formulation with non-degradable alginate AF350 leads to microgel composites with controllable degradation and LV release rates as observed in promoted GFP expression in HEK-293T cells by merged phase-contrast/fluorescent photomicrographs (scale bar: 100 μm). Adapted with permission from ref. 262. Copyright 2018 Elsevier. B. Illustration depicting the diffusion controlled release of AAV and degradation limited release of LV from alginate hydrogels. Merged phase-contrast/fluorescent images show gen-expression achieved after first day for AAV and LV in slow degradable alginate hydrogels (scale bar: 100 μm). Adapted with permission from ref. 263. Copyright 2019 The Royal Society of Chemistry. C. Schematic diagram shows the formation of VPs complexed with colloidal cellulose nanocrystals (CNC). TEM micrographs show CCMV complexed with CNC-g-P(QDMAEMA) (scale bar: 100 nm). Adapted with permission from ref. 274. Copyright 2014 The Royal Society of Chemistry. | ||
Mannose receptors were highly expressed in liver tissue and endocytosis-mediated cell entry is enhanced in the presence of mannose.264 In order to target hepatocellular carcinoma, polymannose was covalently attached to Ad surface either by reductive amination265 or by oxidation with sodium periodate.266 The polymannose–Ad conjugate showed enhanced gene delivery to hepatocellular carcinoma cells both in vitro and in vivo.266
Chitosan is a non-toxic, biodegradable cationic polymer, that shows neuroprotective effects after spinal cord injury by sealing nerve cell membranes267 and promotes peripheral nerve regeneration.268 Cross-linked with β-glycerol phosphate, chitosan yields compact fibrous hydrogels with increased charge density and binding to anionic particles as well as long-term gene expression over a period of seven days of encapsulated LV to dorsal root ganglia neurons.127 Furthermore, chitosan has been used to replace the function of the viral envelope in non-infectious murine leukemia virus (MLV), which led to increased infectivity and transduction.269 Hyaluric acid (HA) is a highly abundant linear polysaccharide in the extracellular matrix. HA applied as an injectable in situ forming scaffold can generate macroporous structures which are attractive vehicles for localized long-term release of viral particles. In a recent report, HA scaffolds of various pore sizes for LV delivery in mice mammary fat were compared with each other. Void spaces in HA scaffolds were created by different fabrication techniques. Nanoporous structures were achieved by cross-linking HA particles with PEG precursors, while macroporous architectures were created through in situ assembly of HA particles with PEG particles or enzymatic degradation of included PEG particles. Open, macroporous HA–PEG hydrogels displayed increased host cell infiltration and yielded higher TE compared to nanoporous hydrogels.162
Cyclodextrins (CD) are composed of 6–8 (α–γ) cyclic arranged glucose subunits forming a hydrophobic interior and a hydrophilic exterior toroid-shaped oligosaccharide, which is commonly used in pharmaceutical applications for drug delivery.270 CDs have been applied as hydrogels in combination with other polymers138,271 or as supramolecular linkers272 for gene delivery as recently reviewed elsewhere.273 Examples include Ad delivery to tumor microenvironment with a responsive polymer design containing PEG, PEI, MMP-sensitive peptide and β-CD.138 Further, polypseudorotaxane gels based on either HA or chondroitin sulfate combined with α-CD were used to encapsulate and release rAAV to hMSCs. α-CD enhanced the viscoelasticity and storage modulus of the gels at physiological temperature and prolonged the permanence at the application site.271 CD were also covalently attached to TMV surface in a supramolecular strategy to enable facile host–guest interactions with adamantyl moieties of imaging agents or chemotherapeutic drugs.272
In order to overcome anionic surface charges of some polysaccharides, modifications and combination approaches with other polymers have been made. For example, cellulose nanocrystals were surface-modified by atom-transfer radical polymerization with poly(N,N-dimethylaminoethyl methacrylate) to yield a brush-like cationic polymer (Fig. 8C). This polymer showed high-affinity virus binding.274
Bioreducible, branched polyaminoglycosides involving the antibiotic aminoglycoside tobramycin were proposed by Xu et al. as a transfection reagent.275 Recently, they reported, LV–polyaminoglycoside complexes, which efficiently induce cell apoptosis to glioma cells by facilitating cellular uptake via endocytosis pathways.276
Polysaccharides are highly suitable biopolymers for viral gene delivery, mainly as hydrogels that adapt to the tissue environment. Release of VPs is achieved by degradation into non-toxic components. Combination of polysaccharides with polymers and functional molecules can further expand the range of material properties and influence controllable and efficient delivery of VPs in future applications.
Several examples of hydrophilic polymers with similar stealth properties to PEG have been reported. For example, poly(2-oxazoline)s are thermoresponsive, hydrophilic polymers which gained increasing attraction for biomedical applications.190,277 Recently, hepatitis B-like viral particles grafted with poly(2-ethyl-2-oxazoline)s were reported to reduce antigenic reaction.278
Similarly, poly(N-(2-hydroxypropyl)methacrylamide) (pHPMA) was conjugated through lysine residues on Ad surface and was able to shield from neutralizing antibodies, enhanced plasma circulation, decreased hepatotoxicity and indirect accumulation in tumor site. However, coating with pHPMA prevented efficient cellular uptake and reduced TE.156,158,279 Francini et al. applied a new conjugation strategy with pHPMA bearing diazonium reactive groups, which can be bioconjugated onto a variety of functional amino acid residues of oncolytic Ad resulting in more efficient coupling and dense coverage of surface. While immunogenicity of conjugated VPs is low, TE is severely reduced due to low cellular uptake and delayed unpackaging of the vector. This study illustrates the necessary balance between efficient screening of the virus from being recognized by the immune system and sufficient cellular uptake in order to achieve effective transduction.280 To take advantage of benefits from different systems, hybrids consisting of polymers and peptides have been investigated for enhancing TE. For example, arginine grafted onto poly(disulfide amine) were able to enhance TE, while being bio reducible and less immunogenic, when coated onto Ad.114,281 AAV in hydrogels from PEG incorporated with poly-L-histidine showed ratio-controlled and pH-dependant swelling and TE (Fig. 9A).282 PEG diacrylate matrices (PEGDA) blended with PLL can improve long-term, localized and efficient LV transduction when implanted in vivo.283 Further, Kwon et al. introduced elaborate viral/non-viral chimeric systems by siRNA or DNA encapsulating polyketals assembling in a core–shell structure around AAV. This design enabled simultaneous gene transduction in a stimuli-responsive fashion by using of polyketals that are degraded in the acidic endosomal environment. Chimeric systems are promising platforms for obtaining synergistic therapeutic effects by simultaneous expression and silencing of multiple genes. In cancer therapy, for example, simultaneous upregulation of pro-apoptotic mediators by AAV delivery and silencing of pro-survival genes by siRNA, can result in significantly more effective treatment.284,285 Beside classical polymer systems, DNA-aptamers have also been utilized for targeted gene delivery. DNA-aptamers were covalently attached to viral capsid and selectively targeted Jurkat T cells and delivered cargo through an endocytic pathway.286 DNA aptamers can further improve biocompatibility of viruses by shielding them from neutralizing antibodies and enhancing in vivo circulation rate.287 Reducible disulfide linkages were utilized to covalently attach AAV to multiple DNA-aptamers, which were cleaved by intracellular glutathione and facilitate release of AAV in the cell, thereby enhancing TE.288
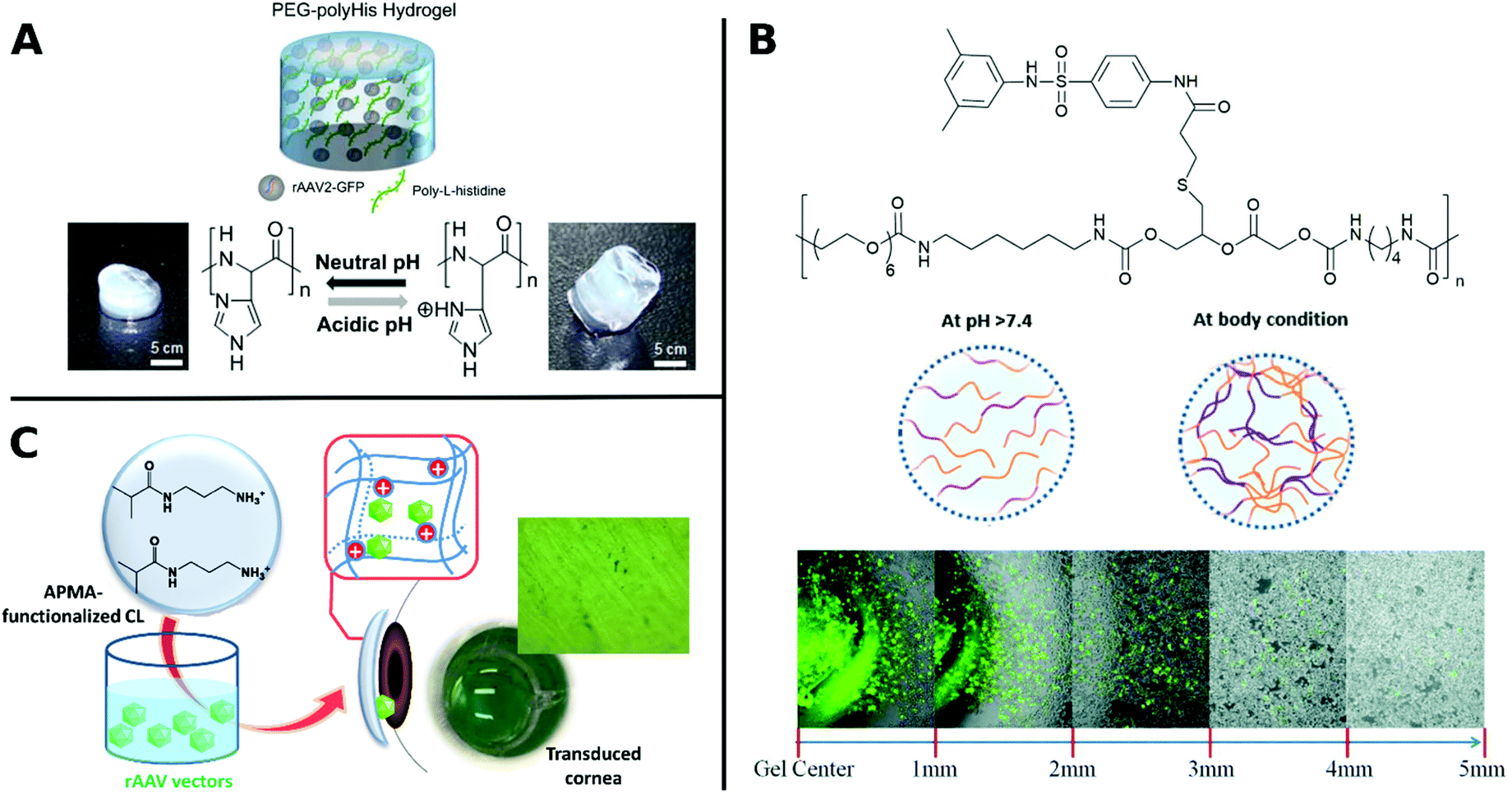 | ||
| Fig. 9 A. Schematic illustration of rAAV embedded into PEG-polyHis hydrogel incubated under neutral (pH 7.4) or acidic (pH 6.0) conditions. Protonation of amine groups in polyHis under acidic conditions result in increased water uptake and swelling of the hydrogel. Adapted with permission from ref. 282. Copyright 2012 Elsevier. B. Structure of PUSMA and schematic illustration of sol–gel phase transition state at physiological conditions. Merged optical/fluorescence image of distance-dependent release of GFP-expressing Ad from PUSMA hydrogels. Adapted with permission from ref. 163. Copyright 2019 The Royal Society of Chemistry. C. Illustration of functionalization of APMA-hydrogel contact lenses for sustained VV delivery to cornea and picture of X-Gal stained bovine cornea after seven days in direct contact with rAAV encapsulated hydrogels. Adapted with permission from ref. 291. Creative Commons BY 4.0. 2020 MDPI. | ||
Polymers can also be used as 2D coatings to promote transduction by colocalization of viruses with host cells. So-called substrate mediated delivery was enabled via AAV capturing adhesive catecholamine surfaces (Fig. 10A).289 Adhesive polydopamine-coated substrates were further improved in an simple upside down arrangement to adhere inverted quasi spherical droplets containing human neural stem cells (hNSCs) and AAVs (Fig. 10B). TE was enhanced by shortened path lengths and increased contact frequencies between cells and vectors due to the large contact angle of droplets on polydopamine coated surfaces.290
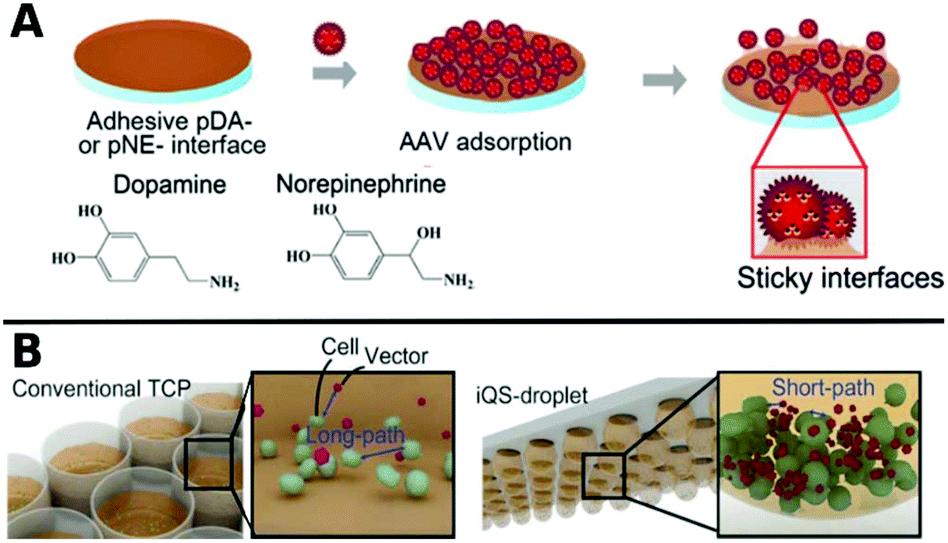 | ||
| Fig. 10 A. Schematic illustrations of adsorption of AAV on catecholamine coated substrates. Adapted with permission from ref. 289. Copyright 2014 American Chemical Society. B. Schematic illustration demonstrating the promotion of cell–AAV interactions in an inverted quasi-spherical (iQS) droplet on polydopamine coated substrates due to shorter pathways compared to conventional tissue culture plates (TCPs). Adapted with permission from ref. 290. Copyright 2017 John Wiley & Sons. | ||
Several hydrophilic polymers have been applied as 3D materials, namely hydrogels, often for slow release of VPs. The biodegradable multiblock sulfamethazine and PEG-containing polyurethane (PUSMA) exhibit pH and thermoresponsive behavior and was used as an injectable hydrogel for in vivo oncolytic Ad delivery. The sol–gel transition occurred below pH 8.0 through a hydrophilic to hydrophobic transition leading to the formation of a porous hydrogel network. This hydrogel enabled spatiotemporal Ad delivery to injection site under physiological conditions (Fig. 9B).163 An interesting example for the rAAV delivery to the cornea used hydrogels from a copolymer of hydroxyethyl methacrylate (HEMA) and aminopropyl methacrylamide (APMA) which can be worn as contact lenses (Fig. 9C). This transparent hydrogel network allowed high vector loading and controlled, long-term gene expression over a period of 14 days while being able to correct refractive errors.291
Microgels from vinyl ether acrylate-functionalized poly(vinyl alcohol) (PVA–VEA) and thiolated PVA–VEA were fabricated by a microfluidic technology and Michael-type cross-linking reaction to yield pH-degradable injectable spheres for efficient Ad delivery to tumor sites. The antitumor treatment could be reinforced by the addition of the bromodomain inhibitor JQ1. Accumulation of oncolytic Ad at the tumor site was enabled by pH-dependent controlled release from microgels. This combination approach paves the way for treatment of tumors by synergistic viral, chemo and immunotherapy as a promising system in clinical applications.292
Polystyrene coated with methyl methacrylate and divinylbenzene were formed into nanocups <500 nm by a template and applied as cavitation mediated carrier for oncolytic VV. Physical stimuli, e.g. ultrasound after intravenous injection resulted in enhanced transport and antitumor activity to treatment site.293
Several new polymeric systems and composites are emerging. Careful choice of the polymer and conjugation strategy allows the user to tailor pharmaceutical properties such as circulation time, controlled release of viral cargo and targeting of cells or tissues for next generation gene therapy.
Peptides
Due to their natural abundance and their ability for interactions with cells and viruses, bioactive peptides are highly interesting auxiliary agents for VV delivery.296 Tailor-made peptides can be easily produced in large scale, are biocompatible and biodegradable.Peptides applied for non-viral gene delivery were summarized recently elsewhere.297 When used as enhancers for viral gene delivery three different classes of peptides have been applied so far: cell penetrating peptides (CPP), fibrils formed from self-assembling peptides and proteins.
The formation of peptide–virion complexes is typically achieved either by attractive electrostatic interactions between negatively charged viral particles and positively charged peptides or by bioconjugation of the respective peptide to the capsid. With the large structural variety offered by peptide sequences, it is not surprising that many different types of peptides have been reported as promoting viral gene delivery. Table 4 provides an overview of the reported peptides, their sequences, and physicochemical properties. Furthermore, the types of VVs that have been reported in combination with these peptides are highlighted.
| Name | Structure | Properties | Fabrication of VP-peptide | Ref. |
|---|---|---|---|---|
| Abbreviations: cationic (c.), hydrophilic (h.), amphiphilic (amph.), anionic (a.), incubation (inc.), covalent conjugation (cov. conj.), adenovirus (Ad), adeno-associated virus (AAV), lentivirus (LV), retrovirus (RV), tobacco mosaic virus (TMV), polyomavirus (Pol). | ||||
| Tat47–57 | YGRKKRRQRRR | h., c. | Inc. (Ad)/cov. conj. (Ad)/bioengineering (Ad) | 311,315,318 |
| Tat48–57 | GRKKRRQRRR | h., c. | Cov. conj. (Ad) | 309 |
| Tat48–60 | GRKKRRQRRRPPQ | h., c. | Cov. conj. (Ad)/bioengineering (Ad) | 310, 316, 320 and 137 |
| Tat HA2 | CRRRQRRKKRGGDIMGEWGNEIFGAIAGFLG | Amph., c. | Bioengineering (AAV)/inc. (AAV) | 322,328 |
| OM-pBAEs | CRRR-PEG-CRRR | h., c. | Inc. (Ad) | 312 |
| R5 | RRRRR | h., c. | Cov. conj. (Ad) | 319 |
| R8 | RRRRRRRR | h., c. | Cov. conj. (Ad5/Ad/TYMV)/inc. (Pol., Ad) | 311,316–318,320,329 |
| R9 | RRRRRRRRR | h., c. | Cov. conj. (Ad)/— | 309 and 327 |
| HP4 | RRRRPRRRTTRRRR | h., c. | Inc. (Ad) | 119 |
| K7 | KKKKKKK | h., c. | Bioengineering (Ad)/— | 137, 365, 366 and 326 |
| Pep1 | KETWWETWWTEWSQPKKKRKV | Amph., c. | Cov. conj. (Ad)/inc. (Ad) | 311,318,320 |
| Pen/Antp62–77 | RQIKIWFQNRRMKWKKGG | Amph., c. | Cov. conj. (Ad)/inc. (Ad) | 308,309,311,318,320,328 |
| Pro | VRLPPPVRLPPPVRLPPP | Amph., c. | Cov. conj. (Ad5) | 316 and 317 |
| n.n | NRPDSAQFWLHHGGGSLLGRMKGA | c. | Cov. conj. (hepatitis B) | 314 |
| 12.51 | TARGEHKEEELI | c. | Bioengineering (Ad) | 313 |
| THR | THRPPMWSPVWP | Amph., c. | Inc. (AAV) | 321 |
| KH27K | KHHHHHHHHHHHHHHHHHHHHHHHHHHHK | h., c. | Inc. (polyomavirus) | 329 |
| FUSO | CGLFEALLELLESLWELLLEA | a. | Inc. (polyomavirus) | 329 |
| LAH4 | KKALLALALHHLAHLALHLALALKKA | Amph., c. | Inc. (AAV2 and AAV8, Pol)/inc. (LV) | 328,329,351 |
| Vectofusin-1 | KKALLHAALAHLLALAHHLLALLKKA | Amph., α-helix | Inc. (LV) | 130 and 354 |
| PAP248–286 | GIHKQKEKSRLQGGVLVNEILNHMKRATQIPSYKKLIMY | Amyloid, c. | Inc. (HIV-1) | 336 |
| PAP85–120 | IRKRYRKFLNESYKHEQVYIRSTDVDRTLMSAMTNL | Amyloid, c. | Inc. (HIV-1) | 337 |
| SEM145–107 | GQHYSGQKGKQQTESKGSFSIQYTYHVDANDHDQSRKSQQYDLNALHKTTKSQRHLGGSQQLL | Amyloid, c. | Inc. (HIV-1) | 338 |
| EF-C | QCKIKQIINMWQ | Amph., c. amyloid | Inc. (RV, LV) | 118 |
| EP2 | QCKIKQIINMWQEVG | Amyloid, c. | Inc. (HIV-1) | 343 |
| EP3 | NITLQCKIKQIINMWQEVG | Amyloid, c. | Inc. (HIV-1) | 344 |
| P13 | Ac-NWFDITNWLWYIK-NH2 | Amyloid, c. | Inc. (HIV-1) | 339 |
| P16 | Ac-NWFDITNWLWYIKKKK-NH2 | Amyloid, c. | Inc. (HIV-1) | 339 |
| P16-D | Ac-NWFAITNWLWYIKKKK-NH2 | Amyloid, c. | Inc. (HIV-1) | 341 |
| CD4bs-M | XGXSGGDPEIVTXKXXLTRDGGN (X = ε-aminohexanoic acid) | Amyloid, c. | Inc. (HIV-1) | 346 |
| Fmoc-SAP | Fmoc-DDIKVAVK | Gel, c. | Coinjection (LV) | 348 |
| DPF1 | INMWQG | Fibrils | Inc. (HIV-1) | 342 |
| RAD16-1 | Ac-RADARADARADARADA-CONH2 | Fibrils | Cogelation (AAV) | 234 |
| KK, KY | KYKGAIIGNIK, KYRSGAITIGY | Amyloid | 347 | |
| α-Syn | 140 amino acids, consecutive KTKEGV | Amyloid | Inc. (RV) | 117 |
| Protamine sulfate | Mixture of polypeptides | c. | Inc. (RV, Ad, LV) | 243, 356 and 357 |
| Retronectin | 574 amino acids, 63 kDa | c. | Inc. on precoated substrates (LV, RV) | 359 and 362–364 |
| PEG–PLL | PEG12000–PLL48/20 kDa PEG dendrimer–PLL | c. | Inc. (RV)/inc. (LV) | 368 and 369 |
| PLL | Poly-L-lysine 150–300 kDa | c. | Inc. (VLP) | 367 |
| Fibrin | Fibrinogen and thrombin mixture | Fibrous gel | Cogelation (LV, Ad/AAV) | 372, 374, 235 and 373 |
| Collagen | Collagen type 1 | Fibrous gel | Cogelation (LV) | 126 |
| Collagen/hydroxyapatite (Ca5(PO4)3(OH)) | Gel | Cogelation (LV) | 126 | |
| Gelatin | Hydrolyzed collagen | Gel | Cov. conj: (Ad)/cogelation (Ad) | 385 and 386 |
| Serum proteins | HSA, LDL, Transferrin | c. | Inc. (AAV8/HIV-1) | 375, 377 and 376 |
| SELP | GAGAGS and GVGVP blocks | Gel | Inc. (Ad/GLV-1h68) | 382, 383 and 384 |
Their ability to transport variable cargo across cellular membranes has made CPPs a facile tool for the delivery of DNA and RNA, liposomes, nanoparticles, proteins, and drugs as reviewed elsewhere303,304 and of viral nanoparticles for efficient transduction as comprehensively reviewed.305,306 In this segment, we highlight the most important developments in CPPs for facilitating transport of VP into target cells.
The most straightforward and easy way to obtain CPP–virus complexes is by co-incubation, making use of electrostatic interactions between positively charged CPPs and negatively charged VP. This method can be applied independently of the virus type, and ratio of CPP to VP can be easily adjusted in solution. However, the formation of CPP–VP complexes cannot be controlled, there is batch-to-batch variability and the highly positively charged CPP–VP complexes are prone to aggregation in physiologic conditions, for example, in the presence of electrolytes or serum (Fig. 11).307 A more stable but demanding approach is to covalently connect CPPs to the vector capsid. Early studies have demonstrated both approaches: the utilization of electrostatically bound Pen and Ad complexes to facilitate viral gene transfer in muscle cells308 and covalently bound Tat–Ad conjugates for delivery to tumor cell lines.309 By covalently attaching Tat to exposed lysine residues of the vector capsid via an MHS linker, transduction efficiency was further improved.310 Similar observations were made for Tat, Pen, R8 and Pep1 when they were covalently attached to PEGylated adenoviruses (Fig. 11).311
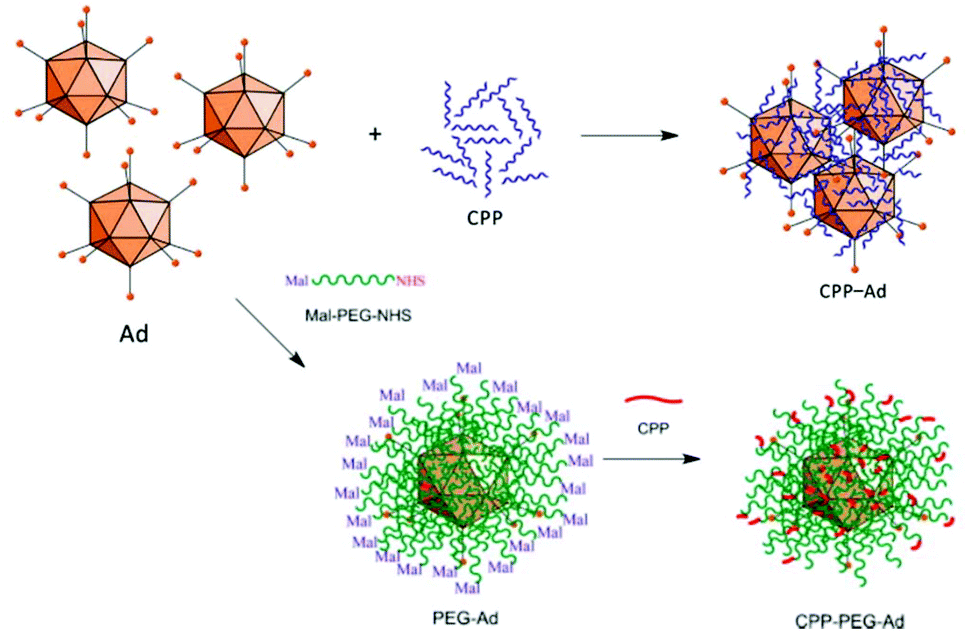 | ||
| Fig. 11 Approaches for producing CPP–Ad complexes (top) and conjugates (bottom). Complex formation results from electrostatic interactions between positively charged CPPs and negatively charged VPs. Covalent conjugations of CPPs are conducted on viral capsids, e.g. via bifunctional PEG linkers. Adapted with permission from ref. 307 and 311. Copyright 2013 and 2015 Elsevier. | ||
Coating of CRRR-co-PEG-co-CRRR onto an oncolytic Ad (Fig. 12A) enhanced transduction in tumor sites with a longer blood circulation time and lower liver sequestration was achieved.312
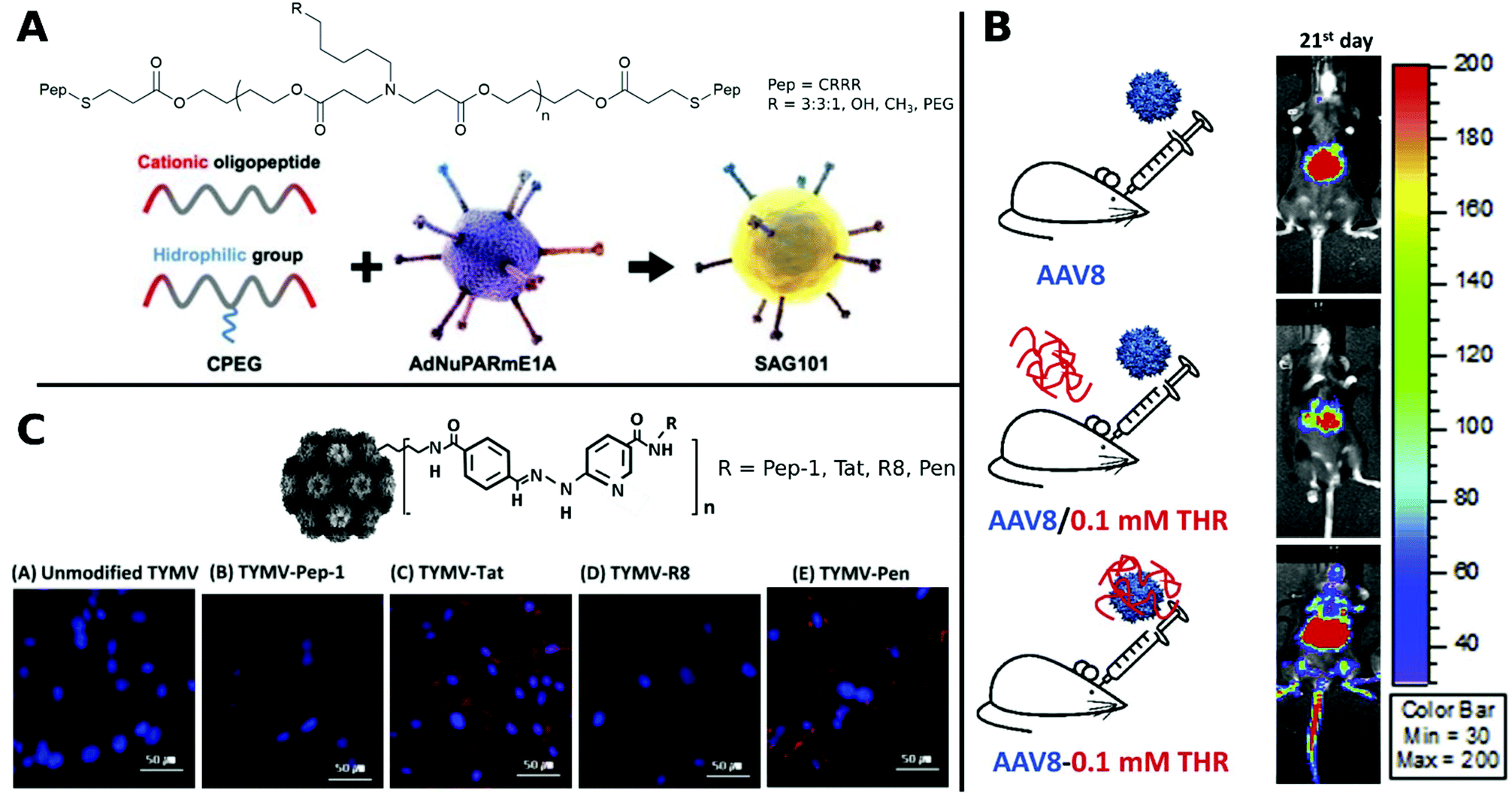 | ||
| Fig. 12 Selection of CPPs. A. Schematic representation of oncolytic Ad coating with oligopeptides. Adapted with permission from ref. 312. Creative Commons BY 4.0. 2020 Ivyspring International Publisher. B. Schematic representation of three groups of AAV8 administration: AAV8 only, AAV8/0.1 mM THR (not incubated) and AAV8–0.1 mM THR (incubated). Luminescence expression images were taken on 21st day after systemic administration. Adapted with permission from ref. 321. Copyright 2018 Elsevier. C. Hydrazone conjugated TYMV–peptide. Confocal analysis of transfected BHK21 cells stained with Hoechst 33258 dye (blue) with CPP modified and unmodified TYMV visualized with AF594 (red) (scale bar: 50 μm). Adapted with permission from ref. 320. Copyright 2018 Elsevier. | ||
In addition to CPPs derived from natural peptides, screening for suitable sequences by phage display has become a powerful method to discover new CPPs for enhanced transduction and targeted virus delivery.313,314
It was further possible to broaden the tropism of Ad by gene transfer to otherwise non-transducable CAR-negative cells. To this end, the surface knobs were modulated with Tat peptides,137,315 Tat peptides were attached to surface bound lysine residues316,317 or simply incubated in solution.318 Other examples for hard-to-transduce cell types that are successfully targeted with CPPs include Ad delivery to resistant stem cells and various cancer cells which was efficiently achieved by addition of arginine-rich HP4 derived from herring protamine.119 Furthermore, by decorating CPPs onto capsids via hydrazone chemistry, plant viruses like CPMV and TYMV were able to transduce otherwise non-infectable mammalian cells.319,320
Specific CPPs capable of crossing the blood brain barrier (BBB), can facilitate the delivery of CPP virus-complexes to the central nervous system after systemic application (Fig. 12B).321 In an interesting approach AAV containing a brain derived neurotrophic factor fused with Tat were delivered intranasally to the central nervous system to act as an antidepressant in mice.322
Frequently used CPPs for VV delivery are various Tat peptide fragments, oligoarginines, and penetratin (Pen). These peptides show different transduction enhancing properties depending on the viral particle, host cell, and CPP concentration.318 This difference in enhancement is believed to result from structural properties of the CPPs as well as their respective cellular entry mechanism. In general, various endocytic and non-endocytic cell entry pathways are controversially discussed in the literature.323,324
Regarding the structure, one requirement for the electrostatic stabilization of the virus peptide complex, as well as adhering to and crossing the negatively charged lipid bilayer of cells, is a high amount of positive charges in the peptide.318 Among peptides with positive charge those rich in arginine showed greater membrane permeability than CPPs with high amount of other cationic amino acids like lysine, histidine or ornithine.325,326 Beside the charge, hydrophilicity can also influence interaction with the membrane. It is believed that the higher performance of hydrophilic peptides like Tat and oligoarginines is due to stronger interactions with heparan sulfate proteoglycans of the cell membrane, whereas amphipathic peptides like Pep1 interfere with electrostatic binding of proteoglycans, thus resulting in lower transduction efficiency.318 Interestingly, a higher CPP concentration for VV delivery did not necessarily lead to a linear increase in transduction efficiency as shown for Tat, Pen, oligoarginine and Pep1.316,318 Chirality also significantly affects cell permeability. A 9-mer of poly-D-arginine showed five-fold higher cellular uptake than poly-L-arginine.327
Mechanistic pathways for the TE of AAV–CPP complexes were investigated by delivering AAV to non-permissive cell lines and blocking specific receptors. The tested peptides Pen, Tat-HA2 and LAH4 facilitate energy dependent and independent endocytosis as well as receptor-mediated pathways for the internalization of AAV.328 Further, Tat–Ad promoted cellular uptake via heparane sulfate receptors on the membrane surface, while the oligoarginine adenovirus conjugate R8-Ad was more dependent on chondroitin sulfate B receptors. This lays the foundation for CPP-dependent virus delivery.316
In a comparative study, TYMV was bioconjugated to Tat, R8, Pen or Pep-1. Improved efficiency was observed for Tat, R8 and Pen, whereas Pep-1 showed no change in transduction. This was traced back to different internalization routes and distributions of CPP–virus complexes in the cytoplasm and visualized by confocal images (Fig. 12C).320
Recently, Váňová et al. reported further mechanistic explanations for different TEs by investigating the influence of various CPPs on the activity and stability of VPs. They found that KH27K, FUSO, R8 and LAH4 affected the stability of VPs in different ways. KH27K promotes the aggregation and enlargement of VPs, while LAH4 destabilizes VPs but still enhances infection by concentrating them onto the cell surfaces (Fig. 13A).329 This study provides an example for enhanced TE via virus disassembly, which was suggested in an earlier report covering Pen, Tat and R8311 and provides the basis for further explanations of observed differences between CPPs.320
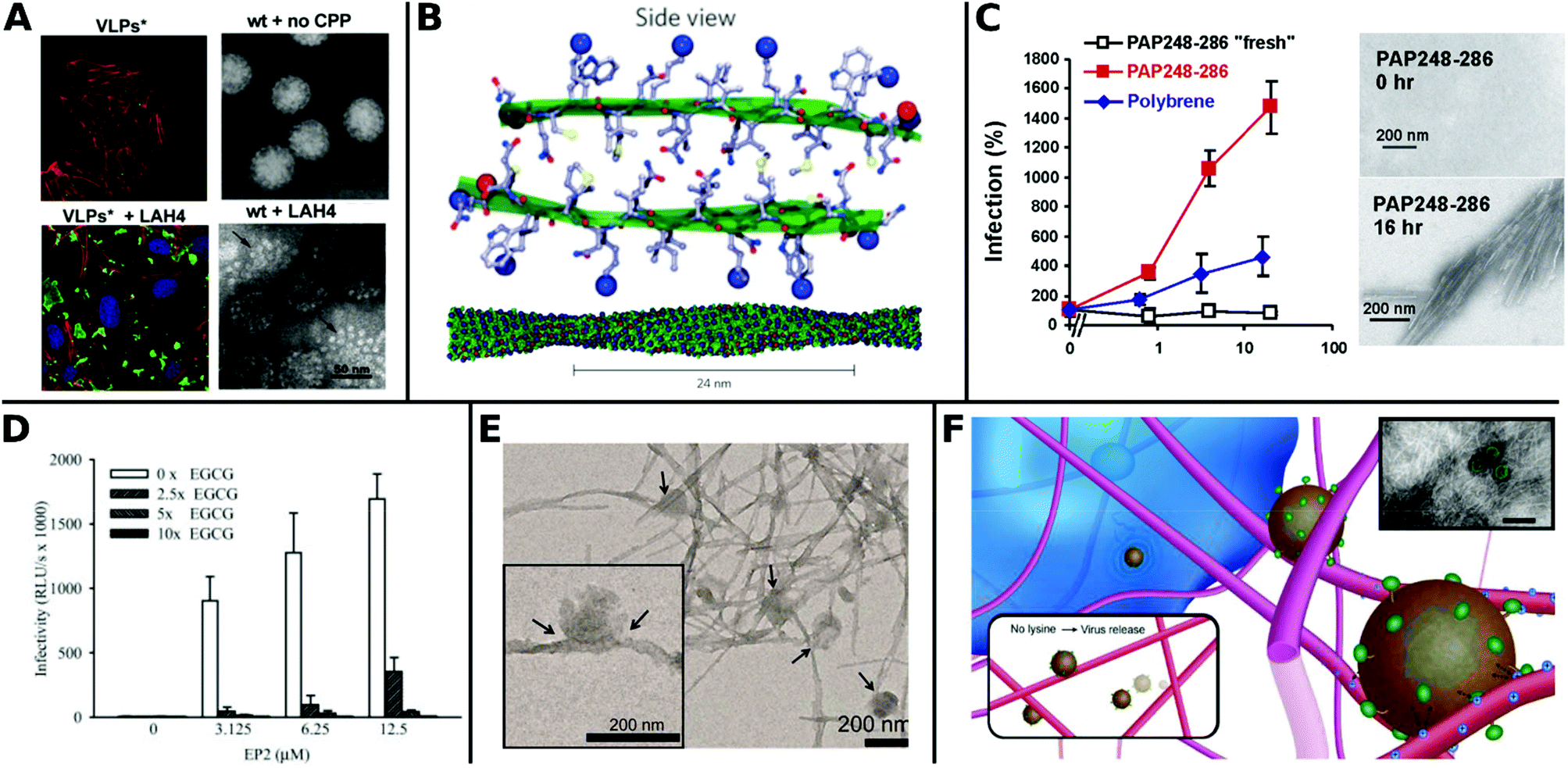 | ||
| Fig. 13 Overview of fibril virus interaction and bioactivity. A. Left panels: Confocal analysis of 3T6 cells (actin = red, nucleus = blue) after association with MPyV virus like particles (green) without (upper image) and with LAH4 peptide (bottom image). Right panels: TEM images of virions in buffer were stable and compact (upper image), whereas coincubation with LAH4 led to virions disassembling into pentamers (bottom image) (scale bar: 50 μm). Adapted with permission from ref. 329. Copyright 2020 Elsevier. B. Molecular model of EF-C amyloid peptide fibril. Adapted with permission from ref. 118. Copyright 2013 Springer Nature. C. Overnight incubated PAP248–286 enhances HIV-1 infection of TZM-bl cells more efficiently than non-incubated fresh PAP248–286 or Polybrene. TEM images show structural change to peptides before and after incubation for 16 h (scale bar = 200 nm). Adapted with permission from ref. 345. Copyright 2007 Elsevier. D. Decrease of HIV-1 R5 infectivity after EGCG addition on EP2 peptides. Adapted with permission from ref. 344. Copyright 2014 John Wiley & Sons. E. TEM image of virions adhered to α-Syn fibrils (scale bar = 200 nm). Adapted with permission from ref. 117. Copyright 2018 American Chemical Society. F. Schematic representation of electrostatic interaction between LV and fibrillar peptide. Charge based immobilization of LV is achieved through peptide functionalization with additional Lysine to give Fmoc-DDIKVAVK. Inset shows TEM image of nanofibrous network with green colored LV (TEM scale bar = 200 nm). Adapted with permission from ref. 348. Copyright 2020 Springer Nature. | ||
In contrast to CPPs, fibrillar peptides typically have a distinct secondary structure and cannot necessarily cross cell membranes. Enhanced viral gene delivery is achieved by colocalization of electrostatically complexed peptides and viral particles with the cellular membrane. Due to the high aspect ratio and rigidity of fibrils it is assumed that they cannot adapt to and cover virus surfaces thoroughly, thus creating excess positive charges in the virus–fibril complex for electrostatic interaction with cell membranes.332
The most thoroughly investigated fibrils for viral gene delivery are amyloids. Amyloid fibrils are highly stable and rigid, which makes them interesting materials for applications where long-term stability or tolerance of harsh conditions are required. For a long time, amyloid-forming peptides have exclusively been associated with diseases like plaque formation in Alzheimer's disease.333,334 However, these structures were also found in non-pathogenic contexts as functional amyloids, for example on bacteria surfaces335 and in seminal fluids.336 In the latter case, prostatic acidic phosphatase (PAP), a semen derived enhancer of virus infection (SEVI) with two distinct regions PAP248–286 and PAP85–120, and semenogelins (SEM) was found to form amyloidal structures and enhance HIV-1 infection.336–338 These very first findings sparked further research in amyloid peptides for viral gene delivery.
Transduction enhancing peptides can also be found in virus envelopes. The optimization of the glycoprotein fragments gp120417–428 (EF-C, commercialized as Protransduzin) and gp41671–682 (P13 and P16) from the HIV-1 envelope and transmembrane protein, respectively, form small amyloid fragments that assemble into cationic nanofibrils of several hundred nanometers in length and a few nanometers in diameter (Fig. 13B). These short fragments showed higher TE compared to Tat, polybrene, DEAE and SEVI in various cell lines including difficult to transduce TZM-bl cells, while being cost effective and convenient in handling.118,339 Fluorescent dyes coupled with free amino groups in EF-C have been introduced as a tool to study plasma stability and in vivo biodistribution while maintaining the structural and functional properties of non-labeled EF-C.340 Point mutations in the peptide sequences had a great impact on TE as shown for P16-D, where a substitution of aspartic acid with alanine resulted in increased activity.341 Sequence variations of gp120417–428 yielded the 6-mer DPF1 and longer sequences EP2 and EP3 which accelerate amyloid formation of SEVI and SEM and enhanced TE.342–344
The fibrillar structure is necessary for enhanced VV delivery (Fig. 13C)345 as shown by the following example: addition of epigallocatechin gallate, a main compound of green tea, which inhibits amyloid formation resulted in lower TE (Fig. 13D).344 Amyloids can also help to broaden the tropism of viruses. For instance HIV-1 was able to transduce in hardly infectable CD4 negative cells with the addition of amyloidal CD4bs-M peptides.346
Moreover, the cofibrillation of amyloid α-Syn with positively charged polymers like poly-L-lysine or chitosan enhanced TE of amyloid fibrils as recently reported by Maji and coworkers. The authors suggested that these positively charged additives increased electrostatic interactions and local density of virions on cell surface, thereby facilitating the transduction (Fig. 13E).117
Besides using derivates of naturally occurring amyloids for vector delivery, amyloids can also be computationally designed as shown for KK and KY, which enhanced DNA delivery to mammalian cells by forming DNA–amyloid complex and overcoming charge repulsion, as discussed for VVs.347 Since synthetic amyloid fibrils have so far only been applied in ex vivo studies, the question of fibril stability and degradability in vivo remains open.
Hydrogels formed from fibrillar peptides are emerging as VV delivery scaffolds, due to their simple handling and spatiotemporal release of viruses. For instance, Fmoc-SAP derived from the epitope IKVAV enabled localized delivery of LV after implantation into the central nervous system of mice. The modification of the sequence with an additional lysine at the C-terminus increased electrostatic interactions and immobilized the VP to obtain focal gene delivery to the site of injection (Fig. 13F).348 The peptide hydrogel RAD16-1 (commercialized as PuraMatrix™) is frequently used in cell culture. It displays favourable nano-structural and biomechanical properties and promotes the proliferation of various mammalian cells.349 Rey-Rico et al. reported RAD16-1 hydrogels for durable genetic modifications to stem cells by enhanced localized AAV delivery over a period of 21 days in vivo.234
The LAH4 peptide family has attracted growing interest in recent years as a DNA transfection reagent as well as for VV delivery.328,350,351 One commercialized derivative of the LAH4 family is Vectofusin-1.351 This 26-mer cell penetrating peptide is the first transduction enhancer with α-helical fibrillar structure. Studies have shown enhanced TE of Vectofusin-1 compared to other delivery agents such as Tat, Pen, LAH4 derivatives, KH27K, R8, FUSO, polybrene, and retronectin.328,351–355 Interestingly, a variation in the histidine sequence order or peptide length resulted in a significant change of bioactivity. A minimum length of 21 amino acids of the LAH4 peptide family was found to be necessary for successful vector delivery.355
Protamine sulfate is an early examples for viral transduction enhancers, originally derived from salmon sperm and clinically used for treatment of heparin overdose. This substance is actually a mixture of several polycationic peptides and facilitates virus–cell interactions by charge neutralization similar to other enhancers like polybrene, DEAE dextran or poly-L-lysine.356,357 In comparative transduction studies of LV delivery to CD34+ stem cells, protamine sulfate showed a TE similar to Vectofusin-1 and higher than that of Retronectin.243 TE could be enhanced even further by combining protamine sulfate with chondroitin sulfate.358
Retronectin is a 574 amino acid long recombinant polypeptide based on the fibronectin fragment CH296. It is one of the first commercially available enhancers for VV delivery and has already been tested in clinical trials.359–361 A typical workflow for transduction involves the coating of Retronectin on a solid surface, the addition of viral particles and cells followed by spin centrifugation.362 To overcome the limitations of the laborious preparation on surfaces and the necessity of centrifugation, various alternative approaches have been developed. The coating of epoxy-modified paramagnetic beads with Retronectin enables the effective capture of retroviral particles from solution and provides access to remote-controllable transduction.363In vivo LV delivery to stem cells via intra-bone marrow injection in mice showed enhanced TE when Retronectin was co-injected with the virus.364
Poly-L-lysine (PLL) is a popular additive in cell cultures for reinforcing adhesion of cells to culture dish. It is used as short oligomers up to polypeptides of several 100 kDa and can either be covalently attached to virus surface or simply coated on surfaces.365–367 PLL has been investigated as a replacement for polybrene in viral delivery by creating the block copolymer PEG12000–PLL48 which was coated on RV through electrostatic interactions. In contrast to polybrene, this copolymer augments RV transduction to delicate primary cultured brain cells without cytotoxic effects.368 Further optimization of PEG–PLL compositions were made by functionalizing four-arm PEG acrylates with PLL of different molecular weights ranging from 1–70 kDa. Increased TE was observed for increased molecular weights of PLL.369 Moreover, PLL has been applied as a mimic of the viral envelope and enhanced cellular internalization and transduction of virus-like particles.367 Recently, modifications of PLL120 with p-toluylsulfonyl arginine resulted in change of conformation from random coil to α-helix, which gave higher DNA-transfection rates due to higher cell-permeability.370 Layer-by-layer deposition – referred to as protein surface precipiation here – of PLL and dextran sulfate sodium salt were applied onto Ad surface to obtain nanospheres that protect Ad from antigenic reactions and thereby enhanced TE (Fig. 14A).371 These recent findings highlight the importance of material deposition techniques and conformational structure for viral gene delivery.
 | ||
| Fig. 14 A. Schematic representation of naked Ad and layer-by-layer PLL/dextran sulfate sodium nanosphere coated Ad (PSP). Fluorescence images show gene expression with percentual transduction rates (scale bar: 15 μm). Adapted with permission from ref. 371. Copyright 2019 Elsevier. B. SEM images of fibrin scaffold with LV incorporated without (left) and with hydroxylapatite (right) (scale bar: 500 nm). Adapted with permission from ref. 374. Copyright 2012 Elsevier. C. Schematic illustration of site-specific antibody conjugation on VP capsid surface. Adapted with permission from ref. 378. Copyright 2020 American Chemical Society. D. SEM images of Ad immobilized on gelatin sponges (scale bar: left 500 μm, right 500 nm). Adapted with permission from ref. 385. Copyright 2013 John Wiley & Sons. | ||
Enhanced viral delivery with spatiotemporal control can be achieved by fibrous protein scaffolds, such as fibrin. For example, when VPs were incorporated in fibrin, a local, long-term release of Ad,372 AAV235,373 and LV374in vitro and in vivo was observed at the injection site up to 14 days. Fibrin scaffolds could be further improved by the addition of hydroxyapatite nanoparticles, resulting in stronger trapping as well as slower release of VPs from scaffolds374 (Fig. 14B) and gels.126
Vupputuri et al. reported that protein impurities in cell culture can lead to increase or decrease of viral TE. By carefully adjusting the amount of protein added to viral particles, they could improve TE of polymer–virus complexes. Proteins bound to VPs can increase TE due to sedimentation of heathier particles, however at the same time cellular uptake pathway is changed by larger VPs thus protein addition has to be carefully balanced.132 This observation have far-reaching consequences for comparability of studies concerning TE, when taking batch-to-batch variability of culture medium and VP purification into account. For example, proteins from the human serum can also augment AAV transduction. Cationic human serum albumin (cHSA), an abundant endogenous transporter protein in blood, increased transduction of an in vivo hemophilia B mouse model fivefold.375 PEGylated cHSA improved TE of HIV-1 to TZM-bl cells while displaying lower immunogenicity.376 Combinations of HSA, low density lipoproteins (LDL) or transferrin yield AAV–serum protein complexes which enhanced transduction to liver in vivo, prevented binding of AAV to other proteins, and suppressed nonspecific binding to cells.377 Finally, site-specific antibody conjugation to TYMV, CPMV and bacteriophage via azide–alkyne click chemistry was reported by Park et al. (Fig. 14C).378
These examples highlight that natural proteins can efficiently enhance viral delivery. However, concerns of potential immunogenicity of xenogeneic materials exist.379 Nature-inspired, synthetic silk elastin-like protein polymers (SELPs) may circumvent this problem. SELPs mainly consist of repeating amino acid blocks based on the typical silk (GAGAGS) and mammalian elastin (GVGVP) motifs and were first reported for Ad release in cancer gene therapy in 2004.380,381 SELPs showed enhanced, local, long-term Ad delivery up to four weeks in various studies.382,383 In a comparative study with poloxamer 407, SELPs showed promising results with a superior TE, lower toxicity and immunoreactivity.384 Furthermore, collagen, an abundant fibrous protein in the extracellular matrix, showed long-term localized gene expression in vivo up to 4 weeks when it was co-gelated with LV.126 Organic–inorganic hybrid systems, such as collagen hydrogels combined with hydroxyapatite nanoparticles retain LV over a period of four weeks in vivo and increase local delivery efficiency even more.126 Moreover, gelatin gels, which consist mainly of hydrolysed collagen, were also applied for viral delivery. Gelatin sponges and Ad were functionalized with biotin to link them after avidin addition. The virus tethered in a virus–biotin–avidin–biotin–gelatin arrangement showed enhanced spatiotemporal TE in vivo after implantation for bone regeneration (Fig. 14D).385 Local long-term gene expression was also shown for oncolytic Ad delivery from gelatin gels in vivo up to 20 days. These gels could further protect Ad from immune response and prevent unwanted delivery to healthy sites.386
Lipids
Lipids are amphiphilic molecules typically consisting of a hydrophilic head group and a hydrophobic tail. Lipids are the main component of cellular membranes, virus envelopes and extracellular vesicles, which are Nature's endogenous nanocarriers to deliver biological information from cell to cell.387 The unique ability of lipids in aqueous media to form bilayers in a vesicular structure (liposomes) and spherical monolayers (micelles) enables them to encapsulate both hydrophilic and hydrophobic cargo.388,389 After encapsulation the cargo is protected from degradation as well as immune reaction390 and can further address specific targets by functionalization at the liposome's outer sphere.391 Since their first discovery in the 1960s,392 liposomes have been extensively used as delivery agents in clinical trials and pharmaceutical industry.393,394In non-viral gene delivery, lipids are currently the gold standard. For example, liposomes were applied as DNA and RNA nanocarriers395,396 as well as mimics of the viral envelope for gene delivery.397 Recent reviews on non-viral application of lipids for gene delivery can be found elsewhere.398,399 Lipids for viral gene delivery, which are discussed in the following section are summarized in Table 5.
| Name | Structure | Properties | Fabrication of VP lipid complexes | Ref. |
|---|---|---|---|---|
| Abbreviations: cationic (c.), hydrophilic (h.), zwitterionic (z.), anionic (a.), incubation (inc.), fusogenic (fuso), adenovirus (Ad), adeno-associated virus (AAV), lentivirus (LV), retrovirus (RV), herpes simplex virus (HSV), murine leukemia viruses (MLV). | ||||
| Chol | Cholesterol | z., fuso. | Inc. (Ad) | 405 |
| DOTAP/Chol | (1,2-Dioleoyloxypropyl)-N,N,N-trimethylammonium chloride/cholesterol | c. | Lipid bilayer wrapping (Ad)/inc. in preformed liposomes (Ad)/self-assembly around VP (Ad) | 409, 390, 53 and 416 |
| DOTAP/DOPE | DOTAP/(1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) | c./fuso., z. | Self-assembly around VP (Ad) | 409 |
| DOTAP/DOPE/Chol | c./fuso. z. | Inc. (dry film or extrusion method) or assembly around VP (RV) | 410 | |
| DMPC/Chol | Dimyristoyl phosphatidylcholine/cholesterol | z. | Self-assembly around VP (Ad) | 53 and 409 |
| DMPC/Chol/apoA-1 | DMPC/Chol/apolipoprotein A-1 | z. | Inc. in preformed liposomes (Ad) | 124 |
| DMPC/Chol/DSPE-PEG | z. | Self-assembly around VP (Ad) | 53 | |
| TMAG/DLPC/DOPE | N-(α-Trimethylammonio-acetyl)didodecyl-D-glutamate chloride/dilauroyl phosphatidylcholine/DOPE | z. | Inc. in preformed liposomes (AAV, rAd) | 407 and 417 |
| DC-Chol/DOPE | 3 beta [N-(N′,N′′-dimethylaminoethane)-carbamoyl]cholesterol/DOPE | z. | Inc. in preformed liposomes (RV, Ad + siRNA) | 401 and 423 |
| Lipofectamine (DOSPA/DOPE (3/1)) | 2,3-Dioleyloxy-N-[2(sperminecarboxamido)ethyl]-N,N-dimethyl-1-propaninium trifluoroacetate/DOPE | c., fuso. | Inc. in preformed lipid-DNA complex (AAV)/inc. in preformed liposomes (RV/HSV/Reovirus) | 415, 400, 418 and 420 |
| Lipofectin (DOTMA/DOPE (1/1)) | N-[1-(2,3-Dioleyloxy)propyl]-N,N,N-trimethylammonium chloride/DOPE | c., fuso. | Inc. in preformed liposomes (RV) | 400 and 414 |
| Lecithin/Chol (4/1) | z., fuso. | Inc. in preformed liposomes (alphavirus) | 421 | |
| Lysolecithin | L-α-Lysophosphatidylcholine | z., fuso | Preconditioning of host cells prior to LV injection | 424–427 |
| PC/PG/Chol | Phosphatidylcholine/phosphatidylglycerol/Chol | z. | Reverse-evaporation unilamellar vesicle (MLV) | 406 |
| PEGDE | [1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly-ethylene glycol)-2000] | Inc. in preformed liposomes (Ad) | 432 and 433 | |
| DSPE-PEG-biotin | (1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[biotinyl(polyethylene glycol)2000]) | Inc. in preformed liposomes (RV) | 435 | |
| PC/CHEMS/Chol-PEG-MMP-peptide | PC/CHEMS/Chol-PEG2000-GPLGIAGQC | a., MMP responsive | Inc. in preformed liposomes with Ca2+ ions (Ad) | 413 |
| DOPE/CHEMS (3/2) | DOPE/cholesteryl hemisuccinate | a., fuso | Inc. in preformed liposomes with Ca2+ ions (Ad) | 412 |
| CHEMS/PC/Chol (4/5/1) | CHEMS/phosphatidylcholine (PC)/Chol | a. | Inc. in preformed liposomes with Ca2+ ions (Ad) | 123 |
| Lecithin/Chol/DSPE-PEG/folate | a. | Inc. in preformed liposomes (Ad) | 422 | |
 | ||
| Chart 2 Overview of most frequently used lipids for viral gene delivery. | ||
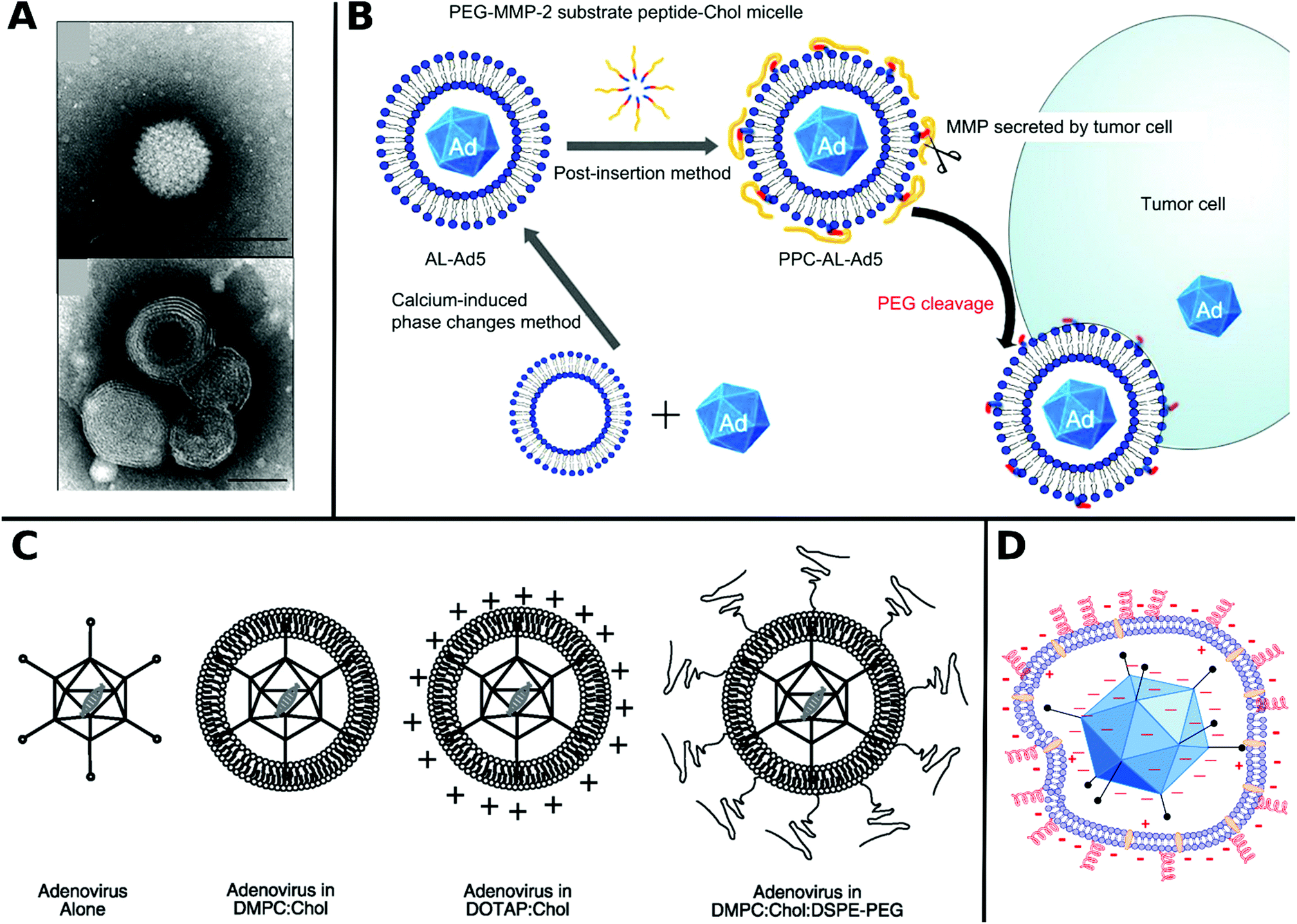 | ||
| Fig. 15 A. TEM images of naked Ad (top) and DOTAP/Chol encapsulated Ad (bottom) (scale bar: 100 nm). Adapted with permission from ref. 409. Copyright 2008 American Chemical Society. B. Schematic illustration of enzyme-responsive liposome-encapsulated Ad5 for specific delivery in tumor cells. Adapted with permission from ref. 413. Copyright 2013 Elsevier. C. Schematic representation of lipid encapsulated Ad. Adapted with permission from ref. 53. Copyright 2008 John Wiley & Sons. D. Schematic diagram of electrostatic interactions between zwitterionic phospholipid lecithin with negatively charged Ad5. Cholesterol is shown between lipid bilayer and PEG chains on the outer sphere as red spirals. Adapted with permission from ref. 422. Copyright 2014 Elsevier. | ||
In general, it is assumed that TE of VPs is increased by accelerated cell entry due to the small size and positive charge of liposomal carriers53,410 and further promoted by the so-called proton sponge effect, which has recently been critically reviewed.195 The proton sponge effect is caused by an increase of cationic charge at endosomal acidic pH, leading to osmotic swelling and accelerated liposome dissociation in the cytoplasm.129,411 Apart from this, various supporting strategies have been developed to facilitate the endosomal escape by creating stimuli-responsive liposomes, for example, by employing pH-triggered release of Ad from DOPE/CHEMS412 or MMP-cleavable enzyme-responsive liposomes (Fig. 15B).413
Most studies were conducted with complexes from cationic liposomes and VPs. Enhanced TE, shielding from immune response and broadened tropism of RV,400,401,410,414 AAV,415 Ad,124,416,417 HSV,418,419 as well as recently for oncolytic applications with reovirus420 and alphavirus421 were reported. Moreover, liposome vesicles from zwitterionic, cationic and PEG-lipid formulations can act as an artificial envelope for non-enveloped viruses and thereby enhance targeted delivery and prolong blood circulation lifetime (Fig. 15C and D).53,412,422 Further co-envelopment of Ad and siRNA yielded a dual active hybrid vector for multiple gene delivery.423
Not all applications with lipids for virus delivery require preformed VP–lipid complexes. Parsons et al. applied lysolecithins as surfactants on host cells prior to LV administration for cystic fibrosis treatment in mice. In this way, a more effective, less immunogenic and persistent transduction was achieved already with a single injection.424–427 Maguire et al. were the first to report and isolate AAV in extracellular vesicles, which were released from AAV producer cells during the reproduction process.428 These naturally encapsulated AAV showed enhanced TE and high biocompatibility at various application sites in vivo and has been reviewed elsewhere.429
Nanoparticles
Nanoparticles (NPs) are nanoscale materials, which have attracted growing interest in recent years due to their versatility and unique size-scaling properties. For example, NPs made from noble metals such as gold display strong surface plasmon absorption, making them an interesting material for local phototherapy and as an imaging agent in vivo.448,449 Among manifold preparation techniques to obtain uniform, functional NPs, template-based strategies are well established. VPs are attractive templates due to the uniform, highly precise 3D structure of the virus capsid which are decorated with chemical functional groups.450NPs have also emerged in recent years as an invaluable asset for viral gene delivery. In general three approaches can be distinguished: (1) NPs and VP administered together without specific association between them; (2) VPs and NPs that are covalently bound, e.g. in a conjugate; and (3) non-covalent interactions such as electrostatic binding between VPs and NPs. Depending on the NP–VP size ratio, various architectures can be achieved. If the NPs size exceed the VPs size e.g. iron oxide NPs combined with AAV120 VPs are decorated on the outer sphere of NPs. However, if the VPs are larger than NPs typically, NPs and VPs are combined either by coating VP surface via incubation and precipitation or by covalent attachment through functional groups on VP surface. For example, mineral shells for VPs can be created by precipitation from a solution of an inorganic salt and exhibit new biological and physical properties impacting stability, host cell adsorption and entry mechanism.451
Covalent attachment of NPs to VPs is accessible by abundant lysine residues on capsid proteins and by genetically engineered functional groups, e.g. thiol- or azide-moieties.
Frequently applied NPs for viral gene delivery are made from calcium phosphate, gold, silica, graphene or iron oxide, are discussed in the following sections and summarized in Table 6.
| Name | Structure | Properties | Fabrication of VP–NP vectors | Ref. |
|---|---|---|---|---|
| Abbreviations: incubation (inc.), adenovirus (Ad), oncolytic adenovirus (oAd), adeno-associated virus (AAV), lentivirus (LV). | ||||
| Ca3(PO4)2 | Calcium phosphate | Shell like encapsulation | Coprecipitation by inc. in CaCl2 and subsequent biomineralization through addition of Na2HPO4 (Ad5) | 112 |
| Ca3(PO4)2, DOPA, PEG-DPPE | Calcium phosphate, dioleoylphosphatydic acid, 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[azido(polyethylene glycol)-2000] | 3 layered encapsulation | Coprecipitation in Ca-rich medium, inc. by assembly of lipid bilayers of DOPE, coassembly of PEG-DPPE with DOPE (oAd) | 434 |
| CaCO3/MnCO3 | Shell like encapsulation | Coprecipitation from CaCl2 and MnCl2 with Na2CO3 (oAd) | 454 | |
| Gold nanorods | NP = ca. 60 × 30 × 1 nm | Coinjection (oAd) | 463 | |
| Gold NPs | Funct. with PEG/RGD or azide-group | NP = 14 nm | Cov. conj. (Ad) | 457 |
| Gold NPs | Au-sulfo-NHS | NP = 1.4 nm | Cov. conj. (Ad) | 456 |
| Silica gel | SiO2 gel polymerized from tetraethoxysilane | Macroporous hydrogel | Cogelation from sol–gel drying process (Ad) | 128 |
| Silica cloak | SiO2 | Anionic | Coprecipitation from silic acid in aqueous media on Ad pretreated with PLL | 125 |
| SPIONs | Fe3O4 (superparamagnetic iron oxide nanoparticles) | NP = 10 nm | Cov. conj. (Ad)/inc. (Ad/measles virus/LV | 498, 490, 505, 494, 484 and 493 |
| SPIONs–COOH | Fe3O4 functionalized with carboxylic acid | NP = 5 nm | Cov. conj. (AAV) | 489, 502 and 503 |
| SPIONs/heparin | Heparin coated SPION | NP = 100 nm | Inc. (AAV) | 120 |
| SPIONs/chitosan | Chitosan coated SPION | NP = 50–150 nm | Inc. (Ad) | 497 |
| SPIONs/PEG2000 | PEG2000 cross-linked SPION | NP = 117 nm | Inc. (Ad) | 492 |
| SPIONs/PEI or polybrene or Si-PEI | PEI/polybrene/Si-PEI coatings on SPION | NP = 28 nm/64 nm/101 nm | Inc. (oAd) | 491 |
| NiNTA-biotin–streptavidin SPIONs | Fe3O4 particles conjugated with streptavidin–biotin–nitrilotriacetic acid–Ni | NP = 120 nm | Hexahistidine coding AAV interact with Ni-ions chelated on NPs. | 115 |
| PEI–GO–PEG–FA | Polyethyleneimine–graphene oxide sheets–polyethylene glycol–folic acid | NP = 25 nm | Inc. (measles virus) | 481 |
| Carbon dots | Carbon dots, citric acid, branched PEI | NP = 11–36 nm | Inc. (rAAV) | 482 |
 | ||
| Fig. 16 Schematic illustration of in situ mineralization of Ad5 by precipitation with Ca3(PO4)2. Adapted with permission from ref. 112. Copyright 2012 John Wiley & Sons. | ||
Inorganic particles from Ca3(PO4)2 can provide stealth biomineral shells by spontaneous coating of VPs surface and thereby have an influence on tropism, TE and biocompatibility. Combinations with other inorganic particles enable precise tuning of the release properties and give access to tailored carriers.
Gold NPs were applied as covalent conjugates with VPs and reported either to retain or enhance TE456,457 or attenuate infectivity,458–461 depending on the capsid modification. Covalent conjugates were mainly achieved by attachment to the abundant lysine residues of capsid proteins. However, modifications with high gold amounts interfered with capsid protein functions, which are important for virus infectivity.456 Nevertheless, enhanced transduction was shown with various positively charged PEGylated Gold NPs functionalized with RGD epitope or azide group and coincubated with Ad, which formed spherical particles and exhibit higher TE to hMSCs compared to PEI or Lipofectamine coated Ad (Fig. 17).457
 | ||
| Fig. 17 Optimization process of Ad conjugated to gold NPs by maintaining gold NP concentration fixed at 0.1 pmol and varying amount of VPs (moi, multiplicity of infectivity, top). The best ratio at 50 moi was kept and gold NP concentration was varied (bottom) to observe enhanced TE at higher gold NP concentrations. Adapted with permission from ref. 457. Copyright 2019 The Royal Society of Chemistry. | ||
A further interesting clinical application of Gold NPs is photothermal therapy,462 which has been efficiently addressed in a combinatorial therapeutic approach with VPs.456 Oncolytic Ad applied in combination with gold nanorods exhibit hyperthermia-induced enhancement of receptor-mediated endocytosis and TE in vivo, enabling locally induced tumor cell lysis.463
Since pioneering works from Kostarelos et al.475,476 research efforts have been focussed on graphene as non-viral transfection agents, often in combination with grafted cationic polymers as adhesion promoters.477–480
Recently, graphene have started to attract interest as viral delivery platforms. Graphene oxide (GO) sheets decorated with PEG-folate were shown to target susceptible cancer cells.468 Based on these results, oncolytic measles virus was encapsulated by GO sheets functionalized with PEI and PEG-folate (Fig. 18). The encapsulated measles virus was protected against neutralizing antibodies, showed high tumor accumulation in vivo, exhibited higher infectivity and TE to cancer cells than virus alone and folate-free GO–PEI–PEG sheets. However, the release mechanism and in vivo stability of the VP–GO sheet cluster remain unexplored.481
 | ||
| Fig. 18 Structure of PEI–GO–PEG–FA and schematic illustration of GOS/measles virus (MV-Edm) complexation for cancer therapy. Adapted with permission from ref. 481. Creative Commons BY 4.0. 2019 Springer Nature. | ||
Another carbon-based material, carbon dots, spherical particles of few nanometer-size diameter, have been applied for viral gene delivery for the first time.482 This relatively new material class, mostly derived from graphene, has been reported to show size-, charge-, and aggregation-dependent toxicity.483 In order to stimulate cartilage repair, rAAV coding for the highly chondroreparative SOX9 transcription factor was complexed with various functionalized carbon dots, which resulted in significant increase of transduction to hMSCs with high cell viability. Especially carbon dots functionalized with 2-citric acid, poly(ethylene glycol) monomethyl ether and N,N-dimethylethylenediamine displayed efficient release and TE of rAAV.482
Magnetite (Fe3O4) as superparamagnetic iron oxide nanoparticles (SPIONs) have been reported in several studies as a vehicle to enhance gene delivery efficiencies, e.g. for delivery of AAV,115,489 Ad,487,490 oncolytic Ad,491,492 LV,484,493 and measles virus.494 They are biocompatible and degradable and allow surface modification with various functionalities.495,496
Coating SPIONs with biopolymers like heparin120 or chitosan497 can further augment TE with lower vector dosage and enable faster transduction (Fig. 19A). Moreover, polymeric coatings such as PEI or PEG on SPIONs as well as liposome encapsulation are reported to be highly beneficial for improving TE of Ad with reduced dose (Fig. 19B).484,492,498 For example, PEI coatings give access to positively charged particles boosting cell interaction and endosomal escape by their proton sponge effect without affecting the superparamagnetic properties.499 In a comparative study PEI, polybrene, or silica–PEI were coated onto iron oxide NPs and resulted in improved potency of oncolytic Ad. The highest antitumor activity in vivo was observed for silica–PEI coated iron oxide NPs, which was attributed to a higher shielding ability against neutralizing antibodies and higher magnetophoretic mobility.491 In another study, various commercially available SPIONs were compared to each other in terms of susceptibility for aggregation and TE of attached alphavirus. Positively-charged NPs displayed rapid and effective isolation and concentration of VPs, resulting in higher TE than negatively charged NPs.500
 | ||
| Fig. 19 A. Schematic illustration of AAV bound to heparin-coated SPIONs. Adapted with permission from ref. 120. Copyright 2011 Elsevier. B. Schematic illustrations of PEG8-functionalized SPION NPs-labeled Ad5 capsids. Adapted with permission from ref. 498. Copyright 2012 Elsevier. C. Schematic illustration of streptavidin functionalized SPIONs chelated with biotin bound hexahistidine/AAV. Adapted with permission from ref. 115. Copyright 2011 Elsevier. D. Schematic representations of the main steps for single-cell virus stamping and colored SEM image of magnetic NP (blue) bound to LV (green) (scale bar: 500 nm, inset: 100 nm). Adapted with permission from ref. 501. Copyright 2018 Springer Nature. E. Tumor bio-reduction-activated NPs incorporating magnetized virus delivery. In the reductive tumor microenvironment the carrier payload dissociates and AAV conjugated SPIONs can be tracked by MRI. Adapted with permission from ref. 503. Copyright 2018 American Chemical Society. | ||
In general, VPs can be coated with modified SPIONs through simple incubation due to electrostatic interactions to obtain hybrid particles.487,492 Beside this, covalent attachment of VPs onto SPIONs is regularly achieved by EDC–NHS chemistry coupling carboxy-functionalized SPIONs to the lysine residues on viral capsid.489,498
Another, more laborious route to attach VPs onto SPIONs surfaces can be achieved by using a chelating approach, with a linker unit consisting of biotin-functionalized nickel ions conjugated to streptavidin modified SPIONs, which further bind to hexahistidine displaying AAV particles (Fig. 19C). This hybrid particle can transduce into otherwise non-permissive hNSCs by employing magnetic force.115 Recently, precise manual virus stamping to single cells in culture as well as to mouse brain was reported by Schubert et al. This technique employs VPs covalently bound to silica-coated SPIONs which are loaded in a micropipette tip and brought into contact with the desired cells by a magnetic field (Fig. 19D). This method enables the delivery of multiple virus types to a single cell for multiple virus engineering or to track genetically modified single cells.501
An interesting example to implement magnetofection with phototherapeutic tumor treatment was shown for AAV encoding a photosensitive apoptosis-inducing protein. AAV conjugated to iron oxide NPs were remotely guided and delivered to the tumor site with microscale precision, where phototoxicity was then triggered by irradiation.489,502,503 Recently, Tseng et al. further developed this approach with an elaborate hybrid design. AAV encoding the photosensitive apoptosis-inducing protein was conjugated to SPIONs and loaded to a nitroimidazole conjugated hyaluronic acid NP containing lactate oxidase. This design employed the lactate-rich tumor microenviroment to subsequently initiate H2O2 production by lactate oxidase, resulting in reduction and disassembly of 2-nitroamidazole-hyaluronic acid matrix and finally release of SPIONs attached to AAV into tumor site (Fig. 19E). This enzymatically-controlled release enabled targeted tumor cell lysis with an combinatorial approach of functional materials.503
SPIONs are approved as contrast agents for magnetic resonance imaging,504 which makes them a clinically promising vehicle for targeted delivery of VPs. One of main advantages of using magnetic NPs for VP delivery is the reduced vector dosage and short exposure times for efficient transduction due to controlled colocalization of cells and VPs. Magnetic NPs enable to deliver VPs to otherwise non-permissible cells and may enable the crossing of biophysical barriers in vivo in the future.
Small molecules
Small molecules are defined as organic compounds with a molecular mass below 900 Da and a physical size below 1 nm in pharmaceutical research. Today, the majority of drugs used as medicine in everyday life are small molecules, which are characterized by their high bioavailability and effective mode of action at the target site.81 Research on small molecules that promote viral gene delivery has become more wide-spread in recent years.The aim of adding small molecules during a transduction procedure is to achieve satisfactory TE with low vector doses to minimize side-effects and to enable transduction to cell types that are difficult to infect. The underlying mode of action of small molecules to achieve this aim is fundamentally different to previously discussed materials, which were interacting with VPs, forming a complex, aggregate or conjugate which in turn facilitates the interaction with cells. Here, small molecules interfere with multiple cellular processes, for example, by affecting gene transcription, cell entry mechanisms or DNA replication and thereby facilitating viral gene expression. The focus in small molecule research for viral gene delivery has been on dual mode drugs, which are promising due to their therapeutic as well as transduction enhancing properties. These molecules are usually simply added to VPs for application, even though covalent conjugation to VPs were also shown exemplarily.506 In the following sections important findings are highlighted and summarized in Table 7 and Chart 3.
 | ||
| Chart 3 Overview of selected drugs as enhancers of viral gene transfer. | ||
| Name | Medical use | Assumed mechanism of action | VPs and cell line | Ref. |
|---|---|---|---|---|
| Camptothecin | Cytostatic drug | Topoisomerase inhibitor/G2/M cell cycle arrest | AAV to fibroblasts/HIV to HeLa | 508 and 135 |
| Taxol | Cytostatic drug | G2/M cell cycle arrest | HIV to HeLa/AAV to HeLa | 135 and 506 |
| Hydroxyurea | Cytostatic drug | DNA-synthesis inhibitor | AAV to fibroblasts | 508 |
| Aphidicolin | Cytostatic drug | DNA-synthesis inhibitor | AAV to fibroblasts/HIV to HeLa | 135 and 508 |
| Verapamil | Calcium channel blocker | Adenosine 5′-triphosphate-binding cassette-inhibitor | Vesicular stomatitis virus-G to hematopoietic progenitor cells | 515 |
| Bortezomib | Cytostatic drug | Proteasome inhibitor | Lymphocytic choriomeningitis virus (LCMV) to T-cells/rAAV to HeLa, U87, HepG2, NHF1 | 513 and 517 |
| MG132 | Proteasome inhibitor | rAAV to human airway epithelia | 514 | |
| Phorbol 12-myristate 13-acetate | Proteinkinase activator | LV to HSC | 516 | |
| Doxorubicin | Cytostatic drug | Proteasome inhibitior | rAAV to neuronal cells/rAAV to human airway epithelia | 510, 509 and 527 |
| Teniposide | Cytostatic drug | Topoisomerase inhibitior | rAAV to HeLa, U87, HepG2, NHF1 | 517 |
| Etoposide | Cytostatic drug | Topoisomerase inhibitor | rAAV to HeLa/AAV to fibroblasts/HIV to HeLa | 517, 508 and 135 |
| Bleomycin | Cytostatic drug, glycopeptide antibiotic | DNA damage | rAAV to HeLa, U87, HepG2, NHF1 | 517 |
| Parthenin | Sesquiterpene lactone | rAAV to HeLa, U87, HepG2, NHF1 | 517 | |
| RH-1 | Diaziridinylbenzoquinone | rAAV to HeLa, U87, HepG2, NHF1 | 517 | |
| Vorinostat | Cytostatic drug | HDAC inhibition | rAAV to HeLa, U87, HepG2, NHF1 | 517 |
| Nanaomycin A | DNMT3B inhibitor | DNMT3B inhibitor | rAAV to HeLa, U87, HepG2, NHF1 | 517 |
| Menogaril | Anthracycline | rAAV to HeLa, U87, HepG2, NHF1 | 517 | |
| Pyrromycin | rAAV to HeLa, U87, HepG2, NHF1 | 517 | ||
| Daunorubicin | DNA intercalation, topoisomerase inhibition, polymerase inhibition, free radical damage to DNA | rAAV to HeLa, U87, HepG2, NHF1 | 517 | |
| Physalin B | rAAV to HeLa, U87, HepG2, NHF1 | 517 | ||
| Siomycin | Thiazole antibiotic | rAAV to HeLa, U87, HepG2, NHF1 | 517 | |
| Tetrocarcin A | Microbial metabolite | BCL-2 inhibitor | rAAV to HeLa, U87, HepG2, NHF1 | 517 |
| 6-Gingerol | Phytoactive material, anticancer activity | Bypassing endocytic receptor mediated pathway, interfering with cell cycle | AAV2- to A375 and 526 malignant melanoma cells | 58 |
| Pyrrol based derivatives | Inhibition of production of IFNβ and various interferon-stimulated genes | Oncolytic Rhabodviruses to resistant cancer cells (e.g. CT26) | 518 | |
| Prostaglandine E2 | Abortifacient agent, labor induction | LV to HSCs | 520–522 | |
| (+)-JQ1 | Cytostatic drug | Bromodomain inhibitor | Ad to A549, HeLa, Jurkat and P388D1 cells | 524 |
| Hydroxychloroquine | Antimalaria medication | TLR9 inhibition | AAV to murine and human tissue | 523 |
| Rosuvastatin | Statin medication | Promoter of low-density lipoprotein-receptor | Vesicular stomatitis virus (LV) to natural killer cells | 526 |
| Embelin | Cytostatic drug and anti-inflammatory effects | Inhibitor of X chromosome-linked inhibitor-of-apoptosis protein XIAP | Oncolytic vaccinia virus to T cells and NK cells | 525 |
In early works, Miller et al. showed that DNA-damaging agents like UV-irradiation or cisplatin increased TE of AAV to non-dividing cells.507 Later they introduced less cytotoxic DNA-synthesis inhibitors like aphidicolin or hydroxyurea and topoisomerase inhibitors such as etoposide or camptothecin as enhancers of AAV transduction to fibroblasts.508 Groschel et al. demonstrated that these cytostatic agents etoposide, camptothecin, taxol, and aphidicolin, interfere with various mechanisms of cell cycle progression and arrest the cell cycle at G2/M phase, which is a checkpoint to control DNA replication before nuclei division. Arresting the cell cycle in this phase boosted the early step of HIV virus replication and thereby enhanced TE.135 Another well-known chemotherapeutic drug, doxorubicin, greatly enhanced rAAV infection to airway cells as well as neuronal cells with only mild cytotoxic effects at tested concentrations. Doxorubicin promoted rAAV accumulation at the nucleus, probably through proteasome inhibiting effects resulting in accelerated intracellular viral nuclear transport, but without affecting cell entry mechanisms.509,510 The exact mechanism remained unclear and it was suggested that enhanced TE is more likely resulting from stimulated capsid uncoating and increased reverse transcription of viral genome than by increased cell entry.135 Further cytokine stimulated downregulation of proteasome activity is believed to enhance TE.511 Evidence for this hypothesis was found by addition of cytokines or proteasome inhibitors, particularly FDA approved bortezomib, resulting in enhanced TE of even low dosed LV to stem cells.511–513
Furthermore, the proteasome inhibitor MG132 (carbobenzoxy-L-leucyl-L-leucyl-L-leucinal) enhanced TE of rAAV to airway epithelia cells by modulating the ubiquitin–proteasome system in the endosomal environment.514 Moreover, adenosine 5′-triphosphate-binding cassette transporter proteins, which efflux chemotherapeutic drugs and HIV protease inhibitors, play a significant role in reducing TE of LV into CD34+ stem cells. Enhanced TE of up to sixfold was observed after the addition of Verapamil, an adenosine 5′-triphosphate-binding cassette-inhibitor.515
However, the identified chemotherapeutic compounds have huge disadvantages including damaging DNA and are therefore not ideal candidates for clinical use to healthy cells. In order to identify additional transduction enhancing drugs, high-throughput screens were conducted.
The first library screening for compounds promoting LV transduction identified 30 potential enhancers of viral transduction from 1280 investigated pharmaceutically active drugs. The non-DNA damaging, protein kinase activator phorbol 12-myristate 13-acetate (PMA) was found as a very promising candidate to enhance TE of LV into hard to transduce stem cells. Similar to previously discovered agents, PMA displayed interference with the cell cycle and inhibition of proliferation, suggesting that the transduction efficiency is influenced by processes within the cell rather than by promoted cell entry.516
Another high-throughput screening with focus on enhancing TE of rAAV to HeLa cells investigated 2396 pharmaceutically known, mostly FDA approved compounds from which 13 compounds were identified as capable potentiators. In this screening, five main enhancer groups were identified: epipodophyllotoxins, inducers of DNA damage, effectors of epigenetic modification, anthracyclines, and proteasome inhibitors. The highest level of transduction, with approx. 50–100 times compared with virus-only transduction, was observed for the chemotherapeutic epipodophyllotoxin teniposide. It was observed that the TE varied significantly for the tested substances depending on cell type, which indicates an underlying correlation between mode-of-action and cell type. In unison with previous studies, the authors suggest that rAAV can be enhanced by two pathways, whose detailed mechanisms remain unknown: the inhibition of proteasomes and the induction of DNA damage.517
A further screening for enhancing interferon sensitized oncolytic virus transduction to resistant cancer cells identified pyrrole-based molecules as potent enhancers. This was also feasible with AAV and Ad. High activities up to thousand-fold enhancement of viral transduction, tolerability and plasma stability were found for systemic administration in mice. The mode of action of these molecules was attributed to an inhibition of interferon-β production, which gives antiviral properties to resistant cancer cells.518,519
In recent years, further pharmaceutically applied drugs were identified as promising viral gene delivery agents.
Prostaglandine E2 enhanced LV transduction to HSCs by acting during the endocytosis phase via a yet unclear mechanism.520–522
Lee et al. reported synergistic effects in terms of inducing apoptosis to malignant melanoma cells when AAV2 encoding a pro-apoptotic protein and the phytoactive compound 6-gingerol were combined. 6-Gingerol was previously reported to be effective against cancer and for the inhibition of angiogenesis. The cytostatic activity has its source in the control of the cell cycle and subsequently inducing apoptosis. The authors proposed that AAV2 can bypass its usual endocytic receptor-mediated pathway in the presence of 6-gingerol and thereby enhancing apoptosis in melanoma cells more efficiently.58
Furthermore, hydroxychloroquine, an approved antimalaria drug, has been applied for promoting AAV transduction in murine and human tissue in vitro and in vivo, e.g. in the retina with no adverse effects. Among the multimodal effects this drug might have on TE, one mode of action was proposed as an inhibition of TLR9 gene expression, which is a known sensor for anti-viral response of cells.523
Intervention in cell regulation processes was similarly shown for Ad infection which was enantioselectively promoted by the bromodomain inhibitor (+)-JQ1. This molecule blocks acetylated lysine interaction with the bromodomain family proteins, which play a significant role in cell regulation and gene transcription. In this way, Ad infectivity and gene expression were elevated by suppression of regulating bromodomains, whose mechanisms still need to be elucidated.524
Embelin, used as an adjuvant in oncolytic virotherapy was recently reported by Wang et al. Lymphoma cell lysis was induced by mitigating antiviral immunity against oncolytic virus upon addition of Embelin. The authors proposed that the infiltration of the oncolytic virus was facilitated by the disruption of Interleukin-6/STAT3 signalling and further promoted by the inherent ability of Embelin to induce apoptotic death to tumor cells.525
An interesting approach was reported by Gong et al. who enhanced TE by increasing low-density lipoprotein-receptors expression with Rosuvastatin. Vesicular stomatitis virus was able to efficiently target and transduce in natural killer cells after addition of Rosuvastatin. Naturally, untreated killer cells lack of sufficient low-density lipoprotein-receptors, which are necessary for virus interaction. Statins, especially Rosuvastatin, can induce low-density lipoprotein expression and thereby facilitate virus–cell interaction. Additionally, geranylgeranyl-pyrophosphate suppresses cytotoxic effects of statins, resulting in a highly effective and powerful tool to enable genetic modification of natural killer cells. Genetically modified natural killer cells can, for example, express tumor antigen-specific receptors, which may pave the way for targeted treatment of malignant cells.526
Small molecules, which are beneficial for disease treatment, eliciting host immune response while enabling efficient transduction are actively being pursued for clinical application. Pharmaceutically active drugs that have been found so far, are paving the way especially for oncolytic gene therapy and can be included easily in ex vivo gene transfer protocols. However, further research is indispensable for investigation of unwanted side effects of drugs combined with viruses during delivery and should be carefully evaluated in therapeutic application.
From development to usage
Most of the introduced materials for promoting viral gene transfer have not yet been translated to the clinic and are often not commercially available. Here, we will highlight materials that are emerging from the development stage to actual clinical usage. Various material classes which have the potential for application in medical formulations are currently investigated in preclinical studies. For example, in vivo studies in animals utilized materials like PLGA,133,155,215,216 PEG,163,181,283 PEI,208 poloxamers,160,229,231,232,238,239 dendrimers,116,251,253 polysaccharides,258,259,262,265 gelatin,385,386 collagen,126 fibrin,374 human serum albumin,375,377 calcium carbonate,454 gold nanorods,454 silica implants,128 graphene,481 magnetic NPs,491,501,505 and small molecules.517,518,523Commercially available kits make transduction enhancers more broadly available and are thus attractive tools for use in research. For instance, several studies about CAR T-cell manufacturing employed kits of the peptidic enhancers Vectofusin®-1 (Miltenyi Biotec) and Protransduzin™ (JPT Peptide Technologies).353,354,528,529 However, popular materials in research cannot seamlessly be transferred to the clinical context. Low cell viability in high concentrations limits the application of Polybrene® (Abbott Laboratories Corp.) in drug formulations.530 More efficient enhancers than polybrene531 are traded as jetPEI® (Polyplus transfection) and Lipofectamine™ (Thermo Fisher Scientific) and can be applied to a broad range of VVs. Kits for specific viruses are available, but their exact composition is a trade secret, e.g.: for LVs (ViraDuctin™ (Cell Biolabs), TransDux™ (System Biosciences), ViralPlus (Applied Biological Materials Inc.), SureENTRY (Qiagen)), as well as for Ad in the form of AdenoBoost™ (Sirion Biotech). High transduction rates with low-titer viruses are enabled with polycationic magnetic beads, commercialized as MACSductin™ (Miltenyi Biotec), ViroMag and AdenoMag (OZ Biosciences), Lenti-X™ Accelerator (Takara Bio) and MISSION® ExpressMag® (Sigma-Aldrich). In recent years, a growing number of such kits were made available in GMP grade addressing the demand for systematic investigation of transduction agents in a clinical context.
Clinical trials use RetroNectin® (Takara Bio) in particular as the gold standard for ex vivo transduction of lenti- and retroviruses, even though a high multiplicity of infection and a time consuming coating protocol is needed.361,532–534 Poloxamers such as LentiBoost™ (Sirion Biotech)535 are commonly used in formulations for in vivo clinical trials as enhancers and to prevent uncontrolled binding of VVs to injection devices.536–539 While the majority of viral gene therapy drugs approved up until 2017 do not use additional transduction enhancers in their formulations, those approved since then have made use of agents such as protamine sulfate (ex vivo, Zynteglo),540,541 Poloxamer 188 (in vivo, Zolgensma and Luxturna),542,543 and human serum albumin (ex vivo, Yescarta, Kymriah).544,545
Conclusions and outlook
Viral gene delivery has come a long way and is now becoming increasingly therapeutically relevant. The rising number of approvals for clinical use in recent years holds a great promise for treatment of currently uncurable diseases. However, the full potential of gene therapy cannot be realized until safe and efficient delivery systems are available. In this review, we highlighted the most important approaches for viral gene delivery from a materials perspective. In particular, the high biocompatibility of peptidic delivery systems, and the superior shielding effects of polymer coatings and lipid encapsulation need to be emphasized. Both hydrogels and magnetic NPs allow spatially defined virus release, often in a time-controlled manner. Especially the local targeted delivery of VVs via magnetic nanoparticles to tumor sites has enormous potential in oncolytic virotherapy. The performance of delivery systems is determined by many experimental parameters, e.g. virus type, cell type, concentration, and incubation time. However, detailed large-scale comparative studies of different delivery agents are still missing. Therefore, it is often difficult or even impossible to draw conclusions on the relative performance of the delivery agents. In current viral gene delivery systems, immunogenicity, native tropism, unspecific delivery, and poor efficiency at low vector doses are remaining hurdles to be overcome. The materials introduced here have the potential to establish hybrid VVs for clinical applications. However, clinical data of administration to humans remains scarce.Gene delivery as a therapeutic approach is one of the major medicinal and biotechnological advances of this century. We expect that novel bioengineered viruses promoted by synthetic materials are destined for future efficient and safe clinical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Nicole Kirsch-Pietz for proof-reading the manuscript. Open Access funding provided by the Max Planck Society.Notes and references
- C. E. Thomas, A. Ehrhardt and M. A. Kay, Nat. Rev. Genet., 2003, 4, 346–358 CrossRef CAS.
- S. Pearson, H. Jia and K. Kandachi, Nat. Biotechnol., 2004, 22, 3–4 CrossRef CAS.
- Gene Therapy Clinical Trials Worldwide, http://www.abedia.com/wiley/years.php, (accessed 15 April 2020).
- R. Goswami, G. Subramanian, L. Silayeva, I. Newkirk, D. Doctor, K. Chawla, S. Chattopadhyay, D. Chandra, N. Chilukuri and V. Betapudi, Front. Oncol., 2019, 9, 297 CrossRef.
- Grand View Research, Gene Therapy Market Size, Share & Trends Analysis Report By Indication (Acute Lymphoblastic Leukemia, Large B-cell Lymphoma), By Vector Type, By Region, And Segment Forecasts, 2020–2027, 2020 Search PubMed.
- K. A. High and M. G. Roncarolo, N. Engl. J. Med., 2019, 381, 455–464 CrossRef CAS.
- C.-C. Ma, Z.-L. Wang, T. Xu, Z.-Y. He and Y.-Q. Wei, Biotechnol. Adv., 2020, 40, 107502 CrossRef CAS.
- FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss, https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss, (accessed 15 April 2020).
- FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality, https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease, (accessed 15 April 2020).
- M. Zheng, J. Huang, A. Tong and H. Yang, Mol. Ther.–Oncolytics, 2019, 15, 234–247 CrossRef.
- C.-H. Lu, Y.-H. Chang, S.-Y. Lin, K.-C. Li and Y.-C. Hu, Biotechnol. Adv., 2013, 31(8), 1695–1706 CrossRef CAS.
- A. M. Thomas and L. D. Shea, J. Controlled Release, 2013, 170, 421–429 CrossRef CAS.
- P. R. Gupta and R. M. Huckfeldt, J. Neural Eng., 2017, 14, 051002 CrossRef.
- M. G. Katz, A. S. Fargnoli, R. D. Williams and C. R. Bridges, Hum. Gene Ther., 2013, 24, 914–927 CrossRef CAS.
- S. Rangarajan, L. Walsh, W. Lester, D. Perry, B. Madan, M. Laffan, H. Yu, C. Vettermann, G. F. Pierce, W. Y. Wong and K. J. Pasi, N. Engl. J. Med., 2017, 377, 2519–2530 CrossRef CAS.
- L. Samaranch, A. Pérez-Cañamás, B. Soto-Huelin, V. Sudhakar, J. Jurado-Arjona, P. Hadaczek, J. Ávila, J. R. Bringas, J. Casas, H. Chen, X. He, E. H. Schuchman, S. H. Cheng, J. Forsayeth, K. S. Bankiewicz and M. D. Ledesma, Sci. Transl. Med., 2019, 11, eaat3738 CrossRef.
- U. Griesenbach, K. M. Pytel and E. W. F. W. Alton, Hum. Gene Ther., 2015, 26, 266–275 CrossRef CAS.
- E. Marshall, Science, 1999, 286, 2244–2245 CrossRef CAS.
- N. A. Helal, A. Osami, A. Helmy, T. McDonald, L. A. Shaaban and M. I. Nounou, Pharmazie, 2017, 72, 627–651 CAS.
- M. Ogris and E. Wagner, Hum. Gene Ther., 2011, 22, 799–807 CrossRef CAS.
- H. Kamiya, H. Tsuchiya, J. Yamazaki and H. Harashima, Adv. Drug Delivery Rev., 2001, 52, 153–164 CrossRef CAS.
- K. Lundstrom, Diseases, 2018, 6, 42 CrossRef.
- C. A. Suttle, Nat. Rev. Microbiol., 2007, 5, 801–812 CrossRef CAS.
- T. Watanabe and Y. Kawaoka, Clin. Transl. Immunol., 2020, 9, e1114 Search PubMed.
- T. Dull, R. Zufferey, M. Kelly, R. J. Mandel, M. Nguyen, D. Trono and L. Naldini, J. Virol., 1998, 72, 8463–8471 CrossRef CAS.
- M. A. Kay, J. C. Glorioso and L. Naldini, Nat. Med., 2001, 7, 33–40 CrossRef CAS.
- W. Lucas, in Encyclopedia of Life Sciences, John Wiley & Sons, Ltd, Chichester, UK, 2010 Search PubMed.
- D. Bouard, N. Alazard-Dany and F.-L. Cosset, Br. J. Pharmacol., 2009, 157, 153–165 CrossRef CAS.
- R. G. Crystal, Hum. Gene Ther., 2014, 25, 3–11 CrossRef CAS.
- T. J. Wickham, P. Mathias, D. A. Cheresh and G. R. Nemerow, Cell, 1993, 73, 309–319 CrossRef CAS.
- M. C. Dechecchi, A. Tamanini, A. Bonizzato and G. Cabrini, Virology, 2000, 268, 382–390 CrossRef CAS.
- V. N. Krasnykh, J. T. Douglas and V. W. van Beusechem, Mol. Ther., 2000, 1, 391–405 CrossRef CAS.
- N. C. Di Paolo, N. van Rooijen and D. M. Shayakhmetov, Mol. Ther., 2009, 17, 675–684 CrossRef CAS.
- N. F. Nidetz, M. C. McGee, L. V. Tse, C. Li, L. Cong, Y. Li and W. Huang, Pharmacol. Ther., 2020, 207, 107453 CrossRef CAS.
- M. F. Naso, B. Tomkowicz, W. L. Perry and W. R. Strohl, BioDrugs, 2017, 31, 317–334 CrossRef CAS.
- D. Escors and K. Breckpot, Arch. Immunol. Ther. Exp., 2010, 58, 107–119 CrossRef CAS.
- A. V. Joglekar and S. Sandoval, Hum. Gene Ther: Methods., 2017, 28, 291–301 CrossRef CAS.
- G. B. Li and G. X. Lu, Mol. Biotechnol., 2009, 43, 250–256 CrossRef CAS.
- O. W. Merten, S. Charrier, N. Laroudie, S. Fauchille, C. Dugué, C. Jenny, M. Audit, M. A. Zanta-Boussif, H. Chautard, M. Radrizzani, G. Vallanti, L. Naldini, P. Noguiez-Hellin and A. Galy, Hum. Gene Ther., 2011, 22, 343–356 CrossRef CAS.
- S. K. Totsch, C. Schlappi, K.-D. Kang, A. S. Ishizuka, G. M. Lynn, B. Fox, E. A. Beierle, R. J. Whitley, J. M. Markert, G. Y. Gillespie, J. D. Bernstock and G. K. Friedman, Oncogene, 2019, 38, 6159–6171 CrossRef CAS.
- D. M. Patel, P. M. Foreman, L. B. Nabors, K. O. Riley, G. Y. Gillespie and J. M. Markert, Hum. Gene Ther.: Clin. Dev., 2016, 27, 69–78 CAS.
- Q. Lan, S. Xia, Q. Wang, W. Xu, H. Huang, S. Jiang and L. Lu, Front. Med., 2020, 14, 160–184 CrossRef.
- Y.-N. Zhang, S.-B. Wang, S.-S. Song, P.-Y. Hu, Y.-C. Zhou, Y.-P. Mou and X.-Z. Mou, Biotechnol. Lett., 2020, 42, 865–874 CrossRef CAS.
- J. W. Choi, Y. S. Lee, C. O. Yun and S. W. Kim, J. Controlled Release, 2015, 219, 181–191 CrossRef CAS.
- N. Martínez-Vélez, M. Garcia-Moure, M. Marigil, M. González-Huarriz, M. Puigdelloses, J. Gallego Pérez-Larraya, M. Zalacaín, L. Marrodán, M. Varela-Guruceaga, V. Laspidea, J. J. Aristu, L. I. Ramos, S. Tejada-Solís, R. Díez-Valle, C. Jones, A. Mackay, J. A. Martínez-Climent, M. J. García-Barchino, E. Raabe, M. Monje, O. J. Becher, M. P. Junier, E. A. El-Habr, H. Chneiweiss, G. Aldave, H. Jiang, J. Fueyo, A. Patiño-García, C. Gomez-Manzano and M. M. Alonso, Nat. Commun., 2019, 10, 1–10 CrossRef.
- T. Shi, X. Song, Y. Wang, F. Liu and J. Wei, Front. Immunol., 2020, 11, 683 CrossRef.
- C. S. Lee, E. S. Bishop, R. Zhang, X. Yu, E. M. Farina, S. Yan, C. Zhao, Z. Zeng, Y. Shu, X. Wu, J. Lei, Y. Li, W. Zhang, C. Yang, K. Wu, Y. Wu, S. Ho, A. Athiviraham, M. J. Lee, J. M. Wolf, R. R. Reid and T.-C. He, Genes Dis., 2017, 4, 43–63 CrossRef CAS.
- K. P. Kicielinski, E. A. Chiocca, J. S. Yu, G. M. Gill, M. Coffey and J. M. Markert, Mol. Ther., 2014, 22, 1056–1062 CrossRef CAS.
- E. Galanis, S. N. Markovic, V. J. Suman, G. J. Nuovo, R. G. Vile, T. J. Kottke, W. K. Nevala, M. A. Thompson, J. E. Lewis, K. M. Rumilla, V. Roulstone, K. Harrington, G. P. Linette, W. J. Maples, M. Coffey, J. Zwiebel and K. Kendra, Mol. Ther., 2012, 20, 1998–2003 CrossRef CAS.
- S. Aref, K. Bailey and A. Fielding, Viruses, 2016, 8, 294 CrossRef.
- K. Harrington, D. J. Freeman, B. Kelly, J. Harper and J.-C. Soria, Nat. Rev. Drug Discovery, 2019, 18, 689–706 CrossRef CAS.
- A. Ricobaraza, M. Gonzalez-Aparicio, L. Mora-Jimenez, S. Lumbreras and R. Hernandez-Alcoceba, Int. J. Mol. Sci., 2020, 21, 3643 CrossRef.
- R. Singh, B. Tian and K. Kostarelos, FASEB J., 2008, 22, 3389–3402 CrossRef CAS.
- J. F. Wright, T. Le, J. Prado, J. Bahr-Davidson, P. H. Smith, Z. Zhen, J. M. Sommer, G. F. Pierce and G. Qu, Mol. Ther., 2005, 12, 171–178 CrossRef CAS.
- J. L. Madrigal, R. Stilhano and E. A. Silva, Tissue Eng., Part B, 2017, 23, 347–361 CrossRef CAS.
- B. Balakrishnan and G. Jayandharan, Curr. Gene Ther., 2014, 14, 86–100 CrossRef CAS.
- M. A. Croyle, S. M. Callahan, A. Auricchio, G. Schumer, K. D. Linse, J. M. Wilson, L. J. Brunner and G. P. Kobinger, J. Virol., 2004, 78, 912–921 CrossRef CAS.
- J. H. Lee, Y. Kim, Y.-E. Yoon, Y.-J. Kim, S.-G. Oh, J.-H. Jang and E. Kim, New Biotechnol., 2017, 37, 194–199 CrossRef CAS.
- O. S. Kumru, Y. Wang, C. W. R. Gombotz, B. Kelley-Clarke, W. Cieplak, T. Kim, S. B. Joshi and D. B. Volkin, J. Pharm. Sci., 2018, 107, 2764–2774 CrossRef CAS.
- Y. Zhang, N. Chirmule, G. P. Gao, R. Qian, M. Croyle, B. Joshi, J. Tazelaar and J. M. Wilson, Mol. Ther., 2001, 3, 697–707 CrossRef CAS.
- D. Ghosh and M. A. Barry, J. Virol., 2005, 79, 13667–13672 CrossRef CAS.
- F. H. E. Schagen, M. Ossevoort, R. E. M. Toes and R. C. Hoeben, Crit. Rev. Oncol. Hematol., 2004, 50, 51–70 CrossRef.
- K. Kawabata, F. Sakurai, N. Koizumi, T. Hayakawa and H. Mizuguchi, Mol. Pharm., 2006, 3, 95–103 CrossRef CAS.
- A. S. Chuck, M. F. Clarke and B. O. Palsson, Hum. Gene Ther., 1996, 7, 1527–1534 CrossRef CAS.
- J. S. Bartlett, R. Wilcher and R. J. Samulski, J. Virol., 2000, 74, 2777–2785 CrossRef CAS.
- J. E. Ziello, Y. Huang and I. S. Jovin, Mol. Med., 2010, 16, 222–229 CAS.
- D. L. Puhl, A. R. D'Amato and R. J. Gilbert, Brain Res. Bull., 2019, 150, 216–230 CrossRef CAS.
- C. K. Yun, J. W. Hwang, T. J. Kwak, W. J. Chang, S. Ha, K. Han, S. Lee and Y. S. Choi, Lab Chip, 2019, 19, 580–588 RSC.
- D. A. Amado, J. M. Rieders, F. Diatta, P. Hernandez-Con, A. Singer, J. T. Mak, J. Zhang, E. Lancaster, B. L. Davidson and A. S. Chen-Plotkin, Mol. Ther., 2019, 27, 465–478 CrossRef CAS.
- N. Moore, J. R. Chevillet, L. J. Healey, C. McBrine, D. Doty, J. Santos, B. Teece, J. Truslow, V. Mott, P. Hsi, V. Tandon, J. T. Borenstein, J. Balestrini and K. Kotz, Sci. Rep., 2019, 9, 1–11 CrossRef CAS.
- Y. Deng, M. Kizer, M. Rada, J. Sage, X. Wang, D. J. Cheon and A. J. Chung, Nano Lett., 2018, 18, 2705–2710 CrossRef CAS.
- C. H. Peng, L. C. Woung, K. H. Lu, C. Y. Tsai, S. D. Lee, C. S. Huang, T. C. Lin, K. H. Chien and D. K. Hwang, J. Chin. Med. Assoc., 2018, 81, 830–836 CrossRef.
- L. Qian, B. Thapa, J. Hong, Y. Zhang, M. Zhu, M. Chu, J. Yao and D. Xu, J. Thorac. Dis., 2018, 10, 1099–1111 CrossRef.
- A. B. Bahnsonz, J. T. Dunigan, B. E. Baysal, T. Mohney, R. Wayne Atchison, M. T. Nimgaonkar, E. D. Ball and J. A. Barranger, J. Virol. Methods, 1995, 54, 131–143 CrossRef.
- Y. Chen, S. Aslanoglou, G. Gervinskas, H. Abdelmaksoud, N. H. Voelcker and R. Elnathan, Small, 2019, 15, 1904819 CrossRef CAS.
- S. Y. Chang, Y. H. Park, N. T. Carpena, T. T. Pham, P. S. Chung, J. Y. Jung and M. Y. Lee, Lasers Med. Sci., 2019, 34, 367–375 CrossRef.
- M. Tsukakoshi, S. Kurata, Y. Nomiya, Y. Ikawa and T. Kasuya, Appl. Phys., B, 1984, 35, 135–140 CrossRef.
- E. Neumann, M. Schaefer-Ridder, Y. Wang and P. H. Hofschneider, EMBO J., 1982, 1, 841–845 CrossRef CAS.
- H. Song, R. A. Bush, Y. Zeng, H. Qian, Z. Wu and P. A. Sieving, Mol. Ther.–Methods Clin. Dev., 2019, 13, 77–85 CrossRef CAS.
- A. L. Coulberson, N. V. Hud, J. M. LeDoux, I. D. Vilfan and M. R. Prausnitz, J. Controlled Release, 2003, 86, 361–370 CrossRef CAS.
- M. P. Stewart, R. Langer and K. F. Jensen, Chem. Rev., 2018, 118, 7409–7531 CrossRef CAS.
- X. Du, J. Wang, Q. Zhou, L. Zhang, S. Wang, Z. Zhang and C. Yao, Drug Delivery, 2018, 25, 1516–1525 CrossRef CAS.
- F. Kreppel, J. Gackowski, E. Schmidt and S. Kochanek, Mol. Ther., 2005, 12, 107–117 CrossRef CAS.
- S. J. White, S. A. Nicklin, H. Büning, M. J. Brosnan, K. Leike, E. D. Papadakis, M. Hallek and A. H. Baker, Circulation, 2004, 109, 513–519 CrossRef CAS.
- J. S. Chandran, P. S. Sharp, E. Karyka, J. M. D. C. Aves-Cruzeiro, I. Coldicott, L. Castelli, G. Hautbergue, M. O. Collins and M. Azzouz, Sci. Rep., 2017, 7, 14766 CrossRef.
- E. J. Lee, C. M. Guenther and J. Suh, Curr. Opin. Biomed. Eng., 2018, 7, 58–63 CrossRef.
- H. Büning and A. Srivastava, Mol. Ther.–Methods Clin. Dev., 2019, 12, 248–265 CrossRef.
- R. E. Kelemen, S. B. Erickson and A. Chatterjee, in Methods in Molecular Biology, Humana Press Inc., New York, NY, 2018, vol. 1728, pp. 313–326 Search PubMed.
- R. Schubert, S. Herzog, S. Trenholm, B. Roska and D. J. Müller, Nat. Protoc., 2019, 14, 3205–3219 CrossRef CAS.
- R. E. Kelemen, R. Mukherjee, X. Cao, S. B. Erickson, Y. Zheng and A. Chatterjee, Angew. Chem., Int. Ed., 2016, 55, 10645–10649 CrossRef CAS.
- N. Gabriel, S. Hareendran, D. Sen, R. A. Gadkari, G. Sudha, R. Selot, M. Hussain, R. Dhaksnamoorthy, R. Samuel, N. Srinivasan, A. Srivastava and G. R. Jayandharan, Hum. Gene Ther: Methods., 2013, 24, 80–93 CrossRef CAS.
- J. A. Sullivan, L. M. Stanek, M. J. Lukason, J. Bu, S. R. Osmond, E. A. Barry, C. R. O'Riordan, L. S. Shihabuddin, S. H. Cheng and A. Scaria, Gene Ther., 2018, 25, 205–219 CrossRef CAS.
- F. Sakurai, H. Mizuguchi and T. Hayakawa, Gene Ther., 2003, 10, 1041–1048 CrossRef CAS.
- M. Nakatake, H. Kurosaki, N. Kuwano, K. Horita, M. Ito, H. Kono, T. Okamura, K. Hasegawa, Y. Yasutomi and T. Nakamura, Mol. Ther.–Oncolytics, 2019, 14, 159–171 CrossRef CAS.
- Z. Chai, J. Sun, K. M. Rigsbee, M. Wang, R. J. Samulski and C. Li, J. Controlled Release, 2017, 262, 348–356 CrossRef CAS.
- M. J. Brun, E. J. Gomez and J. Suh, J. Controlled Release, 2017, 267, 80–89 CrossRef CAS.
- Y. Wang, S. Li, Z. Tian, J. Sun, S. Liang, B. Zhang, L. Bai, Y. Zhang, X. Zhou, S. Xiao, Q. Zhang, L. Zhang, C. Zhang and D. Zhou, Nucleic Acids Res., 2019, 47, e114 CrossRef CAS.
- E. J. Gomez, K. Gerhardt, J. Judd, J. J. Tabor and J. Suh, ACS Nano, 2016, 10, 225–237 CrossRef CAS.
- N. N. Thadani, J. Yang, B. Moyo, C. M. Lee, M. Y. Chen, G. Bao and J. Suh, ACS Synth. Biol., 2020, 9, 461–467 CrossRef CAS.
- R. C. Münch, A. Muth, A. Muik, T. Friedel, J. Schmatz, B. Dreier, A. Trkola, A. Plückthun, H. Büning and C. J. Buchholz, Nat. Commun., 2015, 6, 6246 CrossRef.
- Y. Lv, F. J. Xiao, Y. Wang, X. H. Zou, H. Wang, H. Y. Wang, L. S. Wang and Z. Z. Lu, BMC Biotechnol., 2019, 19, 23 CrossRef.
- C. Li and R. J. Samulski, Nat. Rev. Genet., 2020, 21, 255–272 CrossRef CAS.
- C. Domenger and D. Grimm, Hum. Mol. Genet., 2019, 28, 1–3 CrossRef.
- A. M. Wen and N. F. Steinmetz, Chem. Soc. Rev., 2016, 45, 4074–4126 RSC.
- P. S. Banerjee, P. Ostapchuk, P. Hearing and I. Carrico, J. Am. Chem. Soc., 2010, 132, 13615–13617 CrossRef CAS.
- C. R. O'Riordan, A. Lachapelle, C. Delgado, V. Parkes, S. C. Wadsworth, A. E. Smith and G. E. Francis, Hum. Gene Ther., 1999, 10, 1349–1358 CrossRef.
- Z. Chen, N. Li, S. Li, M. Dharmarwardana, A. Schlimme and J. J. Gassensmith, WIREs Nanomed. Nanobiotechnol., 2016, 8, 512–534 CrossRef.
- F. Kreppel and S. Kochanek, Mol. Ther., 2008, 16, 16–29 CrossRef CAS.
- M. A. Bruckman, G. Kaur, L. A. Lee, F. Xie, J. Sepulveda, R. Breitenkamp, X. Zhang, M. Joralemon, T. P. Russell, T. Emrick and Q. Wang, ChemBioChem, 2008, 9, 519–523 CrossRef CAS.
- Y. Chu, Y. H. Oum and I. S. Carrico, Virology, 2016, 487, 95–103 CrossRef CAS.
- I. Yildiz, I. Tsvetkova, A. M. Wen, S. Shukla, M. H. Masarapu, B. Dragnea and N. F. Steinmetz, RSC Adv., 2012, 2, 3670–3677 RSC.
- X. Wang, Y. Deng, S. Li, G. Wang, E. Qin, X. Xu, R. Tang and C. Qin, Adv. Healthcare Mater., 2012, 1, 443–449 CrossRef CAS.
- H. Zhou, G. Wang, X. Wang, Z. Song and R. Tang, Angew. Chem., Int. Ed., 2017, 56, 12908–12912 CrossRef CAS.
- P. H. Kim, T. il Kim, J. W. Yockman, S. W. Kim and C. O. Yun, Biomaterials, 2010, 31, 1865–1874 CrossRef CAS.
- E. Kim, J. S. Oh, I. S. Ahn, K. I. Park and J. H. Jang, Biomaterials, 2011, 32, 8654–8662 CrossRef CAS.
- Y. Wu, L. Li, L. Frank, J. Wagner, P. Andreozzi, B. Hammer, M. D'Alicarnasso, M. Pelliccia, W. Liu, S. Chakrabortty, S. Krol, J. Simon, K. Landfester, S. L. Kuan, F. Stellacci, K. Müllen, F. Kreppel and T. Weil, ACS Nano, 2019, 13, 8749–8759 CrossRef CAS.
- S. Kirti, K. Patel, S. Das, P. Shrimali, S. Samanta, R. Kumar, D. Chatterjee, D. Ghosh, A. Kumar, P. Tayalia and S. K. Maji, ACS Biomater. Sci. Eng., 2019, 5, 126–138 CrossRef CAS.
- M. Yolamanova, C. Meier, A. K. Shaytan, V. Vas, C. W. Bertoncini, F. Arnold, O. Zirafi, S. M. Usmani, J. A. Müller, D. Sauter, C. Goffinet, D. Palesch, P. Walther, N. R. Roan, H. Geiger, O. Lunov, T. Simmet, J. Bohne, H. Schrezenmeier, K. Schwarz, L. Ständker, W.-G. Forssmann, X. Salvatella, P. G. Khalatur, A. R. Khokhlov, T. P. J. Knowles, T. Weil, F. Kirchhoff and J. Münch, Nat. Nanotechnol., 2013, 8, 130–136 CrossRef CAS.
- J. I. Youn, S. H. Park, H. T. Jin, C. G. Lee, S. H. Seo, M. Y. Song, C. W. Lee and Y. C. Sung, Cancer Gene Ther., 2008, 15, 703–712 CrossRef CAS.
- J.-H. Hwang, S. Lee, E. Kim, J.-S. Kim, C.-H. Lee, I.-S. Ahn and J.-H. Jang, Int. J. Pharm., 2011, 421, 397–404 CrossRef CAS.
- D. Xia, L. B. Feng, X. L. Wu, G. D. Xia and L. Xu, Mol. Med. Rep., 2015, 12, 2336–2342 CrossRef CAS.
- P. Díaz-Rodríguez, A. Rey-Rico, H. Madry, M. Landin and M. Cucchiarini, Int. J. Pharm., 2015, 496, 614–626 CrossRef.
- Z. Zhong, S. Shi, J. Han, Z. Zhang and X. Sun, Mol. Pharm., 2010, 7, 105–115 CrossRef CAS.
- K.-H. Park, C.-O. Yun, O.-J. Kwon, C.-H. Kim, J.-R. Kim and K.-H. Cho, Hum. Gene Ther., 2010, 21, 579–587 CrossRef CAS.
- A. A. Sapre, G. Yong, Y. san Yeh, L. E. Ruff, J. S. Plaut, Z. Sayar, A. Agarwal, J. Martinez, T. N. Nguyen, Y. T. Liu, B. T. Messmer, S. C. Esener and J. M. Fischer, J. Controlled Release, 2019, 297, 48–59 CrossRef CAS.
- S. Shin and L. D. Shea, Mol. Ther., 2010, 18, 700–706 CrossRef CAS.
- S. S. McMahon, N. Nikolskaya, S. N. Choileáin, N. Hennessy, T. O'Brien, P. M. Strappe, A. Gorelov and Y. Rochev, J. Gene Med., 2011, 13, 591–601 CrossRef CAS.
- L. Kangasniemi, M. Koskinen, M. Jokinen, M. Toriseva, R. Ala-Aho, V. M. Kähäri, H. Jalonen, S. Ylä-Herttuala, H. Moilanen, U. H. Stenman, I. Diaconu, A. Kanerva, S. Pesonen, T. Hakkarainen and A. Hemminki, Gene Ther., 2009, 16, 103–110 CrossRef CAS.
- A. Elouahabi and J. M. Ruysschaert, Mol. Ther., 2005, 11, 336–347 CrossRef CAS.
- D. Fenard, D. Ingrao, A. Seye, J. Buisset, S. Genries, S. Martin, A. Kichler and A. Galy, Mol. Ther.–Nucleic Acids, 2013, 2, e90 CrossRef.
- N. Landázuri and J. M. Le Doux, J. Gene Med., 2004, 6, 1304–1319 CrossRef.
- S. Vupputuri, S. Karode, B. J. Neely and J. D. Ramsey, J. Virol. Methods, 2013, 192, 1–11 CrossRef CAS.
- S. Shin, D. M. Salvay and L. D. Shea, J. Biomed. Mater. Res., Part A, 2010, 93, 1252–1259 Search PubMed.
- C. A. Gersbach, S. R. Coyer, J. M. Le Doux and A. J. García, Biomaterials, 2007, 28, 5121–5127 CrossRef CAS.
- B. Groschel and F. Bushman, J. Virol., 2005, 79, 5695–5704 CrossRef CAS.
- J. T. Douglas, M. Kim, L. A. Sumerel, D. E. Carey and D. T. Curiel, Cancer Res., 2001, 61, 813–817 CAS.
- S. Kurachi, K. Tashiro, F. Sakurai, H. Sakurai, K. Kawabata, K. Yayama, H. Okamoto, S. Nakagawa and H. Mizuguchi, Gene Ther., 2007, 14, 1160–1165 CrossRef CAS.
- G. Fan, M. Fan, Q. Wang, J. Jiang, Y. Wan, T. Gong, Z. Zhang and X. Sun, Acta Biomater., 2016, 30, 94–105 CrossRef CAS.
- K. Singarapu, I. Pal and J. D. Ramsey, J. Biomed. Mater. Res., Part A, 2013, 101, 1857–1864 CrossRef.
- A. Vetter, K. S. Virdi, S. Espenlaub, W. Rödl, E. Wagner, P. S. Holm, C. Scheu, F. Kreppel, C. Spitzweg and M. Ogris, Mol. Pharm., 2013, 10, 606–618 CrossRef CAS.
- S. M. Moghimi, P. Symonds, J. C. Murray, A. C. Hunter, G. Debska and A. Szewczyk, Mol. Ther., 2005, 11, 990–995 CrossRef CAS.
- J. Raymond, A. Metcalfe, I. Salazkin and A. Schwarz, Biomaterials, 2004, 25, 3983–3989 CrossRef CAS.
- J. S. Pagano and A. Vaheri, Arch. Gesamte Virusforsch., 1965, 17, 456–464 CrossRef CAS.
- P. K. Vogt, Virology, 1967, 33, 175–177 CrossRef CAS.
- J. H. McCutchan and J. S. Pagano, J. Natl. Cancer Inst., 1968, 41, 351–357 CAS.
- J. S. Manning, A. J. Hackett and N. B. Darby, Appl. Microbiol., 1971, 22, 1162–1163 CrossRef CAS.
- H. E. Davis, J. R. Morgan and M. L. Yarmush, Biophys. Chem., 2002, 97, 159–172 CrossRef CAS.
- C. Zhao, N. Wu, F. Deng, H. Zhang, N. Wang, W. Zhang, X. Chen, S. Wen, J. Zhang, L. Yin, Z. Liao, Z. Zhang, Q. Zhang, Z. Yan, W. Liu, D. Wu, J. Ye, Y. Deng, G. Zhou, H. H. Luu, R. C. Haydon, W. Si and T.-C. He, PLoS One, 2014, 9, e92908 CrossRef.
- K. Toyoshima and P. K. Vogt, Virology, 1969, 38, 414–426 CrossRef CAS.
- A. Fasbender, J. Zabner, M. Chillón, T. O. Moninger, A. P. Puga, B. L. Davidson and M. J. Welsh, J. Biol. Chem., 1997, 272, 6479–6489 CrossRef CAS.
- A. Leydet, C. Moullet, J. P. Roque, M. Witvrouw, C. Pannecouque, G. Andrei, R. Snoeck, J. Neyts, D. Schols and E. De Clercq, J. Med. Chem., 1998, 41, 4927–4932 CrossRef CAS.
- M. Baba, R. Pauwels, J. Balzarini, J. Arnout, J. Desmyter and E. De Clercq, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 6132–6136 CrossRef CAS.
- N. Landázuri, D. Krishna, M. Gupta and J. M. Le Doux, Biotechnol. Prog., 2007, 23, 480–487 CrossRef.
- M. Chillón, J. H. Lee, A. Fasbender and M. J. Welsh, Gene Ther., 1998, 5, 995–1002 CrossRef.
- S. J. Beer, C. B. Matthews, C. S. Stein, B. D. Ross, J. M. Hilfinger and B. L. Davidson, Gene Ther., 1998, 5, 740–746 CrossRef CAS.
- K. D. Fisher, Y. Stallwood, N. K. Green, K. Ulbrich, V. Mautner and L. W. Seymour, Gene Ther., 2001, 8, 341–348 CrossRef CAS.
- Y. Eto, J. Q. Gao, F. Sekiguchi, S. Kurachi, K. Katayama, H. Mizuguchi, T. Hayakawa, Y. Tsutsumi, T. Mayumi and S. Nakagawa, Biol. Pharm. Bull., 2004, 27, 936–938 CrossRef CAS.
- N. K. Green, C. W. Herbert, S. J. Hale, A. B. Hale, V. Mautner, R. Harkins, T. Hermiston, K. Ulbrich, K. D. Fisher and L. W. Seymour, Gene Ther., 2004, 11, 1256–1263 CrossRef CAS.
- S. K. Seidlits, R. M. Gower, J. A. Shepard and L. D. Shea, Expert Opin. Drug Delivery, 2013, 10, 499–509 CrossRef CAS.
- H.-F. Wu, J.-S. Cen, Q. Zhong, L. Chen, J. Wang, D. Y. B. Deng and Y. Wan, Biomaterials, 2013, 34, 1686–1700 CrossRef CAS.
- J. A. Shepard, F. R. Virani, A. G. Goodman, T. D. Gossett, S. Shin and L. D. Shea, Biomaterials, 2012, 33, 7412–7421 CrossRef CAS.
- A. Ehsanipour, T. Nguyen, T. Aboufadel, M. Sathialingam, P. Cox, W. Xiao, C. M. Walthers and S. K. Seidlits, Cell. Mol. Bioeng., 2019, 12, 399–413 CrossRef CAS.
- T. M. D. Le, B.-K. Jung, Y. Li, H. T. T. Duong, T. L. Nguyen, J. W. Hong, C.-O. Yun and D. S. Lee, Biomater. Sci., 2019, 7, 4195–4207 RSC.
- I.-C. Liao, S. Chen, J. B. Liu and K. W. Leong, J. Controlled Release, 2009, 139, 48–55 CrossRef CAS.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2012, 64, 116–127 CrossRef.
- A. L. Klibanov, K. Maruyama, V. P. Torchilin and L. Huang, FEBS Lett., 1990, 268, 235–237 CrossRef CAS.
- Y. Inada, M. Furukawa, H. Sasaki, Y. Kodera, M. Hiroto, H. Nishimura and A. Matsushima, Trends Biotechnol., 1995, 13, 86–91 CrossRef CAS.
- F. Martin, A. Huang, B. Uziely, B. Kaufman and T. Safra, Cancer Res., 1994, 54, 987–992 Search PubMed.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. van Es and F. F. Davis, J. Biol. Chem., 1977, 252, 3582–3586 CAS.
- P. Wonganan and M. A. Croyle, Viruses, 2010, 2, 468–502 CrossRef CAS.
- R. Alemany, K. Suzuki and D. T. Curiel, J. Gen. Virol., 2000, 81, 2605–2609 CrossRef CAS.
- H. Romanczuk, C. E. Galer, J. Zabner, G. Barsomian, S. C. Wadsworth and C. R. O'Riordan, Hum. Gene Ther., 1999, 10, 2615–2626 CrossRef CAS.
- K. I. Ogawara, M. G. Rots, R. J. Kok, H. E. Moorlag, A. M. Van Loenen, D. K. F. Meijer, H. J. Haisma and G. Molema, Hum. Gene Ther., 2004, 15, 433–443 CrossRef CAS.
- G. K. Lee, N. Maheshri, B. Kaspar and D. V. Schaffer, Biotechnol. Bioeng., 2005, 92, 24–34 CrossRef CAS.
- H. T. Le, Q. C. Yu, J. M. Wilson and M. A. Croyle, J. Controlled Release, 2005, 108, 161–177 CrossRef CAS.
- T. Yao, X. Zhou, C. Zhang, X. Yu, Z. Tian, L. Zhang and D. Zhou, Molecules, 2017, 22, 1155 CrossRef.
- J. Q. Gao, Y. Eto, Y. Yoshioka, F. Sekiguchi, S. Kurachi, T. Morishige, X. Yao, H. Watanabe, R. Asavatanabodee, F. Sakurai, H. Mizuguchi, Y. Okada, Y. Mukai, Y. Tsutsumi, T. Mayumi, N. Okada and S. Nakagawa, J. Controlled Release, 2007, 122, 102–110 CrossRef CAS.
- K. Doronin, E. V. Shashkova, S. M. May, S. E. Hofherr and M. A. Barry, Hum. Gene Ther., 2009, 20, 975–988 CrossRef CAS.
- S. E. Hofherr, E. V. Shashkova, E. A. Weaver, R. Khare and M. A. Barry, Mol. Ther., 2008, 16, 1276–1282 CrossRef CAS.
- A. Wortmann, S. Vöhringer, T. Engler, S. Corjon, R. Schirmbeck, J. Reimann, S. Kochanek and F. Kreppel, Mol. Ther., 2008, 16, 154–162 CrossRef CAS.
- H. Mok, D. J. Palmer, P. Ng and M. A. Barry, Mol. Ther., 2005, 11, 66–79 CrossRef CAS.
- M. Lyons, D. Onion, N. K. Green, K. Aslan, R. Rajaratnam, M. Bazan-Peregrino, S. Phipps, S. Hale, V. Mautner, L. W. Seymour and K. D. Fisher, Mol. Ther., 2006, 14, 118–128 CrossRef CAS.
- S. E. Hofherr, H. Mok, F. C. Gushiken, J. A. Lopez and M. A. Barry, Hum. Gene Ther., 2007, 18, 837–848 CrossRef CAS.
- H. Hatakeyama, H. Akita and H. Harashima, Adv. Drug Delivery Rev., 2011, 63, 152–160 CrossRef CAS.
- J. W. Choi, S. J. Jung, D. Kasala, J. K. Hwang, J. Hu, Y. H. Bae and C. O. Yun, J. Controlled Release, 2015, 205, 134–143 CrossRef CAS.
- K. Nam, H. Y. Nam, P. H. Kim and S. W. Kim, Biomaterials, 2012, 33, 8122–8130 CrossRef CAS.
- D. Kasala, S. H. Lee, J. W. Hong, J. W. Choi, K. Nam, Y. H. Chung, S. W. Kim and C. O. Yun, Biomaterials, 2017, 145, 207–222 CrossRef CAS.
- J. J. F. Verhoef, J. F. Carpenter, T. J. Anchordoquy and H. Schellekens, Drug Discovery Today, 2014, 19, 1945–1952 CrossRef CAS.
- Y. Zhao, C. Wang, L. Wang, Q. Yang, W. Tang, Z. She and Y. Deng, Eur. J. Pharm. Biopharm., 2012, 81, 506–513 CrossRef CAS.
- T. T. Hoang Thi, E. H. Pilkington, D. H. Nguyen, J. S. Lee, K. D. Park and N. P. Truong, Polymer, 2020, 12, 298 Search PubMed.
- S.-M. Zou, P. Erbacher, J.-S. Remy and J.-P. Behr, J. Gene Med., 2000, 2, 128–134 CrossRef CAS.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Controlled Release, 1999, 60, 149–160 CrossRef CAS.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS.
- L. M. P. Vermeulen, S. C. De Smedt, K. Remaut and K. Braeckmans, Eur. J. Pharm. Biopharm., 2018, 129, 184–190 CrossRef CAS.
- J. W. Choi, J. P. Nam, K. Nam, Y. S. Lee, C. O. Yun and S. W. Kim, Biomacromolecules, 2015, 16, 2132–2143 CrossRef CAS.
- T. Blessing, M. Kursa, R. Holzhauser, R. Kircheis and E. Wagner, Bioconjugate Chem., 2001, 12, 529–537 CrossRef CAS.
- S. H. Pun, N. C. Bellocq, A. Liu, G. Jensen, T. Machemer, E. Quijano, T. Schluep, S. Wen, H. Engler, J. Heidel and M. E. Davis, Bioconjugate Chem., 2004, 15, 831–840 CrossRef CAS.
- H. Yao, S.-C. Chen, Z. Shen, Y.-C. Huang, X. Zhu, X. Wang, W. Jiang, Z.-F. Wang, X.-W. Bian, E.-A. Ling, H. Kung and M. C. Lin, Curr. Med. Chem., 2013, 20, 2601–2608 CrossRef CAS.
- J. Han, D. Zhao, Z. Zhong, Z. Zhang, T. Gong and X. Sun, Nanotechnology, 2010, 21, 105106 CrossRef.
- C. Yoshihara, K. Hamada and Y. Koyama, Oncol. Rep., 2010, 23, 733–738 CAS.
- K. Nosaki, K. Hamada, Y. Takashima, M. Sagara, Y. Matsumura, S. Miyamoto, Y. Hijikata, T. Okazaki, Y. Nakanishi and K. Tani, Mol. Ther.–Oncolytics, 2016, 3, 16022 CrossRef CAS.
- Y. Na, J. P. Nam, J. W. Hong, E. Oh, H. C. Shin, H. S. Kim, S. W. Kim and C. O. Yun, J. Controlled Release, 2019, 305, 75–88 CrossRef CAS.
- S. Y. Chae, H. J. Kim, M. S. Lee, Y. L. Jang, Y. Lee, S. H. Lee, K. Lee, S. H. Kim, H. T. Kim, S.-C. Chi, T. G. Park and J. H. Jeong, Macromol. Biosci., 2011, 11, 1169–1174 CrossRef CAS.
- A. Bajaj, P. Kondaiah and S. Bhattacharya, Bioconjugate Chem., 2008, 19, 1640–1651 CrossRef CAS.
- D. Lee, D. Kim, H. Mok, J. H. Jeong, D. Choi and S. H. Kim, Pharm. Res., 2012, 29, 2213–2224 CrossRef CAS.
- C.-H. Lee, D. Kasala, Y. Na, M. S. Lee, S. W. Kim, J. H. Jeong and C.-O. Yun, Biomaterials, 2014, 35, 5505–5516 CrossRef CAS.
- H. Pan, P. Li, G. Li, W. Li, B. Hu, H. He, Z. Chen, F. Wang, L. Liu, Y. Gong, Y. Han, Y. Luo, M. Zheng, Y. Ma, L. Cai and Y. Jin, Adv. Funct. Mater., 2019, 29, 1807528 CrossRef.
- E. Kim, I. T. Song, S. Lee, J.-S. Kim, H. Lee and J.-H. Jang, Angew. Chem., Int. Ed., 2012, 51, 5598–5601 CrossRef CAS.
- S.-H. Kim, S. J. Yu, I. Kim, J. Choi, Y. H. Choi, S. G. Im and N. S. Hwang, Chem. Commun., 2019, 55, 2317–2320 RSC.
- H. K. Makadia and S. J. Siegel, Polymer, 2011, 3, 1377–1397 CAS.
- M. N. V. Ravi Kumar, U. Bakowsky and C. M. Lehr, Biomaterials, 2004, 25, 1771–1777 CrossRef CAS.
- C. B. Matthews, G. Jenkins, J. M. Hilfinger and B. L. Davidson, Gene Ther., 1999, 6, 1558–1564 CrossRef CAS.
- H. Mok, J. W. Park and T. G. Park, Pharm. Res., 2007, 24, 2263–2269 CrossRef CAS.
- Y. Zhu, D. Li, K. Zhang, L. Jiang, C. Shi, J. Fangteng, C. Zheng, B. Yang and H. Sun, J. Biomed. Nanotechnol., 2017, 13, 437–446 CrossRef CAS.
- N. Badrinath, Y. Il Jeong, H. Y. Woo, S. Y. Bang, C. Kim, J. Heo, D. H. Kang and S. Y. Yoo, Int. J. Pharm., 2018, 552, 437–442 CrossRef CAS.
- P. W. Lee, S. Shukla, J. D. Wallat, C. Danda, N. F. Steinmetz, J. Maia and J. K. Pokorski, ACS Nano, 2017, 11, 8777–8789 CrossRef CAS.
- K. Deshmukh, M. Basheer Ahamed, R. R. Deshmukh, S. K. Khadheer Pasha, P. R. Bhagat and K. Chidambaram, in Biopolymer Composites in Electronics, Elsevier, Amsterdam, NL, 2017, pp. 27–128 Search PubMed.
- S. Stojanov and A. Berlec, Front. Bioeng. Biotechnol., 2020, 8, 130 CrossRef.
- Y. Zheng, Y. Wu, Y. Zhou, J. Wu, X. Wang, Y. Qu, Y. Wang, Y. Zhang and Q. Yu, ACS Appl. Mater. Interfaces, 2020, 12, 7905–7914 CrossRef CAS.
- Z. M. Huang, C. L. He, A. Yang, Y. Zhang, X. J. Han, J. Yin and Q. Wu, J. Biomed. Mater. Res., Part A, 2006, 77, 169–179 CrossRef.
- X. Gu, Y. Matsumura, Y. Tang, S. Roy, R. Hoff, B. Wang and W. R. Wagner, Biomaterials, 2017, 133, 132–143 CrossRef CAS.
- S. Lee, J. S. Kim, H. S. Chu, G. W. Kim, J. I. Won and J. H. Jang, Acta Biomater., 2011, 7, 3868–3876 CrossRef CAS.
- S. H. Kim, S. Lee, H. Lee, M. Cho, D. V. Schaffer and J. H. Jang, Mol. Ther.–Nucleic Acids, 2019, 18, 432–443 CrossRef CAS.
- J. K. Venkatesan, C. Falentin-Daudré, A. Leroux, V. Migonney and M. Cucchiarini, Tissue Eng., Part A, 2020, 26, 450–459 CrossRef CAS.
- Wyandotte Chemicals Corporation, U.S, 2674619, 1954 Search PubMed.
- V. C. McLain, Int. J. Toxicol., 2008, 27, 93–128 CrossRef.
- K. W. Chun, J. B. Lee, S. H. Kim and T. G. Park, Biomaterials, 2005, 26, 3319–3326 CrossRef CAS.
- Y. Wang, S. Liu, C. Y. Li and F. Yuan, Cancer Res., 2005, 65, 7541–7545 CrossRef CAS.
- K. L. March, J. E. Madison and B. C. Trapnell, Hum. Gene Ther., 1995, 6, 41–53 CrossRef CAS.
- L. J. Feldman, C. J. Pastore, N. Aubailly, M. Kearney, D. Chen, M. Perricaudet, P. G. Steg and J. M. Isner, Gene Ther., 1997, 4, 189–198 CrossRef CAS.
- P. M. Strappe, D. W. Hampton, B. Cachon-Gonzalez, J. W. Fawcett and A. Lever, Eur. J. Pharm. Biopharm., 2005, 61, 126–133 CrossRef CAS.
- A. Rey-Rico, J. K. Venkatesan, J. Frisch, I. Rial-Hermida, G. Schmitt, A. Concheiro, H. Madry, C. Alvarez-Lorenzo and M. Cucchiarini, Acta Biomater., 2015, 27, 42–52 CrossRef CAS.
- A. Rey-Rico, J. K. Venkatesan, J. Frisch, G. Schmitt, A. Monge-Marcet, P. Lopez-Chicon, A. Mata, C. Semino, H. Madry and M. Cucchiarini, Acta Biomater., 2015, 18, 118–127 CrossRef CAS.
- H. H. Lee, A. M. Haleem, V. Yao, J. Li, X. Xiao and C. R. Chu, Tissue Eng., Part A, 2011, 17, 1969–1978 CrossRef CAS.
- A. Rey-Rico, J. Frisch, J. K. Venkatesan, G. Schmitt, I. Rial-Hermida, P. Taboada, A. Concheiro, H. Madry, C. Alvarez-Lorenzo and M. Cucchiarini, ACS Appl. Mater. Interfaces, 2016, 8, 20600–20613 CrossRef CAS.
- A. Rey-Rico, J. K. Venkatesan, G. Schmitt, S. Speicher-Mentges, H. Madry and M. Cucchiarini, Mol. Pharm., 2018, 15, 2816–2826 CrossRef CAS.
- H. Madry, L. Gao, A. Rey-Rico, J. K. Venkatesan, K. Müller-Brandt, X. Cai, L. Goebel, G. Schmitt, S. Speicher-Mentges, D. Zurakowski, M. D. Menger, M. W. Laschke and M. Cucchiarini, Adv. Mater., 2020, 32, 1906508 CrossRef CAS.
- J. M. Caronia, D. W. Sorensen, H. M. Leslie, J. H. van Berlo and S. M. Azarin, Biotechnol. Bioeng., 2019, 116, 2353–2363 CrossRef CAS.
- I. Höfig, M. J. Atkinson, S. Mall, A. M. Krackhardt, C. Thirion and N. Anastasov, J. Gene Med., 2012, 14, 549–560 CrossRef.
- I. Hauber, N. Beschorner, S. Schrödel, J. Chemnitz, N. Kröger, J. Hauber and C. Thirion, Hum. Gene Ther: Methods., 2018, 29, 104–113 CrossRef CAS.
- Y. Jang, Y.-S. Kim, M. M. Wielgosz, F. Ferrara, Z. Ma, J. Condori, L. E. Palmer, X. Zhao, G. Kang, D. J. Rawlings, S. Zhou and B. Y. Ryu, Gene Ther., 2020, 1–12 Search PubMed.
- J. W. Schott, D. León-Rico, C. B. Ferreira, K. F. Buckland, G. Santilli, M. A. Armant, A. Schambach, A. Cavazza and A. J. Thrasher, Mol. Ther.–Methods Clin. Dev., 2019, 14, 134–147 CrossRef CAS.
- M. Delville, T. Soheili, F. Bellier, A. Durand, A. Denis, C. Lagresle-Peyrou, M. Cavazzana, I. Andre-Schmutz and E. Six, Mol. Ther.–Methods Clin. Dev., 2018, 10, 341–347 CrossRef CAS.
- B. Simon, D. C. Harrer, C. Thirion, B. Schuler-Thurner, G. Schuler and U. Uslu, J. Immunol. Methods, 2019, 472, 55–64 CrossRef CAS.
- E. Abbasi, S. Aval, A. Akbarzadeh, M. Milani, H. Nasrabadi, S. Joo, Y. Hanifehpour, K. Nejati-Koshki and R. Pashaei-Asl, Nanoscale Res. Lett., 2014, 9, 247 CrossRef.
- J. Yang, Q. Zhang, H. Chang and Y. Cheng, Chem. Rev., 2015, 115, 5274–5300 CrossRef CAS.
- Z. Mhlwatika and B. Aderibigbe, Molecules, 2018, 23, 2205 CrossRef.
- M. A. Kostiainen, O. Kasyutich, J. J. L. M. Cornelissen and R. J. M. Nolte, Nat. Chem., 2010, 2, 394–399 CrossRef CAS.
- G. Navarro, G. Maiwald, R. Haase, A. L. Rogach, E. Wagner, C. T. de Ilarduya and M. Ogris, J. Controlled Release, 2010, 146, 99–105 CrossRef CAS.
- A. R. Yoon, D. Kasala, Y. Li, J. Hong, W. Lee, S. J. Jung and C. O. Yun, J. Controlled Release, 2016, 231, 2–16 CrossRef CAS.
- R. E. Bauer, C. G. Clark and K. Müllen, New J. Chem., 2007, 31, 1275–1282 RSC.
- R. Stangenberg, Y. Wu, J. Hedrich, D. Kurzbach, D. Wehner, G. Weidinger, S. L. Kuan, M. I. Jansen, F. Jelezko, H. J. Luhmann, D. Hinderberger, T. Weil and K. Müllen, Adv. Healthcare Mater., 2015, 4, 377–384 CrossRef CAS.
- J. Wagner, L. Li, J. Simon, L. Krutzke, K. Landfester, V. Mailänder, K. Müllen, D. Y. W. Ng, Y. Wu and T. Weil, Angew. Chem., Int. Ed., 2020, 59, 5712–5720 CrossRef CAS.
- L. M. Kasman, S. Barua, P. Lu, K. Rege and C. Voelkel-Johnson, Mol. Pharm., 2009, 6, 1612–1619 CrossRef CAS.
- D. Steinhauff and H. Ghandehari, Bioconjugate Chem., 2019, 30, 384–399 CrossRef CAS.
- J. A. Rowley and D. J. Mooney, J. Biomed. Mater. Res., 2002, 60, 217–223 CrossRef CAS.
- G. Sailaja, H. HogenEsch, A. North, J. Hays and S. K. Mittal, Gene Ther., 2002, 9, 1722–1729 CrossRef CAS.
- J. W. Choi, E. Kang, O. J. Kwon, T. J. Yun, H. K. Park, P. H. Kim, S. W. Kim, J. H. Kim and C. O. Yun, Gene Ther., 2013, 20, 880–892 CrossRef CAS.
- R. S. Stilhano, J. L. Madrigal, K. Wong, P. A. Williams, P. K. M. Martin, F. S. M. Yamaguchi, V. Y. Samoto, S. W. Han and E. A. Silva, J. Controlled Release, 2016, 237, 42–49 CrossRef CAS.
- J. L. Madrigal, R. S. Stilhano, C. Siltanen, K. Tanaka, S. N. Rezvani, R. P. Morgan, A. Revzin, S. W. Han and E. A. Silva, J. Mater. Chem. B, 2016, 4, 6989–6999 RSC.
- J. L. Madrigal, S. N. Sharma, K. T. Campbell, R. S. Stilhano, R. Gijsbers and E. A. Silva, Acta Biomater., 2018, 69, 265–276 CrossRef CAS.
- J. L. Madrigal, S. Shams, R. S. Stilhano and E. A. Silva, Biomater. Sci., 2019, 7, 645–656 RSC.
- S. Magnusson and T. Berg, Biochem. J., 1989, 257, 651–656 CrossRef CAS.
- S. Espenlaub, A. Wortmann, T. Engler, S. Corjon, S. Kochanek and F. Kreppel, J. Gene Med., 2008, 10, 1303–1314 CrossRef CAS.
- Z. Liu, F. Ke, C. Duan, H. Lan, J. Li, C. Gao, J. Li and Z. Zhong, Bioconjugate Chem., 2013, 24, 1387–1397 CrossRef CAS.
- Y. Cho, R. Shi and R. B. Borgens, J. Exp. Biol., 2010, 213, 1513–1520 CrossRef CAS.
- Q. Guo, C. Liu, B. Hai, T. Ma, W. Zhang, J. Tan, X. Fu, H. Wang, Y. Xu and C. Song, J. Biomed. Mater. Res., Part B, 2018, 106, 787–799 CrossRef CAS.
- R. Keswani, K. Su and D. W. Pack, J. Controlled Release, 2014, 192, 40–46 CrossRef CAS.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1753 CrossRef CAS.
- A. Rey-Rico, H. Babicz, H. Madry, A. Concheiro, C. Alvarez-Lorenzo and M. Cucchiarini, Int. J. Pharm., 2017, 531, 492–503 CrossRef CAS.
- L. Chen, X. Zhao, Y. Lin, Y. Huang and Q. Wang, Chem. Commun., 2013, 49, 9678–9680 RSC.
- A. Rey-Rico and M. Cucchiarini, Polymer, 2019, 11, 514 CAS.
- H. Rosilo, J. R. McKee, E. Kontturi, T. Koho, V. P. Hytönen, O. Ikkala and M. A. Kostiainen, Nanoscale, 2014, 6, 11871–11881 RSC.
- Y. Huang, X. Ding, Y. Qi, B. Yu and F. J. Xu, Biomaterials, 2016, 106, 134–143 CrossRef CAS.
- W. Fan, M. Shao, J. Zhang, G. Jin, F. Liu and F.-J. Xu, Adv. Funct. Mater., 2019, 29, 1807104 CrossRef.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12, 986–998 CrossRef CAS.
- S. Y. Fam, C. F. Chee, C. Y. Yong, K. L. Ho, A. R. Mariatulqabtiah, H. Y. Lau and W. S. Tan, Int. J. Mol. Sci., 2019, 20, 4903 CrossRef CAS.
- K. D. Fisher, N. K. Green, A. Hale, V. Subr, K. Ulbrich and L. W. Seymour, J. Drug Targeting, 2007, 15, 546–551 CrossRef CAS.
- N. Francini, D. Cochrane, S. Illingworth, L. Purdie, G. Mantovani, K. Fisher, L. W. Seymour, S. G. Spain and C. Alexander, Bioconjugate Chem., 2019, 30, 1244–1257 CrossRef CAS.
- T. Kim, M. Ou, M. Lee and S. W. Kim, Biomaterials, 2009, 30, 658–664 CrossRef CAS.
- Y. F. Zeng, S. J. Tseng, I. M. Kempson, S. F. Peng, W. T. Wu and J. R. Liu, Biomaterials, 2012, 33, 9239–9245 CrossRef CAS.
- P. Shrimali, M. Peter, A. Singh, N. Dalal, S. Dakave, S. V. Chiplunkar and P. Tayalia, Biomater. Sci., 2018, 6, 3241–3250 RSC.
- S. K. Cho and Y. J. Kwon, Biomaterials, 2012, 33, 3316–3323 CrossRef CAS.
- C. A. Hong, S. K. Cho, J. A. Edson, J. Kim, D. Ingato, B. Pham, A. Chuang, D. A. Fruman and Y. J. Kwon, ACS Nano, 2016, 10, 8705–8714 CrossRef CAS.
- G. J. Tong, S. C. Hsiao, Z. M. Carrico and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 11174–11178 CrossRef CAS.
- D. Muharemagic, M. Labib, S. M. Ghobadloo, A. S. Zamay, J. C. Bell and M. V. Berezovski, J. Am. Chem. Soc., 2012, 134, 17168–17177 CrossRef CAS.
- Y. Wu, L. Zhang, C. Cui, S. Cansiz, H. Liang, C. Wu, I.-T. Teng, W. Chen, Y. Liu, W. Hou, X. Zhang and W. Tan, J. Am. Chem. Soc., 2018, 140, 2–5 CrossRef CAS.
- E. Kim, S. Lee, S. Hong, G. Jin, M. Kim, K. I. Park, H. Lee and J.-H. Jang, ACS Appl. Mater. Interfaces, 2014, 6, 8288–8294 CrossRef CAS.
- S.-H. Kim, M. Lee, M. Cho, I.-S. Kim, K. I. Park, H. Lee and J.-H. Jang, Macromol. Biosci., 2017, 17, 1700148 CrossRef.
- F. Alvarez-Rivera, A. Rey-Rico, J. K. Venkatesan, L. Diaz-Gomez, M. Cucchiarini, A. Concheiro and C. Alvarez-Lorenzo, Pharmaceutics, 2020, 12, 335 CrossRef.
- H. Qiao, X. Chen, Q. Wang, J. Zhang, D. Huang, E. Chen, H. Qian, Y. Zhong, Q. Tang and W. Chen, Biomater. Sci., 2020, 8, 2472–2480 RSC.
- R. Myers, C. Coviello, P. Erbs, J. Foloppe, C. Rowe, J. Kwan, C. Crake, S. Finn, E. Jackson, J. M. Balloul, C. Story, C. Coussios and R. Carlisle, Mol. Ther., 2016, 24, 1627–1633 CrossRef CAS.
- J. L. Madrigal, S. Shams, R. S. Stilhano and E. A. Silva, Biomater. Sci., 2019, 7, 645–656 RSC.
- L. M. Kasman, S. Barua, P. Lu, K. Rege and C. Voelkel-Johnson, Mol. Pharm., 2009, 6, 1612–1619 CrossRef CAS.
- M. Rabenstein and Y.-K. Shin, Biochemistry, 1995, 34, 13390–13397 CrossRef CAS.
- Z. Kang, Q. Meng and K. Liu, J. Mater. Chem. B, 2019, 7, 1824–1841 RSC.
- K. Numata, Y. Horii, K. Oikawa, Y. Miyagi, T. Demura and M. Ohtani, Sci. Rep., 2018, 8, 1–17 CrossRef CAS.
- F. Milletti, Drug Discovery Today, 2012, 17, 850–860 CrossRef CAS.
- A. D. Frankel and C. O. Pabo, Cell, 1988, 55, 1189–1193 CrossRef CAS.
- M. Green and P. M. Loewenstein, Cell, 1988, 55, 1179–1188 CrossRef CAS.
- P. Agrawal, S. Bhalla, S. S. Usmani, S. Singh, K. Chaudhary, G. P. S. Raghava and A. Gautam, Nucleic Acids Res., 2016, 44, D1098–D1103 CrossRef CAS.
- B. Gupta, T. Levchenko and V. Torchilin, Adv. Drug Delivery Rev., 2005, 57, 637–651 CrossRef CAS.
- S. M. Farkhani, A. Valizadeh, H. Karami, S. Mohammadi, N. Sohrabi and F. Badrzadeh, Peptides, 2014, 57, 78–94 CrossRef CAS.
- J. Váňová, A. Hejtmánková, M. H. Kalbáčová and H. Španielová, Materials, 2019, 12, 2671 CrossRef.
- R. E. Taylor and M. Zahid, Pharmaceutics, 2020, 12, 225 CrossRef CAS.
- A. S. Nigatu, S. Vupputuri, N. Flynn, B. J. Neely and J. D. Ramsey, J. Pharm. Sci., 2013, 102, 1981–1993 CrossRef CAS.
- J. P. Gratton, J. Yu, J. W. Griffith, R. W. Babbitt, R. S. Scotland, R. Hickey, F. J. Giordano and W. C. Sessa, Nat. Med., 2003, 9, 357–362 CrossRef CAS.
- F. Kühnel, B. Schulte, T. Wirth, N. Woller, S. Schäfers, L. Zender, M. Manns and S. Kubicka, J. Virol., 2004, 78, 13743–13754 CrossRef.
- Y. Yoshioka, R. Asavatanabodee, Y. Eto, H. Watanabe, T. Morishige, X. Yao, S. Kida, M. Maeda, Y. Mukai, H. Mizuguchi, K. Kawasaki, N. Okada and S. Nakagawa, Life Sci., 2008, 83, 747–755 CrossRef CAS.
- A. S. Nigatu, S. Vupputuri, N. Flynn and J. D. Ramsey, Biomed. Pharmacother., 2015, 71, 153–160 CrossRef CAS.
- P. Brugada-Vilà, A. Cascante, M. Á. Lázaro, C. Castells-Sala, C. Fornaguera, M. Rovira-Rigau, L. Albertazzi, S. Borros and C. Fillat, Theranostics, 2020, 10, 2744–2758 CrossRef.
- T. V. Nguyen, S. S. Anguiano-Zarate, W. E. Matchett, M. E. Barry and M. A. Barry, Virology, 2018, 514, 118–123 CrossRef CAS.
- B. K. Gan, C. Y. Yong, K. L. Ho, A. R. Omar, N. B. Alitheen and W. S. Tan, Sci. Rep., 2018, 8, 8499 CrossRef.
- T. Han, Y. Tang, H. Ugai, L. E. Perry, G. P. Siegal, J. L. Contreras and H. Wu, Virol. J., 2007, 4, 103 CrossRef.
- Y. Eto, Y. Yoshioka, R. Asavatanabodee, S. Kida, M. Maeda, Y. Mukai, H. Mizuguchi, K. Kawasaki, N. Okada and S. Nakagawa, Peptides, 2009, 30, 1548–1552 CrossRef CAS.
- S. Kida, Y. Eto, Y. Yoshioka, S. Nakagawa, K. Kawasaki and M. Maeda, Protein Pept. Lett., 2010, 17, 164–167 CrossRef CAS.
- A. S. Nigatu, S. Vupputuri, N. Flynn, B. J. Neely and J. D. Ramsey, J. Pharm. Sci., 2013, 102, 1981–1993 CrossRef CAS.
- Z. Wu, K. Chen, I. Yildiz, A. Dirksen, R. Fischer, P. E. Dawson and N. F. Steinmetz, Nanoscale, 2012, 4, 3567 RSC.
- D. Kim, Y. Lee, T. W. Dreher and T.-J. Cho, Virus Res., 2018, 252, 13–21 CrossRef CAS.
- X. Zhang, T. He, Z. Chai, R. J. Samulski and C. Li, Biomaterials, 2018, 176, 71–83 CrossRef CAS.
- X. Ma, P. Liu, X. Zhang, W. Jiang, M. Jia, C. Wang, Y. Dong, Y. Dang and C. Gao, Sci. Rep., 2016, 6, 22404 CrossRef CAS.
- E. Koren and V. P. Torchilin, Trends Mol. Med., 2012, 18, 385–393 CrossRef CAS.
- S. Pujals, J. Fernández-Carneado, C. López-Iglesias, M. J. Kogan and E. Giralt, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 264–279 CrossRef CAS.
- S. Futaki, T. Suzuki, W. Ohashi, T. Yagami, S. Tanaka, K. Ueda and Y. Sugiura, J. Biol. Chem., 2001, 276, 5836–5840 CrossRef CAS.
- D. J. Mitchell, L. Steinman, D. T. Kim, C. G. Fathman and J. B. Rothbard, J. Pept. Res., 2000, 56, 318–325 CrossRef CAS.
- P. A. Wender, D. J. Mitchell, K. Pattabiraman, E. T. Pelkey, L. Steinman and J. B. Rothbard, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13003–13008 CrossRef CAS.
- Y. Liu, Y. J. Kim, M. Ji, J. Fang, N. Siriwon, L. I. Zhang and P. Wang, Mol. Ther.–Methods Clin. Dev., 2014, 1, 12 CrossRef.
- J. Váňová, A. Hejtmánková, J. Žáčková Suchanová, P. Sauerová, J. Forstová, M. Hubálek Kalbáčová and H. Španielová, Int. J. Pharm., 2020, 576, 119008 CrossRef.
- S. Das, R. S. Jacob, K. Patel, N. Singh and S. K. Maji, Biomacromolecules, 2018, 19, 1826–1839 CrossRef CAS.
- J. Chen and X. Zou, Bioact. Mater., 2019, 4, 120–131 CrossRef.
- C. Meier, T. Weil, F. Kirchhoff and J. Münch, WIREs Nanomed. Nanobiotechnol., 2014, 6, 438–451 CrossRef CAS.
- G. G. Glenner and C. W. Wong, Biochem. Biophys. Res. Commun., 1984, 122, 1131–1135 CrossRef CAS.
- W. M. Wojtowicz, M. Farzan, J. L. Joyal, K. Carter, G. J. Babcock, D. I. Israel, J. Sodroski and T. Mirzabekov, J. Biol. Chem., 2002, 277, 35019–35024 CrossRef CAS.
- D. Romero, C. Aguilar, R. Losick and R. Kolter, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2230–2234 CrossRef CAS.
- J. Münch, E. Rücker, L. Ständker, K. Adermann, C. Goffinet, M. Schindler, S. Wildum, R. Chinnadurai, D. Rajan, A. Specht, G. Giménez-Gallego, P. C. Sánchez, D. M. Fowler, A. Koulov, J. W. Kelly, W. Mothes, J. C. Grivel, L. Margolis, O. T. Keppler, W. G. Forssmann and F. Kirchhoff, Cell, 2007, 131, 1059–1071 CrossRef.
- F. Arnold, J. Schnell, O. Zirafi, C. Sturzel, C. Meier, T. Weil, L. Standker, W.-G. Forssmann, N. R. Roan, W. C. Greene, F. Kirchhoff and J. Münch, J. Virol., 2012, 86, 1244–1249 CrossRef CAS.
- N. R. Roan, J. A. Müller, H. Liu, S. Chu, F. Arnold, C. M. Stürzel, P. Walther, M. Dong, H. E. Witkowska, F. Kirchhoff, J. Münch and W. C. Greene, Cell Host Microbe, 2011, 10, 541–550 CrossRef CAS.
- L. Zhang, C. Jiang, H. Zhang, X. Gong, L. Yang, L. Miao, Y. Shi, Y. Zhang, W. Kong, C. Zhang and Y. Shan, J. Pept. Sci., 2014, 20, 46–54 CrossRef CAS.
- S. Rode, M. Hayn, A. Röcker, S. Sieste, M. Lamla, D. Markx, C. Meier, F. Kirchhoff, P. Walther, M. Fändrich, T. Weil and J. Münch, Bioconjugate Chem., 2017, 28, 1260–1270 CrossRef CAS.
- H. Zhang, X. He, Y. Shi, Y. Yu, S. Guan, X. Gong, H. Yin, Z. Kuai and Y. Shan, RSC Adv., 2016, 6, 82082–82087 RSC.
- J. Chen, R. Ren, F. Yu, C. Wang, X. Zhang, W. Li, S. Tan, S. Jiang, S. Liu and L. Li, Biophys. J., 2017, 113, 1425–1439 CrossRef CAS.
- J. Chen, R. Ren, S. Tan, W. Zhang, X. Zhang, F. Yu, T. Xun, S. Jiang, S. Liu and L. Li, PLoS One, 2015, 10, e0144522 CrossRef.
- S. Tan, L. Li, L. Lu, C. Pan, H. Lu, Y. Oksov, X. Tang, S. Jiang and S. Liu, FEBS Lett., 2014, 588, 1515–1522 CrossRef CAS.
- J. Münch, E. Rücker, L. Ständker, K. Adermann, C. Goffinet, M. Schindler, S. Wildum, R. Chinnadurai, D. Rajan, A. Specht, G. Giménez-Gallego, P. C. Sánchez, D. M. Fowler, A. Koulov, J. W. Kelly, W. Mothes, J.-C. Grivel, L. Margolis, O. T. Keppler, W.-G. Forssmann and F. Kirchhoff, Cell, 2007, 131, 1059–1071 CrossRef.
- A. Groß, K. Rödel, B. Kneidl, N. Donhauser, M. Mössl, E. Lump, J. Münch, B. Schmidt and J. Eichler, ChemBioChem, 2015, 16, 446–454 CrossRef.
- C. Kokotidou, S. V. R. Jonnalagadda, A. A. Orr, G. Vrentzos, A. Kretsovali, P. Tamamis and A. Mitraki, Biomolecules, 2020, 10, 1–31 Search PubMed.
- A. L. Rodriguez, T.-Y. Wang, K. F. Bruggeman, R. Li, R. J. Williams, C. L. Parish and D. R. Nisbet, Nano Res., 2016, 9, 674–684 CrossRef CAS.
- S. Zhang, T. C. Holmes, C. M. DiPersio, R. O. Hynes, X. Su and A. Rich, Biomaterials, 1995, 16, 1385–1393 CrossRef CAS.
- A. Kichler, C. Leborgne, J. Marz, O. Danos and B. Bechinger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1564–1568 CrossRef CAS.
- D. Fenard, S. Genries, D. Scherman, A. Galy, S. Martin and A. Kichler, J. Virol. Methods, 2013, 189, 375–378 CrossRef CAS.
- C. Piovan, V. Marin, C. Scavullo, S. Corna, E. Giuliani, S. Bossi, A. Galy, D. Fenard, C. Bordignon, G. P. Rizzardi and C. Bovolenta, Mol. Ther.–Methods Clin. Dev., 2017, 5, 22–30 CrossRef CAS.
- C. Radek, O. Bernadin, K. Drechsel, N. Cordes, R. Pfeifer, P. Sträßer, M. Mormin, A. Gutierrez-Guerrero, F. Cosset, A. D. Kaiser, T. Schaser, A. Galy, E. Verhoeyen and I. C. D. Johnston, Hum. Gene Ther., 2019, 30, 1477–1493 CrossRef CAS.
- A. Jamali, L. Kapitza, T. Schaser, I. C. D. Johnston, C. J. Buchholz and J. Hartmann, Mol. Ther.–Methods Clin. Dev., 2019, 13, 371–379 CrossRef CAS.
- S. Majdoul, A. K. Seye, A. Kichler, N. Holic, A. Galy, B. Bechinger and D. Fenard, J. Biol. Chem., 2016, 291, 2161–2169 CrossRef CAS.
- K. Cornetta and W. F. Anderson, J. Virol. Methods, 1989, 23, 187–194 CrossRef CAS.
- S. M. Arcasoy, J. D. Latoche, M. Gondor, B. R. Pitt and J. M. Pilewski, Gene Ther., 1997, 4, 32–38 CrossRef CAS.
- J. Y. Lee and H. H. Lee, Cytotechnology, 2018, 70, 193–201 CrossRef CAS.
- H. Hanenberg, X. L. Xiao, D. Dilloo, K. Hashino, I. Kato and D. A. Williams, Nat. Med., 1996, 2, 876–882 CrossRef CAS.
- A. Quintás-Cardama, R. K. Yeh, D. Hollyman, J. Stefanski, C. Taylor, Y. Nikhamin, G. Imperato, M. Sadelain, I. Rivière and R. J. Brentjens, Hum. Gene Ther., 2007, 18, 1253–1260 CrossRef.
- G. Enblad, H. Karlsson, G. Gammelgård, J. Wenthe, T. Lövgren, R. M. Amini, K. I. Wikstrom, M. Essand, B. Savoldo, H. Hallböök, M. Höglund, G. Dotti, M. K. Brenner, H. Hagberg and A. Loskog, Clin. Cancer Res., 2018, 24, 6185–6194 CrossRef CAS.
- K. Dodo, H. Chono, N. Saito, Y. Tanaka, K. Tahara, I. Nukaya and J. Mineno, PLoS One, 2014, 9, e86275 CrossRef.
- B. Heemskerk, A. Jorritsma, R. Gomez-Eerland, M. Toebes, J. B. A. G. Haanen and T. N. M. Schumacher, Hum. Gene Ther., 2010, 21, 1335–1342 CrossRef CAS.
- H.-J. Lee, Y.-S. Lee, H.-S. Kim, Y.-K. Kim, J.-H. Kim, S.-H. Jeon, H.-W. Lee, S. Kim, H. Miyoshi, H.-M. Chung and D.-K. Kim, Biologicals, 2009, 37, 203–209 CrossRef CAS.
- H. Mizuguchi, T. Sasaki, K. Kawabata, F. Sakurai and T. Hayakawa, Biochem. Biophys. Res. Commun., 2005, 332, 1101–1106 CrossRef CAS.
- K. Tashiro, A. Kondo, K. Kawabata, H. Sakurai, F. Sakurai, K. Yamanishi, T. Hayakawa and H. Mizuguchi, Biochem. Biophys. Res. Commun., 2009, 379, 127–132 CrossRef CAS.
- J. D. Ramsey, H. N. Vu and D. W. Pack, J. Controlled Release, 2010, 144, 39–45 CrossRef CAS.
- H. Katakura, A. Harada, K. Kataoka, M. Furusho, F. Tanaka, H. Wada and K. Ikenaka, J. Gene Med., 2004, 6, 471–477 CrossRef CAS.
- M. Skoumal, S. Seidlits, S. Shin and L. Shea, Biotechnol. Bioeng., 2016, 113, 2033–2040 CrossRef CAS.
- H. Fang, Z. Guo, L. Lin, J. Chen, P. Sun, J. Wu, C. Xu, H. Tian and X. Chen, J. Am. Chem. Soc., 2018, 140, 11992–12000 CrossRef CAS.
- H. Ran, G. Quan, Y. Huang, C. Zhu, C. Lu, W. Liu, X. Pan and C. Wu, Biochem. Biophys. Res. Commun., 2019, 508, 791–796 CrossRef CAS.
- A. Breen, P. Dockery, T. O'Brien and A. Pandit, J. Biomed. Mater. Res., Part A, 2009, 89, 876–884 CrossRef.
- C. Schmidt, D. Bezuidenhout, P. Zilla and N. H. Davies, J. Biomater. Appl., 2014, 28, 1408–1418 CrossRef CAS.
- M. E. Kidd, S. Shin and L. D. Shea, J. Controlled Release, 2012, 157, 80–85 CrossRef CAS.
- M. Wang, J. Sun, A. Crosby, K. Woodard, M. L. Hirsch, R. J. Samulski and C. Li, Gene Ther., 2017, 24, 49–59 CrossRef CAS.
- D. Palesch, F. Boldt, J. A. Müller, K. Eisele, C. M. Stürzel, Y. Wu, J. Münch and T. Weil, ChemBioChem, 2016, 17, 1504–1508 CrossRef CAS.
- X. Pei, T. He, N. E. Hall, D. Gerber, R. J. Samulski and C. Li, Virology, 2018, 518, 95–102 CrossRef CAS.
- J. Park, P. L. Chariou and N. F. Steinmetz, Bioconjugate Chem., 2020, 31, 1408–1416 CrossRef CAS.
- J. A. Gustafson and H. Ghandehari, Adv. Drug Delivery Rev., 2010, 62, 1509–1523 CrossRef CAS.
- J. Cappello, J. Crissman, M. Dorman, M. Mikolajczak, G. Textor, M. Marquet and F. Ferrari, Biotechnol. Prog., 1990, 6, 198–202 CrossRef CAS.
- Z. Megeed, M. Haider, D. Li, B. W. O'Malley, J. Cappello and H. Ghandehari, J. Controlled Release, 2004, 94, 433–445 CrossRef CAS.
- A. Hatefi, J. Cappello and H. Ghandehari, Pharm. Res., 2007, 24, 773–779 CrossRef CAS.
- D. L. Price, P. Li, C.-H. Chen, D. Wong, Z. Yu, N. G. Chen, Y. A. Yu, A. A. Szalay, J. Cappello, Y. Fong and R. J. Wong, Head Neck, 2016, 38, 237–246 CrossRef.
- R. Price, J. Gustafson, K. Greish, J. Cappello, L. McGill and H. Ghandehari, Int. J. Pharm., 2012, 427, 97–104 CrossRef CAS.
- W.-W. Hu, Z. Wang and P. H. Krebsbach, J. Tissue Eng. Regener. Med., 2016, 10, E63–E72 CrossRef CAS.
- B. K. Jung, E. Oh, J. W. Hong, Y. Lee, K. D. Park and C. O. Yun, Biomaterials, 2017, 147, 26–38 CrossRef CAS.
- R. A. Haraszti, M. C. Didiot, E. Sapp, J. Leszyk, S. A. Shaffer, H. E. Rockwell, F. Gao, N. R. Narain, M. DiFiglia, M. A. Kiebish, N. Aronin and A. Khvorova, J. Extracell. Vesicles, 2016, 5, 32570 CrossRef.
- D. Bitounis, R. Fanciullino, A. Iliadis and J. Ciccolini, ISRN Pharm., 2012, 2012, 1–11 Search PubMed.
- A. D. Miller, Angew. Chem., Int. Ed., 1998, 37, 1768–1785 CrossRef.
- P. Yotnda, D. H. Chen, W. Chiu, P. A. Piedra, A. Davis, N. S. Templeton and M. K. Brenner, Mol. Ther., 2002, 5, 233–241 CrossRef CAS.
- Y. L. Tseng, J. J. Liu and R. L. Hong, Mol. Pharmacol., 2002, 62, 864–872 CrossRef CAS.
- A. D. Bangham and R. W. Horne, J. Mol. Biol., 1964, 8, 660–668 CrossRef CAS.
- G. Bozzuto and A. Molinari, Int. J. Nanomed., 2015, 10, 975 CrossRef CAS.
- B. Balakrishnan and E. David, J. Biosci., 2019, 44, 1–8 CrossRef.
- I. Tariq, S. R. Pinnapireddy, L. Duse, M. Y. Ali, S. Ali, M. U. Amin, N. Goergen, J. Jedelská, J. Schäfer and U. Bakowsky, Eur. J. Pharm. Biopharm., 2019, 135, 72–82 CrossRef CAS.
- E. Baghdan, S. R. Pinnapireddy, B. Strehlow, K. H. Engelhardt, J. Schäfer and U. Bakowsky, Int. J. Pharm., 2018, 535, 473–479 CrossRef CAS.
- B. Malaekeh-Nikouei, L. Gholami, F. Asghari, S. Askarian, S. Barzegar, M. Rezaee and R. Kazemi Oskuee, Colloids Surf., B, 2018, 165, 252–261 CrossRef CAS.
- J. Buck, P. Grossen, P. R. Cullis, J. Huwyler and D. Witzigmann, ACS Nano, 2019, 13, 3754–3782 CrossRef CAS.
- I. A. Khalil, M. A. Younis, S. Kimura and H. Harashima, Biol. Pharm. Bull, 2020, 584, 584–595 CrossRef.
- C. P. Hodgson and F. Solaiman, Nat. Biotechnol., 1996, 14, 339–342 CrossRef CAS.
- C. D. Porter, K. V. Lukacs, G. Box, Y. Takeuchi and M. K. L. Collins, J. Virol., 1998, 72, 4832 CrossRef CAS.
- S. Fletcher, A. Ahmad, E. Perouzel, M. R. Jorgensen and A. D. Miller, Org. Biomol. Chem., 2006, 4, 196–199 RSC.
- P. L. G. Chong, W. Zhu and B. Venegas, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 2–11 CrossRef CAS.
- N. Imelli, O. Meier, K. Boucke, S. Hemmi and U. F. Greber, J. Virol., 2004, 78, 3089–3098 CrossRef CAS.
- S. Worgall, T. S. Worgall, K. Kostarelos, R. Singh, P. L. Leopold, N. R. Hackett and R. G. Crystal, Mol. Ther., 2000, 1, 39–48 CrossRef CAS.
- D. V. Faller and D. Baltimore, J. Virol., 1984, 49, 269–272 CrossRef CAS.
- M. Mizuno and J. Yoshida, J. Cancer Res., 1998, 89, 352–354 CAS.
- C. Qiu, M. B. de Young, A. Finn and D. A. Dichek, Hum. Gene Ther., 1998, 9, 507–520 CrossRef CAS.
- R. Singh, K. T. Al-Jamal, L. Lacerda and K. Kostarelos, ACS Nano, 2008, 2, 1040–1050 CrossRef CAS.
- R. K. Keswani, I. M. Pozdol and D. W. Pack, Mol. Pharm., 2013, 10, 1725–1735 CrossRef CAS.
- R. Mo, Q. Sun, N. Li and C. Zhang, Biomaterials, 2013, 34, 2773–2786 CrossRef CAS.
- J. Van den Bossche, W. T. Al-Jamal, A. Yilmazer, E. Bizzarri, B. Tian and K. Kostarelos, Biomaterials, 2011, 32, 3085–3093 CrossRef CAS.
- Y. Wan, J. Han, G. Fan, Z. Zhang, T. Gong and X. Sun, Biomaterials, 2013, 34, 3020–3030 CrossRef CAS.
- C. L. Innes, P. B. Smith, R. Langenbach, K. R. Tindall and L. R. Boone, J. Virol., 1990, 64, 957–961 CrossRef CAS.
- C. Meunier-Durmort, R. Picart, T. Ragot, M. Perricaudet, B. Hainque and C. Forest, Biochim. Biophys. Acta, Biomembr., 1997, 1330, 8–16 CrossRef CAS.
- H. S. Shi, L. P. Yang, W. Wei, X. Q. Su, X. P. Li, M. Li, S. T. Luo, H. L. Zhang, L. Lu, Y. Q. Mao, B. Kan and L. Yang, J. Transl. Med., 2013, 11, 86 CrossRef CAS.
- A. Natsume, M. Mizuno, Y. Ryuke and J. Yoshida, Jpn. J. Cancer Res., 2000, 91, 363–367 CrossRef CAS.
- T. Shikano, H. Kasuya, T. T. Sahin, N. Nomura, A. Kanzaki, M. Misawa, Y. Nishikawa, T. Shirota, S. Yamada, T. Fujii, H. Sugimoto, N. Kanazumi, S. Nomoto, S. Takeda and A. Nakao, Curr. Cancer Drug Targets, 2011, 11, 111–122 CrossRef CAS.
- X. Fu and X. Zhang, Mol. Ther., 2001, 4, 447–453 CrossRef CAS.
- F. Sakurai, S. Inoue, T. Kaminade, T. Hotani, Y. Katayama, E. Hosoyamada, Y. Terasawa, M. Tachibana and H. Mizuguchi, Int. J. Pharm., 2017, 524, 238–247 CrossRef CAS.
- Y. Wang, H. Huang, H. Zou, X. Tian, J. Hu, P. Qiu, H. Hu and G. Yan, Mol. Pharm., 2019, 16, 779–785 CrossRef CAS.
- N. Mendez, V. Herrera, L. Zhang, F. Hedjran, R. Feuer, S. L. Blair, W. C. Trogler, T. R. Reid and A. C. Kummel, Biomaterials, 2014, 35, 9554–9561 CrossRef CAS.
- A. Yilmazer, B. Tian and K. Kostarelos, PLoS One, 2014, 9, e114985 CrossRef.
- M. Limberis, D. S. Anson, M. Fuller and D. W. Parsons, Hum. Gene Ther., 2002, 13, 1961–1970 CrossRef CAS.
- A. G. Stocker, K. L. Kremer, R. Koldej, D. S. Miller, D. S. Anson and D. W. Parsons, J. Gene Med., 2009, 11, 861–867 CrossRef CAS.
- P. Cmielewski, D. S. Anson and D. W. Parsons, Respir. Res., 2010, 11, 84 CrossRef.
- P. Cmielewski, N. Farrow, S. Devereux, D. Parsons and M. Donnelley, Exp. Lung Res., 2017, 43, 426–433 CrossRef CAS.
- C. A. Maguire, L. Balaj, S. Sivaraman, M. H. W. Crommentuijn, M. Ericsson, L. Mincheva-Nilsson, V. Baranov, D. Gianni, B. A. Tannous, M. Sena-Esteves, X. O. Breakefield and J. Skog, Mol. Ther., 2012, 20, 960–971 CrossRef CAS.
- B. György and C. A. Maguire, WIREs Nanomed. Nanobiotechnol., 2018, 10, e1488 CrossRef.
- T. Ishida, H. Harashima and H. Kiwada, Biosci. Rep., 2002, 22, 197–224 CrossRef CAS.
- M. L. Immordino, F. Dosio and L. Cattel, Int. J. Nanomed., 2006, 1, 297–315 CrossRef CAS.
- S. Y. Han, Y. J. Lee, H. I. Jung, S. W. Lee, S. J. Lim, S. H. Hong and J. S. Jeong, Exp. Mol. Med., 2008, 40, 427–434 CrossRef CAS.
- L. Yang, L. Wang, X. Q. Su, L. Wang, X. C. Chen, D. Li, S. T. Luo, H. S. Shi, L. J. Chen and Y. S. Wang, Cancer Gene Ther., 2010, 17, 49–57 CrossRef CAS.
- J. Chen, P. Gao, S. Yuan, R. Li, A. Ni, L. Chu, L. Ding, Y. Sun, X. Y. Liu and Y. Duan, ACS Nano, 2016, 10, 11548–11560 CrossRef CAS.
- N. G. Mukherjee, L. Andrew Lyon and J. M. Le Doux, Nanotechnology, 2009, 20, 065103 CrossRef.
- V. P. Torchilin, R. Rammohan, V. Weissig and T. S. Levchenko, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8786–8791 CrossRef CAS.
- Z. Zhong, J. Han, Y. Wan, Z. Zhang and X. Sun, Mol. Pharm., 2011, 8, 673–682 CrossRef CAS.
- K. K. Son, D. Tkach and D. H. Patel, Biochim. Biophys. Acta, Biomembr., 2000, 1468, 11–14 CrossRef CAS.
- L. F. Neves, J. Duan, A. Voelker, A. Khanal, L. McNally, J. Steinbach-Rankins and B. P. Ceresa, J. Microencapsulation, 2016, 33, 391–399 CrossRef CAS.
- S. Resina, P. Prevot and A. R. Thierry, PLoS One, 2009, 4, e6058 CrossRef.
- Y. Nie, L. Ji, H. Ding, L. Xie, L. Li, B. He, Y. Wu and Z. Gu, Theranostics, 2012, 2, 1092–1103 CrossRef CAS.
- M. Kapoor and D. J. Burgess, Int. J. Pharm., 2012, 432, 80–90 CrossRef CAS.
- L. Billiet, J. P. Gomez, M. Berchel, P. A. Jaffrès, T. Le Gall, T. Montier, E. Bertrand, H. Cheradame, P. Guégan, M. Mével, B. Pitard, T. Benvegnu, P. Lehn, C. Pichon and P. Midoux, Biomaterials, 2012, 33, 2980–2990 CrossRef CAS.
- M. Jedynak, R. Worch, M. Podsiadła-Białoskórska, J. Chroboczek and E. Szołajska, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 2215–2223 CrossRef CAS.
- M. Mazzon and J. Mercer, Cell. Microbiol., 2014, 16, 1493–1502 CrossRef CAS.
- R. F. A. Zwaal, P. Comfurius and E. M. Bevers, Cell. Mol. Life Sci., 2005, 62, 971–988 CrossRef CAS.
- S. Ran and P. E. Thorpe, Int. J. Radiat. Oncol., Biol., Phys., 2002, 54, 1479–1484 CrossRef CAS.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS.
- W. Chen, G. Wang and R. Tang, Nano Res., 2014, 7, 1404–1428 CrossRef CAS.
- X. Wang, X. Liu, Y. Xiao, H. Hao, Y. Zhang and R. Tang, Chem. – Eur. J., 2018, 24, 11518–11529 CrossRef CAS.
- H. L. Jang, K. Jin, J. Lee, Y. Kim, S. H. Nahm, K. S. Hong and K. T. Nam, ACS Nano, 2014, 8, 634–641 CrossRef CAS.
- G. Wang, R. Y. Cao, R. Chen, L. Mo, J. F. Han, X. Wang, X. Xu, T. Jiang, Y. Q. Deng, K. Lyu, S. Y. Zhu, E. De Qin, R. Tang and C. F. Qin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7619–7624 CrossRef CAS.
- L.-L. Huang, X. Li, J. Zhang, Q. R. Zhao, M. J. Zhang, A.-A. Liu, D.-W. Pang and H.-Y. Xie, Nano Lett., 2019, 19, 8002–8009 CrossRef CAS.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS.
- M. Everts, V. Saini, J. L. Leddon, R. J. Kok, M. Stoff-Khalili, M. A. Preuss, C. L. Millican, G. Perkins, J. M. Brown, H. Bagaria, D. E. Nikles, D. T. Johnson, V. P. Zharov and D. T. Curiel, Nano Lett., 2006, 6, 587–591 CrossRef CAS.
- Y. Hernandez, R. González-Pastor, C. Belmar-Lopez, G. Mendoza, J. M. De La Fuente and P. Martin-Duque, RSC Adv., 2019, 9, 1327–1334 RSC.
- J. Kim, M. Yeom, T. Lee, H.-O. Kim, W. Na, A. Kang, J.-W. Lim, G. Park, C. Park, D. Song and S. Haam, J. Nanobiotechnol., 2020, 18, 54 CrossRef CAS.
- I. Papp, C. Sieben, K. Ludwig, M. Roskamp, C. Böttcher, S. Schlecht, A. Herrmann and R. Haag, Small, 2010, 6, 2900–2906 CrossRef CAS.
- M. Sametband, S. Shukla, T. Meningher, S. Hirsh, E. Mendelson, R. Sarid, A. Gedanken and M. Mandelboim, MedChemComm, 2011, 2, 421 RSC.
- M.-C. Bowman, T. E. Ballard, C. J. Ackerson, D. L. Feldheim, D. M. Margolis and C. Melander, J. Am. Chem. Soc., 2008, 130, 6896–6897 CrossRef CAS.
- V. P. Zharov, J. W. Kim, D. T. Curiel and M. Everts, Nanomedicine, 2005, 1, 326–345 CrossRef CAS.
- B. K. Jung, Y. K. Lee, J. Hong, H. Ghandehari and C. O. Yun, ACS Nano, 2016, 10, 10533–10543 CrossRef CAS.
- Y. Wang, Q. Zhao, N. Han, L. Bai, J. Li, J. Liu, E. Che, L. Hu, Q. Zhang, T. Jiang and S. Wang, Nanomedicine, 2015, 11, 313–327 CrossRef CAS.
- R. Peters, E. Kramer, A. G. Oomen, Z. E. Herrera Rivera, G. Oegema, P. C. Tromp, R. Fokkink, A. Rietveld, H. J. P. Marvin, S. Weigel, A. A. C. M. Peijnenburg and H. Bouwmeester, ACS Nano, 2012, 6, 2441–2451 CrossRef CAS.
- M. Ekkapongpisit, A. Giovia, C. Follo, G. Caputo and C. Isidoro, Int. J. Nanomed., 2012, 7, 4147–4158 CAS.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- J. Tian, Y. Luo, L. Huang, Y. Feng, H. Ju and B. Y. Yu, Biosens. Bioelectron., 2016, 80, 519–524 CrossRef CAS.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. Sanchez Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS.
- K. Yang, L. Hu, X. Ma, S. Ye, L. Cheng, X. Shi, C. Li, Y. Li and Z. Liu, Adv. Mater., 2012, 24, 1868–1872 CrossRef CAS.
- L. Feng, X. Yang, X. Shi, X. Tan, R. Peng, J. Wang and Z. Liu, Small, 2013, 9, 1989–1997 CrossRef CAS.
- F. Liu, K. S. Choi, T. J. Park, S. Y. Lee and T. S. Seo, BioChip J., 2011, 5, 123–128 CrossRef CAS.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS.
- D. Pantarotto, R. Singh, D. McCarthy, M. Erhardt, J.-P. Briand, M. Prato, K. Kostarelos and A. Bianco, Angew. Chem., Int. Ed., 2004, 43, 5242–5246 CrossRef CAS.
- R. Singh, D. Pantarotto, D. McCarthy, O. Chaloin, J. Hoebeke, C. D. Partidos, J. P. Briand, M. Prato, A. Bianco and K. Kostarelos, J. Am. Chem. Soc., 2005, 127, 4388–4396 CrossRef CAS.
- Z. Wang, H. Shen, S. Song, L. Zhang, W. Chen, J. Dai and Z. Zhang, J. Nanosci. Nanotechnol., 2017, 18, 2286–2293 CrossRef.
- M. Vincent, I. De Lázaro and K. Kostarelos, Gene Ther., 2017, 24, 123–132 CrossRef CAS.
- J. Chen, Q. Wang, J. Zhou, W. Deng, Q. Yu, X. Cao, J. Wang, F. Shao, Y. Li, P. Ma, M. Spector, J. Yu and X. Xu, Nanoscale, 2017, 9, 10820–10831 RSC.
- H. N. Abdelhamid, M. Dowaidar and Ü. Langel, Microporous Mesoporous Mater., 2020, 302, 110200 CrossRef CAS.
- M. Xia, D. Luo, J. Dong, M. Zheng, G. Meng, J. Wu and J. Wei, J. Exp. Clin. Cancer Res., 2019, 38, 408 CrossRef.
- W. Meng, A. Rey-Rico, M. Claudel, G. Schmitt, S. Speicher-Mentges, F. Pons, L. Lebeau, J. K. Venkatesan and M. Cucchiarini, Nanomaterials, 2020, 10, 855 CrossRef CAS.
- J. Fan, M. Claudel, C. Ronzani, Y. Arezki, L. Lebeau and F. Pons, Int. J. Pharm., 2019, 569, 118521 CrossRef CAS.
- F. Scherer, M. Anton, U. Schillinger, J. Henke, C. Bergemann, A. Krüger, B. Gänsbacher and C. Plank, Gene Ther., 2002, 9, 102–109 CrossRef CAS.
- S. Huth, J. Lausier, S. W. Gersting, C. Rudolph, C. Plank, U. Welsch and J. Rosenecker, J. Gene Med., 2004, 6, 923–936 CrossRef CAS.
- H. Mok and M. Zhang, Expert Opin. Drug Delivery, 2013, 10, 73–87 CrossRef CAS.
- C. Sapet, C. Pellegrino, N. Laurent, F. Sicard and O. Zelphati, Pharm. Res., 2012, 29, 1203–1218 CrossRef CAS.
- P. G. Kyrtatos, P. Lehtolainen, M. Junemann-Ramirez, A. Garcia-Prieto, A. N. Price, J. F. Martin, D. G. Gadian, Q. A. Pankhurst and M. F. Lythgoe, JACC Cardiovasc. Interv., 2009, 2, 794–802 CrossRef.
- S. J. Tseng, K. Y. Huang, I. M. Kempson, S. H. Kao, M. C. Liu, S. C. Yang, Z. X. Liao and P. C. Yang, ACS Nano, 2016, 10, 10339–10346 CrossRef CAS.
- S. M. Shalaby, M. K. Khater, A. M. Perucho, S. A. Mohamed, I. Helwa, A. Laknaur, I. Lebedyeva, Y. Liu, M. P. Diamond and A. A. Al-Hendy, Fertil. Steril., 2016, 105, 1638–1648 CrossRef CAS.
- N. Tresilwised, P. Pithayanukul, P. S. Holm, U. Schillinger, C. Plank and O. Mykhaylyk, Biomaterials, 2012, 33, 256–269 CrossRef CAS.
- J. W. Choi, J. W. Park, Y. Na, S. J. Jung, J. K. Hwang, D. Choi, K. G. Lee and C. O. Yun, Biomaterials, 2015, 65, 163–174 CrossRef CAS.
- S. Castellani, C. Orlando, A. Carbone, S. Di Gioia and M. Conese, Genes, 2016, 7, 103 CrossRef.
- S. I. Kadota, T. Kanayama, N. Miyajima, K. Takeuchi and K. Nagata, J. Virol. Methods, 2005, 128, 61–66 CrossRef CAS.
- T. N. Britos, C. E. Castro, B. M. Bertassoli, G. Petri, F. L. A. Fonseca, F. F. Ferreira and P. S. Haddad, Mater. Sci. Eng., C, 2019, 99, 171–179 CrossRef CAS.
- A. A. Belanova, N. Gavalas, Y. M. Makarenko, M. M. Belousova, A. V. Soldatov and P. V. Zolotukhin, Oncol. Res. Treat., 2018, 41, 139–143 CrossRef CAS.
- S. R. Bhattarai, S. Y. Kim, K. Y. Jang, K. C. Lee, H. K. Yi, D. Y. Lee, H. Y. Kim and P. H. Hwang, Nanomedicine, 2008, 4, 146–154 CrossRef CAS.
- J. Yun, A. M. Sonabend, I. V. Ulasov, D. H. Kim, E. A. Rozhkova, V. Novosad, S. Dashnaw, T. Brown, P. Canoll, J. N. Bruce and M. S. Lesniak, J. Clin. Neurosci., 2012, 19, 875–880 CrossRef CAS.
- J. W. Park, K. H. Bae, C. Kim and T. G. Park, Biomacromolecules, 2011, 12, 457–465 CrossRef CAS.
- B. Kurena, A. Vežāne, D. Skrastiņa, O. Trofimova and A. Zajakina, J. Virol. Methods, 2017, 245, 28–34 CrossRef CAS.
- R. Schubert, S. Trenholm, K. Balint, G. Kosche, C. S. Cowan, M. A. Mohr, M. Munz, D. Martinez-Martin, G. Fläschner, R. Newton, J. Krol, B. G. Scherf, K. Yonehara, A. Wertz, A. Ponti, A. Ghanem, D. Hillier, K. K. Conzelmann, D. J. Müller and B. Roska, Nat. Biotechnol., 2018, 36, 81–88 CrossRef CAS.
- Z. X. Liao, I. M. Kempson, Y. C. Fa, M. C. Liu, L. C. Hsieh, K. Y. Huang and L. F. Wang, Bioconjugate Chem., 2017, 28, 1702–1708 CrossRef CAS.
- S.-J. Tseng, I. M. Kempson, K.-Y. Huang, H.-J. Li, Y.-C. Fa, Y.-C. Ho, Z.-X. Liao and P.-C. Yang, ACS Nano, 2018, 12, 9894–9902 CrossRef CAS.
- P. Reimer and T. Balzer, Eur. Radiol., 2003, 13, 1266–1276 CrossRef.
- F. Scherer, M. Anton, U. Schillinger, J. Henke, C. Bergemann, A. Krüger, B. Gänsbacher and C. Plank, Gene Ther., 2002, 9, 102–109 CrossRef CAS.
- F. Wei, K. I. McConnell, T. K. Yu and J. Suh, Eur. J. Pharm. Sci., 2012, 46, 167–172 CrossRef CAS.
- I. E. Alexander, D. W. Russell and A. Dusty Miller, J. Virol., 1994, 68, 8282–8287 CrossRef CAS.
- D. W. Russell, I. E. Alexander and A. D. Miller, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5719–5723 CrossRef CAS.
- L. N. Zhang, P. Karp, C. J. Gerard, E. Pastor, D. Laux, K. Munson, Z. Yan, X. Liu, S. Godwin, C. P. Thomas, J. Zabner, H. Shi, C. W. Caldwell, R. Peluso, B. Carter and J. F. Engelhardt, Mol. Ther., 2004, 10, 990–1002 CrossRef CAS.
- T. Zhang, J. Hu, W. Ding and X. Wang, Neurochem. Int., 2009, 55, 521–528 CrossRef CAS.
- F. R. Santoni De Sio, P. Cascio, A. Zingale, M. Gasparini and L. Naldini, Blood, 2006, 107, 4257–4265 CrossRef CAS.
- V. Leuci, G. Mesiano, L. Gammaitoni, C. Cammarata, S. Capellero, M. Todorovic, N. Jordaney, P. Circosta, A. Elia, M. Lesnikova, G. E. Georges, W. Piacibello, F. Fagioli, A. Cignetti, M. Aglietta and D. Sangiolo, J. Biotechnol., 2011, 156, 218–226 CrossRef CAS.
- M. Basler, C. Lauer, U. Beck and M. Groettrup, J. Immunol., 2009, 183, 6145–6150 CrossRef CAS.
- D. Duan, Y. Yue, Z. Yan, J. Yang and J. F. Engelhardt, J. Clin. Invest., 2000, 105, 1573–1587 CrossRef CAS.
- B. M. Davis, L. Humeau, V. Slepushkin, G. Binder, L. Korshalla, Y. Ni, E. Oluwakemi Ogunjimi, L. F. Chang, X. Lu and B. Dropulic, Blood, 2004, 104, 364–373 CrossRef CAS.
- J. M. Johnston, G. Denning, R. Moot, D. Whitehead, J. Shields, J. M. Le Doux, C. B. Doering and H. T. Spencer, Gene Ther., 2014, 21, 1008–1020 CrossRef CAS.
- S. C. Nicolson, C. Li, M. L. Hirsch, V. Setola and R. J. Samulski, J. Virol., 2016, 90, 7019–7031 CrossRef CAS.
- M. H. Dornan, R. Krishnan, A. M. Macklin, M. Selman, N. El Sayes, H. H. Son, C. Davis, A. Chen, K. Keillor, P. J. Le, C. Moi, P. Ou, C. Pardin, C. R. Canez, F. Le Boeuf, J. C. Bell, J. C. Smith, J.-S. Diallo and C. N. Boddy, Sci. Rep., 2016, 6, 26786 CrossRef CAS.
- J. S. Diallo, F. Le Boeuf, F. Lai, J. Cox, M. Vaha-Koskela, H. Abdelbary, H. MacTavish, K. Waite, T. Falls, J. Wang, R. Brown, J. E. Blanchard, E. D. Brown, D. H. Kirn, J. Hiscott, H. Atkins, B. D. Lichty and J. C. Bell, Mol. Ther., 2010, 18, 1123–1129 CrossRef CAS.
- E. Zonari, G. Desantis, C. Petrillo, F. E. Boccalatte, M. R. Lidonnici, A. Kajaste-Rudnitski, A. Aiuti, G. Ferrari, L. Naldini and B. Gentner, Stem Cell Rep., 2017, 8, 977–990 CrossRef CAS.
- K. E. Masiuk, R. Zhang, K. Osborne, R. P. Hollis, B. Campo-Fernandez and D. B. Kohn, Mol. Ther.–Methods Clin. Dev., 2019, 13, 390–398 CrossRef CAS.
- G. C. Heffner, M. Bonner, L. Christiansen, F. J. Pierciey, D. Campbell, Y. Smurnyy, W. Zhang, A. Hamel, S. Shaw, G. Lewis, K. A. Goss, O. Garijo, B. E. Torbett, H. Horton, M. H. Finer, P. D. Gregory and G. Veres, Mol. Ther., 2018, 26, 320–328 CrossRef CAS.
- L. C. Chandler, A. R. Barnard, S. L. Caddy, M. I. Patrício, M. E. McClements, H. Fu, C. Rada, R. E. MacLaren and K. Xue, Mol. Ther.–Methods Clin. Dev., 2019, 14, 77–89 CrossRef CAS.
- B. Lv, J. Li, M. Li, Y. Zhuo, K. Ren, E. Li and G. Yang, Sci. Rep., 2018, 8, 1–10 CrossRef CAS.
- P. Wang, Y. Wu, C. Yang, G. Zhao, Y. Liu, G. Cheng and S. Wang, OncoTargets Ther., 2020, 13, 1421–1429 CrossRef CAS.
- Y. Gong, R. G. J. Klein Wolterink, I. Janssen, A. J. Groot, G. M. J. Bos and W. T. V. Germeraad, Mol. Ther.–Methods Clin. Dev., 2020, 17, 634–646 CrossRef CAS.
- Z. Yan, R. Zak, Y. Zhang, W. Ding, S. Godwin, K. Munson, R. Peluso and J. F. Engelhardt, J. Virol., 2004, 78, 2863–2874 CrossRef CAS.
- L. S. Vermeer, L. Hamon, A. Schirer, M. Schoup, J. Cosette, S. Majdoul, D. Pastré, D. Stockholm, N. Holic, P. Hellwig, A. Galy, D. Fenard and B. Bechinger, Acta Biomater., 2017, 64, 259–268 CrossRef CAS.
- A. Castro, M. Drosch, M. Liesenfeld, T. Kaan, A. Schilz, H. Wenschuh, W. Forsmann and K. Kuhlke, Cytotherapy, 2019, 21, e14 CrossRef.
- P. Lin, D. Correa, Y. Lin and A. I. Caplan, PLoS One, 2011, 6, e23891 CrossRef CAS.
- M. Themis, S. Forbes, L. Chan, R. Cooper, C. Etheridge, A. Miller, H. Hodgson and C. Coutelle, Gene Ther., 1998, 5, 1180–1186 CrossRef CAS.
- P. F. Kelly, S. Radtke, C. von Kalle, B. Balcik, K. Bohn, R. Mueller, T. Schuesler, M. Haren, L. Reeves, J. A. Cancelas, T. Leemhuis, R. Harris, A. D. Auerbach, F. O. Smith, S. M. Davies and D. A. Williams, Mol. Ther., 2007, 15, 211–219 CrossRef CAS.
- A. Aiuti, Science, 2002, 296, 2410–2413 CrossRef CAS.
- K. L. Shaw, E. Garabedian, S. Mishra, P. Barman, A. Davila, D. Carbonaro, S. Shupien, C. Silvin, S. Geiger, B. Nowicki, E. M. Smogorzewska, B. Brown, X. Wang, S. de Oliveira, Y. Choi, A. Ikeda, D. Terrazas, P.-Y. Fu, A. Yu, B. C. Fernandez, A. R. Cooper, B. Engel, G. Podsakoff, A. Balamurugan, S. Anderson, L. Muul, G. J. Jagadeesh, N. Kapoor, J. Tse, T. B. Moore, K. Purdy, R. Rishi, K. Mohan, S. Skoda-Smith, D. Buchbinder, R. S. Abraham, A. Scharenberg, O. O. Yang, K. Cornetta, D. Gjertson, M. Hershfield, R. Sokolic, F. Candotti and D. B. Kohn, J. Clin. Invest., 2017, 127, 1689–1699 CrossRef.
- S. S. De Ravin, S. Anaya O'Brien, N. Kwatemaa, N. Theobald, S. Liu, J. Lee, L. Kardava, T. Liu, F. Goldman, S. Moir, J. Bleesing, B. Neven, J. Puck, M. J. Cowan, E. Mamcarz, S. Gottschalk, M. M. Meagher, L. Notarangelo, E. Kang, X. Wu and H. L. Malech, Blood, 2019, 134, 608–608 CrossRef.
- R. E. MacLaren, M. Groppe, A. R. Barnard, C. L. Cottriall, T. Tolmachova, L. Seymour, K. R. Clark, M. J. During, F. P. M. Cremers, G. C. M. Black, A. J. Lotery, S. M. Downes, A. R. Webster and M. C. Seabra, Lancet, 2014, 383, 1129–1137 CrossRef CAS.
- A. M. Maguire, F. Simonelli, E. A. Pierce, E. N. Pugh, F. Mingozzi, J. Bennicelli, S. Banfi, K. A. Marshall, F. Testa, E. M. Surace, S. Rossi, A. Lyubarsky, V. R. Arruda, B. Konkle, E. Stone, J. Sun, J. Jacobs, L. Dell'Osso, R. Hertle, J. Ma, T. M. Redmond, X. Zhu, B. Hauck, O. Zelenaia, K. S. Shindler, M. G. Maguire, J. F. Wright, N. J. Volpe, J. W. McDonnell, A. Auricchio, K. A. High and J. Bennett, N. Engl. J. Med., 2008, 358, 2240–2248 CrossRef CAS.
- L. A. George, S. K. Sullivan, A. Giermasz, J. E. J. Rasko, B. J. Samelson-Jones, J. Ducore, A. Cuker, L. M. Sullivan, S. Majumdar, J. Teitel, C. E. McGuinn, M. V. Ragni, A. Y. Luk, D. Hui, J. F. Wright, Y. Chen, Y. Liu, K. Wachtel, A. Winters, S. Tiefenbacher, V. R. Arruda, J. C. M. van der Loo, O. Zelenaia, D. Takefman, M. E. Carr, L. B. Couto, X. M. Anguela and K. A. High, N. Engl. J. Med., 2017, 377, 2215–2227 CrossRef CAS.
- J. R. Mendell, Z. Sahenk, V. Malik, A. M. Gomez, K. M. Flanigan, L. P. Lowes, L. N. Alfano, K. Berry, E. Meadows, S. Lewis, L. Braun, K. Shontz, M. Rouhana, K. R. Clark, X. Q. Rosales, S. Al-Zaidy, A. Govoni, L. R. Rodino-Klapac, M. J. Hogan and B. K. Kaspar, Mol. Ther., 2015, 23, 192–201 CrossRef CAS.
- A. A. Thompson, M. C. Walters, J. Kwiatkowski, J. E. J. Rasko, J.-A. Ribeil, S. Hongeng, E. Magrin, G. J. Schiller, E. Payen, M. Semeraro, D. Moshous, F. Lefrere, H. Puy, P. Bourget, A. Magnani, L. Caccavelli, J.-S. Diana, F. Suarez, F. Monpoux, V. Brousse, C. Poirot, C. Brouzes, J.-F. Meritet, C. Pondarré, Y. Beuzard, S. Chrétien, T. Lefebvre, D. T. Teachey, U. Anurathapan, P. J. Ho, C. von Kalle, M. Kletzel, E. Vichinsky, S. Soni, G. Veres, O. Negre, R. W. Ross, D. Davidson, A. Petrusich, L. Sandler, M. Asmal, O. Hermine, M. De Montalembert, S. Hacein-Bey-Abina, S. Blanche, P. Leboulch and M. Cavazzana, N. Engl. J. Med., 2018, 378, 1479–1493 CrossRef CAS.
- European Medicines Agency Committee for Medicinal Products for Human Use, Assessment Report Zynteglo, 2019 Search PubMed.
- AveXis, Package Insert Zolgensma, https://www.fda.gov/media/126109/download, (accessed 13 September 2020).
- Spark Therapeutics, Package Insert LUXTURNA, https://www.fda.gov/media/109906/download, (accessed 13 September 2020).
- Kite Pharma, Package Insert YESCARTA, https://www.fda.gov/media/108377/download, (accessed 13 September 2020).
- Novartis, Package Insert KYMRIAH, https://www.fda.gov/media/107296/download, (accessed 13 September 2020).
| This journal is © The Royal Society of Chemistry 2020 |